Congenital and Neonatal Abnormalities
Esophageal Atresia with and Without Tracheoesophageal Fistula
Overview: First described in 1670, esophageal atresia was a universally fatal anomaly until late in the fifth decade of the twentieth century, when pediatric surgeons began successfully treating these children. Esophageal atresia with and without TEF is part of a heterogeneous spectrum of anomalies that ranges from isolated esophageal atresia to isolated TEF. Lesions are typically classified according to the association and location of the fistulous connection between the esophagus and the airway (Fig. 97-1). The incidence of esophageal atresia is approximately 1 in 4500 births in the United States and 1 in 3500 births worldwide, with a tendency to occur more commonly in boys, although the gender difference varies with the type of lesion. Maternal risk factors include white race, first pregnancy, and advanced age. There is also an increased risk in the siblings of patients with esophageal atresia.1,2
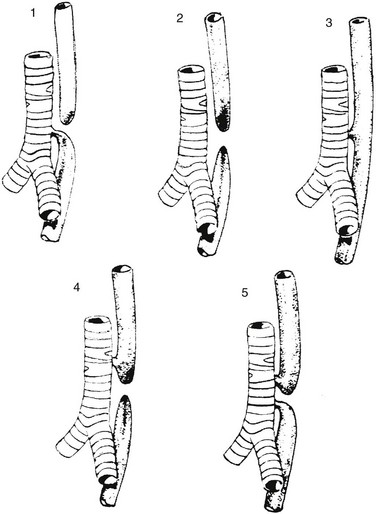
Figure 97-1 Types of esophageal atresia, with and without tracheoesophageal fistula.
Type 1 represents esophageal atresia with a distal fistula and is the most common form, occurring in approximately 84% of patients. Type 2 illustrates esophageal atresia without a fistula, seen in about 6%. Type 3 is an isolated H-type fistula (4%), and type 4 describes esophageal atresia with the fistula arising from the proximal pouch (5%); type 5, with fistulae arising from both the proximal and distal pouch, is the least common (~1%).1
Etiology: The development of esophageal atresia is poorly understood and still debated. Dominant theories in the past have been based on the model of a tracheoesophageal septum, a lateral ridge of tissue believed to separate the foregut into a dorsal component (the esophagus) from a ventral component (the trachea) in a caudocephalic direction. However, further investigation has suggested that this process does not occur in humans.1,3 Kluth and Fiegel3 suggest that the formation of the esophagus and trachea is the result of “a system of folds” in the cranial and caudal ends of the tracheoesophageal space that move toward each other, thus separating the space into the ventral trachea and dorsal esophagus. Following this theory, esophageal atresia with fistula results from an abnormally ventral position of the dorsal fold; isolated esophageal atresia is attributed to a vascular accident rather than abnormal separation of the alimentary and respiratory canals.3,4 The association of the esophageal atresia complex with conotruncal anomalies and anomalies associated with DiGeorge syndrome suggests that abnormal development of pharyngeal arches may be related to this malformation.1
Further research with animal models concerns the investigation of genes that influence the temporal and spatial sequence of expression of molecular and genetic pathways, including retinoic acid receptors, and sonic hedgehog pathway effectors. Human teratogens that have been associated with this anomaly include abnormal maternal hormonal milieu (including estrogen, progesterone, and thyroid hormones), intrauterine exposure to thalidomide and diethylstilbestrol, and maternal diabetes.1
Clinical Presentation: Patients with esophageal atresia with or without fistula typically come to medical attention prenatally or soon after birth. Patients with isolated esophageal atresia are most likely to be seen with maternal polyhydramnios and to be identified at prenatal evaluation. Esophageal atresia and TEF may have been suspected during pregnancy, but neonates generally come to medical attention in the first few hours of life with excessive salivation and drooling, choking and regurgitation during feeds, and sometimes with cyanosis and/or respiratory distress.1
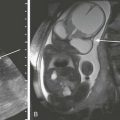
The incidence of associated congenital anomalies has been reported to be between 50% and 70% and is most common in infants with isolated esophageal atresia and least common in children with isolated TEF.5 The most common associated anomalies, in descending order of frequency, include cardiac, genitourinary, gastrointestinal, musculoskeletal, and neurologic anomalies. Approximately half of the children with esophageal atresia who have associated anomalies can eventually be classified as having a known syndrome, either chromosomal or named, such as VACTERL (vertebral anomaly, anorectal atresia, cardiac lesion, tracheoesophageal fistula, renal anomaly, limb defect), (Fig. 97-2) CHARGE (coloboma, heart malformation, choanal atresia, mental retardation, genitourinary and ear malformations), Fanconi anemia, Opitz and Goldenhar syndromes.6,7 In particular, patients with isolated esophageal atresia have an increased incidence of trisomy 21 (11%) and duodenal atresia (10%).
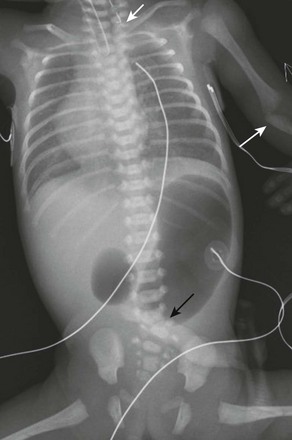
Figure 97-2 VACTERL (vertebral anomaly, anorectal atresia, cardiac lesion, tracheoesophageal fistula, renal anomaly, limb defect) syndrome.
Newborn girl born at 32 weeks gestation demonstrates the abnormalities seen with the VACTERL association. The patient is intubated because of the lung disease of prematurity. The enteric tube (short white arrow) ends just above thoracic inlet in the proximal esophageal pouch, and abdominal bowel gas indicate a distal tracheoesophageal fistula, whereas a “double bubble” sign, and absence of distal bowel gas indicate duodenal atresia. There are 13 pairs of ribs, and a hemivertebra between L5 and S1 (black arrow). Note the absent radius in the left arm (white arrow points to the ulna).
Imaging: Imaging is used in both the prenatal and neonatal diagnosis of esophageal atresia. Esophageal atresia is a cause of polyhydramnios. Fetuses with esophageal atresia may also have an absent or small stomach bubble. Unfortunately, these ultrasound findings alone lack specificity, with a reported 20% to 40% positive predictive value.8 Accuracy in diagnosis is improved when a blind-ending pouch is visualized in the fetal neck, and newer data suggest that fetal magnetic resonance imaging (MRI) may play an increasingly important role in prenatal diagnosis.9,10 More commonly, the diagnosis is made in the immediate neonatal period. If the diagnosis of esophageal atresia is suspected in the first few hours of life, placement of a flexible feeding tube or enteric tube should be attempted. Introduction of contrast into the pouch is not typically necessary for confirmation of the diagnosis, although introduction of air into the pouch may improve its identification. Introduction of contrast into the pouch may reveal the rare patient who has a proximal fistula with or without atresia, although today this is typically diagnosed endoscopically.
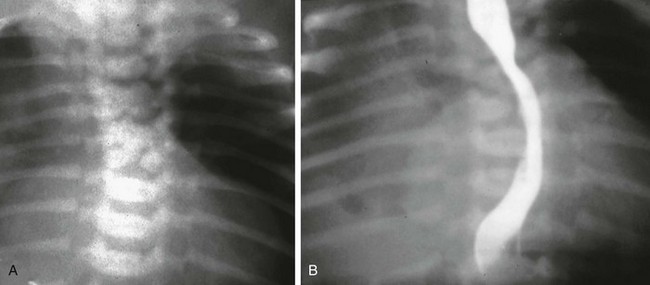
On radiographs of the abdomen, absence of bowel gas indicates esophageal atresia without a distal TEF (Fig. 97-3). In this setting, a TEF from the proximal pouch cannot be excluded with chest radiographs and requires endoscopy for diagnosis. Likewise, the presence of bowel gas on abdominal radiographs in a patient with esophageal atresia indicates the presence of a distal TEF (Fig. 97-2 and 97-4).1 In patients who are intubated, air may be forced into the stomach through the fistula, resulting in gastric overdistension, which may result in perforation. In addition, approximately 2.5% of patients will have a right-sided aortic arch that should be identified on preoperative echo or ultrasound since esophageal repair is performed on the side opposite the arch.1,11,12
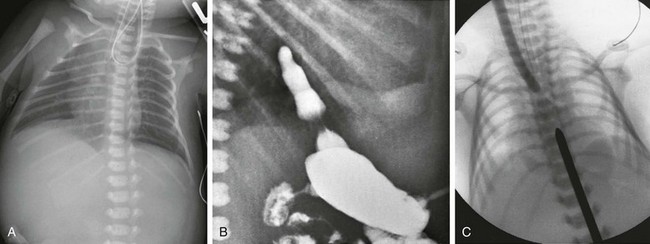
Figure 97-3 Isolated esophageal atresia.
A, Newborn boy born at 37 weeks gestation with isolated esophageal atresia. The enteric tube turns cephalad in the blind-ending proximal esophageal pouch, and abdominal bowel gas is absent. B, Gastrostomy injection with reflux into the short distal esophageal segment in a patient with esophageal atresia without a distal fistula. C, Esophageal atresia with the gap between the segments confirmed at surgery. Frontal view reveals bougies in both the proximal and distal segments, showing a long gap between them.
Treatment/Follow-up: Surgical repair is typically undertaken within 1 to 2 days, after the clinical workup is complete, including treatment of more emergent medical issues. Open or thoracoscopic division/ligation of the fistula and primary esophageal anastomosis is the treatment of choice. For long-gap atresia, a gastrostomy tube can be placed for palliation, while mechanical efforts are made to elongate the proximal and distal pouches prior to primary repair. Alternatively, if these procedures fail, an esophageal substitute (usually colon or ileocolon) can be interposed between the proximal and distal segments. Isolated esophageal atresia is typically associated with a small proximal pouch and almost no thoracic esophagus (see Fig. 97-3), and patients are typically palliated with a gastrostomy tube in the first 24 to 48 hours of life, followed by delayed repair.1
Imaging also plays an important role in the evaluation of early and late complications of esophageal atresia repair. Complications include anastomotic leak, anastomotic stricture, and recurrent TEF; these are evaluated with esophagram (e-Fig. 97-5). Because standard esophageal contrast studies miss a reported 50% of recurrent fistulae, bronchoscopy may also be required if recurrent fistula is suspected.1
Isolated Tracheoesophageal Fistula
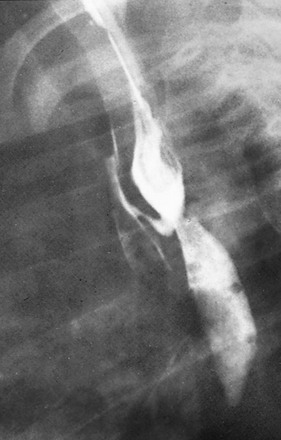
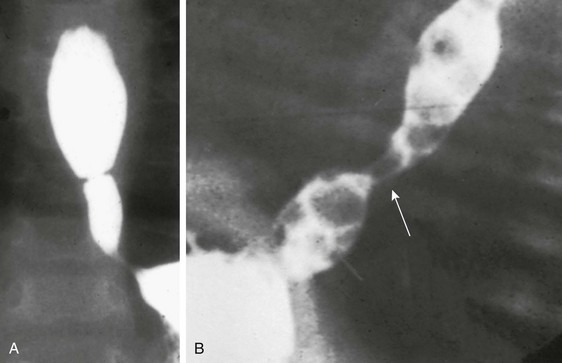
Overview: Congenital, isolated TEF without atresia (type 3 in Fig. 97-1) is often termed an H-type fistula, and it differs significantly from the other types of tracheoesophageal malformations in its presenting spectrum and features. This type of fistula can be difficult to identify, both clinically and radiographically.13 Large fistulae usually present very early in life and are relatively easy to see on an esophagram (Fig. 97-6). More commonly, the fistulae are small, inconstantly patent, and may require repeated examinations to identify. A major reason for inconstant patency of a fistula is that normal esophageal mucosa can be redundant, which can transiently occlude the esophageal side of the fistula.14 Although rare, multiple fistulae can be present.15
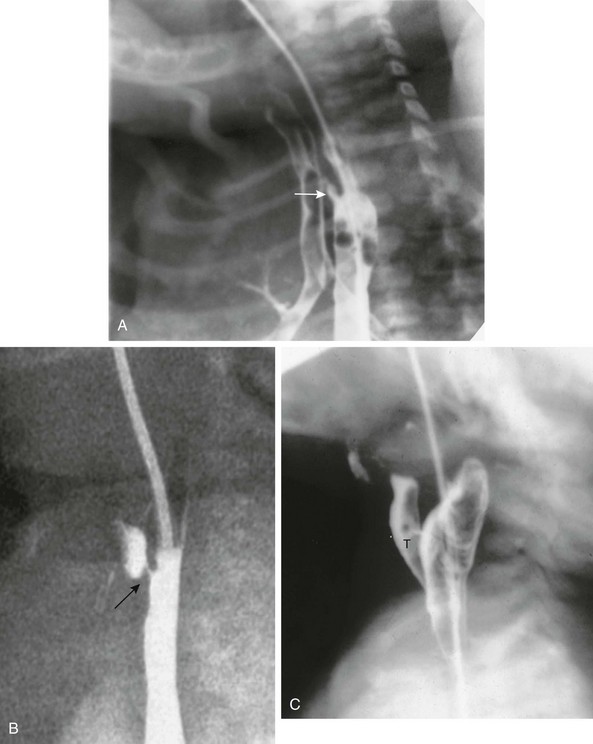
Figure 97-6 H-type fistula.
A, Tracheoesophageal fistula (arrow) in a 7-day-old boy with imperforate anus. B, Demonstration of another H-type fistula (arrow) from the upper cervical esophagus to the trachea, using the technique of contrast injection through a feeding tube with very careful volume control. C, Large H-type fistula from the upper cervical esophagus to the trachea (T).
Rarely, a bronchus originates directly from the esophagus, resulting in an anomaly termed esophagotrachea or esophageal bronchus (Fig. 97-7). This anomaly leads to severe respiratory distress with feeding and may be associated with esophageal atresia, TEF, or both.16 Bronchoesophageal fistula between the esophagus and a normal bronchial tree has also been described.17
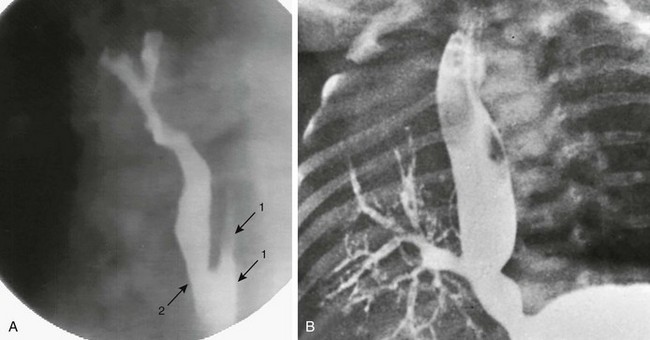
Figure 97-7 Connections between esophagus and airway.
A, Congenital esophagobronchial fistula. Oblique view shows esophagus (arrows with 1) and bronchus to right upper lobe (arrow with 2). B, Esophageal bronchus. Frontal view from an esophagram demonstrates the origin of the right main bronchus from the distal esophagus.
Clinical Presentation: These patients typically come to medical attention with recurrent episodes of coughing or choking and may progress to recurrent pneumonias, either in infancy or later in childhood.18,19 Patients with unexplained, intermittent respiratory distress and recurrent pneumonia should always be considered at risk for the presence of a TEF, and radiographic examination is warranted.
Imaging: This is often termed an H-type fistula because of its appearance on an esophagram: it connects to the trachea posteriorly and to the esophagus anteriorly as it courses cephalad from the esophagus to the trachea (see Fig. 97-6).
The examination should begin with a single contrast esophagram using digital, pulsed fluoroscopy in the right lateral to slightly right anterior oblique position. The position should be optimized during the examination to allow the best view of the anterior wall of the esophagus and posterior wall of the trachea. One of the keys of this examination is achieving full distension of the esophagus, because this is important to optimize filling and visualization of the fistula. If no abnormality is detected while the patient is swallowing, an esophageal catheter may be placed under fluoroscopic guidance, particularly in patients with normal prior examinations, in whom a fistula remains suspect, or in those in whom contrast is seen within the trachea from an unclear source.20 The examiner slowly withdraws the catheter from the distal esophagus in the cephalic direction, while carefully injecting contrast material with enough velocity and volume to distend the esophagus maximally under constant fluoroscopic monitoring. The catheter should have an end hole, without more proximal side holes, for adequate control of the contrast infusion. Even if a fistula is noted, the injection should continue until the catheter is withdrawn into the hypopharynx, but care must be taken not to allow contrast material to spill over into the trachea. Although there is a danger of spilling contrast material into the airway with injection of the high cervical esophagus, it is important to examine this area, because many fistulae occur at the level of the lower cervical or upper thoracic spine (see Fig. 97-6, C).21 Slow retraction of the catheter and careful fluoroscopic monitoring allow constant visualization and help to prevent overflow into the trachea.22
Treatment/Follow-up: Surgical correction of the fistula is the treatment of choice. Endoscopic, thoracoscopic, and open thoracotomy surgical techniques can be used.23,24
Fistula identification can be elusive at times, and repeat examination can be undertaken if clinical suspicion remains high. This also applies to those children who have been previously repaired, because a previously unseen fistula can be present, or the child may have failed primary therapy.15
Laryngotracheoesophageal Clefts
Overview: Laryngotracheoesophageal clefts occur in approximately 1 in 10,000 to 20,000 live births and consist of fistulous communications between the alimentary canal and the airway, either confined to the hypopharynx and larynx (Fig. 97-8) or extending inferiorly to include the esophagus.25 Several related classifications have been proposed based on the extent of the communication between the respiratory and alimentary pathways.
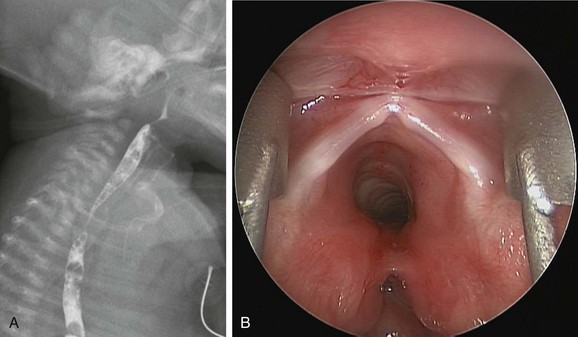
Figure 97-8 Laryngotracheal cleft.
A, Laryngotracheoesophageal cleft. Contrast swallow demonstrates large-volume aspiration from a posterior laryngeal wall defect. B, Endoscopic photograph from the same patient shows a posterior laryngeal wall defect and type 2 or 3 laryngotracheal cleft.
Pettersson26 originally described three types in 1955, with a fourth type identified by Ryan and colleagues27 in 1991: type I is limited to the larynx, type II extends beyond the cricoid lamina to include the cervical trachea, type III forms an esophagotrachea and extends to the carina; and type IV extends beyond the carina to involve part of one or both mainstem bronchi.
More recently, other investigators have made modifications to the original classifications, to include therapeutic implications. The classification of Benjamin and Inglis,28 as modified by Sandu and Monnier,29 takes into account the degree of involvement of the cricoid cartilage in a cleft and extension into the cervical trachea and beyond. In this classification, type 0 refers to a submucosal cleft; type I is a cleft confined to the supraglottic region, type II extends below the vocal cords into the cricoid cartilage, type IIIa extends through the cricoid cartilage, type IIIb extends beyond the cricoid cartilage into the cervical trachea, and type IV extends into the thoracic trachea for a variable length.25,28,29
Etiology: The larynx forms from the endoderm of the foregut caudally and from the mesenchyme of branchial arches four and six cranially.25 Incomplete separation and/or incomplete midline fusion of these concurrent processes during early gestation is believed to result in the persistent midline defect with varying extent and severity.
Clinical Presentation: The clinical presentation varies with the extent of the cleft, with varying degrees of respiratory distress aggravated during feeding, that begins early in infancy with choking, stridor, aspiration, recurrent pneumonia, and cyanosis.25 In some patients, esophageal mucosa herniates into the defect and offers some protection against aspiration, but symptoms of respiratory distress secondary to compromise of the airway increase.25 Anomalies such as esophageal atresia and VACTERL association may occur in patients with laryngotracheoesophageal clefts and other abnormalities, such as anal atresia, bronchial or tracheal stenosis and pulmonary hypoplasia, hypospadias, and coarctation of the aorta. Laryngotracheoesophageal clefts also occur as part of syndromes such as CHARGE, Opitz G/BBB (laryngeal malformations, craniofacial anomalies, genitourinary anomalies, and ventral midline anomalies), and Pallister-Hall (laryngeal, GI, cardiopulmonary, limb, and neurologic malformations) syndromes.25
Imaging: Diagnosis is usually made by laryngoscopy. On contrast esophagram (see Fig. 97-8, A), contrast is seen within the airway, although this can be confused with aspiration or with TEF. If computed tomography (CT) is performed, a common tracheoesophagus may be seen in extensive lesions, containing both the endotracheal and orogastric tubes.30
Treatment Follow-up: The goal of treatment is to maintain the airway and ventilation while minimizing aspiration and allowing adequate nutrition. Endoscopic surgical therapy can be used to repair some of the less extensive clefts, and it has been reported with success in type IIIa and IIIb clefts. Types III and IV are usually corrected with an open surgical approach,29 and extensive clefts may require both cervical and thoracic approaches.1,25 Postoperative survival rates range between 50% and 75%, depending on the extent of the cleft and severity of associated anomalies. Late complications include anastomotic leaks, pharyngeal and esophageal dysfunction, and gastroesophageal reflux.1
Acquired Pharyngoesophageal Perforation
Overview: Perforation of the pharynx or upper esophagus occurs in approximately 0.1% of neonatal intensive care unit discharges and is typically secondary to manipulation, such as attempted intubation; the imaging findings of this injury overlap with those of esophageal atresia.
Etiology: This injury is more common among premature infants. The mechanism of injury is believed to relate to passage of a catheter, or even the obstetrician’s finger, against reflexive contraction of the cricopharyngeus muscle and narrowing of the esophageal introitus as it is compressed against the cervical vertebrae with hyperextension of the infant’s neck. This leads to formation of a retroesophageal tract, which descends along the posterior mediastinum. The path of the catheter, or that of subsequently inserted catheters, may follow the perforation and enter the blind-ending tract in the prevertebral space; this resembles the appearance of esophageal atresia on radiographs.
Imaging: On chest radiographs, the findings of air or of a coiled enteric tube over the cervicothoracic junction resemble the findings in esophageal atresia. Distinguishing features include an unusually long blind pouch and irregularity of the outline of the false lumen. On esophagram, contrast extravasates into the newly created retroesophageal space (e-Fig. 97-9). Introduced contrast material is not easily aspirated into the syringe, and with time, the contrast dissipates along adjacent tissue planes (contrast should be water soluble and nearly iso-osmolal). Coexisting pneumomediastinum and pneumothorax or hydropneumothorax are helpful signs in detecting this complication and in differentiating it from esophageal atresia (see e-Fig. 97-9).
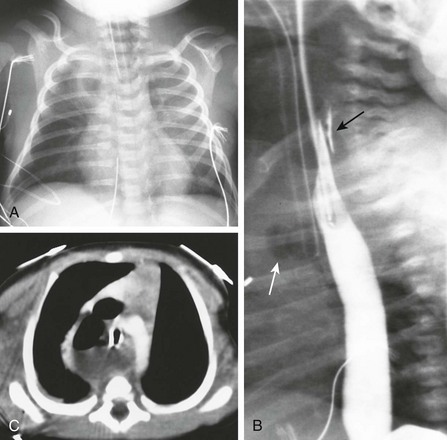
e-Figure 97-9 Traumatic pharyngoesophageal perforation.
A, Frontal chest radiograph reveals an opacity in the right upper lobe with air in its center, consistent with hydropneumothorax. Note the hyperlucency in the lower neck, resembling the findings of an esophageal pouch in esophageal atresia. B, Esophagram shows air in the mediastinum (white arrow), and contrast extending behind the esophagus (black arrow). C, Computed tomography scan shows posterior mediastinal fluid and air.
Clinical Findings: The initial clinical presentation may be relatively silent, but patients manifest increased secretions with episodes of choking and cyanosis associated with feeding, and the development of crepitus and clinical deterioration follows, along with the inability to easily pass a feeding catheter.
Treatment/Follow-up: Once the injury has been recognized, the majority of these patients can be managed conservatively, with withdrawal of enteral feedings, institution of antibiotics, and drainage of pleural effusion or pneumothorax if present. This management is successful in the vast majority of patients, and surgical treatment is reserved for complications, such as mediastinal abscess.
Esophageal Stenosis
Overview: Congenital esophageal stenosis is a rare condition that occurs in 1 in 25,000 to 50,000 live births.1,31 The three histologic subtypes are fibromuscular stenosis, membranous web, or tracheobronchial remnants.
Membranous stenosis is considered one of the rarest forms of this rare lesion and is usually located in the middle or distal third of the esophagus; it typically demonstrates an eccentric opening and is covered with squamous epithelium. Fibromuscular stenosis demonstrates subepithelial proliferation of smooth muscle and fibrosis. Stenosis associated with tracheobronchial remnants demonstrates cartilaginous tissue proliferation that partially or completely encircles the esophagus—usually in the lower third, within 3 cm of the gastroesophageal junction—and is often associated with esophageal atresia and TEF.1,31,32
Etiology: The etiology of fibromuscular stenoses is uncertain, whereas that of membranous stenoses may be similar to that of membranous stenosis elsewhere in the GI tract. Stenoses as a result of tracheobronchial rests are thought to arise from incomplete separation of the tracheoesophageal septum during gestation with sequestration of the tracheal cartilage in the esophageal wall; this is carried distally by the growth of the esophagus.1,32,33
Clinical Presentation: The typical presentation is that of dysphagia later in infancy, especially after introduction of solids,34 or in older patients who may present with regurgitation, choking, vomiting, and failure to thrive.35 The child may also come to medical attention with retention of a foreign body outside of the cervicothoracic junction, the aortic arch, the left bronchial crossing, or the gastroesophageal junction, which are the typical locations in which foreign bodies lodge in the normal esophagus.
Imaging: Esophagram in patients with esophageal stenosis demonstrate an area of narrowing with partial obstruction (Fig. 97-10). Stenoses secondary to web or to fibromuscular hypertrophy may be seen in the upper or mid portion of the esophagus. Stenoses secondary to tracheobronchial remnants appear as a discrete narrowing of the esophagus along its distal portion, typically within 3 cm of the gastroesophageal junction and particularly in a patient with esophageal atresia. There may be impaction of food material, therefore a filling defect appears on esophagram proximal to the stenotic site. The diagnosis may be confirmed by endoscopy and histopathology.34
Because of the rarity of congenital esophageal stenosis, the findings at esophagram may be misinterpreted as stricture, such as secondary to gastroesophageal reflux, or as achalasia, if located very close to the gastroesophageal junction.33,35 In such cases, manometric studies may be helpful in excluding the diagnosis of achalasia.35
If the diagnosis is delayed, cross-sectional imaging may demonstrate marked thickening of the wall of the esophagus, resembling esophageal leiomyomatosis.35
Treatment/Follow-up: It is essential that a stricture as a result of gastroesophageal reflux be excluded prior to the diagnosis of esophageal stenosis.1 Endoscopic balloon dilatation can be used as a primary and therapeutic procedure and is most successful in those patients without a true cartilaginous component of the stenosis.36,37 Membranous stenoses are amenable to endoscopic dilation and resection, although surgical resection of the stenotic segment is reserved for strictures that are not amenable to one or more treatments with dilation or that recur after dilation.38 Follow-up esophagram can be performed to assess for perforation, recurrent stenosis, or stricture after dilation or surgical resection.
Esophageal Duplication Cysts
Overview: Duplication cysts of the esophagus represent part of the spectrum of foregut duplication cysts: they include bronchogenic cysts, which typically include mural cartilage or respiratory glands39; neurenteric cysts, which communicate with the spinal canal through a vertebral defect; and esophageal duplication cysts that may occur in the neck, associated with the cervical esophagus, or in the thorax, or they may involve the thorax and the upper abdomen as in a thoracoabdominal duplication cyst. Approximately two thirds occur in the right side of the mediastinum secondary to dextrorotation of the stomach during embryogenesis.40
Esophageal duplication cysts comprise approximately 20% of alimentary tract duplications and represent the most common site of duplications after the ileum41,42 and approximately 10% of mediastinal masses in children.43 Diagnostic criteria met by esophageal duplication cysts include attachment to the esophagus, two muscular layers enveloping the cyst, and lining by squamous, columnar, cuboidal, pseudostratified or ciliated epithelium, or epithelium that “demonstrates some level of the gastrointestinal tract.”40 True tubular duplications of the esophagus are rare. These cysts may extend below the diaphragm to involve the stomach, and they may communicate with the stomach or esophagus.
Neurenteric cysts are duplication cysts that communicate with the spinal canal, and they differ in etiology from the classic type of duplication cyst. The archetype of this spectrum of anomalies is the dorsal enteric fistula, a patent communication between the gut and the dorsal midline skin surface that traverses the vertebral body (or interspace), the spinal canal and its contents, and the posterior vertebral elements. These are discussed in Chapter 43.
Etiology: Esophageal duplication cysts are believed to be the result of a budding error of the dorsal portion of the foregut between the third and sixth weeks of gestation. These cysts arise from the posterior portion of the foregut and contain mucosa that, by histologic examination, is usually gastric or, more rarely, intestinal.40 True tubular esophageal duplications likely develop from faulty recanalization of the esophageal lumen during the tenth week of gestation.39,41,44 They may or may not communicate with the esophageal lumen.
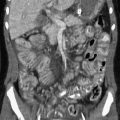
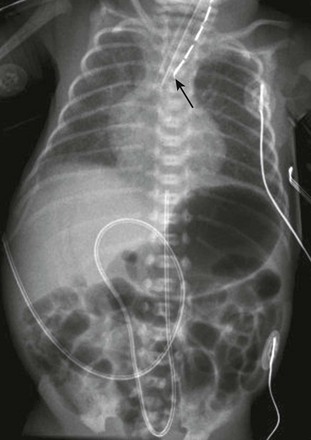
Clinical Presentation: Cervical duplication cysts are usually symptomatic very early in life, and patients come to medical attention with respiratory distress. Potential for respiratory compromise may be evident in fetal life, and an ex utero intrapartum treatment may be needed at delivery.40 Large thoracic duplication cysts that occur in the superior mediastinum may manifest with respiratory distress or dysphagia because of compression of adjacent structures, or they may be found incidentally (e-Fig. 97-11).42 Duplication cysts that occur in the lower esophagus are often asymptomatic and are discovered incidentally (Fig. 97-12), although they may become symptomatic as a result of inflammation, if they contain gastric mucosa, or as a result of superinfection or hemorrhage.46
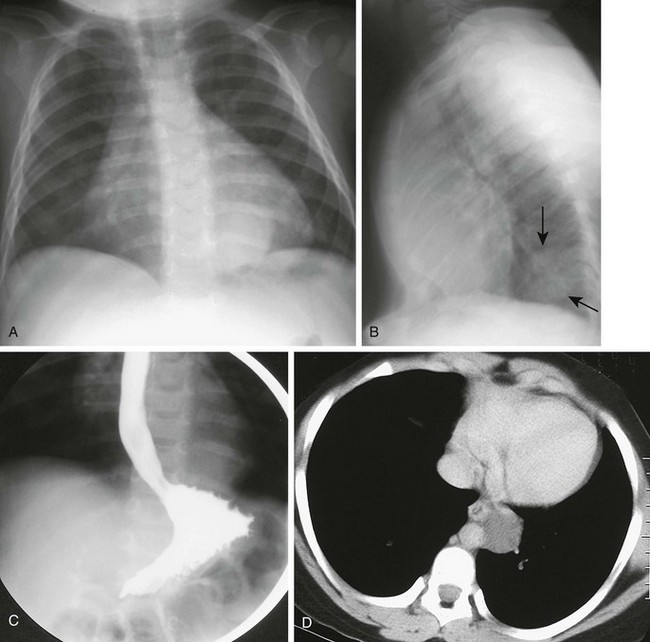
Figure 97-12 Esophageal duplication cysts.
A, Frontal chest radiograph reveals a paraspinal retrocardiac mass at the left base. B, Lateral image confirms the posterior location of the lesion (arrows) with a positive spine sign. C, Esophagram shows deviation of the esophagus by the mass, confirming its posterior mediastinal location. D, Computed tomographic scan with intravenous contrast material demonstrates the persistently low attenuation of the duplication and its relationship to the esophagus.
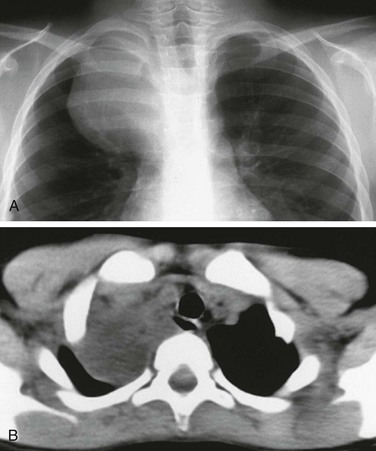
e-Figure 97-11 Esophageal duplication cysts.
A, Chest radiograph demonstrates a large mass projecting over the right thoracic apex, without evidence of effect upon the adjacent ribs. B, Computed tomography demonstrates the low-attenuation cystic lesion and its relationship to the mediastinum and the adjacent, air-filled esophagus seen posterior to the airway. At surgery the diagnosis of esophageal duplication was confirmed.
Neurenteric cysts may present with back pain and progressive neurologic deficit if not diagnosed prenatally or in early infancy.47 Infants may be brought to medical attention with meningitis. These defects are typically associated with midline vertebral lesions such as spina bifida, other dysraphic defects, or scoliosis (e-Figs. 97-13 and 97-14 and Fig. 97-15).
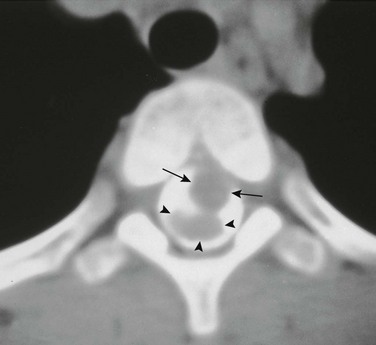
Figure 97-15 Intraspinal neurenteric cyst.
Axial computed tomographic section with intrathecal contrast demonstrates an intraspinal mass (arrows) anterior to the compressed spinal cord (arrowheads) surgically proven to represent an intraspinal neurenteric cyst. Note the associated congenital cleft in the vertebral body.
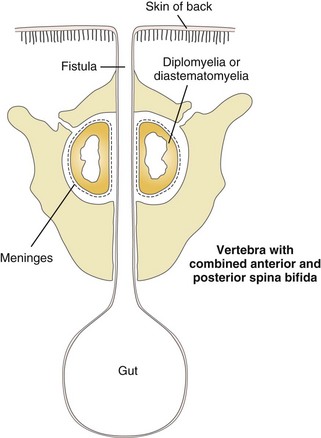
e-Figure 97-14 Neurenteric cysts.
A, Frontal chest radiograph demonstrates a right-sided mass lesion associated with a midthoracic vertebral segmentation anomaly. B, Esophagram demonstrates the vertebral body malformations and paravertebral soft tissue mass and further outlines displacement of the esophagus. These findings in conjunction are suggestive of a neurenteric cyst.
The combination of esophageal duplication with other anomalies, such as spinal defects, should prompt a search for additional duplications along the alimentary tract, which can be found in up to one third of cases.42
Imaging: Radiographs may demonstrate a sharply demarcated middle or posterior mediastinal mass and associated vertebral anomalies in cases of neurenteric cyst (see e-Fig. 97-14, A).
Esophagrams (see e-Fig. 97-14, B) will suggest an intramural mass, but typically no connection between the two structures is seen, except in cases of communication of the lumen in long, tubular duplications.44 Ultrasound can confirm the cystic nature of the mass in utero but is rarely utilized in these chest lesions after birth.48 CT and MRI are the modalities of choice to confirm the cystic nature of the mass and to best delineate the abnormality. The cyst fluid typically has the attenuation and signal characteristics of water, although this can be affected by hemorrhage or infection.
For neurenteric cysts, CT myelography (see Fig. 97-15) can demonstrate the extent of the lesion and any associated vertebral anomaly. MRI, however, is the study of choice to evaluate the presence and extent of intraspinal abnormalities.49 These lesions are usually intradural and extramedullary.50 Those enteric cysts that contain gastric mucosa with acid and pepsin secretion can be identified by nuclear imaging and are at risk for hemorrhage.51
Berrocal, T, Torres, I, Gutiérrez, J, et al. Congenital anomalies of the upper gastrointestinal tract. Radiographics. 1999;19(4):855–872.
Ioannides, AS, Copp, AJ. Embryology of oesophageal atresia. Semin Pediatr Surg.. 2009;18(1):2–11.
Jones, DW, Kunisaki, SM, Teitelbaum, DH, et al. Congenital esophageal stenosis: the differential diagnosis and management. Pediatr Surg Int. 2010;26(5):547–551.
Kluth, D, et al. The embryology of foregut malformations. J Pediatr Surg. 1987;22:389–393.
Laffan, EE, Daneman, A, Ein, SH, et al. Tracheoesophageal fistula without esophageal atresia: are pull-back tube esophagograms needed for diagnosis? Pediatr Radiol. 2006;36(11):1141–1147.
Leboulanger, N, Garabedian, EN. Laryngo-tracheo-oesophageal clefts. Orphanet J Rare Dis. 2011;6:81.
References
1. Harmon, C, Coran, A. Congenital anomalies of the esophagus. In: Coran A, et al, eds. Coran: Pediatric Surgery. Philadelphia: Elsevier, 2012.
2. Oddsberg, J, et al. Influence of maternal parity, age, and ethnicity on risk of esophageal atresia in the infant in a population-based study. J Pediatr Surg. 2008;43(9):1660–1665.
3. Kluth, D, Fiegel, H. The embryology of the foregut. Semin Pediatr Surg. 2003;12(1):3–9.
4. Ioannides, AS, Copp, AJ. Embryology of oesophageal atresia. Semin Pediatr Surg. 2009;18(1):2–11.
5. Holder, TM, et al. Esophageal atresia and tracheoesophageal fistula. A survey of its members by the Surgical Section of the American Academy of Pediatrics. Pediatrics. 1964;34:542–549.
6. Stoll, C, et al. Associated malformations in patients with esophageal atresia. Eur J Med Genet. 2009;52(5):287–290.
7. Burjonrappa, S, et al. Type A esophageal atresia: a critical review of management strategies at a single center. J Pediatr Surg. 2010;45(5):865–871.
8. Sparey, C, et al. Esophageal atresia in the Northern Region Congenital Anomaly Survey, 1985-1997: prenatal diagnosis and outcome. Am J Obstet Gynecol. 2000;182(2):427–431.
9. Shulman, A, et al. Prenatal identification of esophageal atresia: the role of ultrasonography for evaluation of functional anatomy. Prenat Diagn. 2002;22(8):669–674.
10. Salomon, LJ, et al. Real-time fetal magnetic resonance imaging for the dynamic visualization of the pouch in esophageal atresia. Ultrasound Obstet Gynecol. 2009;34(4):471–474.
11. Hernanz-Schulman, M. Vascular rings: a practical approach to imaging diagnosis. Pediatr Radiol. 2005;35(10):961–979.
12. Lillehei, CW, Colan, S. Echocardiography in the preoperative evaluation of vascular rings. J Pediatr Surg. 1992;27(8):1118–1120.
13. Karnak, I, et al. The diagnosis and treatment of H-type tracheoesophageal fistula. J Pediatr Surg. 1997;32(12):1670–1674.
14. Yamamoto, N, Imai, S, Motohiro, K. Congenital tracheoesophageal fistula with symptoms commencing at the onset of trauma: report of a case. Surg Today. 1996;26(9):744–746.
15. Guo, W, et al. Tracheoesophageal fistula after primary repair of type C esophageal atresia in the neonatal period: recurrent or missed second congenital fistula. J Pediatr Surg. 2010;45(12):2351–2355.
16. Lallemand, D, Quignodon, JF, Courtel, JV. The anomalous origin of bronchus from the esophagus: report of three cases. Pediatr Radiol. 1996;26(3):179–182.
17. Kemp, JL, Sullivan, LM. Bronchoesophageal fistula in an 11-month-old boy. Pediatr Radiol. 1997;27(10):811–812.
18. Crabbe, DC. Isolated tracheo-oesophageal fistula. Paediatr Respir Rev. 2003;4(1):74–78.
19. Biechlin, A, Delattre, A, Fayoux, P. [Isolated congenital tracheoesophageal fistula. Retrospective analysis of 8 cases and review of the literature]. Rev Laryngol Otol Rhinol (Bord). 2008;129(3):147–152.
20. Laffan, EE, et al. Tracheoesophageal fistula without esophageal atresia: are pull-back tube esophagograms needed for diagnosis? Pediatr Radiol. 2006;36(11):1141–1147.
21. Bax, KN, Roskott, AM, van der Zee, DC. Esophageal atresia without distal tracheoesophageal fistula: high incidence of proximal fistula. J Pediatr Surg. 2008;43(3):522–525.
22. Gopal, M, Woodward, M. Potential hazards of contrast study diagnosis of esophageal atresia. J Pediatr Surg. 2007;42(6):E9–E10.
23. Richter, GT, et al. Endoscopic management of recurrent tracheoesophageal fistula. J Pediatr Surg. 2008;43(1):238–245.
24. Tzifa, KT, et al. Endoscopic treatment of congenital H-Type and recurrent tracheoesophageal fistula with electrocautery and histoacryl glue. Int J Pediatr Otorhinolaryngol. 2006;70(5):925–930.
25. Leboulanger, N, Garabedian, EN. Laryngo-tracheo-oesophageal clefts. Orphanet J Rare Dis. 2011;6:81.
26. Pettersson, G. Inhibited separation of larynx and the upper part of trachea from oesophagus in a newborn; report of a case successfully operated upon. Acta Chir Scand. 1955;110(3):250–254.
27. Ryan, DP, et al. Laryngotracheoesophageal cleft (type IV): management and repair of lesions beyond the carina. J Pediatr Surg. 1991;26(8):962–969. [discussion 969-970].
28. Benjamin, B, Inglis, A. Minor congenital laryngeal clefts: diagnosis and classification. Ann Otol Rhinol Laryngol. 1989;98(6):417–420.
29. Sandu, K, Monnier, P. Endoscopic laryngotracheal cleft repair without tracheotomy or intubation. Laryngoscope. 2006;116(4):630–634.
30. Wilkinson, AG, Mackenzie, S, Hendry, GM. Complete laryngotracheoesophageal cleft: CT diagnosis and associated abnormalities. Clin Radiol. 1990;41(6):437–438.
31. Takamizawa, S, et al. Congenital esophageal stenosis: Therapeutic strategy based on etiology. J Pediatr Surg. 2002;37(2):197–201.
32. Nemolato, S, et al. Oesophageal tracheobronchial remnants. Gastroenterol Clin Biol. 2008;32(8-9):779–781.
33. Neilson, IR, et al. Distal congenital esophageal stenosis associated with esophageal atresia. J Pediatr Surg. 1991;26(4):478–481. [discussion 481-482].
34. Pumberger, W, Geissler, W, Horcher, E. [Congenital oesophageal stenosis due to tracheobronchial remnants]. Chirurg. 1999;70(9):1031–1035.
35. Jones, DW, et al. Congenital esophageal stenosis: the differential diagnosis and management. Pediatr Surg Int. 2010;26(5):547–551.
36. Deiraniya, AK. Congenital oesophageal stenosis due to tracheobronchial remnants. Thorax. 1974;29(6):720–725.
37. Ibrahim, AH, et al. Congenital esophageal stenosis associated with esophageal atresia: new concepts. Pediatr Surg Int. 2007;23(6):533–537.
38. Elhalaby, EE, et al. Congenital esophageal stenosis: to dilate or to resect. Ann Pediatr Surg. 2006;2(1):2–9.
39. Martin, ND, et al. Intra-abdominal esophageal duplication cysts: a review. J Gastrointest Surg. 2007;11(6):773–777.
40. Nayan, S, et al. Cervical esophageal duplication cyst: case report and review of the literature. J Pediatr Surg. 2010;45(9):e1–e5.
41. Berrocal, T, et al. Congenital anomalies of the upper gastrointestinal tract. Radiographics. 1999;19(4):855–872.
42. Lund, D. Alimentary Tract Duplications. In: Coran A, et al, eds. Coran: Pediatric Surgery. Philadelphia: Elsevier, 2012.
43. Wootton-Gorges, SL, et al. Duplication of the cervical esophagus: a case report and review of the literature. Pediatr Radiol. 2002;32(7):533–535.
44. Fitch, SJ, Tonkin, IL, Tonkin, AK. Imaging of foregut duplication cysts. Radiographics. 1986;6(2):189–201.
45. Savage, JJ, et al. Neurenteric cysts of the spine. J Craniovertebr Junction Spine. 2010;1(1):58–63.
46. Al-Majali, R, et al. Mediastinal foregut duplication cyst. JRMS. 2004;11(1):48–50.
47. Garg, N, et al. Is total excision of spinal neurenteric cysts possible? Br J Neurosurg. 2008;22(2):241–251.
48. Sherer, DM, et al. Prenatal sonographic findings of an isolated cervical esophageal duplication cyst. J Ultrasound Med. 2009;28(3):405–407.
49. Cai, C, et al. Intraspinal neurenteric cysts in children. Can J Neurol Sci. 2008;35(5):609–615.
50. Menezes, AH, Traynelis, VC. Spinal neurenteric cysts in the magnetic resonance imaging era. Neurosurgery. 2006;58(1):97–105.
51. Kumar, R, et al. Diagnosis of ectopic gastric mucosa using 99Tcm-pertechnetate: spectrum of scintigraphic findings. Br J Radiol. 2005;78(932):714–720.