CHAPTER 8 COMA AND BRAIN DEATH
Assessing the level of consciousness and diagnosing the cause of coma are fundamental aspects of medical practice. Consciousness consists of two main components: the level of alertness or arousal and the content of thought. Impairment of arousal is a continuum from drowsiness to stupor and then to coma. In stupor, the patient awakens briefly in response to stimulation and then slips back into a sleeplike state. There is no verbal response. Coma is the inability of the patient to be aroused (i.e., to obey commands, speak, or open the eyes in response to painful stimuli). This is obviously different from a sleep state from which the individual can be aroused. Stupor and coma of recent onset are medical emergencies. Speedy diagnosis and treatment are imperative for achieving the optimal outcome; however, a discussion of the treatment of the many causes of coma is beyond the scope of this chapter.
CONSCIOUSNESS
Qualia are the high-order perceptions of qualities, such as the warmth of warm or the redness of red, that are experienced in the normal conscious state.1 Free will, conscience, meta-memory (knowledge and beliefs about the functioning of one’s own memory systems), the analysis of one’s own thoughts, and imagination are all integral components of human higher order consciousness that remain mysterious. Much of the planning and execution that humans perform is unconscious or preconscious in the “zombie mode.” An example is sensory processing and gating. The automatisms of complex partial seizures are an extended pathological example of this. Penfield (1937) showed that electrical stimulation of the cortex could alter the content of consciousness and produce “experiential responses” that the patient usually realized were unreal. The “déjà vu” phenomenon is a natural example of this.
The neural correlates of consciousness are slowly yielding to the study of individual and group neuronal electrical activity, through the use of surface cortical electrodes and implanted microelectrodes, and to functional brain imaging. Although there is a modularity to brain function, it is the integration of all the modules that is necessary for consciousness. There also seems to be competition between different cortical areas and neurons to choose the best fit for a set of perceptual inputs. There are “essential nodes” for particular perceptions such as face recognition or color perception. Clearly, multiple nodes and regions of the brain, functioning in concert, are necessary for consciousness to exist.2,3
Both the cerebral cortex and subcortical structures such as the thalamus are involved in consciousness. Dandy described temporary loss of consciousness after removal of both frontal lobes with sacrifice of the anterior cerebral arteries at the genu of the corpus callosum and speculated that the striatal damage was responsible for the coma.4 The reticular nucleus that surrounds the thalamus acts as a switch or “gate” to particular thalamic nuclei. It results in different patterns of activity of the thalamic nuclei and therefore in different weighting of sensory input. The intralaminar nucleus of the thalamus sets thresholds for the cortical response to the thalamic input. Thalamic gating is also influenced by feedback from the prefrontal cortex.5 The filtering of sensory information is done at a preconscious level so that attention is selectively directed, although it can also be altered at will.
Moruzzi and Magoun6 in 1949 produced electroencephalographic arousal by stimulating the brainstem reticular formation rostral to the mid-pons. They termed this system, including its rostral projection, the ascending reticular activating system (ARAS). This is an alerting or arousal system that also indirectly influences sensory processing in the cerebral cortex. It projects rostrally through the midline, intralaminar nucleus, and other nuclei of the thalamus, and via these structures to the cerebral cortex. Attention enables an awake and alert individual to select a task or a stimulus to process from a number of alternatives and to select a cognitive strategy to carry it out.5 The ARAS is thought to facilitate this process by enhancing the perception of differences between competing stimuli. The anterior cingulate gyrus is involved in a wide range of attentional and discriminatory activity and is involved in higher order motor control of many tasks. The parietal cortex is involved in attention in the visual field and the pulvinar of the thalamus is involved in selecting information for attention. The dorsolateral prefrontal cortex is also involved in attention, intention, and working memory; working memory is the term applied to the holding and manipulation of the current content of consciousness. The hippocampal system—including the fornices, the mammillary bodies and mammillothalamic tracts, the amygdala, the anterior thalamic nuclei, the medial dorsal thalamic nuclei, and the entorhinal cortex—are all involved with establishing new anterograde episodic memory. The amygdala, hippocampus, and associated limbic and nonlimbic structures are involved in the generation of internal feelings, emotions, and motivation.5
The γ-amino butyric acid (GABA) inhibitory neurons are widely dispersed throughout the central nervous system and are involved with the selection of sensory information. Barbiturates increase GABAergic activity in the ARAS. Glutamate and aspartate are the excitatory neurotransmitters that play a key role in cortical interplay.5 The N-methyl-D-aspartate (NMDA) receptor may be the main target for the action of general anesthetic agents that produce a pharmacological coma. The corollary, that the NMDA receptor is essential for consciousness, constitutes the Flohr hypothesis, about which there is considerable debate.7 Clearly, many other peptides and receptors are also involved in cortical function and consciousness.
Single neurons in the human entorhinal cortex have very specific responses (e.g., only to faces or only to different types of animal), and some temporal lobe neurons function at a different hierarchical level by responding to the extensively processed perception or imagining of an object and not to the raw retinal input.2 Different components of consciousness therefore seem to exist from the level of individual neurons to that of different brain regions and, indeed, that of the whole brain.
Edelman (2004) proposed a theory called neural Darwinism, or neuronal group selection, to explain consciousness, as opposed to an instructive model in which the brain has computer-like properties with a set of programs and algorithms. The three tenets of neural Darwinism are that (1) developmental selection leads to a highly diverse group of circuits, (2) experiential selection leads to changes in the connection strength of synapses, and (3) reentrant mapping occurs, in which brain maps are coordinated in space and time through reentrant signaling across reciprocal connections. This coordination leads to widespread synchronization of widely dispersed neuronal groups, which integrates information such as the color and orientation of visual objects and which Edelman proposed is central to the understanding of consciousness. This is a possible solution to the binding problem, which is the seamless reintegration of separately processed aspects of a sensory percept at a preconscious level. According to neural Darwinism, the brain is a selectional system. Other examples of such systems in nature are evolution and the immune system. Another important characteristic of the brain is degeneracy, in which different elements of the brain can perform the same function and one element can carry out different functions in different neuronal networks at different times. This creates great diversity of brain function. There is no need in the theory of neural Darwinism for a homunculus in the head directing the brain and being the seat of consciousness.1
The principal neural structures of consciousness according to Edelman’s (2004) theory are the cerebral cortex, the thalamus, and the reentrant loops between the two, which he called the dynamic thalamocortical core. He proposed that this neural activity generates the qualia of consciousness. The gamma, or 40-Hz, rhythm of the electroencephalogram (EEG) is believed to be produced by thalamocortical circuits during attention and sensory processing tasks. Attention is directed partly by the reticular nucleus of the thalamus and partly by the relationship of the basal ganglia to the frontal and parietal cortices. Higher order consciousness is based in part on episodic memory, which depends on the hippocampi, and in part on semantic and linguistic ability, which depend on the language cortices of Broca and Wernicke and associated areas (for further reading, see Edelman, 2004; Jasper et al, 1998; John, 2002; Metzinger, 2002; Young et al, 1998; Zeman, 2001).
COMA
The Etiology of Coma
The fundamental causes of coma are structural, including the mechanical deformation or disruption of neural tissue and ischemia, and metabolic or toxic derangement of neural tissue, inducing hypoxia. A detailed list of the causes is presented in Table 8-1. A detailed history from bystanders or relatives is vital in determining the cause of the coma.
ADEM, acute disseminated encephalomyelitis; PaO2, partial pressure of arterial oxygen; PVS, persistent vegetative state.
The Anatomy and Pathophysiology of Coma
Diffuse lesions of both cerebral hemispheres (cortical and subcortical white matter) may cause coma. Bilateral diencephalic damage (especially to the paramedian dorsal thalamus) may also cause coma (Fig. 8-1). The extension of the thalamic lesions into the midbrain tegmentum has an even greater propensity for causing coma or severe neurological deficit, apathy, and impaired attention. Damage to the paramedian gray matter anywhere from the posterior hypothalamus to the tegmentum of the lower pons causes coma.8,9 When the respiratory centers in the lower medulla are damaged, apnea ensues. The testing of brainstem reflexes and for apnea is the clinical means of confirming the destruction of theses critical areas. The irreversible destruction of critical brainstem areas usually follows catastrophic supratentorial events that cause brain herniation and subsequent compression and ischemia of the brainstem.
The sequence of cardiovascular changes resulting from progressive mechanical compression and/or ischemia of the brainstem begins with vagal stimulation, which causes decreases in heart rate, mean arterial pressure, and cardiac output. As the pons becomes ischemic, sympathetic stimulation occurs, which results in hypertension with persistence of the bradycardia (Cushing’s reflex). As the medulla becomes ischemic, there is unopposed sympathetic stimulation with tachycardia, increased mean arterial pressure, and increased cardiac output. This sequence has been called an “autonomic storm” and has not been well recognized in intensive care unit patients, partly because it may have occurred by the time the patient is admitted to intensive care unit and may be affected by treatment or the presence of other injuries.10
Clinical Assessment of the Comatose Patient
A full neurological and general examination of the patient must be undertaken. Some of the pertinent components of the examination are described as follows.5,9
Assessment of the Conscious State
The Glasgow Coma Scale (GCS) was devised to provide a simple, reliable, and reproducible method of assessment of conscious state so as to avoid misinterpretation and blurring of terms such as stupor, semicoma, and confusion.11 It has become a universally accepted scale for neurological observation, prognostication, and grading severity and has been found to have good interrater reliability. It was originally a 14-point scale but has been extended to 15 points by splitting limb flexion into two tiers: flexion-withdrawal and flexion-abnormal. Abnormal flexion is defined as any two elements of stereotyped flexion posture, extreme wrist flexion, adduction of the upper arm, and fisting of the fingers over the thumb.12,13 The 15-point scale is the preferred scale for research studies. The GCS is scored on the best response in each of the three categories: eye opening, vocalization, and limb movement (Table 8-2). Testing nail bed pressure with a pencil is the recommended method of applying a painful stimulus. Patients who do not open their eyes, do not speak, and are not obeying commands are said to be comatose. Many clinicians regard a maximum GCS score of 8 as the cutoff for coma. Some limitations to the usefulness of the GCS do exist, however. For example, periorbital swelling and endotracheal intubation, both common conditions in the trauma patient, prevent the accurate assessment of eye opening and verbal response, respectively. Some centers record a “T” next to the score when the patient is intubated and the verbal score cannot be assessed. The GCS is commonly used to monitor neurological progress, and a drop in the GCS of 2 points or more is a sensitive measure of neurological deterioration and necessitates action to halt the progression. Age and depth of coma are the principal predictors of poor outcome or death after traumatic brain injury. Repeated GCS assessments and the recognition of confounding factors are required for any confidence in prediction of outcome.
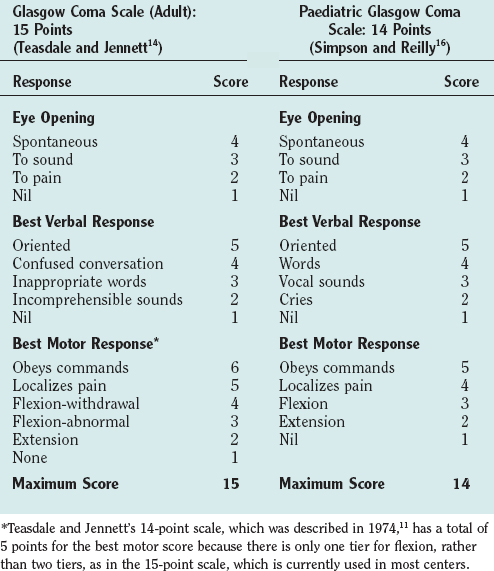
The conscious state is more difficult to assess in infants and young children, and special pediatric coma scales have been devised.14 The Paediatric Glasgow Coma Scale is a simple system based on the GCS with age-related norms for verbal and motor responses (see Table 8-2).15
Respiratory Pattern
Pupil Examination
Eye Motion: Extraocular Muscle Function (Fig. 8-2)
Deviation of the Ocular Axes
 Frontal lobe lesion (frontal center for contralateral gaze): the eyes look toward the side of the lesion.
Frontal lobe lesion (frontal center for contralateral gaze): the eyes look toward the side of the lesion.


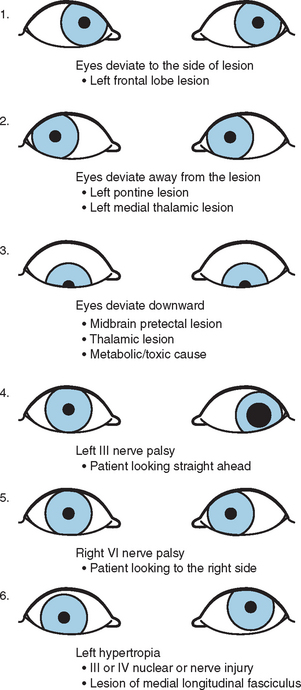
Unilateral outward deviation with dilated pupil (nerve III palsy)
Reflex Eye Movement
Oculocephalic reflex (doll’s-eye reflex)
In the awake patient, the eyes follow the movement of the head or, if the movement is performed slowly, there is contraversive conjugate eye movement if the eyes fixate on a stationary target. In the comatose patient with intact brainstem and cranial nerves, the reflex is preserved and the eyes move in the opposite direction when the head is turned laterally or the neck flexed and extended; that is, the conjugate eye movement is contraversive. When the brainstem is damaged, the reflex is lost, and the eyes follow the head movement. Cervical spine injury should be ruled out before this test is performed.
Herniation Syndromes (Coning)
Cerebral herniation is the displacement of cerebral tissue as a result of intracranial mass lesions. This displacement may disturb the conscious state by direct or indirect pressure on the diencephalon and brainstem. Indirect pressure means displacement of tissue at a distance from the mass. Coning is the name given to progressive cerebral herniation with secondary compression of the brainstem and resultant deepening coma. Computed tomography (CT) and magnetic resonance imaging have helped significantly to elucidate the anatomy of herniation. Plum and Posner (1982) in their classic treatise described progressive and predictable stages of herniation with cephalad-caudal progression. There are five types of supratentorial and infratentorial herniation (Fig. 8-3), described as follows.
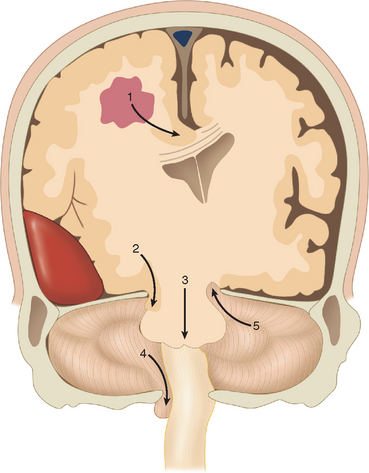
Supratentorial Herniation
Central (Transtentorial) Herniation
The classic description is of a sequential rostral-caudal failure from diencephalon to the midbrain, the pons, and finally the medulla. Central herniation is often caused by a tumor in the frontal, parietal, or occipital lobes that reaches the point of decompensation. The diencephalon may herniate through the tentorial incisura, damaging the pituitary stalk and causing diabetes insipidus. Vertical downward shift of the brainstem may be a critical factor in the development of the features of central herniation.16 The brainstem may become ischemic as a consequence of shearing of perforating arteries from the basilar artery. Resultant hemorrhages in brainstem are called Duret hemorrhages. CT may show a downward displacement of the pineal gland and effacement of the perimesencephalic (ambient) cisterns. The posterior cerebral arteries may be kinked at the tentorial edge as they pass onto the inferior surface of the occipital lobe, which results in occipital infarction and cortical blindness. This may further increase the intracranial pressure.
The clinical stages of central herniation are outlined in Table 8-3. It is difficult to discern these various stages (and those of the other types of coning) in many patients because of the speed of deterioration, the tendency of the stages to merge, and the mixed and complex pathology that may affect the supratentorial and infratentorial compartments, and because patients often receive early intervention with paralysis, endotracheal intubation, and ventilation that obscures or retards the clinical progression. Nevertheless, it is useful to think conceptually in these terms in contemplating the patient’s condition and the appropriate treatment.
| Diencephalon Stage | May be caused by displacement or ischemia of the diencephalon |
| This stage is reversible | |
| Consciousness | Altered alertness is first sign |
| Usually lethargy is present | |
| Some patients have agitation | |
| Later stupor, then coma | |
| Respiration | Sighs, yawns, occasional pauses |
| Pupils | Small (1-3 mm), reacting |
| Oculomotor | Conjugate or slightly divergent roving eyes |
| Doll’s-eye reflex present | |
| Parinaud’s syndrome with impaired upward gaze may be present | |
| Conjugate ipsilateral response to cold water calorics | |
| Motor | Early: appropriate response to painful stimulus |
| Contralateral hemiparesis may worsen | |
| Bilateral Babinski reflex; later, grasp reflexes | |
| Decorticate (contralateral to the lesion initially) | |
| Midbrain–Upper Pons Stage | Prognosis is very poor when midbrain signs have developed; <5% having a good outcome if treatment is undertaken |
| Respiration | Cheyne-Stokes respiration → sustained tachypnea |
| Pupils | Moderately dilated: midposition (3-5 mm), fixed |
| Oculomotor | Doll’s-eye reflex impaired |
| May be disconjugate | |
| May be internuclear ophthalmoplegia with medial moving eye moving less than the laterally moving eye) | |
| Response to calorics impaired | |
| Motor | Decorticate → bilaterally decerebrate |
| Lower Pons–Medulla Stage | |
| Respiration | Regular, shallow, and rapid (20-40/minute) |
| Pupils | Midposition (3-5 mm), fixed |
| Oculomotor | No doll’s-eye reflex |
| Response to calorics absent | |
| Motor | Flaccid, bilateral Babinski reflex |
| May be lower limb flexion in response to pain | |
| Medullary Stage (Terminal Stage) | |
| Respiration | Slow, irregular; sighs, gasps, occasional hyperpnea alternating with apnea |
| Pupils | Widely dilated and fixed |
Adapted from Greenberg MS: Coma. In Greenberg MS, ed: Handbook of Neurosurgery, 5th ed. New York: Thieme, 2001, pp 118-127. Also see Plum and Posner (1982).
Uncal herniation
The uncus of the temporal lobe herniates over the free edge of the tentorium, compressing the midbrain and the oculomotor nerve. This is usually caused by a rapidly expanding middle cranial fossa epidural, subdural, or intratemporal lobe hematoma. The earliest sign of uncal herniation is the ipsilateral dilation of the pupil. A depressed state of consciousness is not a reliable early sign, but the patient may be confused or agitated. Once the brainstem is compromised, the conscious state may deteriorate rapidly to a deep coma. The mass lesion causing the uncal herniation usually causes a contralateral hemiparesis, but as the pressure increases, the opposite cerebral peduncle is compressed against the tentorium, which causes an ipsilateral hemiparesis (Kernohan’s sign). This is recognized at autopsy as Kernohan’s notch. As in central herniation, the posterior cerebral arteries may be compressed against the edge of the tentorium, which may cause occipital lobe infarcts. The early CT findings of uncal herniation are early unilateral encroachment on the suprasellar cistern and later brainstem displacement and flattening, flattening of the contralateral cerebral peduncle, and rotation of the midbrain. Contralateral hydrocephalus may occur. The clinical stages of uncal herniation are outlined in Table 8-4.
| Early Third Nerve Stage | |
| Pupils | Unilateral dilating pupil, ipsilateral to lesion in 85% of patients; often sluggish |
| Oculomotor | Doll’s-eye reflex normal or disconjugate (failure of adduction of ipsilateral eye) |
| Caloric reflex may be disconjugate | |
| Respiration | Normal |
| Motor | Appropriate response to pain |
| Contralateral Babinski reflex may be present | |
| Late Third Nerve Stage | |
| Consciousness | Stupor → coma |
| Pupil | Fully dilated (unilateral) |
| Oculomotor | Complete third nerve palsy with pupil dilatation (>6 mm) and ophthalmoplegia |
| Respiration | Hyperventilation sustained; in rare cases, Cheyne-Stokes respiration |
| Motor | Usually contralateral weakness |
| If opposite cerebral peduncle is compressed against the tent, an ipsilateral hemiparesis (Kernohan’s sign) and then bilateral decerebrate posture develop | |
| Midbrain–Upper Pons Stage | |
| Pupils | Contralateral pupil dilates initially to midposition (5-6 mm) |
| Oculomotor | Palsy |
| Respirations | Sustained hyperpnea |
| Motor | Bilateral decerebrate rigidity |
Adapted from Greenberg MS: Coma. In Greenberg MS, ed: Handbook of Neurosurgery, 5th ed. New York: Thieme, 2001, pp 118-127. Also see Plum and Posner (1982).
The distinction between uncal and central herniation is difficult when the compression has reached the midbrain and below. Prediction of the location of the pathology on the basis of the type of herniation syndrome is unreliable, inasmuch as central and uncal herniation may occur together. Decrease in consciousness occurs early in the sequence of central herniation but later in the sequence of uncal herniation. Cushing’s reflex, consisting of hypertension, bradycardia, and respiratory irregularity, which is a feature of medullary compression, is often absent with slowly progressive supratentorial mass lesions. The cause of the coma and the third nerve palsy in cases of presumed uncal herniation is not always uncal compression, because the degree of uncal herniation on imaging or at autopsy may not be correlated with clinical state. The anatomy varies among patients, and in some cases, there is no uncal herniation or it is the contralateral pupil that dilates first, so that the mechanism of the third nerve palsy is likely to be central (i.e., intrinsic to the midbrain) in these cases. Horizontal displacement of the central supratentorial structures may also contribute to or cause the depressed conscious state (see Investigation of Coma).17
Differential Diagnosis of Coma
The differential diagnosis of coma includes (1) the “locked-in” syndrome, which may occur with ventral pontine infarction (see Chapter 9); (2) psychiatric disorder with catatonia or hysterical conversion reaction; (3) neuromuscular weakness with unreversed neuromuscular blockade, which may be encountered in the intensive care unit; (4) myasthenia gravis; and (5) Guillain-Barré syndrome.
The Investigation of Coma
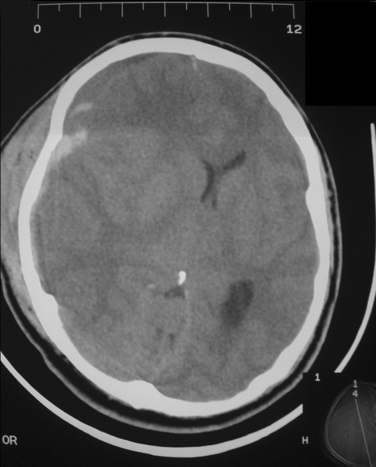
CT should be performed urgently to establish the presence of a structural lesion, determine whether there are any radiological signs of brain herniation or midline shift, and determine whether urgent neurosurgical intervention is required. Midline shift is the degree of horizontal shift of midline cerebral structures as seen on axial images and is correlated with the conscious state. Anteriorly, the midline is the septum pellucidum between the two frontal horns of the lateral ventricles. Posteriorly, it is the pineal gland, which can be identified on CT if it is calcified. A patient with midline shift is generally alert with 0- to 3-mm shift, drowsy with 3- to 4-mm shift, stuporous with 6- to 8-mm shift, and comatose with 8- to 13-mm shift (Fig. 8-4).17 This correlation of the extent of midline shift with level of consciousness is variable and also depends on the rapidity over which the shift occurred. In our experience, the rapid development of a large midline shift (>2 cm) usually results in a poor outcome or death. A slowly enlarging mass may produce marked midline shift without disturbing the conscious state. The location, as well as the size, of the mass lesion is also an important factor. A large frontal tumor may not cause significant shift, whereas a smaller temporal lobe mass might.
Other investigations that directly concern the neurologist are as follows:
Electroencephalogram
The EEG slows in traumatic coma; the amount of slowing is proportional to the depth of coma. There may be some lag before the slowing occurs. There is also a loss of electroencephalographic reactivity to external stimuli, such as noise or eye opening, and a loss of spontaneous variability of the EEG patterns. The prognosis may be better when this reactivity is not completely lost and when there are periodic sleep patterns (“spindle pattern coma”). Burst suppression is the worst pattern short of electrical silence and, unless it is drug induced (e.g., by barbiturates), it is a preterminal finding.18 Alpha coma is widespread alpha (8- to 12-Hz) activity in the presence of coma. This is present over the entire scalp and does not vary with external stimuli. This is in contrast to normal alpha rhythm, which is seen over the occipital lobes of relaxed subjects with their eyes closed and is abolished by their becoming alert. Patients with alpha coma usually have a poor prognosis. The grades of electroencephalographic abnormality have been correlated with prognosis.19,20 The use of phase and coherence data improves the accuracy of the EEG. The EEG is easily perturbed by drugs, and so the clinician must be very careful in interpreting the EEG findings in the intensive care environment.18 Subclinical seizure activity may continue after the cessation of clinical seizures and is a possible cause of persistent coma. This seizure activity may be identified through continuous electroencephalographic monitoring with a single channel with two electrodes, one of which is usually a reference electrode. Repeated or continuous multiple-lead EEGs are obtained routinely in some intensive care units.
Power Spectral Analysis
Real-time power spectral analysis of the EEG is achieved with a fast Fourier transform algorithm, resolves the EEG into its individual frequency components, and can be displayed over time.21 Power spectral analysis simplifies the interpretation of the EEG, but a display of the EEG is still necessary to detect seizures. Power spectral analysis has some predictive value; variable spectral patterns are associated with a better prognosis. An unvarying pattern with the frequency component in the delta range (1 to 3 Hz) carries a poor prognosis.22,23 The overall trend in background frequency mirrors the course of the patient over a number of days. Evoked potentials have less variability over time than does power spectral analysis and therefore are more useful.
Evoked Potentials
Evoked potentials measure the response of the cerebral hemispheres or brainstem to a sensory stimulus. Signal-averaging techniques are necessary to eliminate the background and preserve the repeated stimuli at fixed intervals. They have some value in prognostication after brain injury. Loss of wave I of the brainstem auditory evoked potentials (BAEPs) after head injury occurs if the inner ear is damaged, and therefore this loss cannot be used for prognostication in these cases. The absence of BAEPs is predictive of a poor outcome. However, the BAEPs may be normal and the outcome poor after traumatic brain injury because the integrity of the cerebral hemispheres is not measured by BAEPs. Somatosensory evoked potentials (SSEPs) have better prognostic value than BAEPs because they test the integrity of the brainstem and the cerebral hemispheres. The absence of any activity beyond wave P15 is highly predictive of death. P15 is the SSEP wave thought to arise from the caudal medulla. N20 is the first cortical peak and is thought to arise from the postcentral gyrus. The presence of SSEP activity beyond 50 to 70 milliseconds appears essential for functional survival. Activity occurring beyond 70 milliseconds has particular prognostic value for quality outcome after anoxic or traumatic brain injury. However, elderly patients may do poorly despite the prediction for a good outcome on the basis of the SSEP. The SSEPs often deteriorate over time after traumatic brain injury and may be absent with high doses of barbiturates.18 Testing for visual evoked responses is not often performed in the comatose patient but may be used to assess the integrity of the visual pathways.
BRAIN DEATH
Concepts of human death have evolved over the centuries. The ancient Greeks believed the heart was the essence of life and that absence of the heartbeat was the principal sign of death. Maimonides, the famous Jewish physician and philosopher in the 12th century, believed that breathing, not heartbeat, was the essence of life and that cessation of breathing defined death. He recognized that the decapitated body was dead: Even though there were muscle spasms, there was no central control. He believed the central control of locomotion was also as essential to life as breathing.24 The modern concept of brain death was developed in the 1950s with the advent of mechanical ventilation because patients with irreparable brain damage and apnea could have their heartbeat temporarily sustained. Mollaret and Goulon25 used the term le coma dépassé (a “state beyond coma”) to describe patients in profound coma, although they did not assert that those patients were dead. There were further reports of the same condition with varying causes, and in 1968 an ad hoc committee of the Harvard Medical School formulated criteria asserting that patients with irreversible apnea, areflexia, and complete unresponsiveness from devastating brain injury were legally dead.26 These concepts and the tests for brain death were further refined over two decades, and the declaration of brain death became a widely accepted practice in industrialized nations by the mid-1990s.24
The Whole Brain Formulation
Bernat24 defined death as the permanent cessation of function of the “organism as a whole,” which includes the coordination and integration of organ subsystems, the generation of vital functions, and the set of physiological homeostatic mechanisms. It is the “whole brain” that subserves all the clinical functions of the organism. The cerebral cortex directs higher mental function, the diencephalon is responsible for gating and initial processing of sensory input, the hypothalamus regulates homeostatic functions, and vital functions such as heart rate and respiratory drive are controlled by the brainstem. Therefore, permanent cessation of function of the whole brain is required for this formulation. Some subsystems may still be functioning under this definition of death, but they are uncoordinated and meaningless to that individual. Likewise, pockets of functioning neurons after death do not contribute to overall brain function or to the person’s function as a whole. This whole brain definition is favored by the President’s Commission for the Study of Ethical Problems in Medicine and Biomedical and Behavioral Research and is accepted by the Law Reform Commission of Canada, many states of the United States, and several European countries.24
Machado27 proposed a new standard of human death that was based on the concept that consciousness is the key human attribute and provides the highest level of control in the hierarchy of integrating functions in the human being. There must be an irreversible destruction of the anatomical and functional substrata of consciousness throughout the whole brain to diagnose brain death in this formulation. There should be unresponsiveness, no arousal to any stimuli, and no cognitive and affective functions. This definition subtly distinguishes it from the standard whole brain formulation.27 This concept has not replaced the current formulation.
The Higher Brain Formulation
The neocortex is essential for consciousness and cognition, which are essential human attributes. The cerebral cortex, not the lower centers or brainstem, subserves awareness, memory, and personal identity. Permanent loss of the neocortex is necessary and sufficient to determine death in the higher brain formulation of death. Continued functioning of the brainstem and diencephalon are irrelevant to the determination of brain death by this definition. This concept was first proposed in 1975 by Veatch28,29 and supported by others.30 According to this definition, patients in a permanent vegetative state (permanent postcoma unresponsiveness) and anencephalic infants are dead. However, there are major conceptual and practical problems with this definition. Patients with loss of neocortex are still breathing spontaneously, and most societies would therefore not declare these individuals dead. There is a “slippery slope” argument to neocortical death in that there are various degrees of cortical and subcortical death, and patients with advanced dementias may manifest a similar situation, although no one would argue that these individuals are brain dead. How would the clinician distinguish these cases? The established diagnosis of permanent vegetative state requires several assessments over a considerable time, but a diagnosis of death cannot be made in this way. The higher brain formulation determines a loss of personhood, not death. Personhood has a spiritual and psychosocial dimension, in contradistinction to the biological dimension of death.24
The Brainstem Formulation
Brain death in the United Kingdom requires the determination of permanent cessation of brainstem function. This concept was developed in the United Kingdom by Pallis8 (see also Pallis, 1983). He recognized that most of the bedside tests for brain death were tests of brainstem function. The term brainstem death indicates that the whole brain is dead, because even if the cortex or basal ganglia, were alive they would not be able to function without a functioning reticular activating system and the body’s vital functions could not be maintained. In other words, brainstem death is equivalent to brain death. This concept is stated in the U.K. Royal Colleges memorandum of 1979.31 The one conceptual flaw in this determination is the rare possibility of a patient’s being “locked-in” with a functioning cerebral cortex and no clinical evidence of brainstem function, which is not an issue with the whole brain formulation. Typically, patients locked because of pontine tegmental pathology differ from patients with brainstem death in that they still have respiratory movements and often have preserved eye opening or vertical eye movements to command. The EEG may also identify this condition, which is discussed further in Chapter 9. (For further reading, see Pallis, 1983.)
The Circulatory Formulation
It held by some conservative theologians, and has been stated by authorities in Japan, Israel, Denmark, and the Islamic countries, that death occurs only when the circulation ceases. Thus, some people and cultures do not recognize the concept of brain death.32
The Legal or Statutory Definition of Brain Death
In 1970, Kansas became the first state to incorporate brain death in a statutory definition of death. By 1993, more than 90% of states in the United States and the majority of industrialized nations had enacted legislation recognizing brain death. The American Bar Association drafted a model statute that stated in 1975, “For all legal purposes, a human body with irreversible cessation of total brain function, according to the usual and customary standards of medical practice, shall be considered dead.”24 This statute does not specify that death can be determined by the irreversible cessation of spontaneous respiratory and circulatory functions in the majority of cases. In 1981, the President’s Commission for the Study of Ethical Problems in Medicine and Biomedical and Behavioral Research developed a model statute called the Uniform Determination of Death Act: “An individual who has sustained either (1) irreversible cessation of circulatory and respiratory functions or (2) irreversible cessation of all functions of the entire brain, including the brainstem, is dead. A determination of death must be made in accordance with acceptable medical standards.”33 Bernat and associates34 proposed a modification to this statement so that the primary definition was that an individual who has sustained irreversible cessation of all functions of the entire brain, including the brainstem, is dead and that this can be determined (1) in the absence of cardiopulmonary support by prolonged absence of respiratory and circulatory function or (2) in the presence of artificial cardiopulmonary support by the tests of brain function.34
The Diagnosis of Brain Death
The United Kingdom Guidelines for Brain Death
The criteria for the diagnosis of brain death were published by the U.K. Conference of the Medical Royal Colleges in 197635,36 and were further confirmed in a memorandum of the U.K. Conference of the Medical Royal Colleges in 1979,31 in which it was stated that death could be declared once the criteria were satisfied. The diagnosis of brain death is clinical and does not require any confirmatory laboratory or imaging tests.
The confirmatory tests for brainstem death are simple to perform and should be carried out by two independent senior medical practitioners no less than half an hour apart. (The Australia and New Zealand Intensive Care Society recommends that the two examinations be done at least 2 hours apart.37) The U.K. criteria specify that one examiner be a consultant and the other a senior registrar or consultant. These tests are outlined in Table 8-5. They are essentially tests of brainstem reflexes and are entirely clinical. The time of death is arbitrarily determined at the completion of the second examination. The Royal College of Physicians reviewed the U.K. criteria in 1995 and endorsed the original recommendations.38 Physicians in Australia follow the U.K. criteria.
TABLE 8-5 Confirmatory Tests for Brainstem Death: The United Kingdom Guidelines
Paco2, arterial partial pressure of carbon dioxide.
The United States Guidelines for Brain Death
The U.S. guidelines for brain death were presented by the medical consultants on the diagnosis of death to the President’s Commission for the Study of the Ethical Problems in Medicine and Biomedical and Behavioral Research in 1981.33,39 These guidelines were updated and clarified in 1995 by the Quality Standards Committee of the American Academy of Neurology40 and have achieved wide acceptance in the United States. Brain death is defined as the irreversible loss of function of the brain, including the brainstem. In summary, there are three parts to the diagnostic criteria for clinical diagnosis of brain death. These are outlined in detail in Table 8-6.

TABLE 8-6 Diagnostic Criteria for Clinical Diagnosis of Brain Death: The United States Guidelines
Rights were not granted to include this table in electronic media. Please refer to the printed book.
From Practice parameters for determining brain death in adults (summary statement). The Quality Standards Subcommittee of the American Academy of Neurology. Neurology 1995; 45:1012-1014.
Prerequisites
Cardinal Findings
The three cardinal findings in brain death are coma, absence of brainstem reflexes, and apnea. The examination of brainstem reflexes and testing for apnea are similar to the U.K. criteria, but the guidelines in Table 8-6 should be referred to for the details.
Apnea Testing
Pitfalls in the diagnosis of brain death in the U.S. guidelines are stated as follows:
Clinical Observations Compatible with the Diagnosis of Brain Death
Spinal reflexes may persist after brainstem death is diagnosed and may include movements of the body in response to light peripheral stimulation or to flexion of the neck or rotation of the body. These tend to occur at the time of the apnea test, during preparation for transport, at the time of abdominal incision for organ transplantation, and in the morgue. These movements involve withdrawal of the lower limbs, raising of the arms independently of each other, abduction or adduction of the arms, head rotation, back arching, and even attempts to sit to 40 to 60 degrees. These are called Lazarus signs and are usually single events. Deep tendon reflexes, abdominal reflexes, and the Babinski sign may persist. There may also be skin flushing, shivering, sweating, and myoclonic twitching of limb muscles (see Wijdicks, 2001). There is also the maintenance of blood pressure and even some hypertensive response during donor nephrectomy, which may in part result from adrenal medullary stimulation by a reflex spinal arc.37,41
Patients with brain death develop gross vascular regulatory disturbance and a diffuse metabolic cellular injury that leads inexorably to organ failure and eventual cardiac arrest.10 The vascular regulatory disturbance results initially from extreme sympathetic stimulation and, in a second phase, from a failure of sympathetic outflow, which leads to hypotension and reduced cardiac output. This results in impaired autoregulation with vasodilation at the organ level. The cellular injury with a global mitochondrial dysfunction may result part from the hormonal deficiency (e.g., triiodothyronine) from loss of hypothalamic control.10 Myocardial and renal ischemia commonly result. Neurogenic pulmonary edema and coagulopathy may also occur. Patients with a high chance of proceeding to brain death frequently develop hypotension, and cardiac arrest may occur despite active support.10 However, brain death does not always rapidly lead to somatic death. If hemodynamic parameters in patients with brain death are maintained with norepinephrine/epinephrine, cardiac standstill usually occurs within 48 hours, but this period can be extended to a mean of 23 days with the addition of arginine vasopressin.42 Anterior pituitary dysfunction occurs variably after brain death with falls in triiodothyronine, thyroxine, cortisol, prolactin and follicle-stimulating hormone levels. However, thyroid-stimulating hormone and adrenocorticotropic hormone levels may remain normal. Posterior pituitary failure causing diabetes insipidus is common after brain death.10 Hypothalamic failure results in hypothermia.
What is the validity of the criteria for the diagnosis of brain death? There has never been a case reported in which a person has recovered when the U.K. criteria were satisfied.43 Some intensive care staff and relatives are confused by the presence of spinal reflexes in the patient who is brain dead. These may increase as the brain-dead patient is maintained on mechanical ventilation. Some explanation to these staff and relatives is required. The diagnosis of brain death is now widely accepted in industrialized countries by both hospital staff and the lay public.
The Confirmatory Laboratory Tests for Brain Death
These investigations are not a substitute for clinical examination except if the full clinical examination cannot be carried out, as in gross facial trauma, and should not precede it. They are used to confirm the diagnosis. The U.S. guidelines state that these confirmatory laboratory tests are desirable for patients in whom specific components of the clinical testing cannot be reliably performed or evaluated. In order of sensitivity, these tests as stated in the U.S. guidelines are conventional angiography, electroencephalography, transcranial Doppler ultrasonography, radionuclide brain scan, and SSEPs.40
Cerebral angiography and radionuclide brain scan (Figs. 8-5 and 8-6)
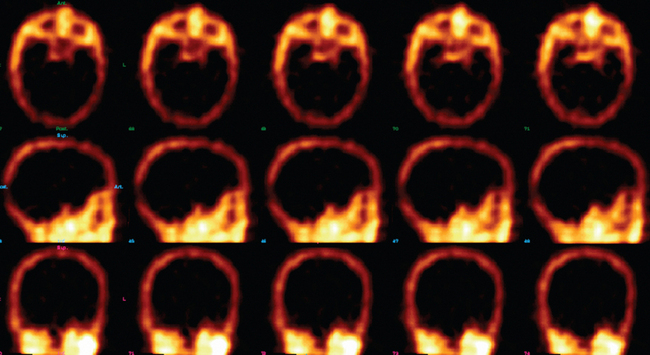
Blood flow tests such as angiography or radionuclide brain scans show no entry of contrast material or isotope into the brain when either is injected systemically into the brain-dead patient. These tests can usually be performed rapidly and are being used in some centers, in cases in which the clinical diagnosis cannot be confirmed, and in some children. Blood flow studies obviate the need for awaiting the elimination of sedatives such as barbiturates, which can linger for days and delay the diagnosis of brain death, and can be performed in patients with metabolic causes of brain death, in whom the etiology of brain death is unclear.44 The confirmation of diagnosis with imaging is of value in optimizing the timing of organ harvesting, but the use of these confirmatory tests is somewhat controversial and is not universally practiced.
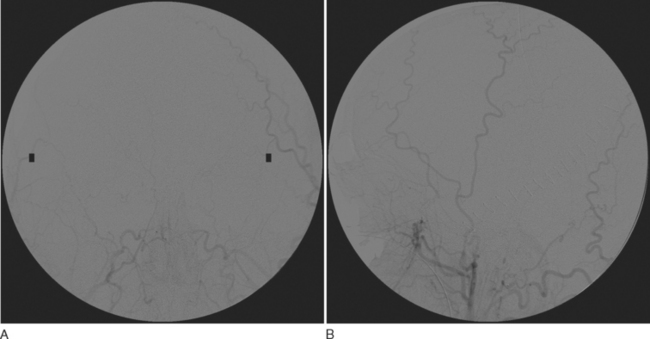
Cerebral angiography has been considered the final determinant for confirming the diagnosis of brain death. Blood flow must be absent from the anterior and posterior circulations. The blood flow in the internal carotid stops abruptly in the petrous carotid at the skull base. The carotid siphon does not fill. The vertebral artery flow stops at the atlanto-occipital junction. The external carotid flow remains patent and fills early. A criticism of the technique is that a subintimal injection may produce a false block to the artery, but this is unlikely in more than one vessel and should not alter a diagnosis made on the basis of a four-vessel angiogram. Correct positioning of the head and correct rate of contrast material injection are important points of technique that, if not adhered to, may introduce some contrast material into cerebral vessels, producing artifactual cerebral blood flow. The contrast material may injure transplantable organs or may reduce remaining cerebral blood flow. The angiogram should be obtained twice at an interval of 20 minutes to make sure the first result is not artifactual. Intracranial blood flow may still be present if the study is done very early after the diagnosis of brain death, particularly if the mechanism did not involve raised intracranial pressure. In cases in which the supratentorial intracranial pressure is raised, the posterior circulation may be present but the carotid flow is absent, because the pressure from the supratentorial compartment has not yet been completely transmitted to the infratentorial compartment.44–46
A radionuclide brain scan showing the brainstem and supratentorial circulation is an alternative to cerebral angiography in diagnosis of brain death.47 The radionuclide test for brain death is reliable, safe, and rapid. Intravenous injection of technetium 99m (Tc-99m) hexamethylpropylene-amine oxime or iodine 123 iodoamphetamine, which cross the blood-brain barrier, results in their accumulation by functioning brain cells, in which they are held for several hours. There is no uptake of these agents when there is widespread neuronal death. These agents are preferred to the blood pool radionuclides (Tc-99m pertechnetate, Tc-99m diethylenetriaminepentaacetic acid, and Tc-99m glucoheptonate), which do not cross the blood-brain barrier, may not demonstrate the state of blood flow in the posterior fossa on standard scans, and appear to produce more artifact. However, the addition of single photon emission computed tomography can give precise regional information and show whether there is any preservation of posterior fossa blood flow.44 Absence of uptake produces a characteristic “hollow skull” or “empty light bulb” appearance (see Wijdicks, 2001).
Electroencephalography
An EEG is not necessary for making the diagnosis of brain death. It is optional39 but is still used in many countries as a confirmatory test of brain death. A 16- to 18- channel instrument should be used for at least 30 minutes of recording. Electrical activity above 2 μV is absent at a sensitivity of 2 μV/mm with the low filter setting at less than 1 Hz and the high filter setting at 70 Hz. The sensitivity and specificity are about 90%. However, if brainstem death is used for diagnosis of brain death, the absence of electrical activity on an EEG is not very helpful because it reflects cortical activity rather than brainstem activity and may show some activity even though all the criteria for brainstem death have been met. The electroencephalographic findings are therefore irrelevant when brainstem death is used to signify brain death8 (see Wijdicks, 2001). It can also be difficult to achieve a flat trace with all the electronic devices around the patient and if high-gain amplification is used. Nonetheless, a flat trace may help the patient’s relatives accept the diagnosis of brain death.
Evoked potentials
Both BAEPs and SSEPs can be used as confirmatory tests for brain death, but they have a rather poor predictive value. An intact auditory nerve (wave I) is required for BAEP interpretation. In addition, BAEPs are not well correlated with the severity of brain injury. Absence of waves II to V indicates profound brainstem dysfunction; however, some waves may be present in patients who are brain dead, with absence of brainstem reflexes (see Wijdicks, 2001).
The median nerve SSEP cortical wave (N20) is typically absent bilaterally in brain death, but it is also absent in 15% to 20% of patients who are comatose but not brain dead. Nevertheless, bilaterally absent SSEPs have an extremely high positive predictive value for failure to recover beyond permanent vegetative state (see Chapter 9). Similar uncertainties arise from the absence of the N18 wave, which is possibly generated in the cuneate nucleus (see Wijdicks, 2001). A single-wave SSEP with a mean latency of 9 milliseconds may be recorded at the level of C2 in patients with brain death. This potential may arise near the cervicomedullary junction and may indicate residual medullary function, but this does not negate the diagnosis of brain death. Erb’s point potentials must be present for the absence of cortical potentials to be interpretable. The evoked potentials may also be contaminated by potentials that are time locked to the stimulus but generated by extracranial sources.
Ultrasonography
Transcranial Doppler ultrasonography is a noninvasive, quick bedside test. The characteristic features of brain death on transcranial Doppler ultrasonography are an oscillating movement of the blood column within arteries, short systolic spikes, and an absence of signal in patients in whom it was previously found to be present. Specificity and sensitivity have been found to be as high as 100% and 90% to 99%, respectively. The anterior and posterior circulations and the extracranial vessels must be studied on both sides to minimize false-positive findings. Patients with a large craniotomy or ventricular drains do not undergo this evaluation44 (also see Wijdicks, 2001). The World Federation of Neurology published a consensus opinion for the use of ultrasonography for the diagnosis of brain death.49
Computed tomography
CT is performed with and without contrast material and may show the cause of the coma. After injection of contrast material, there is no increase in brain attenuation in the brain parenchyma of patients with brain death, nor is there any significant opacification of the vessels of the circle of Willis. Identification of contrast material in the extracranial soft tissue, the cervical great vessels, or the kidneys should be seen in order to confirm that enough contrast material has been administered. CT angiography can be used to show an absence of flow in the intracranial vessels. Xenon inhalation has been added to CT to detect cerebral blood flow; in the patient with brain death, there is minimal (<1 to 5 mL/100 g/minute) or no cerebral blood flow.44
Magnetic resonance imaging
It is not often practicable to obtain magnetic resonance imaging in patients with brain death. Such imaging reveals transtentorial and tonsillar herniation, lack of intracranial flow void, poor gray-white differentiation, and marked contrast enhancement of the nose and scalp. Magnetic resonance angiography, similar to conventional angiography, shows no flow in the intracranial vessels in brain-dead patients. Magnetic resonance spectroscopy reveals high lactate and choline levels and decreased N-acetyl aspartate levels44 (also see Wijdicks, 2001).
The Differential Diagnosis of Brain Death
Disorders that mimic brain death include hypothermia, acute poisoning, and acute metabolic encephalopathies. It should be possible to rule out these conditions after a detailed assessment of the patient. In rare cases, the coning process or pathology such as hemorrhage spares the medulla, and there is retained respiratory and cardiovascular function in the absence of other brainstem reflexes. Such patients have “isolated medulla oblongata” and do not meet criteria for brain death (see Wijdicks, 2001).
The related neurological disorders akinetic mutism, persistent or permanent vegetative state (postcoma unresponsiveness), and the “locked-in” syndrome caused by pontine tegmental disruption have some of the features of brain death, but respiration is preserved. These conditions are discussed further in Chapter 9.
Brain Death in Neonates and Children
The tests for brain death in young children and neonates have not been as thoroughly validated as in adults. The diagnosis of brain death in children should not be made in the first 7 days after birth. Confirmatory tests, in addition to the clinical tests, are required in children up to 12 months of age. From 7 days to 2 months of age, two isoelectric EEGs or two radionuclide studies showing absence of intracranial uptake, 48 hours apart, are recommended. From 2 to 12 months of age, this interval need be only 24 hours. In children older than 12 months, the adult criteria can be used with up to 12 hours of observation without the need for electroencephalographic confirmation.50,51 There remains controversy in determination of brain death in the infant younger than 2 months because the clinical test results are difficult to interpret: the blood pressure and the duration and severity of the insult are often uncertain, and the degree of brain damage is difficult to determine on imaging.52 The determination of brain death in the infant is of relevance only for organ donation. Cessation of treatment for the neonate or infant with irreparable brain damage and ahopeless prognosis is common practice.
There is controversy as to whether anencephalic newborns are brain dead and can proceed to multiple-organ donation.24 These infants have no cerebral hemispheres but do have a variably functioning brainstem and therefore, according to the definitions and concepts of brain death described previously, are technically not brain dead. Approximately 65% of anencephalic fetuses die in utero, and most anencephalic liveborn infants die in the first few days. Only about 5% are still alive at 1 week.53 Few of these infants die of brain death; most succumb to respiratory failure, cardiac arrhythmia, or sepsis. Our opinion (and that of Bernat24) is that it is not ethically acceptable to take the organs if these infants are still alive. These infants should be declared dead before their organs can be procured.
Maternal Brain Death and Live Birth
It is possible to keep a brain-dead pregnant women alive on a ventilator many weeks until her fetus is mature enough for delivery. Is it ethically acceptable to do this in order to save the child? In this situation, there is a conflict of interest between the mother and the fetus. Usually the mother’s interests are placed before those of the fetus, so that abortion is permitted if the mother’s health is in jeopardy. However, in this case, the mother is dead; therefore, even though there is indignity to the mother, delay in determination of maternal death and burial, prolonged emotional trauma for the family, and financial costs, it is ethically justified for the interests of the fetus to be placed first. This applies only if the obstetrician believes there is a reasonable chance that the fetus will reach maturity. The closer to term the fetus is, the more likely there will be a healthy delivery. The father should be able make the final decision on the basis of all the information provided. The mother’s wishes, if stated in advance, also need to be considered.24
Research and Teaching with Brain-Dead Patients
The research team should not be involved in the brain death determination. LaPuma54 has formulated ethical guidelines for experimentation that, in summary, are as follows: The dignity and humanity of the body should not be violated. The experiment should be well designed and brief. The patient must be declared brain dead. Voluntary and knowledgeable consent must be given by the next of kin. There should be a clear medical importance to the experiment that will yield valuable information, such as a safe or efficacious treatment for a lethal disease. The institutional ethics review board must sanction the research. The investigators pay for the extra time that the patient spends in the intensive care unit.24,54
Religious and Family Attitudes toward Brain Death
Although a religious definition of death may depend on the loss of the soul from the body, this concept is not suitable for medical purposes.24 Judeo-Christian religions have generally accepted the concept of brain death. However, some Catholic and Orthodox Jewish scholars have not accepted it, arguing that brain death is not equivalent to death determined by cardiopulmonary criteria because brain life is not equivalent to human life.24 There is differing opinion among rabbis; some say that according to ancient Jewish law (the Halachah), death can be determined only by prolonged cessation of the breathing and heartbeat, which represent life itself, the only exception being decapitation. Other rabbis have argued that brain death is consistent with Halachic law because brain death is the functional equivalent of decapitation.24 Muslims assert that they do not have the right to determine the timing of death—that is, when to terminate support. The Third International Conference of Islamic Jurists considered brain death to be death of a person. Legal death and Sharia principles apply when the following signs are established (see Wijdicks, 2001, page 136):
The family may firmly believe that, on religious grounds, brain death is not the true death of their relative and that the ventilator should not be turned off. If this objection is not based on emotional or psychological difficulty in accepting the diagnosis and prognosis, the physician should accept the family’s objections. This does not exclude the withdrawal of some support, such as vasopressor agents, in which case asystole will occur within hours or days. Some states, such as New Jersey, have enacted laws to provide personal religious exemption for the family that does not accept the diagnosis of brain death and desires that ventilation be continued.24
The Management of Brain Death
The family must be fully informed of the likely outcome, and the doctor should try to establish that the relatives are making decisions on the basis of the patient’s wishes rather than their own. It is best if the patient has made known a directive about brain death, as well as his or her wishes regarding organ donation, before death, but this does not often happen.55
Organ Procurement
The demand for organ donors has been a main driver for the acceptance and legalization of brain death. Families consenting to organ donation may derive considerable benefit in knowing that some good has come from the tragedy of the death of their loved one. However, there is a common fear held by family members that the diagnosis of brain death will be made prematurely and that organs will be procured while the patient is still alive. There should be no perception by the family of a conflict of interest by the staff. The family should be given sufficient time to accept the diagnosis of brain death before any discussion of organ donation. It is essential that the staff members who raise the issue of organ donation with the family are independent of the physicians treating the patient and that they not approach the family until the diagnosis of brain death is made by the treating physicians. The next of kin has the right to refuse the organ donation even if the patient has declared previously that he or she wishes to be a donor. It must be made very clear to the family that there can be no organ donation procedure unless the diagnosis of brain death is absolutely confirmed and that even if the family chooses not to proceed with organ donation, the ventilation and support will still cease. If the organ donation does not proceed, the patient is extubated after the second set of brain death tests. It is considered ethically acceptable to continue intravenous fluids and to maintain the blood pressure and ventilation of the patient who is declared brain dead until the organs are procured, as long as the delay is not excessive. A model policy on organ donation has been developed by the Australia and New Zealand Intensive Care Society.37
Edelman GM. Wider than the Sky. The Phenomenal Gift of Consciousness. New Haven, CT: Yale University Press, 2004.
Jasper HH, Descarries L, Castellucci VF, et al, editors. Advances in Neurology, Vol 77: Consciousness: At the Frontiers of Neuroscience. Philadelphia: Lippincott Raven, 1998.
John ER. The neurophysics of consciousness. Brain Res Rev. 2002;39:1-28.
Metzinger T, editor. Neural Correlates of Consciousness. Empirical and Conceptual Questions. A Bradford Book. Cambridge, MA: MIT Press, 2002.
Pallis C. ABC of Brainstem Death. London: British Medical Journal Publishers, 1983.
Penfield W. The cerebral cortex and consciousness. The Harvey Lectures 1937, 35–39. In: Wilkins RH, editor. Neurosurgical Classics. American Association of Neurological Surgeons; 1992:226-241.
Plum F, Posner JB. The Diagnosis of Stupor and Coma, 3rd ed. Philadelphia: FA Davis, 1982.
Wijdicks EFM. Brain Death. Philadelphia: Lippincott, Williams & Wilkins, 2001.
Young GB, Ropper AH, Bolton CF. Coma and Impaired Consciousness. A Clinical Perspective. New York: McGraw-Hill, 1998.
Zeman A. Consciousness [Invited review]. Brain. 2001;124:1263-1289.
1 Edelman GM. Wider than the Sky. The Phenomenal Gift of Consciousness. New Haven, CT: Yale University Press, 2004.
2 Crick F, Koch C, Kreiman G, Fried I. Consciousness and neurosurgery. Neurosurgery. 2004;55:273-282.
3 Valatx JL. Disorders of consciousness: anatomical and physiological mechanisms. Adv Tech Standards Neurosurg. 2004;29:3-22.
4 Dandy W. The location of the conscious center in the brain: The corpus striatum. Bull Johns Hopkins Hosp. 1946;79:34-58.
5 Young GB. Consciousness. In: Young GB, Ropper AH, Bolton CF, editors. Coma and Impaired Consciousness. A Clinical Perspective. New York: McGraw-Hill; 1998:3-37.
6 Moruzzi G, Magoun HW. Brain stem reticular formation and activation of the EEG. Electroencephalogr Clin Neurophysiol. 1949;1:455.
7 Franks NP, Lieb WR. The role of NMDA receptors in consciousness: what can we learn from anesthetic mechanisms? In: Metzinger T, editor. Neural Correlates of Consciousness. Empirical and Conceptual Questions. A Bradford Book. Cambridge, MA: MIT Press; 2002:265-269.
8 Pallis C. Further thoughts on brainstem death. Anaesth Intensive Care. 1995;23:20-23.
9 Plum F, Posner JB. The Diagnosis of Stupor and coma, 3rd ed. Philadelphia: FA Davis, 1982.
10 Power BM, Van Heerden PV. The physiological changes associated with brain death—current concepts and implications for treatment of the brain dead donor. Anaesth Intensive Care. 1995;23:26-36.
11 Teasdale G, Jennett B. Assessment of coma and impaired consciousness. A practical scale. Lancet. 1974;2:81-84.
12 Jennett B, Teasdale G, Galbraith J, et al. Severe head injuries in three countries. J Neurol Neurosurg Psychiatry. 1977;40:291-298.
13 Teasdale G, Jennett B. Assessment and prognosis of coma after head injury. Acta Neurochir (Vienna). 1976;34:45-55.
14 Simpson DA, Cockington RA, Hanieh A, et al. Head injuries in young children: the value of the Paediatric Coma Scale. Review of literature and report on a study. Childs Nerv Syst. 1991;7:183-190.
15 Simpson DA, Reilly PL. Paediatric Coma Scale [Letter]. Lancet. 1982;2:450.
16 Ropper AH. Transtentorial herniation. In: Young GB, Ropper AH, Bolton CF, editors. Coma and Impaired Consciousness. A Clinical Perspective. New York: McGraw-Hill; 1998:119-129.
17 Ropper AH. Lateral displacement of the brain and level of consciousness in patients with an acute hemispheral mass. N Engl J Med. 1986;314:953-958.
18 Moulton RJ. Electrical function monitoring. In: Reilly PL, Bullock R, editors. Head Injury. Pathophysiology and Management of Severe Closed Head Injury. London: Chapman & Hall; 1997:229-240.
19 Synek VM. EEG abnormality grades and subdivisions of prognostic importance in traumatic and anoxic coma in adults. Clin Electroencephalogr. 1988;19:160-166.
20 Synek VM. Revised EEG coma scale in diffuse acute head injuries in adults. Clin Exp Neurol. 1990;27:99-111.
21 Bickford RG. Computer analysis of background activity. In: Remond A, editor. Electroencephalography Informatics: A Didactic Review of Methods and Applications of Electroencephalography Data Processing. Amsterdam: Elsevier; 1977:215-232.
22 Bricolo A, Turazzi S, Faccioli F, et al. Clinical application of compressed spectral array in long-term EEG monitoring of comatose patients. Electroencephalogr Clin Neurophysiol. 1978;45:211-225.
23 Sironi VA, Ravagnati L, Signoroni G, et al. Diagnostic and prognostic value of EEG compressed spectral analysis in post-traumatic coma. In: Villani RJ, Papo I, Giovanelli M, et al, editors. Advances in Neurotraumatology. Amsterdam: Excerpta Medica; 1982:328-330.
24 Bernat JL. Brain death. In: Bernat JL, editor. Ethical Issues in Neurology. Newton, MA: Butterworth-Heinemann; 1994:113-143.
25 Mollaret P, Goulon M. Le coma dépassé. Rev Neurol. 1959;101:3-15.
26 A definition of irreversible coma. Report of the Ad Hoc Committee of the Harvard Medical School to Examine the Definition of Brain Death. J Am Med Assoc. 1968;205:337-340.
27 Machado C. Is the concept of brain death secure? In: Zeman A, Emanuel L, editors. Ethical Dilemmas in Neurology. London: WB Saunders; 2000:193-212.
28 Veatch RM. Defining death: the role of brain function [Editorial]. JAMA. 1979;242:2001-2002.
29 Veatch RM. The whole-brain oriented concept of death: an outmoded philosophical formulation. J Thanatol. 1975;3:13-30.
30 Shann F. A personal comment: whole brain death versus cortical death. Anaesth Intensive Care. 1995;23:14-15.
31 Conference of Medical Royal Colleges and their Faculties in the UK. Memorandum on the diagnosis of death. BMJ. 1979;1:322.
32 Pallis C. The position in the USA and elsewhere. ABC of brain stem death. BMJ. 1983;286:209-210.
33 President’s Commission for the Study of Ethical Problems in Medicine and Biomedical and Behavioral Research. Defining Death. Medical, Ethical and Legal Issues in the Determination of Death. Washington, DC: U.S. Government Printing Office, 1981;160.
34 Bernat JL, Culver CM, Gert B. On the definition and criterion of death. Ann Intern Med. 1981;94:389-394.
35 Conference of Medical Royal Colleges and their Faculties of the UK. Diagnosis of death. Statement issued by the honorary secretary of the Conference of Medical Royal Colleges and their Faculties in the United Kingdom on 11 Oct 1976. BMJ. 1976;1:322.
36 Conference of Medical Royal Colleges and their Faculties of the UK. Diagnosis of brain death. BMJ. 1976;2:1187-1188.
37 Pearson IY. Australia and New Zealand Intensive Care Society Statement and Guidelines on brain death and model policy on organ donation. Anaesth Intensive Care. 1995;23:104-108.
38 Royal College of Physicians. Criteria for the diagnosis of brainstem death. J R Coll Physicians. 1995;29:381-382.
39 Guidelines for the determination of death. Report of the medical consultants on the diagnosis of death to the President’s Commission for the Study of Ethical Problems in Medicine and Biomedical and Behavioral Research. JAMA. 1981;246:2184-2186.
40 Practice parameters for determining brain death in adults (summary statement). The Quality Standards Subcommittee the American Academy of Neurology. Neurology. 1995;45:1012-1014.
41 Wetzel R, Setzer N, Stiff JL, et al. Hemodynamic responses in brain dead organ donor patients. Anesth Analg. 1985;64:125-128.
42 Yoshioka T, Sugimoto H, Uenishi M, et al. Prolonged hemodynamic maintenance by the combined administration of vasopressin and epinephrine in brain death. Neurosurgery. 1986;18:565-567.
43 Pallis C. Brain stem death. In: Braakman R, editor. Handbook of Clinical Neurology, Volume 13: Head Injury. Amsterdam: Elsevier; 1990:441-496.
44 Monsein LH. The imaging of brain death. Anaesth Intensive Care. 1995;23:44-50.
45 Rosenklint A, Jorgensen PB. Evaluation of angiographic methods in the diagnosis of brain death. Correlation with local and systemic blood pressure and intracranial pressure. Neuroradiology. 1974;7:215-219.
46 Vatne K, Nakstad P, Lundar T. Digital subtraction angiography (DSA) in the diagnosis of brain death. A comparison of conventional cerebral angiography with intravenous and intraarterial DSA. Neuroradiology. 1985;27:155-157.
47 Reid RH, Gulenchyn KY, Ballinger JR. Clinical use of technetium-99m HM-PAO for determination of brain death. J Nucl Med. 1989;30:1621-1626.
48 Chatrian GE. Electrophysiological evaluation of brain death: a critical appraisal. In: Aminoff MJ, editor. Electrodiagnosis in Clinical Neurology. Edinburgh: Churchill Livingstone, 1980. chapter 7.
49 Durocq X, Hassler W, Moritake K, et al. Consensus opinion on the diagnosis of cerebral circulatory arrest using Dopplersonography: Task Force Group on Cerebral Death of the Neurosonology Research Group of the World Federation of Neurology. J Neurol Sci. 1998;159:145-150.
50 Conference of Medical Royal Colleges and their Faculties in the UK. Report of the Working Party on Organ Transplantation in Neonates. London: Department of Health and Social Security, 1988.
51 Task Force for the Determination of Brain Death in Children. Guidelines for the determination of brain death in children. Arch Neurol. 1987;44:587-588.
52 Volpe JJ. Brain death determination in the newborn. Pediatrics. 1987;80:293-297.
53 Medical Task Force on Anencephaly. The infant with anencephaly. N Engl J Med. 1990;322:669-674.
54 La Puma J. Discovery and disquiet: research on the brain dead. Ann Intern Med. 1989;109:606-608.
55 Emanuel LL, Barry MI, Stoeckle JD, et al. Advance directives for medical care: a case for greater use. N Engl J Med. 1991;324:889-895.






