[level-membership-for-orthopaedics-category]
CHAPTER 31 Congenital Anomalies of the Spinal Cord
Myelomeningocele
Myelomeningocele is the most common significant birth defect involving the spine. The condition is manifest at birth and is characterized by herniation of a malformed spinal cord through a defect in the bony canal and skin. It almost always results in permanent disability regardless of medical intervention and often deprives the victim of “those qualities held in high esteem by our society—independence, physical powers and intelligence.”1 In the past, it was assumed that all of the neurologic dysfunction that occurs with myelomeningocele arose from disordered embryogenesis. More recent work suggests, however, that at least some of the spinal cord damage is acquired in utero,2 and fetal surgery to close the spinal defect is now being performed at selected centers.
Embryology
By 18 days of development, the embryo is a flattened oval disc with all three germ layers present. A longitudinal depression, the neural groove, appears in the neural plate, which is destined to become the brain and spinal cord. By 22 days, the neural groove has deepened, and fusion of the adjacent tissue begins the transformation of the flat neural plate into a hollow neural tube. The entire process is called neurulation, which begins in the dorsal midline and simultaneously progresses cephalad and caudad. The final portions of the tube to close are the rostral opening (the anterior neuropore, at 24 days) and the caudal opening (the posterior neuropore, at 28 days). By the 1st month of gestation, the entire process has been completed. The development of the meninges begins after closure of the posterior neuropore, as does formation of the bony laminae.3
Myelomeningocele presumably occurs when the posterior neuropore fails to close or if it reopens as the result of distention of the central canal of the spinal cord with cerebrospinal fluid (CSF). The spinal abnormality is only one component of a complex of central nervous system abnormalities, which include the Chiari II malformation, hydrocephalus, and brain anomalies such as partial agenesis of the corpus callosum and gyral malformations. McLone and Naidich4 attempted to explain these associations with their “unified theory.” It is hypothesized that the open spinal defect allows excessive drainage of CSF in utero and that this results in collapse of the rhombencephalic vesicle, resulting in a small posterior fossa volume. Growth of the cerebellum and brainstem within a small posterior fossa results in downward herniation and caudal displacement of the cerebellar vermis and brainstem into the cervical spinal canal (the Chiari II malformation). Because the outlet of the fourth ventricle is occluded by impacted brain tissue, obstructive hydrocephalus develops either in the fetal period or in the newborn period after closure of the myelomeningocele eliminates the spinal defect as a drainage pathway.
Epidemiology
The incidence of myelomeningocele ranges from less than 1 case per 1000 live births in the United States to almost 9 cases per 1000 in areas of Ireland. The etiology of myelodysplasia is unknown, and evidence exists for environmental and multifactorial genetic influences. A role for genetic risk factors is supported by numerous studies documenting familial aggregation of this condition. In addition, several lines of evidence point to the potential importance of maternal nutritional status as a determinant of the risk for having a child with spina bifida. Indirect support is provided by studies that indicate that season of conception, socioeconomic status, and degree of urbanization may be related to the risk of spina bifida. Several micronutrients (vitamins C and B12, zinc, and folic acid) have been implicated as potential risk factors as well.5
Folic acid is the most extensively studied micronutrient. In August 1991 after the Medical Research Council Vitamin Study Group report was published, the U.S. Centers for Disease Control and Prevention (CDC) advised that women with a history of an affected pregnancy should take 4 mg of folic acid daily, starting at the time they planned to become pregnant.6 A dose of 0.4 mg was later recommended for all women of childbearing age capable of becoming pregnant. It was anticipated that these recommendations would have a substantial impact on the risk of neural tube defects in the offspring of such women. Most affected pregnancies (approximately 95%) occur in women with no history of a prior affected fetus or child, however.7 It has been suggested that fortification of the food supply may be more effective than individual supplementation for the prevention of neural tube defects.
Prenatal Diagnosis
More recent developments in prenatal diagnosis of fetal anomalies have made antenatal recognition of myelomeningocele commonplace. Families at risk are routinely offered amniocentesis for amniotic α-fetoprotein and acetylcholinesterase, which are important in separating open lesions from skin-covered masses such as myelocystocele. Screening ultrasonography performed on mothers when the fetus is at 16 to 20 weeks of gestation has also proved valuable.8 Other anomalies may be detected, and ultrasonography may detect skin-covered lesions such as lipomyelomeningocele. The cardinal finding is splaying of the posterior elements of the spine in the axial plane in the lumbosacral region. Indirect signs are the lemon sign, which refers to a bilateral concave contour of the frontal bones of the skull, and the banana sign, which describes anterior curvature of the cerebellar hemispheres. Hydrocephalus is also readily detected. In referral centers, diagnostic sensitivity is close to 100% in diagnosing spina bifida.9
Prenatal magnetic resonance imaging (MRI), using ultrafast T2-weighted sequences, may also be used to characterize the Chiari II malformation and other associated anomalies.10 Fetal MRI may detect spinal cord abnormalities in instances where ultrasonography can detect only bony abnormalities.11 Studies indicate that such prenatal imaging studies can help to determine prognosis. Specifically, lesion level determined by prenatal imaging studies seems to predict neurologic deficit and ambulatory potential, but the degree of fetal ventriculomegaly and the extent of hindbrain deformity are not predictive.12 Families can be professionally counseled regarding the expected prognosis and offered conventional treatment, abortion, or the possibility of fetal closure. Patients of low socioeconomic class, who are most at risk for neural tube defects, often do not present for prenatal care until after 24 weeks of gestation, when screening is no longer of value.
The value of prenatal diagnosis of spinal dysraphism is that an opportunity is provided to terminate the pregnancy, to evaluate the fetus for possible fetal surgery, and to determine the best mode of delivery. It has been proposed that children with myelomeningocele sustain traumatic damage to the placode and nerves when they are delivered vaginally and that children electively delivered by cesarean section before the onset of labor have improved motor levels.13 Other investigators have found little benefit to this strategy,12 but in the United States cesarean delivery has become routine.
Initial Evaluation
The initial assessment of a newborn infant with a myelomeningocele begins with a detailed examination to evaluate general well-being and to seek associated anomalies. Fatal urologic or cardiac anomalies may be evident that would favor nonoperative treatment. Some infants may have abnormal facies suggesting Down syndrome, and although chromosomal studies should be obtained, these are most often normal, and the facial appearance becomes normal with age. Approximately 85% of infants with myelomeningocele present with hydrocephalus or develop it within the newborn period.14 A large head circumference or bulging fontanel suggests the need for early head ultrasonography via the fontanel. Stridor, apnea, or bradycardia in the absence of overt intracranial hypertension suggests a symptomatic Chiari II malformation and hindbrain dysfunction, which carries a poor prognosis.
The neurologic examination is difficult in neonates, and it is easy to mistake reflex motion for voluntary movement. Fixed contractures and foot deformity suggest paralysis of the spinal segments innervating the joints. Any movement in response to painful stimulation of the same extremity must be viewed as potentially reflexive. Crying in response to a painful stimulus suggests intact sensation at that level. Table 31–1 lists the segmental innervation of the lower extremities and may be used as a guide in assigning a functional level. It is best, however, to document function of the individual muscle groups rather than simply to record a spinal level.
| Hip flexion | L1-3 |
| Hip adduction | L2-4 |
| Knee extension | L2-4 |
| Ankle inversion | L4 |
| Toe extension | L5-S1 |
| Hip abduction | L5-S1 |
| Hip extension | L5-S1 |
| Knee flexion | L5-S2 |
| Ankle plantar flexion | S1-2 |
Data from Sharrard WJW: The segmental innervation of the lower limb muscles in man. Ann R Coll Surg 35:106-122, 1964.
Management of an Unrepaired Newborn
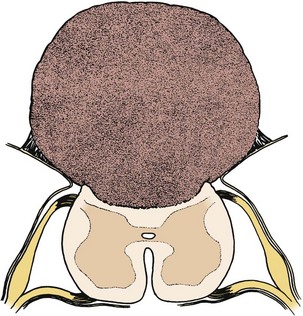
Pending plans for definitive care, the infant is nursed in the prone position with a sterile saline-soaked gauze dressing loosely applied to the sac or placode. Broad-spectrum antibiotics (ampicillin and cefotaxime) are begun intravenously pending discussion with the parents. If there is no sign of overt hydrocephalus, the back is closed initially, and hydrocephalus is treated with a ventriculoperitoneal shunt at a separate procedure if needed. In patients with hydrocephalus and intracranial hypertension at birth, it may be advisable to perform both procedures at the same time because failure to treat the hydrocephalus may allow continued leakage of CSF into the back and threaten the closure and because the infant is exposed to a single anesthetic. The goal of back closure is to seal, using multiple tissue layers, the spinal cord and subarachnoid space against entry of bacteria from the skin. At the same time, the surgeon must preserve whatever neurologic function remains and attempt to prevent tethering of the spinal cord. The cross-sectional anatomy of a typical myelomeningocele is shown in Figure 31–1.
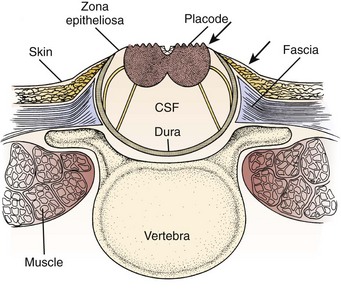
FIGURE 31–1 Cross-sectional anatomy of typical lumbosacral myelomeningocele. CSF, cerebrospinal fluid.
The infant is positioned in the prone position under general anesthesia. Rolls are placed under the chest and hips to allow the abdomen to hang freely and minimize epidural bleeding (Fig. 31–2A). If the sac is intact, fluid is aspirated and sent for culture. The surgeon gently attempts to approximate the base of the sac or defect vertically and then horizontally to determine which direction would produce the smallest skin defect. An elliptical incision is made, oriented along that axis, outside the junction of the normal, full-thickness skin and the thin, pearly zona epithelioserosa. Full-thickness skin forming the base of the sac is viable and should not be excised. This incision is carried through the subcutaneous tissue until the glistening layer of everted dura or fascia is encountered. The base of the sac is mobilized medially until it is seen to enter the fascial defect (Fig. 31–2B).
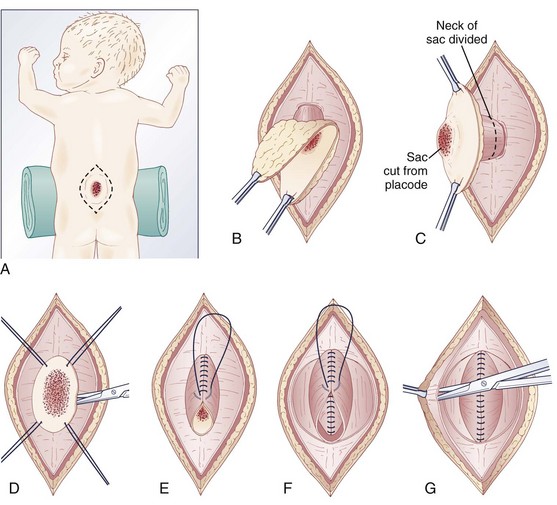
The sac is entered by radially incising the cuff of skin surrounding the placode. This skin is sharply excised circumferentially around the placode and discarded, with care being taken to avoid damaging the placode (Fig. 31–2C). All of the zona epithelioserosa is removed to prevent later development of an epidermoid cyst. At this point, the placode is floating freely inside the everted dura (Fig. 31–2D). In some thoracic myelomeningoceles, the placode is large and “thinned out,” and there is complete paraplegia below the level of the defect. In these instances, it may be appropriate to excise the placode to prevent spasticity and high-pressure bladder dysfunction.
Attention is now directed toward the dura, which is everted and loosely attached to the underlying fascia. It is undermined and reflected medially on each side until enough has been mobilized to effect closure (Fig. 31–2E). The dura is very thin anteriorly where the root sleeves exit and is easily torn. When it is free, the dura is closed in a watertight fashion with 4-0 nonabsorbable suture material.
Particularly if the dural closure is suboptimal, it is desirable also to close the fascia as a separate layer. The fascia is incised laterally in a semicircular fashion on either side, elevated from the underlying muscle, and reflected medially (Fig. 31–2F). It is closed with 4-0 suture over the underlying dural closure. The fascia is poor at the caudal end of a lumbar myelomeningocele or with sacral lesions, and the closure may be incomplete.
Mobilization of the skin is by blunt dissection with scissors or a finger; it may be necessary to free it anteriorly to the abdomen (Fig. 31–2G). In most instances, the closure is easiest in the mid-sagittal (vertical) plane, but occasionally less tension is required for a horizontal closure. A two-layer closure is performed. The epidermis may be closed with absorbable suture material and tissue glue if there is little tension, but interrupted nonabsorbable suture material is preferred for large defects.
Very large lesions require special techniques. There is often an associated gibbous deformity, and it is helpful to use a rongeur to remove the everted lamina, which, if left in place, would produce pressure points on the skin closure. Large circular defects may be closed by an S-shaped skin opening, allowing the use of rotation flaps (Fig. 31–3). Alternatively, a Z-rhomboid flap, tissue expanders, or latissimus dorsi myocutaneous flaps may be employed. Lateral relaxing incisions with split-thickness skin grafting have been described, but should be avoided if possible because the cosmetic result is poor. Alternatively, allogeneic skin grafts such as Alloderm (Lifecell, Branchburg, NJ) can be sewn directly to the edges of an approximated closure and may offer a better cosmetic result.15 In rare cases where patients are referred late for surgery, significant scarring of the placode can occur. Tissue expansion is particularly useful in such cases.16
Large defects occasionally undergo dehiscence, owing to tension on the skin closure or superficial infection. This dehiscence is usually evident by 7 days postoperatively. In most cases, the underlying fascial and dural closures remain intact. The dehiscence may be treated by removing the sutures to the point of good skin closure, débriding the devitalized tissue, and beginning a program of saline wet-to-dry dressings. When a good granulation bed is observed, the wound can simply be allowed to granulate and contract or may be secondarily closed or repaired with a skin graft with or without tissue expansion.17 For persistent wound breakdown, the pedicled “propeller” flap can be used as a vascularized salvage procedure.18 CSF leakage is treated by a ventricular diversion procedure, even if the ventricles are small.
Closure Before Birth
In a few centers, a fetus with spina bifida may be a candidate for in utero treatment because this condition is routinely detected before 20 weeks of gestation (Fig. 31–4). There is evidence that neurologic deterioration occurs during gestation. Normal lower extremity movement can be seen on ultrasound studies of affected fetuses before 17 and 20 weeks of gestation, whereas most late-gestation fetuses and newborns have some degree of deformity and paralysis. Such deterioration could be the result of exposure of neural tissue to amniotic fluid and meconium or direct trauma as the exposed neural placode impacts against the uterine wall. Theoretically, such deterioration could be reduced or eliminated by in utero closure of the lesion.
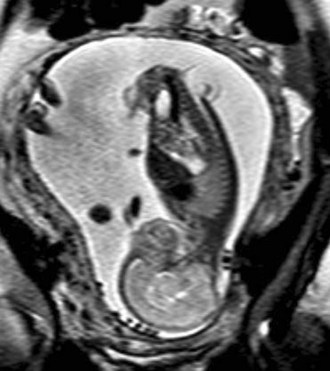
FIGURE 31–4 Fast spin-echo T2-weighted fetal MRI of a 20-week fetus with myelomeningocele. The sac is intact.
Some animal studies (in which a model for spina bifida is created by laminectomy and exposure of the fetal spinal cord to the amniotic fluid) have shown improved leg function if the lesion is closed before birth.19 Although other animal studies have failed to show behavioral improvement, they have documented the presence of somatosensory evoked potentials (SSEPs) in the treated myelomeningocele group compared with an absence of SSEPs in the untreated group.20 There is also evidence that the Chiari II malformation, which occurs in most individuals with spina bifida, is acquired and could potentially be prevented by in utero closure.21
In 1997, in utero repair of spina bifida was performed by hysterotomy at Vanderbilt University and at The Children’s Hospital of Philadelphia.22,23 Fetuses treated in utero were subsequently delivered by cesarean section, ideally at around 36 weeks of gestation. The early experience at both institutions suggested that compared with infants treated postnatally, fetuses treated in utero had a decreased incidence of hindbrain herniation and possibly a decreased need for shunting.24,25 The combined experience at the Children’s Hospital of Philadelphia and Vanderbilt University indicates that the incidence of hydrocephalus requiring shunting in patients treated in utero is less than that of historical controls stratified by spinal level who received standard postnatal care.26,27 It is hypothesized that fetal closure of the spinal lesion reduces the need for shunting by eliminating the leakage of CSF, which puts back-pressure on the hindbrain. The hindbrain hernia can be reduced, and the obstruction of the outflow from the fourth ventricle is relieved.28
It is now estimated that more than 330 in utero spina bifida closures have been performed.29 The procedure seems to be generally well tolerated by the expectant mothers. The fetal death rate is about 5% and is due to uncontrollable labor and premature birth. One analysis of leg function in children treated prenatally revealed no significant difference from a set of historical controls treated with conventional postnatal repair. Many of the children evaluated in this series had lower limb paralysis at the time of the surgery, however, which may have diluted any possible benefit.30 Data from the Children’s Hospital of Philadelphia suggested possibly better leg function in patients who already had intact leg movement shown by ultrasound before fetal sugery.31 Problems with delayed development of dermoid inclusion cysts and tethered cord may adversely affect long-term outcome.32 The preliminary experience suggests that children treated in utero have the same urodynamic abnormalities that are seen in conventionally treated children with spina bifida.33,34
To date, outcomes for spina bifida infants treated in utero have been assessed relative to outcomes in conventionally treated, historical controls.14 Such comparisons are prone to substantial bias, however, because fetuses who undergo in utero closure represent a highly selected subset of cases. In addition, the medical management of spina bifida is continuously improving, making comparisons with historical controls particularly problematic. Definitive answers about the benefits of fetal closure can be obtained only by a properly designed and conducted randomized trial.
Surgical Complications
Latex Allergy
An increased incidence of allergy to latex, as found in surgical gloves, balloons, and urinary catheters, has been well documented in children with spinal dysraphism.35 Allergy to latex is a type I, immediate, IgE-mediated reaction that can lead to anaphylaxis and death. Sensitization presumably arises from repeated exposure, at home and in the operating room. In many centers, all children with spina bifida are managed in a latex-free environment, and surgical procedures are preceded by prophylactic medications.
Late Deterioration
Tethered Cord
Approximately 20% to 30% of patients with myelomeningocele experience spinal cord tethering, which may result in progressive loss of function similar to that seen with occult spinal dysraphism.36,37 This phenomenon does not seem to be reduced by fetal repair, in which inclusion cysts have been found in 19% of patients who underwent fetal repair.38 Signs and symptoms usually appear during the 1st decade of life when growth is rapid and include back and leg pain, decrease in urinary control, gait difficulty and leg weakness, hip dislocation, progressive foot deformity, and possibly scoliosis. Urinary and gait difficulties are complaints confined to patients with low-level lesions, who are already functioning quite well neurologically and have the most to lose. In more severely affected patients who may already be wheelchair bound, pain may be the only manifestation of cord tethering and may be confused with appendicitis, hernia, urinary tract infection, or traumatic arthritis associated with abnormal posture or gait.
As with lipomyelomeningocele, the aim of surgery is to prevent further deterioration of function, but occasionally improvement is seen, even in preexisting deficits of long-standing duration. Reigel39 reported improvement in bladder function in five of nine patients with recent deterioration and in 77% of patients with gait difficulty and motor weakness. Pain almost invariably is relieved. Retethering can occur, and if new symptoms suggest this as an etiology, reoperation is indicated. An evidence-based review of the literature concluded that aggressive tethered cord release should be performed in adults within 5 years of symptom onset.40 Post–myelomeningocele repair patients tend to fare worse, however, than adult patients with closed dysraphism.
Hydromyelia
The routine availability of MRI has heightened awareness of this entity, which may be found in 68% of myelomeningocele patients.36 Hydromyelia presumably is the result of hydrodynamic forces arising from persistent fetal or untreated hydrocephalus that forces CSF down the central canal of the spinal cord.4 This explanation accounts for the frequency of symptomatic hydromyelia in patients with unshunted ventriculomegaly and patients considered to have “compensated” hydrocephalus, whose nonfunctional shunts were not revised because they lacked overt signs or symptoms of intracranial hypertension.41 An alternative explanation has been proposed by Oldfield and colleagues,42 who used dynamic MRI to study CSF flow. They suggested that a systolic pressure wave in the cranial compartment is transmitted in a pistonlike manner to the cerebellar tonsils, causing a systolic spinal CSF pressure wave that pushes CSF into the cord through its outer surface.
Symptoms of hydromyelia in spina bifida differ from symptoms in classic syringomyelia. The latter entity typically produces a dissociated sensory loss (relative loss of pain and temperature modalities with preservation of light touch), atrophy, and fasciculations primarily affecting the shoulder girdle and hands. When found in association with myelomeningocele, hydromyelia typically results in progressive bladder dysfunction, spasticity of upper and lower extremities, quadriparesis, and generally preserved sensation. The myelomeningocele repair may become swollen and tender. Symptoms may be due in part to the direct effects of the hydrocephalus stretching the cortical motor fibers. Hydromyelia may also be seen with progressive scoliosis. Spinal cord tethering may be the primary underlying etiology, however, of scoliosis and hydromyelia in these cases.43
Workup consists of MRI of the brain, followed by MRI of the entire spine to visualize the extent of the hydromyelia and the associated Chiari malformation and tethered spinal cord (Fig. 31–5). The hydromyelia cavity may be localized to a few spinal segments or extend throughout the length of the spinal cord. There may be multiple cavities and septations.
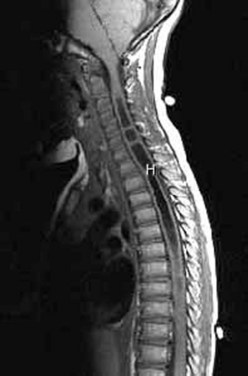
FIGURE 31–5 T1-weighted mid-sagittal MRI of a patient with Chiari malformation and large septated hydromyelia (H).
Other procedures have been described. Terminal ventriculostomy was advocated by Gardner and colleagues44 for classic syringomyelia, but the myelotomy tends to scar closed with time. In addition, the negative pressure provided by the pleural end of a shunt provides more decompression than simply allowing the hydromyelia to communicate with the subarachnoid space. Park and colleagues45 advocated posterior fossa decompression and plugging of the obex to prevent CSF fluid from the fourth ventricle from entering the central canal in patients in whom ventriculoperitoneal shunt revisions do not alleviate symptoms. This is a formidable procedure, however, and recurrent vomiting may occur from irritation or compression of the brainstem at the obex.
Chiari Malformation
MRI of the brain and spine is the procedure of choice for initial evaluation because the lesion is readily visualized and associated abnormalities are easily excluded. Mid-sagittal views show the tongue of cerebellum extending below the foramen magnum in type I lesions (see Fig. 31–5) and the “vermian pseudotumor,” consisting of the fused brainstem and cerebellum, in type II lesions.
Chiari type II lesions manifesting in an infant with myelomeningocele are particularly difficult to manage. When symptoms are severe, the brainstem nuclei may be irreversibly damaged, and the patient would not improve with surgical decompression.46 This situation has prompted a trend toward earlier intervention, when improvement may still be possible.48 If shunt function is shown to be optimal with shunt tap or imaging, Chiari decompression should be undertaken.
Patients are operated on in the prone position. A midline incision is used to expose the suboccipital bone and the cervical spine down to the level of the cerebellar hernia as seen on MRI, and a laminectomy is performed. The tonsillar hernia can usually be seen through the translucent dura, which is opened in the midline, beginning at the lowest point of the laminectomy and progressing cephalad. In this way, MRI-compatible clips or a running suture can be used to control bleeding from the anomalous venous sinuses in the posterior fossa. The dural opening should include the fibrous band usually present at the foramen magnum; however, because the foramen magnum is typically large and the venous sinuses are caudally displaced, the dural incision should rarely be carried above the level of the foramen magnum. It may be possible to open the fourth ventricle and establish CSF flow to the subarachnoid space, but dense adhesions and distorted anatomy often may make this unwise. As in Chiari I repair, the dura is left open, or a dural graft is employed, and the muscles, fascia, and skin are closed as usual.49
Shunt Malfunction and Arrested Hydrocephalus
Any neurologic deterioration in a patient with myelomeningocele should prompt a thorough investigation of the shunt. Some patients have nonfunctioning shunts and ventriculomegaly yet have no overt symptoms of intracranial hypertension or may have never received shunts and have baseline ventriculomegaly. These patients have been described as having “compensated” or “arrested” hydrocephalus. It has been suggested that some of these patients might benefit from shunting or shunt revision, which in some instances may result in improved neuropsychological functioning.50
Outcome
Significant progress has been made in the understanding and management of myelomeningocele over the past 25 years, particularly in the widespread use of multidisciplinary teams of specialists to manage children with this condition. More recent population-based data indicate that 1-year survival of individuals with spina bifida is approximately 92%51 and that 78% of all individuals with spina bifida survive to age 17 years.52 Mortality continues into the adult years53: 57% of patients die by the 4th decade of life.54 Death is due to problems associated with the Chiari II malformation, restrictive lung disease secondary to chest deformity, shunt malfunction, and urinary sepsis. Perineal sensation and urinary continence may be predictive of survival.55
Approximately 75% of young children with myelomeningocele are ambulatory to some extent, although most require braces and crutches. The likelihood that a child will ambulate is related to the level of the lesion; virtually all patients with sacral and lumbosacral lesions walk, but only half of patients with thoracic or thoracolumbar lesions walk with the use of braces and crutches. Patients with high-level lesions often can walk as young children but become wheelchair users as they get older, gain weight, or simply discover that wheelchair locomotion requires less energy and is faster than ambulation with crutches. A more recent report compared ambulatory potential between patients delivered by cesarean section and patients who underwent a trial of labor. Compared with the elective cesarean group, patients in the trial of labor group were more likely to be ambulatory at 2 years (independently ambulant 7% vs. 28%, ambulant with assistance 63% vs. 65%, or wheelchair bound 30% vs. 7%) and at 10 years (independently ambulant 5% vs. 21%, ambulant with assistance 30% vs. 54%, or wheelchair bound 65% vs. 25%). There was no statistical difference based on mode of delivery.56
Overall, approximately 75% of surviving infants have normal intelligence (IQ >80). This percentage decreases slightly to approximately 70% for surviving adults.57 Numerous factors predict poor intellectual outcome. In one study, the mean IQ of infants who were not shunted was 104; of infants shunted without complications, it was 91; and of infants shunted who had complications such as ventriculitis or anoxia, it was 70.58 Intelligence is also related to the level of the lesion: Approximately 55% of infants with thoracic lesions have significant developmental delay compared with only 25% of infants with lesions at lower levels.59
The cause of mental retardation in children with myelodysplasia remains controversial. McLone and colleagues60 attributed it to ventriculitis, but this does not account for all cases, and it is likely that in most cases forebrain dysfunction is simply part of the complex of anomalies associated with myelodysplasia. With respect to fetal surgery, preliminary evidence from the Children’s Hospital of Philadelphia showed that two thirds of surviving patients are within normal cognitive range at 2 years of age.27 Because cognitive function seems to be related to shunting, it is hoped that a decrease in overall shunting rate may improve overall intelligence in this population.
Although virtually all children with myelomeningocele have abnormal bladder function, urinary continence with the use of clean intermittent catheterization approaches 90% in children 5 to 9 years old. Ongoing urologic care is essential.61 Scoliosis is present in 49%, with 43% eventually requiring spinal fusion, for which the procedural complication rate is extremely high.62 Of children, 23% have at least one seizure. A tethered cord release is performed in 32%, with 97% having improvement or stability of preoperative symptoms.53
Occult Spinal Dysraphism and Tethered Cord
Embryology
After closure of the neural tube, the fetal spine is covered by ectoderm, and the low lumbar and sacrococcygeal segments have not developed.3 At this point, the caudal end of the neural tube blends with a large aggregate of undifferentiated cells, the caudal cell mass. A series of vacuoles in this mass coalesce and achieve continuity with the central canal of the previously formed neural tube (at roughly 29 days’ gestation), a process called canalization. The third phase involves retrogressive differentiation, during which the previously formed tail structures undergo a precise, ordered necrosis, leaving only the filum terminale, the coccygeal ligament, and the terminal ventricle of the conus as remnants by 11 weeks. Cell rests with potential for differentiation may be left within these structures, accounting for the development of lipomas, hamartomas, ectopic renal tissue, and the rare malignancy occasionally found in association with occult spinal dysraphism. Failure of the caudal cell mass to regress presumably gives rise to the hypertrophied filum terminale.
Pathogenesis
Symptoms may be caused in several ways. First, abnormal formation of the spinal cord and roots during embryogenesis may result in permanent deficits manifest at birth, as seen in myelomeningocele. Second, local masses growing within the rigid bony spinal canal may compress the conus medullaris or cauda equina and cause mechanical distortion or ischemia, with consequent dysfunction of these neural elements in a progressive fashion.63 This mechanical distortion may account for the acceleration of symptoms seen in some patients with significant weight gain in whom a lipoma enlarges in proportion to other body fat stores.
Finally, symptoms may be produced by traction on the spinal cord. Early in embryonic development, there is a progressive and rapid ascent of the conus within the bony spinal canal owing to faster growth of the vertebral bodies compared with the spinal cord. This “ascent of the conus” in children is slight, being only one segment, from L3 to L2 in the period from the 26th week of intrauterine life until maturity.64 Nonetheless, if the conus is tethered to the bony spinal canal, as often occurs in dysraphism, there is loss of mobility of the conus during spinal flexion and extension. Experimentally, spinal cord tethering has been shown to interfere reversibly with spinal cord oxidative metabolism65 and is probably the major cause of progressive neural damage in older children and adults with dysraphism lesions.
Clinical Manifestations
Bony spina bifida occulta at L5-S1 is a common radiologic finding in children and adults and is usually associated with no symptoms or signs. Unless other findings are present, no further evaluation or treatment is required. Signs of clinically significant spina bifida occulta may be in the form of cutaneous, neurologic, orthopaedic, urologic, or rectal abnormalities (Table 31–2).
TABLE 31–2 Presenting Symptoms and Signs of Occult Spinal Dysraphism
| % | |
|---|---|
| Leg weakness | 48 |
| Cutaneous abnormality | 48 |
| Foot deformity | 39 |
| Urinary incontinence | 36 |
| Fecal incontinence | 32 |
| Sensory abnormality | 32 |
| Recurrent urinary tract infection | 20 |
| Gait abnormality | 16 |
| Scoliosis | 14 |
Adapted from James H, Walsh J: Spinal dysraphism. Curr Prob Pediatr 11:1-25, 1981.
Cutaneous Syndrome
Cutaneous abnormalities indicative of an underlying spinal dysraphism are situated on or near the midline, usually in the lumbrosacral region, but occasionally in the thoracic region or the neck.66 A wide variety of abnormalities are seen.
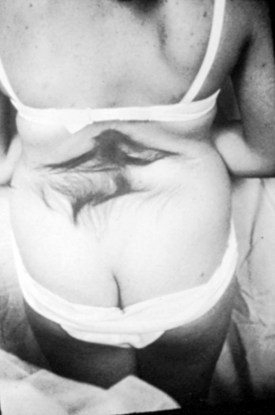
A striking finding is the hairy patch (hypertrichosis), also known as “faun’s tail,” which always occurs in the midline. It may be small but more commonly is a wide, diamond-shaped patch in the lumbar or lower thoracic area (Fig. 31–6). Frequently, the patient’s mother has trimmed the hair for years before presentation to the physician. If the dysraphism is in the cervical or upper thoracic region, there is usually a smaller patch of silky hair. The underlying abnormality may not be confined to the level of the hairy patch, and it is important to image the entire spine. Hypertrichosis may occur with any of the types of dysraphism but is particularly associated with split cord malformations.67
Subcutaneous lipomas at or near the midline of the lumbosacral spine may indicate an underlying intradural lipoma. These are nontender, poorly circumscribed soft masses of fat that are continuous with the normal subcutaneous tissue and with the intraspinal portion of a lipomyelomeningocele. The overlying skin is normal, hairy, or dimpled, and there may be an associated angioma or skin tag (Fig. 31–7). Other conditions may manifest as skin-covered masses overlying the caudal spine (Table 31–3). Often, an older patient gives a history of having had the superficial mass removed for cosmetic reasons.
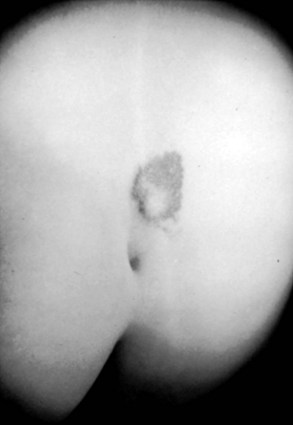
TABLE 31–3 Differential Diagnosis of Skin-Covered Lesions Overlying the Spine
Atretic meningoceles may also be seen, consisting of a central area of thin, pearly skin surrounded by a halo of red, pink, or brown (spinal aplasia cutis). These represent myelomeningoceles or meningoceles that have partially healed spontaneously. Dimples at the tip of the coccyx (coccygeal pits) are frequent findings in normal newborns and usually have no significance.68 Deep dimplelike depressions higher in the spine may be the external stigma of an epithelialized tract that connects with the filum terminale, spinal cord, or intraspinal dermoid cyst and require further investigation. The meningocele manqué is a skin blemish sometimes referred to as a “cigarette burn” because it resembles a small scar with loss of skin in the midline. When this blemish is obvious at birth, it may cause concern of CSF leakage, but this seldom occurs. Other cutaneous lesions that may be associated with a tethered cord include rudimentary caudal appendages (“tails”) and asymmetrical gluteal folds.
Neurologic Syndrome
Muscle weakness and gait disturbance are usually apparent at 2 years of age when a child begins to walk. On examination, there may be striking muscular atrophy and leg-length asymmetry. The deep tendon reflexes may be normal, increased, or absent, giving the pattern of a mixed upper and lower motor neuron lesion. Patchy sensory loss is present, particularly in the distal leg and perineum. Pain in the back with a radicular component is frequent in older children or adults.69
Orthopaedic Syndrome
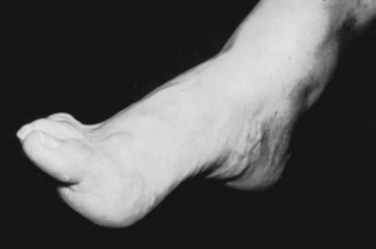
The most frequent orthopaedic finding is unilateral or bilateral cavovarus deformity of the foot, with or without leg-length discrepancy (Fig. 31–8). There may also be “claw toes.” The abnormal gait is the result of orthopaedic deformity and muscular weakness. It is presumed that the foot deformity is due to lack of, or weak innervation of, antagonistic muscles of the lower extremity.70 Scoliosis, especially if found in a young child or a boy or if noted to be rapidly progressive or accompanied by pain, also suggests an underlying spinal cause.
Urologic Syndrome
Occult spinal dysraphism should always be considered when one encounters infants with an abnormal voiding pattern, new-onset urinary or fecal incontinence in a previously toilet-trained child, or urinary tract infection in a child of any age. As opposed to older children, most infants with occult spinal dysraphism have normal results on urodynamic testing.71 This fact underscores the importance of recognizing the syndrome as early as possible so that prophylactic surgery can be performed.
Anorectal Anomalies
Anorectal anomalies, including vesicointestinal fissures, cloacal extrophy, rectovesical fistulas, and imperforate anus, are frequently associated with an underlying tethered cord.72 The overall incidence of tethered cord with imperforate anus is 35% and may be increased in boys.73 In many cases, no cutaneous abnormality is seen, and radiographic screening is indicated in these patients.
Radiologic Investigation
Spine MRI is the imaging study of choice to screen for occult spinal dysraphism. Bony anatomy is poorly seen but is of secondary importance in planning an operative approach for most lesions. A fibrous tract extending from a cutaneous coccygeal pit to the bony coccyx represents a persistent coccygeal ligament and is of no clinical significance. The presence of fat in the filum is a frequent incidental finding on MRI. Generally, if the conus is at the normal level (at or above the L2-3 interspace) and if the patient has no clinical signs or symptoms of a tethered cord, surgery is not warranted. Fat occurring very close to the conus may represent a special case, however, because this finding is associated with clinical tethering.74 A tethered cord may be suspected when the conus lies at the L2-3 level or below in a child or adult or if the conus is dorsally displaced.
Diffusion-weighted imaging may differentiate epidermoid cysts from arachnoid cysts.75 Postoperative studies may be difficult to interpret. The conus rarely ascends significantly after successful surgery, and even in asymptomatic patients the conus is shown to be dorsally displaced postoperatively. Although MRI is currently the best screening test for spinal cord tethering, it may not be useful for ruling out postoperative retethering. Some authors have proposed that imaging in the prone position to evaluate cord motion might be useful, but this has not been of value clinically.76,77
Ultrasonography through the infant spine or through congenital spina bifida or laminectomy defects has also been used to screen for occult spinal dysraphism.76,77 Although the level of the conus is readily seen, it is difficult to assign a specific spinal level to its termination. Sometimes one can visualize the tethering element, such as a hypertrophied filum or lipoma. Spinal cord motion has also been used to assess tethering.78 Ultrasonography may be useful as a screening test when the probability of a tethered cord is low, but MRI is preferable if an occult dysraphic lesion is strongly suspected.
Cases of tethered spinal cord with the conus in normal position on imaging studies have been described.79 Tethering is suspected on the basis of the typical clinical syndrome and confirmed at surgery. Mechanisms included hypertrophied filum terminale, meningocele manqué, and split cord malformation. This situation is uncommon, but when typical symptoms are present, surgical exploration is justified.
Specific Entities
Lipomyelomeningocele
Lipomyelomeningocele occurs at a rate of 1 in 4000 live births and in contemporary series is the most common cause of tethered cord syndrome.80 The term lipomyelomeningocele is misleading, in that it suggests herniation of neural elements through a spina bifida defect into a meningeal sac. The neural elements remain within the spinal canal, and only the lipoma itself may protrude through the spina bifida defect to manifest as a subcutaneous mass. Some authors have used the term lipoma of the cauda equina, but the fatty mass is invariably attached to the conus medullaris or filum terminale rather than to the cauda equina. Chapman81 classified lipomyelomeningoceles as lesions that attach to the caudal end of the conus, lesions that insert dorsally, and lesions that are transitional.
If the lipoma inserts onto the dorsal surface of the conus, there is usually a substantial subcutaneous mass, which may be asymmetrical. Along the lateral interface of the attachment of the lipoma to the spinal cord, the dura and pia are also fused (Fig. 31–9). Sensory roots emerge just anterior to this lateral line of fusion. As a result, neither the sensory roots nor the motor roots are actually within the lipoma. Alternatively, the lipoma may join the conus at its caudal end. The remaining mass may lie entirely within the spinal canal or extend dorsally through the spina bifida defect (Fig. 31–10). The fatty tumor may replace the filum terminale, or there may be a separate filum that lies anteriorly. The nerve roots usually lie ventral to the fatty mass but may lie within the fibrous ventral portion of the mass itself. Transitional forms may occur, which are often the most difficult to manage surgically. Probably the rarest subtype is the chaotic lipomyelomeningocele, described by Pang and colleagues,82 which has a prominent ventral component. MRI often defines the type of lipomyelomeningocele, which is helpful in understanding the anatomy that is encountered at operation.
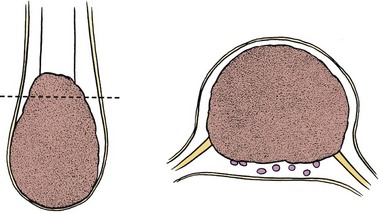
Lipomyelomeningocele is only one form of occult spinal dysraphism, and elements of other forms may coexist. Some patients may have associated anorectal or urogenital anomalies,83 and the pathologic anatomy may include elements of myelocystocele, diastematomyelia, or thickened filum.80 Despite the superficial resemblance of lipomyelomeningocele to open dysraphism (myelomeningocele), it is rare for patients to have associated hydrocephalus, gyral anomalies, or Chiari malformations. The relationship of Chiari type I malformation found in association with lipomyelomeningocele has been controversial, but a more recent study found a 13% incidence of Chiari I malformations in patients with lipomyelomeningocele. There was no difference, however, in hindbrain herniation or posterior fossa volumes of lipomyelomeningocele patients with or without Chiari I malformation.84 It is unclear that CSF overdrainage occurring during untethering procedures has any relationship to tonsillar herniation.85 Cadaver experimentation has not shown downward tonsillar descent with conus medullaris traction.86 As with myelomeningocele, there is an association with hydromyelia, particularly in the terminal spinal segments.87 The cognitive impairments characteristic of children with myelomeningocele seem to be absent, however, in children with spinal lipomas.88
The specific cause of lipomyelomeningocele is unknown. Familial occurrence has been reported in two siblings but seems to be extraordinarily rare.89 Other neural tube defects such as myelomeningocele may be found in 2% to 5% of siblings, suggesting a possible genetic predisposition to open and closed neural tube defects.90 An environmental cause has not been established, although a case of maternal use of valproic acid and a child with lipomyelomeningocele has been reported.91
The usual presentation of lipomyelomeningocele varies according to the age of the patient. Newborns and infants up to age 1 year typically present with a skin-covered lumbosacral mass (Fig. 31–11). There may be associated hair, sinuses, tags, or nevi. Children 1 year of age or older present with progressive signs or symptoms of tethered spinal cord, as previously described. Adults may present with back pain and sciatica or acute neurologic deterioration associated with lifting, falls, exercise, or assuming positions of spinal flexion. It has been suggested that the lipoma may enlarge or shrink with overall body fat.92
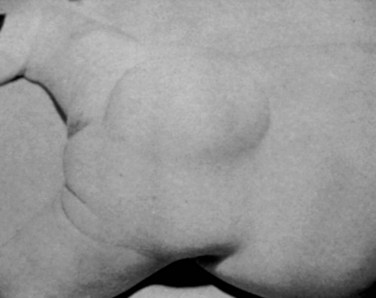
MRI should be performed in sagittal and axial planes. Fatty tumors are readily visualized on T1-weighted images as areas of increased signal intensity because of their short relaxation times. The conus is low, extending to the caudal portion of the thecal sac. The lipoma may insert dorsally (Fig. 31–12) or caudally (Fig. 31–13) and may or may not include the filum. In some instances, the conus may be at the normal level (at or above the L1-2 disc space), but the filum may be thickened and of fatty density. If these patients have other clinical signs or symptoms of tethered cord syndrome, they should be considered to have a tethered cord, and exploration should be carried out.79 An associated syrinx or hydromyelic cavity frequently may be seen within the terminal spinal cord, corresponding to a dilated terminal ventricle (Fig. 31–14). A large neurogenic bladder may also be seen. Lipomyelomeningocele must be distinguished from epidural lipomatosis, which is a rare condition in which fat accumulates in the dorsal epidural space, usually in the thoracic region, as the result of the use of exogenous corticosteroids or from corticosteroid-secreting tumors.
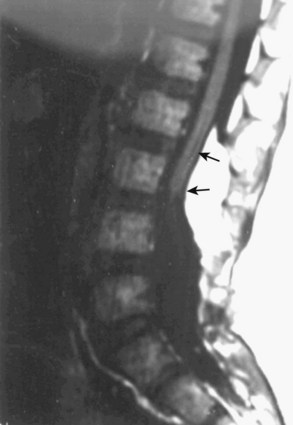
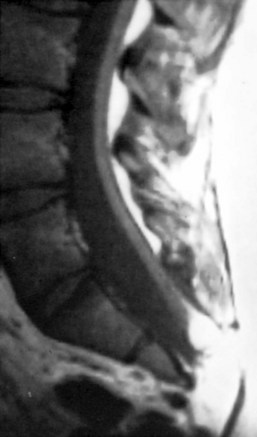
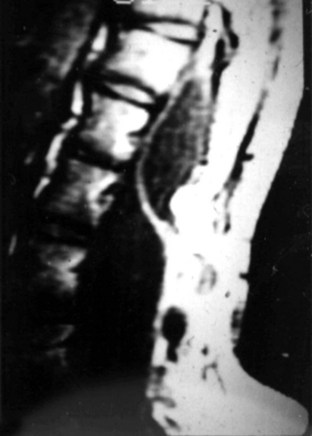
Urologic evaluation is recommended and should include ultrasonography of the bladder and urodynamic studies. More recent work has indicated that the presence of lipomyelomeningocele is predictive of a worse urologic outcome, in contrast to tethered cord syndrome without dysraphism, in which abnormal urodynamic studies alone are not predictive of poor urologic outcome.93
Most neurosurgeons recommend that all children with skin-covered lumbosacral masses, with or without associated neurologic, urologic, or orthopaedic abnormalities, should undergo exploration, preferably in infancy. The surgeon must be prepared to manage a lipomyelomeningocele properly before undertaking the exploration of any midline spinal mass, to avoid damaging neural structures. The first operative procedure stands the best chance of success because scar formation and distorted anatomy found at reoperation compound the difficulties inherent in the procedure.94 Although some authors have questioned the value of prophylactic surgery,95 most believe that infants and children with tethered cord syndrome should have prophylactic surgery at the time the lesion is discovered, preferably in the first 6 months of life.95–98 Early surgery is most likely to untether the cord effectively and, because tethered cord syndrome is progressive, is most likely to maximize functional outcome. An adult patient with a lipomyelomeningocele and nonprogressive symptoms may be followed nonoperatively, although this situation is uncommon. Adult patients who become symptomatic should be offered surgery.99 The patient should be warned of the possibility that permanent worsening may occur.
Magnification with loupes is helpful, and the operative microscope may be required for portions of the dissection. An elliptical skin incision surrounding the subcutaneous mass is made along a vertical axis, with hemostasis being obtained by applying straight mosquito hemostats. The subcutaneous tissue is incised in a circumferential fashion down to the lumbodorsal fascia (Fig. 31–15). The ellipse must be narrow enough to allow skin closure at the end of the procedure but broad enough to resect most of the subcutaneous mass. The subcutaneous fat is incised in a circumferential fashion down to the lumbodorsal fascia using the monopolar cautery. When an anatomic cleavage plane becomes evident, the lipoma is mobilized medially by blunt dissection with the use of a sponge and periosteal elevator so that the stalk can be visualized as it traverses the fascial defect. A self-retaining retractor is inserted, and the lowest intact laminar arch above the bony defect is palpated. The fascia over this spinous process is opened with the cutting cautery, and the laminae are exposed.

A laminectomy of this segment is done, exposing the underlying normal dura. It may be helpful at this point to amputate the large subcutaneous mass with the skin attached at the level of its stalk. Starting at the level of normal dura cephalad to the mass, the epidural fat is melted with the bipolar cautery until the dural defect with fatty tissue extruding through it is encountered. A midline dural opening is made above the defect, exposing the spinal cord (Fig. 31–16). As the dural opening is carried inferiorly toward the defect, a transverse band of thick fibrous tissue is noted at the rostral end of the lipoma stalk, which acts to kink the spinal cord. This is opened widely along with the dura. The dural opening is extended caudad on either side of the exiting lipoma circumferentially.
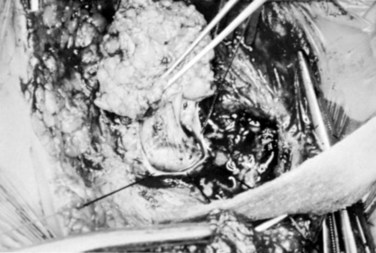
At this point, the lipoma usually is found to correspond to one of the types described by Chapman.81 Lipomas that insert into the conus dorsally can be removed from the dorsal aspect of the cord in a plane superficial to the lateral lines of fusion of lipoma, conus, and meninges, with the nerve roots emerging anteriorly. Simultaneously, the lines of fusion are divided laterally, first on one side and then on the other, with bipolar cautery and microscissors or a knife blade over a dissector (Fig. 31–17). The CO2 laser has been helpful in shaving down the mass of the lipoma. The filum is identified and divided.
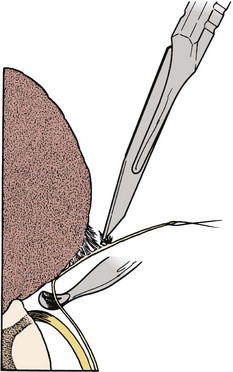
FIGURE 31–17 Lateral attachment between lipoma and dura is sharply divided, while dissector protects underlying nerve roots.
Lipomas that insert caudally into the conus must be sectioned distal to any functional roots, which may be identified using electrical stimulation and EMG. It is unnecessary to remove all the gross lipoma; to attempt this risks damage to the conus and nerve roots. Simple division of the lipoma releases the point of tethering and fulfills the goal of the operation. Pang and colleagues100 reported a decreased rate of retethering, however, after a very aggressive sharp excision of the lipoma.
After the lipoma has been largely removed from the spinal cord, it is sometimes possible to reapproximate the pial edges of the cord to reconstitute the normal tubular configuration (Fig. 31–18); this does not improve neurologic function but may serve to deter retethering. If the thecal sac is capacious, a fine suture may be used to anchor the conus to the ventral dura to discourage dorsal tethering. If a terminal syrinx is seen on preoperative MRI, a midline myelotomy may be performed to provide communication with the subarachnoid space.
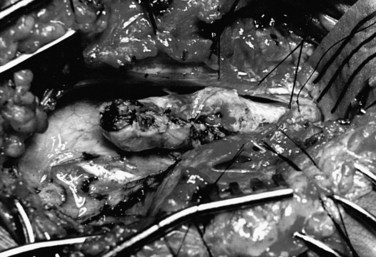
When the cord is free of adhesions, the dura is closed. In some cases, the dura may be approximated and closed with a running locked suture of 4-0 nylon. In many situations, this closure results in stricture of the canal, however, with the likelihood of scar formation and retethering. If there is any doubt, an elliptical graft is sutured to the dural edges. This may be technically very difficult if the dural edge is anteriorly located in the spinal canal. Acellular human dermis (Alloderm) is a convenient graft material. Alternatively, artificial substances such as Gore-Tex (Gore, Newark, DE)101 may be used to deter scar formation, but it is difficult to obtain a watertight closure with these materials, and tethering often occurs to the pseudodura that forms beneath the graft. Often the superficial subcutaneous tissue layers in the closure are inadequate for a truly watertight closure, and the dural repair becomes of primary importance to avoid a CSF fistula. The patient is placed in steep reverse Trendelenburg position, and a Valsalva maneuver is performed to ensure the closure is adequate. It may be useful to seal the suture line with tissue glue.
Surgery is relatively safe. The major postoperative complications are CSF leaks and wound breakdown because it is difficult sometimes to obtain good tissue for surgical repair. A persistent subcutaneous collection or frank leak requires re-exploration of the operative site and meticulous closure. An external spinal drainage catheter carefully inserted above the lamina defect through a Tuohy needle for 1 week is useful in protecting the closure after re-exploration. Persistent fever with sterile CSF cultures and a cellular reaction postoperatively may reflect a mild allergic reaction to the graft. This allergic reaction may persist for several months if untreated but usually resolves with a short course of corticosteroids. The incidence of a new permanent neurologic deficit after initial surgery for lipomyelomeningocele is 3% to 6% in experienced hands and is usually confined to a lower root.80
Surgical treatment for lipomyelomeningocele is basically prophylactic and is aimed at preventing progression of fixed deficits or development of new deficits in infants and asymptomatic children or adults. Data documenting the progressive nature of untreated lipomyelomeningocele are compelling. Hoffman and colleagues102 reported 12 patients who initially were neurologically and functionally intact and were untreated or inappropriately treated. Only one of these patients remained intact, whereas the others experienced deterioration over time. After appropriate repair, there was a halt in further deterioration, but few patients improved in function. Patients who are discovered at a younger age (because of cutaneous abnormality) are more likely to remain symptom-free if they undergo appropriate surgery than patients in whom surgery is deferred.80 Despite these data, other investigators have pointed out that retethering results in late deterioration in 46% of patients with conus lipomas who underwent prophylactic surgery, which is no different from the natural history of the disease.95
Lipomas that asymmetrically interface with the cord present a particular challenge because of their lack of “safe space” at the placode-dural interface. Cochrane and colleagues103,104 reported that an asymmetrically located lipoma inserting laterally or ventrally into dura is more likely associated with earlier neurologic deterioration after surgery compared with symmetrically located placodes.
In symptomatic patients, 20% of patients who undergo an appropriate operation note improvement in stance and gait105; in about 39%, there may be improved bowel or bladder function.98 Deformities of the feet do not improve and may continue to worsen even after successful surgery because abnormal muscular innervation and gait lead to progressive deformity. Continued orthopaedic surveillance and physical therapy are indicated.
Hydromyelia is a frequent accompaniment of lipomas and was seen in 6 of 43 patients evaluated at the authors’ institution by MRI and in 19% of patients reported by Iskandar and colleagues.106 If noted on the preoperative scan, hydromyelia may be addressed at the same time as the lipomyelomeningocele procedure by terminal ventriculostomy, which connects the hydromyelia with the subarachnoid space. If a symptomatic hydromyelia recurs, treatment is best accomplished by a syringopleural or syringosubarachnoid shunt.107
Assuming that no other pathologic process is found, one must be concerned about the possibility of spinal cord retethering. The incidence of this phenomenon is unknown. Various authors have reported symptomatic retethering to occur in 20% of cases,108 but the incidence may be much higher. It is likely that virtually all patients experience anatomic retethering within a few weeks of surgery for lipomyelomeningocele but that the dorsal reattachment is at a higher spinal level, and symptoms are relieved because the spinal cord is no longer stretched. Symptomatic retethering can occur at virtually any time after initial surgery, from weeks to more than 10 years.
Developments in MRI technology have allowed assessment of spinal cord pulsations using phase-motion imaging techniques. McCullough and colleagues109 reported in a small group of patients that cervical spinal cord pulsation peak velocity is decreased in patients with tethering and that it improves after successful surgery. Such techniques would be valuable in diagnosing retethering in patients in whom static images are nondiagnostic. Experience with this technique at the authors’ institution has been disappointing, in that the range of cervical and lumbar cord pulsation velocities in normal individuals is so broad that there is considerable overlap with patients with obviously tethered cords. Another technique that is helpful in some instances is prone-supine metrizamide CT-myelography. In contrast to MRI, it is possible to perform an adequate image with the patient in the prone position using this technique, and ventral migration of the conus with contrast material seen dorsal to the cord rules out retethering.
Despite the technical challenges inherent in the procedure, reoperation for recurrent tethering is usually rewarding, at least in the short-term. Successful repeat untethering can be performed in approximately 90% of patients.94 Pain is relieved in most patients, and gait and motor dysfunction improve in about half of patients. The prognosis for improvement in genitourinary dysfunction and scoliosis is less optimistic.
Sacral Agenesis and Caudal Regression Malformations
Numerous anomalies involving the caudal spine have been described, ranging in severity from simple agenesis of the coccyx to complex malformations of the thoracic and sacral spine, lower extremities, spinal cord, genitourinary system, and gastrointestinal tract. The most common of these include the VACTERL syndrome110 (vertebral anomalies, anal atresia, cardiac anomalies, tracheoesophageal fistula, esophageal atresia, renal and limb anomalies), the OEIS complex (omphalocele, cloacal exstrophy, imperforate anus, and spinal deformities), and sacral agenesis. The most severe form includes fusion of the legs, a condition that has been termed sirenomelia, or the mermaid syndrome.111 A subset of patients with these anomalies are at risk for late deterioration, and detailed imaging should be followed by prophylactic surgery in some cases.112
Severe multiorgan cases of caudal agenesis presumably arise from an embryonic insult to the caudal eminence. Failure of somite differentiation causes failure of corresponding spinal cord and notochord segments. Failure of retrogressive differentiation gives rise to lipomas, myelocystoceles, and related conditions. Related hindgut and urogenital anomalies arise from disturbances of the cloaca. Early dehiscence of the cloacal membrane can result in cloacal exstrophy, in which the posterior wall of the cloaca herniates through a pelvic defect, everting the central bowel field between two hemibladder fields. Because the caudal eminence induces some mesodermal structures, including the metanephros, renal anomalies may follow as well.112,113
Evidence suggests extrinsic and genetic factors in the causation of caudal regression syndromes. A history of preexisting or gestational maternal diabetes mellitus is obtained in 16% to 49% of cases,112,114 although caudal spine anomalies occur in only 1% of infants born to diabetic mothers.115 Most cases are sporadic, although rare familial forms have been reported, and an association with chromosome 7q has been described.116 Drugs have been implicated in some cases.117
The severity of the defect determines the presentation. Newborns may have simple imperforate anus or severe anomalies, including omphalocele, cloacal exstrophy, ambiguous genitalia, and tracheoesophageal fistula. The likelihood of a tethered cord is related to the number of anomalies that are present. Overall, tethering is present in 24% of children with anorectal malformations and in 43% of children with complex malformations.118 Sacral agenesis is suspected by flattening of the buttocks, shortening of the intergluteal cleft, and prominence of the iliac crests. A soft, skin-covered mass overlying the lumbosacral area suggests a myelocystocele or lipomyelomeningocele with tethered spinal cord. The lower extremities may exhibit abnormalities consistent with the involved vertebral segments. Hammer toes, calcaneovalgus or equinovarus ankle deformities, and contractures of involved joints are similar to findings in myelomeningocele or other forms of tethered cord. Paralysis, atrophy, and incompetent sphincter muscles reflect the myotomes of involvement. Sensation is relatively spared, and the sensory level may be several spinal segments below the motor level.112 Anomalies higher in the nervous system generally are absent, but a case of Chiari I malformation has been reported.119
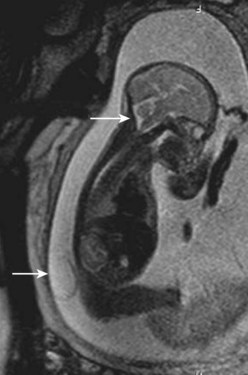
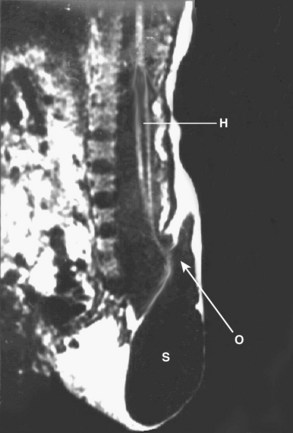
MRI of the lower spine is the most useful study and should be obtained in suspected cases. Fetal diagnosis of myelocystocele has been described, and it is vital to distinguish this lesion from a myelomeningocele with an intact sac (Fig. 31–19).120 From the neurosurgical standpoint, the goal is to determine if spinal cord tethering is present and, if so, to delineate the mechanism. Pang112 classified the appearance of lumbosacral agenesis into five types, based primarily on the appearance of the sacrum. From a practical standpoint, one can divide these anomalies into anomalies with a high conus and anomalies with a low conus. High symmetrical sacral agenesis is correlated with a truncated, club-shaped conus ending around T11 or T12. Tethering is not present, although dural canal stenosis has been reported as the cause of delayed deterioration in a few patients. Paradoxically, the lower, asymmetrical forms of sacral dysgenesis are more likely to have a low-lying tethered cord. Mechanisms of tethering include myelocystocele, typical hypertrophied filum terminale, and lipomyelomeningocele. Myelocystocele is particularly associated with these anomalies and is characterized by a trumpetlike cystic dilation of the caudal central canal, which is extensively tethered to the caudal spinal canal (Fig. 31–20).
Split Cord Anomalies
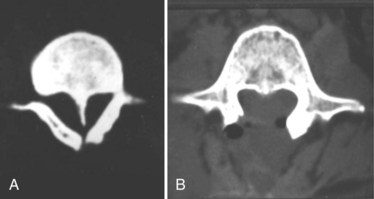
Split cord malformations are anomalies in which the spinal cord is divided along a portion of its length to form a double tube. Classically, two distinct forms, diastematomyelia and diplomyelia, have been described. The term diastematomyelia derives from the Greek words diastema (“cleft”) and myelos (“medulla”) and refers to the split in the spinal cord and not, as frequently misapplied, to the intervening bony or cartilaginous spur that often separates the two hemicords. It has been distinguished from diplomyelia (from the Greek diplos, meaning “double”), which, strictly speaking, refers to a condition in which the spinal cord is completely duplicated (i.e., two complete sets of anterior and posterior horns, with anterior and posterior roots emerging from medial and lateral surfaces of both hemicords). Investigators working more recently maintain that diplomyelia and diastematomyelia are different manifestations of the same pathologic entity and prefer the unifying term split cord malformation.67,121,122 The two hemicords in split cord malformation may exist within a single dural envelope (type II) or, more commonly, are enclosed within separate dural tubes (type I).67,123 The defect may be at any spinal level, but is most common in the lower thoracic or upper lumbar areas.124–126
The embryogenesis of split cord malformation is not fully understood, but the most logical explanation is that suggested by Bremer127 and amplified by others.121,128 Any viable theory must explain the frequent association of diastematomyelia with vertebral anomalies, spina bifida, and gut malformations and the equal involvement of the ventral and the dorsal aspects of the neural tube.
Clinically, split cord malformation occurs predominantly in girls.123–125 The largest series reported a female-to-male ratio of 1.5:1.129 The condition may be asymptomatic, or patients may present with symptoms of spinal cord dysfunction or scoliosis. The clinical findings are no different from findings in other cases of spina bifida occulta. Most patients have a midline thoracic or lumbar cutaneous abnormality. The incidence is 50% to 80%.63,124,125,130 The level of the skin lesion does not correspond to the level of the split cord malformation. The most common finding is a hairy patch (40%), but other abnormalities include dimples, hemangiomas, lipomas, sinus tracts, or myelomeningocele or meningocele.125
Historically, virtually all patients were reported to have significant congenital spinal deformity124,126 averaging 60 degrees during the growth years. Some modern series report a lower incidence, perhaps because of more widespread imaging.129,131 The incidence increases with age throughout childhood and is more likely to occur with higher lesions.123,126 Scoliosis is widely attributed to associated vertebral anomalies such as hemivertebrae, spina bifida, or spinal canal widening rather than to spinal cord dysfunction. In instances of tethering, neural traction may also influence curve development, however.132 The incidence of split cord malformation among patients with congenital scoliosis is estimated to be 4.9%.124 The type of scoliosis is nonspecific, apart from the equal incidence of right-sided or left-sided curves, and some authors have recommended obtaining imaging studies of the spinal cord before undertaking surgical treatment of patients with congenital scoliosis.124,125
Neurologic symptoms occur as a result of spinal cord tethering and may not arise until adulthood. Symptoms are typical of the tethered cord syndrome and include back pain, gait disturbance, muscular atrophy or deformity of the calf or foot, and spasticity or sensory abnormalities below the level of the lesion with trophic skin changes. Urologic abnormalities occur in 30% to 50% of cases.129,133 These abnormalities are nonspecific, and other disease entities, such as spinal cord tumor, Friedreich ataxia, and syringomyelia, must be considered. The traction force exerted on the spinal cord may result in brainstem or spinal cord symptoms as the result of an acquired Chiari malformation.134 Similarly, the spinal traction associated with corrective casts for scoliosis or operative intervention may lead to precipitous paraparesis.135 Late deterioration in adults has been reported after a blow to the back,136 after saddle block anesthesia,133 or simply with associated spondylosis.133 Adult patients may also show progressive myelopathy without a precipitating event, which may recover after surgery.70 Pain is present in some children and in virtually all adults. It is usually sharp and localizes to the level of the affected spinal segment.
Split cord malformations are now diagnosed in the fetal period using ultrasonography and fetal MRI.137 Associated visceral anomalies occur and include horseshoe or ectopic kidney, utero-ovarian malformation, and anorectal malformations. Associated spine anomalies include myelomeningocele and Klippel-Feil syndrome.138 The classic radiographic appearance of split cord malformation is a fusiform interpedicular widening of the spinal canal on the anteroposterior view with a midline oval bony mass projecting posteriorly from the vertebral body. The spike may be invisible on lateral views. Particularly in early infancy, the spur may be invisible and may not be in continuity with the vertebral body or laminae, supporting the view that the septum is calcified from a separate ossification center.126 Even in childhood and adult cases, the septum may be cartilaginous and invisible on plain radiographs. In such cases, widening of the neural canal should alert the clinician to the possibility of the diagnosis, and generally the adjacent pedicles should not be widened, as would be seen with an expanding intraspinal tumor.125 In addition to the widened canal, spina bifida is frequently associated, as are other vertebral anomalies. Scoliosis and kyphosis are common, and their incidence increases with age.125 This observation reinforces the importance of early detection because the chance of scoliosis developing increases the longer that patients are left without treatment.
MRI is the current procedure of choice for evaluation of split cord malformation (Fig. 31–21). A coronal study shows the split nicely, but sagittal views may not show each hemicord separately, and the study may mistakenly be considered normal. Severe scoliosis may make MRI difficult to interpret because the twisted cord weaves in and out of the imaging plane. In these cases, myelography with axial CT slices after the injection of contrast material is the procedure of choice. The entire spine should be evaluated for the possibility of secondary lesions, such as an associated tight filum terminale, a lipoma, a dermoid cyst or teratoma,139 or a second spur.
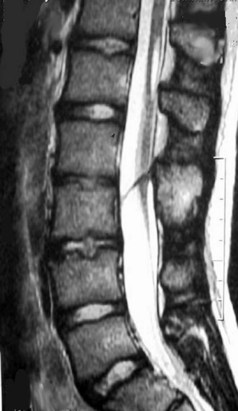
FIGURE 31–21 T2-weighted MRI in sagittal plane shows bony spike penetrating through spinal cord (type I split cord malformation).
Indications for surgery include progressive neurologic deficit and prophylactically before treatment of scoliosis. Application of a cast or halo pelvic traction for severe kyphoscoliosis may result in paraplegia if the point of tethering is not relieved.135 Similarly, it is advisable to remove the bony spike as a separate procedure before operative correction of scoliosis. Although it is technically feasible to perform both procedures at a single sitting,140 removal of the septum may result in early loss of evoked potentials owing to spinal cord manipulation, which reduces the safety of the corrective procedure. In addition, instrumentation of the spine is best avoided with intradural procedures when there is a possibility of a CSF leak.
Management of an asymptomatic patient with split cord malformation is controversial. Most investigators working more recently have urged that asymptomatic children and children who are neurologically stable should undergo prophylactic surgery within the first 2 years of life and cited tethering as the cause of progressive symptoms.67,122 The older literature states that surgery carries potential risk of neurologic worsening and noted that many patients with split cord malformation remain asymptomatic (or with fixed deficit) throughout growth, favoring a conservative approach.130,141 A compromise would be to consider the relationship of the septum to the intermedullary cleft in deciding the likelihood of progression owing to tethering. An asymptomatic patient may be followed nonoperatively if the septum is separated from the inferior portion of the cleft, but if the septum is at the caudal end of the cleft or higher, prophylactic surgery is advisable. To stratify risk of neurologic deterioration after surgery, Mahapatra and Gupta129 proposed a classification scheme based on the amount of bony septum that straddles the intramedullary cleft. Logically, the highest risk cases are cases in which the bony septum maximally occupies the region of cord split.
The role of surgery in a patient with a cleft but no apparent septum is also unclear. In such patients, there seems to be no reason for tethering, but Pang67,122 found that at operation these patients essentially always have an unrecognized midline fibrous point of tethering and recommended that these lesions be surgically explored. Finally, the indications for surgery in a patient with myelomeningocele and associated split cord malformation are unclear, although it seems logical to explore such a patient and untether the cord in the face of progressive symptoms referable to the lesion.
The surgical technique described by Matson and colleagues142 and Meacham143 is employed. Intraoperative evoked potential recording and rectal manometry have been described,144 but their value is unproved. In the thoracic spine, the level of the lesion is marked radiographically before the procedure. The patient is positioned as for a standard laminectomy. The paraspinal muscles on each side of the midline are freed and retracted laterally as in any standard laminectomy, but vigorous blunt dissection with a periosteal elevator is avoided because a spina bifida may coexist with the bony septum. The laminectomy is initiated at least one segment above and below the septum and carried out around the bony spike itself, exposing the dural cleft. The cleft usually extends cephalad to the spur but hugs it tightly caudad. A septal elevator is used to free the septum from the surrounding dura, and the superficial portion of the septum is removed by a rongeur or high-speed drill with a diamond bur within the investing dural sheath, which serves to protect the spinal cord. The inferior portion of the spur may extend anterior to the spinal cord at the inferior portion of the cleft, and in this case final removal is deferred until the dural compartment has been opened.
The dura is opened circumferentially around the cleft, and all intradural adhesions at the cleft side are divided (Fig. 31–22). The island of dura and the remainder of the spike are removed to the level of the anterior spinal canal (Fig. 31–23). A small midline myelotomy at the caudal portion of the cleft may be required to accomplish this. Type II lesions, in which the two hemicords reside within a common dural sleeve, are managed initially by laminectomy over the region of the abnormality. The dura is opened in a paramedian manner because the hemicords are often tethered dorsally at the midline. Fibrous bands arise from the caudal end of the cleft and are displaced inferiorly as they tether to the dorsal dura. These are sharply divided. Median rootlets are often present and are nonfunctional. They may be sectioned if necessary to release the hemicords.122 It is neither necessary nor desirable to close the anterior dura. The posterior dura is closed with a graft if necessary. If there is an associated tethering lesion apart from the split cord malformation, this should be addressed at the same time. A separate laminectomy or laminotomy may be required to section a filum terminale.

The procedure should be considered largely prophylactic, although there may be improvement in half of symptomatic patients.99 Long-standing deficits and scoliosis are unlikely to improve, but pain is almost always alleviated. Bladder and autonomic symptoms are least likely to improve.129 Complications include CSF leakage and worsening of neurologic status. Cases of type II malformation in which there is an oblique septum and a small, delicate hemicord seem to have a higher risk of cord injury.67 Failure to improve or late deterioration after operation may be due to failure to remove the tethered spur entirely, failure to address a second lesion such as a hypertrophied filum, or (rarely) regrowth of the septum.145
Anterior Sacral Meningocele and Currarino Triad
Since its original description in 1837 by “a distinguished surgeon” who preferred to remain anonymous,146 several hundred cases of anterior sacral meningocele have been reported. In 1981, Currarino and colleagues147 described the association of anorectal anomalies, sacral bony anomalies, and a presacral mass, which has been termed the Currarino triad. The presacral mass in these cases is an anterior sacral meningocele, a teratoma, or both. The triad is a form of split notochord malformation and is inherited as an autosomal dominant trait in 50% of patients.148 The varied presentations may bring patients to the attention of a wide range of specialists, including urologists, colorectal surgeons, gynecologists, pediatric surgeons, orthopaedic surgeons, and neurosurgeons. When the condition is properly diagnosed and treated, the cure rate is high; if it is improperly managed, meningitis may result in death.
Most anterior sacral meningoceles are congenital, as evidenced by their frequent appearance in infants and young children, the associated anomalies of pelvic organs, and familial incidence.149 In contrast to the typical posterior myelomeningocele, there is no accompanying hydrocephalus or association with Chiari malformations. In the Currarino triad, there is an abnormal adhesion between endoderm and ectoderm, or there are abnormalities of the notochord that cause a failure of the mesoderm to fuse in the midline. The notochord and somites fuse to form vertebral bodies, separating the endoderm and ectoderm. If the notochord is split, anterior fusion of the vertebral body fails, and a fistula between the gut and the spinal canal is formed. Partial resorption of this connection forms an anterior sacral meningocele or a retrorectal enteric cyst. The combination of mesodermal tissue with these enteric and neuroectodermal elements leads to formation of a presacral teratoma.150 Genes in the terminal region of chromosome 7q have been linked with sacral dysgenesis116,151 and may play a role in these malformations.
The lesion is detected far more commonly in women than in men, but in children the sex incidence is approximately equal. Most likely, there is really no sex predilection, and the lesion simply remains undetected in many men.152 Most clinical manifestations are caused by pressure of the sac on adjacent pelvic structures such as the rectum, bladder, uterus, and sacral nerve roots. Constipation is the most common symptom, particularly in infants and children. Many patients report use of laxatives and enemas. Urinary difficulties may be due to direct pressure of the mass or to pressure on sacral nerve roots. Midline lumbosacral back pain occurs in some patients and may radiate to the perineal area or inner thighs. Some patients report relief after a bowel movement. Headache may be due to either high or low intracranial pressure. Classically, the headache begins in childhood with sudden onset with Valsalva maneuver, as during straining or defecation. In later life, it may accompany coitus.153 The headache is reproduced when the pelvic mass is compressed on digital examination. When the patient stands, the meningocele fills with fluid, which may cause a secondary low-pressure headache.
Meningitis may be either septic or aseptic and may be recurrent.154 Bacterial meningitis may be spontaneous owing to microperforations of the rectum allowing passage of bacteria into the sac, or may result from traumatic rupture of the sac during childbirth. Meningitis may also result from attempted needle aspiration of the sac. Aseptic meningitis has been described, presumably from irritation secondary to an associated dermoid.155 In women of childbearing age, anterior meningocele may result in obstructed labor or rupture of the sac during delivery.153
The cardinal sign is a smooth, cystic mass palpable on rectal or pelvic examination. It is usually adherent to the sacrum. The mass is detectable on routine abdominal examination only when it is enormous. The differential diagnosis of masses includes dermoids, chordomas, and osseous or metastatic tumors.156
Radiologic evaluation of the sacrum usually shows some abnormality, classically the “scimitar” sacrum. The anteroposterior radiograph shows a well-circumscribed lucent defect on one side of the sacrum corresponding to the sac, and there is no surrounding bony destruction.153 Urologic contrast studies often show compression of the bladder and uterus and duplication of the ureters or renal pelvis. CT-myelography of the pelvis may be indicated in some cases and is performed with water-soluble contrast medium to show a small communication between the subarachnoid space and the meningocele sac. MRI is often sufficient to show the communication (Fig. 31–24). Heterogeneous signal intensity on T1-weighted and T2-weighted sequences may suggest a teratoma.157
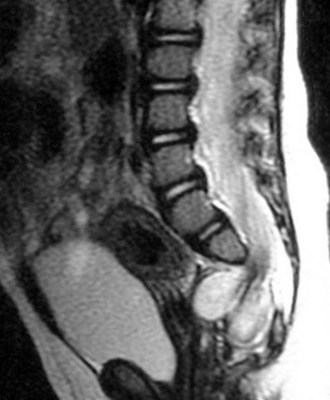
FIGURE 31–24 Mid-sagittal MRI of anterior sacral meningocele in a child. The communication was oversewn by posterior approach.
Surgical treatment of symptomatic lesions is generally advisable because there is no possibility of spontaneous regression, and in untreated patients there is a 30% mortality rate owing to pelvic obstruction at the time of labor or to erosion into the rectum followed by meningitis.152,153 An asymptomatic lesion may be followed without the need for surgery if there is no possibility of pregnancy and if the lesion does not enlarge on repeated rectal examination.
Surgical treatment via laparotomy has been described historically and has been advocated more recently.152,158 Most modern authors favor sacral laminectomy as the initial approach, however.159 This approach allows visualization of the intraspinal contents and resection of adhesions or division of an abnormal filum terminale. Nerve roots can be protected, and there is usually good visualization of the opening into the meningocele to allow suturing. The goal of surgery is to untether the spinal cord, decompress the sac, and obliterate the CSF fistula. It is unnecessary to remove the lining of the pelvic cyst, which may be densely adherent to the rectum. If the dura is fragile or cannot be mobilized sufficiently to allow suture even with a fascial graft, a posterior pelvic operation or laparotomy should be performed with the assistance of an experienced abdominal surgeon.160
The technique of sacral laminectomy has been reviewed.161 Antibiotic coverage and bowel preparation are begun 48 hours before surgery in case the rectum is inadvertently entered during the procedure. Under general anesthesia, the patient is placed in the prone position with the chest and abdomen supported, and the anus is draped out of the field with a plastic sheet. A lumbosacral laminectomy is performed from L5 to S4, and the posterior dura is opened longitudinally.
The lithotomy position is used in case an abdominal procedure is required.152 After a urethral catheter is placed, a midline incision is extended from the symphysis pubis to above the umbilicus. The procedure is carried out similar to an abdominoperineal resection of the rectum. The wall of the meningocele is exposed by opening the peritoneum on the right side of the rectum and dissected between the wall of the cyst and the fibrous capsule, keeping the sac intact. When the base has been identified, the sac is opened, and nerve roots are identified. The meningocele is resected at its base, leaving enough membrane to effect a watertight closure. The incision is closed without drains.
Surgical results reported in the recent literature have been generally good.158,162 Complications include meningitis or neurologic deterioration in cases in which nerve roots have been found within the sac.
Congenital Dermal Sinus and Dermoid Cysts
In early development, the terminal portion of the neural tube and the coccygeal vertebrae are intimately related and perhaps fused to the overlying epithelium. As the spine begins to elongate, the sacrococcygeal ligament applies traction on the overlying skin, producing a dimple in the skin overlying the sacrococcygeal region. There is no connection with the spinal canal, but MRI may show the fibrous connection to the coccyx. These coccygeal pits are reported to occur in about 4% of children.163 Some authors have suggested that a “traction test” to determine the rostral or caudal direction of the tract may be useful,164 but a more useful test is simply to feel the tip of the coccyx directly beneath the dimple. No treatment is necessary except local cleanliness.
More superficial sinus tracts that do not penetrate to the spinal canal are explained as failure of fusion of the cutaneous ectoderm after it has separated from the neuroectoderm. Focal enlargement of the tract may occur at any point to form dermoid or epidermal inclusion cysts. The dermal elements are typically intramedullary in the thoracic region and adherent to multiple nerve roots of the cauda equina in the lumbosacral region. Similar dermal inclusions occur in myelomeningoceles165 and after lumbar puncture. Pilonidal sinuses are acquired lesions in adults and are believed to result from repeated trauma or an inflammatory reaction secondary to penetration of broken strands of hair. It is possible that some pilonidal sinuses in adults represent chronic infection within a superficial congenital dermal sinus, and the distinction between these entities may be blurred. In practice, the term pilonidal sinus should be reserved for acquired lesions in adults.
Congenital dermal sinuses may be detected during routine examination of an infant or child or deliberately sought after recurrent bouts of meningitis or local infection at the level of the skin.166 The sinus occurs in the skin as a deep depression, usually in the lumbar region, which may be accompanied by nevi or hypertrichosis. Sinus tracts may occur in the cervical or thoracic regions.167 Occasionally, multiple sinuses are encountered. There may be a history of an accompanying purulent or clear discharge. Local infection of the sinus tract with staphylococci or Escherichia coli is common. The opening of the sinus tract is invariably in the midline and may be extremely small. Such a sinus opening occurring above the sacrococcygeal region should be explored and excised. Probing and injection of dyes are inadvisable.
Dimples below the top of the intergluteal crease require no investigation beyond physical examination. Ultrasound may be a useful screening test,168 but MRI is indicated in higher lesions to follow the depth of the tract, to determine the level of the conus, and to evaluate the spinal canal for inclusion cysts or abscesses (Fig. 31–25).169 The tract may be difficult to visualize, and surgical exploration of pores above the sacral region may be indicated even if radiologic studies do not clearly show a communication with the thecal sac. Sinus tracts in the cervical region should be evaluated with cranial MRI because the dermoid tumor and tract may extend into the posterior fossa.
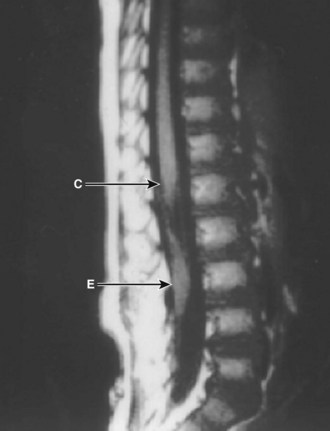
FIGURE 31–25 MRI of spinal congenital dermal sinus tract and lumbar dermoid inclusion cyst. C, conus; E, epidermoid.
Prophylactic surgery is performed as early as possible to excise the entire tract.166,167 In asymptomatic patients, the lesions may be explored and the tract followed to its termination. A surgeon undertaking such an operation must be prepared to perform an extensive intradural dissection because the tract may extend for a considerable distance. The operation is begun with an elliptical skin incision surrounding the sinus opening and any abnormal skin surrounding it. The tract is sharply dissected and followed through the fascia. A laminectomy is performed if the tract appears to continue to the dura. If the tract attaches to the dura, the dura is opened lateral to the entrance point, and the intradural contents are inspected. Any intradural tract must be followed to its termination even if this involves an extensive laminectomy to the conus because remaining tissue has the capacity to grow into a large dermoid inclusion. Occasionally, it may be unclear if an intradural tract is a hypertrophied filum or a true sinus tract, and frozen section may be useful.
Intradural cysts are completely removed without violating the capsule if possible. If the cyst has ruptured or is infected, a dense arachnoiditis with scarred nerve roots prevents complete excision.170 In this event, judicious intracapsular removal of purulent material and dermoid elements is performed, but no attempt is made to remove the scarred capsule wall from the nerve roots. If a cervical or thoracic tract terminates in a neuroglial mass, the tract is amputated at the mass, and no attempt is made to remove it. A watertight dural closure is made except in patients in whom closure would compress residual infected dermoid cyst material, in which case the dura may be left open, and the muscle and fascia are closed.
Filum Terminale Syndrome
Patients present with a typical tethered cord syndrome. Cutaneous abnormalities are noted in approximately half of the patients171 and may consist of a sinus tract, a hairy nevus, a hemangioma, an accessory appendage (tail), or an area of atretic skin. Other symptoms include orthopaedic deformity (scoliosis, leg or foot atrophy), progressive weakness, and urologic dysfunction (urinary incontinence, bladder distention, loss of perineal sensation). Back pain is often prominent in teenage and adult patients. The condition is rarely familial.171,172 This condition is often suspected in association with imperforate anus, and an incidence of 35% has been reported.73 The condition is also suspected in children with voiding dysfunction, particularly if there is associated impaired bladder sensation or poor bladder emptying. The value of screening MRI is controversial in these patients.173,174
Radiologic investigation usually shows a simple posterior lumbosacral spina bifida but may reveal other vertebral anomalies. Further workup consists of studies to evaluate the level of the conus and may include MRI or supine myelography with CT. Barson175 measured the level of conus termination in normal infants and children and reported that on average the adult level (L1-2) is reached by 2 months of age. A conus tip below the L2-3 interspace in a child older than 5 years is definitely abnormal. A filum thicker than 2 mm at myelography is also abnormal. Although MRI may suggest the diagnosis with the finding of a low conus on sagittal and axial views, the filum itself may be poorly seen. If the clinical history is suggestive, myelography in the supine position with a follow-up metrizamide CT scan may be worthwhile even in the face of a normal MRI study. A few patients have been reported who have the typical skin and neurologic manifestations of tethered cord syndrome yet have the conus at the normal spinal level on imaging studies. At operation, these patients had thickened or fatty fila and evidence of tethering.79,176
The term minimal tethered cord syndrome is sometimes applied to such a scenario,177 in which the patient exhibits urologic dysfunction but lacks the MRI evidence of tethering. In these cases, division of the filum to relieve urinary symptoms must be guided by strong clinical suspicion. In adults, fat in the filum within 13 mm of the conus has been correlated with neurologic dysfunction, independent of conus position.74 Conversely, the presence of an asymptomatic fatty filum terminale on MRI is a normal variant, occurring in 0.24% of patients. If the conus is at a normal level, no further workup or treatment is required.178
If there is evidence of spinal cord tethering (a low conus or clinical symptoms), prophylactic surgery is recommended at the time the entity is discovered to prevent progression of symptoms and to relieve chronic back pain. A few children with spinal cord tethering have been reported to show no deterioration over a period of a few years,179 but most investigators believe that most patients eventually show progression of symptoms and that prophylactic surgery is indicated. A laminectomy is performed above or below the bifid segment, and the dura is opened. The filum is coagulated and sectioned as low as possible (Fig. 31–26). In some reported cases, clips placed at the cut ends of the filum have separated by 2.5 cm on postoperative radiographs.171 If it proves difficult to identify the filum clearly, it can be electrically stimulated or followed to its termination by performing an extensive sacral laminectomy. Often, small but functional rootlets are intimately attached to the filum and must be identified and dissected away before sectioning.

Terminal syringohydromyelia is reported with meningocele manqué and with tight filum anomalies and may be symptomatic.180,181 This condition is usually treated by untethering the cord and repeating imaging a few months later. If the terminal syrinx persists and is believed to be the cause of symptoms, shunting the cyst to the subarachnoid space, pleura, or peritoneum may be required.
The results of surgery are favorable in relieving back pain and preventing the progression of existing deficits, and often some long-standing neurologic deficits may improve. In the series by Anderson,105 urinary incontinence improved in 14 of 33 patients, and weakness and numbness of the legs lessened postoperatively in one third. Back and leg pains were relieved in virtually all children and adults in several series.69,105,182 Retethering is rare but is reported.183
Neurenteric Cyst
Although neurenteric cysts are quite rare, they are well-described entities consisting of cystic structures within the spinal canal or anterior to the vertebral bodies in the mediastinum, abdomen, or neck. Ventral cysts may have connections via a stalk to the meninges and spinal cord through a tunnel-like defect in the vertebral bodies. The cyst wall resembles foregut tissue histologically, and in the literature these lesions are also referred to as enterogenous cysts.184 They may occur in isolation or as part of a more complex group of spinal anomalies, including Klippel-Feil syndrome, spinal lipoma, and syringomyelia.185,186 They may occur at any spinal level, but most are between C3 and T7.187
Early in the development of the human embryo there is a communication between the yolk sac (destined to become the gut) and the dorsal surface of the embryo called the neurenteric canal. This fistulous tract transiently connects the future enteric cavity with the neural groove in the region of the coccyx. Bremer188 pointed out that abnormal accessory neurenteric canals may persist cephalad to the coccygeal tip and can give rise to cysts along the persistent tract. The tract also gives rise to associated vertebral anomalies: a widened vertebral body resulting from bony proliferation in an attempt to fill the gap after disappearance of the tract or a circular defect in the vertebral body produced by the persistent tract that forces bone to form around it. Because the accessory neurenteric tract can be elongated and stretched during growth, the canal may become divided into noncommunicating diverticula that lie at some distance from each other (e.g., in the thoracic cavity and spinal canal). The tendency for the cysts to lie to the right of the vertebral column has been ascribed to the developmental process of gastric and gut rotation.189 Persistence of only a portion of the tract results in a cyst only within the spinal canal.190 The cysts are usually anteriorly located within the canal (Fig. 31–27), although dorsally located ones are described.191
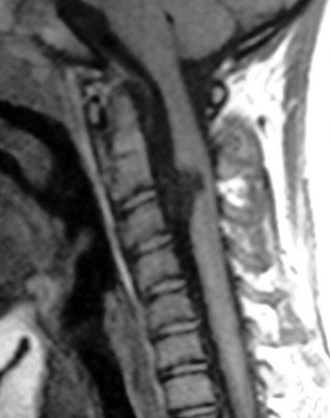
FIGURE 31–27 MRI of cervical spine shows anteriorly placed neurenteric cyst compressing spinal cord and causing myelopathy.
Neurenteric cysts within the spinal canal or ventral masses are similar pathologically. They may be thick, well-defined capsules or delicate, clear walls that may be confused with spinal arachnoid cysts. They are described as strawberry-colored192 or whitish190 and when in the spinal canal may be intradural-extramedullary193 or intramedullary.194 The cyst fluid is usually described as clear or milky and quite gelatinous. There may or may not be a ventral dural defect.
Histologically, the cyst wall typically is formed of endodermal ciliated columnar epithelium resting on a basement membrane and may contain gastric parietal and chief cells or mucin-producing goblet cells (Fig. 31–28).193 The epithelial cells are positive for keratin markers. Some investigators have made a distinction between neurenteric cysts, with well-differentiated gastrointestinal epithelium and a clear connection of the spinal cyst and the chest and abdomen, and “teratomatous cysts,” which have less differentiated epithelium and no ventral connection.195,196
FIGURE 31–28 Light microscopy with hematoxylin and eosin staining of neurenteric cyst. Epithelium resembles gut.
Neurenteric cysts are usually detected in childhood,190,193,194,197,198 although adult patients have been reported.197,199,200 Neurenteric cysts have been detected by prenatal MRI or ultrasound.201 The diagnosis is suggested by the combination of a thoracic cystic mass with vertebral anomalies. In early childhood, the lesion may manifest as a chest or abdominal mass with cardiorespiratory compromise202,203 or as a neck mass with tracheal compression or may be detected because of vertebral anomalies seen on a radiograph. Adults and older children tend to present with signs of spinal cord compression in the cervical or thoracic regions,204 which may be acute or follow trivial trauma.205 In addition to motor weakness, burning dysesthesias may occur when aggravated by coughing or sneezing or recumbency. Rarely, a neurenteric fistula may lead to meningitis with bowel organisms.204 Associated malrotation of the gut206 and diaphragmatic hernia207 may be found.
Plain radiographs may show widening of the neural canal, cleft vertebrae, or fusion of vertebral segments.207 An anterior soft tissue mass may be seen in the chest, neck, or abdomen, and the association of such a mass with vertebral anomalies should suggest the diagnosis. The presence of the classic circular defect through the vertebral body, although diagnostic, is not essential.207 Patients in whom the diagnosis is suspected should undergo MRI208 and possibly myelography with water-soluble contrast medium and postmyelogram CT to define the bony anatomy and establish the presence or absence of a communication between the spinal canal and a ventral mass. MRI shows a cystic mass and its relationship with the spinal cord.190
The treatment of neurenteric cysts varies with location and presentation. An intraspinal lesion can be corrected first, at a separate procedure, or as part of a combined procedure, such as thoracotomy, to excise the prevertebral lesion. Because the spinal mass is largely cystic, the traditional treatment has been a wide laminectomy with cyst drainage and conservative resection of cyst wall.197 Some authors have advocated radical resection of the entire cyst wall, however, which may require a lateral approach200 or corpectomy189,199 to approach lesions located ventral to the spinal cord. A cyst-subarachnoid shunt has been described,206 but the thick nature of the fluid and its possible irritation of the meninges make this unlikely to be useful in most cases. If there is an anterior dural defect, this may be plugged with a muscle or fascial graft from a posterior approach or ligated or sutured if a thoracotomy is performed.
Patients generally recover neurologic function if the deficit is not severe at the time of operation. Good outcomes can still be attained with subtotal resection.209 Recurrence has been noted after incomplete resection, but the reported recurrence rate in subtotally resected cysts ranges from 11% to 75%.204,209 In children in particular, delayed kyphosis or swan neck deformity may follow extensive laminectomy.
Intraoperative Neurophysiologic Monitoring
The routine use of intraoperative neurophysiologic monitoring to help guide the surgical management of tethered cord syndrome is controversial, and consequently this monitoring is perhaps underused. Although there have been few published studies on the efficacy of intraoperative neurophysiologic monitoring during release of tethered spinal cord, the authors’ clinical experience over 2 decades has been positive, as has been the experience of others.210,211 Application of a multimodality neuromonitoring protocol has been helpful in (1) identifying functional neural elements that may otherwise have been sacrificed, particularly in the case of a thickened filum terminale; (2) identifying the level of exiting nerve root when faced with ambiguous anatomy; (3) warning of excessive surgical traction on nerve root or spinal cord during manipulation and untethering; (4) assessing the functional properties of each hemicord in patients with diastematomyelia; (5) identifying emerging spinal cord injury; and (6) providing an overall sense of security to the neurosurgeon.
The techniques used in the authors’ practice to assess spinal cord and nerve root function and integrity include spontaneous and stimulated EMG, multipulse transcranial motor evoked potentials, and SSEPs. The first two EMG monitoring modalities are used to identify nerve root presence and assess their function in real time. Transcranial motor evoked potentials and SSEPs provide information about the functional integrity of the efferent motor and afferent sensory spinal cord tracts. Transcranial motor evoked potentials are of additional value in verifying nerve root function, serving as a cross-check to spontaneous and stimulated EMG. The details of this technique have been published.212–215
Neurophysiologic monitoring of spinal cord and nerve root function is most applicable to operations for spina bifida occulta and least beneficial in cases of spina bifida cystica. Children with myelomeningocele often have severe neurologic deficits that preclude reliable monitoring; SSEPs are invariably absent in high-level dysraphism. Neuromonitoring of myelomeningocele patients with low-level lesions undergoing untethering procedures is potentially helpful, however. A detailed description of the specific stimulus and recording parameters for each of these neurophysiologic monitoring modalities may be found elsewhere (see Chapter 14). EMG is briefly discussed here.
The preferred EMG monitoring technique is 10-channel recording from five bipolar subdermal needle electrode pairs inserted into the left and right adductor, quadriceps, tibialis anterior, gastrocnemius, and external anal sphincter muscles. In infants and young children, limited perianal space may preclude two distinct bipolar electrode pairs for the left and right sides; the left electrode is referenced to the right and vice versa. This electrode montage provides nerve root coverage from L2 to S4 segments. Although manometric monitoring of anal sphincter tone has been described,144,216 the combination of spontaneous and stimulated EMG is sufficient by itself.217 Urinary bladder manometry has been advocated as an additional technique to assess urinary sphincter tone because the parasympathetic fibers innervating the detrusor muscle do not travel with the pudendal nerves.218 This method is cumbersome, however, and not routinely implemented. A sixth EMG channel, rectus abdominis-iliopsoas, is sometimes added when it is necessary to monitor T12-L1 nerve roots.
To achieve focal stimulation for valid stimulated EMG recordings, a hand-held concentric bipolar stimulating electrode is used. Monopolar stimulators such as those often used for cranial nerve stimulation produce too much current spread, precluding individual nerve root identification. The authors employ a gradual “stair-step” approach to nerve root stimulation beginning at a low constant current and continuing until a compound muscle action potential is recorded. Generally, compound muscle action potentials for sensory nerve roots are elicited between 1 mA and 4 mA, depending on the vertebral level, whereas motor roots have significantly larger compound muscle action potentials and depolarize at much lower current levels (0.1 to 0.2 mA) at short stimulus durations (50 µsec). Nerve root fascicles often require higher current intensity (e.g., 5 to 8 mA) to depolarize and present responses of small amplitude compared with their root trunk counterparts. When attempting to confirm the absence of neural element within the filum, the stimulation intensity should be increased to 10 to 20 mA (depending on stimulus duration) to ensure that no root fascicle is present on the dorsal, ventral, or medial surfaces. If using voltage parameters, the threshold should be up to 100 V for filum sectioning.219
Key Points
1 Luthy D, Wardinsky T, Shurtleff D. Cesarean section before the onset of labor and subsequent motor function in infants with myelomeningocele diagnosed antenatally. N Engl J Med. 1991;324:662-666.
2 McLone D, Naidich T. Developmental morphology of the subarachnoid space, brain vasculature, and contiguous structures, and the cause of the Chiari II malformation. AJNR Am J Neuroradiol. 1992;13:463-482.
3 Sutton L, Adzick N, Bilaniuk L, et al. Improvement in hindbrain herniation demonstrated by serial fetal magnetic resonance imaging following fetal surgery for myelomeningocele. JAMA. 1999;282:1826-1831.
4 Bruner J, Tulipan N, Paschall R, et al. Fetal surgery for myelomeningocele and the incidence of shunt-dependent hydrocephalus. JAMA. 1999;282:1819-1825.
5 Tubbs R, Elton S, Grabb P, et al. Analysis of the posterior fossa in children with the Chiari 0 malformation. Neurosurgery. 2001;48:1050-1054.
6 Chapman P. Congenital intraspinal lipomas: Anatomic considerations and surgical treatment. Childs Brain. 1982;9:37-47.
7 Kulkarni A, Pierre-Kahn A, Zerah M. Conservative management of asymptomatic spinal lipomas of the conus. Neurosurgery. 2004;54:868-875.
8 Pang D, Dias M, Ahab-Barmada M. Split cord malformation: I. A unified theory of embryogenesis for double cord malformations. Neurosurgery. 1992;31:451-480.
9 Pang D. Split cord malformation: II. Clinical syndrome. Neurosurgery. 1992;31:481-500.
10 Warder D, Oakes W. Tethered cord syndrome and the conus in a normal position. Neurosurgery. 1993;33:374-378.
1 French B. Midline fusion defects and defects in formation. In: Youmans J, editor. Neurological Surgery. 3rd ed. Philadelphia: WB Saunders; 1990:1081-1235.
2 Stiefel D, Meuli M. Scanning electron microscopy of fetal murine myelomeningocele reveals growth and development of the spinal cord in early gestation and neural tissue destruction around birth. J Pediatr Surg. 2007;42:1561-1565.
3 Lemire RJ. Normal and Abnormal Development of the Human Nervous System. Hagerstown, MD: Harper & Row; 1975.
4 McLone D, Naidich T. Developmental morphology of the subarachnoid space, brain vasculature, and contiguous structures, and the cause of the Chiari II malformation. AJNR Am J Neuroradiol. 1992;13:463-482.
5 Little J, Elwood J. Epidemiology of neural tube defects. In: Kiely M, editor. Reproductive and Perinatal Epidemiology. Boca Raton, FL: CRC Press; 1991:251-336.
6 Prevention of neural tube defects: Results of the Medical Research Council Vitamin Study. MRC Vitamin Study Research Group. Lancet. 1991;338:131-137.
7 Botto L, Moore C, Khoury M, et al. Neural tube defects. N Engl J Med. 1999;341:1509-1519.
8 Babcook C. Ultrasound evaluation of prenatal and neonatal spina bifida. Neurosurg Clin N Am. 1995;6:203-218.
9 Nadel A, Green J, Holmes L, et al. Absence of need for amniocentesis in patients with elevated levels of maternal serum alpha-fetoprotein and normal ultrasonographic examinations. N Engl J Med. 1990;323:557-561.
10 Simon E, Goldstein R, Coakley F, et al. Fast MR imaging of fetal CNS anomalies in utero. AJNR Am J Neuroradiol. 2000;21:1688-1698.
11 von Koch CS, Glenn OA, Goldstein RB, et al. Fetal magnetic resonance imaging enhances detection of spinal cord anomalies in patients with sonographically detected bony anomalies of the spine. J Ultrasound Med. 2005;24:781-789.
12 Cochrane D, Aronyk K, Sawatzky B, et al. The effect of labor and delivery on spinal cord function and ambulation in patients with meningomyelocele. Child Nerv Syst. 1991;7:312-315.
13 Luthy D, Wardinsky T, Shurtleff D. Cesarean section before the onset of labor and subsequent motor function in infants with myelomeningocele diagnosed antenatally. N Engl J Med. 1991;324:662-666.
14 Rintoul N, Sutton L, Hubbard A, et al. A new look at myelomeningoceles: Functional level, vertebral level, shunting, and the implications for fetal intervention. Pediatrics. 2002;109:409-413.
15 Danish SF, Samdani AF, Storm PB, et al. Use of allogeneic skin graft for the closure of large meningomyeloceles: Technical case report. Neurosurgery. 2006;58(4 Suppl 2):ONS-E376. discussion ONS-E376
16 Mowatt DJ, Thomson DN, Dunaway DJ. Tissue expansion for the delayed closure of large myelomeningoceles. J Neurosurg. 2005;103(6 Suppl):544-548.
17 Arnell K. Primary and secondary tissue expansion gives high quality skin and subcutaneous coverage in children with a large myelomeningocele and kyphosis. Acta Neurochir (Wien). 2006;148:293-297. discussion 297
18 Murakami M, Hyakusoku H, Ogawa R. The multilobed propeller flap method. Plast Reconstr Surg. 2005;116:599-604.
19 Meuli M, Meuli-Simmen C, Yingling C, et al. In utero surgery rescues neurological function at birth in sheep with spina bifida. Nat Med. 1995;1:342-347.
20 Julia V, Sancho MA, Albert A, et al. Prenatal covering of the spinal cord decreases neurologic sequelae in a myelomeningocele model. J Pediatr Surg. 2006;41:1125-1129.
21 Osaka K, Tanimura T, Hirayama A, et al. Myelomeningocele before birth. J Neurosurg. 1978;49:711-724.
22 Adzick N, Sutton L, Crombleholme T, et al. Successful fetal surgery for spina bifida. Lancet. 1998;352:1675-1676.
23 Tulipan N, Bruner J. Myelomeningocele repair in utero: A report of three cases. Pediatr Neurosurg. 1998;28:177-180.
24 Sutton L, Adzick N, Bilaniuk L, et al. Improvement in hindbrain herniation demonstrated by serial fetal magnetic resonance imaging following fetal surgery for myelomeningocele. JAMA. 1999;282:1826-1831.
25 Bruner J, Tulipan N, Paschall R, et al. Fetal surgery for myelomeningocele and the incidence of shunt-dependent hydrocephalus. JAMA. 1999;282:1819-1825.
26 Tulipan N, Sutton L, Bruner J, et al. The effect of intrauterine myelomeningocele repair on the incidence of shunt-dependent hydrocephalus. Pediatr Neurosurg. 2003;38:27-33.
27 Johnson MP, Gerdes M, Rintoul N, et al. Maternal-fetal surgery for myelomeningocele: Neurodevelopmental outcomes at 2 years of age. Am J Obstet Gynecol. 2006;194:1145-1150. discussion 1150-1152
28 Sutton L, Sun P, Adzick N. Fetal neurosurgery. Neurosurgery. 2001;48:124-142.
29 Fichter MA, Dornseifer U, Henke J, et al. Fetal spina bifida repair—current trends and prospects of intrauterine neurosurgery. Fetal Diagn Ther. 2008;23:271-286.
30 Tubbs R, Chambers M, Smyth M, et al. Late gestational intrauterine myelomeningocele repair does not improve lower extremity function. Pediatr Neurosurg. 2003;38:128-132.
31 Johnson M, Sutton L, Rintoul N, et al. Fetal myelomeningocele repair: Short term clinical outcomes. Am J Obstet Gynecol. 2003;189:482-487.
32 Mazzola C, Albright A, Sutton L, et al. Dermoid inclusion cysts and early spinal cord tethering after fetal surgery for myelomeningocele. N Engl J Med. 2002;347:256-259.
33 Holzbeierlein J, Pope JI, Adams MC, et al. The urodynamic profile of myelodysplasia in childhood with spinal closure during gestation. J Urol. 2000;164:1336-1339.
34 Holmes NM, Nguyen HT, Harrison MR, et al. Fetal intervention for myelomeningocele: Effect on postnatal bladder function. J Urol. 2001;166:2383-2386.
35 Ellsworth P, Mergueria P, Klein R, et al. Evaluation and risk factors of latex allergy in spina bifida patients: Is it preventable. J Urol. 1993;150:691-693.
36 Talamonti G, D’Aliberti G, Collice M. Myelomeningocele: Long-term neurosurgical treatment and follow-up in 202 patients. J Neurosurg. 2007;107(5 Suppl):368-386.
37 Phuong L, Schoeberl K, Raffel C. Natural history of tethered cord in patients with meningomyelocele. Neurosurgery. 2002;50:989-993. discussion 993-995
38 Danzer E, Adzick NS, Rintoul NE, et al. Intradural inclusion cysts following in utero closure of myelomeningocele: Clinical implications and follow-up findings. J Neurosurg Pediatr. 2008;2:406-413.
39 Reigel D. Tethered spinal cord. Concepts Pediatr Neurosurg. 1983;4:142-150.
40 George TM, Fagan LH. Adult tethered cord syndrome in patients with postrepair myelomeningocele: An evidence-based outcome study. J Neurosurg. 2005;102(2 Suppl):150-156.
41 Hall P, Campbell R, Kalsbeck J. Meningomyelocele and progressive hydromyelia: Progressive paresis in myelodysplasia. J Neurosurg. 1975;43:457-463.
42 Oldfield E, Murasko K, Shawker T, et al. Pathophysiology of syringomyelia associated with Chiari I malformation of the cerebellar tonsils: Implications for diagnosis and treatment. J Neurosurg. 1994;80:3-15.
43 Dias MS. Neurosurgical causes of scoliosis in patients with myelomeningocele: An evidence-based literature review. J Neurosurg. 2005;103(1 Suppl):24-35.
44 Gardner W, Bell H, Poolos P, et al. Terminal ventriculostomy for syringomyelia. J Neurosurg. 1977;46:609-617.
45 Park T, Cail W, Maggio W, et al. Progressive spasticity and scoliosis in children with myelomeningocele: Radiologic investigation and surgical treatment. J Neurosurg. 1985;62:367-375.
46 Bell W, Charney E, Bruce D, et al. Symptomatic Arnold-Chiari malformation: Review of experience with 22 cases. J Neurosurg. 1987;66:812-816.
47 Tubbs R, Elton S, Grabb P, et al. Analysis of the posterior fossa in children with the Chiari 0 malformation. Neurosurgery. 2001;48:1050-1054.
48 Pollack I, Pang D, Albright A, et al. Outcome following hindbrain decompression of symptomatic Chiari malformations in children previously treated with myelomeningocele closure and shunts. J Neurosurg. 1992;77:881-888.
49 Danish SF, Samdani A, Hanna A, et al. Experience with acellular human dura and bovine collagen matrix for duraplasty after posterior fossa decompression for Chiari malformations. J Neurosurg. 2006;104(1 Suppl):16-20.
50 Mataro M, Poca M, Sahuquillo J, et al. Cognitive changes after cerebrospinal fluid shunting in young adults with spina bifida and assumed arrested hydrocephalus. J Neurol Neurosurg Psychiatry. 2000;68:615-621.
51 Bol KA, Collins JS, Kirby RS. Survival of infants with neural tube defects in the presence of folic acid fortification. Pediatrics. 2006;117:803-813.
52 Hunt G. Open spina bifida: Outcome for a complete cohort treated unselectively and followed into adulthood. Dev Med Child Neurol. 1990;32:108-188.
53 Bowman R, McLone D, Grant J, et al. Spina bifida outcome: A 25 year prospective. Pediatr Neurosurg. 2001;34:114-120.
54 Hunt GM, Oakeshott P. Outcome in people with open spina bifida at age 35: Prospective community based cohort study. BMJ. 2003;326:1365-1366.
55 Oakeshott P, Hunt GM, Whitaker RH, et al. Perineal sensation: An important predictor of long-term outcome in open spina bifida. Arch Dis Child. 2007;92:67-70.
56 Lewis D, Tolosa J, Kaufmann M, et al. Elective cesarean delivery and long-term motor function or ambulation status in infants with meningomyelocele. Obstet Gynecol. 2004;103:469-473.
57 Oakeshott P, Hunt GM. Long-term outcome in open spina bifida. Br J Gen Pract. 2003;53:632-636.
58 Mapstone T, Rekate H, Nulsen F, et al. The mean IQ of infants who were not shunted was 104, of those shunted without complications it was 91, and of those shunted who had complications it was 70. Childs Brain. 1984;11:112-118.
59 Sutton L, Charney E, Bruce D. Myelomeningocele—the question of selection. Clin Neurosurg. 1986;33:371-382.
60 McLone D, Czyzewski D, Raimondi A, et al. Central nervous system infection as a limiting factor in the intelligence of children with myelomeningocele. Pediatrics. 1982;70:338-342.
61 Stone A. Neurourologic evaluation and urologic management of spinal dysraphism. Neurosurg Clin N Am. 1995;6:269-277.
62 Ko AL, Song K, Ellenbogen RG, et al. Retrospective review of multilevel spinal fusion combined with spinal cord transection for treatment of kyphoscoliosis in pediatric myelomeningocele patients. Spine. 2007;32:2493-2501.
63 James C, Lassman L. Spinal dysraphism: Spinal cord lesions associated with spina bifida occulta. Physiotherapy. 1962;48:154-157.
64 Till K. A study of congenital malformations of the lower back. J Bone Joint Surg. 1969;51:415-422.
65 Yamada S, Iacono R, Andrade T, et al. Pathophysiology of tethered cord syndrome. Neurosurg Clin N Am. 1995;6:311-324.
66 Steinbok P, Cochrane D. The nature of congenital posterior cervical or cervicothoracic midline cutaneous mass lesions. J Neurosurg. 1991;75:206-212.
67 Pang D. Split cord malformation: Part II. Clinical syndrome. Neurosurgery. 1992;31:481-500.
68 Gibson P, Britton J, Hall D, et al. Lumbosacral skin markers and identification of occult spinal dysraphism in neonates. Acta Paediatr. 1995;84:208-209.
69 Pang D, Wilberger J. Tethered cord syndrome in adults. J Neurosurg. 1982;57:32-47.
70 Sharrard W. The mechanism of paralytic deformity in spina bifida. Cerebr Palsy Bull. 1962;4:310.
71 Keating M, Rink R, Bauer S, et al. Neurourological implications of the changing approach in management of occult spinal lesions. J Urol. 1988;140:1299-1301.
72 Morimoto K, Takemoto O, Wakayama A. Tethered cord associated with anorectal malformation. Pediatr Neurosurg. 2003;38:79-82.
73 Golonka N, Haga L, Keating R, et al. Routine MRI evaluation of low imperforate anus reveals unexpected high incidence of tethered spinal cord. J Pediatr Surg. 2002;37:966-969.
74 Bulsara K, Zomorodi A, Enterline D, et al. The value of magnetic resonance imaging in the evaluation of fatty filum terminale. Neurosurgery. 2004;54:375-379.
75 Kukreja K, Manzano G, Ragheb J, et al. Differentiation between pediatric spinal arachnoid and epidermoid-dermoid cysts: Is diffusion-weighted MRI useful? Pediatr Radiol. 2007;37:556-560.
76 Vernet O, O’Gorman A, Farmer J, et al. Use of the prone position in the MRI evaluation of spinal cord retethering. Pediatr Neurosurg. 1996;25:286-294.
77 Witkamp T, Vandertop W, Beek F, et al. Medullary cone movement in subjects with a normal spinal cord and in patients with a tethered cord. Radiology. 2001;220:208-212.
78 Lam W, Ai V, Wong V, et al. Ultrasound measurement of lumbosacral spine in children. Pediatr Neurol. 2004;30:115-121.
79 Warder D, Oakes W. Tethered cord syndrome and the conus in a normal position. Neurosurgery. 1993;33:374-378.
80 Kanev P, Bierbrauer K. Reflections on the natural history of lipomyelomeningocele. J Neurosurg. 1995;22:137-140.
81 Chapman P. Congenital intraspinal lipomas: Anatomic considerations and surgical treatment. Childs Brain. 1982;9:37-47.
82 Pang D, Zovickian J, Oviedo A. Long term outcome of total and near total resection of spinal cord lipomas and radical reconstruction of the neural placode: Part I. Surgical technique. Neurosurgery. 2009;65:511-528.
83 Appignani B, Jaramillo D, Barmes P, et al. Dysraphic myelodysplasias associated with urogenital and anorectal anomalies: Prevalence and types seen with MR imaging. AJR Am J Roentgenol. 1994;163:1199-1203.
84 Tubbs RS, Bui CJ, Rice WC, et al. Critical analysis of the Chiari malformation Type I found in children with lipomyelomeningocele. J Neurosurg. 2007;106(3 Suppl):196-200.
85 Waldau B, Grant G, Fuchs H. Development of an acquired Chiari malformation Type I in the setting of an untreated lipomyelomeningocele: Case report. J Neurosurg Pediatr. 2008;1:164-166.
86 Tubbs RS, Loukas M, Shoja MM, et al. Observations at the craniocervical junction with simultaneous caudal traction of the spinal cord. Childs Nerv Syst. 2007;23:367-369.
87 Brophy J, Sutton L, Zimmerman R, et al. Magnetic resonance imaging of lipomyelomeningocele and tethered cord. Neurosurgery. 1989;25:336-340.
88 Friedrich W, Shurtleff D, Shaffer J. Cognitive abilities and lipomyelomeningocele. Psychol Rep. 1993;73:467-470.
89 Seeds J, Powers S. Early prenatal diagnosis of familial lipomyelomeningocele. Obstet Gynecol. 1988;72:469-471.
90 Sebold CD, Melvin EC, Siegel D, et al. Recurrence risks for neural tube defects in siblings of patients with lipomyelomeningocele. Genet Med. 2005;7:64-67.
91 Carter B, Stewart J. Valproic acid prenatal exposure: Association with lipomyelomeningocele. Clin Pediatr. 1989;28:81-85.
92 Endoh M, Iwasaki Y, Koyanagi I, et al. Spontaneous shrinkage of lumbosacral lipoma in conjunction with a general decrease in body fat: Case report. Neurosurgery. 1998;43:150-151.
93 Macejko AM, Cheng EY, Yerkes EB, et al. Clinical urological outcomes following primary tethered cord release in children younger than 3 years. J Urol. 2007;178(4 Pt 2):1738-1742. discussion 1742-1743
94 Herman J, McLone D, Storrs B, et al. Analysis of 153 patients with myelomeningocele or spinal lipoma reoperated upon for a tethered cord. Pediatr Neurosurg. 1993;19:243-249.
95 Kulkarni A, Pierre-Kahn A, Zerah M. Conservative management of asymptomatic spinal lipomas of the conus. Neurosurgery. 2004;54:868-875.
96 Wu H, Kogan B, Baskin L, et al. Long-term benefits of early neurosurgery for lipomyelomeningocele. J Urol. 1998;160:511-514.
97 Xenos C, Sgouros S, Walsh R, et al. Spinal lipomas in children. Pediatr Neurosurg. 2000;32:295-307.
98 Kang J, Lee K, Jeun S, et al. Role of surgery for maintaining urological function and prevention of retethering in the treatment of lipomeningomyelocele: Experience recorded in 75 lipomeningomyelocele patients. Child Nerv Syst. 2003;19:23-29.
99 Huttmann S, Krauss J, Collmann H, et al. Surgical management of tethered spinal cord in adults: Report of 54 cases. J Neurosurg. 2001;95(Suppl):173-178.
100 Pang D, Zovickian J, Oviedo A. Long term outcome of total and near total resection of spinal cord lipomas and radical reconstruction of the neural placode: Part II: Outcome analysis and preoperative profiling. Neurosurgery. 2010;66:253-272.
101 Aliredjo R, de Vries J, Menovsky T, et al. The use of Gore-Tex membrane for adhesion prevention in tethered spinal cord surgery: Technical case reports. Neurosurgery. 1999;44:674-677.
102 Hoffman H, Taecholarn C, Hendrick E, et al. Lipomyelomeningoceles and their management. Concepts Pediatr Neurosurg. 1985;5:107-117.
103 Cochrane DD, Finley C, Kestle J, et al. The patterns of late deterioration in patients with transitional lipomyelomeningocele. Eur J Pediatr Surg. 2000;10(Suppl 1):13-17.
104 Cochrane DD. Cord untethering for lipomyelomeningocele: Expectation after surgery. Neurosurg Focus. 2007;23:1-7.
105 Anderson R. Occult spinal dysraphism: A series of 73 cases. Pediatrics. 1975;55:826-834.
106 Iskandar B, Oakes W, McLaughlin C, et al. Terminal syringohydromyelia and occult spinal dysraphism. J Neurosurg. 1994;81:513-519.
107 Chapman P, Frim D. Symptomatic syringomelia following surgery to treat retethering of lipomyelomeningoceles. J Neurosurg. 1995;82:752-755.
108 Colak A, Pollack I, Albright A. Recurrent tethering: A common long-term problem after lipomyelomeningocele repair. Pediatr Neurosurg. 1998;29:184-190.
109 McCullough D, Levy L, DiChiro G, et al. Toward the prediction of neurological injury from tethered spinal cord: Investigation of cord motion with magnetic resonance. Pediatr Neurosurg. 1990-1991;16:3-7.
110 Quinn T, Adzick N. Fetal surgery. Obstet Gynecol Clin North Am. 1997;24:143-157.
111 Valenzano M, Paoletti R, Rossi A, et al. Sirenomelia: Pathological features, antenatal ultrasonographic clues, and a review of current embryogenic theories. Hum Reprod Update. 1999;5:82-86.
112 Pang D. Sacral agenesis and caudal spinal cord malformations. Neurosurgery. 1993;32:755-778.
113 Estin D, Cohen A. Caudal agenesis and associated caudal spinal cord malformations. Neurosurg Clin N Am. 1995;6:377-391.
114 O’Neill O, Piatt J, Pitchell P, et al. Agenesis and dysgenesis of the sacrum: Neurosurgical implications. Pediatr Neurosurg. 1995;22:20-28.
115 Banta J, Nichols O. Sacral agenesis. J Bone Joint Surg Am. 1969;51:693-703.
116 Schrander-Stumpel C, Schrander J, Fryns J, et al. Caudal deficiency sequence in 7q terminal deletion. Am J Med Genet. 1988;30:757-761.
117 Rojansky N, Fasouliotis S, Ariel I, et al. Extreme caudal agenesis: Possible drug-related etiology? J Reprod Med. 2002;47:241-245.
118 Levitt M, Patel M, Rodriguez G, et al. The tethered spinal cord in patients with anorectal malformation. J Pediatr Surg. 1997;32:462-468.
119 Tubbs R, Smyth M, Oakes W. Chiari malformation and caudal regression syndrome: A previously unreported association. Clin Dysmorphol. 2003;12:147-148.
120 Midrio P, Silberstein H, Bilaniuk L, et al. Prenatal diagnosis of terminal myelocystocele in the fetal surgery era: Case report. Neurosurgery. 2002;50:1152-1155.
121 Pang D, Dias M, Ahab-Barmada M. Split cord malformation: Part I: A unified theory of embryogenesis for double cord malformations. Neurosurgery. 1992;31:451-480.
122 Dias M, Pang D. Split cord malformations. Neurosurg Clin N Am. 1995;6:339-358.
123 Ersahin Y, Mutluer S, Kocaman S, et al. Split cord malformations in children. J Neurosurg. 1998;88:57-65.
124 Winter R, Haven J, Moe JH, et al. Diastematomyelia and congenital spinal deformities. J Bone Joint Surg Am. 1974;56:27-39.
125 Keim H. Diastematomyelia and scoliosis. J Bone Joint Surg Am. 1973;55:1425-1435.
126 Hilal S, Marton K, Pollack E. Diastematomyelia in children: Radiographic study of 34 cases. Neuroradiology. 1974;112:609-621.
127 Bremer J. Developmental posterior enteric remnants and spinal malformations: The split notocord syndrome. Arch Dis Child. 1960;35:76-86.
128 Dias M, Walker J. The embryogenesis of complex dysraphic malformations: A disorder of gastrulation? Pediatr Neurosurg. 1992;18:229-253.
129 Mahapatra AK, Gupta DK. Split cord malformations: A clinical study of 254 patients and a proposal for a new clinical-imaging classification. J Neurosurg. 2005;103(6 Suppl):531-536.
130 Eid K, Hochberg J, Saunders D. Skin abnormalities of the back in diastematomyelia. Plast Reconstr Surg. 1979;63:534-539.
131 Akay K, Izci Y, Baysefer A, et al. Split cord malformation in adults. Neurosurg Rev. 2004;27:99-105.
132 Tubbs RS, Oakes WJ, Heimburger RF. The relationship of the spinal cord to scoliosis. J Neurosurg. 2004;101(2 Suppl):228-233.
133 English W, Maltby G. Diastmatomyelia in adults. J Neurosurg. 1967;27:260-264.
134 Davis E. Diastematomyelia with early Arnold-Chiari syndrome and congenital dysplastic hip. Clin Orthop. 1967;52:179-185.
135 Shorey W. Diastematomyelia associated with dorsal kyphosis producing paraplegia. J Neurosurg. 1955;12:300-305.
136 Hamby W. Pilonidal cyst, spina bifida occulta and bifid spinal cord: Report of a case with review of the literature. Arch Pathol. 1963;21:831-838.
137 Sonigo-Cohen P, Schmit P, Zerah M, et al. Prenatal diagnosis of diastematomyelia. Child Nerv Syst. 2003;19:555-560.
138 David K, Copp A, Stevens J, et al. Split cervical spinal cord with Klippel-Feil syndrome: Seven cases. Brain. 1996;119:1859-1872.
139 Uzum N, Dursun A, Baykaner K, et al. Split-cord malformation and tethered cord associated with immature teratoma. Child Nerv Syst. 2005;21:77-80.
140 Samdani AF, Asghar J, Pahys J, et al. Concurrent spinal cord untethering and scoliosis correction: Case report. Spine. 2007;32:E832-E836.
141 James C, Lassman L. Diastematomyelia: A critical survey of 24 cases submitted to laminectomy. Arch Dis Child. 1964;39:125-130.
142 Matson D, Woods R, Campbell J, et al. Diastematomyelia (congenital clefts of the spinal cord): Diagnosis and surgical treatment. Pediatrics. 1950;6:98-112.
143 Meacham W. Surgical treatment of diastematomyelia. J Neurosurg. 1967;27:78-85.
144 Pang D, Casey K. Use of an anal sphincter pressure monitor during operations on the sacral spinal cord and nerve roots. Neurosurgery. 1983;13:562-568.
145 Pang D, Parrish R. Regrowth of diastematomyelic bone spur after extradural resection. J Neurosurg. 1983;59:887-890.
146 Bryant T. Case of deficiency of the anterior part of the sacrum with a thecal sac in the pelvis, similar to the tumour of spina bifida. Lancet. 1837;1:358.
147 Currarino G, Coln D, Votteler T. Triad of anorectal, sacral and presacral anomalies. AJR Am J Roentgenol. 1981;137:395-398.
148 Bentley JF, Smith JR. Developmental posterior enteric remnants and spinal malformations: The split notochord syndrome. Arch Dis Child. 1960;35:76-86.
149 Chatkupt S, Speer M, Ding Y, et al. Linkage analysis of a candidate locus (HLA) in autosomal dominant sacral defect with anterior meningocele. Am J Med Genet. 1994;52:1-4.
150 Gegg C, Vollmer D, Tullous M, et al. An unusual case of complete Currarino triad: Case report, discussion of the literature and embryogenic implications. Neurosurgery. 1999;44:658-662.
151 Wang J, Spitz L, Hayward R, et al. Sacral dysgenesis associated with terminal deletion of chromosome 7q: A report of two families. Eur J Pediatr. 1999;158:902-905.
152 Amacher A, Drake C, McLaughlin A. Anterior sacral meningocele. Surg Gynecol Obstet. 1968;126:986-994.
153 Oren M, Lorber B, Lee S, et al. Anterior sacral meningocele: Report of 5 cases and review of the literature. Dis Colon Rectum. 1977;20:492-505.
154 Haga Y, Cho H, Shinoda S, et al. Recurrent meningitis associated with complete Currarino triad in an adult—case report. Neurol Med Chir (Tokyo). 2003;43:505-508.
155 Quigley M, Schinco F, Brown J. Anterior sacral meningocele with an unusual presentation. J Neurosurg. 1984;61:790-792.
156 Vogel EH. Anterior sacral meningocele as a gynecologic problem: Report of a case. Obstet Gynecol. 1970;36:766-768.
157 Vliegen RF, Beets-Tan RG, van Heurn LW, et al. High resolution MRI of anorectal malformation in the newborn: Case reports of Currarino syndrome and anocutaneous fistula. Abdom Imaging. 2002;27:344-346.
158 Ashley WWJr, Wright NM. Resection of a giant anterior sacral meningocele via an anterior approach: Case report and review of literature. Surg Neurol. 2006;66:89-93. discussion 93
159 Lee SC, Chun YS, Jung SE, et al. Currarino triad: Anorectal malformation, sacral bony abnormality, and presacral mass—a review of 11 cases. J Pediatr Surg. 1997;32:58-61.
160 Samuel M, Hosie G, Holmes K. Currarino triad—diagnostic dilemma and a combined surgical approach. J Pediatr Surg. 2000;35:1790-1794.
161 Smith HP, Davis CHJr. Anterior sacral meningocele: Two case reports and discussion of surgical approach. Neurosurgery. 1980;7:61-67.
162 Sanchez AA, Iglesias CD, Lopez CD, et al. Rectothecal fistula secondary to an anterior sacral meningocele. J Neurosurg Spine. 2008;8:487-489.
163 Haworth JC, Zachary RB. Congenital dermal sinuses in children: Their relation to pilonidal sinuses. Lancet. 1955;269:10-14.
164 Humphreys R. Clinical evaluation of cutaneous lesions of the back: Spinal signatures that do not go away. In: Loftus C, editor. Clinical Neurosurgery, vol 43. Baltimore: Williams & Wilkins; 1995:175-187.
165 Martinez-Lage JF, Masegosa J, Sola J, et al. Epidermoid cyst occurring within a lumbosacral myelomeningocele: Case report. J Neurosurg. 1983;59:1095-1097.
166 Ackerman LL, Menezes AH. Spinal congenital dermal sinuses: A 30-year experience. Pediatrics. 2003;112(3 Pt 1):641-647.
167 Ackerman LL, Menezes AH, Follett KA. Cervical and thoracic dermal sinus tracts: A case series and review of the literature. Pediatr Neurosurg. 2002;37:137-147.
168 Lin K, Wang H, Chou M, et al. Sonography for detection of spinal dermal sinus tracts. J Ultrasound Med. 2002;21:903-907.
169 Barkovich AJ, Edwards M, Cogen PH. MR evaluation of spinal dermal sinus tracts in children. AJNR Am J Neuroradiol. 1991;12:123-129.
170 van Aalst J, Beuls EA, Cornips EM, et al. Anatomy and surgery of the infected dermal sinus of the lower spine. Childs Nerv Syst. 2006;22:1307-1315.
171 Fitz CR, Harwood Nash DC. The tethered conus. Am J Roentgenol Radium Ther Nucl Med. 1975;125:515-523.
172 Love JG, Daly DD, Harris LE. Tight filum terminale: Report of condition in three siblings. JAMA. 1961;176:31-33.
173 Wraige E, Borzyskowski M. Investigation of daytime wetting: When is spinal cord imaging indicated? Arch Dis Child. 2002;87:151-155.
174 Pippi Salle JL, Capolicchio G, Houle AM, et al. Magnetic resonance imaging in children with voiding dysfunction: Is it indicated? J Urol. 1998;160(3 Pt 2):1080-1083.
175 Barson AJ. The vertebral level of termination of the spinal cord during normal and abnormal development. J Anat. 1970;106(Pt 3):489-497.
176 Selcuki M, Vatansever S, Inan S, et al. Is a filum terminale with a normal appearance really normal? Childs Nerv Syst. 2003;19:3-10.
177 Selden NR. Minimal tethered cord syndrome: What’s necessary to justify a new surgical indication? Neurosurg Focus. 2007;23:1-5.
178 Uchino A, Mori T, Ohno M. Thickened fatty filum terminale: MR imaging. Neuroradiology. 1991;33:331-333.
179 Jamil M, Bannister CM. A report of children with spinal dysraphism managed conservatively. Eur J Pediatr Surg. 1992;2(Suppl 1):26-28.
180 Sade B, Beni-Adani L, Ben-Sira L, et al. Progression of terminal syrinx in occult spina bifida after untethering. Childs Nerv Syst. 2003;19:106-108.
181 Erkan K, Unal F, Kiris T. Terminal syringomyelia in association with the tethered cord syndrome. Neurosurgery. 1999;45:1351-1359. discussion 1359-1360
182 van Leeuwen R, Notermans NC, Vandertop WP. Surgery in adults with tethered cord syndrome: Outcome study with independent clinical review. J Neurosurg. 2001;94(2 Suppl):205-209.
183 Souweidane MM, Drake JM. Retethering of sectioned fibrolipomatous filum terminales: Report of two cases. Neurosurgery. 1998;42:1390-1393.
184 Lea ME, Sage MR, Bills D, et al. Enterogenous cyst of the cervical spinal canal. Australas Radiol. 1992;36:327-329.
185 Puca A, Cioni B, Colosimo C, et al. Spinal neurenteric cyst in association with syringomyelia: Case report. Surg Neurol. 1992;37:202-207.
186 Whiting DM, Chou SM, Lanzieri CF, et al. Cervical neurenteric cyst associated with Klippel-Feil syndrome: A case report and review of the literature. Clin Neuropathol. 1991;10:285-290.
187 Agnoli AL, Laun A, Schonmayr R. Enterogenous intraspinal cysts. J Neurosurg. 1984;61:834-840.
188 Bremer JL. Dorsal intestinal fistula; accessory neurenteric canal; diastematomyelia. AMA Arch Pathol. 1952;54:132-138.
189 Schmidbauer M, Reinprecht A, Schuster H, et al. Atypical vertebral artery in a patient with an intra- and extraspinal cervical neurenteric cyst. Acta Neurochir (Wien). 1991;109(3-4):150-153.
190 Devkota UP, Lam JM, Ng H, et al. An anterior intradural neurenteric cyst of the cervical spine: Complete excision through central corpectomy approach—case report. Neurosurgery. 1994;35:1150-1153. discussion 1153-1154
191 Macdonald RL, Schwartz ML, Lewis AJ. Neurenteric cyst located dorsal to the cervical spine: Case report. Neurosurgery. 1991;28:583-587. discussion 587-588
192 Levin P, Antin SP. Intraspinal neurenteric cyst in the cervical area. Neurology. 1964;14:727-730.
193 Kumar R, Jain R, Rao KM, et al. Intraspinal neurenteric cysts—report of three paediatric cases. Childs Nerv Syst. 2001;17:584-588.
194 Agrawal D, Suri A, Mahapatra AK, et al. Intramedullary neurenteric cyst presenting as infantile paraplegia: A case and review. Pediatr Neurosurg. 2002;37:93-96.
195 Hes R. Neurenteric cyst or teratomatous cyst. J Neurosurg. 1994;80:179-180. author reply 180-181
196 Daszkiewicz P, Roszkowski M, Przasnek S, et al. Teratoma or enterogenous cyst? The histopathological and clinical dilemma in co-existing occult neural tube dysraphism. Folia Neuropathol. 2006;44:24-33.
197 Kim CY, Wang KC, Choe G, et al. Neurenteric cyst: Its various presentations. Childs Nerv Syst. 1999;15(6-7):333-341.
198 Rizk T, Lahoud GA, Maarrawi J, et al. Acute paraplegia revealing an intraspinal neurenteric cyst in a child. Childs Nerv Syst. 2001;17:754-757.
199 Takase T, Ishikawa M, Nishi S, et al. A recurrent intradural cervical neurenteric cyst operated on using an anterior approach: A case report. Surg Neurol. 2003;59:34-39. discussion 39
200 Song JK, Burkey BB, Konrad PE. Lateral approach to a neurenteric cyst of the cervical spine: Case presentation and review of surgical technique. Spine. 2003;28:E81-E85.
201 Fernandes ET, Custer MD, Burton EM, et al. Neurenteric cyst: Surgery and diagnostic imaging. J Pediatr Surg. 1991;26:108-110.
202 Gilchrist BF, Harrison MW, Campbell JR. Neurenteric cyst: Current management. J Pediatr Surg. 1990;25:1231-1233.
203 Superina RA, Ein SH, Humphreys RP. Cystic duplications of the esophagus and neurenteric cysts. J Pediatr Surg. 1984;19:527-530.
204 de Oliveira RS, Cinalli G, Roujeau T, et al. Neurenteric cysts in children: 16 consecutive cases and review of the literature. J Neurosurg. 2005;103(6 Suppl):512-523.
205 Midha R, Gray B, Becker L, et al. Delayed myelopathy after trivial neck injury in a patient with a cervical neurenteric cyst. Can J Neurol Sci. 1995;22:168-171.
206 Silvernail WIJr, Brown RB. Intramedullary enterogenous cyst: Case report. J Neurosurg. 1972;36:235-238.
207 Neuhauser EB, Harris GB, Berrett A. Roentgenographic features of neurenteric cysts. Am J Roentgenol Radium Ther Nucl Med. 1958;79:235-240.
208 Ellis AM, Taylor TK. Intravertebral spinal neurenteric cysts: A unique radiographic sign—“the hole-in-one vertebra.”. J Pediatr Orthop. 1997;17:766-768.
209 Garg N, Sampath S, Yasha TC, et al. Is total excision of spinal neurenteric cysts possible? Br J Neurosurg. 2008;22:241-251.
210 Sala F, Krzan MJ, Deletis V. Intraoperative neurophysiological monitoring in pediatric neurosurgery: Why, when, how? Childs Nerv Syst. 2002;18(6-7):264-287.
211 von Koch CS, Quinones-Hinojosa A, Gulati M, et al. Clinical outcome in children undergoing tethered cord release utilizing intraoperative neurophysiological monitoring. Pediatr Neurosurg. 2002;37:81-86.
212 Schwartz D, Wierzbowski L, Fan D, et al. Intraoperative neurophysiological monitoring during spine surgery. In: Vaccaro A, Betz R, Zeidman S, editors. Principles and Practices of Spine Surgery. St. Louis: Mosby; 2003:115-126.
213 Schwartz D, Vacarro A. Neurophysiologic monitoring during cervical spine surgery. In: Clark C, editor. The Cervical Spine. 4th ed. Philadelphia: Lippincott Williams & Wilkins; 2005:238-244.
214 Hilibrand AS, Schwartz DM, Sethuraman V, et al. Comparison of transcranial electric motor and somatosensory evoked potential monitoring during cervical spine surgery. J Bone Joint Surg Am. 2004;86:1248-1253.
215 Fan D, Schwartz DM, Vaccaro AR, et al. Intraoperative neurophysiologic detection of iatrogenic C5 nerve root injury during laminectomy for cervical compression myelopathy. Spine. 2002;27:2499-2502.
216 Ikeda K, Kubota T, Kashihara K, et al. Anorectal pressure monitoring during surgery on sacral lipomeningocele: Case report. J Neurosurg. 1986;64:155-156.
217 James HE, Mulcahy JJ, Walsh JW, et al. Use of anal sphincter electromyography during operations on the conus medullaris and sacral nerve roots. Neurosurgery. 1979;4:521-523.
218 Krassioukov AV, Sarjeant R, Arkia H, et al. Multimodality intraoperative monitoring during complex lumbosacral procedures: Indications, techniques, and long-term follow-up review of 61 consecutive cases. J Neurosurg Spine. 2004;1:243-253.
219 Khealani B, Husain AM. Neurophysiologic intraoperative monitoring during surgery for tethered cord syndrome. J Clin Neurophysiol. 2009;26:76-81.
[/level-membership-for-orthopaedics-category][not-level-membership-for-orthopaedics-category]
CHAPTER 31 Congenital Anomalies of the Spinal Cord
Myelomeningocele
Myelomeningocele is the most common significant birth defect involving the spine. The condition is manifest at birth and is characterized by herniation of a malformed spinal cord through a defect in the bony canal and skin. It almost always results in permanent disability regardless of medical intervention and often deprives the victim of “those qualities held in high esteem by our society—independence, physical powers and intelligence.”1 In the past, it was assumed that all of the neurologic dysfunction that occurs with myelomeningocele arose from disordered embryogenesis. More recent work suggests, however, that at least some of the spinal cord damage is acquired in utero,2 and fetal surgery to close the spinal defect is now being performed at selected centers.
Embryology
By 18 days of development, the embryo is a flattened oval disc with all three germ layers present. A longitudinal depression, the neural groove, appears in the neural plate, which is destined to become the brain and spinal cord. By 22 days, the neural groove has deepened, and fusion of the adjacent tissue begins the transformation of the flat neural plate into a hollow neural tube. The entire process is called neurulation, which begins in the dorsal midline and simultaneously progresses cephalad and caudad. The final portions of the tube to close are the rostral opening (the anterior neuropore, at 24 days) and the caudal opening (the posterior neuropore, at 28 days). By the 1st month of gestation, the entire process has been completed. The development of the meninges begins after closure of the posterior neuropore, as does formation of the bony laminae.3
Myelomeningocele presumably occurs when the posterior neuropore fails to close or if it reopens as the result of distention of the central canal of the spinal cord with cerebrospinal fluid (CSF). The spinal abnormality is only one component of a complex of central nervous system abnormalities, which include the Chiari II malformation, hydrocephalus, and brain anomalies such as partial agenesis of the corpus callosum and gyral malformations. McLone and Naidich4 attempted to explain these associations with their “unified theory.” It is hypothesized that the open spinal defect allows excessive drainage of CSF in utero and that this results in collapse of the rhombencephalic vesicle, resulting in a small posterior fossa volume. Growth of the cerebellum and brainstem within a small posterior fossa results in downward herniation and caudal displacement of the cerebellar vermis and brainstem into the cervical spinal canal (the Chiari II malformation). Because the outlet of the fourth ventricle is occluded by impacted brain tissue, obstructive hydrocephalus develops either in the fetal period or in the newborn period after closure of the myelomeningocele eliminates the spinal defect as a drainage pathway.
Epidemiology
The incidence of myelomeningocele ranges from less than 1 case per 1000 live births in the United States to almost 9 cases per 1000 in areas of Ireland. The etiology of myelodysplasia is unknown, and evidence exists for environmental and multifactorial genetic influences. A role for genetic risk factors is supported by numerous studies documenting familial aggregation of this condition. In addition, several lines of evidence point to the potential importance of maternal nutritional status as a determinant of the risk for having a child with spina bifida. Indirect support is provided by studies that indicate that season of conception, socioeconomic status, and degree of urbanization may be related to the risk of spina bifida. Several micronutrients (vitamins C and B12, zinc, and folic acid) have been implicated as potential risk factors as well.5
Folic acid is the most extensively studied micronutrient. In August 1991 after the Medical Research Council Vitamin Study Group report was published, the U.S. Centers for Disease Control and Prevention (CDC) advised that women with a history of an affected pregnancy should take 4 mg of folic acid daily, starting at the time they planned to become pregnant.6 A dose of 0.4 mg was later recommended for all women of childbearing age capable of becoming pregnant. It was anticipated that these recommendations would have a substantial impact on the risk of neural tube defects in the offspring of such women. Most affected pregnancies (approximately 95%) occur in women with no history of a prior affected fetus or child, however.7 It has been suggested that fortification of the food supply may be more effective than individual supplementation for the prevention of neural tube defects.
Prenatal Diagnosis
More recent developments in prenatal diagnosis of fetal anomalies have made antenatal recognition of myelomeningocele commonplace. Families at risk are routinely offered amniocentesis for amniotic α-fetoprotein and acetylcholinesterase, which are important in separating open lesions from skin-covered masses such as myelocystocele. Screening ultrasonography performed on mothers when the fetus is at 16 to 20 weeks of gestation has also proved valuable.8 Other anomalies may be detected, and ultrasonography may detect skin-covered lesions such as lipomyelomeningocele. The cardinal finding is splaying of the posterior elements of the spine in the axial plane in the lumbosacral region. Indirect signs are the lemon sign, which refers to a bilateral concave contour of the frontal bones of the skull, and the banana sign, which describes anterior curvature of the cerebellar hemispheres. Hydrocephalus is also readily detected. In referral centers, diagnostic sensitivity is close to 100% in diagnosing spina bifida.9
Prenatal magnetic resonance imaging (MRI), using ultrafast T2-weighted sequences, may also be used to characterize the Chiari II malformation and other associated anomalies.10 Fetal MRI may detect spinal cord abnormalities in instances where ultrasonography can detect only bony abnormalities.11 Studies indicate that such prenatal imaging studies can help to determine prognosis. Specifically, lesion level determined by prenatal imaging studies seems to predict neurologic deficit and ambulatory potential, but the degree of fetal ventriculomegaly and the extent of hindbrain deformity are not predictive.12 Families can be professionally counseled regarding the expected prognosis and offered conventional treatment, abortion, or the possibility of fetal closure. Patients of low socioeconomic class, who are most at risk for neural tube defects, often do not present for prenatal care until after 24 weeks of gestation, when screening is no longer of value.
The value of prenatal diagnosis of spinal dysraphism is that an opportunity is provided to terminate the pregnancy, to evaluate the fetus for possible fetal surgery, and to determine the best mode of delivery. It has been proposed that children with myelomeningocele sustain traumatic damage to the placode and nerves when they are delivered vaginally and that children electively delivered by cesarean section before the onset of labor have improved motor levels.13 Other investigators have found little benefit to this strategy,12 but in the United States cesarean delivery has become routine.
Initial Evaluation
The initial assessment of a newborn infant with a myelomeningocele begins with a detailed examination to evaluate general well-being and to seek associated anomalies. Fatal urologic or cardiac anomalies may be evident that would favor nonoperative treatment. Some infants may have abnormal facies suggesting Down syndrome, and although chromosomal studies should be obtained, these are most often normal, and the facial appearance becomes normal with age. Approximately 85% of infants with myelomeningocele present with hydrocephalus or develop it within the newborn period.14 A large head circumference or bulging fontanel suggests the need for early head ultrasonography via the fontanel. Stridor, apnea, or bradycardia in the absence of overt intracranial hypertension suggests a symptomatic Chiari II malformation and hindbrain dysfunction, which carries a poor prognosis.
The neurologic examination is difficult in neonates, and it is easy to mistake reflex motion for voluntary movement. Fixed contractures and foot deformity suggest paralysis of the spinal segments innervating the joints. Any movement in response to painful stimulation of the same extremity must be viewed as potentially reflexive. Crying in response to a painful stimulus suggests intact sensation at that level. Table 31–1 lists the segmental innervation of the lower extremities and may be used as a guide in assigning a functional level. It is best, however, to document function of the individual muscle groups rather than simply to record a spinal level.
| Hip flexion | L1-3 |
| Hip adduction | L2-4 |
| Knee extension | L2-4 |
| Ankle inversion | L4 |
| Toe extension | L5-S1 |
| Hip abduction | L5-S1 |
| Hip extension | L5-S1 |
| Knee flexion | L5-S2 |
| Ankle plantar flexion | S1-2 |
Data from Sharrard WJW: The segmental innervation of the lower limb muscles in man. Ann R Coll Surg 35:106-122, 1964.
Management of an Unrepaired Newborn
Pending plans for definitive care, the infant is nursed in the prone position with a sterile saline-soaked gauze dressing loosely applied to the sac or placode. Broad-spectrum antibiotics (ampicillin and cefotaxime) are begun intravenously pending discussion with the parents. If there is no sign of overt hydrocephalus, the back is closed initially, and hydrocephalus is treated with a ventriculoperitoneal shunt at a separate procedure if needed. In patients with hydrocephalus and intracranial hypertension at birth, it may be advisable to perform both procedures at the same time because failure to treat the hydrocephalus may allow continued leakage of CSF into the back and threaten the closure and because the infant is exposed to a single anesthetic. The goal of back closure is to seal, using multiple tissue layers, the spinal cord and subarachnoid space against entry of bacteria from the skin. At the same time, the surgeon must preserve whatever neurologic function remains and attempt to prevent tethering of the spinal cord. The cross-sectional anatomy of a typical myelomeningocele is shown in Figure 31–1.
FIGURE 31–1 Cross-sectional anatomy of typical lumbosacral myelomeningocele. CSF, cerebrospinal fluid.
The infant is positioned in the prone position under general anesthesia. Rolls are placed under the chest and hips to allow the abdomen to hang freely and minimize epidural bleeding (Fig. 31–2A). If the sac is intact, fluid is aspirated and sent for culture. The surgeon gently attempts to approximate the base of the sac or defect vertically and then horizontally to determine which direction would produce the smallest skin defect. An elliptical incision is made, oriented along that axis, outside the junction of the normal, full-thickness skin and the thin, pearly zona epithelioserosa. Full-thickness skin forming the base of the sac is viable and should not be excised. This incision is carried through the subcutaneous tissue until the glistening layer of everted dura or fascia is encountered. The base of the sac is mobilized medially until it is seen to enter the fascial defect (Fig. 31–2B).
The sac is entered by radially incising the cuff of skin surrounding the placode. This skin is sharply excised circumferentially around the placode and discarded, with care being taken to avoid damaging the placode (Fig. 31–2C). All of the zona epithelioserosa is removed to prevent later development of an epidermoid cyst. At this point, the placode is floating freely inside the everted dura (Fig. 31–2D). In some thoracic myelomeningoceles, the placode is large and “thinned out,” and there is complete paraplegia below the level of the defect. In these instances, it may be appropriate to excise the placode to prevent spasticity and high-pressure bladder dysfunction.
Attention is now directed toward the dura, which is everted and loosely attached to the underlying fascia. It is undermined and reflected medially on each side until enough has been mobilized to effect closure (Fig. 31–2E). The dura is very thin anteriorly where the root sleeves exit and is easily torn. When it is free, the dura is closed in a watertight fashion with 4-0 nonabsorbable suture material.
Particularly if the dural closure is suboptimal, it is desirable also to close the fascia as a separate layer. The fascia is incised laterally in a semicircular fashion on either side, elevated from the underlying muscle, and reflected medially (Fig. 31–2F). It is closed with 4-0 suture over the underlying dural closure. The fascia is poor at the caudal end of a lumbar myelomeningocele or with sacral lesions, and the closure may be incomplete.
Mobilization of the skin is by blunt dissection with scissors or a finger; it may be necessary to free it anteriorly to the abdomen (Fig. 31–2G). In most instances, the closure is easiest in the mid-sagittal (vertical) plane, but occasionally less tension is required for a horizontal closure. A two-layer closure is performed. The epidermis may be closed with absorbable suture material and tissue glue if there is little tension, but interrupted nonabsorbable suture material is preferred for large defects.
Very large lesions require special techniques. There is often an associated gibbous deformity, and it is helpful to use a rongeur to remove the everted lamina, which, if left in place, would produce pressure points on the skin closure. Large circular defects may be closed by an S-shaped skin opening, allowing the use of rotation flaps (Fig. 31–3). Alternatively, a Z-rhomboid flap, tissue expanders, or latissimus dorsi myocutaneous flaps may be employed. Lateral relaxing incisions with split-thickness skin grafting have been described, but should be avoided if possible because the cosmetic result is poor. Alternatively, allogeneic skin grafts such as Alloderm (Lifecell, Branchburg, NJ) can be sewn directly to the edges of an approximated closure and may offer a better cosmetic result.15 In rare cases where patients are referred late for surgery, significant scarring of the placode can occur. Tissue expansion is particularly useful in such cases.16
Large defects occasionally undergo dehiscence, owing to tension on the skin closure or superficial infection. This dehiscence is usually evident by 7 days postoperatively. In most cases, the underlying fascial and dural closures remain intact. The dehiscence may be treated by removing the sutures to the point of good skin closure, débriding the devitalized tissue, and beginning a program of saline wet-to-dry dressings. When a good granulation bed is observed, the wound can simply be allowed to granulate and contract or may be secondarily closed or repaired with a skin graft with or without tissue expansion.17 For persistent wound breakdown, the pedicled “propeller” flap can be used as a vascularized salvage procedure.18 CSF leakage is treated by a ventricular diversion procedure, even if the ventricles are small.
Closure Before Birth
In a few centers, a fetus with spina bifida may be a candidate for in utero treatment because this condition is routinely detected before 20 weeks of gestation (Fig. 31–4). There is evidence that neurologic deterioration occurs during gestation. Normal lower extremity movement can be seen on ultrasound studies of affected fetuses before 17 and 20 weeks of gestation, whereas most late-gestation fetuses and newborns have some degree of deformity and paralysis. Such deterioration could be the result of exposure of neural tissue to amniotic fluid and meconium or direct trauma as the exposed neural placode impacts against the uterine wall. Theoretically, such deterioration could be reduced or eliminated by in utero closure of the lesion.
FIGURE 31–4 Fast spin-echo T2-weighted fetal MRI of a 20-week fetus with myelomeningocele. The sac is intact.
Some animal studies (in which a model for spina bifida is created by laminectomy and exposure of the fetal spinal cord to the amniotic fluid) have shown improved leg function if the lesion is closed before birth.19 Although other animal studies have failed to show behavioral improvement, they have documented the presence of somatosensory evoked potentials (SSEPs) in the treated myelomeningocele group compared with an absence of SSEPs in the untreated group.20 There is also evidence that the Chiari II malformation, which occurs in most individuals with spina bifida, is acquired and could potentially be prevented by in utero closure.21
In 1997, in utero repair of spina bifida was performed by hysterotomy at Vanderbilt University and at The Children’s Hospital of Philadelphia.22,23 Fetuses treated in utero were subsequently delivered by cesarean section, ideally at around 36 weeks of gestation. The early experience at both institutions suggested that compared with infants treated postnatally, fetuses treated in utero had a decreased incidence of hindbrain herniation and possibly a decreased need for shunting.24,25 The combined experience at the Children’s Hospital of Philadelphia and Vanderbilt University indicates that the incidence of hydrocephalus requiring shunting in patients treated in utero is less than that of historical controls stratified by spinal level who received standard postnatal care.26,27 It is hypothesized that fetal closure of the spinal lesion reduces the need for shunting by eliminating the leakage of CSF, which puts back-pressure on the hindbrain. The hindbrain hernia can be reduced, and the obstruction of the outflow from the fourth ventricle is relieved.28
It is now estimated that more than 330 in utero spina bifida closures have been performed.29 The procedure seems to be generally well tolerated by the expectant mothers. The fetal death rate is about 5% and is due to uncontrollable labor and premature birth. One analysis of leg function in children treated prenatally revealed no significant difference from a set of historical controls treated with conventional postnatal repair. Many of the children evaluated in this series had lower limb paralysis at the time of the surgery, however, which may have diluted any possible benefit.30 Data from the Children’s Hospital of Philadelphia suggested possibly better leg function in patients who already had intact leg movement shown by ultrasound before fetal sugery.31 Problems with delayed development of dermoid inclusion cysts and tethered cord may adversely affect long-term outcome.32 The preliminary experience suggests that children treated in utero have the same urodynamic abnormalities that are seen in conventionally treated children with spina bifida.33,34
To date, outcomes for spina bifida infants treated in utero have been assessed relative to outcomes in conventionally treated, historical controls.14 Such comparisons are prone to substantial bias, however, because fetuses who undergo in utero closure represent a highly selected subset of cases. In addition, the medical management of spina bifida is continuously improving, making comparisons with historical controls particularly problematic. Definitive answers about the benefits of fetal closure can be obtained only by a properly designed and conducted randomized trial.
Surgical Complications
Latex Allergy
An increased incidence of allergy to latex, as found in surgical gloves, balloons, and urinary catheters, has been well documented in children with spinal dysraphism.35 Allergy to latex is a type I, immediate, IgE-mediated reaction that can lead to anaphylaxis and death. Sensitization presumably arises from repeated exposure, at home and in the operating room. In many centers, all children with spina bifida are managed in a latex-free environment, and surgical procedures are preceded by prophylactic medications.
Late Deterioration
Tethered Cord
Approximately 20% to 30% of patients with myelomeningocele experience spinal cord tethering, which may result in progressive loss of function similar to that seen with occult spinal dysraphism.36,37 This phenomenon does not seem to be reduced by fetal repair, in which inclusion cysts have been found in 19% of patients who underwent fetal repair.38 Signs and symptoms usually appear during the 1st decade of life when growth is rapid and include back and leg pain, decrease in urinary control, gait difficulty and leg weakness, hip dislocation, progressive foot deformity, and possibly scoliosis. Urinary and gait difficulties are complaints confined to patients with low-level lesions, who are already functioning quite well neurologically and have the most to lose. In more severely affected patients who may already be wheelchair bound, pain may be the only manifestation of cord tethering and may be confused with appendicitis, hernia, urinary tract infection, or traumatic arthritis associated with abnormal posture or gait.
As with lipomyelomeningocele, the aim of surgery is to prevent further deterioration of function, but occasionally improvement is seen, even in preexisting deficits of long-standing duration. Reigel39 reported improvement in bladder function in five of nine patients with recent deterioration and in 77% of patients with gait difficulty and motor weakness. Pain almost invariably is relieved. Retethering can occur, and if new symptoms suggest this as an etiology, reoperation is indicated. An evidence-based review of the literature concluded that aggressive tethered cord release should be performed in adults within 5 years of symptom onset.40 Post–myelomeningocele repair patients tend to fare worse, however, than adult patients with closed dysraphism.
Hydromyelia
The routine availability of MRI has heightened awareness of this entity, which may be found in 68% of myelomeningocele patients.36 Hydromyelia presumably is the result of hydrodynamic forces arising from persistent fetal or untreated hydrocephalus that forces CSF down the central canal of the spinal cord.4 This explanation accounts for the frequency of symptomatic hydromyelia in patients with unshunted ventriculomegaly and patients considered to have “compensated” hydrocephalus, whose nonfunctional shunts were not revised because they lacked overt signs or symptoms of intracranial hypertension.41 An alternative explanation has been proposed by Oldfield and colleagues,42 who used dynamic MRI to study CSF flow. They suggested that a systolic pressure wave in the cranial compartment is transmitted in a pistonlike manner to the cerebellar tonsils, causing a systolic spinal CSF pressure wave that pushes CSF into the cord through its outer surface.
Symptoms of hydromyelia in spina bifida differ from symptoms in classic syringomyelia. The latter entity typically produces a dissociated sensory loss (relative loss of pain and temperature modalities with preservation of light touch), atrophy, and fasciculations primarily affecting the shoulder girdle and hands. When found in association with myelomeningocele, hydromyelia typically results in progressive bladder dysfunction, spasticity of upper and lower extremities, quadriparesis, and generally preserved sensation. The myelomeningocele repair may become swollen and tender. Symptoms may be due in part to the direct effects of the hydrocephalus stretching the cortical motor fibers. Hydromyelia may also be seen with progressive scoliosis. Spinal cord tethering may be the primary underlying etiology, however, of scoliosis and hydromyelia in these cases.43
Workup consists of MRI of the brain, followed by MRI of the entire spine to visualize the extent of the hydromyelia and the associated Chiari malformation and tethered spinal cord (Fig. 31–5). The hydromyelia cavity may be localized to a few spinal segments or extend throughout the length of the spinal cord. There may be multiple cavities and septations.
FIGURE 31–5 T1-weighted mid-sagittal MRI of a patient with Chiari malformation and large septated hydromyelia (H).
Other procedures have been described. Terminal ventriculostomy was advocated by Gardner and colleagues44 for classic syringomyelia, but the myelotomy tends to scar closed with time. In addition, the negative pressure provided by the pleural end of a shunt provides more decompression than simply allowing the hydromyelia to communicate with the subarachnoid space. Park and colleagues45 advocated posterior fossa decompression and plugging of the obex to prevent CSF fluid from the fourth ventricle from entering the central canal in patients in whom ventriculoperitoneal shunt revisions do not alleviate symptoms. This is a formidable procedure, however, and recurrent vomiting may occur from irritation or compression of the brainstem at the obex.
Chiari Malformation
MRI of the brain and spine is the procedure of choice for initial evaluation because the lesion is readily visualized and associated abnormalities are easily excluded. Mid-sagittal views show the tongue of cerebellum extending below the foramen magnum in type I lesions (see Fig. 31–5) and the “vermian pseudotumor,” consisting of the fused brainstem and cerebellum, in type II lesions.
Chiari type II lesions manifesting in an infant with myelomeningocele are particularly difficult to manage. When symptoms are severe, the brainstem nuclei may be irreversibly damaged, and the patient would not improve with surgical decompression.46 This situation has prompted a trend toward earlier intervention, when improvement may still be possible.48 If shunt function is shown to be optimal with shunt tap or imaging, Chiari decompression should be undertaken.
Patients are operated on in the prone position. A midline incision is used to expose the suboccipital bone and the cervical spine down to the level of the cerebellar hernia as seen on MRI, and a laminectomy is performed. The tonsillar hernia can usually be seen through the translucent dura, which is opened in the midline, beginning at the lowest point of the laminectomy and progressing cephalad. In this way, MRI-compatible clips or a running suture can be used to control bleeding from the anomalous venous sinuses in the posterior fossa. The dural opening should include the fibrous band usually present at the foramen magnum; however, because the foramen magnum is typically large and the venous sinuses are caudally displaced, the dural incision should rarely be carried above the level of the foramen magnum. It may be possible to open the fourth ventricle and establish CSF flow to the subarachnoid space, but dense adhesions and distorted anatomy often may make this unwise. As in Chiari I repair, the dura is left open, or a dural graft is employed, and the muscles, fascia, and skin are closed as usual.49
Shunt Malfunction and Arrested Hydrocephalus
Any neurologic deterioration in a patient with myelomeningocele should prompt a thorough investigation of the shunt. Some patients have nonfunctioning shunts and ventriculomegaly yet have no overt symptoms of intracranial hypertension or may have never received shunts and have baseline ventriculomegaly. These patients have been described as having “compensated” or “arrested” hydrocephalus. It has been suggested that some of these patients might benefit from shunting or shunt revision, which in some instances may result in improved neuropsychological functioning.50
Outcome
Significant progress has been made in the understanding and management of myelomeningocele over the past 25 years, particularly in the widespread use of multidisciplinary teams of specialists to manage children with this condition. More recent population-based data indicate that 1-year survival of individuals with spina bifida is approximately 92%51 and that 78% of all individuals with spina bifida survive to age 17 years.52 Mortality continues into the adult years53: 57% of patients die by the 4th decade of life.54 Death is due to problems associated with the Chiari II malformation, restrictive lung disease secondary to chest deformity, shunt malfunction, and urinary sepsis. Perineal sensation and urinary continence may be predictive of survival.55
Approximately 75% of young children with myelomeningocele are ambulatory to some extent, although most require braces and crutches. The likelihood that a child will ambulate is related to the level of the lesion; virtually all patients with sacral and lumbosacral lesions walk, but only half of patients with thoracic or thoracolumbar lesions walk with the use of braces and crutches. Patients with high-level lesions often can walk as young children but become wheelchair users as they get older, gain weight, or simply discover that wheelchair locomotion requires less energy and is faster than ambulation with crutches. A more recent report compared ambulatory potential between patients delivered by cesarean section and patients who underwent a trial of labor. Compared with the elective cesarean group, patients in the trial of labor group were more likely to be ambulatory at 2 years (independently ambulant 7% vs. 28%, ambulant with assistance 63% vs. 65%, or wheelchair bound 30% vs. 7%) and at 10 years (independently ambulant 5% vs. 21%, ambulant with assistance 30% vs. 54%, or wheelchair bound 65% vs. 25%). There was no statistical difference based on mode of delivery.56
Overall, approximately 75% of surviving infants have normal intelligence (IQ >80). This percentage decreases slightly to approximately 70% for surviving adults.57 Numerous factors predict poor intellectual outcome. In one study, the mean IQ of infants who were not shunted was 104; of infants shunted without complications, it was 91; and of infants shunted who had complications such as ventriculitis or anoxia, it was 70.58 Intelligence is also related to the level of the lesion: Approximately 55% of infants with thoracic lesions have significant developmental delay compared with only 25% of infants with lesions at lower levels.59
The cause of mental retardation in children with myelodysplasia remains controversial. McLone and colleagues60 attributed it to ventriculitis, but this does not account for all cases, and it is likely that in most cases forebrain dysfunction is simply part of the complex of anomalies associated with myelodysplasia. With respect to fetal surgery, preliminary evidence from the Children’s Hospital of Philadelphia showed that two thirds of surviving patients are within normal cognitive range at 2 years of age.27 Because cognitive function seems to be related to shunting, it is hoped that a decrease in overall shunting rate may improve overall intelligence in this population.
Although virtually all children with myelomeningocele have abnormal bladder function, urinary continence with the use of clean intermittent catheterization approaches 90% in children 5 to 9 years old. Ongoing urologic care is essential.61 Scoliosis is present in 49%, with 43% eventually requiring spinal fusion, for which the procedural complication rate is extremely high.62 Of children, 23% have at least one seizure. A tethered cord release is performed in 32%, with 97% having improvement or stability of preoperative symptoms.53
Occult Spinal Dysraphism and Tethered Cord
Embryology
After closure of the neural tube, the fetal spine is covered by ectoderm, and the low lumbar and sacrococcygeal segments have not developed.3 At this point, the caudal end of the neural tube blends with a large aggregate of undifferentiated cells, the caudal cell mass. A series of vacuoles in this mass coalesce and achieve continuity with the central canal of the previously formed neural tube (at roughly 29 days’ gestation), a process called canalization. The third phase involves retrogressive differentiation, during which the previously formed tail structures undergo a precise, ordered necrosis, leaving only the filum terminale, the coccygeal ligament, and the terminal ventricle of the conus as remnants by 11 weeks. Cell rests with potential for differentiation may be left within these structures, accounting for the development of lipomas, hamartomas, ectopic renal tissue, and the rare malignancy occasionally found in association with occult spinal dysraphism. Failure of the caudal cell mass to regress presumably gives rise to the hypertrophied filum terminale.
Pathogenesis
Symptoms may be caused in several ways. First, abnormal formation of the spinal cord and roots during embryogenesis may result in permanent deficits manifest at birth, as seen in myelomeningocele. Second, local masses growing within the rigid bony spinal canal may compress the conus medullaris or cauda equina and cause mechanical distortion or ischemia, with consequent dysfunction of these neural elements in a progressive fashion.63 This mechanical distortion may account for the acceleration of symptoms seen in some patients with significant weight gain in whom a lipoma enlarges in proportion to other body fat stores.
Finally, symptoms may be produced by traction on the spinal cord. Early in embryonic development, there is a progressive and rapid ascent of the conus within the bony spinal canal owing to faster growth of the vertebral bodies compared with the spinal cord. This “ascent of the conus” in children is slight, being only one segment, from L3 to L2 in the period from the 26th week of intrauterine life until maturity.64 Nonetheless, if the conus is tethered to the bony spinal canal, as often occurs in dysraphism, there is loss of mobility of the conus during spinal flexion and extension. Experimentally, spinal cord tethering has been shown to interfere reversibly with spinal cord oxidative metabolism65 and is probably the major cause of progressive neural damage in older children and adults with dysraphism lesions.
Clinical Manifestations
Bony spina bifida occulta at L5-S1 is a common radiologic finding in children and adults and is usually associated with no symptoms or signs. Unless other findings are present, no further evaluation or treatment is required. Signs of clinically significant spina bifida occulta may be in the form of cutaneous, neurologic, orthopaedic, urologic, or rectal abnormalities (Table 31–2).
TABLE 31–2 Presenting Symptoms and Signs of Occult Spinal Dysraphism
| % | |
|---|---|
| Leg weakness | 48 |
| Cutaneous abnormality | 48 |
| Foot deformity | 39 |
| Urinary incontinence | 36 |
| Fecal incontinence | 32 |
| Sensory abnormality | 32 |
| Recurrent urinary tract infection | 20 |
| Gait abnormality | 16 |
| Scoliosis | 14 |
Adapted from James H, Walsh J: Spinal dysraphism. Curr Prob Pediatr 11:1-25, 1981.
Cutaneous Syndrome
Cutaneous abnormalities indicative of an underlying spinal dysraphism are situated on or near the midline, usually in the lumbrosacral region, but occasionally in the thoracic region or the neck.66 A wide variety of abnormalities are seen.
A striking finding is the hairy patch (hypertrichosis), also known as “faun’s tail,” which always occurs in the midline. It may be small but more commonly is a wide, diamond-shaped patch in the lumbar or lower thoracic area (Fig. 31–6). Frequently, the patient’s mother has trimmed the hair for years before presentation to the physician. If the dysraphism is in the cervical or upper thoracic region, there is usually a smaller patch of silky hair. The underlying abnormality may not be confined to the level of the hairy patch, and it is important to image the entire spine. Hypertrichosis may occur with any of the types of dysraphism but is particularly associated with split cord malformations.67
Subcutaneous lipomas at or near the midline of the lumbosacral spine may indicate an underlying intradural lipoma. These are nontender, poorly circumscribed soft masses of fat that are continuous with the normal subcutaneous tissue and with the intraspinal portion of a lipomyelomeningocele. The overlying skin is normal, hairy, or dimpled, and there may be an associated angioma or skin tag (Fig. 31–7). Other conditions may manifest as skin-covered masses overlying the caudal spine (Table 31–3). Often, an older patient gives a history of having had the superficial mass removed for cosmetic reasons.
TABLE 31–3 Differential Diagnosis of Skin-Covered Lesions Overlying the Spine
Atretic meningoceles may also be seen, consisting of a central area of thin, pearly skin surrounded by a halo of red, pink, or brown (spinal aplasia cutis). These represent myelomeningoceles or meningoceles that have partially healed spontaneously. Dimples at the tip of the coccyx (coccygeal pits) are frequent findings in normal newborns and usually have no significance.68 Deep dimplelike depressions higher in the spine may be the external stigma of an epithelialized tract that connects with the filum terminale, spinal cord, or intraspinal dermoid cyst and require further investigation. The meningocele manqué is a skin blemish sometimes referred to as a “cigarette burn” because it resembles a small scar with loss of skin in the midline. When this blemish is obvious at birth, it may cause concern of CSF leakage, but this seldom occurs. Other cutaneous lesions that may be associated with a tethered cord include rudimentary caudal appendages (“tails”) and asymmetrical gluteal folds.
Neurologic Syndrome
Muscle weakness and gait disturbance are usually apparent at 2 years of age when a child begins to walk. On examination, there may be striking muscular atrophy and leg-length asymmetry. The deep tendon reflexes may be normal, increased, or absent, giving the pattern of a mixed upper and lower motor neuron lesion. Patchy sensory loss is present, particularly in the distal leg and perineum. Pain in the back with a radicular component is frequent in older children or adults.69
Orthopaedic Syndrome
The most frequent orthopaedic finding is unilateral or bilateral cavovarus deformity of the foot, with or without leg-length discrepancy (Fig. 31–8). There may also be “claw toes.” The abnormal gait is the result of orthopaedic deformity and muscular weakness. It is presumed that the foot deformity is due to lack of, or weak innervation of, antagonistic muscles of the lower extremity.70 Scoliosis, especially if found in a young child or a boy or if noted to be rapidly progressive or accompanied by pain, also suggests an underlying spinal cause.
Urologic Syndrome
Occult spinal dysraphism should always be considered when one encounters infants with an abnormal voiding pattern, new-onset urinary or fecal incontinence in a previously toilet-trained child, or urinary tract infection in a child of any age. As opposed to older children, most infants with occult spinal dysraphism have normal results on urodynamic testing.71 This fact underscores the importance of recognizing the syndrome as early as possible so that prophylactic surgery can be performed.
Anorectal Anomalies
Anorectal anomalies, including vesicointestinal fissures, cloacal extrophy, rectovesical fistulas, and imperforate anus, are frequently associated with an underlying tethered cord.72 The overall incidence of tethered cord with imperforate anus is 35% and may be increased in boys.73 In many cases, no cutaneous abnormality is seen, and radiographic screening is indicated in these patients.
Radiologic Investigation
Spine MRI is the imaging study of choice to screen for occult spinal dysraphism. Bony anatomy is poorly seen but is of secondary importance in planning an operative approach for most lesions. A fibrous tract extending from a cutaneous coccygeal pit to the bony coccyx represents a persistent coccygeal ligament and is of no clinical significance. The presence of fat in the filum is a frequent incidental finding on MRI. Generally, if the conus is at the normal level (at or above the L2-3 interspace) and if the patient has no clinical signs or symptoms of a tethered cord, surgery is not warranted. Fat occurring very close to the conus may represent a special case, however, because this finding is associated with clinical tethering.74 A tethered cord may be suspected when the conus lies at the L2-3 level or below in a child or adult or if the conus is dorsally displaced.
Diffusion-weighted imaging may differentiate epidermoid cysts from arachnoid cysts.75 Postoperative studies may be difficult to interpret. The conus rarely ascends significantly after successful surgery, and even in asymptomatic patients the conus is shown to be dorsally displaced postoperatively. Although MRI is currently the best screening test for spinal cord tethering, it may not be useful for ruling out postoperative retethering. Some authors have proposed that imaging in the prone position to evaluate cord motion might be useful, but this has not been of value clinically.76,77
Ultrasonography through the infant spine or through congenital spina bifida or laminectomy defects has also been used to screen for occult spinal dysraphism.76,77 Although the level of the conus is readily seen, it is difficult to assign a specific spinal level to its termination. Sometimes one can visualize the tethering element, such as a hypertrophied filum or lipoma. Spinal cord motion has also been used to assess tethering.78 Ultrasonography may be useful as a screening test when the probability of a tethered cord is low, but MRI is preferable if an occult dysraphic lesion is strongly suspected.
Cases of tethered spinal cord with the conus in normal position on imaging studies have been described.79 Tethering is suspected on the basis of the typical clinical syndrome and confirmed at surgery. Mechanisms included hypertrophied filum terminale, meningocele manqué, and split cord malformation. This situation is uncommon, but when typical symptoms are present, surgical exploration is justified.
Specific Entities
Lipomyelomeningocele
Lipomyelomeningocele occurs at a rate of 1 in 4000 live births and in contemporary series is the most common cause of tethered cord syndrome.80 The term lipomyelomeningocele is misleading, in that it suggests herniation of neural elements through a spina bifida defect into a meningeal sac. The neural elements remain within the spinal canal, and only the lipoma itself may protrude through the spina bifida defect to manifest as a subcutaneous mass. Some authors have used the term lipoma of the cauda equina, but the fatty mass is invariably attached to the conus medullaris or filum terminale rather than to the cauda equina. Chapman81 classified lipomyelomeningoceles as lesions that attach to the caudal end of the conus, lesions that insert dorsally, and lesions that are transitional.
If the lipoma inserts onto the dorsal surface of the conus, there is usually a substantial subcutaneous mass, which may be asymmetrical. Along the lateral interface of the attachment of the lipoma to the spinal cord, the dura and pia are also fused (Fig. 31–9). Sensory roots emerge just anterior to this lateral line of fusion. As a result, neither the sensory roots nor the motor roots are actually within the lipoma. Alternatively, the lipoma may join the conus at its caudal end. The remaining mass may lie entirely within the spinal canal or extend dorsally through the spina bifida defect (Fig. 31–10). The fatty tumor may replace the filum terminale, or there may be a separate filum that lies anteriorly. The nerve roots usually lie ventral to the fatty mass but may lie within the fibrous ventral portion of the mass itself. Transitional forms may occur, which are often the most difficult to manage surgically. Probably the rarest subtype is the chaotic lipomyelomeningocele, described by Pang and colleagues,82 which has a prominent ventral component. MRI often defines the type of lipomyelomeningocele, which is helpful in understanding the anatomy that is encountered at operation.
[/not-level-membership-for-orthopaedics-category]
