[level-membership-for-pediatrics-category]Chapter 88
Congenital Hepatobiliary Anomalies
Fibropolycystic liver disorders are a group of associated congenital anomalies of the liver caused by malformation of the embryonic ductal plates (Fig. 88-1). They include choledochal cysts, Caroli disease, hepatic fibrosis, biliary hamartomas, and cystic liver disease. The specific type of fibropolycystic disorder depends on the size of the embryonic duct with faulty development (Table 88-1).
Table 88-1
Fibropolycystic Liver Disorder and Size of Ducts with Faulty Embryogenesis
| Disorder | Duct Size |
| Congenital hepatic fibrosis | Small |
| Biliary hamartoma | Small |
| Polycystic liver disease | Medium |
| Choledochal cyst | Large extrahepatic |
| Caroli disease | Large intrahepatic |
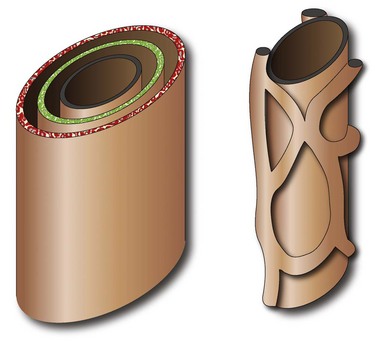
Figure 88-1 Schema of ductal plate.
A bilayer of hepatocytes (green and red layers) surrounds the portal venous structures (central black), forming the lumina of primitive bile ducts (left). A process of organized resorption leads to the normal organization of biliary ducts within the portal triads. Failure of this process leads to ductal plate malformations.
Choledochal Cyst
Overview: Choledochal cysts, which are among the most frequent of congenital hepatobiliary anomalies, are a fusiform or saccular dilation of the bile ducts. The Todani classification system is commonly used to categorize these anomalies into five general types (with several subtypes) that differ based on etiology, pathogenesis, appearance, and presentation (Fig. 88-2).1
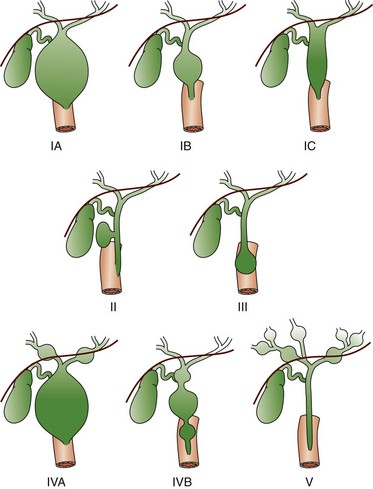
Figure 88-2 Schematic of Todani classification of choledochal cysts based on cholangiographic morphology.
Type IA and IB involve cystic dilatation of the common duct, with IB limited to the area below the insertion of the cystic duct; IC is fusiform dilatation of the duct. Type II is a saccular diverticulum from the common bile duct, and type III is a choledochocele at the ampulla of Vater. Types IVA and IVB are multiple cystic dilations of the intrahepatic and extrahepatic biliary tree, and type V is equivalent to Caroli disease with numerous intrahepatic bile lakes throughout the biliary tree and liver. (From Kim OH, Chung HJ, Choi BG. Imaging of the choledochal cyst. Radiographics. 1995;15:69-88.)
The most common choledochal cyst is the Todani type I, found in 80% to 90% of cases.2 It involves variable lengths and degrees of common bile duct dilatation and is further classified into subtypes.2 Todani type IA, IB, and IC describe cystic dilation of the common bile duct, segmental dilation below the cystic duct, and fusiform dilation, respectively. Todani type II choledochal cysts are found in only 2% of cases and consist of one or more diverticula of the common bile duct.3 Todani type III occurs in 1.5% to 5% of cases and involves dilation of the intraduodenal portion of the duct, forming a cystlike mass, termed a “choledochocele,” with both the common bile and pancreatic ducts emptying into it. Todani type IVA consists of multiple intrahepatic and extrahepatic biliary dilatations and occurs in 10% of patients with choledochal cysts.1,3 Type IVB, which involves multiple extrahepatic biliary cysts without intrahepatic involvement, is rare. Todani type V is synonymous with Caroli disease.1 Todani type IVA also has been described as a type I cyst with intrahepatic involvement, and controversy exists about whether it is actually a type V (Caroli disease) cyst with common duct enlargement.
Etiology: Several theories for the pathogenesis of the type I choledochal cysts exist. In addition to the ductal plate malformation described previously, other theories have suggested cyst formation as a result of obstruction of the distal biliary duct and/or reflux of pancreatic enzymes into the biliary tree because of anomalous proximal insertion of the pancreatic duct into the common bile duct (ductal malunion).4,5 This ductal malunion could permit reflux of pancreatic enzymes into the common bile duct, with subsequent inflammation and weakening of the wall, a pathologic mechanism that occurs in about 60% of patients.5 This anomalous ductal connection has been demonstrated on endoscopic retrograde cholangiopancreatography (ERCP) (Fig. 88-3). Type I choledochal cysts are more common in girls than in boys in Western countries, but the sex ratio is equal in Asia.2,3 About 65% of all reported cases are from Japan.2
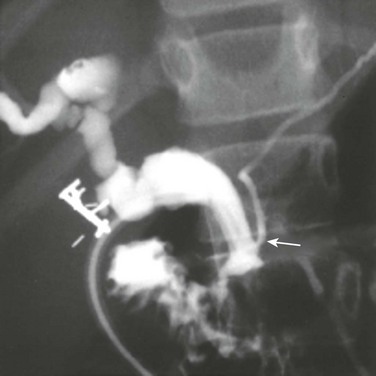
It has been theorized that type II choledochal cysts result from prenatal rupture of the common bile duct with subsequent healing.3 Type III choledochal cysts may be the sequelae of ampullary obstruction or congenital duplication of the duodenum at the ampulla. The intrahepatic cysts seen in types IV and V are thought to be due to primary ductal ectasia resulting from ductal plate malformation.
Clinical Presentation: Choledochal cysts may present in infancy with cholestatic jaundice, which clinically is inseparable from neonatal hepatitis or biliary atresia.2 In older children and young adults, the clinical presentation is variable. A characteristic triad of abdominal pain, obstructive jaundice, and fever has been reported, but few patients present with all three components. Abdominal pain is the most characteristic presentation, followed by obstructive jaundice, fever, pale stools, splenomegaly, hepatomegaly, and a palpable mass.6 The most common complication of a choledochal cyst is ascending cholangitis.7 Long-term complications, such as hepatic cirrhosis with subsequent portal hypertension, can occur.6 Carcinoma of the biliary tree has a twentyfold increased incidence in patients with choledochal cysts. This risk is low in the first decade but increases with advancing age.8 Spontaneous cyst rupture also has been reported.6
Imaging: When the clinical presentation points to a hepatobiliary problem, sonography usually is the initial imaging study requested. The markedly dilated common bile duct is readily discernible in types I and IV choledochal cysts. The gallbladder usually can be identified adjacent to the dilated common duct (Fig. 88-4). Most frequently, the intrahepatic ducts are normal, but varying degrees of dilation may be present as a result of obstruction. Sludge or stones may be identified within the dilated ducts.9 Type III cysts may cause mass effect at the ampulla of Vater (Fig. 88-5, A and B).
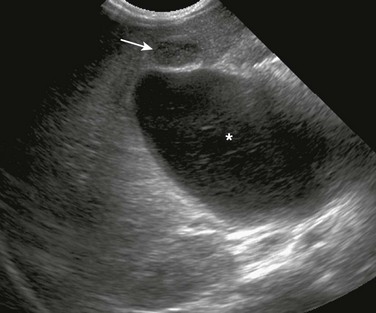
Figure 88-4 Choledochal cyst (Todani type I) in an 8-day-old girl with jaundice.
Transverse sonogram shows a large fusiform cyst in the porta hepatis (asterisk) just beneath a small, contracted sludge filled gallbladder (arrow).
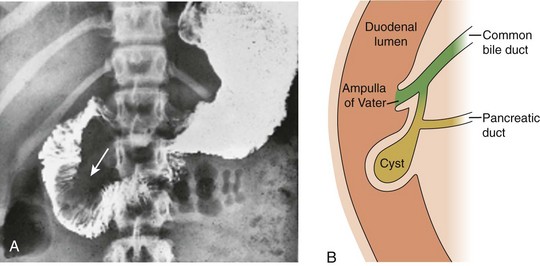
Figure 88-5 Choledochocele (Todani type III) in a 12-year-old girl with abdominal pain.
A, Upper gastrointestinal tract image shows a large filling defect (arrow) coincident with the site of the Ampulla of Vater. B, Drawing of findings of choledochocele confirmed at surgery.
Abdominal magnetic resonance imaging (MRI) with cholangiopancreatography (MRCP) often is performed to delineate anatomy (Fig. 88-6).10–12 Hepatobiliary scintigraphy can prove useful in difficult cases by demonstrating communication between the choledochal cyst and the hepatobiliary ducts (Fig. 88-7).9 Computed tomography (CT) does not demonstrate the biliary ductal anatomy as well as ultrasound and MRI, although it can be useful along with sonography to guide abscess drainage and to evaluate hepatic anatomy.10,11
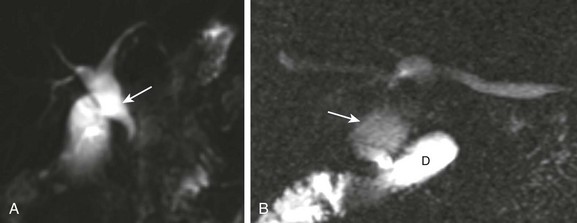
Figure 88-6 A choledochal cyst (Todani type I).
A, Magnetic resonance imaging with cholangiopancreatography (MRCP) in a 10-day-old boy with jaundice demonstrates dilatation of the common bile duct (arrow). B, A choledochal cyst (Todani type I) in a 6-year-old boy with abdominal pain. MRCP reveals dilation of the common bile duct (arrow) draining into the normal duodenum (D).
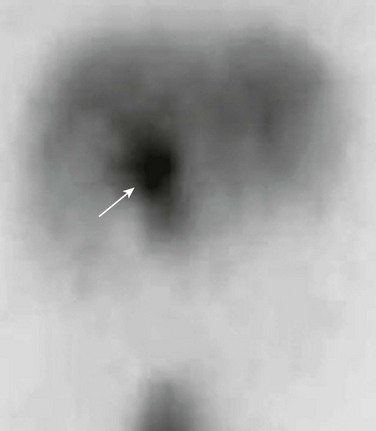
Percutaneous transhepatic cholangiography (PTC) and ERCP are useful when placement of a percutaneous biliary drain may be helpful (Fig. 88-8). Some investigators prefer PTC to ERCP because it carries a lower risk of iatrogenic cholangitis.7
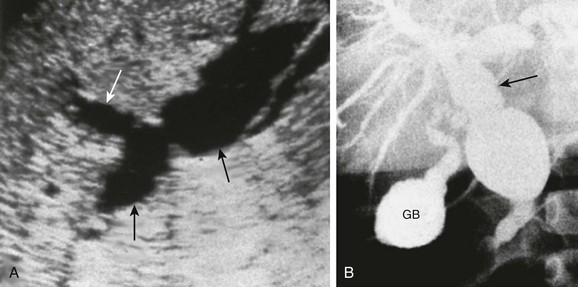
Figure 88-8 Choledochal cyst (Todani type IV) in a 1-year-old boy.
A, A longitudinal sonogram shows marked saccular dilation of the intrahepatic and extrahepatic bile ducts (arrows). B, An operative cholangiogram confirms dilation of the intrahepatic biliary tree and common bile duct (arrow). The gallbladder (GB) is noted.
Treatment: Because of the potential long-term sequelae, surgical excision and hepatojejunostomy are performed as definitive treatments. Patients with intrahepatic cysts are closely monitored for cholangitis, cholestasis, and stone formation. Antibiotics are administered in the setting of acute cholangitis. Medical therapy can be instituted to reduce the risk of cholestasis and promote bile flow; however, when extensive liver damage and cirrhosis occurs, a liver transplant may be necessary.
Caroli Disease and Caroli Syndrome
Overview: Caroli disease, also classified as Todani type V choledochal cyst, is a segmental nonobstructive dilation of the intrahepatic bile ducts. It is characterized by multiple hepatic cysts in continuity with the biliary system, representing ectatic intrahepatic ducts,7 and can be associated with stone formation, cholangitis, and hepatic abscesses.13 In Caroli syndrome, which is discussed further below, hepatic fibrosis is also present.
Etiology: Leading theories on the etiology of Caroli disease include maldevelopment of the large intrahepatic ductal plates. Other postulated mechanisms include occlusion of the hepatic artery in the neonatal period with associated ischemia of the bile ducts versus abnormal growth rate of the biliary epithelium and supporting connective tissues with lack of the normal involution of ductal plates. This process leads to ectatic biliary cysts surrounding the portal triads. In Caroli syndrome, which involves large and small ductal plate abnormalities, associated hepatic fibrosis is present. Caroli hepatic ductal ectasia also may be associated with extrahepatic ductal plate abnormalities and choledochal cysts (Todani IVA choledochal cyst), as discussed previously. An association also exists with renal disorders, including autosomal recessive polycystic kidney disease, medullary sponge kidney, and nephronopthisis.7 Although it typically is a diffuse process, monolobar Caroli disease has been reported, with 88% of cases involving the left lobe.14
Clinical Presentation: Although the disease is present from birth, most patients do not present until later in life when abdominal pain resulting from cholangitis and cholestasis leads them to seek medical attention. The abdominal pain also may be related to hepatic abscesses (a consequence of cholangitis) or biliary stones (a consequence of cholestasis). Patients with associated hepatic fibrosis may present with symptoms and signs of portal hypertension as a result of the disease process.13
Imaging: In most patients, sonography reveals very large and irregular ectatic ducts.7,15 To distinguish Caroli disease from hepatic cysts, one must recognize that the ectatic ducts connect with one another and with the ductal system (Fig. 88-9). The ectatic ducts surround portal vein radicles that produce the central dot sign, which has been considered pathognomonic for Caroli disease.16 Doppler interrogation can be used to confirm blood flow within these portal venous branches.16 Biliary sludge and calculi are common findings within the dilated bile ducts.7 If an abscess develops, one or more cysts will show mixed echogenicity rather than the anechoic appearance of uncomplicated cysts.
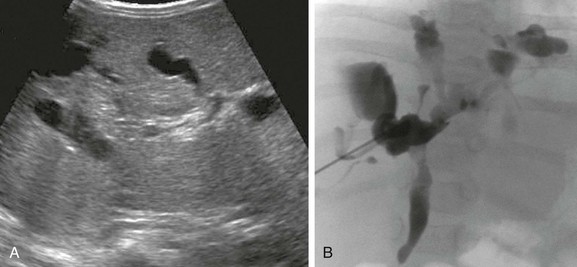
Figure 88-9 Caroli disease (type V choledochal cyst) in a 6-month-old girl with failure to thrive.
A, A transverse hepatic sonogram shows multiple, large, hypoechoic, well-defined, variably shaped bile lakes scattered throughout the liver. B, A percutaneous transhepatic cholangiogram confirms variably shaped patulous bile lakes connected to the biliary tree.
The kidneys should be examined in all patients with confirmed Caroli disease. Kidneys may be normal or polycystic, or they may show increased medullary echogenicity with loss of corticomedullary differentiation (Fig. 88-10). MRI and CT are excellent modalities to show the extent of disease. An enhancing “central dot” may be present that corresponds to the portal vein radicles seen sonographically.17 If an abscess is present, the affected cyst may show higher attenuation or peripheral enhancement compared with adjacent uninfected cysts.7 Biliary patency HIDA (hepato-iminodiacetic acid) scans have been used to demonstrate a connection to the biliary tree. Occasionally, PTC is used during abscess drainage.
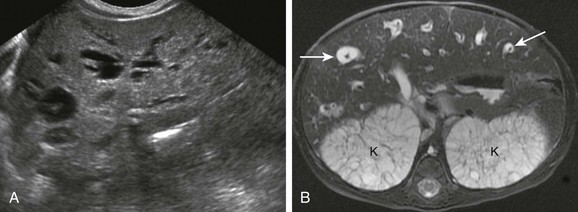
Figure 88-10 Caroli (type V choledochal cyst) disease in a 3-day-old girl with organomegaly and associated autosomal recessive polycystic kidney disease.
A, Longitudinal sonograms through the liver show focal dilatation of the biliary ducts throughout the liver. B, Magnetic resonance imaging with cholangiopancreatography shows the central dot sign, seen as numerous T2 bright distended bile ducts each surrounding a portal vessel or “dot” (arrows). Associated enlarged, polycystic T2 bright kidneys (K) also are seen.
Treatment: Ursodeoxycholic acid can decrease complications that occur as a result of cholelithiasis. Broad-spectrum antibiotic coverage can be used to prevent and treat cholangitis. Supplementation of fat-soluble vitamins may help alleviate symptoms of cholestasis. In addition, serologic screening for carcinogenic antigen (CA) 19-9 and carcinoembryogenic antigen (CEA) are helpful. Surgical resection has been used carcinogenic antigen successfully in patients with monolobar disease. For patients with diffuse involvement and progression beyond medical management, orthotopic liver transplant is the definitive treatment.18
Congenital Hepatic Fibrosis
Overview: Congenital hepatic fibrosis is a progressive condition that leads to portal hypertension. Autosomal recessive polycystic kidney disease is invariably associated with this condition.19,20 The hepatic abnormality is expressed later in life and therefore depends on less severe renal involvement that affords sufficient longevity for the hepatic abnormalities to become manifest clinically.
Etiology: Congenital hepatic fibrosis is a ductal plate malformation involving the small interlobular ducts.19 Periportal fibrosis occurs from scarring between adjacent biliary tracts and ductal plate remnants, resembling bile ducts. Hepatic fibrosis (due to malformation of small intrahepatic ductal plates) can be associated with Caroli disease, which is a large ductal plate anomaly; this association has been termed Caroli syndrome.21
Clinical Presentation: The onset of congenital hepatic fibrosis is variable, with less severe disease presenting later in childhood or in adulthood, frequently with splenomegaly and portal hypertension.7,19,22 Major complications of congenital hepatic fibrosis include ascending cholangitis, hepatic failure, and an increased risk of hepatocellular carcinoma. An increased incidence of hepatocellular carcinoma is found in patients with congenital hepatic fibrosis as well.7,23,24
Imaging: Ultrasound is the initial imaging modality typically requested (Fig. 88-11, A). Increased hepatic echogenicity and poorly defined portal vessels are seen, with increased echogenicity of the portal triads. If associated with Caroli syndrome, large ectatic biliary cysts are seen encircling irregular portal vein radicals. Associated renal lesions also should be investigated with ultrasound.7 Portal hypertension, supervening splenomegaly, and multiple collaterals bypassing the hepatic sinusoids can be seen with ultrasound, but they are more easily and completely revealed with contrast CT or MRI. CT and MRI reveal areas of hepatic parenchymal heterogeneity with variable CT attenuation (Fig. 88-11, B) and T1 and T2 MRI parenchymal signal.7 Magnetic resonance angiography can demonstrate the vascular sequelae of portal hypertension, and MRCP is useful to ascertain the anatomy of the biliary tree.19,21
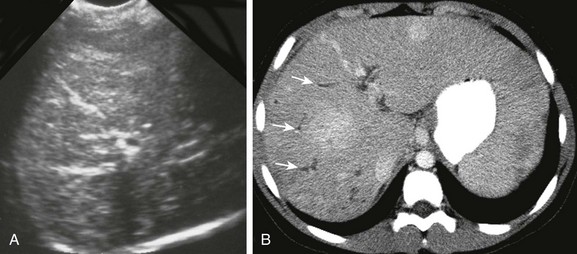
Figure 88-11 Congenital hepatic fibrosis complicated by portal hypertension in a 12-year-old girl.
A, A transverse sonogram shows heterogeneous echogenicity of the liver with linear bands of hyperechogenicity due to advanced fibrosis. B, Axial contrast-enhanced computed tomography images show heterogeneous hepatic enhancement and linear branching hypoattenuating tubular structures indicating mild ductal dilation (arrows). Extensive collateral vessels caused by portal hypertension were present (not shown).
Treatment: Medical management of hepatic fibrosis and portal hypertension consists of diuretics as first-line pharmacotherapy used to reduce ascites and increase glomerular filtration. When hepatic cirrhosis and portal hypertension become severe, orthotopic hepatic transplant is a more definitive treatment.
Biliary Hamartomas
Overview: Biliary hamartomas, also known as microhamartomas and Von Meyenburg complex, are rare.25 Patients usually have many well-defined, similar-sized lesions measuring less than 15 mm; their uniform size helps distinguish these lesions from metastatic disease, although a biopsy ultimately is required.7,26 Because microhamartomas are small, they usually are asymptomatic and are identified at pathologic examination. A small increased risk of hepatobiliary carcinomas exists.7
Etiology: Biliary hamartomas result from ductal plate malformations of the small bile ducts. They usually are multiple and typically do not have associated abnormalities, although at times they can coexist with simple hepatic cysts or polycystic kidney and liver disease.24,27
Imaging: Biliary hamartomas are seen with ultrasound; they are predominantly hypoechoic with comet tail echoes and can have mild cyst wall irregularity. Increased heterogeneity of the hepatic architecture often is seen.26 The lesions have low attenuation on CT, and on MRI they have a decreased signal with T1 weighting and an increased signal with T2 weighting (Fig. 88-12).7
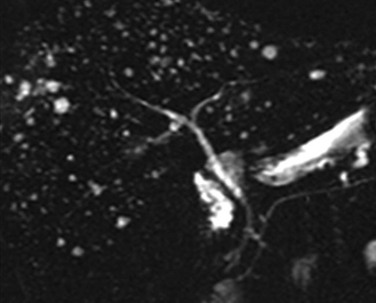
Figure 88-12 Biliary hamartomas.
Magnetic resonance imaging with cholangiopancreatography demonstrates innumerable small, well-defined, bright foci throughout the liver. Note the normal size ductal system. (From Kim OH, Chung HJ, Choi BG. Imaging of the choledochal cyst. Radiographics. 1995;15(1):69-88. Reprinted with permission.)
Biliary Atresias
Overview: Biliary atresia (BA) is the most common cause of neonatal cholestasis and is the primary indication for pediatric liver transplantation. It is more common in Japan than in the United States.28
Etiology: The etiology of BA remains unknown; viral, genetic, and autoimmune origins have been suggested as possibilities. Recent theories suggest that BA and hepatitis are inflammatory cholangiopathies on two ends of a spectrum.2 With severe inflammation, bile ducts are obliterated and BA develops. Less severe inflammatory cholangiopathy does not cause duct obliteration, and thus hepatitis ensues. Distinguishing between BA and hepatitis is important to determine therapy.29
Clinical Presentation: BA usually presents in the first month of life with jaundice, light-colored stools, and direct hyperbilirubinemia.2 If untreated, BA progresses to end-stage liver disease and death within the first 3 years of life.28
Imaging: Ultrasound is the initial study for neonatal jaundice; it is used to exclude surgical lesions such as choledochal cysts. Key findings in children with BA include an absent or small gallbladder (<15 mm) and the presence of a triangular cord sign (>4 mm echogenic tissue at the porta hepatitis following the portal veins) (Fig. 88-13, A).30 Identification of a triangular cord sign has been cited as 96% accurate for a diagnosis of BA. A small (measuring <15 mm) or absent gallbladder has been cited as 73% accurate for a diagnosis of BA compared with neonatal hepatitis; 90% of infants with neonatal hepatitis have a normal gallbladder.31 Accuracy can be improved to 98% when ultrasound findings are combined.32,33
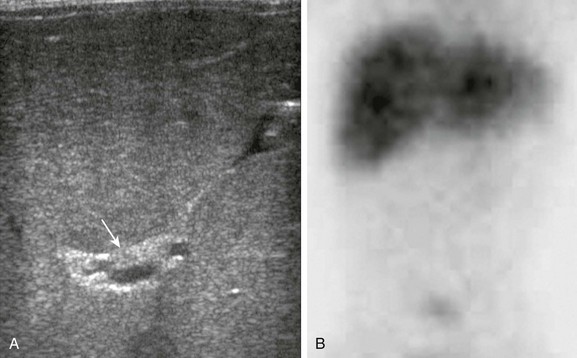
Figure 88-13 Biliary atresia in a 2-month-old boy with persistent jaundice.
A, A transverse image at the porta hepatis shows increased echogenicity along the expected course of the common bile duct and common hepatic ducts (arrow). A small (9 mm) gallbladder was present (not shown). B, A delayed image from a technetium-99m–labeled hepatoiminodiacetic acid shows normal radiotracer uptake in the liver but lack of excretion into the biliary tree. This finding persisted on all images.
Biliary scintigraphy with technetium-99m iminodiacetic acid and phenobarbital pretreatment has been used to distinguish BA and neonatal hepatitis (Fig. 88-13, B). A study without excretion is difficult to interpret, but if excretion of a radiopharmaceutical agent into the bowel occurs, BA is excluded.34,35 Postoperative use of nuclear scintigraphy has been advocated to assess restoration of biliary drainage after the portoenterostomy (Kasai) procedure.36 MRCP may be useful in infants to further delineate biliary tree anatomy before surgery and after ultrasound.3 ERCP typically is performed only when percutaneous intervention is needed. Despite all of the aforementioned imaging techniques, making a definitive diagnosis of BA can be challenging. In some cases, even a biopsy is nondiagnostic, and diagnosis is confirmed via an intraoperative cholangiogram.37,38
Treatment: BA is managed surgically with the Kasai procedure (hepatic portoenterostomy),39 and neonatal hepatitis is managed medically. It is important to diagnose BA early because infants younger than 3 months who are treated with the Kasai procedure tend to have a good long-term prognosis compared with infants older than 3 months, who often require liver transplantation.33
References
1. Todani, T, Watanabe, Y, Narusue, M, et al. Congenital bile duct cysts: classification, operative procedures, and review of thirty-seven cases including cancer arising from choledochal cyst. Am J Surg. 1977;134(2):263–269.
2. Gubernick, JA, Rosenberg, HK, Llaslan, H, et al. US approach to jaundice in infants and children. Radiographics. 2000;20(1):173–195.
3. Carneiro, RC, Fordham, LA, Semelka, RC. MR imaging of the pediatric liver. Magn Reson Imaging Clin North Am. 2002;10(1):137–164.
4. Kim, OH, Chung, HJ, Choi, BG. Imaging of the choledochal cyst. Radiographics. 1995;15(1):69–88.
5. Wiedmeyer, DA, Stewart, ET, Dodds, WJ, et al. Choledochal cyst: findings on cholangiopancreatography with emphasis on ectasia of the common channel. AJR Am J Roentgenol. 1989;153(5):969–972.
6. de Vries, JS, de Vries, S, Aronson, DC, et al. Choledochal cysts: age of presentation, symptoms, and late complications related to Todani’s classification. J Pediatr Surg. 2002;37(11):1568–1573.
7. Brancatelli, G, Federle, MP, Vilgrain, V, et al. Fibropolycystic liver disease: CT and MR imaging findings. Radiographics. 2005;25(3):659–670.
8. Yoshida, H, Itai, Y, Minami, M, et al. Biliary malignancies occurring in choledochal cysts. Radiology. 1989;173(2):389–392.
9. Han, BK, Babcock, DS, Gelfand, MH. Choledochal cyst with bile duct dilatation: sonography and 99mTc IDA cholescintigraphy. AJR Am J Roentgenol. 1981;136(6):1075–1079.
10. Arcement, CM, Towbin, RB, Meza, MP, et al. Intrahepatic chemoembolization in unresectable pediatric liver malignancies. Pediatr Radiol. 2000;30(11):779–785.
11. Irie, H, Honda, H, Jimi, M, et al. Value of MR cholangiopancreatography in evaluating choledochal cysts. AJR Am J Roentgenol. 1998;171(5):1381–1385.
12. Vitellas, KM, Keogan, MT, Freed, KS, et al. Radiologic manifestations of sclerosing cholangitis with emphasis on MR cholangiopancreatography. Radiographics. 2000;20(4):959–975. [quiz 1108-1109, 1112].
13. Dodd, GD, 3rd., Baron, RL, Oliver, JH, 3rd., et al. End-stage primary sclerosing cholangitis: CT findings of hepatic morphology in 36 patients. Radiology. 1999;211(2):357–362.
14. Terada, T, Moriki, T. Monolobar ductal plate malformation disease of the liver. Pathol Int. 2010;60(5):407–412.
15. Marchal, GJ, Desmet, VJ, Proesmans, WC, et al. Caroli disease: high-frequency US and pathologic findings. Radiology. 1986;158(2):507–511.
16. Toma, P, Lucigrai, G, Pelizza, A. Sonographic patterns of Caroli’s disease: report of 5 new cases. J Clin Ultrasound. 1991;19(3):155–161.
17. Choi, BI, Yeon, KM, Kim, SH, et al. Caroli disease: central dot sign in CT. Radiology. 1990;174(1):161–163.
18. Bockhorn, M, Malagó, M, Lang, H, et al. The role of surgery in Caroli’s disease. J Am Coll Surg. 2006;202(6):928–932.
19. Akhan, O, Karaosmanoglu, AD, Ergen, B. Imaging findings in congenital hepatic fibrosis. Eur J Radiol. 2007;61(1):18–24.
20. Desmet, V. Pathogenesis of ductal plate malformation. J Gastroenterol Hepatol. 2004;19:s356–s360.
21. Ernst, O, Gottrand, F, Calvo, M, et al. Congenital hepatic fibrosis: findings at MR cholangiopancreatography. AJR Am J Roentgenol. 1998;170(2):409–412.
22. Lowe, L, Schlesinger, A. Hepatobiliary system, congenital abnormalaties. In: Slovis T, ed. Caffey’s pediatric diagnostic imaging. Philadelphia: Mosby Elsevier, 2007.
23. Lowe, LH. Imaging hepatobiliary disease in children. Semin Roentgenol. 2008;43(1):39–49.
24. Veigel, MC, Prescott-Focht, J, Rodriguez, MG, et al. Fibropolycystic liver disease in children. Pediatr Radiol. 2009;39(4):317–327. [quiz 420-421].
25. Karahan, OI, Kahriman, G, Soyuer, I, et al. Hepatic von Meyenburg complex simulating biliary cystadenocarcinoma. Clin Imaging. 2007;31(1):50–53.
26. Zheng, RQ, Zhang, B, Kudo, M, et al. Imaging findings of biliary hamartomas. World J Gastroenterol. 2005;11(40):6354–6359.
27. Yönem, O, Ozkayar, N, Balkanci, F, et al. Is congenital hepatic fibrosis a pure liver disease? Am J Gastroenterol. 2006;101(6):1253–1259.
28. Superina, R, Magee, JC, Brandt, ML, et al. The anatomic pattern of biliary atresia identified at time of Kasai hepatoportoenterostomy and early postoperative clearance of jaundice are significant predictors of transplant-free survival. Ann Surg. 2011;254(4):577–585.
29. Kirks, DR, Coleman, RE, Filston, HC, et al. An imaging approach to persistent neonatal jaundice. AJR Am J Roentgenol. 1984;142(3):461–465.
30. Lee, HJ, Lee, SM, Park, WH, et al. Objective criteria of triangular cord sign in biliary atresia on US scans. Radiology. 2003;229(2):395–400.
31. Kanegawa, K, Akasaka, Y, Kitamura, E, et al. Sonographic diagnosis of biliary atresia in pediatric patients using the “triangular cord” sign versus gallbladder length and contraction. AJR Am J Roentgenol. 2003;181(5):1387–1390.
32. Humphrey, TM, Stringer, MD. Biliary atresia: US diagnosis. Radiology. 2007;244(3):845–851.
33. Sun, Y, Zheng, S, Qian, Q. Ultrasonographic evaluation in the differential diagnosis of biliary atresia and infantile hepatitis syndrome. Pediatr Surg Int. 2011;27(7):675–679.
34. Majd, M, Reba, RC, Altman, RP. Hepatobiliary scintigraphy with 99mTc-PIPIDA in the evaluation of neonatal jaundice. Pediatrics. 1981;67(1):140–145.
35. Park, WH, Choi, SO, Lee, HJ, et al. A new diagnostic approach to biliary atresia with emphasis on the ultrasonographic triangular cord sign: comparison of ultrasonography, hepatobiliary scintigraphy, and liver needle biopsy in the evaluation of infantile cholestasis. J Pediatr Surg. 1997;32(11):1555–1559.
36. Castagnetti, M, Davenport, M, Tizzard, S, et al. Hepatobiliary scintigraphy after Kasai procedure for biliary atresia: clinical correlation and prognostic value. J Pediatr Surg. 2007;42(6):1107–1113.
37. Sokol, RJ, Mack, C, Narkewicz, MR, et al. Pathogenesis and outcome of biliary atresia: current concepts. J Pediatr Gastroenterol Nutr. 2003;37(1):4–21.
38. Schwartz, MZ. An alternate method for intraoperative cholangiography in infants with severe obstructive jaundice. J Pediatr Surg. 1985;20(4):440–442.
39. Kasai, M, Suzuki, H, Ohashi, E, et al. Technique and results of operative management of biliary atresia. World J Surg. 1978;2(5):571–579.
[/level-membership-for-pediatrics-category][not-level-membership-for-pediatrics-category]Chapter 88
Congenital Hepatobiliary Anomalies
Fibropolycystic liver disorders are a group of associated congenital anomalies of the liver caused by malformation of the embryonic ductal plates (Fig. 88-1). They include choledochal cysts, Caroli disease, hepatic fibrosis, biliary hamartomas, and cystic liver disease. The specific type of fibropolycystic disorder depends on the size of the embryonic duct with faulty development (Table 88-1).
Table 88-1
Fibropolycystic Liver Disorder and Size of Ducts with Faulty Embryogenesis
| Disorder | Duct Size |
| Congenital hepatic fibrosis | Small |
| Biliary hamartoma | Small |
| Polycystic liver disease | Medium |
| Choledochal cyst | Large extrahepatic |
| Caroli disease | Large intrahepatic |
Figure 88-1 Schema of ductal plate.
A bilayer of hepatocytes (green and red layers) surrounds the portal venous structures (central black), forming the lumina of primitive bile ducts (left). A process of organized resorption leads to the normal organization of biliary ducts within the portal triads. Failure of this process leads to ductal plate malformations.
Choledochal Cyst
Overview: Choledochal cysts, which are among the most frequent of congenital hepatobiliary anomalies, are a fusiform or saccular dilation of the bile ducts. The Todani classification system is commonly used to categorize these anomalies into five general types (with several subtypes) that differ based on etiology, pathogenesis, appearance, and presentation (Fig. 88-2).1
Figure 88-2 Schematic of Todani classification of choledochal cysts based on cholangiographic morphology.
Type IA and IB involve cystic dilatation of the common duct, with IB limited to the area below the insertion of the cystic duct; IC is fusiform dilatation of the duct. Type II is a saccular diverticulum from the common bile duct, and type III is a choledochocele at the ampulla of Vater. Types IVA and IVB are multiple cystic dilations of the intrahepatic and extrahepatic biliary tree, and type V is equivalent to Caroli disease with numerous intrahepatic bile lakes throughout the biliary tree and liver. (From Kim OH, Chung HJ, Choi BG. Imaging of the choledochal cyst. Radiographics. 1995;15:69-88.)
The most common choledochal cyst is the Todani type I, found in 80% to 90% of cases.2 It involves variable lengths and degrees of common bile duct dilatation and is further classified into subtypes.2 Todani type IA, IB, and IC describe cystic dilation of the common bile duct, segmental dilation below the cystic duct, and fusiform dilation, respectively. Todani type II choledochal cysts are found in only 2% of cases and consist of one or more diverticula of the common bile duct.3 Todani type III occurs in 1.5% to 5% of cases and involves dilation of the intraduodenal portion of the duct, forming a cystlike mass, termed a “choledochocele,” with both the common bile and pancreatic ducts emptying into it. Todani type IVA consists of multiple intrahepatic and extrahepatic biliary dilatations and occurs in 10% of patients with choledochal cysts.1,3 Type IVB, which involves multiple extrahepatic biliary cysts without intrahepatic involvement, is rare. Todani type V is synonymous with Caroli disease.1 Todani type IVA also has been described as a type I cyst with intrahepatic involvement, and controversy exists about whether it is actually a type V (Caroli disease) cyst with common duct enlargement.
Etiology: Several theories for the pathogenesis of the type I choledochal cysts exist. In addition to the ductal plate malformation described previously, other theories have suggested cyst formation as a result of obstruction of the distal biliary duct and/or reflux of pancreatic enzymes into the biliary tree because of anomalous proximal insertion of the pancreatic duct into the common bile duct (ductal malunion).4,5 This ductal malunion could permit reflux of pancreatic enzymes into the common bile duct, with subsequent inflammation and weakening of the wall, a pathologic mechanism that occurs in about 60% of patients.5 This anomalous ductal connection has been demonstrated on endoscopic retrograde cholangiopancreatography (ERCP) (Fig. 88-3). Type I choledochal cysts are more common in girls than in boys in Western countries, but the sex ratio is equal in Asia.2,3 About 65% of all reported cases are from Japan.2
It has been theorized that type II choledochal cysts result from prenatal rupture of the common bile duct with subsequent healing.3 Type III choledochal cysts may be the sequelae of ampullary obstruction or congenital duplication of the duodenum at the ampulla. The intrahepatic cysts seen in types IV and V are thought to be due to primary ductal ectasia resulting from ductal plate malformation.
Clinical Presentation: Choledochal cysts may present in infancy with cholestatic jaundice, which clinically is inseparable from neonatal hepatitis or biliary atresia.2 In older children and young adults, the clinical presentation is variable. A characteristic triad of abdominal pain, obstructive jaundice, and fever has been reported, but few patients present with all three components. Abdominal pain is the most characteristic presentation, followed by obstructive jaundice, fever, pale stools, splenomegaly, hepatomegaly, and a palpable mass.6 The most common complication of a choledochal cyst is ascending cholangitis.7 Long-term complications, such as hepatic cirrhosis with subsequent portal hypertension, can occur.6 Carcinoma of the biliary tree has a twentyfold increased incidence in patients with choledochal cysts. This risk is low in the first decade but increases with advancing age.8 Spontaneous cyst rupture also has been reported.6
Imaging: When the clinical presentation points to a hepatobiliary problem, sonography usually is the initial imaging study requested. The markedly dilated common bile duct is readily discernible in types I and IV choledochal cysts. The gallbladder usually can be identified adjacent to the dilated common duct (Fig. 88-4). Most frequently, the intrahepatic ducts are normal, but varying degrees of dilation may be present as a result of obstruction. Sludge or stones may be identified within the dilated ducts.9 Type III cysts may cause mass effect at the ampulla of Vater (Fig. 88-5, A and B).
Figure 88-4 Choledochal cyst (Todani type I) in an 8-day-old girl with jaundice.
Transverse sonogram shows a large fusiform cyst in the porta hepatis (asterisk) just beneath a small, contracted sludge filled gallbladder (arrow).
Figure 88-5 Choledochocele (Todani type III) in a 12-year-old girl with abdominal pain.
A, Upper gastrointestinal tract image shows a large filling defect (arrow) coincident with the site of the Ampulla of Vater. B, Drawing of findings of choledochocele confirmed at surgery.
Abdominal magnetic resonance imaging (MRI) with cholangiopancreatography (MRCP) often is performed to delineate anatomy (Fig. 88-6).10–12 Hepatobiliary scintigraphy can prove useful in difficult cases by demonstrating communication between the choledochal cyst and the hepatobiliary ducts (Fig. 88-7).9 Computed tomography (CT) does not demonstrate the biliary ductal anatomy as well as ultrasound and MRI, although it can be useful along with sonography to guide abscess drainage and to evaluate hepatic anatomy.10,11
Figure 88-6 A choledochal cyst (Todani type I).
A, Magnetic resonance imaging with cholangiopancreatography (MRCP) in a 10-day-old boy with jaundice demonstrates dilatation of the common bile duct (arrow). B, A choledochal cyst (Todani type I) in a 6-year-old boy with abdominal pain. MRCP reveals dilation of the common bile duct (arrow) draining into the normal duodenum (D).
Percutaneous transhepatic cholangiography (PTC) and ERCP are useful when placement of a percutaneous biliary drain may be helpful (Fig. 88-8). Some investigators prefer PTC to ERCP because it carries a lower risk of iatrogenic cholangitis.7
Figure 88-8 Choledochal cyst (Todani type IV) in a 1-year-old boy.
A, A longitudinal sonogram shows marked saccular dilation of the intrahepatic and extrahepatic bile ducts (arrows). B, An operative cholangiogram confirms dilation of the intrahepatic biliary tree and common bile duct (arrow). The gallbladder (GB) is noted.
Treatment: Because of the potential long-term sequelae, surgical excision and hepatojejunostomy are performed as definitive treatments. Patients with intrahepatic cysts are closely monitored for cholangitis, cholestasis, and stone formation. Antibiotics are administered in the setting of acute cholangitis. Medical therapy can be instituted to reduce the risk of cholestasis and promote bile flow; however, when extensive liver damage and cirrhosis occurs, a liver transplant may be necessary.
Caroli Disease and Caroli Syndrome
Overview: Caroli disease, also classified as Todani type V choledochal cyst, is a segmental nonobstructive dilation of the intrahepatic bile ducts. It is characterized by multiple hepatic cysts in continuity with the biliary system, representing ectatic intrahepatic ducts,7
[/not-level-membership-for-pediatrics-category]
