[level-membership-for-neurology-category]
Chapter 13 Congenital Cerebral Impairments
Cerebral Palsy
Preventable obstetric injuries, such as anoxia, account for less than 10% of cases. In contrast, unalterable antepartum factors account for more than 70%. For example, CP is often a manifestation of genetic or congenital malformations, such as microgyria (small cerebral gyri), pachygyria (thickened gyri), hydrocephalus, and porencephaly (see Fig. 20-4). Also, because 5% of CP children have a first-degree relative with a similar condition, as yet undetermined genetic factors undoubtedly determine or at least contribute to many cases.
Several conditions mimic CP closely enough to represent diagnostic pitfalls. Several of the disorders included in this chapter cause motor impairments that physicians may mistake for CP. In addition, insidiously advancing leukoencephalopathies (see Chapter 15) may produce spastic paresis almost identical to spastic CP. Dopa-responsive dystonia gives rise to a disorder similar to choreoathetotic CP (see later and Chapter 18). Deafness, which may occur alone or be accompanied by other neurologic disabilities, may mimic CP or mental retardation.*
Neurologists often divide CP into four varieties. Each one has a characteristic motor impairment, such as spastic paresis or choreoathetosis (Fig. 13-1), and a correlation with epilepsy and mental retardation. Neurologists usually do not diagnose CP in infants until they are at least 4 months old and, in some cases, not until they are 4 years old. Moreover, once children have an established motor deficit attributable to a perinatal cerebral injury, it must not progress as the affected child grows. In fact, impairments may seem to recede as children learn compensatory strategies and benefit from various therapies.
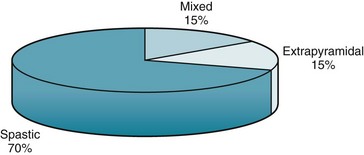
FIGURE 13-1 Most cases of cerebral palsy (CP) are varieties of spastic CP: hemiplegic, diplegic, and quadriplegic. Because studies vary, the percentages are approximations.
Although CP-induced motor impairments and associated mental retardation remain stable, comorbid epilepsy may further impair the CP child. Although epilepsy may not appear during infancy, it is usually evident before 5 years of age. Its incidence roughly corresponds to the severity of physical impairments and mental retardation (Fig. 13-2).
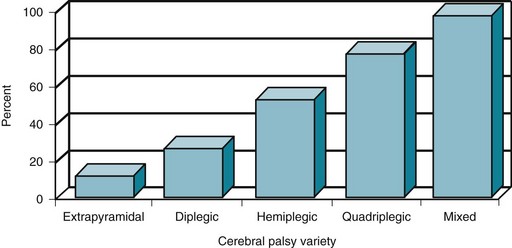
FIGURE 13-2 The proportion of cerebral palsy (CP) patients with mental retardation and epilepsy increases with more extensive cerebral disease. The incidence of those complications in choreoathetosis or extrapyramidal CP is only approximately 10%; however, the incidence in diplegic CP is 25%; hemiplegic CP 50%; quadriplegic CP 75%; and mixed CP 95%.
Spastic Cerebral Palsy
Diplegic CP (spastic diplegia) consists of bilateral symmetric paresis characteristically involving the legs more than the arms (Fig. 13-3). This CP variety usually forces children to hold their legs straight, drawn together (adducted), and crossed over each other (“scissored”). It also forces them to keep their feet and toes pointed downward (extended). When children begin to walk, this posture obligates them to stand on their toes with their legs brought closely together.
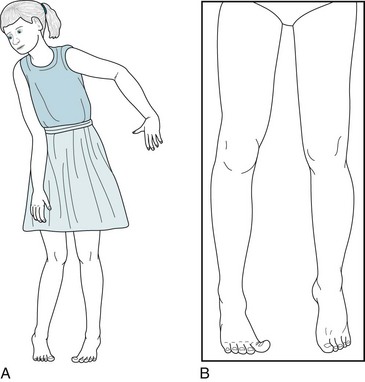
FIGURE 13-3 A, Spastic diplegia in this 10-year-old girl with low-normal intelligence causes straightening, inturning, and adduction of her legs; a tiptoe stance; and scissor-like gait. Her uncoordinated, awkward arm movements (posturing) also reflect her cerebral palsy. B, Another patient with spastic diplegia, an 18-year-old college engineering student, has typical increased muscle tone, adduction of his legs, and “toe-walking.” He also has a subtle, sustained right-sided Babinski sign.
Hemiplegic CP consists of spastic hemiparesis that typically affects the face and arm more than the leg (Fig. 13-4). The motor impairment of children and adults with hemiplegic CP resembles adults with strokes from middle cerebral artery occlusions, but they differ in three respects. While normal infants younger than 2 year old do not show hand preference, infants with hemiplegic CP show premature handedness. For example, unequivocal right-handedness in infants younger than 1 year old may mean that the left hand, if not the entire left arm, is paretic. Because left hemisphere injury during the perinatal period forces the right hemisphere to assume dominance, children and adults with congenital right hemiparesis maintain dominance in the right hemisphere and have no language impairment (aphasia). Their lack of aphasia accompanying right hemiparesis contrasts starkly with the results of a left middle cerebral artery stroke, where aphasia is often the most devastating result of damage to the mature left-sided perisylvian language arc. Finally, older children and adults with hemiplegic CP show growth arrest of the affected limbs. Compared to their normal limbs, affected ones are shorter, less muscular, and weaker.
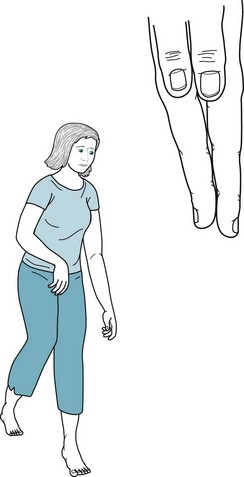
FIGURE 13-4 Hemiparetic since birth, this 28-year-old woman with normal intelligence holds her spastic and weak arm, wrist, and fingers in a flexed posture. Growth arrest of her right hand has led to shortened fingers and a less broad thumb nail bed. Similarly, her right leg, especially the heel (Achilles) tendon, is short. The growth arrest causes her to walk on her right toes and circumduct that leg. Her posture and gait are similar to that of adults after a left middle cerebral artery infarction (see Figs. 2-3 to 2-5).
As a general rule, the underlying cerebral damage in quadriplegic CP is worse than in spastic hemiparesis and much worse than in spastic diplegia. Thus, epilepsy and mental retardation occur more frequently in quadriplegic than in hemiplegic CP, and occur much more frequently than in diplegic CP. Physical and occupational therapy, bracing, and orthotics may all help these children. Neurologists often reduce spasticity by recommending surgery that transposes or lengthens tendons; prescribing oral antispasticity medications, such as baclofen and tizanidine; and administering intramuscular injections of botulinum toxin (see Chapter 18). However, epilepsy in these children resists treatment. Seizure control often requires two or more antiepileptic drugs (AEDs), which in turn may produce undesirable side effects, particularly sedation, cognitive impairments, paradoxical hyperactivity, and other behavioral disturbances.
Extrapyramidal Cerebral Palsy
Involuntary writhing movements (athetosis) of the face, tongue, hands, and feet punctuated by jerking movements (chorea) of the trunk, arms, and legs – embraced by the term choreoathetosis – define extrapyramidal CP (Fig. 13-5). Although choreoathetosis may remain subtle throughout a patient’s lifetime, it often interferes with fine hand movements, walking, and even sitting. Another manifestation – involuntary larynx, pharynx, and diaphragm movements – may lead to incomprehensible dysarthria.
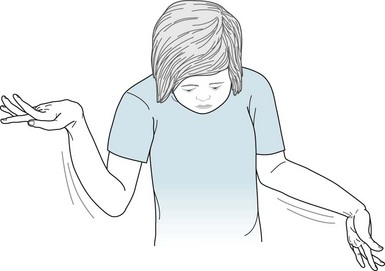
FIGURE 13-5 The slow sinuous movements (athetosis) of her wrists, hands, and fingers, present since age 3 years, signify that this 13-year-old girl has congenital cerebral palsy-induced choreoathetosis. The athetosis intermittently forces her hands into flexion at the wrist and her fingers into extension with overlapping positions.
Physicians should distinguish choreoathetotic CP from dopamine-responsive dystonia, which produces similar involuntary movements in young children (see Chapter 18). In short, unlike CP, dopamine-responsive dystonia is progressive (albeit slowly), fluctuating in a characteristic diurnal pattern at its onset, and, most important, responsive to small doses of levodopa (L-dopa). Despite the differences, the clinical similarity can be so great that many neurologists insist on a therapeutic trial of L-dopa before accepting a diagnosis of choreoathetotic CP.
Neural Tube Closure Defects
During the third and fourth weeks of gestation, dorsal ectoderm normally invaginates to form a closed, midline neural tube that eventually forms the brain and spinal cord and seals itself at both ends (Fig. 13-6, A). While ectoderm thus gives rise to the CNS as well as the skin, mesoderm forms the coverings of the CNS – the meninges, vertebrae, and skull.
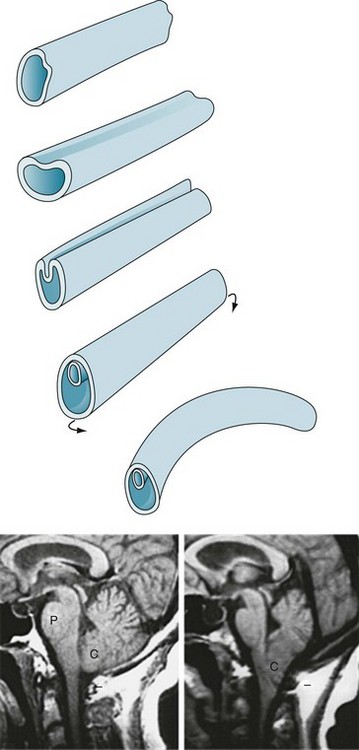
FIGURE 13-6 Top, The neural tube forms during the third and fourth weeks of gestation. Its formation begins when the embryo’s external layer, the ectoderm, invaginates to shape a distinct, midline neural tube that eventually closes at both ends. Once closed, the embryo begins to bend into a curved fetus with the tube on the convex surface. Failure to complete this process results in defects, most commonly at the upper and lower ends of the spinal cord. Bottom left, The magnetic resonance imaging (MRI) scan shows the normal relationship of several of the structures contained in the posterior fossa: the pons (P), medulla (unmarked), cerebellum (C), and the fourth ventricle (the black, cerebrospinal fluid-filled, triangular area between the pons and middle of the cerebellum). Note that the lower portion of the cerebellum remains above the foramen magnum (indicated by a short horizontal line). Bottom right, This MRI shows an Arnold–Chiari abnormality. The lower portion of the cerebellum, which includes the tonsils, and the medulla protrude below the foramen magnum. In addition, in severe cases, aqueductal stenosis causes hydrocephalus.
Upper Neural Tube Closure Defects
A group of malformations, collectively termed the Arnold–Chiari malformation, constitute a variety of upper neural tube closure defects. Usually not obvious by external appearances, the Arnold–Chiari malformation consists of downward displacement of the lower portion of the medulla and cerebellum through the foramen magnum (Figs 13-6 and 20-22). In older children and adults, who may previously have escaped detection, this malformation may produce headaches (especially when bending), bulbar palsy, and neck pain. Patients with compression of the medulla or cerebellum require “unroofing” of the upper cervical spine and occipital portion of the skull.
Lower Neural Tube Closure Defects
In meningocele, a more serious variety, the meninges and skin protrude through a lumbosacral spine defect to form a large, CSF-filled bulge. Although this condition may remain asymptomatic, it frequently causes symptoms originating in dysfunction of the lumbar and sacral nerves, such as leg weakness, gait impairment, and bladder-emptying problems. Thus progressive hydronephrosis often complicates the deficit. Meningomyelocele (myelomeningocele), which occurs far more frequently than meningocele, is the most serious variety. It consists of a tangle of a rudimentary lower spinal cord, lumbar and sacral nerve roots, and meninges protruding into a saclike structure overlying the lumbosacral spine (Fig. 13-7). The disrupted nerve tissue causes paraparesis, areflexia, and incontinence. In addition, hydrocephalus and other brain abnormalities are comorbid in about 25% of cases. Approximately 10% of infants born with meningomyelocele die from the defect.
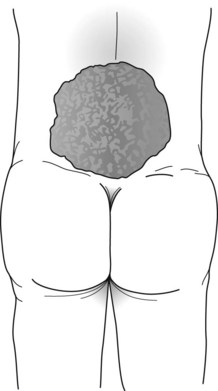
FIGURE 13-7 The meningomyelocele of this newborn infant has a typical broad-based, loose, translucent sac of thin, friable skin arising from the lumbar area. It contains rudiments of a spinal cord and lumbosacral nerves. Its surface weeps a mixture of serum and cerebrospinal fluid. The infant’s legs, lacking innervation, remain weak, flaccid, and areflexic. Similarly, the bladder, also lacking innervation, distends.
Neurocutaneous Disorders
Tuberous Sclerosis
Tuberous sclerosis usually causes conspicuous smooth and firm nodules, facial angiofibromas (adenoma sebaceum), on the malar surface of the face (Fig. 13-8), but this illness-defining skin lesion usually fails to appear until adolescence. However, during infancy and childhood, the skin shows several other characteristics: subtle hypopigmented macules (ash-leaf spots); shagreen patches, which are leathery, scaly areas, on the lower trunk and buttocks; and periungual fibromas of the fingers.
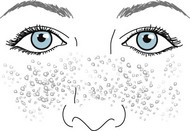
FIGURE 13-8 Adenoma sebaceum (facial angiofibromas), the cutaneous component of tuberous sclerosis, consists of nodules several millimeters in diameter, firm, and uniformly pale. They spread over the malar surface of the face. Although adenoma sebaceum may resemble acne, acne pimples have a liquid (pus) center surrounded by inflammation. Also acne pimples accumulate on the trunk as well as the face.
In another important aspect of the illness, some tuberous sclerosis children display autistic behavior. Thus, neurologists consider tuberous sclerosis as one of several neurologic causes of autism-like symptoms (Box 13-1).
Neurofibromatosis
Café-au-lait spots, the signature of neurofibromatosis, are areas of uniformly light brown, oval, and flat skin (Fig. 13-9). Although individual café-au-lait spots are found in at least 10% of normal individuals, the presence of more than six of them, each larger than 5 mm in children and 1.5 cm in adults, strongly suggests a diagnosis of neurofibromatosis. Freckling in the axilla and groin – two skin surfaces sheltered from sun exposure – often accompanies NF1-related café-au-lait spots.
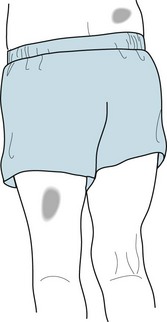
FIGURE 13-9 Café-au-lait spots are flat, light-brown skin lesions. The diagnosis of neurofibromatosis requires six or more lesions, each measuring at least 1.5 cm in adults and 0.5 cm in children.
Neurofibromas consist of soft, palpable, subcutaneous growths, each a few millimeters to several centimeters in size, that emerge along peripheral nerves (Figs 13-10 and 13-11). They can also grow from nerve roots within the spinal canal and compress the spinal cord or cauda equina. They occasionally reach grotesque proportions or induce extraordinary growth of an affected limb. However, the famous nineteenth-century “elephant man,” Joseph Merrick, commonly cited as an example of neurofibromatosis, probably suffered from a related condition, Proteus syndrome.
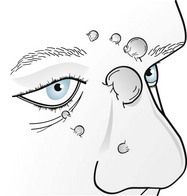
FIGURE 13-10 Neurofibromas often grow to several centimeters of disfiguring protuberances on the face.
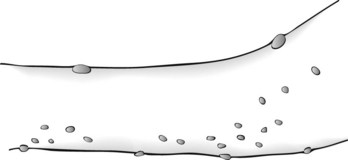
FIGURE 13-11 Neurofibromas are often subtle, multiple, subcutaneous, soft, and typically less than 0.5 cm in size.
Lisch nodules, the least obvious but most common manifestation, are multiple, asymptomatic, macroscopic, yellow to brown nodules (melanocytic hamartomas) situated on the iris (Fig. 13-12). Although a slit-lamp examination may be required to detect Lisch nodules and then differentiate them from inconsequential pigment collections, they are almost pathognomonic of the disorder.
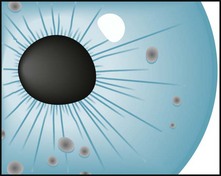
FIGURE 13-12 Lisch nodules, virtually pathognomonic of neurofibromatosis type 1, are pigmented aggregations on the iris that are often visible with the unaided eye.
Neurofibromatosis type 2 (NF2), which occurs only 10% as frequently as NF1, is an almost completely different disorder. NF2, also called familial acoustic neuroma or “central-type” neurofibromatosis, is characterized by bilateral acoustic neuromas (vestibular schwannomas) that steadily impair hearing until deafness ensues. It may induce a few neurofibromas and large, pale café-au-lait spots, but its hallmark remains the acoustic neuromas (see Fig. 20-27). In fact, NF2 is usually unrecognized until acoustic neuromas are discovered.
Sturge–Weber Syndrome
The facial angioma consists of a deep red discoloration (“port-wine stain”) in the distribution of one or more divisions of the trigeminal nerve (Fig. 13-13). Its extent does not correlate with the size of the cerebral abnormality. Clinicians must distinguish it from completely benign, more common skin abnormalities, such as small forehead angiomas (“strawberry nevi”). Also, port-wine stains, even in the trigeminal nerve distribution, are associated with Sturge–Weber syndrome in only 8% of cases. Whether or not facial angiomas are a manifestation of Sturge–Weber syndrome, laser therapy can bleach them.
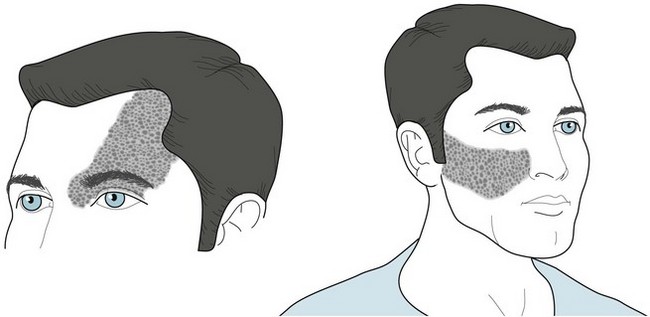
FIGURE 13-13 The cutaneous angioma (port-wine stain) of Sturge–Weber syndrome encompasses one or more divisions of the distribution of the trigeminal nerve (see Fig. 4-12). The commonest sites are the anterior scalp, forehead, and upper eyelid, i.e., the first division of the trigeminal nerve. One-third of patients have bilateral involvement.
Other Genetic Neurologic Disorders
Autosomal Chromosomal Disorders
Phenylketonuria (PKU) (Chromosome 12)
1. The deficiency would prevent the normal metabolism of phenylalanine to tyrosine. Thus, affected untreated individuals have elevated plasma concentrations of phenylalanine and little or no tyrosine.
2. The deficiency would also prevent the normal synthesis of “downstream” neurotransmitters, including dopamine, norepinephrine, and melatonin (see Chapter 21).
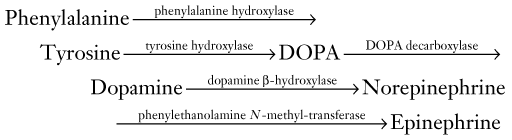
3. A deficiency of phenylalanine hydroxylase would divert phenylalanine metabolism to secondary metabolic pathways. Those pathways yield phenylpyruvic acid and eventually phenylketones, which would be excreted in the urine. Untreated affected individuals thus characteristically have PKU.
Homocystinuria (Chromosome 21)
Cystathionine beta-synthase, along with vitamin B6, converts homocysteine to cystathionine (see Fig. 5-8). A deficiency of this enzyme leads to accumulation not only of homocysteine but also its precursor, methionine. The genetic disorder, homocystinuria, is attributable to a mutation on chromosome 21. Other conditions that lead to accumulation of homocysteine and possibly some of the same clinical manifestations include vitamin B12 deficiency, exposure to nitrous oxide, and use of certain AEDs (such as carbamazepine and phenytoin).
Homocystinuria leads primarily to vascular thrombotic events, particularly strokes in young and middle-aged adults, and mental retardation. The relationship between homocystinuria and strokes is so strong that an elevated serum homocysteine level is a risk factor for stroke (see Chapter 11). The other features of homocystinuria, which reflect malformation of multiple organs, include dislocation of the ocular lens, pectus excavatum or carinatum, and a tall, Marfan-like stature. In addition to mental retardation, which is almost universal, homocystinuria patients often have behavioral disturbances, obsessive-compulsive symptoms, and personality disorders.
Prader–Willi and Angelman Syndromes (Chromosome 15)
Beyond their striking obesity and other physical anomalies (Fig. 13-14), Prader–Willi children have low normal to below normal IQ. They comprise about 1% of children with mental retardation. They also tend toward affective disorders that may, after adolescence, be accompanied by psychotic features. The severity of the obesity does not correlate with either the nature or severity of the neuropsychiatric disturbances, but, like obesity in general, it carries the comorbidities of hypoventilation, hypersomnia, hypertension, diabetes, stroke, and osteoporosis.
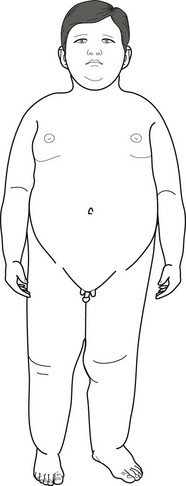
FIGURE 13-14 An 8-year-old boy with Prader–Willi syndrome shows the distinctive obesity, which can reach grotesque proportions, small penis and testicles, short stature, small hands, and short feet. Girls with the syndrome, who also have obesity and hypogonadism, usually have small labia majora and no labia minora. Because both boys and girls with Prader–Willi syndrome also have intellectual disabilities, neurologists remember the manifestations of the illness as the “three Hs”: hyperphagia, hypomentia, and hypogonadism.
Down Syndrome (Trisomy 21)
At 1 in 600 births, Down syndrome is the most frequently occurring disorder in this group. Affected children have distinctive physical features (Fig. 13-15) and mild to moderate mental retardation, with a median IQ of 40–50. They are also plagued by hearing loss, congenital cardiac anomalies, and gastrointestinal disease, and, as adults, the development of hypothyroidism and leukemia.

FIGURE 13-15 Children with Down syndrome are short with low-set ears that have small lobes. Their eyes’ epicanthal folds are widened and the lids appear to slant upward – thus the outdated term “mongolism.” The bridge of the nose is depressed. The tongue, characteristically large, tends to protrude over a slack jaw. Children’s palms are broad with a single midline crease, and their fingers are short and stubby.
Another, almost uniform complication is that, by age 50 years, Down syndrome leads to an Alzheimer-like dementia (see Chapter 7). In fact, one theory holds that both Down syndrome and Alzheimer disease result from a common genetic abnormality on chromosome 21.
Williams Syndrome (Chromosome 7)
Lifelong neuropsychologic oddities and a distinctive, “elfin” facial appearance (Fig. 13-16) characterize Williams syndrome. Children with this disorder are slow to acquire motor milestones, have strikingly poor sense of visuospatial relationships, and cannot perform construction or copying tasks. The majority have ADHD, phobias, or both. In general, they perform in the mild to moderate intellectual disability range on testing, with an average IQ of approximately 65 points. As adults, Williams syndrome individuals rarely find steady employment and develop memory impairment at a greater rate than controls.

FIGURE 13-16 This 10-year-old girl shows the characteristic elfin (elf-like) appearance of Williams syndrome. She is short. Her forehead is broad and her cheeks are prominent. Her nose has a flat bridge, and its nostrils are full and turned slightly upward. Her teeth are hypoplastic and widely spaced.
Velocardiofacial (VCF) Syndrome (Chromosome 22)
A cleft palate or velopharyngeal dysfunction is the most obvious characteristic. The deformity causes palate insufficiency that gives the child’s voice a nasal tone. Hearing the nasality, physicians may further suspect the diagnosis by inspection of the palate at rest and during voluntary retraction. In addition, VCF children may have external ear malformations that cause hearing impairment and worsen the speech impediment. The facial appearance of children with VCF has several characteristic, if not pathognomonic, features (Fig. 13-17).
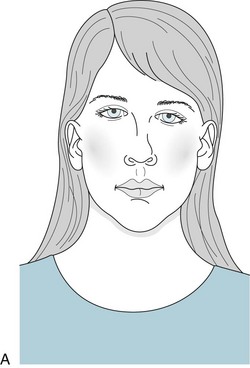
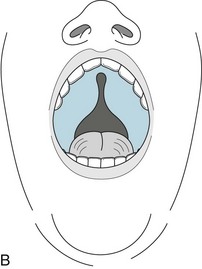
FIGURE 13-17 A, The face of children with velocardiofacial syndrome typically appears long, tapering to a small lower jaw (micrognathia). A tubular nose with a broad tip and small nasal alae, and deformed ears are also typical. B, The cleft palate, another hallmark of the disorder, may not be as overt as in this figure.
Sex-Linked Chromosomal Disorders
Fragile X Syndrome
The fragile X syndrome, like many other genetic disorders, consists of mental retardation, behavioral disturbances, and distinctive nonneurologic physical stigmata (Fig. 13-18). About 70% of boys who inherit the entire or “full” mutation have moderate to severe mental retardation. In contrast, most females carrying the full mutation – typically the mother and sisters of affected boys – show none of the syndrome’s physical stigmata. However, about one-third of them have borderline IQs and one-quarter have IQs of 70 or less.

FIGURE 13-18 This fragile X syndrome boy, with the full mutation resulting in an IQ of 65, has the syndrome’s typical features. He has a prominent forehead and jaw; long, thin face; and large, low-set, “seashell-shaped” ears. Although his penis will remain normal in size for his age, his testicles will grow during puberty to a remarkably large size – two- to threefold greater in volume than normal. This macro-orchidism occurs in almost all fragile X syndrome boys and represents the single most consistent physical anomaly.
The mutation (FMR1) consists of excessive repetitions of the CGG trinucleotide in the X chromosome.*
Rett Syndrome
Rett syndrome girls display two striking neurologic abnormalities: stereotypies and acquired microcephaly (Fig. 13-19). Their stereotypies consist of incessant hand movements, particularly hand wringing, hair pulling, clapping, or flapping. As these stereotypies progress, Rett children often lose hand function. In addition, most of them also have stereotypies that do not involve the hands, such as bruxism, mouthing, and body twisting. Stereotypies usually first appear at about 18 months of age. Although different stereotypies emerge, the first one that appears persists through life.
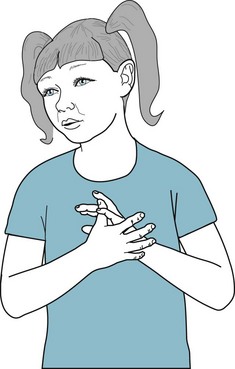
FIGURE 13-19 A 6-year-old girl with Rett syndrome, displaying stereotypies, incessantly moves her hands as though she were washing or clapping. In addition she pulls her hair and has bruxism. In another hallmark of the disease, her head circumference is only 48 cm, which would be normal for a 3-year-old girl, but 2 standard deviations below the mean for her age (51 cm). Because her head circumference had been normal during her first 2–3 years, neurologists determined that she had developed acquired microcephaly. She has also progressively lost her language ability and now cannot speak in a meaningful manner. When her symptoms first appeared, her pediatrician understandably suggested the erroneous diagnosis of autism.
Lesch–Nyhan Syndrome
Another mutation carried on the X chromosome causes the infamous Lesch–Nyhan syndrome, which consists of mental retardation, a distinctive behavioral phenotype of self-mutilation, and dystonia. This disorder is transmitted in a recessive sex-linked pattern and therefore appears only in boys. Because the dystonia is the most salient feature, this book includes Lesch–Nyhan syndrome among the involuntary movement disorders (see Chapter 18).
Turner Syndrome (XO)
Individuals with Turner syndrome have a mutation in or, more usually, absence of one of their sex chromosomes. With a complement of only 45 fully functioning chromosomes, these individuals are usually described as having an “XO” chromosome pattern. From infancy, they are outwardly phenotypically female, but with readily identifiable dysmorphic features (Fig. 13-20). In addition, they do not undergo puberty.
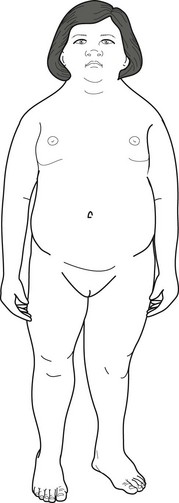
FIGURE 13-20 This 16-year-old girl with Turner syndrome (XO) has mental retardation and the distinctive short stature and webbed neck. As with several other genetic disorders, her ears are low-set (but hidden by a low hairline), her nose is flat, and its bridge spreads into broad epicanthal folds. Because she characteristically failed to undergo puberty, she lacks breast development and other secondary sexual characteristics. Also, her elbows’ carrying angles are relatively straight, which is the male pattern.
Klinefelter Syndrome (XXY)
With an additional X chromosome, Klinefelter syndrome boys are tall but eunuchoid (Fig. 13-21). As young men, their unusual height and lack of secondary sexual characteristics may draw medical attention, but physicians most often diagnose Klinefelter syndrome only after the boys fail to go through puberty. In fact, physicians sometimes first discover the disorder when an infertility evaluation shows that the man has testicular dysgenesis and lacks sperm.
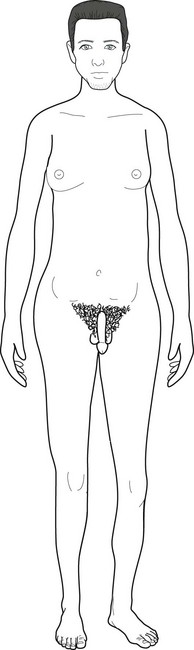
FIGURE 13-21 After a delayed and then incomplete puberty, this 30-year-old man with Klinefelter syndrome (XXY) has grown taller than 6.5 feet (198 cm), largely due to his disproportionately long legs. His body has assumed a eunuchoid habitus with gynecomastia, sparse facial hair, female pattern of pubic hair, and small testicles. In addition to the XXY syndrome, conditions characterized by excessive height include the XYY and Marfan syndromes and homocystinuria.
Adzick NS, Thom EA, Spong CY, et al. A randomized trial of prenatal versus postnatal repair of myelomeningocele. N Engl J Med. 2011;364:993–1004.
Ashwal S, Russman BS, Blaso PA, et al. Practice parameter: Diagnostic assessment of the child with cerebral palsy. Report of the Quality Standards Subcommittee of the American Academy of Neurology and the Practice Committee of the Child Neurology Society. Neurology. 2004;62:851–863.
Bailey DB, Raspa M, Holiday D, et al. Functional skills of individuals with fragile X syndrome: A lifespan cross-sectional analysis. Am J Intellect Dev Disabil. 2009;114:289–303.
Barkovich AJ, Moore KR, Grant E, et al. Diagnostic Imaging: Pediatric Neuroradiology. Philadelphia: Elsevier; 2007.
Belzeaux R, Lacon C. Neurofibromatosis type 1. Psychiatric disorders and quality of life impairment. Presse Med. 2006;35:277–280.
Ben Zeev Ghidoni B. Rett syndrome. Child Adolesc Psychiatric Clin North Am. 2007;16:723–743.
Bourgeois JA, Coffey SM, Rivera SM, et al. A review of fragile X premutation disorders: Expanding the psychiatric perspective. J Clin Psychiatry. 2009;6:852–862.
Canfield RL, Henderson CR, Cory-Slechta DA, et al. Intellectual impairment in children with blood lead concentrations below 10 µg per deciliter. N Engl J Med. 2003;348:1517–1526.
Capone G, Goyal P, Ares W, et al. Neurobehavioral disorders in children, adolescents, and young adults with Down syndrome. Am J Med Genet C Semin Med Genet. 2006;142C:158–172.
Cassidy SB, Driscoll DJ. Prader–Willi syndrome. Eur J Hum Genet. 2009;17:3–13.
Coffey DE, Brumback RA. Pediatric Neuropsychiatry. Philadelphia: Lippincott Williams & Wilkins; 2006.
Crino PB, Nathanson KL, Henske EP. The tuberous sclerosis complex. N Engl J Med. 2006;355:1345–1356.
Detrait ER, George TM, Etchevers HC, et al. Human neural tube defects: Developmental biology, epidemiology, and genetics. Neurotoxicol Teratol. 2005;27:515–524.
Elison S, Stinton C, Howlin P. Health and social outcomes in adults with Williams syndrome. Res Dev Disabil. 2010;31:587–599.
Feinstein C, Chahal L. Psychiatric phenotypes associated with neurogenetic disorders. Psychiatr Clin North Am. 2009;32:15–37.
Green T, Gothelf D, Glaser B, et al. Psychiatric disorders and intellectual functioning throughout development in velocardiofacial (22q11.2 deletion) syndrome. J Am Acad Child Adolesc Psychiatry. 2009;48:1060–1068.
Hadders-Algra M. The Neurological Examination of the Child with Minor Neurological Dysfunction, 3rd ed. London: Mac Keith Press; 2010.
Hagerman RJ, Berry-Kravis E, Kaufman W, et al. Advances in the treatment of fragile X syndrome. Pediatrics. 2008;123:378–390.
Hankins GD, Speer M. Defining the pathogenesis and pathophysiology of neonatal encephalopathy and cerebral palsy. Obstet Gynecol. 2003;102:628–636.
Hiraiwa R, Maegaki Y, Oka A, et al. Behavioral and psychiatric disorders in Prader–Willi syndrome. Brain Dev. 2007;29:535–542.
Howlin P, Udwin O. Outcome in adult life for people with Williams syndrome – results from a survey of 239 families. J Intellect Disabil Res. 2006;50:151–160.
Jacquemont S, Hagerman RJ, Hagerman PJ, et al. Fragile-X syndrome and fragile X-associated tremor/ataxia syndrome: two faces of FMR1. Lancet Neurol. 2007;6:45–55.
Jones KL. Smith’s Recognizable Patterns of Human Malformation, 6th ed. Philadelphia: Elsevier; 2006.
Kobrynski LJ, Sullivan KE. Velocardiofacial syndrome, DiGeorge syndrome: The chromosome 22q11.2 deletion syndromes. Lancet. 2007;370:1443–1452.
Krakovsky G, Huth MM, Lin L, et al. Functional changes in children, adolescents, and young adults with cerebral palsy. Res Dev Disabil. 2007;28:331–340.
Leyfer OT, Woodruff-Borden J, Klein-Tasman B, et al. Prevalence of psychiatric disorders in 4 to 16-year-olds with Williams syndrome. Am J Med Genet. 2006;141B:615–622.
McCarthy J. Behavioral problems and adults with Down syndrome: Childhood risk factors. J Intellect Disabil Res. 2008;52:877–882.
Muzykewicz DA, Newberry P, Danforth N, et al. Psychiatric comorbid conditions in a clinic population of 241 patients with tuberous sclerosis. Epilepsy Behav. 2007;11:506–513.
Neul JL, Kaufman WE, Glaze DG, et al. Rett syndrome: Revised diagnostic criteria and nomenclature. Ann Neurol. 2010;68:944–950.
Pelc K, Cheron G, Dan B. Behavior and neuropsychiatric manifestations in Angelman syndrome. Neuropsychiatric Dis Treat. 2008;4:577–584.
Scheimann AO, Butler MG, Gourash L, et al. Critical analysis of bariatric procedures in Prader–Willi syndrome. J Pediatr Gastroenterol Nutr. 2008;46:80–83.
Siegel MS, Smith WE. Psychiatric features in children with genetic syndromes: Toward functional phenotypes. Child Adolesc Psychiatr Clin North Am. 2010;19:229–261.
Stinton C, Elison S, Howlin P. Mental health problems in adults with Williams syndrome. Am J Intellect Dev Disabil. 2010;115:3–18.
Temudo T, Ramos E, Dias K, et al. Movement disorders in Rett syndrome: An analysis of 60 patients with detected MECP2 mutation and correlation with mutation type. Mov Disord. 2008;23:1384–1390.
Urv TK, Zigman WB, Silverman W. Psychiatric symptoms in adults with Down syndrome and Alzheimer’s disease. Am J Intellect Dev Disabil. 2010;115:265–276.
Vignoli A, LaBriola F, Canevini MP. Evolution of stereotypies in adolescents and women with Rett syndrome. Mov Disord. 2009;24:1379–1383.
Walker A, Kaufman DM, Solomon G, et al. Child and Adolescent Neurology for Psychiatrists. Philadelphia: Lippincott Williams & Wilkins; 2008.
Wiedemann HR, Kunze J. Clinical Syndromes, 3rd ed. London: Times Mirror International Publishers; 1997.
Willimas CA. The behavioral phenotypes of the Angelman syndrome. Am J Med Genet C Semin Med Genet. 2010;154C:432–437.
Winterkorn EB, Pulsifer MB, Thiele EA. Cognitive prognosis of patients with tuberous sclerosis. Neurology. 2007;68:62–64.
Chapter 13 Questions and Answers
1–11. Match the neurocutaneous disorders (a–d) with their primary manifestation (1–11).
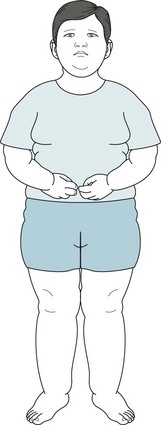
19. An 8-year-old boy, a son of college professors, has slowly and incompletely acquired milestones. Unlike his parents and sibs, he has suffered childhood obesity. His pediatrician recently found that he developed diabetes. He eats incessantly and disregards any limits that his parents place on his calorie consumption. After he broke away from his classmates on a school trip to the children’s zoo and stole food from the rabbit cage, his teachers sent him for a psychiatric evaluation. Which abnormality explains his appearance and behavior?
33. When asked to assess a 9-year-old girl for poor social interactions, a psychiatrist learns that she had late acquisition of her developmental milestones. Her IQ is 88 and she has poor arithmetic and visual-spatial skills. Her handwriting is sloppy and she has impaired fine motor skills. On the other hand, she is articulate and verbal. In an entirely one-sided conversation, the child explained that she has learned two foreign languages, which she speaks with a natural accent, and plays two musical instruments. She has a cute upturned nose and her teeth are hypoplastic and wide-spaced. Which is the most likely disorder?
a. In premature infants, periventricular leukomalacia, the destruction of the white matter surrounding the lateral ventricles, is closely associated with the subsequent development of cerebral palsy. Kernicterus, bilirubin staining of basal ganglia associated with the subsequent development of athetosis, is uncommon because of the prevention of hemolysis from Rhesus factor incompatibility and effective treatments of hyperbilirubinemia with exchange transfusion and phototherapy. Microgyria is small gyri of unknown cause throughout the entire cerebrum or in a limited area. Neurologists or neuropathologists sometimes find this condition in children with mental retardation. Porencephaly is essentially a hole in the brain that might have resulted from an in utero arterial occlusion or simply maldevelopment (see Fig. 20-4).
51. Asked to see a 19-year-old woman who is recovering from ventricular septal defect surgery, a psychiatrist learns from her parents that she has had depressive episodes at least since she was 16 years old and a cleft palate that required surgical repair in infancy. The psychiatrist finds that the woman has nasal speech, external ear deformities, and mild to moderate intellectual disability. Which of the following disorders or syndromes is most likely to underlie the woman’s defects?
52. A 6-year-old girl, born after a normal gestation and delivery to neurologically normal unrelated parents, began to lose her developmental milestones at age 3 years. Her language suffered the most, but then she stopped playing and simply clapped her hands for hours at a time. Looking back at her head circumference determinations, her pediatricians calculated that she had acquired microcephaly. Which mutation most likely explains her appearance, behavior, and development?
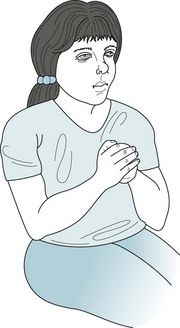
56. Testing shows that this 7-year-old boy, with lifelong slow development, has an IQ score of 60. He has no seizures, physical neurologic deficits, or general medical illness. His parents and two sisters have average intelligence. His X chromosomes tend to break in certain culture media. Which of the following is most likely to occur?

*Neurologists, other physicians, and the public persist in using the term “mental retardation” despite at least the preliminary version of the Diagnostic and Statistical Manual, 5th edition (DSM-5) offering the new but cumbersome designation, Intellectual Developmental Disorder.
*Excessive trinucleotide repeats in other genes produce Friedreich ataxia and other spinocerebellar degenerations, myotonic dystrophy, and Huntington disease (see Chapters 2, 6, and 18, respectively, and Appendix 3D). Neurologists refer to illnesses caused by excessive trinucleotide DNA repeats as polyglutamine illnesses.
[/level-membership-for-neurology-category][not-level-membership-for-neurology-category]
Chapter 13 Congenital Cerebral Impairments
Cerebral Palsy
Preventable obstetric injuries, such as anoxia, account for less than 10% of cases. In contrast, unalterable antepartum factors account for more than 70%. For example, CP is often a manifestation of genetic or congenital malformations, such as microgyria (small cerebral gyri), pachygyria (thickened gyri), hydrocephalus, and porencephaly (see Fig. 20-4). Also, because 5% of CP children have a first-degree relative with a similar condition, as yet undetermined genetic factors undoubtedly determine or at least contribute to many cases.
Several conditions mimic CP closely enough to represent diagnostic pitfalls. Several of the disorders included in this chapter cause motor impairments that physicians may mistake for CP. In addition, insidiously advancing leukoencephalopathies (see Chapter 15) may produce spastic paresis almost identical to spastic CP. Dopa-responsive dystonia gives rise to a disorder similar to choreoathetotic CP (see later and Chapter 18). Deafness, which may occur alone or be accompanied by other neurologic disabilities, may mimic CP or mental retardation.*
Neurologists often divide CP into four varieties. Each one has a characteristic motor impairment, such as spastic paresis or choreoathetosis (Fig. 13-1), and a correlation with epilepsy and mental retardation. Neurologists usually do not diagnose CP in infants until they are at least 4 months old and, in some cases, not until they are 4 years old. Moreover, once children have an established motor deficit attributable to a perinatal cerebral injury, it must not progress as the affected child grows. In fact, impairments may seem to recede as children learn compensatory strategies and benefit from various therapies.
FIGURE 13-1 Most cases of cerebral palsy (CP) are varieties of spastic CP: hemiplegic, diplegic, and quadriplegic. Because studies vary, the percentages are approximations.
Although CP-induced motor impairments and associated mental retardation remain stable, comorbid epilepsy may further impair the CP child. Although epilepsy may not appear during infancy, it is usually evident before 5 years of age. Its incidence roughly corresponds to the severity of physical impairments and mental retardation (Fig. 13-2).
FIGURE 13-2 The proportion of cerebral palsy (CP) patients with mental retardation and epilepsy increases with more extensive cerebral disease. The incidence of those complications in choreoathetosis or extrapyramidal CP is only approximately 10%; however, the incidence in diplegic CP is 25%; hemiplegic CP 50%; quadriplegic CP 75%; and mixed CP 95%.
Spastic Cerebral Palsy
Diplegic CP (spastic diplegia) consists of bilateral symmetric paresis characteristically involving the legs more than the arms (Fig. 13-3). This CP variety usually forces children to hold their legs straight, drawn together (adducted), and crossed over each other (“scissored”). It also forces them to keep their feet and toes pointed downward (extended). When children begin to walk, this posture obligates them to stand on their toes with their legs brought closely together.
FIGURE 13-3 A, Spastic diplegia in this 10-year-old girl with low-normal intelligence causes straightening, inturning, and adduction of her legs; a tiptoe stance; and scissor-like gait. Her uncoordinated, awkward arm movements (posturing) also reflect her cerebral palsy. B, Another patient with spastic diplegia, an 18-year-old college engineering student, has typical increased muscle tone, adduction of his legs, and “toe-walking.” He also has a subtle, sustained right-sided Babinski sign.
Hemiplegic CP consists of spastic hemiparesis that typically affects the face and arm more than the leg (Fig. 13-4). The motor impairment of children and adults with hemiplegic CP resembles adults with strokes from middle cerebral artery occlusions, but they differ in three respects. While normal infants younger than 2 year old do not show hand preference, infants with hemiplegic CP show premature handedness. For example, unequivocal right-handedness in infants younger than 1 year old may mean that the left hand, if not the entire left arm, is paretic. Because left hemisphere injury during the perinatal period forces the right hemisphere to assume dominance, children and adults with congenital right hemiparesis maintain dominance in the right hemisphere and have no language impairment (aphasia). Their lack of aphasia accompanying right hemiparesis contrasts starkly with the results of a left middle cerebral artery stroke, where aphasia is often the most devastating result of damage to the mature left-sided perisylvian language arc. Finally, older children and adults with hemiplegic CP show growth arrest of the affected limbs. Compared to their normal limbs, affected ones are shorter, less muscular, and weaker.
FIGURE 13-4 Hemiparetic since birth, this 28-year-old woman with normal intelligence holds her spastic and weak arm, wrist, and fingers in a flexed posture. Growth arrest of her right hand has led to shortened fingers and a less broad thumb nail bed. Similarly, her right leg, especially the heel (Achilles) tendon, is short. The growth arrest causes her to walk on her right toes and circumduct that leg. Her posture and gait are similar to that of adults after a left middle cerebral artery infarction (see Figs. 2-3 to 2-5).
As a general rule, the underlying cerebral damage in quadriplegic CP is worse than in spastic hemiparesis and much worse than in spastic diplegia. Thus, epilepsy and mental retardation occur more frequently in quadriplegic than in hemiplegic CP, and occur much more frequently than in diplegic CP. Physical and occupational therapy, bracing, and orthotics may all help these children. Neurologists often reduce spasticity by recommending surgery that transposes or lengthens tendons; prescribing oral antispasticity medications, such as baclofen and tizanidine; and administering intramuscular injections of botulinum toxin (see Chapter 18). However, epilepsy in these children resists treatment. Seizure control often requires two or more antiepileptic drugs (AEDs), which in turn may produce undesirable side effects, particularly sedation, cognitive impairments, paradoxical hyperactivity, and other behavioral disturbances.
Extrapyramidal Cerebral Palsy
Involuntary writhing movements (athetosis) of the face, tongue, hands, and feet punctuated by jerking movements (chorea) of the trunk, arms, and legs – embraced by the term choreoathetosis – define extrapyramidal CP (Fig. 13-5). Although choreoathetosis may remain subtle throughout a patient’s lifetime, it often interferes with fine hand movements, walking, and even sitting. Another manifestation – involuntary larynx, pharynx, and diaphragm movements – may lead to incomprehensible dysarthria.
FIGURE 13-5 The slow sinuous movements (athetosis) of her wrists, hands, and fingers, present since age 3 years, signify that this 13-year-old girl has congenital cerebral palsy-induced choreoathetosis. The athetosis intermittently forces her hands into flexion at the wrist and her fingers into extension with overlapping positions.
Physicians should distinguish choreoathetotic CP from dopamine-responsive dystonia, which produces similar involuntary movements in young children (see Chapter 18). In short, unlike CP, dopamine-responsive dystonia is progressive (albeit slowly), fluctuating in a characteristic diurnal pattern at its onset, and, most important, responsive to small doses of levodopa (L-dopa). Despite the differences, the clinical similarity can be so great that many neurologists insist on a therapeutic trial of L-dopa before accepting a diagnosis of choreoathetotic CP.
Neural Tube Closure Defects
During the third and fourth weeks of gestation, dorsal ectoderm normally invaginates to form a closed, midline neural tube that eventually forms the brain and spinal cord and seals itself at both ends (Fig. 13-6, A). While ectoderm thus gives rise to the CNS as well as the skin, mesoderm forms the coverings of the CNS – the meninges, vertebrae, and skull.
FIGURE 13-6 Top, The neural tube forms during the third and fourth weeks of gestation. Its formation begins when the embryo’s external layer, the ectoderm, invaginates to shape a distinct, midline neural tube that eventually closes at both ends. Once closed, the embryo begins to bend into a curved fetus with the tube on the convex surface. Failure to complete this process results in defects, most commonly at the upper and lower ends of the spinal cord. Bottom left, The magnetic resonance imaging (MRI) scan shows the normal relationship of several of the structures contained in the posterior fossa: the pons (P), medulla (unmarked), cerebellum (C), and the fourth ventricle (the black, cerebrospinal fluid-filled, triangular area between the pons and middle of the cerebellum). Note that the lower portion of the cerebellum remains above the foramen magnum (indicated by a short horizontal line). Bottom right, This MRI shows an Arnold–Chiari abnormality. The lower portion of the cerebellum, which includes the tonsils, and the medulla protrude below the foramen magnum. In addition, in severe cases, aqueductal stenosis causes hydrocephalus.
Upper Neural Tube Closure Defects
A group of malformations, collectively termed the Arnold–Chiari malformation, constitute a variety of upper neural tube closure defects. Usually not obvious by external appearances, the Arnold–Chiari malformation consists of downward displacement of the lower portion of the medulla and cerebellum through the foramen magnum (Figs 13-6 and 20-22). In older children and adults, who may previously have escaped detection, this malformation may produce headaches (especially when bending), bulbar palsy, and neck pain. Patients with compression of the medulla or cerebellum require “unroofing” of the upper cervical spine and occipital portion of the skull.
Lower Neural Tube Closure Defects
In meningocele, a more serious variety, the meninges and skin protrude through a lumbosacral spine defect to form a large, CSF-filled bulge. Although this condition may remain asymptomatic, it frequently causes symptoms originating in dysfunction of the lumbar and sacral nerves, such as leg weakness, gait impairment, and bladder-emptying problems. Thus progressive hydronephrosis often complicates the deficit. Meningomyelocele (myelomeningocele), which occurs far more frequently than meningocele, is the most serious variety. It consists of a tangle of a rudimentary lower spinal cord, lumbar and sacral nerve roots, and meninges protruding into a saclike structure overlying the lumbosacral spine (Fig. 13-7). The disrupted nerve tissue causes paraparesis, areflexia, and incontinence. In addition, hydrocephalus and other brain abnormalities are comorbid in about 25% of cases. Approximately 10% of infants born with meningomyelocele die from the defect.
FIGURE 13-7 The meningomyelocele of this newborn infant has a typical broad-based, loose, translucent sac of thin, friable skin arising from the lumbar area. It contains rudiments of a spinal cord and lumbosacral nerves. Its surface weeps a mixture of serum and cerebrospinal fluid. The infant’s legs, lacking innervation, remain weak, flaccid, and areflexic. Similarly, the bladder, also lacking innervation, distends.
Neurocutaneous Disorders
Tuberous Sclerosis
Tuberous sclerosis usually causes conspicuous smooth and firm nodules, facial angiofibromas (adenoma sebaceum), on the malar surface of the face (Fig. 13-8), but this illness-defining skin lesion usually fails to appear until adolescence. However, during infancy and childhood, the skin shows several other characteristics: subtle hypopigmented macules (ash-leaf spots); shagreen patches, which are leathery, scaly areas, on the lower trunk and buttocks; and periungual fibromas of the fingers.
FIGURE 13-8 Adenoma sebaceum (facial angiofibromas), the cutaneous component of tuberous sclerosis, consists of nodules several millimeters in diameter, firm, and uniformly pale. They spread over the malar surface of the face. Although adenoma sebaceum may resemble acne, acne pimples have a liquid (pus) center surrounded by inflammation. Also acne pimples accumulate on the trunk as well as the face.
In another important aspect of the illness, some tuberous sclerosis children display autistic behavior. Thus, neurologists consider tuberous sclerosis as one of several neurologic causes of autism-like symptoms (Box 13-1).
Neurofibromatosis
Café-au-lait spots, the signature of neurofibromatosis, are areas of uniformly light brown, oval, and flat skin (Fig. 13-9). Although individual café-au-lait spots are found in at least 10% of normal individuals, the presence of more than six of them, each larger than 5 mm in children and 1.5 cm in adults, strongly suggests a diagnosis of neurofibromatosis. Freckling in the axilla and groin – two skin surfaces sheltered from sun exposure – often accompanies NF1-related café-au-lait spots.
FIGURE 13-9 Café-au-lait spots are flat, light-brown skin lesions. The diagnosis of neurofibromatosis requires six or more lesions, each measuring at least 1.5 cm in adults and 0.5 cm in children.
Neurofibromas consist of soft, palpable, subcutaneous growths, each a few millimeters to several centimeters in size, that emerge along peripheral nerves (Figs 13-10 and 13-11). They can also grow from nerve roots within the spinal canal and compress the spinal cord or cauda equina. They occasionally reach grotesque proportions or induce extraordinary growth of an affected limb. However, the famous nineteenth-century “elephant man,” Joseph Merrick, commonly cited as an example of neurofibromatosis, probably suffered from a related condition, Proteus syndrome.
FIGURE 13-10 Neurofibromas often grow to several centimeters of disfiguring protuberances on the face.
FIGURE 13-11 Neurofibromas are often subtle, multiple, subcutaneous, soft, and typically less than 0.5 cm in size.
Lisch nodules, the least obvious but most common manifestation, are multiple, asymptomatic, macroscopic, yellow to brown nodules (melanocytic hamartomas) situated on the iris (Fig. 13-12). Although a slit-lamp examination may be required to detect Lisch nodules and then differentiate them from inconsequential pigment collections, they are almost pathognomonic of the disorder.
FIGURE 13-12 Lisch nodules, virtually pathognomonic of neurofibromatosis type 1, are pigmented aggregations on the iris that are often visible with the unaided eye.
Neurofibromatosis type 2 (NF2), which occurs only 10% as frequently as NF1, is an almost completely different disorder. NF2, also called familial acoustic neuroma or “central-type” neurofibromatosis, is characterized by bilateral acoustic neuromas (vestibular schwannomas) that steadily impair hearing until deafness ensues. It may induce a few neurofibromas and large, pale café-au-lait spots, but its hallmark remains the acoustic neuromas (see Fig. 20-27). In fact, NF2 is usually unrecognized until acoustic neuromas are discovered.
Sturge–Weber Syndrome
The facial angioma consists of a deep red discoloration (“port-wine stain”) in the distribution of one or more divisions of the trigeminal nerve (Fig. 13-13). Its extent does not correlate with the size of the cerebral abnormality. Clinicians must distinguish it from completely benign, more common skin abnormalities, such as small forehead angiomas (“strawberry nevi”). Also, port-wine stains, even in the trigeminal nerve distribution, are associated with Sturge–Weber syndrome in only 8% of cases. Whether or not facial angiomas are a manifestation of Sturge–Weber syndrome, laser therapy can bleach them.
FIGURE 13-13 The cutaneous angioma (port-wine stain) of Sturge–Weber syndrome encompasses one or more divisions of the distribution of the trigeminal nerve (see Fig. 4-12). The commonest sites are the anterior scalp, forehead, and upper eyelid, i.e., the first division of the trigeminal nerve. One-third of patients have bilateral involvement.
Other Genetic Neurologic Disorders
Autosomal Chromosomal Disorders
Phenylketonuria (PKU) (Chromosome 12)
1. The deficiency would prevent the normal metabolism of phenylalanine to tyrosine. Thus, affected untreated individuals have elevated plasma concentrations of phenylalanine and little or no tyrosine.
[/not-level-membership-for-neurology-category]
