Coagulopathies and Sickle Cell Disease
Biochemistry and Physiology of Hemostasis
The stimulus that initially causes clot formation occurs as a consequence of disruption of endothelial cells. This leads to exposure of collagen and subendothelial tissues. The hemostatic response to tissue injury consists of four stages. First, vasoconstriction by the contraction of smooth muscle in the injured vessel wall reduces blood flow. Second, platelets adhere to the exposed endothelium, aggregate, and release their granular contents. This activity stimulates further vasoconstriction and recruits more platelets. This action results in ‘primary hemostasis’ that occludes the gap in the blood vessel and stops blood loss through the vessel. Third, the extrinsic and intrinsic coagulation systems are activated to form fibrin, which stabilizes the platelets and prevents disaggregation. Fourth, fibrinolysis results from the release of plasminogen activators from the injured vessel wall. These activators limit the coagulation process. Once healing has taken place, they begin dissolution of formed clot so that vascular patency can be restored.1
Endothelial Cells
Endothelial cells maintain the integrity of the blood vessel and prevent extravasation of blood into the surrounding tissue.1 Passive thromboresistance is provided by endothelial proteoglycans, primarily endogenous heparin sulfate. Active thromboresistance is achieved through several mechanisms, including the synthesis and release of prostacyclin, a potent vasodilator and an inhibitor of platelet adhesion and aggregation.1,2
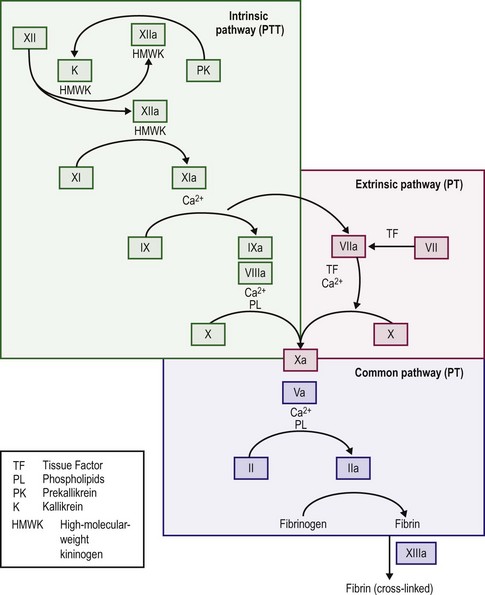
When the endothelium is injured, tissue factor (thromboplastin) is produced and rapidly promotes local thrombin formation.3 Tissue factor binds factor VII and converts it to factor VIIa (Fig. 5-1), which is the first step in activation of the extrinsic coagulation pathway. It also activates factor IX, which is the major activator of the common pathway, resulting in fibrin formation.4 Capillaries seal with little dependence on the hemostatic system, but arterioles and venules require the presence of platelets to form an occluding plug. In arteries and veins, hemostasis depends on both vascular contraction and clot formation around an occluding primary hemostatic plug.5
Platelets
In the resting state, platelets circulate as disk-shaped, anuclear cells that have been released from megakaryocytes in the bone marrow. They are 2–3 µm in size and remain in circulation for approximately five to nine days unless they participate in coagulation reactions or are removed by the spleen. Normal blood contains 150,000–400,000 platelets/µL.5 In the resting state platelets do not bind to intact endothelium.
Platelet Adhesion
Once platelets bind to injured tissue and are activated, their discoid shape changes. They spread on the subendothelial connective tissue and degranulate releasing serotonin, adenosine diphosphate, adenosine triphosphate, and calcium, and alpha granules release factor V, fibrinogen, von Willebrand factor (FVIII : vWF), fibronectin, platelet factor 4, β-thromboglobulin, and platelet-derived growth factor.6,7 This recruits and aggregates more platelets from the circulation onto the already adherent platelets.7
When a vessel is disrupted, platelet adhesion occurs through the binding of collagen and vWF (found in the subendothelium) to the platelet membrane (Fig. 5-2). For platelet adhesion to occur, platelets must express specific glycoprotein Ib receptors on their surface to bind the vWF complex. If this specific glycoprotein is missing, platelets are unable to adhere to areas of injury.8 Platelets in Bernard–Soulier syndrome lack glycoprotein Ib and are unable to adhere and form the initial hemostatic plug.9 If the vWF is defective or deficient, platelets do not adhere to sites of vascular injury. The result is von Willebrand disease, of which several specific types and subtypes have been defined.10–12 Very high concentrations of prostacyclin also can inhibit platelet adhesion to exposed subendothelium.5
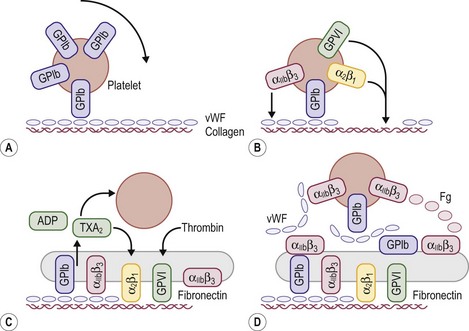
FIGURE 5-2 Schematic representation of platelet adhesion and aggregation under flow conditions. (A) Rolling of platelets over collagen-bound vWF mediated by GPIb. (B) Firm attachment mediated by α2β1 and glycoprotein VI (GP VI) binding to collagen, and by αIIbβ3) binding tocollagen-bound vWF. (C) Platelet activation, secretion, and spreading. (D) Aggregate formation.
Platelet Aggregation
Aggregation is a complex reaction that involves platelet granule release, cleavage of membrane phospholipids by phospholipases A2 and C, alterations in intracellular cyclic adenosine monophosphate levels, mobilization of intracellular calcium, and the expression of fibrinogen receptors on the platelet surface. If fibrinogen receptors (glycoproteins IIb and IIIa) or fibrinogen are missing, platelets do not aggregate.13,14 This results in Glanzmann thrombasthenia causing patients to have a serious, life-long bleeding disorder.6
After aggregation, platelets function to enhance thrombin formation. The platelet membrane provides specific binding sites for factors Xa and V causing effective assembly of the prothrombinase complex making thrombin.7 Thrombin formation makes a stable hemostatic plug of adherent platelets surrounded by a network of fibrin strands.
Generation of Thrombin
The majority of thrombin production results from the activation of the intrinsic coagulation system, not the extrinsic system. Exposed subendothelium converts factor XII to factor XIIa and thereby activates the intrinsic pathway, although deficits in factor XII do not cause a bleeding disorder. Activation of factors XI and IX follows, and activated factor IX in combination with factor VIII, calcium, and platelet phospholipid activates factor X. Activated factor VII, complexed with tissue factor, activates factor IX. Factor Xa with factor V then cleaves prothrombin into the active molecule thrombin, which can convert fibrinogen into fibrin.4,15
Formation of Fibrin
When thrombin acts on fibrinogen, fibrin monomers result after the proteolytic release of fibrinopeptides A and B. The monomeric fibrin then polymerizes into a gel.4,15 With additional stabilization of the fibrin gel provided by factor XIII, fibrin surrounds and stabilizes the platelet plug. This process makes the multimeric fibrin more resistant to plasmin digestion and completes the formation and stabilization of the blood clot.16
Several regulatory proteins serve to localize thrombin formation to the surface of the blood vessel. Endothelial cells have receptors for protein C. Protein S is a co-factor for the activation of protein C. Thrombomodulin is an endothelial surface protein that acts in combination with thrombin to activate the bound protein C. Activated protein C then degrades factors Va and VIIIa, which inhibit thrombin formation.17
Heparin-like anticoagulant molecules, present on endothelial cells, act in combination with antithrombin III to inhibit factors XIIa, XIa, IXa, and Xa and thrombin. Inhibition of these factors prevents the spread of clot to uninjured adjacent vessels and the blockage of large vessels by excessive clot formation.15,17 Endothelial cells, as mentioned previously, produce PGI2 (prostacyclin), a potent vasodilator and inhibitor of platelet aggregation and adhesion.
Fibrinolysis
The regulatory process that dissolves fibrin and preserves vessel patency is called fibrinolysis. Circulating plasminogen is converted into plasmin by tissue plasminogen activators. These activators are released from the vessel walls at the site of blood clotting. They bind to the fibrin clot and convert plasminogen to plasmin. Plasmin enzymatically degrades fibrin, fibrinogen, and other plasma proteins, and this process results in the dissolution of formed clot.15,17
Clinical Evaluation
There is currently no completely reliable screening test available to evaluate hemostasis in the preoperative patients. A careful history, including a full family history, remains the best means of uncovering mild bleeding problems, including von Willebrand disease or qualitative platelet abnormalities.18 These disorders may easily escape standard laboratory screening procedures, such as prothrombin time (PT), activated partial thromboplastin time (aPTT), platelet count, and bleeding time. aPTT screening yields many false-positive results caused by both analytical problems and detection of clinically insignificant disorders. In addition, a normal aPTT may lead to a false sense of safety because it does not exclude all serious bleeding disorders. Because no method can reliably predict all bleeding complications, postoperative monitoring remains important for all patients.19 Likewise, patients with mild disorders who have not previously undergone an operation may have no history of bleeding problems and might be identified preoperatively only if screening tests are performed.18 It is important to consider the history as the most important component of a diagnostic strategy and to investigate thoroughly any story of unusual bleeding, even if the screening tests are normal.20 Conversely, studies examining the utility of a screening preoperative PT and aPTT in patients undergoing tonsillectomy and adenoidectomy concluded that routine screening with a PT and aPTT for all patients regardless of history cannot be recommended.19,21 In obtaining a history from the patient and parents, positive answers to the questions posed in Box 5-1 indicate the need for further evaluation.18,22–24
If there is a history of abnormal bleeding, the following points must be established. The type of bleeding (i.e., petechiae, purpura, ecchymosis, and single or generalized bleeding sites) can give an indication of the underlying defect. Petechiae and purpura are most frequently associated with platelet abnormalities, either of function or numbers. Von Willebrand disease is most frequently associated with mucosal bleeding, including epistaxis, whereas hemophilia is most often associated with bleeding into joints or soft tissue ecchymosis, or both. Bleeding when the umbilical cord separates is most often associated with a deficiency in factor XIII, as is unexplained bleeding of the central nervous system.16,25 A single bleeding site, such as repetitive epistaxis from the same nostril, is frequently indicative of a localized, anatomic problem and not a system-wide coagulation defect.
Any previous or current drug therapy must be fully documented, and a search is made for over-the-counter medications that the patient might be taking but does not consider ‘medicine’ and has therefore not mentioned. Aspirin, ibuprofen, cough medications containing guaifenesin, and antihistamines can lead to platelet dysfunction or uncover a previously undiagnosed bleeding disorder such as von Willebrand disease.26,27 The presence of other medical problems including renal failure with uremia, hepatic failure, malignancies, gastrointestinal malabsorption, vascular malformations, cardiac anomalies with or without repair, and autoimmune disorders is essential to elicit because these may have associated coagulopathies.
Laboratory Evaluation
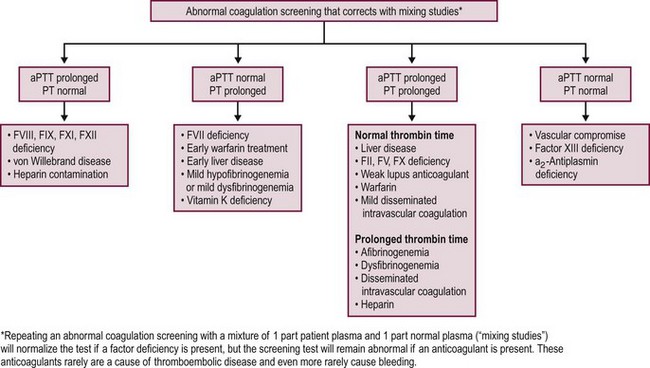
When the bleeding history and/or family history suggest the possibility of a bleeding disorder, or if it is impossible to obtain a history due to family or social circumstances, it is customary to proceed with a series of laboratory investigations to look for a possible bleeding diagnosis. Generally, screening tests are performed first and should include a blood cell count, PT, and aPTT (Fig. 5-3).20 Additional tests can measure fibrinogen levels, assess the thrombin time, screen for inhibitors of specific coagulation factors, measure specific factor levels, and test for platelet function and von Willebrand disease.20,28 Patients also can be evaluated for evidence of DIC by using multiple assays to test for the presence of various fibrinopeptides and products from the breakdown of fibrin or fibrinogen.
Platelet Count
The platelet count measures the adequacy of platelet numbers to provide initial hemostasis. Thrombocytopenia (a platelet count of <150,000/µL) is one of the most common problems that occur in hospitalized patients. As stated previously, typical manifestations include mucocutaneous bleeding. The risk of bleeding is inversely proportional to the platelet count. When the platelet count is <50,000/µL, minor bleeding occurs easily and the risk of major bleeding increases. Counts between 20,000–50,000/µL predispose to bleeding with even minor trauma; with counts <20,000/µL, spontaneous bleeding may occur; with counts <5000/µL, severe spontaneous bleeding is more likely. However, patients with counts <10,000/µL may be asymptomatic for years.29 Surgical bleeding does not usually occur until the platelet count is <50,000 platelets/µL.30 A platelet count of <50,000/µL is considered a cut-off criterion for transfusions, and the prophylactic use of platelet transfusion is indicated for any invasive procedure. Patients with significant clinical bleeding should also be transfused with platelets.30
Bleeding Time and the PFA-100 Analyzer
The bleeding time is defined as the length of time required for a standardized incision to stop oozing blood that can be absorbed onto filter paper. A variety of procedures have been used, but all have variable sensitivity and have been difficult to reproduce accurately, leading many centers to drop the bleeding time from the list of approved laboratory tests.31 The PFA-100 Analyzer (Siemans Healthcare Diagnostics, Deerfield, IL) is now widely used as a replacement for the bleeding time. It creates an in vitro high shear stress condition that results in the activation of platelet-dependent and vWF-dependent attachment and aggregation of platelets to a collagen–ADP or collagen–epinephrine surface. In most cases, the PFA-100 closure time is superior to the bleeding time in the detection of von Willebrand disease, aspirin effect, or platelet dysfunction.31 However, test results can be influenced by the sample’s hematocrit. Although the PFA-100 does not detect all platelet dysfunctions or cases of von Willebrand disease, when used in conjunction with a standardized questionnaire, it will likely detect impaired hemostasis in most cases.31,32
Prothrombin Time
The PT is a measure of the function of the extrinsic and common coagulation pathways. It represents the time (in seconds) for the patient’s plasma to clot after the addition of calcium and thromboplastin (an activator of the extrinsic pathway).33,34 Isolated prolongation of the PT is seen most commonly in patients who are deficient in vitamin K due to previous antibiotic treatment. It also occurs with factor VII deficiency, mild hypofibrinogenemia, dysfibrinogenemia, and warfarin therapy. The PT may also be prolonged with significant liver dysfunction.33,34
Partial Thromboplastin Time
The aPTT measures the function of the intrinsic and common coagulation pathways. The aPTT represents the time (in seconds) for the patient’s plasma to clot after the addition of phospholipid, calcium and an intrinsic pathway activator. The aPTT detects deficiencies in factors XII, XI, IX, and VIII and in the common pathway, but mild factor deficiencies may be missed. The aPTT also is used to monitor anticoagulation with heparin.33,34
Several inherited disorders of coagulation are not detected by the preceding tests. Results from standard hemostatic screening tests, such as the aPTT and international normalized ratio (INR) assessments, are normal in factor XIII (FXIII) deficiency. Therefore, assessment of clot stability is the most common screening test used for FXIII deficiency with a quantitative assay required to confirm the diagnosis of FXIII deficiency.35 Von Willebrand disease patients may have normal or prolonged aPTTs, and patients with a deficiency in α2-antiplasmin have a normal aPTT. Both the PT and aPTT are prolonged in patients with deficiencies of factors X and V, prothrombin, and fibrinogen and in patients with DIC or severe liver disease.34,36
Fibrinogen
The standard method for fibrinogen determination measures clottable fibrinogen by using a kinetic assay. Normal levels of fibrinogen are 150–350 mg/dL. As fibrinogen is the substrate for the final reaction in the formation of a clot and all plasma-based screening tests depend on the formation of a clot as the end point of the reaction, fibrinogen levels below 80 mg/dL prolong the PT, aPTT, and thrombin time and therefore make the results uninterpretable. Large amounts of fibrin degradation products interfere with the formation of fibrin and cause an artificially low level of measured fibrinogen. An immunologic-based assay for fibrinogen is used to measure both clottable and nonclottable fibrinogen. This test is most often used in identifying patients with a dysfibrinogenemia in whom the functional level of fibrinogen is low and the immunologic level is normal.34,36
Inhibitor Screening Tests
Repeating the PT or aPTT by using a 1 : 1 mix of patient plasma with normal plasma is a useful procedure for investigating a prolonged PT or aPTT. Normal plasma has, by definition, 100% levels of all factors. When mixed with an equal volume of patient plasma, if there is a minimum of 50% of any given factor present, the PT or aPTT should normalize. Correction of the clotting time suggests the presence of a factor deficiency whereas lack of normalization suggests the presence of an inhibitor that interferes with either thrombin or fibrin formation.33,34
Two types of acquired inhibitors prolong the aPTT. One blocks or inactivates one of the intrinsic factors, whereas the other is a lupus-like inhibitor that interferes with phospholipid-based clotting reactions. The first type of inhibitor occurs in 10–15% of hemophilia A patients and can occur spontaneously, but it is extremely rare in nonhemophiliac children.37 The lupus-like inhibitor is associated not with bleeding problems but rather with an increased risk of thrombotic problems in adults. Lupus-like inhibitors are mentioned because they commonly cause prolongations of the aPTT.38 Specific investigation of either of these situations should be referred to a coagulation reference laboratory.
Platelet Function Studies
Platelet function studies measure in vitro platelet aggregation. In this procedure, platelet-rich plasma is incubated with an agonist and then changes are noted in the amount of light transmitted through the platelet suspension. Agonists used to induce platelet aggregation include collagen, epinephrine, ADP, thrombin, and ristocetin. Three distinct phases are seen in the reaction. The first is an initial change in the shape of the platelets, leading to a temporary decrease in light transmission. Next is the first wave of aggregation, which is a reversible platelet-platelet interaction. With additional stimulation, the final phase—the second wave of aggregation—occurs and produces irreversible platelet aggregation. The second wave of aggregation is due to release of the platelet granules and thromboxane A2 synthesis. The release reaction is blocked by aspirin and is absent in patients with an inherited storage pool defect, congenital deficiency in thromboxane A2 synthesis, or cyclooxygenase deficiency.7 The PFA-100 has become the test of choice to replace the bleeding time and is used to screen for a variety of disorders, but full characterization of platelet function requires traditional platelet aggregation studies in a specialized laboratory.
Specific Factor Assays
Specific factor assays are available for all known coagulation, fibrinolysis, and anticoagulation factors to quantify their levels in plasma. These tests are not indicated unless a screening test result is abnormal. The only exception involves the patient with a history that is suggestive of von Willebrand disease, factor XIII deficiency, or dysfibrinogenemia. In these cases, the aPTT may not be sensitive enough to detect the disorder. Further testing may be justified on clinical suspicion based on the patient’s history.33,34 For von Willebrand disease, the workup consists of measuring factor VIII levels, vWF antigen levels, ristocetin co-factor activity, and ristocetin-induced platelet aggregation. Analysis of the distribution of vWF multimers can be useful to the hematologist in identifying the specific type of von Willebrand disease.10–12
Tests for Disseminated Intravascular Coagulation
The tests that are available in most hospital laboratories for identification of DIC are semiquantitative fibrin or fibrinogen degradation product assays, which involve a slide agglutination procedure or a D-dimer assay. An increased amount of these degradation products suggests that either plasmin has circulated to lyse fibrin and fibrinogen or the patient’s hepatic function is insufficient to clear the small amounts of regularly produced degradation products. The D-dimer test is a slide agglutination procedure that tests for the presence of two D subunits of fibrin that are cross-linked by factor XIII. This test provides specific evidence that plasmin has digested the fibrin clot and not fibrinogen. It is positive in patients with DIC, in patients with resolving large intravascular clots, and in patients with hepatic insufficiency. Specific assays to demonstrate the presence of soluble fibrin monomer complexes or fibrinopeptides produced by the conversion of prothrombin to thrombin are also useful in some situations and available in specialized laboratories.34,39
Hemophilia A and B
Hemophilia A and B are X-linked recessive bleeding disorders caused by decreased levels of functional procoagulant factor VIII (FVIII) and factor IX (FIX), respectively. Approximately 80% of all hemophilia patients have FVIII deficiency, which is classic hemophilia. The remaining 20% have FIX deficiency, which is called Christmas disease. These are rare disorders, with a prevalence of only 13.4/100,000 males.41 Until 1964, the treatment of hemophilia was limited by volume restrictions imposed by the use of whole blood or fresh frozen plasma. At that time, the FVIII-rich fraction of fresh frozen plasma (known as cryoprecipitate) was discovered.42 Specific lyophilized FVIII concentrates have since been developed, as have prothrombin concentrates containing factors II, VII, IX, and X. Concentrates containing only FIX for the treatment of hemophilia B patients and factor VIII/vWF concentrates for the treatment of von Willebrand disease have also been developed.43,44 The lyophilized factor concentrates have allowed storage of the clotting factor using standard refrigeration and have permitted the outpatient treatment of bleeding episodes plus the development of home self-infusion programs.45 This treatment, combined with the development of comprehensive hemophilia treatment centers, has produced a remarkable change in the outlook for these patients who previously developed significant joint deformities in their teens to 20s and were frequently wheelchair bound in adult life. Home therapy has decreased the damage caused by hemarthroses, with hemophiliac children born since the mid-1970s having far fewer joint deformities than do older hemophilia patients. These factor concentrates have allowed operations to be performed with much less risk, even to the point that orthopedic procedures can be readily accomplished.46 Moreover, the comprehensive hemophilia treatment system has shown a 40% reduction in mortality for hemophilia patients.47
Viral infections transmitted by cryoprecipitate and factor concentrates were the major problem faced by hemophilia patients in the late 1970s to mid-1980s. Approximately 60% of all hemophilia patients became human immunodeficiency virus (HIV) positive in the 1980s.48 Hepatitis C is the other major viral infection that was transmitted by plasma-derived factor concentrates used to treat hemophilia. Estimates from the mid-1980s are that more than 90% of multiply transfused hemophiliacs were positive for non-A, non-B hepatitis and that more than 95% had been infected with hepatitis B.49 A different study showed that 75% of HIV-negative hemophilia patients, treated with earlier plasma-derived factor concentrates, have evidence of hepatitis C infection.50 No documented cases of HIV or hepatitis C transmission by clotting factor concentrates after 1987 are known.48 Since 1993, recombinant-produced FVIII concentrates have been available. At present, only recombinant FVIII and FIX concentrates are used for the treatment of patients with newly diagnosed hemophilia A and B.51
In the future, long-acting FVIII as replacement therapy for hemophilia would significantly improve treatment options for patients. To develop factors with an extended circulating half-life, but without a reduction in activity, engineered FVIII variants with surface-exposed cysteine residues to which a polyethylene glycol polymer is conjugated are being studied in animals.52
Hemophilia patients are classified into three categories based on their level of circulating factor. Severe hemophiliacs (factor levels below 1%) have a high risk of bleeding and are managed with factor prophylaxis to prevent joint damage.41,53 Bleeding occurs in areas subject to minor trauma. Hemarthroses, hematomas, and ecchymoses are common. Recurrent hemarthroses can cause pseudotumors of the bone, whereas hematomas can cause ischemic compartment syndromes. In moderate hemophiliacs (factor levels of 1–5%), spontaneous hemorrhage occurs infrequently but relatively minor trauma can cause bleeding into joints or soft tissues.
Mild hemophiliacs, with levels >5%, rarely have bleeding problems and typically have problems only with major trauma or surgical procedures.37,41 Some mild hemophiliacs may not be diagnosed until late childhood or adulthood. Therefore, the history may not be helpful in alerting the pediatric surgeon about the risk of bleeding. Moreover, because one-third of all cases of hemophilia are caused by new mutations, there also may not be a family history to arouse suspicion of a bleeding problem.37 Preoperative laboratory testing may be the only means by which mild hemophilia is diagnosed.
The need for surgical intervention in hemophilia patients most frequently center on areas of damage secondary to bleeding episodes. In 1985, the results of a review of 350 consecutive operations performed at the Orthopedic Hospital in Los Angeles were published.54 As the study represented patients from before the start of home therapy and comprehensive care, it was expected that this group would have significant orthopedic problems secondary to multiple hemarthroses. Of the 350 procedures reviewed, 312 were characterized as serious and 38 of lesser intensity; 318 operations were on hemophiliacs with moderate and severe hemophilia; and 30 were on patients with mild hemophilia. As expected, musculoskeletal procedures made up two-thirds of all operations on moderate and severe hemophiliacs and half of all operations on mild hemophiliacs. Bleeding problems during operation were not observed, but 23% of all serious operations were complicated by postoperative hemorrhages. Only operations on the knee had significantly more postoperative hemorrhages (40%). Operations on other joints and soft tissue areas had similar rates of complications (15%). Most of the postoperative hemorrhages occurred with plasma factor levels greater than 30%, which is the minimum level that is considered hemostatic. The authors also noted that the incidence of postoperative hemorrhage decreased after postoperative day 11. Interestingly, other studies have found that vigorous physical therapy may cause postoperative hemorrhage and have therefore recommended the continuation of factor replacement throughout the period of physical therapy.54,55
The management of the hemophilic patient requires close cooperation among surgeons, hematologists, and personnel in the hemophilia center, the coagulation laboratory, and the pharmacy or blood bank. Careful preoperative planning is essential to the success of the intervention, and an adequate supply of clotting factor concentrate must be available to cover the child’s needs before admission. The patient also must be screened for the presence of an inhibitor to either FVIII or FIX during the two to four weeks before the operation. A low-titer inhibitor may be overcome with increased doses of factor, but high-titer inhibitors may require the use of activated prothrombin complex concentrate (FEIBA) or recombinant activated factor VII (rFVII) to ‘bypass’ the effect of the antibody against either FVIII or FIX. These patients have been desensitized with daily doses of human factor concentrate over a period of months to years, restoring their response to regular infusions of factor VIII or IX.46,56,57
At our institution, on the day of surgery the hemophilia patient receives a bolus dose of factor (usually 50 units/kg of FVIII in hemophilia A patients), and a continuous infusion of 4–8 units/kg/h of FVIII (for the hemophilia A patient) is started to maintain a factor level greater than 80% for the next one to two days.58 The factor level is checked immediately before the operation and is the final screen for the presence of an inhibitor. The infusion is maintained throughout the procedure and is then reduced on the second or third postoperative day to allow the plasma levels to decrease to 50%. Replacement is continued for a full ten to 14 days. Daily factor levels are necessary to ensure appropriate levels. For neurosurgical or orthopedic procedures, much longer periods of factor coverage—even four to six weeks—are utilized, especially if significant physical therapy is planned.41,46
Previously, the hemophilia B patient undergoing an operation had specific problems because of the thrombogenic risk inherent in the use of older FIX concentrates. Since the advent of newer, more purified plasma-derived and recombinant-produced FIX concentrates, operative interventions in hemophilia B patients have been performed without excess thrombotic problems.59,60
Clotting Factor Dosing
Factor VIII is dosed differently from FIX, based on their half-lives. FVIII has an eight- to 12-hour half-life, and the infusion of 1 unit/kg of body weight increases the plasma level by 2%. If a severe hemophilia A patient weighs 50 kg, an infusion of 25 units/kg, or 1250 units, of FVIII will raise his factor level to 50%. FIX has a half-life of 24 hours and must be infused in larger amounts than FVIII to raise the plasma level. Infusion of 1 unit/kg of FIX will raise the plasma level only by 1%. Continuous infusion of highly purified FIX, as well as FVIII, has been shown to prevent excessive peaks and troughs of factor levels, is simpler to manage, and decreases the cost by decreasing the overall amount of factor used. It has not shown to cause any problems with excess thrombosis.61 Recombinant FIX has a marked variability in dose response to infusions, and individual recovery studies may be needed before it is used for surgical hemostasis. Often, a 20% increase in dose is needed to achieve the same factor levels as obtained by use of plasma-derived FIX.62
Neonatal Hemostasis
The newborn’s coagulation system is not fully mature until 6 months after birth. The lower levels of procoagulant, fibrinolytic, and anticoagulant proteins in neonatal patients complicate both operations and the care of sick and preterm infants. Platelet counts are within the usual adult normal ranges. These platelets have a lower function than those of adults, but enough to produce a normal bleeding time.62 Circulating coagulation factors do not cross the placenta, and infants with inherited deficiencies of clotting factors, fibrinolytic proteins, or natural anticoagulants may initially be seen in the neonatal period. Levels of fibrinogen, factor V, factor VIII, and vWF are within the adult normal range at birth.63 All other procoagulants are at reduced levels, depending on gestational age. Vitamin K-dependent factors may become further depressed in infants who are breast fed and not given vitamin K at birth.62
Of more concern are the low levels of anticoagulant and fibrinolytic proteins. Very low levels of protein C have been associated with purpura fulminans in newborns. In sick infants, levels of antithrombin III and plasminogen may be inadequate to deal with increased levels of clot-promoting activity in the blood. Sick infants with indwelling catheters are at significant risk of thrombotic complications.64
Disseminated Intravascular Coagulation
DIC is the inappropriate activation of both thrombin and fibrin. It may follow sepsis, hypotension, hypoxemia, trauma, malignancy, burns, and extracorporeal circulation. Hemorrhage due to the depletion of clotting factors as well as thrombosis due to the excess formation of clot are seen, and the end-organ damage caused by ischemia and impairment of blood flow causes irreversible disease and death.65
Acute DIC is associated with the consumption of factors II, V, VIII, X, and XIII, as well as fibrinogen, antithrombin III, plasminogen, and platelets. Review of the peripheral smear usually shows a microangiopathic hemolytic anemia. The PT and aPTT may both be prolonged, and the fibrinogen level ultimately decreases as the DIC worsens. The presence of D-dimers may indicate the presence of DIC, but they may also be elevated due to thrombus or hepatic dysfunction. Antithrombin III levels may be low, and the use of antithrombin III concentrates in septic shock may play a role in the future treatment of DIC. However, adult studies have not shown any improvement in mortality for patients with septicemia treated with antithrombin III.66 At present, the major therapy for DIC is correction of the underlying disorder with fresh frozen plasma and platelet transfusions as indicated to support hemostasis. Low-dose heparin infusions have not been shown to appreciably improve the outcome.65,67
Management of Quantitative and Qualitative Platelet Disorders
Thrombocytopenias are caused by either inadequate production of platelets by the bone marrow, or by increased destruction or sequestration of the platelets in the circulation. The history and physical examination may be suggestive of a diagnosis that can be confirmed by laboratory testing. Medication use, a family history of blood disorders, a history of recent viral infection, short stature, absent thumbs or radii, or a congenital malformation may indicate a defect in platelet production. The destruction may be immunologic, as in immune thrombocytopenic purpura (ITP); mechanical, as in septicemia; or drug induced, as in patients with sensitivity to heparin or cimetidine. Establishing the cause of the thrombocytopenia determines the therapy needed to restore the platelet count in preparing the patient for operation. The clinical response to therapeutic modalities, such as a platelet transfusion, can be important tests and can help direct further investigations. In patients with immune-based platelet consumptions, such as ITP, usually no response is found to platelet transfusion. Moreover, only a very short response may be seen in patients with other causes of increased consumption. Management of the patient is then aimed at reducing the consumption and should involve consultation with a hematologist about the use of corticosteroids, the use of intravenous immunoglobulin or anti-D immunoglobulin, the discontinuation of medications, and other treatment modalities.68
If the thrombocytopenia is caused by a lack of production of platelets, due to either aplastic anemia, malignancy, or chemotherapy, transfusion with platelet concentrates to increase the platelet count above a minimum of 50,000/µL will allow minor procedures to be performed safely. Most surgeons and anesthesiologists prefer for the platelet count to be greater than 100,000/µL before major surgery. Continued monitoring of platelet counts is vital because further transfusions may be needed to keep the platelet count above 50,000/µL for three to five days after operation.69
Qualitative platelet defects can be caused by rare congenital defects such as Bernard–Soulier syndrome, Glanzmann thrombasthenia, or platelet storage pool disease. Alternatively, they can be caused by drug ingestions such as an aspirin-induced cyclooxygenase deficiency. In these situations, transfusion of normal donor platelets provides adequate hemostasis for the operation. Discontinuation of all aspirin-containing products one week before operation permits correction of the cyclooxygenase deficiency as new platelets are produced.6,70
Disorders of Thrombin Generation and Fibrin Formation
Patients with rare deficiencies of other clotting factors, such as factors XI, X, VII, V, prothrombin, and fibrinogen, can have clinical bleeding depending on the level of deficiency. Most of these disorders are inherited in an autosomal recessive manner and can therefore affect both male and female patients. Replacement therapy with fresh frozen plasma or, in certain situations, with prothrombin complex concentrates corrects the deficiency and should be conducted under the direction of a hematologist.46,71
Vitamin K deficiency, both in the neonatal period and from malabsorption, can cause deficiencies of factors II, VII, IX, and X. Treatment with 1–2 mg of intravenous vitamin K may begin to correct the deficiencies within four to six hours. However, if a procedure is contemplated, fresh frozen plasma (15 mL/kg) should be given with the vitamin K, and repeated. Also, the PT should be monitored for correction of the coagulopathy before the operation. Laboratory monitoring should be maintained during the postoperative period to ensure continuation of the appropriate factor levels.5
Patients with factor XIII deficiency often present with delayed bleeding from the umbilical cord, rebleeding from wounds, intracranial hemorrhage, and poor wound healing. These problems may be treated with relatively small amounts of fresh frozen plasma (5–10 mL/kg). Because factor XIII has a half-life of six days, this treatment is usually needed only once to stop bleeding or at the time of operation.16,46 Patients with dysfibrinogenemia or afibrinogenemia may be given fresh frozen plasma or cryoprecipitate.46 There are ongoing trials looking at a fibrinogen concentrate preparation infusion for these conditions.
Fibrinolytic and Thrombotic Disorders
Failure to control excess fibrinolysis can result in a bleeding problem, and deficiencies of the naturally occurring anticoagulants may result in excess clot formation. A severe hemorrhagic disorder due to a deficiency of α2-antiplasmin has responded to treatment with aminocaproic acid or tranexamic acid, both antifibrinolytic agents.37 Congenital antithrombin III, protein S, and protein C deficiencies are associated with recurrent thrombosis and are usually controlled with oral anticoagulants.37 Factor V Leiden, prothrombin G20210A, and other activated protein C resistance gene mutations will cause or add additional risk for thrombosis in proportion to their homozygous or heterozygous states.72–74 Discontinuation of the anticoagulation is needed before an operation. The patients will require replacement therapy during the procedure and in the postoperative period until the anticoagulation can be restarted. Depending on the deficiency, antithrombin III concentrates or fresh frozen plasma can be used for replacement therapy.
Recombinant Activated Factor VII
Recombinant activated factor VII (rFVIIa) was developed for the treatment of bleeding in patients with hemophilia A or B who had inhibitors, and was approved by the ultrasound Food and Drug Administration for this indication in 1999.75–77 Good hemostasis with few side effects has been found in patients with intracranial hemorrhage, postlaparotomy and postpartum hemorrhage, hemorrhage into the gluteal muscles (as a complication after cholecystectomy), and for surgical prophylaxis for major and minor procedures.78–80 Home treatment programs for those hemophilia patients who have inhibitors now use rFVIIa as front-line therapy for bleeding.81 Children have a more rapid rate of clearance (elimination mean half-life, 1.32 hours in children vs 2.74 hours in adults).82 They also seem to have fewer side effects with this treatment.77,83 Although various dosages and schedules have been studied, initial recommended therapy in hemophilia A or B with inhibitors is 90 mg/kg intravenously every two hours until the bleeding is controlled.84
The off-label use of rFVIIa has been reported in therapy-resistant severe bleeding from other conditions such as congenital factor VII deficiency, chronic liver disease, and inherited platelet disorders.85–87 Successes in patients without a known bleeding disorder who have trauma or postoperative hemorrhage also are described.83,85,88,89 These reports should be interpreted with caution because rFVIIa is currently not the standard of care in any of these off-label uses and exceptional circumstances impelled its use. It is highly recommended that rFVIIa be administered under the supervision of a physician experienced in its use who can anticipate the risks and respond to the complications, particularly risks of thrombosis, which are reported in 1–3% of patients.75,90,91 rFVIIa shows great promise in the emergency treatment of uncontrolled hemorrhage for many situations, and is becoming the standard of care for the treatment of intracranial hemorrage.92
Sickle Cell Disease
Sickle cell disease (SCD) is the most common disorder identified by neonatal blood screening with approximately 2000 affected infants born in the ultrasound each year. Overall, the incidence of SCD exceeds that of most other serious genetic disorders, including cystic fibrosis and hemophilia.93,94 SCD is caused by a genetic mutation that results in an amino acid change in the β-globin and the production of sickle hemoglobin (Hb S) instead of normal hemoglobin. The sickle cell gene, in combination with any other abnormal β-globin gene, results in SCD. There are many types of SCD. The most common include sickle cell anemia (Hb SS), the sickle β-thalassemias (Hb Sβ0 and Hb Sβ+), hemoglobin SC disease (Hb SC) and sickle cell/hereditary persistence of fetal hemoglobin (S/HPFH). Sickle cell anemia is the most common and, in general, the most severe form of SCD. Sickle β0-thalassemia patients have clinical manifestations similar to patients with Hb SS disease. Sickle-C disease is the second most common form of SCD and generally has a more benign clinical course than does Hb SS or sickle β0-thalassemia. Sickle β+-thalassemia and S/HPFH patients also usually have a more benign clinical course. Patients with Hb SS disease and sickle β0-thalassemia generally have lower hemoglobin levels and present a greater risk under general anesthesia than do patients with Hb SC disease and sickle β+-thalassemia. Patients with S/HPFH may actually have hemoglobin levels that approach or are normal.
The red cell membrane is abnormal in patients with SCD, and the red cell life span is shortened by hemolysis. Intermittent episodes of vascular occlusion cause tissue ischemia, which results in acute and chronic organ dysfunction.95 Consequently, patients with SCD require special considerations to prevent perioperative complications.
Because of the nature of the complications of SCD, people with this disorder are more likely than the general population to need an operation during their lifetime.96 According to one study, the most common procedures were cholecystectomy; ear, nose, and throat procedures; orthopedic procedures; splenectomy; or herniorrhaphy.97 Cholecystectomy, splenectomy, and orthopedic procedures are often required to treat complications of SCD.
The differential diagnosis for acute abdominal pain in a patient with SCD includes an uncomplicated sickle cell acute pain episode (‘crisis’), cholelithiasis, appendicitis, pancreatitis, ulcer, constipation, pneumonia, pericarditis, and splenic sequestration. Whereas 50% of painful crises include abdominal pain, they are usually associated with pain in the chest, back, and joints. However, although previous episodes of pain that are similar in character suggest an acute painful episode, the incidence of gallstones, peptic ulcer disease, and pyelonephritis is increased in these patients. Complications such as splenic or hepatic sequestration are unique problems in patients with this disease. Abdominal pain as a solitary symptom, especially when accompanied by fever, leukocytosis, and localized abdominal tenderness, is suggestive of pathology other than that which occurs with a sickle cell acute painful episode. A study that reviewed the presentation and management of acute abdominal conditions in adults with SCD suggested that a surgical condition is more likely if the pain does not resemble previous painful episodes and if no precipitating event is found.98 Acute painful episodes were relieved within 48 hours with hydration and oxygen in 97% of patients, whereas no patient with a surgical disease achieved pain relief over the same period with these modalities. The leukocyte count and serum bilirubin were not helpful in establishing the correct diagnosis.
Vaso-occlusive episodes can also produce bone pain and fever, symptoms that are difficult to differentiate from those of osteomyelitis. The majority of bone pain in SCD is due to vaso-occlusion, but osteomyelitis secondary to Salmonella species or Staphylococcus aureus is not infrequent.99,100 The presence of an immature leukocyte count or elevation of the sedimentation rate, C-reactive protein, or leukocyte alkaline phosphatase is suggestive of a bone infection and may be an indication for aspiration of the bone lesion. Radiographic studies including plain films, bone scan, or magnetic resonance imaging are generally less helpful but may be useful in arriving at the proper diagnosis when positive and combined with the appropriate clinical findings.100
Preoperative Assessment and Management
An optimal outcome requires careful preoperative, intraoperative, and postoperative management by a team consisting of a surgeon, anesthesiologist, and hematologist. Potential sickle cell-related complications include acute chest syndrome, pain episodes, hyperhemolytic crisis, aplastic crisis, alloimmunization with delayed transfusion reactions, and infections. The outcome of children with SCD requiring a procedure is improved by careful attention to the cardiorespiratory, hemodynamic, hydration, infectious, neurologic, and nutritional status of the child.96,101–103 If possible, procedures should be performed when the child is in his or her usual state of health with regard to the SCD. Attention should be directed toward chronic manifestations of disease because predictors of a poor postoperative outcome include increased age, recent exacerbations of the disease, and preexisting infection and pregnancy.104 Particular attention should be directed toward any recent history of acute chest syndrome, pneumonia, wheezing, and alloimmunization. Special efforts must be made to avoid perioperative hypoxia, hypothermia, acidosis, and dehydration because any of these events can result in serious morbidity.
Many centers perform preoperative transfusions with the aim of reducing the complications of surgery and anesthesia.105 The largest study that examined the role of transfusion in the preoperative management of sickle cell anemia was a randomized study that compared exchange transfusion (with a goal of achieving an Hb value of >10 g/dL and Hb S value of <30%) versus simple transfusion (to achieve an Hb value of >10 g/dL).97 This study concluded that not only was simple transfusion as effective as exchange transfusion in preventing perioperative complications, but it also provided a significantly lower rate of transfusion-related complications. The question about which procedures are safe to perform in children with SCD without preoperative transfusion remains controversial because there is a lack of randomized controlled trials to answer this question. However, transfusion only to increase the Hb level to 10 g/dL for major procedures and blood replacement for both profound anemia of less than 5 g/dL and intraoperative hemorrhage appear appropriate.106 Several studies suggest that minor procedures can be safely undertaken without transfusion.96,105,107 Alloimmunization is minimized by using antigen-matched blood (matched for K, C, E, S, Fy, and Jk antigens).103 Regardless of transfusion status, strong multidisciplinary collaboration is vital throughout the perioperative period.
Intraoperative Management
Anesthetic considerations are based more on the type of operation than on the presence of SCD because no single anesthetic technique has been shown to be the gold standard. However, regional anesthetic techniques may allow for opioid sparing postoperatively.104 The goals of anesthetic management are to avoid factors that predispose the patient to sickling (e.g., hypoxemia, hypothermia, dehydration, and acidosis). Careful monitoring for hypoxia, hypothermia, acidosis, and dehydration is essential. Monitoring should include arterial blood gases, digital oxygen saturation, end-tidal carbon dioxide, temperature, electrocardiogram, blood pressure, and urine output.104,108
Postoperative Management
Appropriate levels of analgesia (preferably by a continuous intravenous line and patient-controlled analgesia, if appropriate) should be provided so the patient is comfortable for ambulation and for vigorous pulmonary toilet. The patient must be monitored closely for the occurrence of pulmonary edema or atelectasis that can progress to acute chest syndrome.109
Specific Surgical Conditions
Adenotonsillectomy
Adenotonsillectomy is a fairly common procedure in children with SCD. Adenotonsillar hypertrophy, which may be associated with early functional hyposplenism and obstructive sleep apnea (OSA) secondary to enlarged adenoids, occurs somewhat frequently.96, 110,111 As with other types of surgery, preoperative transfusions should be performed before operation.112 Clinicians should be aware that postoperative complications may be greater in patients who are younger or have OSA.96,113
Cholelithiasis and Cholecystectomy
Abdominal operations such as cholecystectomy and splenectomy are the most frequent abdominal procedures in patients with SCD.96,106 Currently, there is no clear consensus regarding the appropriate management for SCD children who have cholelithiasis. The reported prevalence of cholelithiasis varies from 4–55%.114,115 This wide variation is dependent on the ages of the study population and the diagnostic modalities used.116 We routinely screen symptomatic children with ultrasonography and serum studies (e.g., total and direct bilirubin, serum glutamic-oxaloacetic transaminase, serum glutamate-pyruvate transaminase, alkaline phosphatase, and γ-glutamyl-transpeptidase). It is our practice to screen all SCD children for gallstones no later than age 12.
A child with SCD and symptomatic cholelithiasis should undergo cholecystectomy after appropriate preoperative preparation to avoid the increased morbidity of an emergency operation on an unprepared patient.116–121 If indicated, intraoperative cholangiography can be performed to assess for common duct stones.122
The utility of laparoscopic cholecystectomy in SCD was first reported in 1990.123 Since that time, laparoscopic cholecystectomy has been performed with increasing frequency in children with SCD.116,124–127 The advantages of laparoscopic cholecystectomy over open cholecystectomy are decreased pain, earlier feeding, earlier discharge, earlier return to school, and improved cosmesis. The presence of common duct stones at times complicates the laparoscopic approach and may require conversion to an open operation for removal. At present, the role of extracorporeal shock wave lithotripsy as a palliative therapeutic modality is uncertain.
Splenic Sequestration and Splenectomy
Before the advent of routine newborn screening for hemoglobinopathies, acute splenic sequestration was the second most common cause of mortality in children younger than age 5 years with sickle cell anemia.128 Splenic sequestration classically was first seen with the acute onset of pallor and listlessness, a precipitate decrease in hemoglobin, thrombocytopenia, and massive splenomegaly.129 It now appears that parental education along with earlier recognition and immediate treatment with volume support (including red blood cell transfusions) has resulted in significantly decreased mortality for this condition. It is rare for an otherwise uncomplicated patient with Hb SS disease who is older than 6 years of age to develop acute splenic sequestration syndrome. However, patients with Hb SC and sickle β+-thalassemia disease can experience splenic sequestration at an older age.130
The management of the SCD child with splenic sequestration can be difficult. The rate of recurrent splenic sequestration is high and greatly influences subsequent management, which may be divided into observation only, chronic transfusion, and splenectomy. Indications for these approaches are not clearly defined.103 The benefit of splenectomy must be balanced with the increased risk of overwhelming bacterial sepsis in the younger asplenic SCD patient.103,131 Partial splenectomy132,133 and, especially, laparoscopic splenectomy134 are being performed more commonly as the number of experienced practitioners grows.
References
1. Thompson, AR, Harker, LA. Manual of Hemostasis and Thrombosis, 3rd ed. Philadelphia: FA Davis; 1983.
2. Moncada, S, Gryglewski, R, Bunting, S, et al. An enzyme isolated from arteries transforms prostaglandin endoperoxides to an unstable substance that inhibits platelet aggregation. Nature. 1976; 263:663–665.
3. Stern, D, Nawroth, P, Handley, D, et al. An endothelial cell-dependent pathway of coagulation. Proc Natl Acad Sci U S A. 1985; 82:2523–2527.
4. Esmon, C. Blood coagulation. In: Nathan D, Orkin S, eds. Nathan and Oski’s Hematology of Infancy and Childhood. Philadelphia: WB Saunders; 1998:1532.
5. Saito, H. Normal hemostatic mechanisms. In: Ratnoff O, Forbes C, eds. Disorders of Hemostasis. 2nd ed. Philadelphia: WB Saunders; 1996:23–52.
6. George, JN, Nurden, AT, Phillips, DR. Molecular defects in interactions of platelets with the vessel wall. N Engl J Med. 1984; 311:1084–1098.
7. Marcus, A. Platelets and their disorders. In Ratnoff OD, Forbes CD, eds.: Disorders of Hemostasis, 3rd ed, Philadelphia: WB Saunders, 1996.
8. Turrito, V, Baumgartner, H. Platelet-surface interactions. In: Coleman R, Hirsh J, Marder V, eds. Hemostasis and Thrombosis: Basic Principles and Clinical Practice. 2nd ed. Philadelphia, JB: Lippincott; 1987:555.
9. Nurden, AT, Didry, D, Rosa, JP. Molecular defects of platelets in Bernard-Soulier syndrome. Blood Cells. 1983; 9:333–358.
10. Castaman, G, Federici, AB, Rodeghiero, F, et al. Von Willebrand’s disease in the year 2003: Towards the complete identification of gene defects for correct diagnosis and treatment. Haematologica. 2003; 88:94–108.
11. Sadler, J. A revised classification of von Willebrand disease. Thromb Haemost. 1994; 71:520–525.
12. Tuddenham, EG. Von Willebrand factor and its disorders: An overview of recent molecular studies. Blood Rev. 1989; 3:251–262.
13. Lisman, T, Weeterings, C, de Groot, P. Platelet aggregation: Involvement of thrombin and fibrin(ogen). Front Biosci. 2005; 10:2504–2517.
14. Coleman, R, Walsh, P. Mechanisms of platelet aggregation. In: Coleman R, Hirsh J, Marder V, eds. Hemostasis and Thrombosis: Basic Principles and Clinical Practice. 2nd ed. Philadelphia, JB: Lippincott; 1987:594.
15. Mackie, IJ, Bull, HA. Normal haemostasis and its regulation. Blood Rev. 1989; 3:237–250.
16. Lorand, L, Losowsky, MS, Miloszewski, KJ. Human factor XIII: Fibrin-stabilizing factor. Prog Hemost Thromb. 1980; 5:245–290.
17. Rosenberg, RD, Rosenberg, JS. Natural anticoagulant mechanisms. J Clin Invest. 1984; 74:1–6.
18. Rodeghiero, F, Tosetto, A, Castaman, G. How to estimate bleeding risk in mild bleeding disorders. J Thromb Haemost. 2007; 5(Suppl 1):157–166.
19. Bidlingmaier, C, Sax, F, Treutwein, J, et al. The PTT is not enough—Preoperative coagulation screening in children. J Thromb Haemost. 2007; 5(Suppl 2):P-S-221.
20. Greaves, M, Watson, HG. Approach to the diagnosis and management of mild bleeding disorders. J Thromb Haemost. 2007; 5(Suppl 1):167–174.
21. Shaw, PH, Reynolds, S, Gunawardena, S, et al. The prevalence of bleeding disorders among healthy pediatric patients with abnormal preprocedural coagulation studies. J Pediatr Hematol Oncol. 2008; 30:135–141.
22. Rapaport, SI. Preoperative hemostatic evaluation: Which tests, if any? Blood. 1983; 61:229–231.
23. Sramek, A, Eikenboom, JC, Briet, E, et al. Usefulness of patient interview in bleeding disorders. Arch Intern Med. 1995; 155:1409–1415.
24. Rodeghiero, F, Castaman, G, Tosetto, A, et al. The discriminate power of bleeding history for the diagnosis of type 1 von Willebrand disease: An international, multicenter study. J Thromb Haemost. 2005; 3:2619–2626.
25. Anwar, R, Minford, A, Gallivan, L, et al. Delayed umbilical bleeding—a presenting feature for factor XIII deficiency: Clinical features, genetics, and management. Pediatrics. 2002; 109:e32.
26. Shen, YM, Frenkel, EP. Acquired platelet dysfunction. Hematol Oncol Clin North Am. 21, 2007. [647–61.vi].
27. George, JNM, Shattil, SJM. The clinical importance of acquired abnormalities of platelet function. N Engl J Med. 1991; 324:27–39.
28. Acosta, M, Edwards, R, Jaffee, IM, et al. A practical approach to pediatric patients referred with an abnormal coagulation profile. Arch Pathol Lab Med. 2005; 129:1011–1016.
29. Merck Manual Online Series. Thrombocytopenia and Platelet Dysfunction. In: Porter R, ed. Hematology and Oncology. Whitehouse Station, NJ: Merck & Co, 2008.
30. Kam, PC. Anaesthetic management of a patient with thrombocytopenia. Curr Opin Anaesthesiol. 2008; 21:369–374.
31. Harrison, P. The role of PFA-100 testing in the investigation and management of haemostatic defects in children and adults. Br J Haematol. 2005; 130:3–10.
32. Koscielny, J, von Tempelhoff, GF, Ziemer, S, et al. A practical concept for preoperative management of patients with impaired primary hemostasis. Clin Appl Thromb Hemost. 2004; 10:155–166.
33. Kamal, AH, Tefferi, A, Pruthi, RK. How to interpret and pursue an abnormal prothrombin time, activated partial thromboplastin time, and bleeding time in adults. Mayo Clin Proc. 2007; 82:864–873.
34. Wicklund, B. The bleeding child: Congenital and acquired disorders. In: Hillyer C, Strauss R, Luban N, eds. Handbook of Pediatric Transfusion Medicine. Boston: Elsevier, 2004.
35. Israels, S. Factor XIII deficiency. emedicine, Omaha: WebMD; 2007.
36. Goodnight S, Hathaway W, eds. Disorders of Hemostasis and Thrombosis. New York: McGraw-Hill, 2001.
37. Lusher, J. Approach to the bleeding patient. In Nathan D, Orkin S, eds.: Nathan and Oski’s Hematology of Infancy and Childhood, 5th ed, Philadelphia, WB: Saunders, 1998.
38. Shapiro, SS, Thiagarajan, P. Lupus anticoagulants. Prog Hemost Thromb. 1982; 6:263–285.
39. Levi, M, Ten Cate, H. Disseminated intravascular coagulation. N Engl J Med. 1999; 341:586–592.
40. Pivalizza, E, Pivalizza, P, Gottschalk, L, et al. Celite-Activated Thrombelastography in Children. J Clin Anesth. 2001; 1:20–23.
41. Soucie, J, Evatt, B, Jackson, D. Occurrence of hemophilia in the United States. The Hemophilia Surveillance System Project Investigators. Am J Hematol. 1998; 59:288–294.
42. Pool, JG, Gershgold, EJ, Pappenhagen, AR. High-potency antihaemophilic factor concentrate prepared from cryoglobulin precipitate. Nature. 1964; 203:312.
43. Kasper, CK, Lusher, JM. Recent evolution of clotting factor concentrates for hemophilia A and B. Transfusion Practices Committee. Transfusion. 1993; 33:422–434.
44. Mannucci, PM, Chediak, J. Treatment of von Willebrand disease with a high-purity factor VIII/von Willebrand factor concentrate: A prospective, multicenter study. Blood. 2002; 99:450–456.
45. Levine, PH. Delivery of health care in hemophilia. Ann N Y Acad Sci. 1975; 240:201–207.
46. Hilgartner, MW. Factor Replacement Therapy. In: Hilgartner MW, Pochedly C, eds. Hemophilia in the Child and Adult. New York: Raven Press; 1989:1–26.
47. Soucie, JM, Nuss, R, Evatt, B, et al. Mortality among males with hemophilia: Relations with source of medical care. The Hemophilia Surveillance System Project Investigators. Blood. 2000; 96:437–442.
48. Report on the Universal Data Collection Program. Atlanta: Centers for Disease Control and Prevention, 2005.
49. Kernoff, PB, Lee, CA, Karayiannis, P, et al. High risk of non-A non-B hepatitis after a first exposure to volunteer or commercial clotting factor concentrates: Effects of prophylactic immune serum globulin. Br J Haematol. 1985; 60:469–479.
50. Troisi, CL, Hollinger, FB, Hoots, WK, et al. A multicenter study of viral hepatitis in a United States hemophilic population. Blood. 1993; 81:412–418.
51. MASAC Recommendations Concerning the Treatment of Hemophilia and Other Bleeding Disorders. National Hemophilia Foundation; revised.
52. Mei, B, Pan, C, et al. Rational design of a fully active, long-acting PEGylated factor VIII for hemophilia A treatment. Blood. 2010; 116(2):270–279.
53. Lusher, JM, Warrier, I. Hemophilia. Pediatr Rev. 1991; 12:275–281.
54. Kasper, CK, Boylen, AL, Ewing, NP, et al. Hematologic management of hemophilia A for surgery. JAMA. 1985; 253:1279–1283.
55. Manco-Johnson, MJ, Abshire, TC, Shapiro, AD, et al. Prophylaxis versus episodic treatment to prevent joint disease in boys with severe hemophilia. N Engl J Med. 2007; 357:535.
56. Scharf, R, Kucharski, W, Nowak, T. Surgery in hemophilia A patients with factor VIII inhibitor: 10-year experience. World J Surg. 1996; 20:1171–1181.
57. Jimenez-Yuste, V, Rodriguez-Merchan, EC, Alvarez, MT, et al. Controversies and challenges in elective orthopedic surgery in patients with hemophilia and inhibitors. Semin Hematol. 2008; 45:S64–S67.
58. Montgomery, RE. Hemophilia and von Willebrand disease. In: Nathan D, Orkin S, eds. Nathan and Oski’s Hematology of Infancy and Childhood. 6th ed. Philadelphia: WB Saunders; 2003:1547.
59. Scharrer, I. The need for highly purified products to treat hemophilia B. Acta Haematol. 1995; 94(Suppl 1):2–7.
60. Shapiro, AD, Di Paola, J, Cohen, A, et al. The safety and efficacy of recombinant human blood coagulation factor IX in previously untreated patients with severe or moderately severe hemophilia B. Blood. 2005; 105:518–525.
61. Kobrinsky, N. Management of hemophilia during surgery. In: Forbes C, Aledort L, Madhok R, eds. Hemophilia. Oxford: Chapman & Hall; 1997:242.
62. Shapiro, AD. Coagulation factor concentrates. In: Goodnight S, Hathaway W, eds. Disorders of Hemostasis and Thrombosis: A Clinical Guide. New York: McGraw-Hill; 2001:505.
63. Andrew, M, Paes, B, Milner, R, et al. Development of the human coagulation system in the full-term infant. Blood. 1987; 70:165–172.
64. Gibson, B. Normal and disordered coagulation. In: Hann I, Gibson B, Letsky E, eds. Fetal and Neonatal Haematology. London: Bailliere Tindall; 1991:123.
65. Bick, RL. Disseminated intravascular coagulation and related syndromes: A clinical review. Semin Thromb Hemost. 1988; 14:299–338.
66. van Beek, EJ, von der Mohlen, MA, ten Cate, JW, et al. Antithrombin III concentrate in the treatment of DIC: A retrospective follow-up study. Neth J Med. 1994; 45:206–210.
67. Albisetti, M, Andrew, M. Hemostatic abnormalities. In: de Alarcon P, Werner E, eds. Neonatal Hematology. Cambridge: Cambridge University Press; 2005:310–348.
68. George, JN. Diagnosis, clinical course, and management of idiopathic thrombocytopenic purpura. Curr Opin Hematol. 1996; 3:335–340.
69. Jackson, D. Management of thrombocytopenia. In: Coleman R, Hirsh J, Marder V, eds. Hemostasis and Thrombosis: Basic Principles and Clinical Practice. 2nd ed. Philadelphia: JB Lippincott; 1987:530.
70. Salzman, E. Hemostatic problems in surgical patients. In Coleman R, Hirsh J, Marder V, eds.: Hemostasis and Thrombosis: Basic Principles and Clinical Practice, 2nd ed, Philadelphia, JB: Lippincott, 1987.
71. Greenberg, C. Hemostasis: Pathophysiology and management of clinical disorders. In: Sabiston DJ, ed. Sabiston’s Essentials of Surgery. Philadelphia: WB Saunders; 1987:79.
72. Bick, RL. Prothrombin G20210A mutation, antithrombin, heparin co-factor II, protein C, and protein S defects. Hematol Oncol Clin North Am. 2003; 17:9–36.
73. Nicolaes, GA, Dahlback, B. Activated protein C resistance (FV[Leiden]) and thrombosis: Factor V mutations causing hypercoagulable states. Hematol Oncol Clin North Am. 2003; 17:37–61.
74. Whiteman, T, Hassouna, HI. Hypercoagulable states. Hematol Oncol Clin North Am. 14, 2000. [355–77.viii].
75. Hay, CR, Negrier, C, Ludlam, CA. The treatment of bleeding in acquired haemophilia with recombinant factor VIIa: A multicentre study. Thromb Haemost. 1997; 78:1463–1467.
76. Hedner, U. Recombinant coagulation factor VIIa: From the concept to clinical application in hemophilia treatment in 2000. Semin Thromb Hemost. 2000; 26:363–366.
77. Hedner, U, Bjoern, S, Bernvil, SS, et al. Clinical experience with human plasma-derived factor VIIa in patients with hemophilia A and high-titer inhibitors. Haemostasis. 1989; 19:335–343.
78. Arkin, S, Cooper, HA, Hutter, JJ, et al. Activated recombinant human coagulation factor VII therapy for intracranial hemorrhage in patients with hemophilia A or B with inhibitors: Results of the NovoSeven emergency-use program. Haemostasis. 1998; 28:93–98.
79. Liebman, HA, Chediak, J, Fink, KI, et al. Activated recombinant human coagulation factor VII (rFVIIa) therapy for abdominal bleeding in patients with inhibitory antibodies to factor VIII. Am J Hematol. 2000; 63:109–113.
80. Shapiro, AD, Gilchrist, GS, Hoots, WK, et al. Prospective, randomised trial of two doses of rFVIIa (NovoSeven) in haemophilia patients with inhibitors undergoing surgery. Thromb Haemost. 1998; 80:773–778.
81. Santagostino, E, Gringeri, A, Mannucci, PM. Home treatment with recombinant activated factor VII in patients with factor VIII inhibitors: The advantages of early intervention. Br J Haematol. 1999; 104:22–26.
82. Erhardtsen, E. Pharmacokinetics of recombinant activated factor VII (rFVIIa). Semin Thromb Hemost. 2000; 26:385–391.
83. Lusher, J, Ingerslev, J, Roberts, H, et al. Clinical experience with recombinant factor VIIa. Blood Coagul Fibrinolysis. 1998; 9:119–128.
84. NovoSeven [package insert]. Princeton, NJ: Novo Nordisk, Inc., 2006.
85. Hedner, U, Erhardtsen, E. Potential role for rFVIIa in transfusion medicine. Transfusion. 2002; 42:114–124.
86. Mariani, G, Testa, MG, Di Paolantonio, T, et al. Use of recombinant, activated factor VII in the treatment of congenital factor VII deficiencies. Vox Sang. 1999; 77:131–136.
87. Poon, MC, d’Oiron, R. Recombinant activated factor VII (NovoSeven) treatment of platelet-related bleeding disorders. International Registry on Recombinant Factor VIIa and Congenital Platelet Disorders Group. Blood Coagul Fibrinolysis. 2000; 11(Suppl 1):S55–S68.
88. Martinowitz, U, Kenet, G, Segal, E, et al. Recombinant activated factor VII for adjunctive hemorrhage control in trauma. J Trauma. 2001; 51:431–439.
89. McMullin, NR, Kauvar, DS, Currier, HM, et al. The clinical and laboratory response to recombinant factor VIIA in trauma and surgical patients with acquired coagulopathy. Curr Surg. 2006; 63:246–251.
90. Mahmoud, A, Al-Ruzzeh, S, McKeague, H, et al. Systemic venous thrombosis after recombinant factor VIIa in the control of bleeding after cardiac surgery. Tex Heart Inst J. 2007; 34:485–488.
91. O’Connell, KA, Wood, JJ, Wise, RP, et al. Thromboembolic adverse events after use of recombinant human coagulation factor VIIa. JAMA. 2006; 295:293–298.
92. Broderick, J, Connolly, S, Feldmann, E, et al. Guidelines for the Management of Spontaneous Intracerebral Hemorrhage in Adults. 2007 Update: A Guideline from the American Heart Association/American Stroke Association Stroke Council, High Blood Pressure Research Council, and the Quality of Care and Outcomes in Research Interdisciplinary Working Group; the American Academy of Neurology affirms the value of this guideline as an educational tool for neurologists. Stroke. 2007; 38:2001–2023.
93. American Academy of Pediatrics. Health supervision for children with sickle cell disease. Pediatrics. 2002; 109:526–535.
94. (CORN) TCoRNfGS. National Newborn Screening Report—1992. New York, 1995.
95. Lane, PA. Sickle cell disease. Pediatr Clin North Am. 1996; 43:639–664.
96. Buck, J, Davies, SC. Surgery in sickle cell disease. Hematol Oncol Clin North Am. 2005; 19:897–902.vii.
97. Vichinsky, EP, Haberkern, CM, Neumayr, L, et al. A comparison of conservative and aggressive transfusion regimens in the perioperative management of sickle cell disease. The Preoperative Transfusion in Sickle Cell Disease Study Group. N Engl J Med. 1995; 333:206–213.
98. Baumgartner, F, Klein, S. The presentation and management of the acute abdomen in the patient with sickle-cell anemia. Am Surg. 1989; 55:660–664.
99. Epps, CH, Jr., Bryant, DD, 3rd., Coles, MJ, et al. Osteomyelitis in patients who have sickle-cell disease: Diagnosis and management. J Bone Joint Surg Am. 1991; 73:1281–1294.
100. Chambers, JB, Forsythe, DA, Bertrand, SL, et al. Retrospective review of osteoarticular infections in a pediatric sickle cell age group. J Pediatr Orthop. 2000; 20:682–685.
101. Sutton, J, et al. Surgical management of patients with sickle cell syndromes. In: Mankad V, Moore R, eds. Sickle Cell Disease: Pathophysiology, Diagnosis, and Management. Westport, CT: Praeger; 1992:364–386.
102. Ware, RE, Filston, HC. Surgical management of children with hemoglobinopathies. Surg Clin North Am. 1992; 72:1223–1236.
103. Management of Sickle Cell Disease. 4th ed, NIH Publication; 2002.
104. Haxby, E, Flynn, F, Bateman, C. Anaesthesia for patients with sickle cell disease or other haemoglobinopathies. Anaesth Intensive Care Med. 2007; 8:217–219.
105. Amrolia, PJ, Almeida, A, Halsey, C, et al. Therapeutic challenges in childhood sickle cell disease: I. Current and future treatment options. Br J Haematol. 2003; 120:725–736.
106. Koshy, M, Weiner, SJ, Miller, ST, et al. Surgery and anesthesia in sickle cell disease. Cooperative Study of Sickle Cell Diseases. Blood. 1995; 86:3676–3684.
107. Hirst, C, Williamson, L. Preoperative blood transfusions for sickle cell disease. Cochrane Database Syst Rev. 2001.
108. Mankad, A. Anesthetic management of patients with sickle cell disease. In: Mankad V, Moore R, eds. Sickle Cell Disease: Pathophysiology, Diagnosis, and Management. Westport, CT: Praeger; 1992:351–363.
109. Castro, O, Brambilla, DJ, Thorington, B, et al. The acute chest syndrome in sickle cell disease: Incidence and risk factors. The Cooperative Study of Sickle Cell Disease. Blood. 1994; 84:643–649.
110. Kemp, JS. Obstructive sleep apnea and sickle cell disease. J Pediatr Hematol Oncol. 1996; 18:104–105.
111. Wali, YA, al Okbi, H, al Abri, R. A comparison of two transfusion regimens in the perioperative management of children with sickle cell disease undergoing adenotonsillectomy. Pediatr Hematol Oncol. 2003; 20:7–13.
112. Duke, RL, Scott, JP, Panepinto, JA, et al. Perioperative management of sickle cell disease children undergoing adenotonsillectomy. Otolaryngol Head Neck Surg. 2006; 134:370–373.
113. Halvorson, DJ, McKie, V, McKie, K, et al. Sickle cell disease and tonsillectomy. Preoperative management and postoperative complications. Arch Otolaryngol Head Neck Surg. 1997; 123:689–692.
114. Lachman, BS, Lazerson, J, Starshak, RJ, et al. The prevalence of cholelithiasis in sickle cell disease as diagnosed by ultrasound and cholecystography. Pediatrics. 1979; 64:601–603.
115. Sarnaik, S, Slovis, TL, Corbett, DP, et al. Incidence of cholelithiasis in sickle cell anemia using the ultrasonic gray-scale technique. J Pediatr. 1980; 96:1005–1008.
116. Suell, MN, Horton, TM, Dishop, MK, et al. Outcomes for children with gallbladder abnormalities and sickle cell disease. J Pediatr. 2004; 145:617–621.
117. Haberkern, CM, Neumayr, LD, Orringer, EP, et al. Cholecystectomy in sickle cell anemia patients: Perioperative outcome of 364 cases from the National Preoperative Transfusion Study. Preoperative Transfusion in Sickle Cell Disease Study Group. Blood. 1997; 89:1533–1542.
118. Pappis, CH, Galanakis, S, Moussatos, G, et al. Experience of splenectomy and cholecystectomy in children with chronic haemolytic anaemia. J Pediatr Surg. 1989; 24:543–546.
119. Stephens, CG, Scott, RB. Cholelithiasis in sickle cell anemia: Surgical or medical management. Arch Intern Med. 1980; 140:648–651.
120. Ware, R, Filston, HC, Schultz, WH, et al. Elective cholecystectomy in children with sickle hemoglobinopathies: Successful outcome using a preoperative transfusion regimen. Ann Surg. 1988; 208:17–22.
121. Miltenburg, DM, Schaffer, R, 3rd., Breslin, T, et al. Changing indications for pediatric cholecystectomy. Pediatrics. 2000; 105:1250–1253.
122. Ware, RE, Schultz, WH, Filston, HC, et al. Diagnosis and management of common bile duct stones in patients with sickle hemoglobinopathies. J Pediatr Surg. 1992; 27:572–575.
123. Dubois, F, Icard, P, Berthelot, G, et al. Coelioscopic cholecystectomy: Preliminary report of 36 cases. Ann Surg. 1990; 211:60–62.
124. Gadacz, TR, Talamini, MA, Lillemoe, KD, et al. Laparoscopic cholecystectomy. Surg Clin North Am. 1990; 70:1249–1262.
125. Tagge, EP, Othersen, HB, Jr., Jackson, SM, et al. Impact of laparoscopic cholecystectomy on the management of cholelithiasis in children with sickle cell disease. J Pediatr Surg. 1994; 29:209–213.
126. Ware, RE, Kinney, TR, Casey, JR, et al. Laparoscopic cholecystectomy in young patients with sickle hemoglobinopathies. J Pediatr. 1992; 120:58–61.
127. Curro, G, Meo, A, Ippolito, D, et al. Asymptomatic cholelithiasis in children with sickle cell disease: Early or delayed cholecystectomy? Ann Surg. 2007; 245:126–129.
128. Gill, FM, Sleeper, LA, Weiner, SJ, et al. Clinical events in the first decade in a cohort of infants with sickle cell disease. Cooperative Study of Sickle Cell Disease. Blood. 1995; 86:776–783.
129. Emond, AM, Collis, R, Darvill, D, et al. Acute splenic sequestration in homozygous sickle cell disease: Natural history and management. J Pediatr. 1985; 107:201–206.
130. Aquino, VM, Norvell, JM, Buchanan, GR. Acute splenic complications in children with sickle cell-hemoglobin C disease. J Pediatr. 1997; 130:961–965.
131. Pegelow, CH, Wilson, B, Overturf, GD, et al. Infection in splenectomized sickle cell disease patients. Clin Pediatr. 1980; 19:102–105.
132. Nouri, A, de Montalembert, M, Revillon, Y, et al. Partial splenectomy in sickle cell syndromes. Arch Dis Child. 1991; 66:1070–1072.
133. Svarch, E, Vilorio, P, Nordet, I, et al. Partial splenectomy in children with sickle cell disease and repeated episodes of splenic sequestration. Hemoglobin. 1996; 20:393–400.
134. Hicks, BA, Thompson, WR, Rogers, ZR, et al. Laparoscopic splenectomy in childhood hematologic disorders. J Laparoendosc Surg. 1996; 6(Suppl 1):S31–S34.