Congenital Bronchopulmonary Malformations
Congenital bronchopulmonary malformations (BPMs) represent a continuum of abnormalities of the bronchopulmonary unit for which classification and management remain in evolution. An improved understanding of the molecular mechanisms underlying the embryologic development of the lung and the pathogenesis of BPMs suggests that these lesions may have similar mechanistic origins that differ in developmental timing or location in the bronchopulmonary tree.1 From a clinical perspective, improvements in prenatal imaging and increasing observational experience have resulted in a better understanding of the natural history of these anomalies, and a better predictive capacity for pre-, peri-, and postnatal events. Finally, postnatal treatment for the majority of lesions has improved with advances in neonatal care, and the development of thoracoscopic surgery. This chapter discusses the prenatal and postnatal management of the major congenital BPMs with an emphasis on utilizing the thoracoscopic approach for management.
Embryology and Development of the Bronchopulmonary Tree
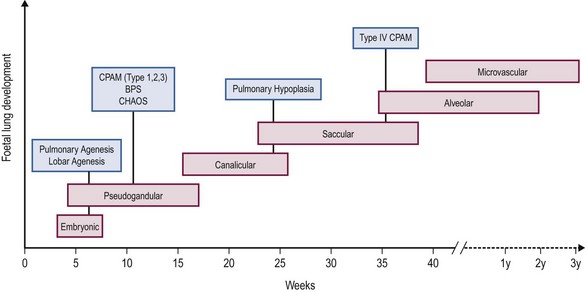
Embryological development of the human lung transitions through six separate stages to form a bronchial tree with greater than 1 × 105 conducting and 1 × 107 respiratory airways.2 These stages include embryonic, pseudoglandular, canalicular, saccular, alveolar, and microvascular. The progression of each stage is a highly coordinated process guided by mesenchymal–epithelial interactions under the influence of a number of regulatory growth factors.
Briefly, the embryonic phase of lung development begins with the formation of the laryngotracheal bud from the anterior portion of the primitive gut. Beginning at week 5 in the pseudoglandular phase, the preacinar airways and blood vessels develop, followed by growth of the bronchial tree until all bronchial divisions are completed by 16 weeks gestation.3 The cannalicular stage follows, and is characterized by capillary growth towards the respiratory epithelium which marks the future blood–air interface.2 The transition to the saccular stage at 24 weeks is marked by the widening of peripheral air spaces distal to the terminal bronchioles with septa formation. The final stages of lung development include the alveolar stage, defined by the formation of secondary septa and budding alveoli, and followed by the microvascular stage with significant alveolar development and maturation. During this complex process, the timing of congenital BPMs and their pathogenesis can be related to specific time points in each of the six developmental stages (Fig. 22-1).
Prenatal Diagnosis and Classification of Congenital Bronchopulmonary Malformations
Malformations
Prenatal diagnosis and fetal therapy for congenital lung malformations have evolved significantly since Adzick et al. described the near universal mortality of congenital pulmonary airway malformation (CPAM)-induced fetal hydrops almost three decades ago.4 Congenital BPMs represent 90% of lung lesions seen in clinical practice and include CPAMs, (formally called congenital cystic adenomatoid malformation or CCAM), bronchopulmonary sequestration (BPS), and congenital lobar emphysema (CLE).5 Other less common malformations are varied and are included in the classification system described by Langston et al.6 (Box 22-1), but will not be discussed in this chapter.
Prenatal ultrasonography (US) functions as a window into fetal development and is the most common mode of prenatal diagnosis of congenital thoracic abnormalities (Box 22-2). We routinely supplement ultrasound with magnetic resonance imaging (MRI) as a complementary method to further define the anatomy of the lesion and overall fetal morphology.7,8 In combination, the two modalities allow accurate prenatal diagnosis of the different types of BPMs and exclude other anatomic anomalies.
Congenital Pulmonary Airway Malformation
CPAMs are the most commonly diagnosed BPM. The best estimate of the incidence of CPAM is 0.66 per 10,000 live births, but better studies are needed based on high quality prenatal imaging of all pregnancies in a study population.9 This heterogeneous group of congenital cystic and noncystic lung masses is characterized by an extensive overgrowth of immature primary bronchioles localized to a segment of the bronchial tree (Fig. 22-2).10 The current classification system defined by Stocker classifies CPAMs into five types that differ by location, cystic structure, size, and epithelial lining (Table 22-1). However, from a practical perspective, the prenatal classification of CPAMs is divided into two categories based on prenatal ultrasound findings: (1) macrocystic lesions containing a single or multiple cysts that are 5.0 mm in diameter or greater; and (2) microcystic lesions presenting as a solid echogenic mass on prenatal ultrasound (Fig. 22-3).4
TABLE 22-1
Stocker Classification: Congenital Pulmonary Airway Malformations
From Stocker JT, Madewell JE, Drake RM. Congenital cystic adenomatoid malformation of the lung. Classification and morphologic spectrum. Hum Pathol 1977;8:155–71.
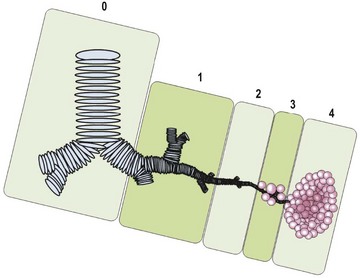
FIGURE 22-2 This CPAM classification schematic is based on the location of the development of the malformation. Type 0, tracheobronchial; 1, bronchial/bronchiolar; 2, bronchiolar; 3, bronchiolar/alveolar; 4, distal acinar. Adapted from Stocker (2009).63
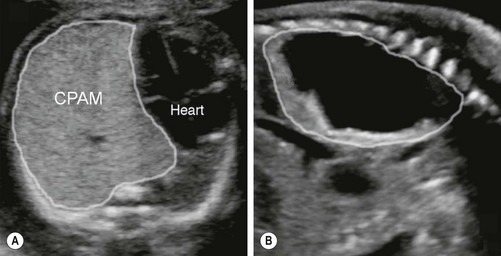
FIGURE 22-3 These two prenatal ultrasounds depict a practical prenatal classification for congenital pulmonary airway malformations (CPAM). (A) The prenatal ultrasound study finds microcystic lesions presenting as a solid echogenic mass. (B) macrocystic lesions contain either a large cyst or multiple cysts that are greater than 5.0 mm diameter.
In the prenatal period, ultrasound will usually demonstrate an area of hyperechogenic tissue with or without hypoechoic cysts that vary in number and size. Signs of mass effect may be seen, such as mediastinal shift, diaphragmatic eversion, and polyhydramnios. With very large lesions, heart failure (hydrops) due to mediastinal shift and cardiac compression can occur.11 CPAMs receive their blood supply from the pulmonary artery and have pulmonary venous drainage. However, there is a subset of CPAMs, known as hybrid lesions, where the blood supply also includes an anomalous systemic artery.12 These lesions demonstrate anatomic and histologic features of both CPAMs and BPS.
The diagnosis of CPAMs by an experienced sonographer is usually straightforward. However, there are specific entities that are commonly misdiagnosed (Table 22-2). A skilled sonographer can usually easily differentiate these lesions by understanding the blood supply (congenital diaphragmatic hernia [CDH], lung agenesis, BPS), observation of bowel peristalsis (CDH), documentation of the absence of one lung (lung agenesis), or visualization of bronchial dilation (bronchial atresia, congenital high airway obstruction syndrome [CHAOS]). MRI is also a useful adjunct for differentiating these entities. In our opinion, it should be applied routinely in fetal diagnostic centers.
TABLE 22-2
Pitfalls in Ultrasonography in the Diagnosis of CPAM
| Anomaly | Misdiagnosis |
| Right-sided congenital diaphragmatic hernia | Large right sided microcystic CPAM: similar echogenicity of liver to microcystic CPAM |
| Lung agenesis | Large microcystic CPAM: appearance of mediastinal shift with a large echogenic lung |
| Congenital high airway obstruction syndrome | Bilateral large microcystic CPAM: bilateral large echogenic lungs with diaphragmatic eversion |
| Main stem bronchial, lobar, or segmental atresia | Microcystic CCAM: hyperplasia of distal lung and increased echogenicity |
Bronchopulmonary Sequestration
Bronchopulmonary sequestrations comprise approximately 10% of prenatally diagnosed BPMs, and are characterized by a portion of the lung that does not connect to the tracheobronchial tree. These lesions have a systemic arterial supply that can arise from the aorta or various systemic arterial branches above or below the diaphragm, and may have systemic or pulmonary venous return. Two different types of sequestration are described: intralobar (ILS) and extralobar (ELS) which differ in their prenatal and postnatal characteristics. An ILS shares visceral pleural investment with normal lung and drains into the pulmonary venous system while an ELS has a separate pleural investment and may have either systemic or pulmonary venous drainage.13,14
ELS is seen as a homogeneous hyperechoic mass in a paraspinal location, most often in the left lower thorax (Fig. 22-4). The pathogenesis is related to a supernumerary lobe developing from abnormal budding early in foregut embryogenesis.15 If the bud arises before the development of the pleura, it is invested with the adjacent lung and becomes an ILS. If the bud develops after visceral pleural formation, it grows separately and acquires its own pleural covering.14 It is important to appreciate that ELS can be found at any level in the pleural space and are also found within or beneath the diaphragm (see Fig. 22-4B).15 The major feature that helps discriminate an ELS from a CPAM is the blood supply derived from the systemic circulation with systemic venous drainage identified by Doppler ultrasound or MRI. However, it is important to appreciate that some anatomic ELSs can contain visible cysts that ultimately are shown to have CPAM histology (Fig. 22-5). In addition, an ELS can occasionally have venous drainage via a large venous channel draining directly into a pulmonary vein that is usually identified as aberrant venous drainage by imaging studies.
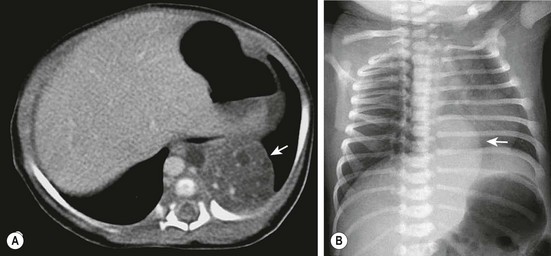
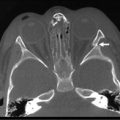
FIGURE 22-4 (A) The CT scan demonstrates an extralobar sequestration in the typical basilar location of the left chest (arrow). (B) In a different patient, the chest radiograph shows a large transdiaphragmatic extralobar sequestration (arrow).
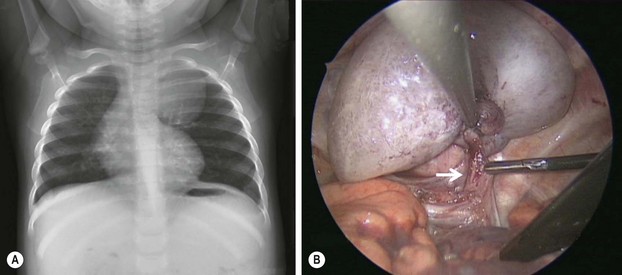
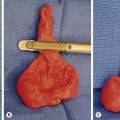
FIGURE 22-5 This 9-month-old developed an upper respiratory infection and a chest radiograph was performed. (A) The chest radiograph shows a left apical mediastinal mass. (B) After her infection resolved, she was found to have this extrapulmonary mass at thoracoscopy, along with a feeding vessel (arrow). The vessel was ligated and the mass removed, and she recovered uneventfully. Histologic examination showed the mass to an extralobar sequestration associated with a microcystic congenital pulmonary airway malformation.
Congenital Lobar Emphysema
CLE is a condition characterized by overinflation and distension of one or more pulmonary lobes with compression of the adjacent lung. In 50% of cases, the cause is unknown. In the remaining 50%, it may result from dysplastic bronchial cartilage, endobronchial obstruction, extrinsic compression from aberrant cardiopulmonary vasculature, or diffuse bronchial abnormalities related to infection.16 In the fetus, amniotic fluid trapping is analogous to air trapping and can lead to lobar expansion. The left upper lobe is the most frequently affected, followed by right middle and upper lobes, with rare bilateral or multifocal involvement. CLE is most commonly diagnosed in the neonate or infant presenting with respiratory distress.
Prenatal discrimination between CLE and CPAM or bronchial atresia may be difficult, but the absence of a systemic vascular supply differentiates this lesion from an ELS. Although complications such as polyhydramnios and hydrops have not been reported with CLE, perinatal respiratory distress correlates with the prenatal size of the lesion as manifest by mediastinal shift or compression of adjacent lung parenchyma.17
Bronchogenic Cysts and Bronchial Atresia/Stenosis
Bronchogenic cysts develop from abnormal budding of the tracheal diverticulum or the ventral aspect of the primitive foregut, which is not followed by bronchial development or branching. The result is a cavity that may or may not communicate with the airway and can be found in a variety of locations depending on the location of abnormal budding during foregut development. Histologically, these lesions are thin walled and have a bronchial epithelial lining, and are filled with mucus.18 Prenatal diagnosis of these lesions is usually made by ultrasound where they may be seen as an isolated cystic structure in the mediastinum, or causing bronchial obstruction with findings of bronchial dilation and lung hyperplasia distal to the point of obstruction. Bronchial atresia without a bronchogenic cyst also results in hyperplasia distal to the level of obstruction and is frequently associated with mucocele formation. The presence of dilated bronchi indicates a diagnosis of atresia rather than microcystic CPAM. The more proximal the atresia, the greater the potential for mass effect manifest by mediastinal shift, and ultimately, fetal hydrops. Segmental bronchial stenosis/atresia is a relatively recently recognized abnormality characterized by an echogenic segment of lung on ultrasound that is indistinguishable from and usually diagnosed as a microcystic CPAM.19
Prenatal and Perinatal Management of Bronchopulmonary Malformations
Congenital Pulmonary Airway Malformation
Experience with serial imaging of large numbers of fetuses with CPAMs has clarified the pre- and perinatal natural history of this anomaly. There is a typical pattern of growth of a CPAM with a period of growth relative to the size of the fetus until approximately 26 weeks gestation at which time growth plateaus. After 28 weeks, the CPAM typically gets smaller relative to the size of the fetus as measured by the CPAM volume ratio or CVR. The CVR is calculated by dividing the volume of the CPAM (length × height × width × 0.52) by the head circumference. In addition, the CVR has proven on retrospective and prospective assessment to be the most useful predictor for the development of hydrops.20 A CVR of <1.6 in a CPAM without a dominant cyst predicts a risk of developing hydrops of less than 3%. If the CVR is >1.6, the risk of hydrops is around 75%. We have found the CVR very useful in counseling parents, determining the intensity of serial follow-up, and determining which patients to preemptively treat with steroids. Other parameters such a mass–thorax ratio, cystic predominance of the lesion, and eventration of the diaphragm, while associated with large lesions, do not add independent predictive value to the CVR.21
Based on the type of CPAM, the prenatal CVR, and gestational age, a management strategy can be formulated to optimize outcome. In recent years, the prenatal treatment of large microcystic CPAMs with a CVR of >1.6 and/or the presence of hydrops at less than 32 weeks gestation has changed. Whereas open fetal surgery and lobectomy were once the primary option at fetal treatment centers, the majority of these patients will respond to steroid treatment, with inhibition of further CPAM growth and/or regression of hydrops. The mechanism for the steroid effect is speculative, but the phenomenon has been documented by multiple fetal treatment centers with very few open resections performed since this strategy was implemented.22–25 In a recent study at our institution, we were able to achieve 100% survival in fetuses either with hydrops (5/5) or a CVR >1.6 at the time of steroid administration.27 This compares to a mortality rate of 100% in fetuses with hydrops and a 56% mortality rate in fetuses with a CVR >1.6 among historical controls. In contrast to microcystic CPAMs, macrocystic CPAMs do not consistently respond to steroid treatment. Also, if hydrops is evolving, the fetus is best treated by thoracoamniotic shunting (Fig. 22-6).
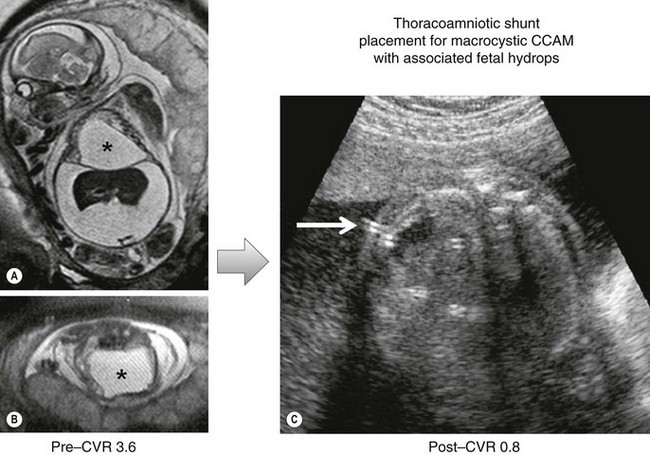
FIGURE 22-6 This fetus was found to have a large cystic mass complicated by a fetal hydrops. (A,B) The mass is marked with an asterisk. Prior to intervention, the CPAM volume ratio (CVR) was calculated at 3.6. This carries almost 100% mortality. (C) A thoracoamniotic shunt (arrow) was placed. After shunting, the CVR was measured at 0.8.
Our algorithm for the prenatal management of CPAM at the Children’s Hospital of Philadelphia (CHOP) is shown in Figure 22-7. Three common clinical scenarios in the management of CPAM are generally apparent by 32 to 34 weeks gestation (Table 22- 3) and guide recommendations for site and mode of delivery. As gestation proceeds, it is not unusual for previously hyperechoic lesions to become isoechoic with surrounding lung parenchyma (the so called ‘disappearing CPAM’). This is due to increasing echogenicity of the surrounding lung tissue and rib shadowing that occurs during the third trimester. However, it is important to realize that, in essentially all such cases, a postnatal CT scan will confirm persistence of the lesion.
TABLE 22-3
Common Clinical Scenarios in The Management of Congenital Pulmonary Airway Malformation
| Scenario | Management |
| CPAM regresses or remains small without mediastinal shift | Delivery at any facility with Level II nursery, discharged home with mother, postnatal CT scan at 4–6 weeks of age and elective resection prior to 3 months of age. |
| CPAM with some mediastinal shift without diaphragmatic eversion or extreme compression of contralateral lung | Delivery at tertiary center with resection performed in first few days of life |
| CPAM that remains massive with extreme mediastinal shift or that induces hydrops after 32 weeks | Transfer to fetal center or tertiary center with EXIT to ECMO capability |
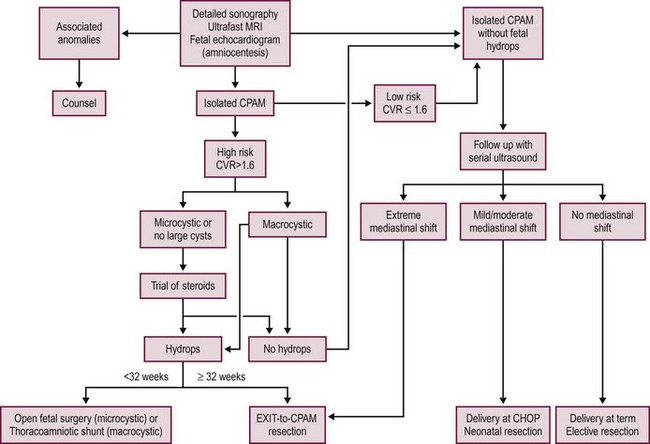
FIGURE 22-7 This algorithm depicts our current management strategy at Children’s Hospital of Philadelphia for a fetus with a congenital pulmonary airway malformation (CPAM). CHOP, Children’s Hospital of Philadelphia; CVR, CPAM volume ratio; MRI, magnetic resonance imaging; US, ultrasound.
Future options for treatment of CPAM in the hydropic fetus may include minimally invasive ablative or vascular occlusion therapy. Thus far, however, techniques such as radiofrequency or laser thermal ablation have been associated with excessive collateral damage due to the difficulty with controlling the energy dispersion in the high fluid content fetus. Sclerotherapy has been described but the patient selection must be questioned and the advisability of injecting highly caustic agents into the fetal bloodstream is concerning and needs further research investigation.26–28
Bronchopulmonary Sequestration
The prenatal manifestations of BPS differ between ELS and ILS. In general, ILS have few if any prenatal manifestations and generally do not cause significant morbidity. Theoretically, the systemic arterial to pulmonary venous circuit could cause high output cardiac failure in the fetus, but the earliest this has been observed is in the neonatal period.29 In contrast, ELS can present as a large mass with a mediastinal shift and frequently an associated pleural effusion. It has been our experience that an ELS that causes a mass effect is usually an edematous lesion with narrow vascular pedicles that presumably develops lymphatic or venous congestion and a secondary pleural effusion. Most ELS will regress in size during the third trimester. The occasional case of ELS-induced fetal hydrops is usually related to mediastinal shift from the associated pleural effusion rather than the ELS itself, and can be treated by thoracoamniotic shunting followed by postnatal resection. However, the vast majority of ELS and ILS cause no fetal compromise and are best treated after birth.
Congenital Lobar Emphysema, Bronchogenic Cysts, Bronchial Atresia
From the perspective of prenatal pathophysiology, these lesions can be grouped under the pathogenesis of bronchial stenosis or atresia. CLE and segmental bronchial stenosis are not associated with significant prenatal pathophysiology. CLE can enlarge prenatally due to fluid trapping, presumably by a similar mechanism to postnatal air trapping, though at most this has been found to cause moderate mediastinal shift. Bronchial atresia, also known as bronchial mucocele, is a condition resulting from focal obliteration or stenosis of a segmental, subsegmental, or lobar bronchus at or near its origin.30 Bronchial atresia can result in hydrops and fetal or neonatal death when located proximally. This is due to the hyperplastic growth response induced by the bronchial obstruction that can lead to massive lung or lobar expansion. Findings of severe mediastinal shift with diaphragmatic eversion and evolution of hydrops have been uniformly fatal in cases of main stem bronchial atresia due to either fetal demise or neonatal death due to the inability to ventilate the infant. Fetuses with lobar hyperplasia resulting in fetal hydrops prior to 30 weeks gestation may be offered open fetal surgery and lobectomy. After 30 weeks gestation, delivery utilizing the ex-utero intrapartum treatment (EXIT) approach followed by resection is performed. In our experience, these lesions have not been responsive to steroid therapy. An interesting recent case of fetal bronchoscopic decompression of a right bronchus intermedius obstruction has been described which may be applicable to a subset of proximal bronchial obstructions.31 Fortunately, cases requiring fetal intervention are rare, and most infants with lobar bronchial atresia can be treated in the early postnatal period.
Postnatal Management of Bronchopulmonary Malformations
Congenital Pulmonary Airway Malformation
The postnatal clinical spectrum of CPAM ranges from the neonate requiring mechanical ventilation, to the child presenting with an infected CPAM, to the asymptomatic adult. The treatment of the neonate with a symptomatic CPAM is immediate resection of the involved lobe by an open or thoracoscopic approach. Lobectomy is preferred over attempts at lung preserving surgery, such as segmental or wedge resection, due to the inability to accurately ascertain the borders of the lesion, the higher complication rate related to segmentectomy, and the compensatory lung growth that occurs in the remaining lobe or lobes that results in normal long-term pulmonary function following infant lobectomy.32 When multiple lobes are involved (1% of CPAMs), segmentectomy should be considered if anatomically feasible. As noted previously, very large lesions with significant mediastinal shift and diaphragmatic displacement should be delivered using the EXIT procedure and resected on placental support. With these very large masses, care must be exercised to avoid rupturing the CPAM or tearing the fragile hilar vessels during attempts at exposure when retracting the CPAM. A thoracoabdominal incision can be helpful with large CPAMs to aid in the safe delivery of the lobe and to provide additional space for the hilar dissection. In contrast to CDH, extracorporeal membrane oxygenation (ECMO) is rarely required for CPAM-related pulmonary hypoplasia, presumably because of the lesion’s slow growth and the relatively late onset of pulmonary compression. Outside the neonatal period, a symptomatic CPAM should be treated by lobectomy when recognized. Babies may present with air trapping in the first few months of life, infection of the CPAM in the first several years of life, and occasionally spontaneous pneumothorax related to the lesion.33,34 Infection in CPAMs may be difficult to clear with antibiotics and resection should be performed once the patient has improved following antibiotic therapy.
In contrast to large and symptomatic CPAMs, there is controversy regarding the treatment of the asymptomatic CPAM. In the era of prenatal diagnosis, the majority of patients are asymptomatic. The arguments for resection are that CPAMs do not completely regress and that they put the patient at risk for future infection, pneumothorax, and malignancy. There is also the risk that the mass is really a pleuropulmonary blastoma (PPB), a lesion that is rare, but indistinguishable from CPAM on pre- and postnatal imaging studies.35 The controversy is related to the belief by some that many lesions regress and disappear, as well as the unknown frequency of infection and malignant deterioration in the asymptomatic patient. Infection in CPAMs is well documented, particularly before the era of prenatal diagnosis. CPAMs can develop infection anytime between a few weeks of age until adulthood. Infections can be severe and life threatening, difficult to clear with antibiotics, associated with complications of abscess, empyema, and other secondary complications, and in neglected or unrecognized cases, may increase the risk of lobectomy. There are no well-designed prospective studies that provide an accurate assessment of the risk of infection in a CPAM during one’s lifetime, much less in the first 10 years of life. In a long-term retrospective study in which 21 patients were initially diagnosed as asymptomatic (including eight that were prenatally diagnosed) and followed, 18 developed symptoms (median age 2 years, range 1 month–13 years) and required operation. Symptoms included pneumonia with or without an infected CPAM (43%), respiratory distress (14%), and spontaneous pneumothorax (14%). Eight patients underwent multiple hospital encounters with complications related to the CPAM.36
There is also a clear association between CPAM and malignancy. A review of children with lung neoplasms demonstrated that 9% had a history of cystic lung malformations.37 The mucinous cells in a type I CPAM produce gastric mucins that are important in the pathogenesis of bronchioloalveolar carcinoma.38 The association with PPB is also clear, although resection of a CPAM does not preclude the development of PPB.39 Type II CPAM has been demonstrated to exhibit skeletal muscle differentiation as found in PPB and rhabdomyosarcoma. As the frequency of malignant transformation of a CPAM is unknown, it is not possible to predict which patients will develop cancer. Moreover, there is no imaging modality that can be safely used to serially image patients during a lifetime. This risk of malignancy may represent the strongest argument for resection of asymptomatic lesions.
While a nonoperative approach to CPAMs was evaluated in a small number of patients demonstrating a low risk of adverse events, the mean follow-up was only three years.40 In addition, a nonoperative approach requires serial surveillance by CT scanning and exposes the patient to a significant risk of cumulative radiation with significant long-term consequences.41 Waiting until the lesion becomes symptomatic places the patient at risk for developing complications when operating in an inflammatory or infectious field. Our data supports lobectomy between the ages of 1–3 months as the optimal management for the asymptomatic CPAM.42,43 We prefer the thoracoscopic approach, but this recommendation is dependent on an experienced surgeon. If that is not the case, then open lobectomy using a muscle sparing thoracotomy is recommended.
Bronchopulmonary Sequestration
An increasing number of BPS are prenatally diagnosed and postnatal therapy can be planned based on the anticipated risk of postnatal complications. For ELS, the treatment depends on the size of the lesion, blood flow, associated pleural effusion, and location. Our indications for resection of ELS are defined in Box 22-3. In both an ELS and ILS that has large systemic feeding vessels, vascular flow can be anticipated to increase, not decrease, over time due to the low resistance of the vascular bed with the end result being high flow cardiac physiology and ultimately cardiac failure as the child grows. Anatomical BPS with cystic areas on imaging likely contain CPAM elements within the BPS,44 and this may be responsible for the sporadic reports of malignancy occurring in both ILS and ELS, but more frequently in ELS. The malignancies that have been reported in association with ELS include lymphoepithelial carcinoma, PPB, squamous cell carcinoma, carcinoid, and mesothelioma.45–48 Within ILS, benign sclerosing hemangioma can be found. Because ELS do not communicate with the tracheobronchial tree, infection is rare. However, in ILS, air enters by tracking through the pores of Kohn. Thus, with stasis of mucous and the absence of bronchial clearance, infection is a frequent complication.
The differential diagnosis of subdiaphragmatic ELS in the fetus or neonate includes neuroblastoma and adrenal hemorrhage, and these lesions can be differentiated on serial ultrasound imaging. Finally, an ELS in close proximity to the esophagus, particularly if difficult to separate, may have an esophageal bronchus that needs to be identified and closed during excision. There is debate regarding the postnatal management of a small intrathoracic or subdiaphragmatic ELS. A small ELS can remain asymptomatic throughout life, have been reported to spontaneously regress, and carry a low risk of infection or malignancy. This has led some surgeons to observe these lesions if the lesion is not visible on plain radiography, or if it is small, or does not have a significant systemic arterial blood supply.49,50 We feel that the ELS treatment should be individualized with specific indications for resection as shown in Box 22-3.
The surgical management of ELS involves division of the systemic blood supply and removal of the mass (see Fig. 22-5). This must be done with care as systemic vessels arising from the abdominal aorta may cross the diaphragm and lead to difficult-to-control bleeding if not adequately ligated. However, ELS are generally straightforward lesions for thoracoscopic resection and patients are usually able to be discharged the next day. Percutaneous coil embolization of the feeding systemic artery to ELS has been performed in children and adults. In most cases there is little reason to favor this approach over resection.51 In contrast to resection, embolization carries the potential for vascular injury at the catheter insertion site, embolic occlusion of other vessels, and the potential for dysplastic residual tissue that may persist with possible future malignant transformation.
In our opinion, all cases of ILS should be resected, because of the risk of infection and evolution of high output physiology. Thoracoscopic lobectomy is our preferred approach. Segmentectomy for ILS has been reported in the literature with success.52,53 However, trying to avoid performing a lobectomy may carry a higher risk as these lesions usually do not have normal segmental anatomy, share pleural investment, can have significant internal blood flow, and can be difficult to distinguish from adjacent lung tissue which risks leaving residual disease.
Congenital Lobar Emphysema
Asymptomatic children with CLE or those with only mild symptoms can be safely managed without resection.54 In neonates or infants presenting with respiratory distress and pulmonary lobar hyperinflation, open lobectomy is our preferred option (Fig. 22-8). These patients have unique and difficult anesthetic challenges from air trapping and cardiovascular compromise that may prevent successful single lung ventilation. Therefore, consideration for thoracoscopic resection should be highly selective and requires careful preoperative review by the anesthetic and surgical team.55 Symptomatic patients usually present with respiratory distress within the first 6 months of life with 25% of patients presenting at birth and 50% presenting prior to one month of age. The severity of disease is dependent on the size of the affected lobe and compression of adjacent lung tissue. The mainstay of management for CLE is resection of the affected lobe. However, whether pre- or intraoperatively, care must be taken to avoid progressive air trapping with resulting lobar tension emphysema from positive pressure ventilation.16
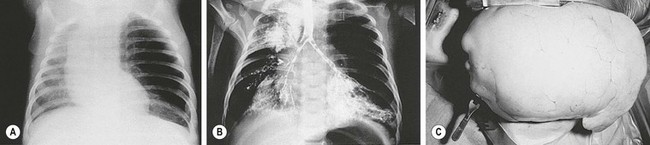
FIGURE 22-8 (A) Chest radiograph demonstrates the characteristic changes in an infant with congenital lobar emphysema (CLE). Marked overdistention of the left upper lobe caused the mediastinal shift, flattening of the left diaphragm, and likely subsequent respiratory distress. (B) Bronchogram shows the absence or occlusion of the left upper lobe bronchus, producing the typical findings in CLE. (C) Operative photograph demonstrates the dramatic herniation of an emphysematous lobe through the thoracotomy incision.
Bronchogenic Cysts and Bronchial Atresia
Bronchogenic cysts are now frequently prenatally diagnosed by ultrasound. Postnatally, they are often diagnosed by a mass on a chest film, either incidentally or in association with respiratory symptoms. CT can confirm the presence of a bronchogenic cyst as well as its relation to adjacent mediastinal structures. Mass effect from these lesions is typically mild, but lesions identified below the carina can cause life threatening airway compromise.56 Complications associated with bronchogenic cysts include infection, malignancy, hemoptysis, hemothorax, and pneumothorax. For these reasons, all bronchogenic cysts should be resected regardless of the clinical presentation or age at diagnosis. Symptomatic neonates or infants may require immediate resection or aspiration of the cysts as a temporizing measure for extreme duress. Asymptomatic newborns with a bronchogenic cyst identified on imaging may be followed as an outpatient and scheduled for resection after 1 month of age. Older patients should undergo resection of the bronchogenic cyst soon after it is identified, though the clinical presentation of pneumonia may complicate resection. These lesions may be safely resected using a thoracoscopic technique with equivalent outcomes to the open approach (see Fig 25-7).57 When the cyst cannot be completely resected, the mucosal layer should be excised leaving behind only peripheral connective tissue to eliminate the risk of recurrence and malignant transformation.58 In our opinion, all cysts associated with bronchial atresias should be excised because of the increased risk of infection. When a bronchogenic cyst is associated with bronchial obstruction, there is inevitably irreversible damage to the bronchus and lobectomy is needed.
Thoracoscopic Lobectomy
The benefits of thoracoscopy include less postoperative pain, more rapid recovery, less hospitalization, and an improved functional and cosmetic result compared to traditional thoracotomy.59,60 However, despite these obvious advantages and the development of appropriate instrumentation for infants, the use of thoracoscopy for congenital lung lesions has been relatively limited. This is likely due to the infrequency of these anomalies, and the technical challenges inherent in learning the technique. Nevertheless, adequate experience has been accumulated in a number of centers to validate the approach and demonstrate safety and efficacy that mimics traditional thoracotomy.59,61
The challenges unique to infant thoracoscopic lobectomy can be separated into anesthesia, anatomic, and technical. The anesthetic challenges relate to single lung ventilation in infants and require a committed anesthesia staff. Main stem bronchial intubation can be easily achieved by the use of a 3-0 micro-cuffed tube introduced using fluoroscopy.62 This approach is much faster than using bronchoscopy to correctly position the endotracheal tube. For left-sided lobectomies in particular, it is important to introduce the tube deep enough in the right main stem bronchus to avoid inadvertent displacement into the left main stem bronchus or trachea with dissection around the hilum during the procedure, but not so deeply to lose right upper lobe ventilation. In general, the cuff does not need to be inflated to achieve single lung ventilation in the less than 3-month-old infant, and bronchial blockers or other methods described for larger patients are not needed. With appropriate tube placement, a transient period of desaturation into the 80–92% range is expected as is resolution when positioning the patient with the ventilated lung down and redistribution of blood flow. Adjustments to the peak ventilator pressure and rate to maintain end tidal CO2 in the 50s Torr range are usually required.
Anatomic challenges relate to the thoracoscopic perspective on pulmonary anatomy and the variations inherent with CBMs. The optimal approach is through the major fissure. Therefore, a detailed understanding of vascular and bronchial anatomy viewed through the fissure is essential (Figs 22-9 and 22-10). Working through the fissure from anterior to posterior, the sequence that structures are encountered are the arteries, bronchus, and vein in that order. The correct anatomic orientation can be significantly altered by the CPAM or lobar hyperplasia from bronchial atresia. A systematic approach in each patient is needed to identify the important structures. The anatomy can also be obscured to varying degrees by incomplete fissure formation which can be problematic for inexperienced surgeons.
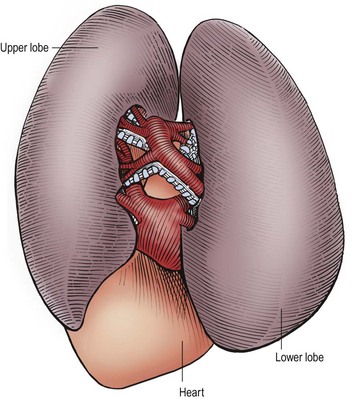
FIGURE 22-9 This is the anatomy seen by the surgeon when the contents of the fissure between the left upper and lower lobes are exposed at thoracoscopy. The arteries are visualized first within the fissure and the bronchial structures are behind the arteries. The pulmonary veins exit from the inferior portion of these lobes and drain into the heart. (From Holcomb GW III, Georgeson KE, Rothenberg SS. Atlas of Pediatric Laparoscopy and Thoracoscopy. Philadelphia: Elsevier; 2008.)
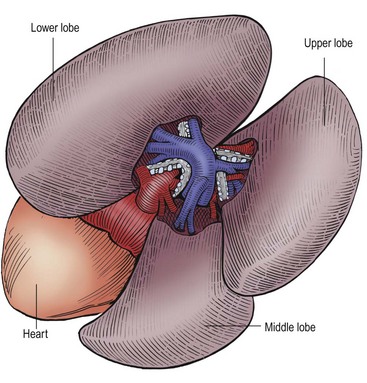
FIGURE 22-10 This is the anatomy as seen by the surgeon when performing a thoracoscopic lobectomy on the right side. Again, the arteries are the first structure seen in the fissure and the bronchial structures are behind the arteries. The pulmonary veins are inferior to the arteries and drain into the heart. (From Holcomb GW III, Georgeson KE, Rothenberg SS. Atlas of Pediatric Laparoscopy and Thoracoscopy. Philadelphia: Elsevier; 2008.)
Technical challenges are primarily related to operating in a much smaller space than is found with infant laparoscopy. When combined with the movement of the structures from ventilation and cardiac activity, thoracoscopic lobectomy can be challenging. Also, the 5 mm vascular sealing devices such as the Ligasure (Covidien, Inc., Mansfield NJ) or endoscopic clips are grossly oversized in the infant chest. It is important to be facile with endocorporeal knot tying in very small spaces. On occasion, under adverse circumstances, it may become the fall back technique at any time. With experience, thoracoscopic lobectomy, even in infants, can become a straightforward procedure that is better in many ways than open thoracotomy. However, it remains the most advanced of infant thoracoscopic procedures and should only be performed by trained individuals in centers with a reasonable volume of these procedures.
References
1. Gonzaga, S, Henriques-Coelho, T, Davey, M, et al. Cystic adenomatoid malformations are induced by localized FGF10 overexpression in fetal rat lung. Am J Respir Cell Mol Biol. 2008; 39:346–355.
2. Correia-Pinto, J, Gonzaga, S, Huang, Y, et al. Congenital lung lesions–underlying molecular mechanisms. Semin Pediatr Surg. 2010; 19:171–179.
3. Biyyam, DR, Chapman, T, Ferguson, MR, et al. Congenital lung abnormalities: Embryologic features, prenatal diagnosis, and postnatal radiologic-pathologic correlation. Radiographics. 2010; 30:1721–1738.
4. Adzick, NS, Harrison, MR, Glick, PL, et al. Fetal cystic adenomatoid malformation: Prenatal diagnosis and natural history. J Pediatr Surg. 1985; 20:483–488.
5. Epelman, M, Kreiger, PA, Servaes, S, et al. Current imaging of prenatally diagnosed congenital lung lesions. Semin Ultrasound CT MR. 2010; 31:141–157.
6. Langston, C. New concepts in the pathology of congenital lung malformations. Semin Pediatr Surg. 2003; 12:17–37.
7. Hubbard, AM, Adzick, NS, Crombleholme, TM, et al. Congenital chest lesions: Diagnosis and characterization with prenatal MR imaging. Radiology. 1999; 212:43–48.
8. Quinn, TM, Hubbard, AM, Adzick, NS. Prenatal magnetic resonance imaging enhances fetal diagnosis. J Pediatr Surg. 1998; 33:553–558.
9. Dolk, H. EUROCAT: 25 years of European surveillance of congenital anomalies. Arch Dis Child Fetal Neonatal Ed. 2005; 90:F355–F358.
10. Stocker, JT, Madewell, JE, Drake, RM. Congenital cystic adenomatoid malformation of the lung. Classification and morphologic spectrum. Hum Pathol. 1977; 8:155–171.
11. Mahle, WT, Rychik, J, Tian, ZY, et al. Echocardiographic evaluation of the fetus with congenital cystic adenomatoid malformation. Ultrasound Obstet Gynecol. 2000; 16:620–624.
12. Cass, DL, Crombleholme, TM, Howell, LJ, et al. Cystic lung lesions with systemic arterial blood supply: A hybrid of congenital cystic adenomatoid malformation and bronchopulmonary sequestration. J Pediatr Surg. 1997; 32:986–990.
13. Savic, B, Birtel, FJ, Tholen, W, et al. Lung sequestration: Report of seven cases and review of 540 published cases. Thorax. 1979; 34:96–101.
14. Stocker, JT. Sequestrations of the lung. Semin Diagn Pathol. 1986; 3:106–121.
15. Rosado-de-Christenson, ML, Frazier, AA, Stocker, JT, et al. From the archives of the AFIP. Extralobar sequestration: Radiologic-pathologic correlation. Radiographics. 1993; 13:425–441.
16. Olutoye, OO, Coleman, BG, Hubbard, AM, et al. Prenatal diagnosis and management of congenital lobar emphysema. J Pediatr Surg. 2000; 35:792–795.
17. Usui, N, Kamata, S, Sawai, T, et al. Outcome predictors for infants with cystic lung disease. J Pediatr Surg. 2004; 39:603–606.
18. Eraklis, AJ, Griscom, NT, McGovern, JB. Bronchogenic cysts of the mediastinum in infancy. N Engl J Med. 1969; 281:1150–1155.
19. Peranteau, WH, Merchant, AM, Hedrick, HL, et al. Prenatal course and postnatal management of peripheral bronchial atresia: Association with congenital cystic adenomatoid malformation of the lung. Fetal Diagn Ther. 2008; 24:190–196.
20. Crombleholme, TM, Coleman, B, Hedrick, H, et al. Cystic adenomatoid malformation volume ratio predicts outcome in prenatally diagnosed cystic adenomatoid malformation of the lung. J Pediatr Surg. 2002; 37:331–338.
21. Vu, L, Tsao, K, Lee, H, et al. Characteristics of congenital cystic adenomatoid malformations associated with nonimmune hydrops and outcome. J Pediatr Surg. 2007; 42:1351–1356.
22. Loh, KC, Jelin, E, Hirose, S, et al. Microcystic congenital pulmonary airway malformation with hydrops fetalis: Steroids vs. open fetal resection. J Pediatr Surg. 2012; 47:36–39.
23. Morris, LM, Lim, FY, Livingston, JC, et al. High-risk fetal congenital pulmonary airway malformations have a variable response to steroids. J Pediatr Surg. 2009; 44:60–65.
24. Peranteau, WH, Wilson, RD, Liechty, KW, et al. Effect of maternal betamethasone administration on prenatal congenital cystic adenomatoid malformation growth and fetal survival. Fetal Diagn Ther. 2007; 22:365–371.
25. Tsao, K, Hawgood, S, Vu, L, et al. Resolution of hydrops fetalis in congenital cystic adenomatoid malformation after prenatal steroid therapy. J Pediatr Surg. 2003; 38:508–510.
26. Bruner, JP, Jarnagin, BK, Reinisch, L. Percutaneous laser ablation of fetal congenital cystic adenomatoid malformation: Too little, too late? Fetal Diagn Ther. 2000; 15:359–363.
27. Milner, R, Kitano, Y, Olutoye, O, et al. Radiofrequency thermal ablation: A potential treatment for hydropic fetuses with a large chest mass. J Pediatr Surg. 2000; 35:386–389.
28. Oepkes, D, Devlieger, R, Lopriore, E, et al. Successful ultrasound-guided laser treatment of fetal hydrops caused by pulmonary sequestration. Ultrasound Obstet Gynecol. 2007; 29:457–459.
29. Millendez, MB, Ridout, E, Pole, G, et al. Neonatal hyperreninemia and hypertensive heart failure relieved with resection of an intralobar pulmonary sequestration. J Pediatr Surg. 2007; 42:1276–1278.
30. Keswani, SG, Crombleholme, TM, Pawel, BR, et al. Prenatal diagnosis and management of mainstem bronchial atresia. Fetal Diagn Ther. 2005; 20:74–78.
31. Martinez, JM, Prat, J, Gomez, O, et al. Decompression through tracheobronchial endoscopy of bronchial atresia presenting as massive pulmonary tumor: A new indication for fetoscopic surgery. Fetal Diagn Ther. 2012.
32. Muller, CO, Berrebi, D, Kheniche, A, et al. Is radical lobectomy required in congenital cystic adenomatoid malformation? J Pediatr Surg. 2012; 47:642–645.
33. Choudhury, SR, Chadha, R, Mishra, A, et al. Lung resections in children for congenital and acquired lesions. Pediatr Surg Int. 2007; 23:851–859.
34. Lujan, M, Bosque, M, Mirapeix, RM, et al. Late-onset congenital cystic adenomatoid malformation of the lung. Embryology, clinical symptomatology, diagnostic procedures, therapeutic approach and clinical follow-up. Respiration. 2002; 69:148–154.
35. Oliveira, C, Himidan, S, Pastor, AC, et al. Discriminating preoperative features of pleuropulmonary blastomas (PPB) from congenital cystic adenomatoid malformations (CCAM): A retrospective, age-matched study. Eur J Pediatr Surg. 2011; 21:2–7.
36. Wong, A, Vieten, D, Singh, S, et al. Long-term outcome of asymptomatic patients with congenital cystic adenomatoid malformation. Pediatr Surg Int. 2009; 25:479–485.
37. Hancock, BJ, Di Lorenzo, M, Youssef, S, et al. Childhood primary pulmonary neoplasms. J Pediatr Surg. 1993; 28:1133–1136.
38. Lantuejoul, S, Nicholson, AG, Sartori, G, et al. Mucinous cells in type 1 pulmonary congenital cystic adenomatoid malformation as mucinous bronchioloalveolar carcinoma precursors. Am J Surg Pathol. 2007; 31:961–969.
39. Papagiannopoulos, KA, Sheppard, M, Bush, AP, et al. Pleuropulmonary blastoma: Is prophylactic resection of congenital lung cysts effective? Ann Thorac Surg. 2001; 72:604–605.
40. Aziz, D, Langer, JC, Tuuha, SE, et al. Perinatally diagnosed asymptomatic congenital cystic adenomatoid malformation: To resect or not? J Pediatr Surg. 2004; 39:329–334.
41. Pearce, MS, Salotti, JA, Little, MP, et al. Radiation exposure from CT scans in childhood and subsequent risk of leukaemia and brain tumours: A retrospective cohort study. Lancet. 2012; 380:499–505.
42. Adzick, NS, Flake, AW, Crombleholme, TM. Management of congenital lung lesions. Semin Pediatr Surg. 2003; 12:10–16.
43. Tsai, AY, Liechty, KW, Hedrick, HL, et al. Outcomes after postnatal resection of prenatally diagnosed asymptomatic cystic lung lesions. J Pediatr Surg. 2008; 43:513–517.
44. Conran, RM, Stocker, JT. Extralobar sequestration with frequently associated congenital cystic adenomatoid malformation, type 2: Report of 50 cases. Pediatr Dev Pathol. 1999; 2:454–463.
45. Hekelaar, N, van Uffelen, R, van Vliet, AC, et al. Primary lymphoepithelioma-like carcinoma within an intralobular pulmonary sequestration. Eur Respir J. 2000; 16:1025–1027.
46. Olgac, G, Peirovi, F, Yilmaz, A, et al. Giant carcinoid tumor mimicking pulmonary sequestration. Ann Thorac Surg. 2007; 84:1375–1376.
47. Priest, JR, McDermott, MB, Bhatia, S, et al. Pleuropulmonary blastoma: A clinicopathologic study of 50 cases. Cancer. 1997; 80:147–161.
48. Westphal, FL, Lima, LC, Lima Netto, JC, et al. Carcinoid tumor and pulmonary sequestration. J Bras Pneumol. 2012; 38:133–137.
49. Laberge, JM, Puligandla, P, Flageole, H. Asymptomatic congenital lung malformations. Semin Pediatr Surg. 2005; 14:16–33.
50. Samuel, M, Burge, DM. Management of antenatally diagnosed pulmonary sequestration associated with congenital cystic adenomatoid malformation. Thorax. 1999; 54:701–706.
51. Chien, KJ, Huang, TC, Lin, CC, et al. Early and late outcomes of coil embolization of pulmonary sequestration in children. Circ J. 2009; 73:938–942.
52. Johnson, SM, Grace, N, Edwards, MJ, et al. Thoracoscopic segmentectomy for treatment of congenital lung malformations. J Pediatr Surg. 2011; 46:2265–2269.
53. Peiry, B, De Buys Roessingh, A, Francini, K, et al. Thoracoscopic segmentectomy: One vessel may hide a second one. J Pediatr Surg. 2012; 47:e11–e13.
54. Mei-Zahav, M, Konen, O, Manson, D, et al. Is congenital lobar emphysema a surgical disease? J Pediatr Surg. 2006; 41:1058–1061.
55. Tempe, DK, Virmani, S, Javetkar, S, et al. Congenital lobar emphysema: Pitfalls and management. Ann Card Anaesth. 2010; 13:53–58.
56. Laje, P, Liechty, KW. Postnatal management and outcome of prenatally diagnosed lung lesions. Prenat Diagn. 2008; 28:612–618.
57. Tolg, C, Abelin, K, Laudenbach, V, et al. Open vs. thoracoscopic surgical management of bronchogenic cysts. Surg Endosc. 2005; 19:77–80.
58. Calzada, AP, Wu, W, Salvado, AR, et al. Poorly differentiated adenocarcinoma arising from a cervical bronchial cyst. Laryngoscope. 2011; 121:1446–1448.
59. Rothenberg, SS. First decade’s experience with thoracoscopic lobectomy in infants and children. J Pediatr Surg. 2008; 43:40–45.
60. Rothenberg, SS, Kuenzler, KA, Middlesworth, W, et al. Thoracoscopic lobectomy in infants less than 10 kg with prenatally diagnosed cystic lung disease. J Laparoendosc Adv Surg Tech A. 2011; 21:181–184.
61. Vu, LT, Farmer, DL, Nobuhara, KK, et al. Thoracoscopic versus open resection for congenital cystic adenomatoid malformations of the lung. J Pediatr Surg. 2008; 43:35–39.
62. Cohen, DE, McCloskey, JJ, Motas, D, et al. Fluoroscopic-assisted endobronchial intubation for single-lung ventilation in infants. Paediatr Anaesth. 2011; 21:681–684.
63. Stocker, JT. Cystic lung disease in infants and children. Fetal Pediatr Pathol. 2009; 28(4):155–184.