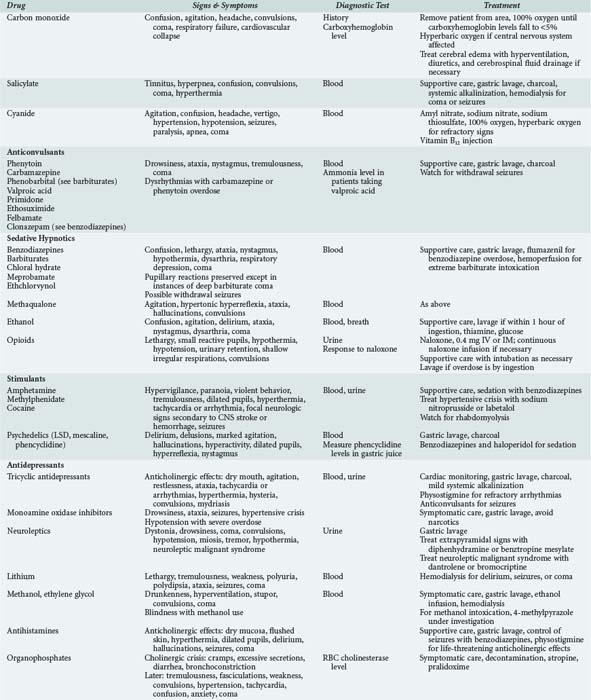
32 Coma
 Anatomy, Pathology, Pathophysiology
Anatomy, Pathology, Pathophysiology
Consciousness depends upon an intact ARAS in the brainstem and adjacent thalamus, which acts as the alerting or awakening element of consciousness, together with a functioning cerebral cortex of both hemispheres, which determines the content of that consciousness.1,2 The ARAS lies within a more or less isodendritic core that extends from the medulla through the tegmentum of the pons to the midbrain and paramedian thalamus. The system is continuous caudally with the reticular intermediate gray matter of the spinal cord and rostrally with the subthalamus, hypothalamus, anterior thalamus, and basal forebrain.3 The ARAS itself arises within the rostral pontine tegmentum and extends across the mesencephalic tegmentum and its adjacent intrathalamic nuclei. ARAS functions and interconnections are considerable and likely contribute more than only a cortical arousal system. The specific role of the various links from the reticular formation to the thalamus has yet to be fully identified.4 Furthermore, the cortex feeds back on the thalamic nuclei to contribute an important loop that amplifies arousal mechanisms.5,6
The ascending arousal system contains cholinergic, monoaminergic, and γ-amino butyric acid (GABA) systems, none of which has been identified as the arousal neurotransmitter.2,7,8 Acute structural damage to, or metabolic/chemical disturbance of, either the ascending brainstem-thalamic activating system or the thalamocorticothalamic loop can alter the aroused attentive state. Consciousness depends on continuous interaction between the mechanisms that provide arousal and awareness. The brainstem and thalamus provide the activating mechanism, and the cerebrum provides full cognition and self-excitation. Content of consciousness is best regarded as the amalgam and integration of all cognitive function that resides in the thalamocortical circuits of both hemispheres. Altered awareness is due to disruption of this cortical activity by diffuse pathology. Focal lesions of the cerebrum can produce profound deficits such as aphasia, alexia, amnesia, and hemianopsia, but only diffuse bilateral damage sparing the ARAS and diencephalon can lead to wakeful unawareness. Thus there are two kinds of altered consciousness: (1) altered arousal due to dysfunction of the ARAS-diencephalon and (2) altered awareness due to bilateral diffuse cerebral hemisphere dysfunction.
Four major pathologic processes can cause such severe global, acute reductions of consciousness.1,9 (1) In the presence of diffuse or extensive multifocal bilateral dysfunction of the cerebral cortex, the cortical gray matter is diffusely and acutely depressed or destroyed. Concurrently, cortical-subcortical physiologic feedback excitatory loops are impaired, with the result that brainstem autonomic mechanisms become temporarily profoundly inhibited, producing the equivalent of acute “reticular shock” below the level of the lesion. (2) Direct damage to a paramedian upper brainstem and posterior-inferior diencephalic ascending arousal system blocks normal cortical activation. (3) Widespread disconnection between the cortex and subcortical activating mechanisms acts to produce effects similar to both conditions 1 and 2. (4) Diffuse disorders, usually metabolic in origin, concurrently affect both the cortical and subcortical arousal mechanisms, although to a different degree according to the cause.
Structural Lesions Causing Coma
Two herniation syndromes demonstrate the mechanism by which supratentorial lesions produce coma. The rate of evolution of a mass dictates whether the anatomic distortion precedes (in slowly evolving lesions) or parallels the patient’s deterioration of wakefulness. Transtentorial herniation can be central or predominantly unilateral. Central herniation results from caudal displacement by deep midline supratentorial masses, large space-occupying hemisphere lesions, or large uni- or bilateral compressive extraaxial lesions with compression of the ARAS. The progressive rostral-caudal pathologic and clinical stages of this herniation syndrome were outlined.1 Pathologically, bilateral symmetric displacement of the supratentorial contents occurs through the tentorial notch into the posterior fossa. Alertness is impaired early, pupils become small (to 3 mm) and reactive, and bilateral upper motor neuron signs develop. Cheyne-Stokes breathing, grasp reflexes, roving eye movements, or depressed escape of oculocephalic reflexes are the clinical manifestations. In the absence of effective therapy at this diencephalic stage, herniation progresses caudally to compress the midbrain, leading to a deep coma and fixed midposition (3-5 mm) pupils, signifying both sympathetic and parasympathetic interruption. Spontaneous eye movements cease, and oculovestibular and oculocephalic reflexes become difficult to elicit. Spontaneous extensor posturing may occur. Once this stage is reached, full recovery becomes unlikely. As the caudal compression-ischemia process advances, pontine and medullary function becomes destroyed, with variable breathing patterns and absent reflex eye movements. Finally, autonomic cardiovascular and respiratory functions cease as medullary centers fail.
Uncus herniation results from laterally placed hemisphere lesions, particularly of the temporal lobes, that cause side-to-side cerebral displacement as well as transtentorial herniation. Focal hemisphere dysfunction (hemiparesis, aphasia, seizures) precedes unilateral (usually ipsilateral) compression paralysis of the third cranial nerve. An early sign of uncus herniation is an ipsilateral (rarely contralateral) enlarged pupil that responds sluggishly to light, followed by a fixed, dilated pupil and an oculomotor palsy (eye turned downward and outward).1 The ipsilateral posterior cerebral artery can become compressed as it crosses the tentorium and causes ipsilateral occipital lobe ischemia. Progressively, the temporal lobe compresses the midbrain, with loss of arousal and bilateral or contralateral extensor posturing. Ipsilateral to the intracranial lesion, a hemiparesis may develop if the opposite cerebral peduncle becomes compressed against the contralateral tentorial edge (Kernohan notch). Abnormal brainstem signs become symmetric, and herniation proceeds in the same pattern seen with central herniation as rostrocaudal brainstem displacement progresses.
Extraaxial posterior fossa lesions cause coma by direct compression of the ARAS in the brainstem, and in the diencephalon by upward transtentorial herniation. Compression of the pons may be difficult to distinguish from intrinsic lesions but is often accompanied by headache, vomiting, and hypertension due to a Cushing reflex. Upward herniation at the midbrain level is initially characterized by coma, reactive miotic pupils, asymmetrical or absent caloric eye responses, and decerebrate posturing; caudal-rostral brainstem dysfunction then occurs, with midbrain failure and midposition fixed pupils.10 Causes of brainstem compression include cerebellar hemorrhage, infarction and abscess, rapidly expanding cerebellar or fourth-ventricle tumors, or less commonly, infratentorial epidural or subdural hematomas. Drainage of the lateral ventricles to relieve obstructive hydrocephalus due to posterior fossa masses can potentially precipitate acute upward transtentorial herniation.11,12
Downward herniation of the cerebellar tonsils through the foramen magnum causes acute medullary dysfunction and abrupt respiratory and circulatory collapse. Less severe impaction of the tonsils in the foramen magnum can lead to obstructive hydrocephalus and consequent bihemispheric dysfunction with altered arousal. Clinical manifestations include headache, nausea, vomiting, lower cranial nerve signs, vertical nystagmus, ataxia, and irregular breathing. Lumbar puncture in this setting carries a risk of catastrophic consequences.11
Nonstructural Causes of Coma
The pathophysiology of other metabolic encephalopathies is less well established and is extensively discussed elsewhere.1 Hepatic encephalopathy is caused not merely by ammonia intoxication but likely also involves accumulation of neurotoxins such as short-chain and medium-chain fatty acids, mercaptans, and phenols. Altered neurotransmission may play a role with accumulation of benzodiazepine-like substances, imbalance of serotonergic and glutaminergic neurotransmission, and accumulation of false neurotransmitters. The identity of the neurotoxin in uremic encephalopathy is uncertain and includes urea itself, guanidine and related compounds, phenols, aromatic hydroxyacids, amines, various peptide “middle molecules,” myoinositol, parathormone, and amino acid imbalance. The cause of the dysequilibrium syndrome may entail more than osmotic water shifts from plasma into brain cells, and reduction is reported in cortical potassium, with intracellular acidosis due to increased production of organic acids in the brain. The pathogenesis of pancreatic encephalopathy may involve patchy demyelination of brain white matter due to liberated enzymes from a damaged pancreas, disseminated intravascular coagulation, or fat embolism.
 Differential Diagnosis
Differential Diagnosis
The vegetative state can be defined as wakefulness without awareness and is the consequence of various diffuse brain insults.1,13 It may be a transient phase through which patients in coma pass as the cerebral cortex recovers more slowly than the brainstem. Clinically, vegetative patients appear to be awake and to have cyclical sleep patterns; however, such individuals do not show evidence of cognitive function or learned behavioral responses to external stimuli. Vegetative patients may exhibit spontaneous eye opening, eye movements, and stereotypic facial and limb movements, but they are unable to demonstrate speech or comprehension, and they lack purposeful activity. Vegetative patients generate normal body temperature and usually have normally functioning cardiovascular, respiratory, and digestive systems, but they are doubly incontinent. The vegetative state should be termed persistent at 1 month after injury and permanent at 3 months after nontraumatic injury or 12 months after traumatic injury.14,15 Extended observation of the patient is required to assess behavioral responses to external stimulation and demonstrate cognitive unawareness. The EEG is never isoelectric but shows various patterns of rhythm and amplitude, inconsistent from one patient to the next. Normal EEG sleep/wake patterns are absent.
In the locked-in syndrome, patients retain or regain arousability and self-awareness, but because of extensive bilateral paralysis (i.e., de-efferentation) can no longer communicate except in severely limited ways. Such patients suffer bilateral ventral pontine lesions with quadriplegia, horizontal gaze palsies, and lower cranial nerve palsies. Voluntarily they are capable only of vertical eye movements and/or blinking.1 Sleep may be abnormal, with marked reduction in non-REM and REM sleep phases. The most common etiology is pontine infarction due to basilar artery thrombosis, but others are pontine hemorrhage, central pontine myelinolysis, and brainstem mass lesions. Neuromuscular causes of locked-in syndrome include severe acute inflammatory demyelinating polyradiculoneuropathies, myasthenia gravis, botulism, and neuromuscular blocking agents. In these peripheral disorders, upward gaze is not selectively spared.
Akinetic mutism describes a rare subacute or chronic state of altered behavior in which an alert-appearing patient is both silent and immobile but not paralyzed.16 External evidence of mental activity is unobtainable. The patient usually lies with eyes opened and retains cycles of self-sustained arousal, giving the appearance of vigilance. Skeletal muscle tone can be normal or hypertonic but usually not spastic. Movements are rudimentary even in response to unpleasant stimuli. Affected patients are usually doubly incontinent. Lesions that cause akinetic mutism may vary widely. One pattern consists of bilateral damage to the frontal lobe or limbic-cortical integration with relative sparing of motor pathways. Vulnerable areas involve both basal medial frontal areas. Somewhat similar behavior also can follow incomplete lesions of the deep gray matter (paramedian reticular formation of the posterior diencephalon and adjacent midbrain), but such patients usually suffer double hemiplegia and act slowly yet are not completely akinetic or noncommunicative.
 Approach to Coma
Approach to Coma
Emergency Management
The key points of the rapid neurologic exam are: hand drop from over the head (to assess for malingering or hysterical loss of consciousness); pupillary size and response to light; abnormal eye movements (active disconjugate, unilaterally paralytic, passively induced, or absent); grimacing/withdrawal from noxious stimulation; and abnormal plantar response (unilateral or bilateral Babinski sign).17 Assisted ventilation should continue during the examination if necessary. Neuromuscular blockade required for patient management and care should be deferred if possible until the neurologic examination is completed (3-5 minutes). Signs of arousal or inadequate sedation include dilated reactive pupils, copious tears, diaphoresis, tachycardia, systemic hypertension, and increased pulmonary artery pressure. Thereafter, monitoring patients neurologically may require head computed tomography (CT) more frequently.
Maintain circulation to assure adequate cerebral perfusion. Appropriate resuscitation fluid is lactated Ringer’s solution; normal saline is also used when intracranial hypertension is suspected. A mean arterial pressure around 100 mm Hg is adequate and safe for most patients. While obtaining venous access, collect blood samples for anticipated tests (Box 32-1). Treat hypotension by replacing any blood volume loss, and use vasoactive agents. Judiciously manage systemic hypertension with hypotensive agents that do not substantially raise ICP by their vasodilating effect (labetalol, hydralazine, or a titrated nitroprusside infusion are the favored agents for managing uncontrollable hypertension). For most situations, systolic blood pressure should not be treated unless it is above 160 mm Hg. Maintain urine output at least 0.5 mL/kg/h; accurate measurement requires bladder catheterization.
Consider specific antidotes: Drug overdose is the largest single cause (30%) of coma in the emergency room. Most drug overdose can be treated by supportive measures alone. However, certain antagonists specifically reverse the effects of coma-producing drugs. Naloxone (0.4-2 mg, IV) is the antidote for opiate coma. The reversal of narcotic effect, however, may precipitate acute withdrawal in an opiate addict. In suspected opiate coma, the minimum amount of naloxone should be administered to establish the diagnosis by pupillary dilatation and to reverse respiratory depression and coma. Do not attempt to reverse completely all drug effects with the first dose. IV flumazenil reverses all benzodiazepine-induced coma. Coma unresponsive to 5 mg flumazenil in divided doses given over 5 minutes is not due to benzodiazepine overdose. Recurrent sedation can be prevented with flumazenil (1 mg IV) every 20 minutes.18 The sedative effects of drugs with anticholinergic properties, particularly tricyclic antidepressants, can be reversed with physostigmine (1-2 mg IV). Pretreatment with 0.5 mg atropine will prevent bradycardia. Only full awakening is characteristic of an anticholinergic drug overdose, as physostigmine has nonspecific arousal properties. Physostigmine has a short duration of action (45-60 minutes), and doses may have to be repeated.
Adjust body temperature: hyperthermia is dangerous because it increases brain metabolic demand and, at extreme levels, denatures brain proteins.19 Hyperthermia greater than 40°C requires nonspecific cooling measures even before the underlying etiology is determined and treated. Hyperthermia most often indicates infection but may be due to intracranial hemorrhage, anticholinergic drug intoxication, or heat exposure. A body temperature of less than 34°C should be slowly increased to above 35°C to prevent cardiac dysrhythmia. Hypothermia accompanies profound sepsis, sedative-hypnotic drug overdose, drowning, hypoglycemia, or Wernicke encephalopathy.
History
Neurologic Profile
Establishing the nature of coma is critical for appropriate management and requires:
TABLE32-1 Correlation Between Levels of Brain Function and Clinical Signs
| Structure | Function | Clinical Sign |
|---|---|---|
| Cerebral cortex | Conscious behavior | Speech (including any sounds) Purposeful movement: Spontaneous To command To pain |
| Brainstem activating and sensory pathways (reticular activating system) | Sleep/wake cycle | Eye opening: Spontaneous To command To pain |
| Brainstem motor pathways | Reflex limb movements | Flexor posturing (decorticate) Extensor posturing (decerebrate) |
| Midbrain CN III | Innervation of ciliary muscle and certain extraocular muscles | Pupillary reactivity |
| Pontomesencephalic MLF | Connects pontine gaze center with CN III nucleus | Internuclear ophthalmoplegia |
| Upper pons: | ||
| CN V | Facial and corneal | Corneal reflex-sensory |
| CN VII | Facial muscle innervation | Corneal reflex-motor response: Blink Grimace |
| Lower pons: | ||
| CN VIII (vestibular portion) connects by brainstem pathways with CN III, IV, VI | Reflex eye movements | Doll’s eyes Caloric responses |
| Ponto-medullary junction | Spontaneous breathing Maintained BP |
Breathing and BP do not require mechanical or chemical support |
| Spinal cord | Primitive protective responses | Deep tendon reflexes Babinski response |
BP, Blood pressure; CN, cranial nerve.
The clinical neurologic functions that provide the most useful information in making a categorical diagnosis are outlined in (Box 32-3). These indices are easily and quickly obtained. Furthermore, they have a high degree of interexaminer consistency, and when applied serially, they accurately reflect the patient’s clinical course. Once the cause of coma can be assigned to one of these categories, specific radiographic, electrophysiologic, or chemical laboratory studies can be used to make a disease-specific diagnosis and detect existing or potential complications.
Specific Management
Infratentorial Lesions
Rapid neurologic deterioration of a patient suspected of harboring an infratentorial lesion sometimes demands emergency treatment before a head CT scan is performed. Treatment of a presumed extrinsic compressive lesion of the brainstem entails measures that decrease ICP as outlined earlier. Patients in stupor or showing signs of progressive brainstem compression from a cerebellar hemorrhage or infarction require urgent evacuation. Intrinsic brainstem lesions are best treated conservatively; an incomplete stroke may benefit from thrombolysis and/or heparin anticoagulation. Posterior fossa tumors are managed initially with osmotic agents and steroids; definitive treatment includes surgery and/or radiation. Placement of a ventricular catheter for acute hydrocephalus must be considered cautiously and in consultation with a neurosurgeon; the danger exists of potentially fatal upward transtentorial herniation.12
Metabolic Toxic Coma
The task of the physician in first contact with the patient in metabolic coma is to preserve and protect the brain from permanent damage. Metabolic and toxicologic studies must be performed on the first blood drawn (see Box 32-1). Treatable conditions that quickly, irreversibly damage the brain include:
Acute Bacterial Meningitis
A lumbar puncture must be considered in any unconscious patient with fever and/or signs of meningeal irritation. If possible, an emergency head CT should be performed before lumbar puncture on a comatose patient to rule out unexpected mass lesions. Increased ICP is present in all cases of bacterial meningitis, but a lumbar puncture is not contraindicated when this diagnosis is suspected. Cerebral herniation seldom, if ever, occurs except in small children.20 Clinical correlates of impending herniation demanding a more cautious approach to lumbar puncture include coma or rapidly deteriorating level of arousal, focal neurologic signs, and tonic or prolonged seizures. Papilledema is rare in acute bacterial meningitis. Should unexpected herniation occur after lumbar puncture, treatment with hyperventilation and IV mannitol is indicated. Appropriate antibiotic treatment can usually await the results of spinal fluid Gram stain. If the Gram stain is negative, yet a bacterial etiology is suspected, empirical broad-spectrum antibiotic treatment with a third-generation cephalosporin and vancomycin is appropriate.
Drug Overdose
Certain general principles apply to all patients suspected of having ingested sedative drugs.21,22 Most drug overdose is treated by emergent and supportive measures (Table 32-2). Once vital signs are stable, attempts should be made to remove, neutralize, or reverse the effects of the drug. Patients in coma from recent drug ingestion require gastric lavage after otracheal intubation. A large (preferably double-lumen) gastric tube must be placed orally. Lavage is performed in the head-down position on the left side, using a 200- to 300-mL bolus of tap water or 0.45% saline and continued until the return is clear. After lavage, 1 or 2 tablespoons of activated charcoal are passed down the lavage tube. With meticulous supportive measures, patients with uncomplicated drug-induced coma should recover without neurologic deficit. The recovery from coma due to massive doses of barbiturates or glutethimide can be hastened by hemodialysis.
 The Role of Special Investigations
The Role of Special Investigations
Neurodiagnostic Imaging
Once the patient with altered mental status is appropriately resuscitated and stabilized, further investigation may be necessary to document the location and type of the lesion and provide guidance for therapeutic intervention. CT and MRI provide an anatomic and/or functional assessment of the CNS and helpful information for defining the localization of lesions that produce coma. Details on the use of these modalities in neurointensive care are provided in Chapter 31.
Cranial CT scan is currently the most expedient imaging technique for evaluating the comatose patient and gives the most rapid information about possible structural lesions with the least risk. The value of CT in demonstrating mass lesions, hemorrhage, and hydrocephalus is well established. The CT scan shows tissue shifts due to intracranial intercompartmental pressure gradients but compared to MRI may underestimate the anatomy of herniation.11 Certain lesions such as early infarction (less than 12 hours duration), encephalitis, and isodense subdural hemorrhage may be difficult to visualize. Posterior fossa pathology may be somewhat obscured by bone artifact inherent in the CT technique. Raised ICP is suggested by effacement of cortical sulci, a narrow third ventricle, and obliteration of the suprasellar or quadrigeminal cisterns but cannot be otherwise quantified.
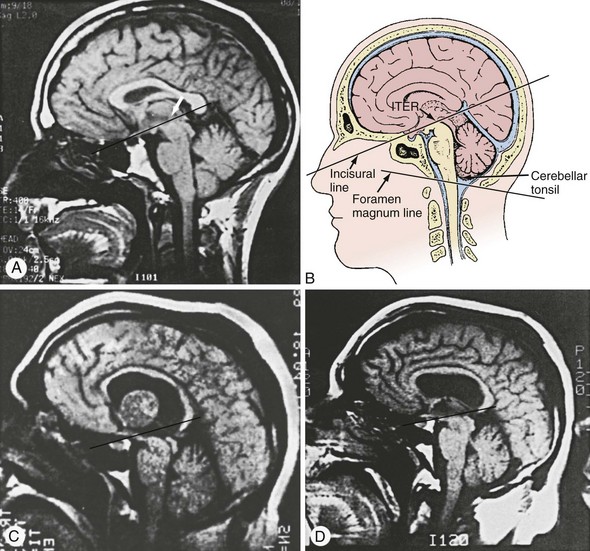
MRI can be performed depending on the clinical setting and stability of the patient’s condition. The use of MRI is limited in the urgent setting of coma evaluation because of the length of time required to perform the imaging, image degradation by even a slight movement of the patient, and the relative inaccessibility of the patient for emergencies that may occur during the imaging process. Nevertheless, MRI provides superb visualization of posterior fossa structures, which is useful when intrinsic brainstem lesions are suspected as the cause of coma.11 MRI images anatomic lesions such as those resulting from acute stroke, encephalitis, central pontine myelinolysis, and traumatic shear injury, with greater resolution and at an earlier time than CT scanning. Injection of the paramagnetic substance, gadolinium, helps delineate areas of blood-brain barrier breakdown and may augment the sensitivity of this scanning technique. Diffusion imaging can demonstrate ischemic brain virtually immediately. Sagittal MRI views are particularly useful in documenting the degree of supratentorial or infratentorial herniations and may enable intervention before clinical deterioration (Figure 32-1).11 Newer MRI techniques allow functional imaging of the CNS by measurement of CBF to a particular region. Future application of this technique may allow rapid determination of diminished CBF, such as occurs in stroke or vasospasm, and will probably be useful in assessing the effect of therapeutic interventions.
Electroencephalogram
The EEG can sometimes give useful additional information in the evaluation of the unresponsive patient. With metabolic and toxic disorders, EEG changes generally reflect the degree and severity of altered arousal or delirium, characterized by decreased frequency of the background rhythm and appearance of diffuse slow activity in the theta (4-7 Hz) and/or delta (1-3 Hz) range. Bilaterally synchronous and symmetric medium- to high-voltage broad triphasic waves are seen in various metabolic encephalopathies, most often in hepatic coma. Rapid beta activity (>13 Hz) in a comatose patient suggests ingestion of sedative hypnotics such as barbiturates and benzodiazepines. Acute focally destructive lesions show focal slow activity. When periodic lateralized epileptiform discharges appear acutely in one or both temporal lobes, herpes simplex encephalitis must be strongly considered. A nonreactive diffuse alpha pattern in a comatose patient usually implies a poor prognosis and is most often seen after anoxic insults to the brain or after acute destructive pontine tegmentum damage.23,24 A normally reactive EEG in an unresponsive patient suggests psychiatric disease, but a relatively normal EEG can accompany the locked-in syndrome, some examples of akinetic mutism, and catatonia—all of which can be caused by structural brain lesions. Attempts to correlate the pattern and frequency spectra of post-resuscitative EEG with neurologic outcome have been unsatisfactory, since its predictive value is at best 88% accurate.25 At present, the most useful information regarding patient prognosis is still obtained by the correct interpretation of physical signs.
Nonconvulsive generalized status epilepticus and repeated complex partial seizures may produce altered levels of awareness or arousal; the EEG is an indispensable tool in diagnosis and management of both these disorders. Continuous EEG monitoring optimizes management of status epilepticus, as clinical assessment is insufficiently sensitive to detect continued electrographic seizures. Furthermore, continuous EEG monitoring in the ICU has shown an unsuspected high incidence of electrographic seizure activity in critically ill neurologic patients.26,27
Jugular Venous Oximetry
Changes in jugular venous oxygen saturation measure the relationship between cerebral metabolic rate and CBF, and this monitoring tool is discussed in Chapter 31.28 This form of monitoring offers the potential to minimize secondary insults after traumatic brain injury (TBI) by providing warning of cerebral ischemia. It should be considered in comatose patients in conjunction with ICP monitoring (discussed later) to provide a logical approach to the treatment of brain injury.
Transcranial Doppler Ultrasonography
Transcranial Doppler ultrasonography (discussed in Chapter 31) allows noninvasive measurement of blood flow velocity in basal cerebral arteries.29 The high dynamic resolution provided and confirmed correlation with other hemodynamic modalities encourages increasing numbers of neurointensivists to adopt the technique. Its importance in coma is in early detection of vasospasm in subarachnoid hemorrhage and at the time of brain death,30 where an oscillating reverbatory movement has been noted in flow-velocity waveforms. The diagnosis is suspected based on the finding of the reflux phenomenon during late systole following anterograde injection of blood into the vascular tree.
Evoked Potentials
Evoked potentials (EPs) are used to follow the level of CNS function in comatose patients.31 Clinical use of brainstem auditory evoked potential (BAEP) and short latency somatosensory evoked potential (SEP) responses stem from the correlation between EP waveform and presumed generators within certain CNS structures. The SEP shows special promise in the ICU field, because EP components generated supratentorially in the thalamus and primary sensory cortex can be identified and followed over time. Shifts of intracranial structures that lead to herniation syndromes are reflected in abnormalities in SEPs, whereas BAEPs are generated entirely at or below the lower midbrain and are less often affected. EPs are less affected than EEG readings by sedative medications and septic or metabolic encephalopathies, factors that frequently confound interpretations in comatose patients. Anatomic specificity and physiologic and metabolic immutability are the basis of clinical utility of EPs. Abnormal test results, however, are etiologically nonspecific and must be carefully integrated into the clinical situation by a physician familiar with their clinical use. Caution is needed in the interpretation of SEPs to insure that absent responses are not due to technical problems. Repeat SEPs are useful in following patients’ progress. A progressive decline in response amplitude appears to be associated with worsening prognosis. Studies have shown that all patients with anoxic coma and bilaterally absent SEPs had died or remained in persistent vegetative state.32 In traumatic coma, absent SEPs may be a less definitive prognostic indicator, as recovery of consciousness has been reported in some patients.33 Furthermore, comatose patients, especially those with motor response of flexor posture or better, with an initial poor prognostic EEG pattern but normal SEPs, may have the potential for recovery and should be supported until their condition has changed to a more prognostically definitive category.34 BAEPs and median SEPs obtained within 24 hours of coma onset had a 3-month predictive outcome (compared to Glasgow Outcome Scale [GOS]) in patients with head injury, brain hemorrhage, or neoplasm.35 Diagnostic sensitivity for an unfavorable outcome was low for both parameters, though specificity and positive predictive value was equally high for abnormal wave VI of BAEPs and median SEPs.
Intracranial Pressure Monitoring
ICP monitoring in neurointensive care is discussed in detail in Chapters 30 and 31. A review of published randomized controlled studies of real-time ICP monitoring by invasive or semi-invasive means in acute coma (traumatic or nontraumatic etiology) versus no ICP monitoring (i.e., clinical assessment of ICP) looked at outcome measures of all-cause mortality and severe disability at the end of a given follow-up period.36 The conclusion drawn is that there are insufficient data to clarify the role of routine ICP monitoring in all severe cases of acute coma. However, it is of value in TBI and should be considered on a case-by-case basis in other cases of coma.
Positron Emission Tomography
Recent studies comparing patients in a minimally conscious state and controls, using oxygen-15 positron emission tomography (PET), revealed activation patterns in key brain regions linked to pain processing that were distinguishable from patterns in patients in a persistent vegetative state. These observations suggest the need for analgesic treatment in the minimally conscious state but not for patients in the persistent vegetative state.37 Additional use of brain PET as a research tool to study patients in comatose states will provide important further insight into this condition.
 Prognosis
Prognosis
Numerous descriptive scoring systems, both pre- and in-hospital, are used to attempt to assess severity of illness and predict patient outcome. A 2-year prospective study compared severity-of-illness scoring systems (Acute Physiology and Chronic Health Evaluation [APACHE] II and Mainz Emergency Evaluation System [MEES]) to mental status measurement (Glasgow Coma Scale [GCS]) in predicting outcome of 286 consecutive adult patients hospitalized for nontraumatic coma.38 There were no statistically significant differences among the scoring systems to correctly predict outcome. APACHE II and MEES should not replace GCS. For prediction of mortality, GCS score also provides the best indicator in nontraumatic comatose patients (simple, less time consuming, and accurate in an emergency situation). Useful factors in determining the outcome of medical coma include cause, depth, and duration of coma. Clinical signs reflecting brainstem, motor, and verbal function are the most helpful and best validated predictors (confidence interval 0.95).39–42 Overall, only 15% of patients in established medical coma for 6 hours will make a good or moderate recovery; others will die (61%), remain vegetative (12%), or become permanently dependent on others for daily living (11%). Prognosis depends on etiology of medical coma. Patients in coma due to a stroke, subarachnoid hemorrhage, or cardiorespiratory arrest have only about a 10% chance of achieving independent function. Some 35% of patients will achieve moderate to good recovery if coma is due to other metabolic reasons including infection, organ failure, and biochemical disturbances. As noted earlier, almost all patients who reach the hospital after sedative overdose or other exogenous agents will recover moderately or completely. Depth of coma affects individual prognosis. Patients who open their eyes in response to noxious stimuli after 6 hours of coma have a 20% chance of making a good recovery, versus 10% if eyes remain closed. The longer coma persists, the less likely the chances for recovery; 15% of patients in coma for 6 hours make a good or moderate recovery compared with only 3% who remain unconscious at 1 week.39,40 Coma following head trauma has a somewhat better prognosis (see later discussion).
The severity of signs of brainstem dysfunction on admission inversely correlates with the chance of good recovery in medical coma. Absent pupillary responses at any time after onset and, except in barbiturate or phenytoin poisoning, absent caloric-vestibular reflexes 1 day after onset indicate a poor prognosis (<2% recovery). Except for sedative drug poisoning, no patient with absent pupillary light reflexes, corneal reflexes, oculocephalic or caloric responses, or lack of a motor response to noxious stimulation at 3 days after onset is likely to ever regain independent function. In a prospective study of 500 patients in medical coma, a uniform group of 210 patients suffered anoxic injury: 52 of these had no pupillary reflex at 24 hours, all of whom died. By the third day, 70 were left with a motor response worse than withdrawal and all died. By the seventh day, the absence of roving eye movements was seen in 16 patients, all of whom died.39,40
Postanoxic convulsive status epilepticus and/or myoclonic status epilepticus reflect a poor prognosis. Occasional patients recover consciousness but remain handicapped. Most die or become vegetative.43,44 Associated clinical findings such as loss of brainstem reflexes or eye opening at the onset of myoclonic jerks, and sinister EEG patterns such as suppression or burst-suppression, confirm a grim neurologic outcome in this group. Autopsy studies show that cerebral and cerebellar damage can be ascribed to the initial ischemic hypoxic event; there is no evidence that status epilepticus further contributes to this damage. We initially treat patients with an IV loading dose of a major anticonvulsant (phenytoin, 13-18 mg/kg at 25 mg/min; and/or phenobarbital, 20 mg/kg at 50 mg/min). Myoclonic status epilepticus is generally resistant to therapy; we give intermittent doses of benzodiazepines (lorazepam, 2-4 mg; or clonazepam, 0.5 mg IV) as needed to suppress particularly severe myoclonus that interferes with ventilatory support. Anesthetic agents are rarely indicated and are unlikely to alter outcome.
A meta-analysis of prognostic studies in anoxic-ischemic coma examined the value of biochemical markers of brain damage in CSF or serum.45 Only concentrations of CSF markers (creatine kinase brain isoenzyme, neuron-specific enolase, lactate dehydrogenase, and glutamate oxaloacetate) reached 0% false-positive rate. Because of small numbers of patients involved in studies (wide confidence levels) and methodological limitations of studies, the results available are not sufficiently accurate to provide a solid basis for management decisions of patients in coma.
The most accurate prediction of outcome in a patient in medical coma is obtained from the use of a combination of clinical signs, and there is little to be added by more sophisticated testing, other than identifying the cause of the coma.39,40 Within the first week, it is hard to justify the withdrawal of therapy from patients in medical coma unless they are already brain dead or lack all signs of brainstem function. After that, the probability of being able to predict the quality of life increases steadily. A multi-society task force of neurologists and neurosurgeons obtained a large number of data concerning the persistent vegetative state that provides guidelines to outcomes in patients remaining vegetative 1 month following severe head trauma or coma-producing medical illness (mostly anoxic).15
The recent and widespread application of mild therapeutic hypothermia after cardiac arrest has raised concern about reduced or altered ability to prognosticate outcome. Clinical variables such as brainstem reflex recovery, myoclonus, and absent motor response to pain showed higher false-positive mortality predictions in comatose survivors of cardiac arrest compared to predictions by the American Academy of Neurology guidelines at 72 hours.46 Greater use of higher doses of sedatives related to hypothermia therapy or direct effects of hypothermia may play a role.47,48 Therefore, caution in prognostication is advised until a better understanding of the effects of this important new therapy emerges.
The outcome of traumatic coma is generally better than medical coma, and prognostic criteria are somewhat different.15,33,49 Many patients with head injury are young; prolonged posttraumatic unconsciousness of up to several months does not always preclude a satisfactory outcome; and compared to the initial degree of neurologic abnormality, patients in traumatic coma improve more than patients in medical coma. Patients in coma for longer than 6 hours after TBI have a 40% chance to recover to moderate disability or better at 6 months. The most reliable predictors of outcome at 6 months are:
| Glasgow Coma Scale Total | |
| 14-15 | 5 |
| 11-13 | 4 |
| 8-10 | 3 |
| 5-7 | 2 |
| 3-4 | 1 |
| Respiratory Rate | |
| 10-24/min | 4 |
| 25-35/min | 3 |
| >35/min | 2 |
| 1-9/min | 1 |
| None | 0 |
| Respiratory Expansion | |
| Normal | 1 |
| None | 0 |
| Systolic Blood Pressure | |
| >89 mm Hg | 4 |
| 70-89 mm Hg | 3 |
| 50-69 mm Hg | 2 |
| 0-49 mm Hg | 1 |
| No pulse | 0 |
| Peripheral Perfusion (Capillary Refill) | |
| Normal | 2 |
| Delayed | 1 |
| None | 0 |
| Total Trauma Score (Sum of Individual Scores)*: | |
* Scores < 10 represent < 60% chance of survival.
An independent poor prognostic indicator is sustained, uncontrollably increased ICP (>20 mm Hg). Additional factors play a role in the eventual outcome from traumatic coma. Specific lesions such as subdural hematoma that result in coma can have less than 10% recovery rate.50 In studies with blunt trauma, comatose patients with increased plasma glucose, hypokalemia, or elevated blood leukocyte counts were associated with lower GCS scores and increased probability of death.51 There are some reports of patients who have suffered coma as a result of TBI in whom an improvement from the vegetative state has been recognized after months, but these anecdotal cases of recovery are difficult to validate. It seems possible that such patients were not truly vegetative but rather in a state of profound disability with minimal cognition at the beginning of observation.52 In a recent case report, however, patient recovery from a 19-year-duration TBI-induced minimally conscious state was associated with improvements in white matter tracts, demonstrated with MRI diffusion tensor technique.53 Novel MRI technology may thus aid in explaining these unusual recoveries; additional studies are warranted.
A systematic review of trials reporting on multisensory stimulation programs in TBI patients in coma or the vegetative state found no reliable evidence of the effectiveness of such techniques when compared to standard rehabilitation.54 Outcome measures included duration of unconsciousness (time between injury and response to verbal commands), level of consciousness (GCS), level of cognitive functioning, functional outcomes (GOS), or by disability rating scale. The overall methodological quality was poor, and studies differed widely in design and conduct. Owing to the diversity in reporting of outcome measures, a meta-analysis was not possible. Recently, continuous subcutaneous apomorphine infusion (to stimulate dopaminergic neurotransmission) has been suggested to facilitate awakening, specifically in traumatic coma.55 Similarly, recent work has suggested possible arousal effects from either prolonged coma or minimally conscious state with zolpidem administration.56 However, larger series are required to confirm or refute these findings.57
Analysis of the SUPPORT (Study to Understand the Prognoses and Preferences for Outcomes and Risks of Treatments) trial was used to estimate the cost-effectiveness of aggressive care for patients in nontraumatic coma.58,59 Patients with reversible metabolic causes of coma were excluded. The incremental cost-effectiveness was calculated for aggressive care versus withholding cardiopulmonary resuscitation and ventilatory support after day 3 of coma. The incremental cost-effectiveness of the more aggressive strategy was $140,000 (1998 dollars) per quality-adjusted life year for high-risk patients and $87,000 per quality-adjusted life year for low-risk patients (five risk factors were age older than 70 years, absent verbal response, absent withdrawal to pain, abnormal brainstem response, and serum creatinine >1.5 mg/dL). From a purely economic standpoint, making earlier decisions to withhold life-sustaining treatments for patients with very poor prognoses may yield considerable cost savings. On moral and ethical grounds, however, many physicians object to having to consider the cost factor when it comes to making treatment decisions for more or less sick patients. But growing financial constraints now imposed on the medical community from the top down by politicians and the business culture may no longer afford such luxury, even in a country like the United States.
Key Points
Plum F, Posner JB. The Diagnosis of Stupor and Coma. Philadelphia: FA Davis; 1980.
Hund EF, Lehman-Horn F. Life-threatening hyperthermic syndromes. In: Hacke W, editor. Neurocritical Care. Berlin: Springer-Verlag; 1994:888-896.
Synek VM. Prognostically important EEG coma patterns in diffuse anoxic and traumatic encephalopathies in adults. J Clin Neurophysiol. 1988;5:161-174.
Levy DE, Bates D, Caronna JJ, et al. Prognosis in non-traumatic coma. Ann Intern Med. 1981;94:293-301.
Levy DE, Caronna JJ, Singer BH, et al. Predicting outcome from hypoxic-ischemic coma. JAMA. 1985;253:1420-1426.
Jennett B, Teasdale G, Braakman R, et al. Prognosis of patients with severe head injury. Neurosurgery. 1979;4:283-301.
Rossetti AO, Oddo M, Logroscino G, Kaplan PW. Prognostication after cardiac arrest and hypothermia: a prospective study. Ann Neurol. 2010;67:301-307.
1 Plum F, Posner JB. The Diagnosis of Stupor and Coma. Philadelphia: FA Davis; 1980.
2 Plum F. Coma and related global disturbances of the human conscious state. In: Peters A, editor. Cerebral Cortex. New York: Plenum Publishing Corp.; 1991:359-425.
3 Brodal A. Neurological Anatomy in relation to Clinical Medicine. Oxford: Oxford University Press; 1981.
4 Steriade M, McCarly RW. Brain Stem Control of Wakefulness and Sleep. New York: Plenum Publishing; 1990.
5 McCormick DA, Von Krosigk M. Corticothalamic activation modulates thalamic firing through glutamate “metabotropic” receptors. Proc Natl Acad Sci U S A. 1992;89:2774-2778.
6 Sejnowski TJ, McCormick DA, Steriade M. Thalamocortical oscillations in sleep and wakefulness. In: Arbib MA, editor. The Handbook of Brain Theory and Neural Networks. Cambridge: Massachusetts, The MIT Press; 1995:976-980.
7 Kales A. Pharmacology of sleep. Handbook of Experimental Pharmacology Series. vol. 116. Berlin: Springer; 1995.
8 Tinuper P. Idiopathic recurring stupor: A case with possible involvement of the gamma aminobutyric acid (GABA) ergic system. Ann Neurol. 1992;31:503-506.
9 Plum F. Coma. In: Adelman G, editor. Encyclopedia of Neuroscience. Boston: Birkhauser Boston Inc.; 1998:446-447.
10 Cuneo RA, Caronna JJ, Pitts L, et al. Upward transtentorial herniation: Seven cases and a literature review. Arch Neurol. 1979;36:618-623.
11 Reich JB, Sierra J, Camp W, et al. Magnetic resonance imaging measurements and clinical changes accompanying transtentorial and foramen magnum brain herniation. Ann Neurol. 1993;33:159-170.
12 Kase CS, Wolf PA. Cerebellar infarction: Upward transtentorial herniation after ventriculostomy. Stroke. 1993;24:1096-1098.
13 Jennett WB, Plum F. The persistent vegetative state: a syndrome in search for a name. Lancet. 1972;1:734-737.
14 Council on Scientific Affairs and Council on Ethical and Judicial Affairs. Persistent Vegetative State and the decision to withdraw or withhold life support. JAMA. 1990;263:426-430.
15 Multi-Society Task Force on PVS. Medical aspects of the persistent vegetative state: statement of a multi-society task force. N Engl J Med. 1994;330:1499-1508.
16 Cairns H. Disturbances of consciousness with lesions of the brain stem and diencephalon. Brain. 1952;75:109-146.
17 Goldberg S. The Four Minute Neurologic Exam. Miami: Medmaster; 1992.
18 Winkler E, Shlomo A, Kriger D, et al. Use of flumazenil in the diagnosis and treatment of patients with coma of unknown etiology. Crit Care Med. 1993;21:538-542.
19 Hund EF, Lehman-Horn F. Life-threatening hyperthermic syndromes. In: Hacke W, editor. Neurocritical Care. Berlin: Springer-Verlag; 1994:888-896.
20 Rennick G, Shann F, de Campo J. Cerebral herniation during bacterial meningitis in children. BMJ. 1993;306:953-955.
21 Howell JM, Altieri M, Jagoda AS, et al. Emergency Medicine. Philadelphia: WB Saunders; 1998. p. 1377-538
22 Ellenhorn MJ, Schonwald S, Ordog G, et al. Ellenhorn’s Medical Toxicology: Diagnosis and Treatment of Human Poisoning. Baltimore: Williams & Wilkins; 1997.
23 Austin EG, Walkus RJ, Longstreth WT. Etiology and prognosis of alpha coma. Neurology. 1988;38:773-777.
24 Synek VM. Prognostically important EEG coma patterns in diffuse anoxic and traumatic encephalopathies in adults. J Clin Neurophysiol. 1988;5:161-174.
25 Edgren E, Hedstrend U, Nordin M, et al. Prediction of outcome after cardiac arrest. Crit Care Med. 1987;15:820-825.
26 Young GB, Jordan KG, Doig GS. An assessment of non-convulsive seizures in the intensive care unit using continuous EEG monitoring: an investigation of variables associated with mortality. Neurology. 1996;47:83-89.
27 Lowenstein DH, Aminoff MJ. Clinical and EEG features of status epilepticus in comatose patients. Neurology. 1992;42:100-104.
28 Souter MJ, Andrews PJD. A review of jugular venous oximetry. Intensive Care World. 1996;13:32-38.
29 DeWitt LD, Wechsler LR. Transcranial doppler. Stroke. 1988;19:915-921.
30 Ropper AH, Kehne SM, Wechsler LR. Transcranial doppler in brain death. Neurology. 1987;37:1733-1735.
31 Chiappa KH, Hoch DB. Electrophysiologic monitoring. In: Roper AH, editor. Neurological and Neurosurgical Intensive Care. New York: Raven Press; 1993:147-183.
32 Chen R, Bolton CF, Young GB. Prediction of outcome in patients with anoxic coma: A clinical and electrophysiologic study. Crit Care Med. 1996;24:672-678.
33 Marshall LF, Gautille T, Klauber MR, et al. The outcome of severe head injury. J Neurosurg. 1991;75(suppl):S28-S36.
34 Lindsay K, Pasaoglu A, Hirst D, et al. Somatosensory and auditory brainstem conduction after head injury: a comparison with clinical features in prediction of outcome. Neurosurgery. 1990;26:278-285.
35 Balogh A, Wedekind C, Klug N. Does wave VI of BAEP pertain to the prognosis of coma? Neurophysiol Clin. 2001;31:406-411.
36 Forsyth R, Baxter P, Elliot T. Routine intracranial pressure monitoring in acute coma. Cochrane Database Syst Rev 2001:pCD002043.
37 Boly M, Faymonville ME, Schnakers C, et al. Perception of pain in the minimally conscious state with PET activation: an observational study. Lancet Neurol. 2008;7:1013-1020.
38 Grmec S, Gasparovic V. Comparison of APACHE II, MEES and Glasgow Coma Scale in patients with non-traumatic coma for prediction of mortality. Crit Care. 2001;5:19-23.
39 Levy DE, Bates D, Caronna JJ, et al. Prognosis in non-traumatic coma. Ann Intern Med. 1981;94:293-301.
40 Levy DE, Caronna JJ, Singer BH, et al. Predicting outcome from hypoxic-ischemic coma. JAMA. 1985;253:1420-1426.
41 Edgren E, Hedstrand U, Sutton-Tyrrel K, Safar P. Assessment of neurological prognosis in comatose survivors of cardiac arrest. Lancet. 1994;343:1055-1059.
42 Longstreth WT, Diehr P, Init S. Prediction of awakening after out of hospital cardiac arrest. N Engl J Med. 1983;308:1378-1382.
43 Young GB, Gilbert JJ, Zochodine DW. The significance of myoclonic status epilepticus in postanoxic coma. Neurology. 1990;40:1843-1848.
44 Wijdecks EF, Parisi JE, Sharbrough FW. Prognostic value of myoclonus status in comatose survivors of cardiac arrest. Ann Neuro. 1994;35:239-243.
45 Zandbergen EG, de Haan RJ, Hijdra A. Systematic review of prediction of poor outcome in anoxic-ischaemic coma with biochemical markers of brain damage. Intensive Care Med. 2001;27:1661-1667.
46 Rossetti AO, Oddo M, Logroscino G, Kaplan PW. Prognostication after cardiac arrest and hypothermia: a prospective study. Ann Neurol. 2010;67:301-307.
47 Samaniego EA, Mlynash M, Caulfield AF, et al. Sedation confounds outcome prediction in cardiac arrest survivors treated with hypothermia. Neurocrit Care. 2010 Aug 3. [Epub ahead of print]
48 Tortorici MA, Kochanek PM, Poloyac SM. Effects of hypothermia on drug disposition, metabolism, and response: a focus of hypothermia-mediated alterations on the cytochrome P450 enzyme system. Crit Care Med. 2007;35:2196-2204.
49 Jennett B, Teasdale G, Braakman R, et al. Prognosis of patients with severe head injury. Neurosurgery. 1979;4:283-301.
50 Gennarelli TA, Spielman GM, Langfitt TW, et al. Influence of the type of intracranial lesion on outcome from severe head injury. J Neurosurg. 1982;56:26-32.
51 Kassum DA, Thomas EJ, Wang CJ. Early determinations of outcome in blunt injury. Can J Surg. 1984;27:64-69.
52 Rosenberg GA, Johnson SF, Brenner RP. Recovery of cognition after prolonged vegetative state. Ann Neurol. 1977;2:167-168.
53 Voss HU, Uluç AM, Dyke JP, et al. Possible axonal regrowth in late recovery from the minimally conscious state. J Clin Invest. 2006;116:2005-2011.
54 Lombardi F, Tarrico M, De Tanti A, et al. Sensory stimulation of brain-injured individuals in coma or vegetative state: results of a Cochrane systematic review. Clin Rehab. 2002;16:464-472.
55 Fridman EA, Krimchansky BZ, Bonetto M, et al. Continuous subcutaneous apomorphine for severe disorders of consciousness after traumatic brain injury. Brain Inj. 2010;24:636-641.
56 Clauss R, Nel W. Drug induced arousal from the permanent vegetative state. NeuroRehabilitation. 2006;21:23-28.
57 Pistoia F, Mura E, Govoni S, et al. Awakening and awareness recovery in disorders of consciousness: is there a role for drugs? CNS Drugs. 2010;24:625-638.
58 The Support Principal Investigators. A controlled trial to improve care for seriously ill hospitalizes patients. The study to understand prognosis and preferences for outcomes and risks for treatments (Support). JAMA. 1995;274:1591-1598.
59 Hamel HB, Phillips R, Teno J, et al. Cost effectiveness of aggressive care for patients with non-traumatic coma. Crit Care Med. 2002;30:1191-1196.
