Congenital Diaphragmatic Hernia and Eventration
The management and treatment of congenital diaphragmatic hernia (CDH) remains a challenge. Despite advances in prenatal diagnosis, operative management, and neonatal critical care, infants born with CDH still have a significant mortality and long-term disability. The relatively rare incidence of the disease and wide spectrum of disease severity results in clinical challenges for individual practitioners and institutions. The large majority of centers will treat less than 10 CDH infants per year.1 Thus, broad clinical experience, especially in cases of the most severe disease, is difficult to achieve.
The morbidity and mortality associated with CDH is largely due to pulmonary hypoplasia and pulmonary vascular hypertension (PHTN).2,3 Over the last 25 years, clinical efforts to avoid lung injury, including extracorporeal membrane oxygenation (ECMO), high-frequency oscillating ventilation (HFOV), and permissive hypercapnia, have improved overall survival, but significant variation remains amongst institutions.2,4 Reported survival ranges from 50–90%.3,5
Epidemiology
The incidence of CDH has been reported as high as 1 in 2000 births.6 In the USA, approximately 1,000 CDH infants are born with a prevalence of 2.4 per 10,000 live births.6,7 One-third of infants with CDH die as stillbirths, often associated with other congenital anomalies.8 However, the overall incidence of CDH may be underestimated. Proportions of fetuses are prenatally diagnosed with CDH and will die in utero or shortly after birth. Thus, many may never been seen or accounted for in a tertiary referral center. Presumed to be the most severe of all CDH infants, these patients contribute to the ‘hidden mortality’ of CDH.9–12
Infants with isolated CDH are typically male and one-third are associated with a major congenital anomaly.7 Approximately 80% are left sided. Bilateral defects are rare and are associated with other major anomalies.13 Although the exact etiology remains unknown, mothers that are thin or underweight may have an increased risk of bearing an infant with CDH.14
Genetics
CDH is no longer thought of as a sporadic isolated anomaly in all cases. A first-degree relative has an expected occurrence rate of approximately 2%.15 All types of structural chromosomal abnormalities including deletions, translocations, and trisomies have been identified.16–21 In addition, a gene distal to the 15q21 locus has been identified for the normal development of the diaphragm.17,18
CDH has been associated with over 70 syndromes.19 In some cases, the diaphragmatic malformation is the predominant defect, as in Fryns and Donnai–Barrow syndromes.20,21 In other syndromes, CDH only occurs in a small percentage, but still greater than the general population, as in Simpson–Golabi–Behmel and Beckwith–Wiedermann syndromes. The inheritance patterns for these syndromes include dominant and recessive as well as autosomal and X-linked variants.22 Identifying the patterns of non-hernia-related anomalies associated with CDH and recognizing genetic syndromes help determine the prognosis, treatments, counseling, and outcomes.16 If the antenatal diagnosis of CDH is made, amniocentesis with karyotype and chromosomal analysis is indicated.
Associated Anomalies
The posterolateral ‘Bochdalek’ hernia accounts for 90% of all diaphragmatic hernia cases.23 The remainder are the anterior ‘Morgagni’ hernia along with defects of the central septum transversum. The majority of posterolateral CDH are left sided (85%), with right sided (13%) and bilateral (2%) accounting for the rest.23–25
Approximately 50% of CDH are isolated defects with the others associated with anomalies of the cardiovascular (27.5%), urogenital (17.7%), musculoskeletal (15.7%), and central nervous (9.8%) systems (CNS).26 Many conditions, such as lung hypoplasia, intestinal malrotation, some cardiac malformations, and patent ductus arteriosus (PDA) are considered to be consequences of the diaphragmatic defect. Non-CDH-related defects are estimated to occur in 40–60% of cases and can involve the cardiovascular, CNS, gastrointestinal, and genitourinary systems, and may be a consequence of an underlying field defect of unknown etiology.27,28
The impact of associated anomalies on prognosis and outcome cannot be overstated. Ninety-five per cent of stillborn infants with CDH have an associated major anomaly.29 Greater than 60% of infants who do not survive the immediate neonatal period have associated anomalies.30 In contrast, infants that survive preoperative stabilization and come to operative repair have less than 10% additional anomalies.30 Although the severity of pulmonary hypoplasia and hypertension are the major determinants of overall survival, there is a significant survival advantage for infants with isolated CDH (43.7% vs 7.1%).27 Due to this dismal outcome, the emphasis on detailed and accurate prenatal diagnosis has influenced the management and treatment of CDH. Twenty per cent of prenatally diagnosed CDH infants have chromosomal anomalies, with 70% having an associated structural malformation. In contrast, only 35% of postnatally diagnosed CDH infants have an associated anomaly.27 This difference may be the result of lethal chromosomal anomalies and/or may reflect parental decisions for termination in high-risk infants with anomalies that portend significant morbidity.
Common cardiac defects include atrial septal defects (ASDs), ventricular septal defects (VSDs), and other outflow tract anomalies (transposition of the great vessels, tetralogy of Fallot, double-outlet right ventricle, and aortic coarctation).30–32 In a review of 4,268 infants with CDH, approximately 18% of infants had an associated cardiac defect. Major cardiac lesions (excluding patent foramen ovale, atrial septal defects, PDA) was 8% with an overall survival of 36% compared to infants with minor anomalies (67% survival) and those without cardiac defects (73% survival).33
Cost
Despite improvements in outcomes and advances in treatment, the cost of caring for CDH infants continues to increase. Data from the Kids’ Inpatient Database has projected the annual national costs to range between $264 and $400 million based on 60% overall survival.34 The utilization of ECMO was the highest single factor associated with cost with a 2.4-fold increase in expenditures from 1997 to 2006. The highest median cost were ECMO patients at $156,499.90/patient, constituting 28.5% of the total national costs.34 Single-center reports have documented expenses as high as $250,000 per CDH case with an estimated annual cost of $230 million, and estimated cost of $98,000 for non-ECMO survivors compared with $365,000 for ECMO survivors.35
Embryology
Diaphragm development and CDH pathogenesis
The development of the human diaphragm is a complex, multicellular, multi-tissue interaction that is poorly understood. Precursors to the diaphragm begin to form during the fourth week of gestation. Historically, the diaphragm was thought to develop from the fusion of four embryonic components: anteriorly by the septum transversum, dorsolaterally by the pleuroperitoneal folds (PPF), dorsally by the crura from the esophageal mesentery, and posteriorly by the body wall mesoderm (Fig. 24-1).36,37 As the embryo begins to form, the septum transversum migrates dorsally and separates the pleuro-pericardial cavity from the peritoneal cavity. At this point, the pleural and peritoneal cavities still communicate. The septum transversum interacts with the PPF and mesodermal tissue surrounding the developing esophagus and other foregut structures, resulting in the formation of primitive diaphragmatic structures. Bound by pericardial, pleural, and peritoneal folds, the paired PPFs now separate the pleuropericardial and peritoneal cavities. Eventually, the septum transversum develops into the central tendon.36,37 As the PPF develops during the sixth week of gestation, concurrently, the pleuroperitoneal membranes close and separate the pleural and abdominal cavities by the eighth week of gestation. Typically, the right side closes before the left. Ultimately, the phrenic axons and myogenic cells destined for neuromuscularization migrate to the PPF and form the mature diaphragm.38 The musculariztion of the primitive diaphragm is a separate, but inter-related process. Historically, the primitive fetal diaphragm is thought to muscularize from the inner thoracic musculature as the diaphragm closes.39 Others have implicated a progressive development of the pleuroperitoneal membrane40 or the posthepatic mesenchymal plate (PHMP) interacting with the PPF.41
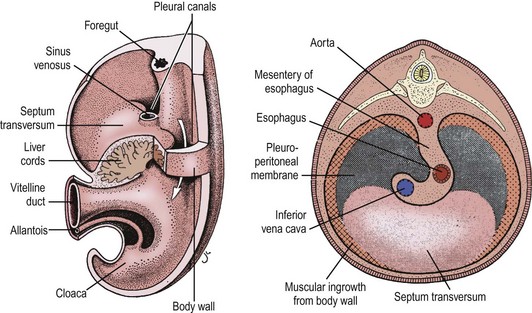
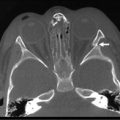
FIGURE 24-1 Historically, the diaphragm has been thought to develop from fusion of its four embryologic components. According to this theory, the septum transversum fuses posteriorly with the mediastinal mesenchyme. The pleuroperitoneal canals (arrow) allow free communication between the pleural and peritoneal cavities. Closure of these canals is completed as the pleuroperitoneal membranes develop. The four embryologic components of the developing diaphragm are shown in cross section. (From Skandalakis IJ, Colborn GL, Skandalakis JE. In: Nyhus LM, Baker RJ, Fischer JE, editors. Mastery of Surgery. 3rd ed. Boston: Little, Brown; 1996.)
Another theory for CDH development is a failure of muscularization of the future diaphragm prior to complete closure of the canal (Fig. 24-2).42 Inadequate closure of the pleuroperitoneal canal allows the abdominal viscera to enter the thoracic cavity when it returns from the extraembryonic coelom as well as the liver to herniate into the chest. Consequently, the limited intrathoracic space, due to the visceral herniation, results in pulmonary hypoplasia.
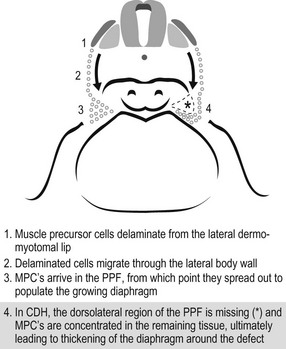
FIGURE 24-2 This schematic depicts a different embryologic pathway for diaphragmatic development and CDH formation than seen in Figure 24-1. On the left side, (1–3) is the proposed normal pathway for diaphragm development. On the right side, (4) is the pathway for CDH formation. MPC, muscle precursor cells; PPF, pleuroperitoneal fold. (From Clugston RD, Greer JJ. Diaphragm development and congenital diaphragmatic hernia. Semin Pediatr Surg 2007;16:94–100.)
Although traditional theories suggest that the lung hypoplasia is secondary to the diaphragmatic malformation, others have postulated that the primary disturbance may be abnormal lung development that causes the diaphragmatic defect.36 According to this theory, disturbances in lung bud formation subsequently impair the PHMP development, and results in failure of diaphragm fusion/muscularization.
The nitrofen murine model has led to improved understanding of abnormal pulmonary development in CDH.43 Nitrofen (2,4-dichloro-phenyl-p-nitrophenyl ether) is an environmental teratogen that is relatively harmless to adult mice. However, if given during pregnancy, it can cause pulmonary, cardiac, skeletal, and diaphragmatic abnormalities.44 Diaphragmatic defects resulting from the administration of nitrofen in mice are very similar to the diaphragmatic defects seen in babies with CDH in regards to size, location, and herniation of abdominal viscera.45,46 The side of the CDH depends on the time of nitrofen exposure during gestation. In nitrofen-exposed fetal mice, a defect is clearly seen in the posterolateral portions of the PPF.47 In addition, nitrofen exposure appears to affect muscularization of the PPF (see Fig. 24-2).48 Finally, the offspring will exhibit features of pulmonary hypoplasia including reduced airway branching, surfactant deficiency, pulmonary vascular abnormalities, and respiratory failure at birth.49,50
Other teratogens structurally similar to nitrofen have been shown to induce CDH in animal models as well. Although the exact etiology of CDH is unknown, these teratogens commonly affect the retinoic acid synthesis pathway by inhibiting retinol dehydrogenase-2 and causing similar diaphragmatic defects.51 Several clinical observations and molecular studies have supported the importance of the retinoic acid pathways in CDH development. Vitamin A-deficient rodents will produce offspring with CDH of variable severity.52 Retinoic acid receptor knockout mice produce fetuses with CDH.53 Failure to convert retinoic acid to retinaldehyde following administration of nitrofen produces posterolateral diaphragmatic defects in rats.51 Lower plasma levels of retinoic acid and retinol binding protein in infants with CDH have been found compared to controls.54
Lung Development and Pulmonary Hypoplasia
Lung development is recognized as a complex programmed event regulated by genetic signals, transcription factors, growth factors, and hormones. These events control the temporal and spatial interactions between epithelium and endothelium. Early transcription signals, such as thyroid transcription factor-1 and hepatocyte nuclear factor-3β, regulate pulmonary development from the primitive foregut mesenchyme. Other pathways of pulmonary development include sonic hedgehog, transforming growth factor-β, Notch-delta pathway, and Wingless-Int.22,55 In addition, glucocorticoids, thyroid hormone, and retinoic acid have all been shown to regulate pulmonary organogenesis.50
Fetal lung development is divided into five overlapping stages.56 (1) The embryonic stage begins during the third week of gestation as a caudal diverticulum from the laryngotracheal groove. The primary lung buds and trachea form from this diverticulum by the fourth week and lobar structures are seen by the sixth week. (2) The pseudoglandular stage occurs between the fifth and 17th weeks of gestation with the formation of formal lung buds as well as the main and terminal bronchi. (3) During the canalicular stage, the pulmonary vessels, respiratory bronchioles, and alveolar ducts develop between weeks 16 and 25 weeks with the appearance of type 1 pneumocytes and type 2 pneumocyte precursors. At this stage, functional gas exchange is possible. (4) The saccular stage continues from 24 weeks to term with the maturation of alveolar sacs. Airway dimensions and surfactant synthesis capabilities continue to mature as well. (5) Finally, the alveolar stage begins after birth with a continued increase and development of functional alveoli.
Concomittantly, fetal pulmonary vascular development occurs in concordance with the associated lung development and follows the pattern of airway and alveolar maturation. A functional unit known as the acinus consists of the alveolus, alveolar ducts, and respiratory bronchioles. The pulmonary vasculature develops as these acinar units multiply and evolve during the canalicular stage. The preacinar structures consist of the trachea, major bronchi, lobar bronchi, and terminal bronchioles. The pulmonary vascular development for the preacinus is typically completed by end of the pseudoglandular stage.57–59 In theory, any impedance to normal pulmonary development will concurrently hinder pulmonary vascular development.
Pulmonary hypoplasia is characterized by a decrease in bronchial divisions, bronchioles, and alveoli. The alveoli and terminal saccules exhibit abnormal septations that impair the air–capillary interface limiting gas exchange.60 At birth, the alveoli are thick-walled with intra-alveolar septations. These immature alveoli have increased glycogen content leading to thickened secretions which further limit gas exchange. Animal models of CDH have demonstrated pulmonary hypoplasia with decreased levels of total lung DNA and protein. In addition, the pulmonary vasculature appears to be less compliant with abnormally thick-walled arterioles.61 Surfactant levels are also decreased which may result in immature functioning lungs.62 Endogenous surfactant synthesis has been found to be decreased in CDH infants that required ECMO compared with non-CDH ventilated neonates without significant lung disease.63 Interestingly, the contralateral lung also exhibits the structural abnormalities of pulmonary hypoplasia.
Pulmonary Vascular Development and Pulmonary Hypertension
Normal fetal cardiopulmonary circulation transitions to it postnatal state rapidly with a tenfold increase in pulmonary blood flow shortly after birth. Fetal pulmonary blood flow is characterized as a low-flow, high-resistance circuit due to medial and adventitial hypertrophy of the vasculature.64 Normally, the pulmonary vascular resistance quickly decreases as the distal small pulmonary arteries and arterioles remodel over the first few months of life, resulting in a low-resistance, high-flow postnatal circulation.65,66 However, this process appears to be arrested in CDH newborns, and the fetal circulation persists resulting in PHTN. In fact, the abnormal fetal pulmonary circulation in CDH fetuses appears to occur in early gestation. The pulmonary arteries exhibit a decrease in density per unit of lung parenchyma as well as an increase in muscularization that extends to the vasculature at the acinar level.67 In fetal lamb models of surgically created CDH as well as human fetuses with CDH, there is a relative decrease in lung parenchyma.68,69 This impaired lung growth and development has been speculated to be related to impaired vascular development.70 As a result, PHTN appears to develop in utero, which may cause a reduction in pulmonary artery growth, proper alveolar development, and normal lung growth.71
Utilizing fetal Doppler ultrasound (US), fetuses with CDH exhibit a decrease in the overall pulmonary blood flow and vascularity.72 The modified McGoon Index, a ratio of the combined diameter of the proximal pulmonary arteries to the descending aorta, may help predict the degree of PHTN that correlates with death. A modified McGoon Index of ≤1.3 has an 85% sensitivity and 100% specificity for mortality.73 An index <1.25 and birth weight <2755 g has been shown to result in 80% mortality, with a sensitivity of 73% and a specificity of 78%.74 The arterial adventitia and medial wall thickness are increased in postmortem CDH infants compared to controls.75,76 In addition, the adventitial thickness and area were greater in CDH stillborns and newborns when compared to live born non-CDH infants, suggesting that increased muscularization of the pulmonary vasculature occurs in utero and fails to appropriately remodel after birth.
In a retrospective study, CDH infants who developed normal pulmonary artery pressures during the first 3 weeks of life were found to have a 100% survival rate.77 In this same study, an intermediate reduction in elevated pulmonary pressures after birth were seen in 34% of infants with a 75% survival. Mortality was 100% in CDH infants that failed to normalize their pulmonary pressures despite therapy. Although contemporary outcomes for infants with pulmonary hypertension have improved, these data underscore the importance of PHTN in CDH.
Diagnosis
Prenatal Diagnosis
Due to the wide discrepancy of disease severity and potential fetal therapies, accurate and timely prenatal diagnosis of CDH is important. The differential diagnosis for CDH include other pulmonary anomalies, such as congenital pulmonary airway malformations (CPAM), bronchogenic cysts, bronchial atresia, or bronchopulmonary sequestrations, as well as mediastinal lesions, including enteric, neuroenteric, or thymic cysts. In these conditions, the normal intra-abdominal anatomy is not disturbed. In addition, diaphragmatic eventration can be misinterpreted for CDH. Although this differentiation from CDH can be difficult, this distinction is important as diaphragmatic eventration portends a much better prognosis and requires different management. Eventration are typically isolated lesions, but may be associated with pleural and/or pericardial effusions.78
The diagnosis of CDH can be made by ultrasound as early as 11 weeks’ gestation, although most are not seen until after 16 weeks (Fig. 24-3).79 Although most CDH can be detected during the second trimester, some sonographic features may not manifest until later in pregnancy.80 Approximately 50–70% of CDH are diagnosed during pregnancy.79,81 However, for some tertiary centers, 90% of CDH newborns may have been diagnosed in utero.82 Fetal ultrasound features include polyhydramnios, bowel loops within the chest, an echogenic chest mass, and/or an intrathoracic stomach. Left-sided CDH typically feature mediastinal/cardiac shift to the right as well as herniation of stomach, intestines, and/or spleen. The liver may herniate but its echogeneity is often similar to the lung, and may be more difficult to differentiate. In right-sided CDH, the right lobe of the liver is herniated, with a left-sided mediastinal shift.
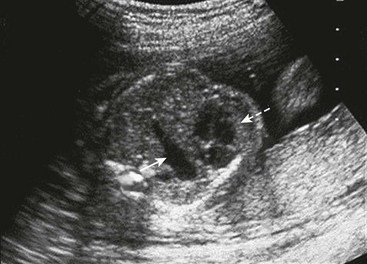
FIGURE 24-3 Fetal ultrasound image at the level of the four-chamber heart (dotted arrow). Gastric bubble (solid arrow) at the level of the four-chamber heart suggests CDH. This is the level used to calculate the lung-to-head ratio.
Ultrasound of the fetal chest is best performed in the axial plane. Fetal lung circumference, used to determine the lung-to-head ratio (LHR), is measured at the level of the four-chambered view of the heart (see Fig. 24-3). However, measurements of the fetal chest require an understanding of the dynamic growth of the fetal thorax which mostly occurs during the first trimester. During this time, the heart-to-thorax ratio is variable. However, after the first trimester, the heart-to-thorax ratio is generally 1 : 3 and remains unchanged thereafter.83 The fetal diaphragm can be seen as early as the first trimester as a thin hypoechogenic line in the sagittal view.
Although independent sonographic features of CDH have not been shown to be accurate predictors for a poor prognosis, severe or advanced CDH may exhibit an intrathoracic ‘liver up’ (liver in the chest), mediastinal shift to the contralateral thoracic cavity, or features consistent with hydrops fetalis.84 Two distinct ultrasound features have been commonly utilized to risk stratify: (1) LHR and (2) liver herniation into the chest. Predicting survival of CDH fetuses based on LHR has been statistically supported: 100% survival with LHR > 1.35, 61% with LHR between 1.35–0.6, and no survival with LHR < 0.6 in one series.85 Survival based on liver herniation alone is 56%, compared to 100% survival without liver herniation.86 The combination of liver herniation and low LHR (LHR < 1.0) has a 60% mortality in prenatally diagnosed CDH.87–89 Although fetal ultrasound is the most reliable modality for prognosis, its inconsistent utilization and inter-rater variability has created variable results.90,91
Although the heart-to-thorax ratio is consistent after the first trimester, the effects of gestational age can influence the LHR. The fetal lung increases 16-fold, compared to a fourfold increase in head circumference, between 12 and 32 weeks gestational age. This gestational age differential can be ameliorated by using the LHR as a function of observed or measured to the expected or mean of the same lung (O/E LHR). The definition of severe CDH as measured by O/E LHR is <25%. In one series, survival for severe CDH using this definition was approximately 10% with liver up and 25% with liver down. There were no survivors when O/E LHR was <15% with liver up.92 The total fetal lung volume (TFLV) as determined by three-dimensional ultrasound may be more accurate than O/E LHR as it includes the contralateral lung.93–95 Similar to the LHR, the effects of gestational age on fetal lung growth can be accounted for by using biometric measurements, total fetal body volume, or normal/expected total fetal lung volume, expressed as O/E TFLV.96–98 Early work suggests that O/E TLFV in utero correlates well with outcome.99,100
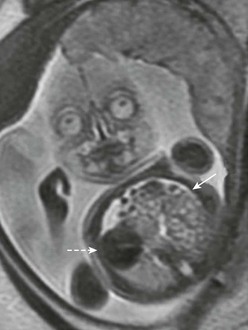
Fetal magnetic resonance imaging (MRI) has become widely utilized in the prenatal diagnosis of CDH (Fig. 24-4). Due to the high water content, fetal lungs exhibit high signal intensity on T2-weighted images which produces a stark contrast to the fetal chest and other organs. In addition, fetal MRI is an excellent modality for morphologic and volumetric measurements of the fetal lung. It is especially advantageous for oligohydramnios and maternal obesity. Utilizing an image thickness between 2–4 mm, the combination of T2– and T1-weighed images can help to differentiate and outline other organs, such as liver and intestine, as well as calculate MRI TFLV. The correlation between ultrasound and MRI-based measurements has been evaluated, but with limited results. The contralateral fetal lung volume (FLV) on ultrasound appears to correlate with TFLV measured by fetal MRI, irrespective of gestational age, side of defect, or liver herniation.101 One study showed that ultrasound O/E LHR correlated well with the O/E contralateral FLV and TFLV by MRI (R2 = 0.44, P < 0.001; R2 = 0.37, P < 0.001, respectively).102 When the MRI-based O/E TFLV is less than 25%, there is a significant decrease in postnatal survival.100
Clinical Presentation
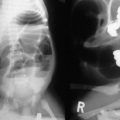
The diagnosis of CDH is typically confirmed by a chest radiograph demonstrating intestinal loops within the thorax (Fig. 24-5). The abdominal cavity may have minimal to no gas. Right-sided CDH is often more difficult to diagnosis (Fig. 24-6). Salient features, such as intestinal and gastric herniation, may not be seen. The herniated right lobe of the liver can be mistaken for a right diaphragmatic eventration. Occasionally, features of lung compression may be the only radiographic sign, which can cause confusion with CPAMs, pulmonary sequestrations, bronchopulmonary cysts, neurogenic cysts, or cystic teratomas.
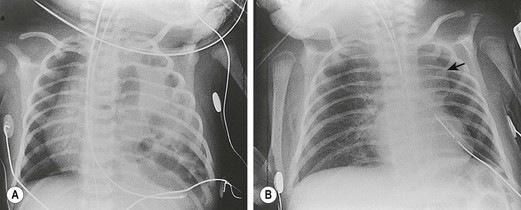
FIGURE 24-5 (A) Anteroposterior chest radiograph in a neonate with a CDH demonstrating air-filled loops of bowel within the left chest. The heart and mediastinum are shifted to the right, and the hypoplastic left lung can be seen medially. (B) Postoperative radiograph demonstrating hyperexpansion of the right lung with shift of the mediastinum to the left. The edge of the severely hypoplastic left lung is again easily visualized (arrow).
Although most infants with CDH will be diagnosed within the first 24 hours of life, as many as 20% may present outside the neonatal period.103 These patients present with milder respiratory symptoms, chronic pulmonary infections, pleural effusions, pneumonias, feeding intolerance, or gastric volvulus.104 As CDH is invariably associated with abnormal intestinal rotation and fixation, some children may present with intestinal obstruction or volvulus. Occasionally, CDH may be completely asymptomatic and is only discovered incidentally.43,105 Older patients who present later in life have a much better prognosis due to milder or absent associated complications, such as pulmonary hypoplasia and hypertension.
Treatment
Prenatal Care
The prenatal diagnosis of CDH has improved with the increased use and refinement of fetal ultrasound examinations. After initial screening, an advanced ultrasound helps to determine discordant size and dates, associated anomalies (cardiovascular and neurological) as well as signs of fetal compromise (i.e., hydrops fetalis). Once diagnosed, chromosomal screening should be performed. Optimally, the mother and fetus should be referred to a tertiary perinatal center with advanced neonatal critical care capabilities, such as ECMO, inhaled nitric oxide (iNO) therapy, and oscillating ventilators.5 A prenatal diagnosis allows the mother and family to be properly informed of treatment options and outcomes.
Prenatal Glucocorticoids
In animal models, the hypoplastic lungs of CDH infants are structurally and functionally immature. Biochemical markers for lung maturity demonstrate decreased total lung DNA, total lung protein, and desaturated phosphatidylcholine in addition to a deficiency of surfactant.61 In one study, prenatal administration of glucocorticoids demonstrated a reduction in alveolar septal thickness, increased DNA synthesis, and increased total lung protein production.106,107 Initial results from small patient series seemed promising in that antenatal administration of glucocorticoids suggested improved lung function.108,109 However, a prospective randomized trial failed to demonstrate any benefit for CDH.110 At this point, no data exist on proper dosing or timing of steroid administration in the setting of CDH. As such, prenatal steroids are not currently recommended for CDH.
Mechanical Ventilation
Mechanical ventilation is a critical component in the care of infants with respiratory failure secondary to CDH. However, the physiologic limits of the hypoplastic lung make mechanical ventilation a challenge. Hypoplastic lungs in CDH infants are characterized by a decreased number of airways and smaller airspaces. Also, the pulmonary vasculature exhibits decreased vascular branching as well as increased adventitia and medial wall thickness.111,112 This combination results in varying degrees of respiratory failure and PHTN. Fortunately, pulmonary and vascular development continues after birth and these pulmonary sequelae of CDH can improve.113,114 Because of this ongoing maturation, mechanical ventilation strategies have trended towards less aggressive approaches with the goal of maintaining oxygenation while limiting the risks of ventilator-induced lung injury, a major contributor to mortality.115
The optimal type of mechanical ventilation in infants with CDH is individual clinician preference, but most cases of CDH can be managed using a pressure-cycled mode. A fractional inspired oxygen (FiO2) of 1.0 is initially utilized to maintain adequate SaO2. Typically, higher respiratory rates and lower peak airway pressures (18–22 cmH2O) are employed while titrating the FiO2 to a preductal PaO2 greater than 60 mmHg, or the preductal SaO2 > 85% and a PaCO2 less than 60 mmHg. Maintaining an acceptable pH and PaCO2 are important in managing PHTN.116,117 The ventilation strategy of induced respiratory alkalosis with hyperventilation to reduce ductal shunting has been abandoned in most centers.118–121 Initial conventional mechanical ventilation settings should include pressure-limited ventilation rates between 40–60 breaths per minute with PIP < 25 cmH2O to minimize barotrauma.118,122 The initial PIP can be weaned to minimal settings, but should be done with caution. While targeting a PaCO2 < 45 mmHg, rapid reduction in mechanical support may allow transient periods of stability, but may also produce potential refractory exacerbations of PHTN. Spontaneous respirations are maintained by avoiding neuromuscular paralysis and minimal ventilation rates. This combination of spontaneous respiration and permissive hypercapnia has been a well-documented preoperative stabilization strategy in some centers with survival rates of almost 90% in patients with isolated CDH.118–120
If conventional ventilation fails to reverse hypercapnia and hypoxemia, HFOV strategies can be tried to avoid ventilatory-induced lung injury by preserving end-expiratory lung volume without overdistending the lung parenchyma. As the understanding of the CDH lungs continues to evolve, HFOV strategies have also changed. Initially used in a high-pressure, lung recruitment mode, this strategy demonstrated no benefit due to the nonrecruitable nature of these hypoplastic lungs.123,124 High-frequency strategies as a rescue therapy for infants with profound hypoxia and refractory hypercapnia on conventional ventilators have also not been successful.4,125 As the concept of preoperative stabilization became better defined, HFOV began to be used as means to avoid barotrauma early in the treatment course prior to refractory respiratory failure. In fact, some institutions have utilized HFOV as primary therapy.5,126,127 These strategies of preoperative stabilization to prevent lung injury along with delayed operation, regardless of ventilator modalities, have resulted in improved survival.5,115,126–128
In order to achieve lower peak airway pressures, HFOV should be considered when PIP exceeds 25 cmH2O with conventional ventilation. Initial HFOV settings include a mean airway pressure between 13–15 cmH2O, 10–12 Hz, amplitude between 30–50 cmH2O, and inspiration to expiration ratio of 50%. The initial PaCO2 should be maintained in the range of 40–55 mmHg and can be weaned by decreasing the amplitude. Eight rib expansion of the contralateral hemithorax can be used as a guide to achieve optimal lung expansion without overdistention.126,127 Tidal volumes are directly related to the amplitude and inversely related to frequency. As such, significant increases in tidal volume can be seen when frequencies are below 10 Hz. This can result in hyperinflation which, in turn, may adversely affect the pulmonary vasculature by impeding venous return. Constant assessment of acid-base status and end-organ perfusion is necessary as lung compliance can change before and after CDH repair.
Surfactant
Animal studies of CDH have demonstrated altered surfactant levels and composition.129,130 Experimentally, surfactant decreases PVR, improves pulmonary blood flow, and reduces ductal shunting.131 However, supporting data are not definite.62 Initial reports demonstrated surfactant administration augmented iNO delivery and improved gas exchange, leading to its empiric utilization.132 However, clinical evidence in term and preterm infants with CDH has not shown benefit.133 Van Meurs et al. suggested that surfactant therapy in term infants with CDH was associated with an increased use of ECMO, increased incidence of chronic lung disease, and higher mortality.134 Similar findings were found in preterm infants despite adjusting for Apgar scores and gestational age.133 Even as a rescue therapy in CDH infants on ECMO, surfactant failed to demonstrate a survival advantage.135,136 Despite the lack of proven efficacy, exogenous surfactant therapy continues to be used with unclear risks and benefits. Proponents of this therapy argue that the clinical evidence is based on a heterogeneous population of disease severity, and that the lack of clinical evidence to support its efficacy is due to its use in the most severe CDH infants. Given the current clinical data, surfactant therapy should only be used in the setting of clinical research.
Pulmonary Vasodilators
PHTN is a common consequence in CDH infants. Even if asymptomatic, the initial management in the CDH newborn is directed at reducing PVR. With increased PVR, patients often display super-systemic pressures leading to right-to-left shunting in both the pre- and postductal circulations. This can cause right heart failure with increases in preductal desaturation, narrowing of the pre- and postductal SaO2, and, ultimately, systemic hypotension and decreased perfusion. In previously stable neonates, signs of poor perfusion may indicate closure of a PDA. In response, prostaglandin E1 (PGE1) can be instituted to re-open the ductus and improve right ventricle (RV) pressures.122 In infants that demonstrate significant PHTN and RV dysfunction, left ventricular filling/function and overall perfusion may improve by off-loading the RV to improve the geometry of the interventricular septum.137 Preductal SaO2 > 85% should be maintained to ensure adequate cerebral perfusion.118 As the RV is unloaded, systolic blood pressure may decrease, especially in the setting of left ventricular dysfunction. Adequate preload should be maintained with volume loading or vasopressor support, such as epinephrine, to ensure ventricular function and coronary artery perfusion. Increasing the systemic pressure may decrease the degree of right-to-left shunting. In a small study, CDH infants with persistent PHTN and right-to-left shunting through the ductus, despite PGE1 use, had an 80% mortality.138
iNO is a potent pulmonary vasodilator that has been shown to have tremendous benefit in the treatment of persistent pulmonary hypertension of the neonate (PPHN).4,139–142 Typically, iNO is utilized in the setting of echocardiographic evidence of right-to-left shunting and RV pressures greater than two-thirds systemic pressures. In clinical studies, iNO has been shown to improve oxygenation and decrease the need for ECMO in infants with respiratory failure secondary to PPHN.142,143 However, the efficacy of iNO as a rescue therapy for PHTN secondary to CDH by either decreasing the need for ECMO or a reduction in mortality has not been supported.144 In the Neonatal Inhaled Nitric Oxide Study Group trial, the CDH subgroup had a greater likelihood for ECMO or death.145 The response to iNO is variable and unpredictable in infants with CDH. The initial improvement in oxygenation in CDH infants suggest that iNO could be utilized as a bridge for transport or until ECMO can be initiated.122 Some infants may demonstrate a rebound PHTN that is more difficult to control than the initial disease.146,147 Also, the effect appears to be transient and does not obviate the need for ECMO.77,148 A meta-analysis of iNO utilization for persistent PHTN did not show benefit in CDH infants, and recommended its use only in hypoxic respiratory failure infants without CDH.149
Phosphodiesterase (PDE) inhibitors are cyclic guanosine monophosphate (cGMP) modulators that target vascular remodeling by limiting smooth muscle cell prolifeation.150 Type 5 PDEs (dipyridamole and sildenafil) are the most active on visceral and vascular smooth muscle and have been utilized with iNO for the treatment of PHTN in patients with and without CDH. In limited case reports, dipyridamole produces a transient improvement in oxygenation which may allow weaning of ventilatory support and may avoid escalation of therapy to ECMO.151–153 Sildenafil has been shown to improve oxygenation and decrease PVR when used independently or in combination with iNO.146,154,155 Sildenafil has been used in oral or intravenous forms for congenital heart disease and CDH-associated PHTN.146,148,156,157 Similar to dipyridamole, utilization of sildenafil may have benefit in improving oxygenation and avoiding ECMO in the setting of refractory iNO.146,158 However, despite its approval for use in adults, the USA Food and Drug Administration has warned against using sildenafil for pediatric pulmonary hypertension between ages 1 and 17 years due to a potential increase in mortality during long-term therapy.159 Other vasodilators have been utilized in infants with CDH, but the experience to date is limited.160–174
Extracorporeal Membrane Oxygenation
CDH accounts for approximately one-quarter of all infants requiring ECMO for respiratory failure.175–178 ECMO utilization for CDH was first introduced as a rescue therapy for infants who had severe ventilator-associated lung injury.179 Prior to the era of preoperative stabilization, ECMO was associated with only a modest improvement in survival in high-risk CDH infants.180–182 Since then, ECMO has evolved as a treatment modality to prevent ventilator-induced barotrauma.183,184 In combination with other adjunctive treatments such as HFOV, ECMO is now routinely used for preoperative stabilization.185 The strategy of stabilization with ECMO and delay in operative correction has been shown to be beneficial with a reported survival of 67% in high-risk patients.8,186
Although results are dependent on patient selection and disease severity, survival has been reported between 60–90% for CDH patients requiring ECMO.5,118,139,175,187 Without ECMO, the predicted mortality in the high-risk cohort reaches 80%.175 Despite these data, the benefits of ECMO for CDH are not universally accepted. Some authors report no survival advantage with ECMO,188,189 while others report as high as 80% survival without using ECMO.190,191
Initially, strict criteria were established as indications for ECMO use in CDH. These included an oxygenation index >40 and persistent alveolar–arterial O2 gradient >610 mmHg.177,192 Today, those criteria have eased and the most common indication for ECMO is a ‘failure to respond’ to other therapy. In efforts to maintain lung-protective ventilation, clinicians have opted for ECMO rather than escalation of positive airway pressures. Relative contraindications include significant congenital anomalies, lethal chromosomal anomalies, intracranial hemorrhage, birth weight <2 kg, and gestational age <34 weeks. See Chapter 6 for more information about ECMO.
Surgery
Timing of Operation
With an improved understanding of its pathophysiology, repair of CDH is no longer considered an emergency procedure. However, the optimal timing for repair remains unclear. Historically, early repair was thought to improve ventilation by reducing intrathoracic pressures after reduction of the herniated viscera. However, this strategy often led to urgent procedures being performed on unstable infants.193 A paradigm shift in management to delay the operative repair until the infant is stable became widely adopted in the early 1990s.121,122,194 Several studies have shown no difference in mortality rate or the need for ECMO in infants undergoing early vs late repairs,195–197 including two randomized trials of early (<12 hours) versus delayed repair (after 24 hours197 and after 96 hours195). Today, repair of CDH is usually delayed until cardiopulmonary stability is achieved, although the definition of physiologic stability remains highly variable and inconsistent amongst centers.127,194,196–200
In 1995, Wung et al. reported advantages with a delayed repair strategy for CDH.121 Comparing three eras of treatment strategies, the study contrasted an early period of emergency surgery to the most recent era of delayed repair and ‘gentle ventilation,’ where infants with CDH were managed with a lung-preserving ventilation strategy. Repair was not performed until the pre- and postductal SpO2 gradient equalized and right-to-left shunting on echocardiogram had resolved. With an average of 4.2 days after birth before operation, survival was 94% with only one patient requiring ECMO. From the same institution in 2002, 120 consecutive patients were treated with permissive hypercapnia, spontaneous respiration, and elective repair after 36 hours of life with an overall survival of 84%.118 Of note, only 13.3% of patients needed ECMO and only 7% of infants required a prosthetic patch, suggesting a relatively small proportion of infants with large diaphragmatic defects. A different group reported similar results from a center that did not offer ECMO.201 In this study, all high-risk patients (defined as assisted ventilation within two hours of life) were divided in three historical cohorts with the most recent group being managed by permissive hypercapnia, gentle ventilation, and delayed repair until hemodynamic stability was achieved (PaCO2 < 60 mmHg, PaO2 > 40 mmHg, SaO2 > 85% with a FiO2 < 50%) for at least 48 consecutive hours. Overall survival in this recent cohort was 90%. Despite the benefits of preoperative stabilization and delayed repair, the specific parameters that define hemodynamic stability and timing of operation remain unanswered.
According to data from the Congenital Diaphragmatic Hernia Registry (CDHR) and Extracorporeal Life Support Organization (ELSO) over a recent 15 year period, one-third of infants with CDH required ECMO during their initial hospitalization, during which 85% of these infants underwent early CDH repair on ECMO.202 In this study, survival was 71% with only 9% requiring operative intervention for bleeding complications following CDH repair on ECMO compared to survival of 49% and a 14.7% bleeding complication rate from the CDH and ELSO registries. The authors attributed the better outcomes after early repair on ECMO to less body wall edema and a quicker recovery following operation which led to fewer total days on ECMO (11.7 vs 13.3 days). In another study, among infants who underwent repair of a unilateral CDH, 47.6% were repaired on ECMO, 45.8% were repaired after ECMO, and only 6.6% were repaired before ECMO.203
Bleeding remains the most significant complication following CDH repair on ECMO including both surgical site bleeding and intracranial hemorrhage. These risks may be minimized with early repair on ECMO prior to the development of a coagulopathy and significant edema,177,202 as well as repair when PHTN has resolved, but prior to decannulation to allow reinstitution of ECMO if respiratory failure and/or PHTN recur.204 Although rare, recurrent PHTN can develop after repair requiring a second ECMO run.205 In addition, use of aminocaproic acid (Amicar®), a fibrinolysis inhibitor, has significantly decreased bleeding complications. One group reported no changes in intracranial hemorrhage rates, but a significant decrease in surgical site bleeding from 12% to 7% (P < 0.05) with Amicar®.176Amicar® should be used prior to operation and for two to three days after repair. Additional strategies include ECMO without heparinization, fresh ECMO circuits, minimal diaphragmatic muscular dissection, fibrin or thrombin sealants, and utilization of recombinant factor VII for established bleeding.
Operative Approach
Open repair of CDH can be performed using a thoracic or abdominal approach. Advantages to laparotomy include easier reduction of intrathoracic viscera, the ability to mobilize the posterior rim of diaphragm, easier management of intestinal rotational anomalies, and avoidance of thoracotomy-associated musculoskeletal sequelae. The vast majority of neonatal repairs for CDH are through a subcostal incision (91%).206,207 Less than 10% are performed via a thoracotomy. The intra-abdominal contents should be reduced out of the thorax with careful attention to the spleen that can be caught and lacerated on the rudimentary rim of diaphragm. A true hernia sac, which is present less than 20% of the time, should be excised.208 The thoracic and abdominal cavities should be inspected for an associated pulmonary sequestration.
Despite this ‘gold standard’ abdominal approach, the morbidity and respiratory sequelae of open CDH repair remain a concern. In addition to pulmonary hypoplasia and hypertension, respiratory compliance is significantly reduced after open repair. Mortality significantly increases when compliance decreases by >50% which can occur as a result of a tight abdominal wall closure.193,209 Careful attention should be paid to the peak airway pressures as the abdominal fascia is closed. Respiratory compromise should alert the surgeon to leave the abdomen open. This approach is more often needed in CDH infants on ECMO.210 Temporary closure can be achieved using just the skin or a prosthetic silo. Delayed closure, especially in those infants on ECMO, should be attempted after the generalized edema has resolved or the intra-abdominal domain has enlarged.211
The routine use of chest tubes after CDH repair to drain pleural fluid has been abandoned.118,121,201 One concern is that the chest tube can cause ipsi- and contralateral lung injury secondary to mediastinal shift, especially if connected to suction. The thoracic space will eventually fill with fluid, and the lung will gradually grow. Tube thoracostomy should only be used for postoperative chylothorax or pleural fluid causing hemodynamic compromise.212 If a chest tube is needed, it is positioned in the thoracic cavity prior to final closure of the diaphragm. Chest tubes should be placed to water seal rather than suction. Symptomatic pleural fluid can be treated with repeated thoracentesis. If used, chest tubes should be removed early to avoid infectious complications.
Minimally Invasive Techniques
The respiratory sequelae and other morbidity seen after open CDH repair has prompted surgeons to adopt minimally invasive surgical (MIS) approaches. Both thoracoscopic and laparoscopic repairs have been performed.207,213–216 Data from the CDHR show that laparoscopic and thoracoscopic strategies are being used worldwide, and have been utilized in 20% of centers since 1995.207 MIS techniques have been used for primary repair as well as prosthetic patch closure with suggested advantages of less postoperative pain, avoidance of thoracotomy-associated complications, and an overall reduction of surgical stress.216,217
The sensitivity of CDH infants to hypercapnia and acidosis has drawn concerns regarding the utilization of MIS. The overall benefits of MIS are questioned because: (1) CDH neonates may absorb the CO2 insufflation;218,219 and (2) insufflation with CO2 can raise intracavity pressures that may limit venous return, end-organ perfusion, and tidal volume. The combination of CDH-related pulmonary hypoplasia, PHTN, and labile pulmonary vascular reactivity may be detrimental during MIS operations. Although increases in CO2 absorption during MIS are generally well tolerated in infants, CDH neonates specifically demonstrate greater changes in end-tidal CO2 (ETCO2) and impaired elimination of CO2 during thoracoscopy and laparoscopy.220,221 Hypercapnia and the associated acidosis may result in increased pulmonary shunting.
Patient selection is paramount for successful completion of an MIS repair as well as for minimizing operative morbidity. Historically, MIS was reserved for stable infants with anticipated small defects. Utilizing anatomic markers such as stomach herniation, surgeons have attempted to predict which defects might be amenable to MIS repairs.215 Initially, the radiographic presence of the nasogastric tube within the abdomen and minimal respiratory compromise (PIP < 24 mmHg) were thought to predict a successful thoracoscopic repair. One group reported 95% success rates with thoracoscopic repairs when patients did not have a significant congenital cardiac anomaly or the need for preoperative ECMO, and had a PIP ≤ 26 cmH2O, and an oxygenation index ≤5 on the day of surgery.222 Another group has advocated for strict preoperative selection criteria for thoracoscopic repair, including minimal ventilatory support (PIP < 24 mmHg), no clinical or sonographic evidence of PHTN, and an intra-abdominal stomach.215 On the other hand, infants requiring preoperative ECMO have undergone successful repair with an MIS approach.218,223 Large defects that require patch repairs218,222,224 and right-sided defects are no longer contraindications to MIS.225 However, patients undergoing MIS repair of CDH should have stringent intraoperative monitoring of ETCO2 and PaCO2.226
Although success with laparoscopic216,227 and thoracoscopic213,224 repairs have been reported, comparative evidence between MIS and open approaches has been limited to single-institution experiences or retrospective analyses.213,228,229 A recent single-institution review of 54 neonates undergoing unilateral CDH repair between 2006–2010 was published.228 Thirty-five neonates underwent attempted thoracoscopic repair with 26 being successfully completed. During the same time interval, 19 open CDH repairs were performed. Recurrence was higher after the thoracoscopic repair (23% vs 0%). There were no individual factors that were found to be predictive of recurrence with the thoracoscopic approach. In another report, a systematic review and meta-analysis of neonatal endosurgical CDH repairs identified only three eligible studies comparing open to endosurgical repair.230 The cumulative risk ratio for death was 0.33 (95% CI:0.10–1.13) in favor of the MIS approach and recurrence was 3.21 (95% CI:1.11–9.29) in favor of the open repair. The overall survival rate for MIS patients was significantly higher compared with patients undergoing open repair (82.9% open and 98.7% MIS, P < 0.01) with a risk-adjusted odds ratio (OR) of survival for MIS repairs of 5.57 (95%CI:1.34–23.14).222 These results suggest a significant survival advantage for the MIS approach, even after risk-adjustment. However, the data are more likely the result of selection bias based on surgeon preference regarding which patients were good candidates for MIS.
Although the ability to perform MIS repair of CDH has been shown, the short- and long-term outcomes regarding the durability and recurrence rates for an MIS approach are less clear. The reported overall recurrence rates for MIS repair range from 5–23.1%,218,222,224,225 with early recurrences as high as 23–33%.228,229 In another study, MIS repairs were performed in only 3.4% infants with CDH, but had a significantly higher in-hospital recurrence rate compared to open (7.9% vs 2.7%, P < 0.05).207 The true risks and benefits of the MIS approach for CDH repair, including the impact of re-operations, remain unclear. At the very least, recurrence seems to be higher.
Robotic CDH repair has been demonstrated to be feasible and safe.231–233 Proponents of robotic CDH repair tout the increased degrees of freedom of the articulating instruments for suturing.
Diaphragmatic Replacements
Repair of large diaphragmatic defects is a challenge, usually requiring diaphragmatic replacement with a prosthetic patch or autologous tissue. In one large study, 48.3% of infants undergoing CDH repair required diaphragmatic replacement.207 Comparative studies between patch and primary repairs have consistently shown increased morbidity and mortality in the patch groups, most likely due to the large defect size and the associated severity of the pulmonary hypoplasia.234,235 In many studies, patch repair has been utilized as a surrogate for defect size and disease severity (i.e., larger the defect = increased severity of disease).
Nonabsorbable Synthetic Patches
Synthetic patches such as polytetrafluoroethylene (PTFE or Gore-Tex®) or composite polypropylene (Marlex®) represent the majority of the mesh replacements used in neonates with a large CDH.236 Advantages to synthetic patches include: (1) immediate availability; (2) minimal preparation time; (3) easily cut to fit the diaphragmatic defect; and (4) less tissue dissection, reducing the risk of hemorrhage, especially during repair on ECMO. However, there are several disadvantages to synthetic patches for CDH repair. PTFE, anchored to the chest wall, can potentially produce a tethering point for creating a pectus-type deformity.237 There is an increased incidence of bowel obstruction, need for splenectomy, patch infections, and abdominal wall deformities.236,238 The overall recurrence rate has been reported to be as high as 50% with a bimodal distribution showing early recurrence in the first months after repair and late recurrence years later.236 Early recurrences for defects requiring a patch are most likely due to lack of tissue adhesion or scarring and an incomplete muscular rim which then requires anchoring the patch to the ribs or esophagus. PTFE tends to scar and retract over time, which may lead to late recurrences in the growing child. In an effort to prevent CDH recurrence, one group described a cone-shaped, double-fixed PTFE patch to allow expansion over time.239 The recurrence rate decreased from 46% to 9% at one year after repair. Similar results have been seen with a mesh plug and patch technique in the setting of a recurrent CDH.240 Another group described a double-sided composite patch consisting of PTFE and type-1 monofilament, macroporous Marlex. Utilizing a pledgeted, nonabsorbable running suture, the recurrence rate was 2.2% with a mean follow-up of 49 months.241
Absorbable Biosynthetic Patches
Absorbable biosynthetic materials have been utilized as an alternative replacement to synthetic patches. They have been reported to decrease complications by offering a lower risk of infection and the ability of the patch to grow with the patient. Surgisis® (SIS) is an acellular, bioengineered porcine intestinal submucosal matrix that consists of a type I collagen lattice with embedded growth factors. This non-crosslinked biological matrix promotes fibroblast migration and cellular differentiation.242 First described for repair of incisional, inguinal, and paraesophageal hernias,243–245 SIS® has also been utilized for CDH repair.216,246 Permacol® is an acellular porcine dermal collagen patch consisting of collagen fibers with cross-linked lysine and hydroxylysine. By promoting an inflammatory response in a manner similar to wound healing, the neodiaphragm is more pliable and, subsequently, less prone to recurrence. One group reported no recurrences observed with a median follow-up of 20 months, while recurrences were noted in 2% of patients with primary repair and 28% with PTFE.247 AlloDerm® is an acellular human cadaveric dermal patch that is cross-linked for rapid revascularization (Fig. 24-7). Animal studies have demonstrated revascularization and cell repopulation within one month.248 Surgimend® is an acellular fetal bovine interwoven dermal collagen that promotes increased type III collagen. Because there is no cross-linking, there is increased collagen resistance leading to greater durability. Poly-lactic-co-glycolic acid (PLGA) is a collagen scaffold that promotes neovascularization and autologous tissue regeneration. Animal studies have demonstrated ingrowth of fibroblasts resulting in a thicker neodiaphragm.249

FIGURE 24-7 A large left posterolateral diaphragmatic hernia was approached through a subcostal incision. After reduction of the abdominal viscera, the large defect was closed using a biosynthetic patch (Alloderm®).
Despite the theoretical advantages, these absorbable biosynthetic patches remain imperfect as diaphragmatic substitutes. Thinning of the patch and incomplete muscular ingrowth, especially in large defects where native diaphragmatic muscle is absent, have been found.250 These biosynthetic patches are also prone to recurrences, similar to nonabsorbable patches.251 In addition, organ adherence may be required for neovascularization, and these organs often include the small bowel, spleen, or liver.238,252 Subsequently, biologic patches can be associated with adhesive bowel obstruction.234,238,251
Autologous Tissue Patches
Complications with synthetic and biosynthetic patches have prompted some surgeons to advocate for primary or staged repair with autologous muscle flaps for large diaphragmatic defects.253–258 Muscle flaps offer the advantage of using a vascularized tissue that will grow with the infant and has a minimal inflammatory response. In 1962, Meeker and Snyder first described using the anterior abdominal wall for CDH repair.259 A few years later came the description of a split abdominal wall muscle flap used to repair a large defect in a newborn.253 More recently, another group described using a split abdominal muscle flap consisting of the internal oblique and transversus abdominis muscles for primary repair of large defects.254 Another approach is to use a lower abdominal incision and the transversus abdominis muscle for repair on ECMO.255 Due to the avascular dissection plane between the muscle layers, this approach may minimize the risk of bleeding while on ECMO. In a recent report, the recurrence rate for split abdominal wall muscle flaps was only 4.3%, while the recurrence rate for patch repair was 50%.256
Chest wall muscles, such as the latissimus dorsi muscle, have also been used as diaphragmatic substitutes.257 For very large defects, such as agenesis of the diaphragm, the combination of the latissimus dorsi and serratus anterior muscles has been described.258,260 The disadvantage with using local chest wall muscle flaps is the resulting body wall deformity.261 Consequently, chest wall muscle flaps have been primarily reserved for patients with a recurrent CDH. Although autologous muscle flaps are vascularized and tend to grow with the child, these diaphragmatic reconstructions with latissimus dorsi/serratus muscle flaps have been shown to atrophy over time due to denervation of the graft.260 In addition, the lack of innervation prevents the natural physiologic movement of the diaphragm. As a result, the reverse latissimus dorsi flap with a microneural anastomosis of the phrenic nerve to the thoracodorsal nerve has been tried to prevent muscle atrophy and to allow physiological muscle movement.260,262
Tissue Engineered Patches
The ideal diaphragmatic replacement remains elusive in the operative treatment of CDH. Advances in regenerative medicine may provide the solution. Tissue engineered muscle may provide a replacement for functional skeletal muscle and does not atrophy. Although the supporting three-dimensional scaffold is a key component of tissue engineering, skeletal muscle regeneration relies on a cell source with myogenic potential.263,264 Amniotic fluid is an abundant source of stem cells with myogenic potential.265 Tissue engineering strategies could utilize amniotic stem cells collected at the time of amniocentesis to develop a muscular patch used during postnatal repair. Fauza and colleagues have developed a fetal tissue-based diaphragm engineered from mesenchymal amniocytes.266 In preclinical studies, these bioengineered diaphragms demonstrated improved mechanical and functional outcomes when compared to acellular bioprosthetic patches.267
Fetal Therapy
The impetus for fetal therapy for CDH coincided with advances in prenatal diagnosis. Fetal ultrasound has allowed a better understanding of the true natural history of CDH. In the late 1980s, open fetal repair for left-sided CDH without liver and stomach herniation before 24 weeks gestation was conceptualized and performed.86 In a more recent prospective study, fetuses undergoing in utero repair had a higher complication rate, including prematurity, with an average gestation of 32 weeks without an improvement in survival.268 Surprisingly, better than expected survival was found in the standard postnatal management patients and there were higher rates of complications in the fetal surgery group. Consequently, open fetal surgery has been abandoned as a therapeutic option for CDH.
The concept of tracheal occlusion originated from the observation that infants with congenital high airway obstructions developed hyperplastic lungs. Several groups have demonstrated increases in total lung protein and DNA, alveolar space, overall lung weight, and cross-sectional area of the pulmonary vasculature as well as better lung compliance following prenatally placed tracheal balloons in sheep.269–272 However, it has been noted that long-term tracheal occlusion decreases the number of type 2 pneumocytes and surfactant production. This finding has led to the concept of temporary in utero tracheal occlusion for CDH.131,273
It is known that liver herniation and LHR < 1.0 are fetal parameters that portend the worst outcomes.274 Current in utero techniques involve endoscopic insertion of an occlusive balloon in the fetal trachea without maternal laparotomy or general anesthesia.275 These balloons are inserted percutaneously with ultrasound guidance between 24–28 weeks’ gestations and are deflated at 34 weeks. This strategy of temporary tracheal occlusion avoids the need for an ex-utero intrapartum treatment (EXIT) procedure at delivery, although emergent airway access may be needed at delivery for any patient who undergoes in utero tracheal occlusion.
In 2003, an NIH-sponsored randomized trial comparing fetal tracheal occlusion versus standard postnatal care was reported.276 Criteria for randomization was LHR < 1.4 and liver in the chest. After 24 cases (11 with tracheal occlusion), the study was terminated early due to comparable survival outcomes (77% by postnatal care, 73% by tracheal occlusion). The hazard ratio (HR) for mortality associated with tracheal occlusion, as compared to conventional postnatal therapy, was 1.2 (95% CI:0.29–4.67). However, when stratified based on LHR, survival was significantly better for LHR greater than 0.9 with a HR for death with tracheal occlusion being 0.13 (95% CI:0.03–0.64).
In 2004, the European Fetal Endoscopic Tracheal Occlusion (FETO) task group reported on 21 fetuses undergoing tracheal occlusion.277 This nonrandomized study of fetuses with a severe CDH (liver up and LHR < 1) showed improved survival (64%) in the last 11 patients compared to the first 10 (30%). In 2009, the FETO group reported their results in 210 patients utilizing a minimal access approach under regional and local anesthesia.278 This FETO study also included fetuses with liver herniation and an O/E LHR < 1.0. The median time to insert the balloon was 10 minutes. Successful balloon placement was achieved in 97% at the first procedure. Ninety-seven per cent of the babies were live born and the overall survival until discharge was 48%. The mean gestational age at birth was 35.3 weeks. When these outcomes were compared to the CDH registry, fetuses with a left-sided CDH, liver up, and an O/E LHR < 1, who were treated with fetal tracheal occlusion, had a survival of 49.1% vs 24.1% in the registry. Survival increased from 0% in the registry to 35% in similar fetuses with a right CDH who underwent fetal tracheal occlusion.278
In 2012, Ruano et al. reported on a randomized controlled trial between FETO and conventional postnatal care for isolated, severe CDH (LHR < 1.0 and liver up) between 22 and 26 weeks.279 With an intention-to-treat analysis, overall survival was 50% in the fetal tracheal occlusion group compared to 4.8% in controls (RR 10.5;95%CI:1.5–74.7). The mean gestational age at delivery was 35.6 ± 2.4 weeks in the treated group and 37.4 ± 1.9 weeks in controls (P < 0.01). Despite these positive results, one limitation of this study was that it did not utilize some of the currently accepted prenatal and postnatal treatment strategies. Tracheal balloons were removed at the time of delivery through an EXIT procedure, thus discarding the temporary tracheal occlusion concept. In addition, ECMO was not available in the study institution for postnatal care in both arms.
Outcomes
Clinical outcomes for the treatment and management of infants with CDH have traditionally been difficult to generalize due to the low incidence of disease and single institutions reports. Patient characteristics and clinical practices are different, and each institution’s ability to offer state-of-the-art neonatal critical care is highly variable. Survival analyses for CDH remain difficult to interpret due to variations in patient disease, management strategies, and operative techniques. Unique to individual institutions are differences in ventilation strategies, availability and entry criteria for ECMO, and operative timing. As a result, survival rates for CDH vary between institutions, even when risk adjusted, and range from 25–83%.6 In an attempt to ameliorate these difference, the CDH Study Group (CDHSG) was the first to publish risk-stratified outcomes.280 Multivariate analysis demonstrated birth weight and 5-minute Apgar scores to be the most significant predictors of outcome.
Volume-Based Outcomes for CDH
Outcomes for CDH appear better in centers that treat a higher volume of CDH infants. The Canadian Pediatric Surgery Network (CAPSNet) grouped centers into low (<12 cases/year) and high (≥12 cases/year) volume.281 Low volume centers had a higher mortality (23% vs 10%). In a recent follow-up report, CAPSNet found that significant differences between centers was found at six cases per year.282 After risk-adjustment, low-volume centers demonstrated 33% mortality compared to 15% in high-volume centers. Recent data from the Pediatric Health Information System (PHIS) administrative database showed the average yearly hospital CDH volume varies from 1.4 to 17.5 cases per year among the 42 institutions.283 After being grouped into low (≤6 cases/year), medium (6–10 cases), and high volume (>10 cases) centers, medium and high volume centers were found to have a significant lower mortality compared to low volume hospitals.
Risk Stratification for CDH
Congenital heart defects occur in 10–35% of CDH infants.33,284,285 Categorized into major (hemodynamically significant) and minor lesions (such asymptomatic ASD, VSD, or PDA), in one study, the overall survival was 73% without any cardiac anomalies, 67% with minor defects, and 36% with major anomalies.33 When compared to infants without structural cardiac anomalies, the adjusted risk in-hospital mortality was 2.2 times higher in infants with major heart defects while there was no statistical difference among infants with minor lesions.
Defined as delivery <38 weeks gestation, preterm birth occurs in approximately 30% of infants with CDH.286,287 The overall survival is approximately 53% for all preterm CDH infants.286 However, mortality significantly increases with younger gestational age. In a recent study, infants born less than 28 weeks gestational age were found to have a survival rate of 31.6%, while survival increased to 73.1% for those born at 37 weeks.286 After adjusting for comorbidities and disease severity, prematurity had an increased OR of 1.68 for death.
Risk stratification by defect size appears to correlate with disease severity. In two reviews, the overall mortality for patients with agenesis of the diaphragm was 43% with an OR of 14.07 compared to defects that could be repaired primarily.288,289 The association between defect size and disease severity has prompted development of a universal grading system to define CDH defect size.1 Based on intraoperative findings, the four classifications range from small defects which could be repaired primarily to total diaphragmatic agenesis (Fig. 24-8). Utilizing other variables of comorbidity and disease severity, the CDHSG is attempting to provide an evidence-based risk-stratification classification for CDH.
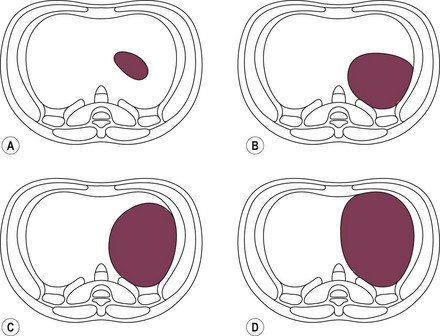
FIGURE 24-8 Classification of CDH based on intraoperative defect size.1 Diagrams are drawn with the diaphragmatic defect on the patient’s left from an abdominal perspective.
Follow-Up Guidelines
Although many survive to discharge, the sequelae and ongoing health needs of CDH infants that require medical care after discharge is significant. Despite many single institution reports, the standard follow-up care for infants with CDH has not been established.290,291 Pulmonary, neurological, gastrointestinal, and musculoskeletal complications necessitate a multidisciplinary team of surgical, medical, and developmental specialists. In 2008, the Section on Surgery and Committee on Fetus and Newborn for the American Academy of Pediatrics established follow-up guidelines for the care of infants with CDH.292 The recommendations begin before discharge and extend through age 16 years (Table 24-1).
TABLE 24-1
Recommended Schedule of Follow-Up for Infants with CDH
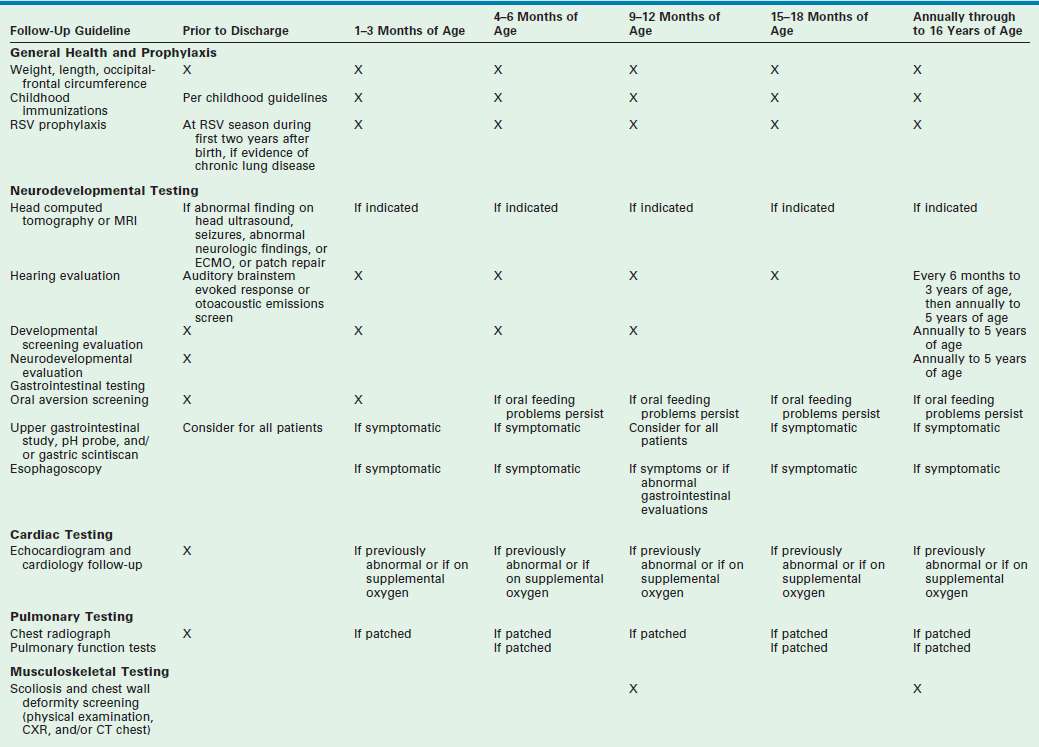
Adapted from Lally KP, Engle W. Post-discharge follow-up of infants with CDH. Pediatrics 2008;121:627–32.
Pulmonary Outcomes
Prior to the era of ECMO and lung-protective ventilation, long-term survivors were usually reported as healthy children without any respiratory disease.59,293 However, with improved care, more severely affected CDH infants are surviving and exhibiting functional and radiographic evidence of chronic lung disease.291,294 Pulmonary function has been reported to be normal in 50–70% of CDH survivors with the remainder exhibiting some form of restrictive or obstructive respiratory symptoms.295,296 Approximately 25% of children beyond the age of 5 have signs of obstructive airway disease. In a review of 100 consecutive CDH survivors, disease severity (defined as the need for ECMO and patch repair) was found to be predictive of pulmonary outcome.295 While only 16% of the infants required oxygen at discharge, 53% required at least transient usage of bronchodilators within the first year. Similarly, 41% were either dependent on or intermittently needing steroids during their first year.
Respiratory infections appear to have a higher prevalence in children with CDH.295,297,298 Respiratory syncytial virus (RSV) is the most common pathogen seen in children less than 3 years of age, suggesting a need for RSV prophylaxis.295 Recurrent pneumonias have been reported in 26–39% of CDH survivors with at least 10 years of follow-up.114,297
Obstructive pulmonary disease is commonly reported in surviving children with CDH. Asthma and general symptoms of bronchospasm and wheezing are well documented.183,298,299 Although symptoms appear to improve with age, most CDH children will exhibit some combination of obstructive and restrictive pulmonary function as well as increased reactivity to pharmacological agents.298,299 This reduced pulmonary function is most likely due to lower functional volumes rather than primary obstruction of the airways.299
Although lung development begins early in gestation, alveolar maturation continues after birth. In CDH, lung hypoplasia affects both lungs with a reduction in airway and pulmonary artery branching.49,300 Based on ventilation/perfusion (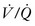 ) radionuclide scans, pulmonary hypoplasia will persist despite continued lung growth.299,301 These
) radionuclide scans, pulmonary hypoplasia will persist despite continued lung growth.299,301 These  mismatches are due to abnormal lung perfusion. Although increases in lung volume continues during early childhood, there is not an associated increase in lung perfusion. In a review of 137 CDH infants, 61% had
mismatches are due to abnormal lung perfusion. Although increases in lung volume continues during early childhood, there is not an associated increase in lung perfusion. In a review of 137 CDH infants, 61% had 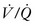 mismatching with the strongest correlation in children that underwent patch repair and ECMO. 302 Even though
mismatching with the strongest correlation in children that underwent patch repair and ECMO. 302 Even though 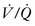 scans in CDH children have provided valuable information, its predictive value for chronic lung disease in CDH survivors remains to be determined.
scans in CDH children have provided valuable information, its predictive value for chronic lung disease in CDH survivors remains to be determined.
Neurological Outcomes
Although most children with CDH may resolve their pulmonary or gastrointestinal conditions, residual neurologic morbidities may be harder to detect. Complications associated with the disease and initial treatment, such as seizure disorders, cerebral palsy, and hemorrhagic or ischemic strokes, occur with a very low prevelance.303,304 However, neurodevelopmental conditions, such as learning disabilities, fine and gross motor skill deficits, behavior problems, and visual-motor coordination may be more common than initially prevalence.290,305,306
Most longitudinal data have been limited to early development (<3 years of age). Overall, only 54–58% of CDH children demonstrate normal neurodevelopment with 23–25% demonstrating suspected delays, and 17–23% showing marked delays.305,307 Utilizing the Bayley Scales of Infant Development (BSID)-II, some neuromotor delay was seen in 77% of infants with a 19 month median follow-up.305 The severity of neurological problems seem to be related to the severity of disease with mild to moderate developmental delay in 37% of CDH survivors, including 72% that had undergone ECMO.291 The largest series of CDH survivors (41 patients) demonstrated mild and severely delayed cognitive and language skills in 17% and 15%, respectively.306 With a median age at 24 months, psychomotor skills were normal, mildly delayed, and severely delayed in 46%, 23%, and 31%, respectively.
Neurodevelopment outcomes beyond 3 years of age are limited. Among preshool children born with CDH, one study showed normal, mildly delayed, and severely delayed neuromotor testing in 58%, 29%, and 13%, respectively.308 In a prospective cohort of ECMO survivors, only 38% of preschool children scored in the normal range.309 Thirty-four per cent demonstrated mild motor delays only, 6% showed mild delay in motor, cognitive, and/or behavior development, while 16% were severely delayed in at least two of the three domains.
Neurological delays may continue beyond preshool age. An age-matched controlled study that compared CDH and normal children for neurocognitive, visual, and fine motor outcomes found significant differences.310 With an average age of 13 years, the mean score for full IQ, verbal IQ, and performance IQ were similar between groups. However, 13% of CDH children were significantly delayed, with 15% scoring below average for Full-IQ and Verbal-IQ while 23% were delayed for performance-IQ. In addition, the CDH cohort had a significantly higher proportion with oral-motor programming and visual-motor deficits compared to normal controls. Furthermore, approximately 45% of CDH children had below average IQ scores at 10 years of age.311
ECMO use, disease severity with prolonged hypoxia, and prolonged hospitalizations have all been implicated in the poor neurologic outcomes. In a study of 82 CDH children that required ECMO, developmental delay at 1 year post-repair was significantly more prevalent compared to children needing ECMO for other indications.303 Abnormal neuroimaging in CDH children with neurodevelopmental delays that underwent ECMO has also been found.312 However, ECMO has not been a significant risk factor for neurological deficits in other studies.313,314 Certainly, other comorbidities, such as low socio-economic status and prematurity, may contribute to the poor neurological outcomes as well.315–317
Hearing impairment is a recognized sequelae of CDH with an overall incidence exceeding those of other neonatal intensive care infants (2%) and the general population (0.1–0.2%).183,318 The prevalence of sensorineural hearing loss (SNHL) ranges from none in some series to 100% in others.304,318 SNHL has been detected in as many as 50% of CDH children that initially tested as normal.318 The etiology of SNHL is unclear. One study suggested a relationship between SNHL and ECMO and hyperventilation.291 While several studies have suggested a causal relationship between ECMO and SNHL,183,318,319 others have suggested that SNHL occurs regardless of ECMO use.317,320 Several ototoxic medications, including diuretics, antibiotics, and muscle relaxants, are routinely used in the acute and chronic care of infants with CDH.319 Another study suggested that severe hypoxia and acidosis may be linked to SNHL.317 Most likely, SNHL is due to a combination of treatments and severity of disease. Because of the increased risk of SNHL in CDH children, audiological testing is warranted as early as 6 months of age.292
Gastrointestinal Outcomes
Gastroesophageal reflux disease (GERD) occurs in two-thirds of all CDH survivors with approximately half requiring fundoplication.320–322 Long-term symptoms can persist with 63% of patients requiring GERD medications after 2 years in one study.323 The etiology of CDH-association GERD is unclear. Certainly, anatomical changes following CDH repair can contribute to GERD. Esophageal ectasia may occur due to the mediastinal shift which results in an abnormal lower esophageal sphincter.324 An intrathoracic stomach due to herniation can cause loss of the angle of His.325,326 The CDH may also cause a differential growth pattern and a shortening of the intra-abdominal esophagus.327 Distortion of the esophageal hiatus and increased intra-abdominal pressure from reducing the viscera have also been implicated as post-surgical causes of GERD.314 Due to the frequency of symptomatic GERD, fundoplication has been recommended at the time of CDH repair by some surgeons, especially in patients with large diaphragmatic defects, but without proven benefit.304
Musculoskeletal Outcomes
Musculoskeletal development and chest wall deformities, such as pectus deformities, chest asymmetry, and scoliosis, are common in CDH children with an estimated incidence between 21–48%.291,296 Scoliosis can be severe and can progress until adulthood. These musculoskeletal abnormalities may be due to tension on the diaphragm after repair, or result from a thoracotomy without muscle sparing techniques, or result from a small hemithorax due to hypoplastic lungs.11,296 Most patients are asymptomatic and do not require operative intervention.
Anterior Hernias of Morgagni
An anterior diaphragmatic Morgagni hernia accounts for less than 2% of all CDH. The foramen of Morgagni hernia results from failure of fusion of the crural and sternal portions of the diaphragm. This can occur on either side at the junction of the septum transversum and thoracic wall where the superior epigastric artery (internal mammary artery, intrathoracically) traverses the diaphragm. These are usually large anterior midline defects. Typically, a hernia sac is present containing omentum, small intestine, and/or colon. Rarely these hernias will contain liver and/or spleen. The majority of children with a Morgagni hernia are asymptomatic and are rarely diagnosed during the neonatal period. Symptoms typically include general epigastric discomfort or sometimes vomiting and coughing due to intermittent obstruction.328,329 An acute presentation may be due to intestinal ischemia with necrosis and perforation as well as gastric volvulus. Herniation into the pericardium causing tamponade has also been described.330
The chest radiograph may exhibit a well-defined air-fluid level in the midline of the chest and visceral herniation in the retrosternal space (Fig. 24-9). Small hernias may require a contrast radiograph or CT scan to confirm the diagnosis. Operative repair usually entails reduction of the herniated viscera, excision of the hernia sac, and approximation of the diaphragm to the posterior rectus sheath at the costal margin. These repairs can be performed laparoscopically (Fig. 24-10).331,332 Although most defects can be repaired primarily, large defects may require patch closure.213,216 The long-term outcome regarding recurrence is yet to be defined.
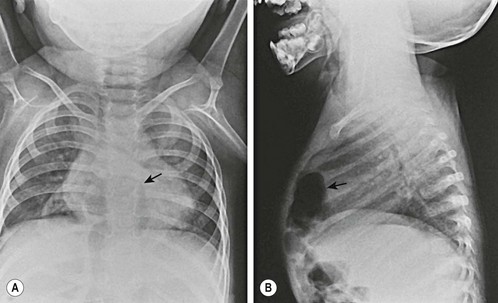
FIGURE 24-9 (A) Chest radiograph in a neonate with a retrosternal hernia. Anteroposterior film demonstrates air-filled loops of bowel above the diaphragm and posterior to the sternum (arrow). (B) Lateral projection confirms the retrosternal position of the herniated viscera (arrow).
FIGURE 24-10 Laparoscopic view of a Morgagni hernia. This anterior hernia (white arrow) is to the left of the falciform ligament (asterisk) with a deep sac that was excised. The large bowel (black arrow) was reduced from the hernia defect, and the defect was closed with a biosynthetic patch. (From Holcomb GW III, Ostlie DJ, Miller KA. Laparoscopic patch repair of diaphragmatic hernias with Surgisis. J Pediatr Surg 2005;40:e1–5.)
An anterior diaphragmatic hernia may be found in association with a pentalogy of Cantrell due to a failure in the development of the septum transversum.333 Pentalogy of Cantrell is a rare cluster of congenital anomalies which includes omphalocele, cardiac defects, ectopic cordis, and an anterior diaphragmatic defect extending into the pericardium. The cardiac defect is the most severe problem and is the main cause of mortality.
Diaphragmatic Eventration
Eventration is an abnormal elevation of the diaphragm which results in a paradoxical motion during respiration and interferes with normal pulmonary mechanics and function.334,335 Congenital eventration results from the incomplete development of the central tendon or muscular portion of the diaphragm. While commonly left-sided, bilateral congenital eventrations have been described.336 The diaphragm muscle is typically present, but does not move in a coordinated fashion. It is usually thin, and may be indistinguishable from a hernia sac seen in CDH. Large eventrations can interfere with lung development due to the paradoxical motion and decreased thoracic space. Similar to CDH, congenital eventration can result in lung hypoplasia, although this is uncommon. Persistent fetal circulation and PHTN are usually not seen with eventration. Acquired diaphragmatic eventration can occur due to paralysis of the phrenic nerve secondary to mediastinal tumors, congenital heart surgery, or birth trauma. Its incidence after congenital cardiac surgery has been reported to be approximately 5% with the highest proportion after Fontan and Blalock–Taussig shunt procedures.337,338
Eventration is typically suspected on a plain chest film showing an elevated hemidiaphragm (Fig. 24-11). The diagnosis is subsequently confirmed by ultrasound or fluoroscopy. Motion studies demonstrate a paradoxical movement of the diaphragm and a mediastinal shift during respiration.339 Occasionally, a CT scan is required to distinguish eventration from pleural effusions, mediastinal tumors, bronchogenic cysts, or pulmonary sequestrations.
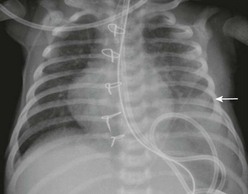
FIGURE 24-11 Anteroposterior chest radiograph of a 4-week-old male neonate demonstrates a left-sided diaphragmatic eventration (arrow) after repair of total anomalous pulmonary–venous return. The abdominal viscera remain beneath an intact left hemidiaphragm.
Small eventrations may be observed as the child will eventually overcome and compensate for the abnormal diaphragmatic dynamics. When needed, initial treatment should include respiratory support, but mechanical ventilation is usually not necessary. However, larger defects that cause functional pulmonary impairment or promote recurring infections require repair. Neonates with large eventrations benefit from early repair rather than a course of conservative treatment.339,340 Acquired eventrations often require repair in order to be weaned from mechanical ventilation.
Repair can be accomplished through the chest or abdomen. The eventrated diaphragm is plicated with a series of nonabsorbable sutures. Sutures should imbricate generous amounts of the diaphragmatic tissue without injuring the phrenic nerve. The edges of the diaphragm should overlap until the plicated muscle is taut. Subsequently, the diaphragm becomes immobilized which results in an increased tidal volume and prevents mediastinal shift. Minimally invasive techniques have been well described using thoracoscopy or laparoscopy.341–343
References
1. Tsao, K, Lally, KP, The Congenital Diaphragmatic Hernia Study Group. A voluntary international registry. Semin Pediatr Surg. 2008; 17:90–97.
2. Moyer, V, Moya, F, Tibboel, R, et al. Late versus early surgical correction for congenital diaphragmatic hernia in newborn infants. Cochrane Database Syst Rev (Online). 2002.
3. Wilson, JM, Lund, DP, Lillehei, CW, et al. Congenital diaphragmatic hernia–a tale of two cities: the Boston experience. J Pediatr Surg. 1997; 32:401–405.
4. Bohn, DJ, Pearl, R, Irish, MS, et al. Postnatal management of congenital diaphragmatic hernia. Clin Perinatol. 1996; 23:843–872.
5. Frenckner, B, Ehren, H, Granholm, T, et al. Improved results in patients who have congenital diaphragmatic hernia using preoperative stabilization, extracorporeal membrane oxygenation, and delayed surgery. J Pediatr Surg. 1997; 32:1185–1189.
6. Langham, MR, Jr., Kays, DW, Ledbetter, DJ, et al. Congenital diaphragmatic hernia. Epidemiology and outcome. Clin Perinatol. 1996; 23:671–688.
7. Dott, MM, Wong, LY, Rasmussen, SA. Population-based study of congenital diaphragmatic hernia: Risk factors and survival in Metropolitan Atlanta, 1968–1999. Birth Defects Res A Clin Mol Teratol. 2003; 67:261–267.
8. Wenstrom, KD, Weiner, CP, Hanson, JW. A five-year statewide experience with congenital diaphragmatic hernia. Am J Obstet Gynecol. 1991; 165:838–842.
9. Ontario Congenital Anomalies Study Group. Apparent truth about congenital diaphragmatic hernia: A population-based database is needed to establish benchmarking for clinical outcomes for CDH. J Pediatr Surg. 2004; 39:661–665.
10. Harrison, MR, Adzick, NS, Estes, JM, et al. A prospective study of the outcome for fetuses with diaphragmatic hernia. Jama. 1994; 271:382–384.
11. Lund, DP, Mitchell, J, Kharasch, V, et al. Congenital diaphragmatic hernia: The hidden morbidity. J Pediatr Surg. 1994; 29:258–262.
12. Stege, G, Fenton, A, Jaffray, B. Nihilism in the 1990s: The true mortality of congenital diaphragmatic hernia. Pediatrics. 2003; 112:532–535.
13. Neville, HL, Jaksic, T, Wilson, JM, et al. Bilateral congenital diaphragmatic hernia. J Pediatr Surg. 2003; 38:522–524.
14. Waller, DK, Tita, AT, Werler, MM, et al. Association between prepregnancy maternal body mass index and the risk of having an infant with a congenital diaphragmatic hernia. Birth Defects Res A Clin Mol Teratol. 2003; 67:73–76.
15. Lipson, AH, Williams, G. Congenital diaphragmatic hernia in half sibs. J Med Genet. 1985; 22:145–147.
16. Witters, I, Legius, E, Moerman, P, et al. Associated malformations and chromosomal anomalies in 42 cases of prenatally diagnosed diaphragmatic hernia. Am J Med Genet. 2001; 103:278–282.
17. Schlembach, D, Zenker, M, Trautmann, U, et al. Deletion 15q24-26 in prenatally detected diaphragmatic hernia: Increasing evidence of a candidate region for diaphragmatic development. Prenat Diagn. 2001; 21:289–292.
18. Aviram-Goldring, A, Daniely, M, Frydman, M, et al. Congenital diaphragmatic hernia in a family segregating a reciprocal translocation t(5;15)(p15.3;q24). Am J Med Genet. 2000; 90:120–122.
19. Slavotinek, AM. Single gene disorders associated with congenital diaphragmatic hernia. Am J Med Genet C Semin Med Genet. 2007; 145C:172–183.
20. Langer, JC, Winthrop, AL, Whelan, D. Fryns syndrome: A rare familial cause of congenital diaphragmatic hernia. J Pediatr Surg. 1994; 29:1266–1267.
21. Neville, HL, Jaksic, T, Wilson, JM, et al. Fryns syndrome in children with congenital diaphragmatic hernia. J Pediatr Surg. 2002; 37:1685–1687.
22. Scott, DA. Genetics of congenital diaphragmatic hernia. Semin Pediatr Surg. 2007; 16:88–93.
23. Torfs, CP, Curry, CJ, Bateson, TF, et al. A population-based study of congenital diaphragmatic hernia. Teratology. 1992; 46:555–565.
24. Pober, BR. Genetic aspects of human congenital diaphragmatic hernia. Clin Genet. 2008; 74:1–15.
25. van Loenhout, RB, Tibboel, D, Post, M, et al. Congenital diaphragmatic hernia: Comparison of animal models and relevance to the human situation. Neonatology. 2009; 96:137–149.
26. Stoll, C, Alembik, Y, Dott, B, et al. Associated malformations in cases with congenital diaphragmatic hernia. Genet Couns. 2008; 19:331–339.
27. Bollmann, R, Kalache, K, Mau, H, et al. Associated malformations and chromosomal defects in congenital diaphragmatic hernia. Fetal Diagn Ther. 1995; 10:52–59.
28. Tibboel, D, Gaag, AV. Etiologic and genetic factors in congenital diaphragmatic hernia. Clin Perinatol. 1996; 23:689–699.
29. Butler, N, Claireaux, AE. Congenital diaphragmatic hernia as a cause of perinatal mortality. Lancet. 1962; 1:659–663.
30. Sweed, Y, Puri, P. Congenital diaphragmatic hernia: Influence of associated malformations on survival. Arch Dis Child. 1993; 69:68–70.
31. Eghtesady, P, Skarsgard, ED, Smith, BM, et al. Congenital diaphragmatic hernia associated with aortic coarctation. J Pediatr Surg. 1998; 33:943–945.
32. Migliazza, L, Xia, H, Alvarez, JI, et al. Heart hypoplasia in experimental congenital diaphragmatic hernia. J Pediatr Surg. 1999; 34:706–710.
33. Menon, SC, Tani, LY, Weng, HY, et al. Clinical characteristics and outcomes of patients with cardiac defects and congenital diaphragmatic hernia. J Pediatr. 2013; 162:114–119.
34. Raval, MV, Wang, X, Reynolds, M, et al. Costs of congenital diaphragmatic hernia repair in the United States-extracorporeal membrane oxygenation foots the bill. J Pediatr Surg. 2011; 46:617–624.
35. Metkus, AP, Esserman, L, Sola, A, et al. Cost per anomaly: What does a diaphragmatic hernia cost? J Pediatr Surg. 1995; 30:226–230.
36. Iritani, I. Experimental study on embryogenesis of congenital diaphragmatic hernia. Anat Embryol (Berl). 1984; 169:133–139.
37. Kluth, D, Keijzer, R, Hertl, M, et al. Embryology of congenital diaphragmatic hernia. Semin Pediatr Surg. 1996; 5:224–233.
38. Babiuk, RP, Zhang, W, Clugston, R, et al. Embryological origins and development of the rat diaphragm. J Comp Neurol. 2003; 455:477–487.
39. Skandalakis, JE, Gray, SE, Ricketts, RR. The Diaphragm. In: Skandalakis JE, Gray SE, eds. Embryology for Surgeons. Baltimore, MD: Williams and Wilkens; 1994:491–539.
40. Jenkinson, EL. Absence of half of the diaphragm (thoracic stomach; diaphragmatic hernia). Am J Roentgenol. 1931; 26:899.
41. Bremer, JL. The diaphragm and diaphragmatic hernia. Arch Pathol. 1943; 36:539.
42. Greer, JJ, Allan, DW, Babiuk, RP, et al. Recent advances in understanding the pathogenesis of nitrofen-induced congenital diaphragmatic hernia. Pediatr Pulmonol. 2000; 29:394–399.
43. Wiseman, NE, MacPherson, RI. ‘Acquired’ congenital diaphragmatic hernia. J Pediatr Surg. 1977; 12:657–665.
44. Noble, BR, Babiuk, RP, Clugston, RD, et al. Mechanisms of action of the congenital diaphragmatic hernia-inducing teratogen nitrofen. Am J Physiol Lung Cell Mol Physiol. 2007; 293:L1079–L1087.
45. Migliazza, L, Otten, C, Xia, H, et al. Cardiovascular malformations in congenital diaphragmatic hernia: Human and experimental studies. J Pediatr Surg. 1999; 34:1352–1358.
46. Migliazza, L, Xia, H, Diez-Pardo, JA, et al. Skeletal malformations associated with congenital diaphragmatic hernia: Experimental and human studies. J Pediatr Surg. 1999; 34:1624–1629.
47. Clugston, RD, Klattig, J, Englert, C, et al. Teratogen-induced, dietary and genetic models of congenital diaphragmatic hernia share a common mechanism of pathogenesis. Am J Pathol. 2006; 169:1541–1549.
48. Allan, DW, Greer, JJ. Pathogenesis of nitrofen-induced congenital diaphragmatic hernia in fetal rats. J Appl Physiol. 1997; 83:338–347.
49. Chinoy, MR. Pulmonary hypoplasia and congenital diaphragmatic hernia: Advances in the pathogenetics and regulation of lung development. J Surg Res. 2002; 106:209–223.
50. Chinoy, MR. Lung growth and development. Front Biosci. 2003; 8:d392–d415.
51. Mey, J, Babiuk, RP, Clugston, R, et al. Retinal dehydrogenase-2 is inhibited by compounds that induce congenital diaphragmatic hernias in rodents. Am J Pathol. 2003; 162:673–679.
52. Wilson, JG, Roth, CB, Warkany, J. An analysis of the syndrome of malformations induced by maternal vitamin A deficiency. Effects of restoration of vitamin A at various times during gestation. Am J Anat. 1953; 92:189–217.
53. Mendelsohn, C, Lohnes, D, Decimo, D, et al. Function of the retinoic acid receptors (RARs) during development (II). Multiple abnormalities at various stages of organogenesis in RAR double mutants. Development. 1994; 120:2749–2771.
54. Major, D, Cadenas, M, Fournier, L, et al. Retinol status of newborn infants with congenital diaphragmatic hernia. Pediatr Surg Int. 1998; 13:547–549.
55. Miniati, D. Pulmonary vascular remodeling. Semin Pediatr Surg. 2007; 16:80–87.
56. Weibel, ER, Gomez, DM. A principle for counting tissue structures on random sections. J Appl Physiol. 1962; 17:343–348.
57. Hislop, A, Reid, L. Intra-pulmonary arterial development during fetal life-branching pattern and structure. J Anat. 1972; 113:35–48.
58. Hislop, A, Reid, L. Pulmonary arterial development during childhood: branching pattern and structure. Thorax. 1973; 28:129–135.
59. Reid, IS, Hutcherson, RJ. Long-term follow-up of patients with congenital diaphragmatic hernia. J Pediatr Surg. 1976; 11:939–942.
60. Dibbins, AW. Congenital diaphragmatic hernia: Hypoplastic lung and pulmonary vasoconstriction. Clin Perinatol. 1978; 5:93–104.
61. Suen, HC, Catlin, EA, Ryan, DP, et al. Biochemical immaturity of lungs in congenital diaphragmatic hernia. J Pediatr Surg. 1993; 28:471–475.
62. Moya, FR, Thomas, VL, Romaguera, J, et al. Fetal lung maturation in congenital diaphragmatic hernia. Am J Obstet Gynecol. 1995; 173:1401–1405.
63. Janssen, DJ, Zimmermann, LJ, Cogo, P, et al. Decreased surfactant phosphatidylcholine synthesis in neonates with congenital diaphragmatic hernia during extracorporeal membrane oxygenation. Intensive Care Med. 2009; 35:1754–1760.
64. Wagenvoort, CA, Neufeld, HN, Edwards, JE. The structure of the pulmonary arterial tree in fetal and early postnatal life. Lab Invest. 1961; 10:751–762.
65. Allen, K, Haworth, SG. Human postnatal pulmonary arterial remodeling. Ultrastructural studies of smooth muscle cell and connective tissue maturation. Lab Invest. 1988; 59:702–709.
66. Haworth, SG. Pulmonary vascular remodeling in neonatal pulmonary hypertension. State of the art. Chest. 1988; 93:S133–S138.
67. Roubliova, X, Verbeken, E, Wu, J, et al. Pulmonary vascular morphology in a fetal rabbit model for congenital diaphragmatic hernia. J Pediatr Surg. 2004; 39:1066–1072.
68. Lipsett, J, Cool, JC, Runciman, SI, et al. Morphometric analysis of preterm fetal pulmonary development in the sheep model of congenital diaphragmatic hernia. Pediatr Dev Pathol. 2000; 3:17–28.
69. Sokol, J, Bohn, D, Lacro, RV, et al. Fetal pulmonary artery diameters and their association with lung hypoplasia and postnatal outcome in congenital diaphragmatic hernia. Am J Obstet Gynecol. 2002; 186:1085–1090.
70. Jakkula, M, Le Cras, TD, Gebb, S, et al. Inhibition of angiogenesis decreases alveolarization in the developing rat lung. Am J Physiol Lung Cell Mol Physiol. 2000; 279:L600–L607.
71. Grover, TR, Parker, TA, Balasubramaniam, V, et al. Pulmonary hypertension impairs alveolarization and reduces lung growth in the ovine fetus. Am J Physiol Lung Cell Mol Physiol. 2005; 288:L648–L654.
72. Ruano, R, Aubry, MC, Barthe, B, et al. Quantitative analysis of fetal pulmonary vasculature by 3-dimensional power Doppler ultrasonography in isolated congenital diaphragmatic hernia. Am J Obstet Gynecol. 2006; 195:1720–1728.
73. Suda, K, Bigras, JL, Bohn, D, et al. Echocardiographic predictors of outcome in newborns with congenital diaphragmatic hernia. Pediatrics. 2000; 105:1106–1109.
74. Casaccia, G, Crescenzi, F, Dotta, A, et al. Birth weight and McGoon Index predict mortality in newborn infants with congenital diaphragmatic hernia. J Pediatr Surg. 2006; 41:25–28.
75. Taira, Y, Yamataka, T, Miyazaki, E, et al. Comparison of the pulmonary vasculature in newborns and stillborns with congenital diaphragmatic hernia. Pediatr Surg Int. 1998; 14:30–35.
76. Taira, Y, Yamataka, T, Miyazaki, E, et al. Adventitial changes in pulmonary vasculature in congenital diaphragmatic hernia complicated by pulmonary hypertension. J Pediatr Surg. 1998; 33:382–387.
77. Dillon, PW, Cilley, RE, Mauger, D, et al. The relationship of pulmonary artery pressure and survival in congenital diaphragmatic hernia. J Pediatr Surg. 2004; 39:307–312.
78. Jeanty, C, Nien, JK, Espinoza, J, et al. Pleural and pericardial effusion: A potential ultrasonographic marker for the prenatal differential diagnosis between congenital diaphragmatic eventration and congenital diaphragmatic hernia. Ultrasound Obstet Gynecol. 2007; 29:378–387.
79. Garne, E, Haeusler, M, Barisic, I, et al. Congenital diaphragmatic hernia: Evaluation of prenatal diagnosis in 20 European regions. Ultrasound Obstet Gynecol. 2002; 19:329–333.
80. Vettraino, IM, Lee, W, Comstock, CH. The evolving appearance of a congenital diaphragmatic hernia. J Ultrasound Med. 2002; 21:85–89.
81. Dillon, E, Renwick, M, Wright, C. Congenital diaphragmatic herniation: Antenatal detection and outcome. Br J Radiol. 2000; 73:360–365.
82. Benjamin, DR, Juul, S, Siebert, JR. Congenital posterolateral diaphragmatic hernia: Associated malformations. J Pediatr Surg. 1988; 23:899–903.
83. DeVore, GR, Horenstein, J, Platt, LD. Fetal echocardiography. VI. Assessment of cardiothoracic disproportion—a new technique for the diagnosis of thoracic hypoplasia. Am J Obstet Gynecol. 1986; 155:1066–1071.
84. Wilcox, DT, Irish, MS, Holm, BA, et al. Prenatal diagnosis of congenital diaphragmatic hernia with predictors of mortality. Clin Perinatol. 1996; 23:701–709.
85. Metkus, AP, Filly, RA, Stringer, MD, et al. Sonographic predictors of survival in fetal diaphragmatic hernia. J Pediatr Surg. 1996; 31:148–151.
86. Harrison, MR, Adzick, NS, Flake, AW, et al. Correction of congenital diaphragmatic hernia in utero: VI. Hard-earned lessons. J Pediatr Surg. 1993; 28:1411–1417.
87. Keller, RL, Glidden, DV, Paek, BW, et al. The lung-to-head ratio and fetoscopic temporary tracheal occlusion: Prediction of survival in severe left congenital diaphragmatic hernia. Ultrasound Obstet Gynecol. 2003; 21:244–249.
88. Laudy, JA, Van Gucht, M, Van Dooren, MF, et al. Congenital diaphragmatic hernia: An evaluation of the prognostic value of the lung-to-head ratio and other prenatal parameters. Prenat Diagn. 2003; 23:634–639.
89. Lipshutz, GS, Albanese, CT, Feldstein, VA, et al. Prospective analysis of lung-to-head ratio predicts survival for patients with prenatally diagnosed congenital diaphragmatic hernia. J Pediatr Surg. 1997; 32:1634–1636.
90. Ba’ath, ME, Jesudason, EC, Losty, PD. How useful is the lung-to-head ratio in predicting outcome in the fetus with congenital diaphragmatic hernia? A systematic review and meta-analysis. Ultrasound Obstet Gynecol. 2007; 30:897–906.
91. Heling, KS, Wauer, RR, Hammer, H, et al. Reliability of the lung-to-head ratio in predicting outcome and neonatal ventilation parameters in fetuses with congenital diaphragmatic hernia. Ultrasound Obstet Gynecol. 2005; 25:112–118.
92. Deprest, JA, Flemmer, AW, Gratacos, E, et al. Antenatal prediction of lung volume and in-utero treatment by fetal endoscopic tracheal occlusion in severe isolated congenital diaphragmatic hernia. Semin Fetal Neonatal Med. 2009; 14:8–13.
93. Peralta, CF, Cavoretto, P, Csapo, B, et al. Lung and heart volumes by three-dimensional ultrasound in normal fetuses at 12–32 weeks’ gestation. Ultrasound Obstet Gynecol. 2006; 27:128–133.
94. Ruano, R, Benachi, A, Martinovic, J, et al. Can three-dimensional ultrasound be used for the assessment of the fetal lung volume in cases of congenital diaphragmatic hernia? Fetal Diagn Ther. 2004; 19:87–91.
95. Ruano, R, Martinovic, J, Dommergues, M, et al. Accuracy of fetal lung volume assessed by three-dimensional sonography. Ultrasound Obstet Gynecol. 2005; 26:725–730.
96. Cannie, MM, Jani, JC, Van Kerkhove, F, et al. Fetal body volume at MR imaging to quantify total fetal lung volume: Normal ranges. Radiology. 2008; 247:197–203.
97. Coakley, FV, Lopoo, JB, Lu, Y, et al. Normal and hypoplastic fetal lungs: Volumetric assessment with prenatal single-shot rapid acquisition with relaxation enhancement MR imaging. Radiology. 2000; 216:107–111.
98. Rypens, F, Metens, T, Rocourt, N, et al. Fetal lung volume: Estimation at MR imaging-initial results. Radiology. 2001; 219:236–241.
99. Cannie, M, Jani, J, Meersschaert, J, et al. Prenatal prediction of survival in isolated diaphragmatic hernia using observed to expected total fetal lung volume determined by magnetic resonance imaging based on either gestational age or fetal body volume. Ultrasound Obstet Gynecol. 2008; 32:633–639.
100. Gorincour, G, Bouvenot, J, Mourot, MG, et al. Prenatal prognosis of congenital diaphragmatic hernia using magnetic resonance imaging measurement of fetal lung volume. Ultrasound Obstet Gynecol. 2005; 26:738–744.
101. Jani, J, Cannie, M, Done, E, et al. Relationship between lung area at ultrasound examination and lung volume assessment with magnetic resonance imaging in isolated congenital diaphragmatic hernia. Ultrasound Obstet Gynecol. 2007; 30:855–860.
102. Sandaite, I, Claus, F, De Keyzer, F, et al. Examining the relationship between the lung-to-head ratio measured on ultrasound and lung volumetry by magnetic resonance in fetuses with isolated congenital diaphragmatic hernia. Fetal Diagn Ther. 2011; 29:80–87.
103. Manning, PB, Murphy, JP, Raynor, SC, et al. Congenital diaphragmatic hernia presenting due to gastrointestinal complications. J Pediatr Surg. 1992; 27:1225–1228.
104. Paut, O, Mely, L, Viard, L, et al. Acute presentation of congenital diaphragmatic hernia past the neonatal period: A life threatening emergency. Can J Anaesth. 1996; 43:621–625.
105. Weber, TR, Tracy, T, Jr., Bailey, PV, et al. Congenital diaphragmatic hernia beyond infancy. Am J Surg. 1991; 162:643–646.
106. Losty, PD, Suen, HC, Manganaro, TF, et al. Prenatal hormonal therapy improves pulmonary compliance in the nitrofen-induced CDH rat model. J Pediatr Surg. 1995; 30:420–426.
107. Oue, T, Shima, H, Guarino, N, et al. Antenatal dexamethasone administration increases fetal lung DNA synthesis and RNA and protein content in nitrofen-induced congenital diaphragmatic hernia in rats. Pediatr Res. 2000; 48:789–793.
108. Kay, HH, Bird, IM, Coe, CL, et al. Antenatal steroid treatment and adverse fetal effects: What is the evidence? J Soc Gynecol Investig. 2000; 7:269–278.
109. Smith, GN, Kingdom, JC, Penning, DH, et al. Antenatal corticosteroids: Is more better? Lancet. 2000; 355:251–252.
110. Lally, KP, Bagolan, P, Hosie, S, et al. Corticosteroids for fetuses with congenital diaphragmatic hernia: Can we show benefit? J Pediatr Surg. 2006; 41:668–674.
111. Levin, DL. Morphologic analysis of the pulmonary vascular bed in congenital left-sided diaphragmatic hernia. J Pediatr. 1978; 92:805–809.
112. Shehata, SM, Sharma, HS, van der Staak, FH, et al. Remodeling of pulmonary arteries in human congenital diaphragmatic hernia with or without extracorporeal membrane oxygenation. J Pediatr Surg. 2000; 35:208–215.
113. Beals, DA, Schloo, BL, Vacanti, JP, et al. Pulmonary growth and remodeling in infants with high-risk congenital diaphragmatic hernia. J Pediatr Surg. 1992; 27:997–1002.
114. Okuyama, H, Kubota, A, Kawahara, H, et al. Correlation between lung scintigraphy and long-term outcome in survivors of congenital diaphragmatic hernia. Pediatr Pulmonol. 2006; 41:882–886.
115. Sakurai, Y, Azarow, K, Cutz, E, et al. Pulmonary barotrauma in congenital diaphragmatic hernia: A clinicopathological correlation. J Pediatr Surg. 1999; 34:1813–1817.
116. Drummond, WH, Gregory, GA, Heymann, MA, et al. The independent effects of hyperventilation, tolazoline, and dopamine on infants with persistent pulmonary hypertension. J Pediatr. 1981; 98:603–611.
117. Rudolph, AM, Yuan, S. Response of the pulmonary vasculature to hypoxia and H+ ion concentration changes. J Clin Invest. 1966; 45:399–411.
118. Boloker, J, Bateman, DA, Wung, JT, et al. Congenital diaphragmatic hernia in 120 infants treated consecutively with permissive hypercapnea/spontaneous respiration/elective repair. J Pediatr Surg. 2002; 37:357–366.
119. Kays, DW, Langham, MR, Jr., Ledbetter, DJ, et al. Detrimental effects of standard medical therapy in congenital diaphragmatic hernia. Ann Surg. 1999; 230:340–348.
120. Langham, MR, Jr., Kays, DW, Beierle, EA, et al. Twenty years of progress in congenital diaphragmatic hernia at the University of Florida. Am Surg. 2003; 69:45–52.
121. Wung, JT, Sahni, R, Moffitt, ST, et al. Congenital diaphragmatic hernia: Survival treated with very delayed surgery, spontaneous respiration, and no chest tube. J Pediatr Surg. 1995; 30:406–409.
122. Bohn, D. Congenital diaphragmatic hernia. Am J Respir Crit Care Med. 2002; 166:911–915.
123. Azarow, K, Messineo, A, Pearl, R, et al. Congenital diaphragmatic hernia–a tale of two cities: The Toronto experience. J Pediatr Surg. 1997; 32:395–400.
124. Paranka, MS, Clark, RH, Yoder, BA, et al. Predictors of failure of high-frequency oscillatory ventilation in term infants with severe respiratory failure. Pediatrics. 1995; 95:400–404.
125. Hirschl, RB. Innovative therapies in the management of newborns with congenital diaphragmatic hernia. Semin Pediatr Surg. 1996; 5:256–265.
126. Desfrere, L, Jarreau, PH, Dommergues, M, et al. Impact of delayed repair and elective high-frequency oscillatory ventilation on survival of antenatally diagnosed congenital diaphragmatic hernia: First application of these strategies in the more ‘severe’ subgroup of antenatally diagnosed newborns. Intensive Care Med. 2000; 26:934–941.
127. Reyes, C, Chang, LK, Waffarn, F, et al. Delayed repair of congenital diaphragmatic hernia with early high-frequency oscillatory ventilation during preoperative stabilization. J Pediatr Surg. 1998; 33:1010–1016.
128. Cacciari, A, Ruggeri, G, Mordenti, M, et al. High-frequency oscillatory ventilation versus conventional mechanical ventilation in congenital diaphragmatic hernia. Eur J Pediatr Surg. 2001; 11:3–7.
129. Cogo, PE, Simonato, M, Danhaive, O, et al. Impaired surfactant protein b synthesis in infants with congenital diaphragmatic hernia. Eur Respir J. 2012.
130. Cogo, PE, Zimmermann, LJ, Rosso, F, et al. Surfactant synthesis and kinetics in infants with congenital diaphragmatic hernia. Am J Respir Crit Care Med. 2002; 166:154–158.
131. O’Toole, SJ, Karamanoukian, HL, Morin, FC, 3rd., et al. Surfactant decreases pulmonary vascular resistance and increases pulmonary blood flow in the fetal lamb model of congenital diaphragmatic hernia. J Pediatr Surg. 1996; 31:507–511.
132. Karamanoukian, HL, Glick, PL, Wilcox, DT, et al. Pathophysiology of congenital diaphragmatic hernia. VIII: Inhaled nitric oxide requires exogenous surfactant therapy in the lamb model of congenital diaphragmatic hernia. J Pediatr Surg. 1995; 30:1–4.
133. Lally, KP, Lally, PA, Langham, MR, et al. Surfactant does not improve survival rate in preterm infants with congenital diaphragmatic hernia. J Pediatr Surg. 2004; 39:829–833.
134. Van Meurs, K. Is surfactant therapy beneficial in the treatment of the term newborn infant with congenital diaphragmatic hernia? J Pediatr. 2004; 145:312–316.
135. Colby, CE, Lally, KP, Hintz, SR, et al. Surfactant replacement therapy on ECMO does not improve outcome in neonates with congenital diaphragmatic hernia. J Pediatr Surg. 2004; 39:1632–1637.
136. Lotze, A, Knight, GR, Anderson, KD, et al. Surfactant (beractant) therapy for infants with congenital diaphragmatic hernia on ECMO: Evidence of persistent surfactant deficiency. J Pediatr Surg. 1994; 29:407–412.
137. Buss, M, Williams, G, Dilley, A, et al. Prevention of heart failure in the management of congenital diaphragmatic hernia by maintaining ductal patency. A case report. J Pediatr Surg. 2006; 41:e9–11.
138. Tanabe, M, Yoshida, H, Iwai, J, et al. Doppler flow patterns through the ductus arteriosus in patients with congenital diaphragmatic hernia. Eur J Pediatr Surg. 2000; 10:92–95.
139. Finer, NN, Barrington, KJ. Nitric oxide in respiratory failure in the newborn infant. Semin Perinatol. 1997; 21:426–440.
140. Kinsella, JP, Abman, SH. Inhalational nitric oxide therapy for persistent pulmonary hypertension of the newborn. Pediatrics. 1993; 91:997–998.
141. Kinsella, JP, Ivy, DD, Abman, SH. Inhaled nitric oxide improves gas exchange and lowers pulmonary vascular resistance in severe experimental hyaline membrane disease. Pediatr Res. 1994; 36:402–408.
142. Roberts, JD, Polaner, DM, Lang, P, et al. Inhaled nitric oxide in persistent pulmonary hypertension of the newborn. Lancet. 1992; 340:818–819.
143. Kinsella, JP, Neish, SR, Shaffer, E, et al. Low-dose inhalation nitric oxide in persistent pulmonary hypertension of the newborn. Lancet. 1992; 340:819–820.
144. Clark, RH, Kueser, TJ, Walker, MW, et al. Low-dose nitric oxide therapy for persistent pulmonary hypertension of the newborn. Clinical Inhaled Nitric Oxide Research Group. N Engl J Med. 2000; 342:469–474.
145. The Neonatal Inhaled Nitric Oxide Study Group (NINOS). Inhaled nitric oxide and hypoxic respiratory failure in infants with congenital diaphragmatic hernia. The Neonatal Inhaled Nitric Oxide Study Group (NINOS). Pediatrics. 1997; 99:838–845.
146. Keller, RL, Hamrick, SE, Kitterman, JA, et al. Treatment of rebound and chronic pulmonary hypertension with oral sildenafil in an infant with congenital diaphragmatic hernia. Pediatr Crit Care Med. 2004; 5:184–187.
147. Ivy, DD, Kinsella, JP, Ziegler, JW, et al. Dipyridamole attenuates rebound pulmonary hypertension after inhaled nitric oxide withdrawal in postoperative congenital heart disease. J Thorac Cardiovasc Surg. 1998; 115:875–882.
148. Kinsella, JP, Ivy, DD, Abman, SH. Pulmonary vasodilator therapy in congenital diaphragmatic hernia: Acute, late, and chronic pulmonary hypertension. Semin Perinatol. 2005; 29:123–128.
149. Finer, NN, Barrington, KJ. Nitric oxide for respiratory failure in infants born at or near term. Cochrane Database Syst Rev (Online). 2006.
150. Bender, AT, Beavo, JA. Cyclic nucleotide phosphodiesterases: Molecular regulation to clinical use. Pharmacol Rev. 2006; 58:488–520.
151. Buysse, C, Fonteyne, C, Dessy, H, et al. The use of dipyridamole to wean from inhaled nitric oxide in congenital diaphragmatic hernia. J Pediatr Surg. 2001; 36:1864–1865.
152. Kinsella, JP, Torielli, F, Ziegler, JW, et al. Dipyridamole augmentation of response to nitric oxide. Lancet. 1995; 346:647–648.
153. Thebaud, B, Saizou, C, Farnoux, C, et al. Dypiridamole, a cGMP phosphodiesterase inhibitor, transiently improves the response to inhaled nitric oxide in two newborns with congenital diaphragmatic hernia. Intensive Care Med. 1999; 25:300–303.
154. Filan, PM, McDougall, PN, Shekerdemian, LS. Combination pharmacotherapy for severe neonatal pulmonary hypertension. J Paediatr Child Health. 2006; 42:219–220.
155. Noori, S, Friedlich, P, Wong, P, et al. Cardiovascular effects of sildenafil in neonates and infants with congenital diaphragmatic hernia and pulmonary hypertension. Neonatology. 2007; 91:92–100.
156. De Luca, D, Zecca, E, Vento, G, et al. Transient effect of epoprostenol and sildenafil combined with iNO for pulmonary hypertension in congenital diaphragmatic hernia. Paediatr Anaesth. 2006; 16:597–598.
157. Steinhorn, RH, Kinsella, JP, Pierce, C, et al. Intravenous sildenafil in the treatment of neonates with persistent pulmonary hypertension. J Pediatr. 2009; 155:841–847.
158. Hunter, L, Richens, T, Davis, C, et al. Sildenafil use in congenital diaphragmatic hernia. Arch Dis Child Fetal Neonatal Ed. 2009; 94:F467.
159. Abman, SH, Kinsella, JP, Rosenzweig, EB, et al. Implications of the FDA warning against the use of Sildenafil for the treatment of pediatric pulmonary hypertension. Am J Respir Crit Care Med. 2013; 187:572–575.
160. Barst, RJ. Recent advances in the treatment of pediatric pulmonary artery hypertension. Pediatr Clin North Am. 1999; 46:331–345.
161. Barst, RJ, Ivy, D, Dingemanse, J, et al. Pharmacokinetics, safety, and efficacy of bosentan in pediatric patients with pulmonary arterial hypertension. Clin Pharmacol Ther. 2003; 73:372–382.
162. Rosenzweig, EB, Barst, RJ. Pulmonary arterial hypertension in children: A medical update. Curr Opin Pediatr. 2008; 20:288–293.
163. Terragno, NA, Terragno, A. Prostaglandin metabolism in the fetal and maternal vasculature. Fed Proc. 1979; 38:75–77.
164. Leffler, CW, Hessler, JR, Green, RS. Mechanism of stimulation of pulmonary prostacyclin synthesis at birth. Prostaglandins. 1984; 28:877–887.
165. Leffler, CW, Hessler, JR, Green, RS. The onset of breathing at birth stimulates pulmonary vascular prostacyclin synthesis. Pediatr Res. 1984; 18:938–942.
166. Yanagisawa, M, Kurihara, H, Kimura, S, et al. A novel potent vasoconstrictor peptide produced by vascular endothelial cells. Nature. 1988; 332:411–415.
167. Buchan, KW, Magnusson, H, Rabe, KF, et al. Characterisation of the endothelin receptor mediating contraction of human pulmonary artery using BQ123 and Ro 46-2005. Eur J Pharmacol. 1994; 260:221–226.
168. Ziegler, JW, Ivy, DD, Kinsella, JP, et al. The role of nitric oxide, endothelin, and prostaglandins in the transition of the pulmonary circulation. Clin Perinatol. 1995; 22:387–403.
169. Ivy, DD, Kinsella, JP, Abman, SH. Physiologic characterization of endothelin A and B receptor activity in the ovine fetal pulmonary circulation. J Clin Invest. 1994; 93:2141–2148.
170. de Lagausie, P, de Buys-Roessingh, A, Ferkdadji, L, et al. Endothelin receptor expression in human lungs of newborns with congenital diaphragmatic hernia. J Pathol. 2005; 205:112–118.
171. Keller, RL, Tacy, TA, Hendricks-Munoz, K, et al. Congenital diaphragmatic hernia: Endothelin-1, pulmonary hypertension, and disease severity. Am J Respir Crit Care Med. 2010; 182:555–561.
172. Kobayashi, H, Puri, P. Plasma endothelin levels in congenital diaphragmatic hernia. J Pediatr Surg. 1994; 29:1258–1261.
173. Ivy, DD, Rosenzweig, EB, Lemarie, JC, et al. Long-term outcomes in children with pulmonary arterial hypertension treated with bosentan in real-world clinical settings. Am J Cardiol. 2010; 106:1332–1338.
174. Rosenzweig, EB, Ivy, DD, Widlitz, A, et al. Effects of long-term bosentan in children with pulmonary arterial hypertension. J Am Coll Cardiol. 2005; 46:697–704.
175. The Congenital Diaphragmatic Hernia Study Group. Does extracorporeal membrane oxygenation improve survival in neonates with congenital diaphragmatic hernia? J Pediatr Surg. 1999; 34:720–725.
176. Downard, C. Impact of Amicar on hemorrhagic complications of ECMO: A ten-year review. J Pediatr Surg. 2003; 38:1212–1216.
177. Lally, KP. Extracorporeal membrane oxygenation in patients with congenital diaphragmatic hernia. Semin Pediatr Surg. 1996; 5:249–255.
178. Somaschini, M, Locatelli, G, Salvoni, L, et al. Impact of new treatments for respiratory failure on outcome of infants with congenital diaphragmatic hernia. Eur J Pediatr. 1999; 158:780–784.
179. German, JC, Gazzaniga, AB, Amlie, R, et al. Management of pulmonary insufficiency in diaphragmatic hernia using extracorporeal circulation with a membrane oxygenator (ECMO). J Pediatr Surg. 1977; 12:905–912.
180. Heiss, K, Manning, P, Oldham, KT, et al. Reversal of mortality for congenital diaphragmatic hernia with ECMO. Ann Surg. 1989; 209:225–230.
181. Sawyer, SF, Falterman, KW, Goldsmith, JP, et al. Improving survival in the treatment of congenital diaphragmatic hernia. Ann Thorac Surg. 1986; 41:75–78.
182. Weber, TR, Connors, RH, Pennington, DG, et al. Neonatal diaphragmatic hernia. An improving outlook with extracorporeal membrane oxygenation. Arch Surg. 1987; 122:615–618.
183. Davis, PJ, Firmin, RK, Manktelow, B, et al. Long-term outcome following extracorporeal membrane oxygenation for congenital diaphragmatic hernia: The UK experience. J Pediatr. 2004; 144:309–315.
184. Langham, MR, Jr., Krummel, TM, Greenfield, LJ, et al. Extracorporeal membrane oxygenation following repair of congenital diaphragmatic hernias. Ann Thorac Surg. 1987; 44:247–252.
185. Lally, KP, Lally, PA, Van Meurs, KP, et al. Treatment evolution in high-risk congenital diaphragmatic hernia: Ten years’ experience with diaphragmatic agenesis. Ann Surg. 2006; 244:505–513.
186. Lally, KP, Paranka, MS, Roden, J, et al. Congenital diaphragmatic hernia. Stabilization and repair on ECMO. Ann Surg. 1992; 216:569–573.
187. Reickert, CA, Hirschl, RB, Atkinson, JB, et al. Congenital diaphragmatic hernia survival and use of extracorporeal life support at selected level III nurseries with multimodality support. Surgery. 1998; 123:305–310.
188. Keshen, TH, Gursoy, M, Shew, SB, et al. Does extracorporeal membrane oxygenation benefit neonates with congenital diaphragmatic hernia? Application of a predictive equation. J Pediatr Surg. 1997; 32:818–822.
189. O’Rourke, PP, Lillehei, CW, Crone, RK, et al. The effect of extracorporeal membrane oxygenation on the survival of neonates with high-risk congenital diaphragmatic hernia: 45 cases from a single institution. J Pediatr Surg. 1991; 26:147–152.
190. Al-Shanafey, S, Giacomantonio, M, Henteleff, H. Congenital diaphragmatic hernia: Experience without extracoporeal membrane oxygenation. Pediatr Surg Int. 2002; 18:28–31.
191. Pusic, AL, Giacomantonio, M, Pippus, K, et al. Survival in neonatal congenital hernia without extracorporeal membrane oxygenation support. J Pediatr Surg. 1995; 30:1188–1190.
192. Skarsgard, ED, Harrison, MR. Congenital diaphragmatic hernia: The surgeon’s perspective. Pediatr Rev. 1999; 20:e71–e78.
193. Harting, M. Surgical management of neonates with congenital diaphragmatic hernia. Semin Pediatr Surg. 2007; 16:109–114.
194. Sakai, H, Tamura, M, Hosokawa, Y, et al. Effect of surgical repair on respiratory mechanics in congenital diaphragmatic hernia. J Pediatr. 1987; 111:432–438.
195. de la Hunt, MN, Madden, N, Scott, JE, et al. Is delayed surgery really better for congenital diaphragmatic hernia?: A prospective randomized clinical trial. J Pediatr Surg. 1996; 31:1554–1556.
196. Langer, JC, Filler, RM, Bohn, DJ, et al. Timing of surgery for congenital diaphragmatic hernia: Is emergency operation necessary? J Pediatr Surg. 1988; 23:731–734.
197. Nio, M, Haase, G, Kennaugh, J, et al. A prospective randomized trial of delayed versus immediate repair of congenital diaphragmatic hernia. J Pediatr Surg. 1994; 29:618–621.
198. Al-Hathal, M, Crankson, SJ, Al-Harbi, F, et al. Congenital diaphragmatic hernia: Experience with preoperative stabilization and delayed surgery without ECMO and inhaled nitric oxide. Am J Perinatol. 1998; 15:487–490.
199. Breaux, CW, Jr., Rouse, TM, Cain, WS, et al. Improvement in survival of patients with congenital diaphragmatic hernia utilizing a strategy of delayed repair after medical and/or extracorporeal membrane oxygenation stabilization. J Pediatr Surg. 1991; 26:333–338.
200. West, KW, Bengston, K, Rescorla, FJ, et al. Delayed surgical repair and ECMO improves survival in congenital diaphragmatic hernia. Ann Surg. 1992; 216:454–462.
201. Bagolan, P, Casaccia, G, Crescenzi, F, et al. Impact of a current treatment protocol on outcome of high-risk congenital diaphragmatic hernia. J Pediatr Surg. 2004; 39:313–318.
202. Dassinger, MS, Copeland, DR, Gossett, J, et al. Early repair of congenital diaphragmatic hernia on extracorporeal membrane oxygenation. J Pediatr Surg. 2010; 45:693–697.
203. Levy, SM, Lally, PA, Lally, KP, et al. The impact of chylothorax on neonates with repaired congenital diaphragmatic hernia. J Pediatr Surg. 2013; 48:724–729.
204. Sigalet, DL, Tierney, A, Adolph, V, et al. Timing of repair of congenital diaphragmatic hernia requiring extracorporeal membrane oxygenation support. J Pediatr Surg. 1995; 30:1183–1187.
205. Meehan, JJ, Haney, BM, Snyder, CL, et al. Outcome following recannulation and a second ECMO course. J Pediatr Surg. 2002; 37:845–850.
206. Clark, RH, Hardin, WD, Jr., Hirschl, RB, et al. Current surgical management of congenital diaphragmatic hernia: a report from the Congenital Diaphragmatic Hernia Study Group. J Pediatr Surg. 1998; 33:1004–1009.
207. Tsao, K, Lally, PA, Lally, KP. Minimally invasive repair of congenital diaphragmatic hernia. J Pediatr Surg. 2011; 46:1158–1164.
208. Puri, P. Congenital diaphragmatic hernia. Curr Probl Surg. 1994; 31:787–846.
209. Kyzer, S, Sirota, L, Chaimoff, C. Abdominal wall closure with a silastic patch after repair of congenital diaphragmatic hernia. Arch Surg. 2004; 139:296–298.
210. Schnitzer, JJ, Kikiros, CS, Short, BL, et al. Experience with abdominal wall closure for patients with congenital diaphragmatic hernia repaired on ECMO. J Pediatr Surg. 1995; 30:19–22.
211. Rana, AR, Khouri, JS, Teitelbaum, DH, et al. Salvaging the severe congenital diaphragmatic hernia patient: Is a silo the solution? J Pediatr Surg. 2008; 43:788–791.
212. Cheah, FC, Noraida, MH, Boo, NY, et al. Chylothorax after repair of congenital diaphragmatic hernia–a case report. Singapore Med J. 2000; 41:548–549.
213. Arca, MJ, Barnhart, DC, Lelli, JL, Jr., et al. Early experience with minimally invasive repair of congenital diaphragmatic hernias: Results and lessons learned. J Pediatr Surg. 2003; 38:1563–1568.
214. Nguyen, TL, Le, AD. Thoracoscopic repair for congenital diaphragmatic hernia: Lessons from 45 cases. J Pediatr Surg. 2006; 41:1713–1715.
215. Yang, EY, Allmendinger, N, Johnson, SM, et al. Neonatal thoracoscopic repair of congenital diaphragmatic hernia: Selection criteria for successful outcome. J Pediatr Surg. 2005; 40:1369–1375.
216. Holcomb, GW, III., Ostlie, DJ, Miller, KA. Laparoscopic patch repair of diaphragmatic hernia with Surgisis. J Pediatr Surg. 2005; 40:E1–E7.
217. Shah, SR, Gittes, GK, Barsness, KA, et al. Thoracoscopic patch repair of a right-sided congenital diaphragmatic hernia in a neonate. Surg Endosc. 2008; 23:215.
218. McHoney, M, Giacomello, L, Nah, SA, et al. Thoracoscopic repair of congenital diaphragmatic hernia: Intraoperative ventilation and recurrence. J Pediatr Surg. 2010; 45:355–359.
219. Pacilli, M, Pierro, A, Kingsley, C, et al. Absorption of carbon dioxide during laparoscopy in children measured using a novel mass spectrometric technique. Br J Anaesth. 2006; 97:215–219.
220. Bliss, D, Matar, M, Krishnaswami, S. Should intraoperative hypercapnea or hypercarbia raise concern in neonates undergoing thoracoscopic repair of diaphragmatic hernia of Bochdalek? J Laparoendosc Adv Surg Tech A. 2009; 19(Suppl. 1):S55–S58.
221. McHoney, M, Corizia, L, Eaton, S, et al. Carbon dioxide elimination during laparoscopy in children is age dependent. J Pediatr Surg. 2003; 38:105–110.
222. Gourlay, DM, Cassidy, LD, Sato, TT, et al. Beyond feasibility: A comparison of newborns undergoing thoracoscopic and open repair of congenital diaphragmatic hernias. J Pediatr Surg. 2009; 44:1702–1707.
223. Kim, AC, Bryner, BS, Akay, B, et al. Thoracoscopic repair of congenital diaphragmatic hernia in neonates: Lessons learned. J Laparoendosc Adv Surg Tech A. 2009; 19:575–580.
224. Cho, SD, Krishnaswami, S, McKee, JC, et al. Analysis of 29 consecutive thoracoscopic repairs of congenital diaphragmatic hernia in neonates compared to historical controls. J Pediatr Surg. 2009; 44:80–86.
225. Shah, SR, Wishnew, J, Barsness, K, et al. Minimally invasive congenital diaphragmatic hernia repair: A 7-year review of one institution’s experience. Surg Endosc. 2009; 23:1265–1271.
226. McHoney, MC, Corizia, L, Eaton, S, et al. Laparoscopic surgery in children is associated with an intraoperative hypermetabolic response. Surg Endosc. 2006; 20:452–457.
227. Taskin, M, Zengin, K, Unal, E, et al. Laparoscopic repair of congenital diaphragmatic hernias. Surg Endosc. 2002; 16:869.
228. Gander, JW, Fisher, JC, Gross, ER, et al. Early recurrence of congenital diaphragmatic hernia is higher after thoracoscopic than open repair: A single institutional study. J Pediatr Surg. 2011; 46:1303–1308.
229. Keijzer, R, van de Ven, C, Vlot, J, et al. Thoracoscopic repair in congenital diaphragmatic hernia: patching is safe and reduces the recurrence rate. J Pediatr Surg. 2010; 45:953–957.
230. Lansdale, N, Alam, S, Losty, PD, et al. Neonatal endosurgical congenital diaphragmatic hernia repair: a systematic review and meta-analysis. Ann Surg. 2010; 252:20–26.
231. Knight, CG, Gidell, KM, Lanning, D, et al. Laparoscopic Morgagni hernia repair in children using robotic instruments. J Laparoendosc Adv Surg Tech A. 2005; 15:482–486.
232. Meehan, JJ, Sandler, A. Robotic repair of a Bochdalek congenital diaphragmatic hernia in a small neonate: Robotic advantages and limitations. J Pediatr Surg. 2007; 42:1757–1760.
233. Slater, BJ, Meehan, JJ. Robotic repair of congenital diaphragmatic anomalies. J Laparoendosc Adv Surg Tech A. 2009; 19(Suppl. 1):S123–S127.
234. Grethel, E, Cortes, R, Wagner, A, et al. Prosthetic patches for congenital diaphragmatic hernia repair: Surgisis vs Gore-Tex. J Pediatr Surg. 2006; 41:29–33.
235. Hajer, GF, vd Staak, FH, de Haan, AF, et al. Recurrent congenital diaphragmatic hernia; which factors are involved? Eur J Pediatr Surg. 1998; 8:329–333.
236. Moss, RL, Chen, CM, Harrison, MR. Prosthetic patch durability in congenital diaphragmatic hernia: A long-term follow-up study. J Pediatr Surg. 2001; 36:152–154.
237. Lally, KP, Cheu, HW, Vazquez, WD. Prosthetic diaphragm reconstruction in the growing animal. J Pediatr Surg. 1993; 28:45–47.
238. St Peter, SD, Valusek, PA, Tsao, K, et al. Abdominal complications related to type of repair for congenital diaphragmatic hernia. J Surg Res. 2007; 140:234–236.
239. Loff, S, Wirth, H, Jester, I, et al. Implantation of a cone-shaped double-fixed patch increases abdominal space and prevents recurrence of large defects in congenital diaphragmatic hernia. J Pediatr Surg. 2005; 40:1701–1705.
240. Saltzman, DA, Ennis, JS, Mehall, JR, et al. Recurrent congenital diaphragmatic hernia: A novel repair. J Pediatr Surg. 2001; 36:1768–1769.
241. St Peter, SD, Shah, SR, Little, DC, et al. Bilateral congenital diaphragmatic hernia with absent pleura and pericardium. Birth Defects Res A Clin Mol Teratol. 2005; 73:624–627.
242. Sandusky, GE, Jr., Badylak, SF, Morff, RJ, et al. Histologic findings after in vivo placement of small intestine submucosal vascular grafts and saphenous vein grafts in the carotid artery in dogs. Am J Pathol. 1992; 140:317–324.
243. Oelschlager, BK, Barreca, M, Chang, L, et al. The use of small intestine submucosa in the repair of paraesophageal hernias: Initial observations of a new technique. Am J Surg. 2003; 186:4–8.
244. Franklin, ME, Jr., Gonzalez, JJ, Jr., Michaelson, RP, et al. Preliminary experience with new bioactive prosthetic material for repair of hernias in infected fields. Hernia. 2002; 6:171–174.
245. St. Peter, SD, Holcomb, GW, III. The use of biosynthetic mesh to enhance hiatal repair at the time of re-do Nissen fundoplication. J Pediatr Surg. 2007; 42:1298–1301.
246. Smith, MJ, Paran, TS, Quinn, F, et al. The SIS extracellular matrix scaffold-preliminary results of use in congenital diaphragmatic hernia (CDH) repair. Pediatr Surg Int. 2004; 20:859–862.
247. Mitchell, IC, Garcia, NM, Barber, R, et al. Permacol: A potential biologic patch alternative in congenital diaphragmatic hernia repair. J Pediatr Surg. 2008; 43:2161–2164.
248. Menon, NG, Rodriguez, ED, Byrnes, CK, et al. Revascularization of human acellular dermis in full-thickness abdominal wall reconstruction in the rabbit model. Ann Plast Surg. 2003; 50:523–527.
249. Urita, Y, Komuro, H, Chen, G, et al. Evaluation of diaphragmatic hernia repair using PLGA mesh-collagen sponge hybrid scaffold: An experimental study in a rat model. Pediatr Surg Int. 2008; 24:1041–1045.
250. Sandoval, JA, Lou, D, Engum, SA, et al. The whole truth: Comparative analysis of diaphragmatic hernia repair using 4-ply vs 8-ply small intestinal submucosa in a growing animal model. J Pediatr Surg. 2006; 41:518–523.
251. Laituri, CA, Garey, CL, Valusek, PA, et al. Outcome of congenital diaphragmatic hernia repair depending on patch type. Eur J Pediatr Surg. 2010; 5.
252. Kimber, CP, Dunkley, MP, Haddock, G, et al. Patch incorporation in diaphragmatic hernia. J Pediatr Surg. 2000; 35:120–123.
253. Simpson, JS, Gossage, JD. Use of abdominal wall muscle flap in repair of large congenital diaphragmatic hernia. J Pediatr Surg. 1971; 6:42–44.
254. Scaife, ER, Johnson, DG, Meyers, RL, et al. The split abdominal wall muscle flap–a simple, mesh-free approach to repair large diaphragmatic hernia. J Pediatr Surg. 2003; 38:1748–1751.
255. Brant-Zawadzki, PB, Fenton, SJ, Nichol, PF, et al. The split abdominal wall muscle flap repair for large congenital diaphragmatic hernias on extracorporeal membrane oxygenation. J Pediatr Surg. 2007; 42:1047–1050.
256. Barnhart, DC, Jacques, E, Scaife, ER, et al. Split abdominal wall muscle flap repair vs patch repair of large congenital diaphragmatic hernias. J Pediatr Surg. 2012; 47:81–86.
257. Bianchi, A, Doig, CM, Cohen, SJ. The reverse latissimus dorsi flap for congenital diaphragmatic hernia repair. J Pediatr Surg. 1983; 18:560–563.
258. Samarakkody, U, Klaassen, M, Nye, B. Reconstruction of congenital agenesis of hemidiaphragm by combined reverse latissimus dorsi and serratus anterior muscle flaps. J Pediatr Surg. 2001; 36:1637–1640.
259. Meeker, IA, Jr., Snyder, WH, Jr. Surgical management of diaphragmatic defects in the newborn infant. A report of twenty infants each less than one week old (1956–1961). Am J Surg. 1962; 104:196–203.
260. Sydorak, R. Reversed latissimus dorsi muscle flap for repair of recurrent congenital diaphragmatic hernia. J Pediatr Surg. 2003; 38:296–300.
261. Bekdash, B, Singh, B, Lakhoo, K. Recurrent late complications following congenital diaphragmatic hernia repair with prosthetic patches: a case series. J Med Case Reports. 2009; 3:7237.
262. Barbosa, RF, Rodrigues, J, Correia-Pinto, J, et al. Repair of a large congenital diaphragmatic defect with a reverse latissimus dorsi muscle flap. Microsurgery. 2008; 28:85–88.
263. Ferrari, G, Cusella-De Angelis, G, Coletta, M, et al. Muscle regeneration by bone marrow-derived myogenic progenitors. Science. 1998; 279:1528–1530.
264. Rossi, CA, Pozzobon, M, Ditadi, A, et al. Clonal characterization of rat muscle satellite cells: Proliferation, metabolism and differentiation define an intrinsic heterogeneity. PLoS One. 2010; 5:e8523.
265. De Coppi, P, Bartsch, G, Jr., Siddiqui, MM, et al. Isolation of amniotic stem cell lines with potential for therapy. Nat Biotechnol. 2007; 25:100–106.
266. Fuchs, JR, Kaviani, A, Oh, JT, et al. Diaphragmatic reconstruction with autologous tendon engineered from mesenchymal amniocytes. J Pediatr Surg. 2004; 39:834–838.
267. Turner, CG, Klein, JD, Steigman, SA, et al. Preclinical regulatory validation of an engineered diaphragmatic tendon made with amniotic mesenchymal stem cells. J Pediatr Surg. 2011; 46:57–61.
268. Harrison, MR, Adzick, NS, Bullard, KM, et al. Correction of congenital diaphragmatic hernia in utero VII: A prospective trial. J Pediatr Surg. 1997; 32:1637–1642.
269. Harrison, MR, Bressack, MA, Churg, AM, et al. Correction of congenital diaphragmatic hernia in utero. II. Simulated correction permits fetal lung growth with survival at birth. Surgery. 1980; 88:260–268.
270. DiFiore, JW, Fauza, DO, Slavin, R, et al. Experimental fetal tracheal ligation reverses the structural and physiological effects of pulmonary hypoplasia in congenital diaphragmatic hernia. J Pediatr Surg. 1994; 29:248–256.
271. Hedrick, MH, Estes, JM, Sullivan, KM, et al. Plug the lung until it grows (PLUG): A new method to treat congenital diaphragmatic hernia in utero. J Pediatr Surg. 1994; 29:612–617.
272. Lipsett, J, Cool, JC, Runciman, SI, et al. Effect of antenatal tracheal occlusion on lung development in the sheep model of congenital diaphragmatic hernia: A morphometric analysis of pulmonary structure and maturity. Pediatr Pulmonol. 1998; 25:257–269.
273. Bin Saddiq, W, Piedboeuf, B, Laberge, JM, et al. The effects of tracheal occlusion and release on type II pneumocytes in fetal lambs. J Pediatr Surg. 1997; 32:834–838.
274. Jani, JC, Nicolaides, KH, Gratacos, E, et al. Fetal lung-to-head ratio in the prediction of survival in severe left-sided diaphragmatic hernia treated by fetal endoscopic tracheal occlusion (FETO). Am J Obstet Gynecol. 2006; 195:1646–1650.
275. Deprest, J, Jani, J, Van Schoubroeck, D, et al. Current consequences of prenatal diagnosis of congenital diaphragmatic hernia. J Pediatr Surg. 2006; 41:423–430.
276. Harrison, MR, Keller, RL, Hawgood, SB, et al. A randomized trial of fetal endoscopic tracheal occlusion for severe fetal congenital diaphragmatic hernia. N Engl J Med. 2003; 349:1916–1924.
277. Deprest, J, Gratacos, E, Nicolaides, KH. Fetoscopic tracheal occlusion (FETO) for severe congenital diaphragmatic hernia: Evolution of a technique and preliminary results. Ultrasound Obstet Gynecol. 2004; 24:121–126.
278. Jani, JC, Nicolaides, KH, Gratacos, E, et al. Severe diaphragmatic hernia treated by fetal endoscopic tracheal occlusion. Ultrasound Obstet Gynecol. 2009; 34:304–310.
279. Ruano, R, Yoshisaki, CT, da Silva, MM, et al. A randomized controlled trial of fetal endoscopic tracheal occlusion versus postnatal management of severe isolated congenital diaphragmatic hernia. Ultrasound Obstet Gynecol. 2012; 39:20–27.
280. The Congenital Diaphragmatic Hernia Study Group. Estimating disease severity of congenital diaphragmatic hernia in the first 5 minutes of life. J Pediatr Surg. 2001; 36:141–145.
281. Javid, PJ, Jaksic, T, Skarsgard, ED, et al. Survival rate in congenital diaphragmatic hernia: The experience of the Canadian Neonatal Network. J Pediatr Surg. 2004; 39:657–660.
282. Grushka, JR, Laberge, JM, Puligandla, P, et al. Effect of hospital case volume on outcome in congenital diaphragmatic hernia: The experience of the Canadian Pediatric Surgery Network. J Pediatr Surg. 2009; 44:873–876.
283. Bucher, BT, Guth, RM, Saito, JM, et al. Impact of hospital volume on in-hospital mortality of infants undergoing repair of congenital diaphragmatic hernia. Ann Surg. 2010; 252:635–642.
284. Cohen, MS, Rychik, J, Bush, DM, et al. Influence of congenital heart disease on survival in children with congenital diaphragmatic hernia. J Pediatr. 2002; 141:25–30.
285. Graziano, JN. Cardiac anomalies in patients with congenital diaphragmatic hernia and their prognosis: A report from the Congenital Diaphragmatic Hernia Study Group. J Pediatr Surg. 2005; 40:1045–1049.
286. Tsao, K, Allison, ND, Harting, MT, et al. Congenital diaphragmatic hernia in the preterm infant. Surgery. 2010; 148:404–410.
287. Levison, J, Halliday, R, Holland, AJ, et al. A population-based study of congenital diaphragmatic hernia outcome in New South Wales and the Australian Capital Territory, Australia, 1992–2001. J Pediatr Surg. 2006; 41:1049–1053.
288. Rygl, M, Pycha, K, Stranak, Z, et al. Congenital diaphragmatic hernia: onset of respiratory distress and size of the defect: Analysis of the outcome in 104 neonates. Pediatr Surg Int. 2007; 23:27–31.
289. Lally, KP, Lally, PA, Lasky, RE, et al. Defect size determines survival in infants with congenital diaphragmatic hernia. Pediatrics. 2007; 120:e651–e657.
290. Danzer, E, Hedrick, HL. Neurodevelopmental and neurofunctional outcomes in children with congenital diaphragmatic hernia. Early Hum Dev. 2011; 87:625–632.
291. Nobuhara, KK, Lund, DP, Mitchell, J, et al. Long-term outlook for survivors of congenital diaphragmatic hernia. Clin Perinatol. 1996; 23:873–887.
292. Lally, KP, Engle, W. Postdischarge follow-up of infants with congenital diaphragmatic hernia. Pediatrics. 2008; 121:627–632.
293. Wohl, ME, Griscom, NT, Strieder, DJ, et al. The lung following repair of congenital diaphragmatic hernia. J Pediatr. 1977; 90:405–414.
294. Falconer, AR, Brown, RA, Helms, P, et al. Pulmonary sequelae in survivors of congenital diaphragmatic hernia. Thorax. 1990; 45:126–129.
295. Muratore, CS, Kharasch, V, Lund, DP, et al. Pulmonary morbidity in 100 survivors of congenital diaphragmatic hernia monitored in a multidisciplinary clinic. J Pediatr Surg. 2001; 36:133–140.
296. Vanamo, K, Rintala, R, Sovijarvi, A, et al. Long-term pulmonary sequelae in survivors of congenital diaphragmatic defects. J Pediatr Surg. 1996; 31:1096–1099.
297. Kamata, S, Usui, N, Kamiyama, M, et al. Long-term follow-up of patients with high-risk congenital diaphragmatic hernia. J Pediatr Surg. 2005; 40:1833–1838.
298. Trachsel, D, Selvadurai, H, Bohn, D, et al. Long-term pulmonary morbidity in survivors of congenital diaphragmatic hernia. Pediatr Pulmonol. 2005; 39:433–439.
299. Marven, SS, Smith, CM, Claxton, D, et al. Pulmonary function, exercise performance, and growth in survivors of congenital diaphragmatic hernia. Arch Dis Child. 1998; 78:137–142.
300. Bargy, F, Beaudoin, S, Barbet, P. Fetal lung growth in congenital diaphragmatic hernia. Fetal Diagn Ther. 2006; 21:39–44.
301. Arena, F, Baldari, S, Centorrino, A, et al. Mid- and long-term effects on pulmonary perfusion, anatomy and diaphragmatic motility in survivors of congenital diaphragmatic hernia. Pediatr Surg Int. 2005; 21:954–959.
302. Hayward, MJ, Kharasch, V, Sheils, C, et al. Predicting inadequate long-term lung development in children with congenital diaphragmatic hernia: An analysis of longitudinal changes in ventilation and perfusion. J Pediatr Surg. 2007; 42:112–116.
303. Bernbaum, J, Schwartz, IP, Gerdes, M, et al. Survivors of extracorporeal membrane oxygenation at 1 year of age: The relationship of primary diagnosis with health and neurodevelopmental sequelae. Pediatrics. 1995; 96:907–913.
304. Jaillard, SM, Pierrat, V, Dubois, A, et al. Outcome at 2 years of infants with congenital diaphragmatic hernia: A population-based study. Ann Thorac Surg. 2003; 75:250–256.
305. Chen, C, Jeruss, S, Chapman, JS, et al. Long-term functional impact of congenital diaphragmatic hernia repair on children. J Pediatr Surg. 2007; 42:657–665.
306. Danzer, E, Gerdes, M, Bernbaum, J, et al. Neurodevelopmental outcome of infants with congenital diaphragmatic hernia prospectively enrolled in an interdisciplinary follow-up program. J Pediatr Surg. 2010; 45:1759–1766.
307. Van Meurs, KP, Robbins, ST, Reed, VL, et al. Congenitaldiaphragmatic hernia: Long-term outcome in neonates treated with extracorporeal membrane oxygenation. J Pediatr. 1993; 122:893–899.
308. van der Cammen-van Zijp, MH, Gischler, SJ, Mazer, P, et al. Motor-function and exercise capacity in children with major anatomical congenital anomalies: An evaluation at 5 years of age. Early Hum Dev. 2010; 86:523–528.
309. Nijhuis-van der Sanden, MW, van der Cammen-van Zijp, MH, Janssen, AJ, et al. Motor performance in five-year-old extracorporeal membrane oxygenation survivors: A population-based study. Crit Care. 2009; 13:R47.
310. Jakobson, LS, Frisk, V, Trachsel, D, et al. Visual and fine-motor outcomes in adolescent survivors of high-risk congenital diaphragmatic hernia who did not receive extracorporeal membrane oxygenation. J Perinatol. 2009; 29:630–636.
311. Bouman, NH, Koot, HM, Tibboel, D, et al. Children with congenital diaphragmatic hernia are at risk for lower levels of cognitive functioning and increased emotional and behavioral problems. Eur J Pediatr Surg. 2000; 10:3–7.
312. McGahren, ED, Mallik, K, Rodgers, BM. Neurological outcome is diminished in survivors of congenital diaphragmatic hernia requiring extracorporeal membrane oxygenation. J Pediatr Surg. 1997; 32:1216–1220.
313. Masumoto, K, Nagata, K, Uesugi, T, et al. Risk factors for sensorineural hearing loss in survivors with severe congenital diaphragmatic hernia. Eur J Pediatr. 2007; 166:607–612.
314. Qi, B, Soto, C, Diez-Pardo, JA, et al. An experimental study on the pathogenesis of gastroesophageal reflux after repair of diaphragmatic hernia. J Pediatr Surg. 1997; 32:1310–1313.
315. Stolar, CJ, Crisafi, MA, Driscoll, YT. Neurocognitive outcome for neonates treated with extracorporeal membrane oxygenation: Are infants with congenital diaphragmatic hernia different? J Pediatr Surg. 1995; 30:366–371.
316. Colvin, J, Bower, C, Dickinson, JE, et al. Outcomes of congenital diaphragmatic hernia: A population-based study in Western Australia. Pediatrics. 2005; 116:e356–e363.
317. Nield, TA, Langenbacher, D, Poulsen, MK, et al. Neurodevelopmental outcome at 3.5 years of age in children treated with extracorporeal life support: Relationship to primary diagnosis. J Pediatr. 2000; 136:338–344.
318. Robertson, CM, Tyebkhan, JM, Hagler, ME, et al. Late-onset, progressive sensorineural hearing loss after severe neonatal respiratory failure. Otol Neurotol. 2002; 23:353–356.
319. Fligor, BJ, Neault, MW, Mullen, CH, et al. Factors associated with sensorineural hearing loss among survivors of extracorporeal membrane oxygenation therapy. Pediatrics. 2005; 115:1519–1528.
320. Bagolan, P, Morini, F. Long-term follow up of infants with congenital diaphragmatic hernia. Semin Pediatr Surg. 2007; 16:134–144.
321. Muratore, CS, Utter, S, Jaksic, T, et al. Nutritional morbidity in survivors of congenital diaphragmatic hernia. J Pediatr Surg. 2001; 36:1171–1176.
322. Vanamo, K, Rintala, RJ, Lindahl, H, et al. Long-term gastrointestinal morbidity in patients with congenital diaphragmatic defects. J Pediatr Surg. 1996; 31:551–554.
323. Cortes, RA, Keller, RL, Townsend, T, et al. Survival of severe congenital diaphragmatic hernia has morbid consequences. J Pediatr Surg. 2005; 40:36–45.
324. Stolar, CJ, Levy, JP, Dillon, PW, et al. Anatomic and functional abnormalities of the esophagus in infants surviving congenital diaphragmatic hernia. Am J Surg. 1990; 159:204–207.
325. Kieffer, J, Sapin, E, Berg, A, et al. Gastroesophageal reflux after repair of congenital diaphragmatic hernia. J Pediatr Surg. 1995; 30:1330–1333.
326. Koot, VC, Bergmeijer, JH, Bos, AP, et al. Incidence and management of gastroesophageal reflux after repair of congenital diaphragmatic hernia. J Pediatr Surg. 1993; 28:48–52.
327. Nagaya, M, Akatsuka, H, Kato, J. Gastroesophageal reflux occurring after repair of congenital diaphragmatic hernia. J Pediatr Surg. 1994; 29:1447–1451.
328. Kimmelstiel, FM, Holgersen, LO, Hilfer, C. Retrosternal (Morgagni) hernia with small bowel obstruction secondary to a Richter’s incarceration. J Pediatr Surg. 1987; 22:998–1000.
329. Sarihan, H, Imamoglu, M, Abes, M, et al. Pediatric Morgagni hernia. Report of two cases. J Cardiovasc Surg (Torino). 1996; 37:195–197.
330. de Fonseca, JM, Davies, MR, Bolton, KD. Congenital hydropericardium associated with the herniation of part of the liver into the pericardial sac. J Pediatr Surg. 1987; 22:851–853.
331. Dutta, S, Albanese, CT. Use of a prosthetic patch for laparoscopic repair of Morgagni diaphragmatic hernia in children. J Laparoendosc Adv Surg Tech A. 2007; 17:391–394.
332. Ponsky, TA, Lukish, JR, Nobuhara, K, et al. Laparoscopy is useful in the diagnosis and management of foramen of Morgagni hernia in children. Surg Laparosc Endosc Percutan Tech. 2002; 12:375–377.
333. Cantrell, JR, Haller, JA, Ravitch, MM. A syndrome of congenital defects involving the abdominal wall, sternum, diaphragm, pericardium, and heart. Surg Gynecol Obstet. 1958; 107:602–614.
334. Symbas, PN, Hatcher, CR, Jr., Waldo, W. Diaphragmatic eventration in infancy and childhood. Ann Thorac Surg. 1977; 24:113–119.
335. Wayne, ER, Campbell, JB, Burrington, JD, et al. Eventration of the diaphragm. J Pediatr Surg. 1974; 9:643–651.
336. Elberg, JJ, Brok, KE, Pedersen, SA, et al. Congenital bilateral eventration of the diaphragm in a pair of male twins. J Pediatr Surg. 1989; 24:1140–1141.
337. Akay, TH, Ozkan, S, Gultekin, B, et al. Diaphragmatic paralysis after cardiac surgery in children: Incidence, prognosis and surgical management. Pediatr Surg Int. 2006; 22:341–346.
338. Joho-Arreola, AL, Bauersfeld, U, Stauffer, UG, et al. Incidence and treatment of diaphragmatic paralysis after cardiac surgery in children. Eur J Cardiothorac Surg. 2005; 27:53–57.
339. Yazici, M, Karaca, I, Arikan, A, et al. Congenital eventration of the diaphragm in children: 25 years’ experience in three pediatric surgery centers. Eur J Pediatr Surg. 2003; 13:298–301.
340. Smith, CD, Sade, RM, Crawford, FA, et al. Diaphragmatic paralysis and eventration in infants. J Thorac Cardiovasc Surg. 1986; 91:490–497.
341. Becmeur, F, Talon, I, Schaarschmidt, K, et al. Thoracoscopic diaphragmatic eventration repair in children: About 10 cases. J Pediatr Surg. 2005; 40:1712–1715.
342. Guvenc, BH, Korkmaz, M, Avtan, L, et al. Thoracoscopic diaphragm plication in children and indications for conversion to open thoracotomy. J Laparoendosc Adv Surg Tech A. 2004; 14:302–305.
343. Huttl, TP, Wichmann, MW, Reichart, B, et al. Laparoscopic diaphragmatic plication: Long-term results of a novel surgical technique for postoperative phrenic nerve palsy. Surg Endosc. 2004; 18:547–551.