CHAPTER 76 CLINICAL SPECTRUM: DEFINITION AND NATURAL PROGRESSION
Once a neurological illness common in the Western and developed world, multiple sclerosis (MS) is now being increasingly reported from most parts of the world, including tropical countries. Marked by ambulatory disabilities caused at a relatively early age, MS is complicated by unpredictability of attacks and progression. We discuss the definition, clinical features, and natural progression of MS in this chapter. The major inputs for natural progression in this chapter are courtesy of the natural history database at the London Multiple Sclerosis Clinic, London Health Sciences Center, University of Western Ontario, London Ontario, Canada, with over 26,000 patient-years of studies.
CLINICAL PHENOTYPES OF MULTIPLE SCLEROSIS
There are no specific clinical or paraclinical markers for MS. Among the many classifications that have been proposed, we chose for the purpose of this chapter the phenomenological classification initially suggested by McAlpine,1 later reviewed by an international survey,2 although there was no unanimity of opinion:
The terms relapsing progressive or progressive relapsing are self-explanatory, but are unnecessary additions to the phenotypes, and hence are best avoided.3–5
An acute attack or exacerbation or relapse, as defined by Schumacher and colleagues,6 is a focal disturbance of function, affecting a white matter tract, and lasting for more than 24 hours. Typically, an acute exacerbation tends to progress over a period of a few days, reaching a maximum in less than 1 week, and then slowly resolving. Complete recovery from an attack is common early in the disease.
The mean annual frequency of relapses has been shown to be between 0.4 and 1.1 relapses per patient per year.7–12 The average attack rate in the first year was found to be 1.5 to 2.3 relapses per patient per year, falling with age and duration13 of disease. The relapse frequency starts to decrease by the second year of the disease.14 The interval between two relapses can be a minimum of 1 month (as defined by Schumacher6) to more than 35 years. A typical relapse lasts for about 6 weeks. Recovery starts by the second week in most patients. The earlier a relapse starts to recover, the better is the total recovery.14
A pseudo-relapse is acute-onset worsening of preexisting symptoms or signs or reappearance of symptoms or signs in the same location as a prior attack, with lesser or equal severity. The usual causes are physiological, primarily fever, urinary tract infection, or vaccination. Table 76-1 compares true and pseudo-relapses.
| Parameter | True Relapses | Pseudo-relapses |
|---|---|---|
| Onset | Acute/subacute | Acute more likely |
| Duration | Weeks likely | Days rather than weeks |
| Previous deficit | Not necessary | Likely |
| Fever associated | Not likely | Usually |
Courtesy of Paty DW, Ebers GC: Multiple Sclerosis. Contemporary Neurology Series (50). Philadelphia: FA Davis, 1997.
Relapses and their associations or precipitating factors have been studied by many. Relapses have been claimed15 but unproved to be more common in warmer months. Large studies have not documented significant reduction in the relapse rates during pregnancy, but the overall relapse rate is higher in the first 6 months postpartum, especially during the first 3 months.16–21 Relapse rates are not influenced by breastfeeding.19
In some instances, relapses have been associated temporally with immunization.22 Large studies failed to confirm increased relapse risk after immunization in the MS population.23 However, in clinical practice, postimmunization relapses have been documented.
On the other hand, infections have been found in most studies to be temporally associated with increased relapse rates in the postinfectious phase,24–26 and it is a common observation that MS patients present typically 3 to 4 weeks after an infection, with a true or pseudo-relapse. Relapse rates were not increased postsurgically or postanesthesia in most studies.27,28 Stress and life adversities have been claimed to be associated with increased relapse risk, but this is difficult to assess objectively.29–31 There are no data to support the notion that physical trauma can precipitate relapses or “unmask” dormant MS that otherwise would have remained silent.15,27,28,32
A small number of patients complain of deterioration in functional capacity in the luteal phase of the menstrual cycle. Slight temperature elevation during this phase may be responsible,33 and tiredness and fatigue may worsen preexisting deficits.
Although the above categories encompass most clinical presentations of MS, individual variations are very common. There may be attacks in PPMS, and sometimes SPMS may begin after one single attack, labeled single attack progressive MS.34
Benign MS: This subcategory of RRMS tends to be common in women with a younger age of onset who had sensory symptoms as the presenting feature. An Expanded Disability Scale Score (EDSS) score of less than 3 for more than 10 years of MS diagnosis was considered as benign in a study by Redekop and Paty,35 and they found 24% of the 536 patients studied in British Columbia belonged to this category. Freedom from either attacks or progression for decades can be considered to be typical of benign MS. It is most easily recognized in hindsight.
Acute malignant MS:14 Some patients often have a polysymptomatic onset and progress rapidly to severe disability, death, or both within months (very rare) or a few years (uncommon). Even this most malignant clinical course cannot be reliably predicted at onset. The distinction between acute malignant MS and acute disseminated encephalomyelitis can be difficult. New lesions manifest clinically or on MRI and progression beyond 2 months are features more consistent with a diagnosis of MS than acute disseminated encephalomyelitis. In addition, in the adult, acute disseminated encephalomyelitis is much less common than MS, so that most patients with the acute malignant progression will turn out to have MS.
Opticospinal MS (Devic’s disease, neuromyelitis optica):14 Reported very frequently from the orient, opticospinal MS predominantly involves attacks and progressive worsening in the optic and spinal systems. This variant is characterized by an earlier age of onset, early progression, minimal or no recovery of neurodeficit after an attack, and poor prognosis.
Lennon and associates36 from the Mayo Clinic, Rochester, Minnesota, studied 102 North American and 12 Japanese patients with neuromyelitis optica against controls, including MS, optic neuritis, myelopathies, and other conditions, for the presence of neuromyelitis optica IgG antibodies. They claim that this is a specific marker autoantibody for neuromyelitis optica, distinguishing the latter from MS. Based on the prevalence of this marker, the group also comments that Asian opticospinal MS is the same entity as neuromyelitis optica.
Progressive and chronic cerebellar MS have both been believed to be variants of PPMS.
Childhood MS is rare (less than 4% of cases). Youngest cases reported include 10-month-old, 24-month-old, and 4-year-old children. This variant is further more frequent in females (3:1). Vertigo is a frequent presenting feature. Most patients will have a remitting sensory onset. Deterioration is slower, and the youngest patients have the best prognosis for disability. About 82% of childhood MS patients have positive oligoclonal banding in the cerebrospinal fluid. In an excellent review of childhood MS, Banwell37 writes that the onset of progression in MS beginning before the age of 16 years is later as compared to adult-onset MS, by an average of 15 years.
CLINICAL FEATURES OF MULTIPLE SCLEROSIS
The common presenting symptoms in MS are summarized in Table 76-2.
Consciousness and Cognition
Cognitive and psychiatric changes in MS have traditionally been underemphasized. Studies have demonstrated that minor deficits in cognition are quite common (up to 70%), even in early MS (up to 50%).38–40 Dementia can be an accompaniment of severe disabling MS of long-term duration. Lesions seen in the corpus callosum on the MRI scan are also well correlated with cognitive defects. Periventricular lesions and the width of the third ventricle are described to be the most frequent MRI correlates with cognitive deficits.41
Euphoria is commonly a manifestation of subtle or obvious cognitive change.42 Focal cortical deficits such as aphasia, apraxia, and agnosias have been reported uncommonly. The most frequent cognitive abnormalities in MS are subtle defects in abstraction and memory,43–46 attention, and word finding.47 These are usually associated with emotional lability and decreased speed of information processing.45,46
Traditional tests for dementia designed for use in neuronal degenerative diseases such as Alzheimer’s disease are not sensitive to the changes seen in MS.47 The most sensitive bedside measures for cognitive defects in MS have been tests such as the repetition of seven numbers forward or backward, serial 7s or 3s, and visual recognition tests.48,49 Verbal working memory is specially susceptible to impairment in MS.50
Mood disorders are frequent in patients with MS. An association may exist between MS and bipolar disorder.51–53 A survey in the University of British Columbia Multiple Sclerosis clinic shows that 38% of patients with MS have been depressed or could have bipolar disorder.52 Other forms of psychosis are rare but can be seen. Some patients develop hypomanic behavior with steroid therapy, commonly used to treat relapses.
Sleep Disturbances
Studies have shown that patients with MS are more likely than control subjects to have sleep disturbances.54 The sleep problems may be due to nocturnal spasms. They may also be a major contributor to fatigue that is so common in MS. Overall, patients with MS have poor sleep efficiency with frequent awakening. Periodic leg movements are also frequent and may contribute to the sleep disturbances.55 Incontinence and other bladder symptoms like nocturia may further deteriorate sleep quality and duration. Narcotic symptoms are common.56
Fatigue
Fatigue, a frequent and disabling feature of MS,57,58 is considered a state of exhaustion distinct from depressed mood or physical weakness.59
Seizures
Convulsive seizures occur in about 2% of MS patients. One half of the seizures are probably due to the MS lesion itself, and the other one half are due to chance association. Seizures in MS are usually easily controlled with anticonvulsants. Patients with seizures tend to have more subcortical or temporal lobe lesions or both than do control MS patients without seizures.60
Headache
Headache is a frequent complaint in MS.61 Occasionally, acute headache occurs in the pseudotumor, acute-onset type of relapse. One type of headache seen sometimes in MS is hemicranial and may be associated with an acute pontine lesion.62 Most patients with MS who have headache probably have tension headache due to muscle spasm in the neck and the scalp muscles or have migraine or are depressed.14 Pain on eye movement due to optic neuritis may also be responsible for headache.63
Cranial Nerve Involvement in Multiple Sclerosis
Pupillary Defects
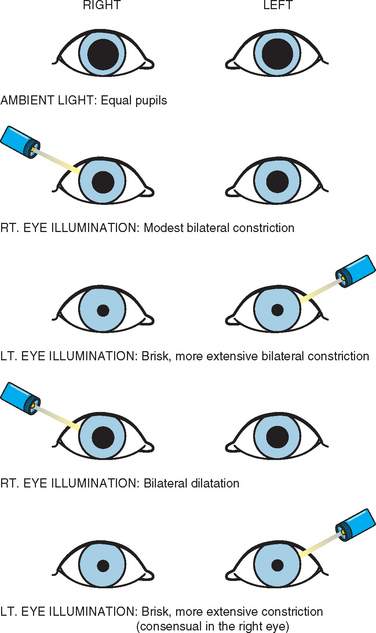
Most of these defects are related to the afferent pupillary defect (or Marcus Gunn pupil) (Fig. 76-1). Fixed dilated pupils associated with or independent of other elements of a third nerve palsy are extremely rare. Central Horner’s syndrome is occasionally seen.64
Loss of Vision
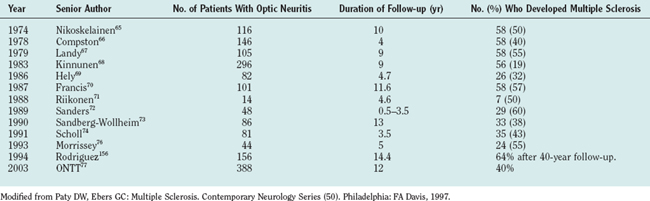
Acute loss of vision due to optic neuritis is a common feature of MS and is mostly unilateral. This is also a common clinical presenting feature. About 50% to 70% patients with optic neuritis proceed to develop MS in future; Table 76-3 shows major trials documenting this.
Optic neuritis may be retrobulbar, chiasmal, or even retrochiasmal. Many patients recover functionally normal vision after optic neuritis, even in the face of severe residual optic atrophy.78,79 Visual field deficits in MS tend to be central, but occasionally a clinician can encounter paracentral or peripheral scotomata or quadrantanopic or hemianopic defects. Homonymous hemianopias can occur in MS but are unusual; when seen, they should raise the possibility of coexistent tumor or vascular disease. Fortunately, legal blindness in MS is unusual (less than 5%).80
Many patients experience a dimming, blurring, or obscuring of vision associated with exercise and heat. Uhthoff first described this symptom.81 The transient appearance or worsening of neurodeficits (most typically, visual impairment, but any functional system can be involved) on exposure to any form of heat (atmospheric, hot shower, exercise, fever, or even hot food or drinks) is known as Uhthoff’s phenomenon, and is a very common accompaniment to relapses.
Pain is usually seen as an accompaniment to acute optic neuritis82 and may be due to traction of the origins of the superior and medial recti on the optic nerve sheath. Besides the blurring of vision and central scotoma, relative afferent pupillary defect (RAPD), color blindness, and sometimes Pulfrich phenomenon are clinical signs found in most MS patients. The Pulfrich phenomenon83 is a subjective correlate of conduction delay in one optic nerve. A pendulum oscillating in front of a normal individual will appear to traverse an ellipse if a neutral density filter or a piece of dark glass is placed over one eye. A patient with unilateral optic neuritis may see this illusion without a filter: the disease delays conduction just as does the filter.
Anosmia may be found on examination in many MS patients,84 although this may not be voluntarily reported by the patient.
Unilateral or bilateral loss of taste is infrequent. Paty and Ebers have observed this in several patients, and this was the presenting complaint in one patient. This symptom always remitted,14 in their observation.
Ocular Motility Disorders in Multiple Sclerosis
The most common in this group is nystagmus.85–88 The frequency of occurrence of nystagmus in MS has been reported to be between 28.3% and 63%. Nystagmus in MS may be acquired pendular, gaze evoked, rebound, torsional, periodic alternating, or another type. Optokinetic nystagmus is never impaired in isolation.14 The most common is the nystagmus that accompanies internuclear ophthalmoplegias.
In many MS patients, inappropriate initiation of saccadic eye movements during fixation or change in gaze position results in saccadic intrusions (square wave jerks, saccadic pulse, and double saccadic pulses) and saccadic oscillations (macro-square wave jerks, macrosaccadic oscillations, and ocular flutter).89 A square wave jerk consists of a small amplitude conjugate saccade away from fixation followed by a saccade back to fixation after a latency delay of about 200 milliseconds.
Oculomotor nerve pareses are uncommon presentations of MS but are known, and Paty and Ebers14 reported it as an initial presentation in two cases.
Paresis of fourth cranial nerve as an isolated feature is rare.14
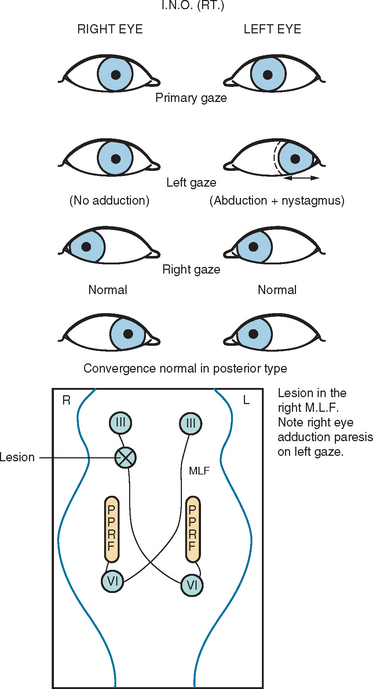
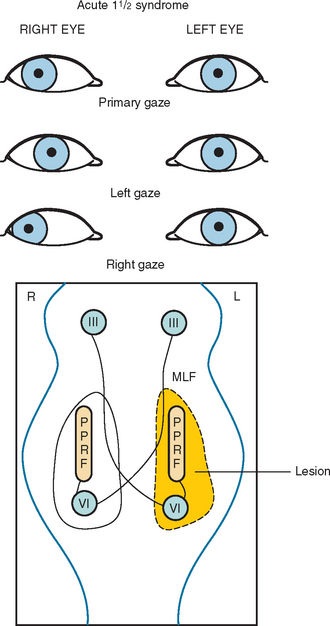
Another common abnormality of eye movements in MS is internuclear ophthalmoplegia, bilateral (most frequent) or unilateral.14 Internuclear ophthalmoplegia manifests as partial or complete paresis of adduction on the side of lesion, with ataxic nystagmus in the other (abducting) eye (Fig. 76-2). Skew deviation and vertical nystagmus (upbeat on gaze up or downbeat on gaze down) may also occur. Bilateral internuclear ophthalmoplegias are associated with vertical nystagmus in one or both directions. One-and-a-half syndrome (Fig. 76-3) and wall eyed bilateral internuclear ophthalmoplegia (Fig. 76-4) have been reported often in the MS literature. Lateral gaze pareses are also a common manifestation of MS. Visual suppression of the vestibulo-ocular reflex is often abnormal.
Trigeminal Motor and Sensory Symptoms
Facial sensory loss can occur in as many as 10% of MS patients.14 This can be a presenting feature as in a relapse or a lingering transient symptom. Trismus has also been reported.90 Trigeminal neuralgia is discussed later in the section on pain.
Facial Palsy
The acute development of a peripheral facial nerve paresis is an uncommon but recognized feature of MS.14 It occurs in less than 4% of patients and almost always recovers spontaneously and completely. Recovery from an MS-associated facial palsy is usually not associated with autonomic abnormalities such as crocodile tears; however, aberrant reinnervation, myokymia, and the phenomenon of intrafacial synkinesis are all commonly seen. Acute facial palsies are accompanied by other brainstem findings such as a sixth nerve palsy, lateral gaze palsy, or deafness. Paty and Ebers have seen one case with remitting bilateral facial myokymia that was disabling because the patient could not open his eyes. The symptom responded to carbamazepine therapy.
Deafness
Central lesions resulting in impaired hearing commonly occur, but persistent complete deafness is unusual in the absence of another etiology.91
Vertigo
This is a relatively common symptom. As many as 50% of patients have intermittent episodes of vertigo. In a review of initial symptoms in MS by Paty and Ebers,14 it was found that onset with vertigo as a symptom favored a long-term, more benign outcome.
Dysphagia
Mild dysphagia is common.14 It usually coexists with dysarthria, often in the setting of pseudobulbar palsy. Dysphagia is usually due to desynchronization of the swallowing mechanism.
Dysarthria
Cerebellar dysarthria is a common component of relapses but is usually late to occur in the progressive phase.14 There is intention tremor of the voice, which can be demonstrated by having the patient sustain a vowel sound for 10 to 15 seconds. The variation in the intensity and sometimes pitch of the sound is easily heard; the frequency is usually 5 to 7Hz, similar to other cerebellar tremors. Patients may show elements of combined cerebellar and pseudobulbar (spastic) dysarthria. Some patients have only spastic speech. Pseudobulbar dysarthria is caused by spastic vocal cords, which result in a high-pitch, low-volume speech, in which consonants are slurred. These patients do not have the oscillating tremor or the explosive variability associated with cerebellar speech. Some patients with MS develop dysarthria after several minutes of sustained speech. This effort-induced symptom tends to recover after a few minutes of rest.
Breathing Disturbance
Rarely, MS patients have a peculiar air hunger, usually associated with severe fatigue. Serious respiratory problems other than pneumonia and bronchitis are uncommon. Howard and colleagues92 described 19 patients who developed respiratory complications an average of 5.9 years after onset. Of these, 12 required mechanical ventilatory support, and 5 recovered. Six patients died after an average of 17.7 months.
Motor and Sensory Findings
Sensory loss is the most frequent of all neurological findings in MS.14 It is present in 90% of patients at some time during the clinical course. The distribution of sensory loss between upper and lower extremities can vary. Isolated transient facial numbness is frequently seen. Unremitting unilateral facial numbness is more likely due to isolated trigeminal neuropathy. It is also unusual for a sensory segmental level of pain or temperature loss to persist, even in advanced cases. However, during acute exacerbations, a sensory level can be seen in 10% to 15% of patients. When persistent abnormalities in touch, pain, and temperature sensation do occur, the pattern of loss is likely to be a patchy one. In contrast, posterior column or discriminatory sensation testing can be very useful. From 85% to 95% of patients with MS have an abnormality in posterior column sensation at some time during their clinical course. The sensation loss is often prominent in the lower extremities, but bilateral predominant upper extremity impairment is common.
The useless hand syndrome (Table 76-4) is a particularly interesting sensory manifestation that is usually seen in patients with predominant upper extremity proprioceptive loss.93 Even though the useless hand syndrome can occur in other disorders, it is highly characteristic of MS. It is common to see young adults with MS develop lack of discriminatory sensation in one upper extremity to the extent that the extremity becomes functionally useless despite normal crude sensation, motor, and cerebellar function. The useless hand syndrome in MS usually resolves spontaneously, although it can be extremely disabling while present. It usually begins with either tingling or lack of fine sensory discrimination in the fingers. The patient notices an inability to recognize objects in the pocket or purse. The patient can then develop impairment of hand function because of the inability to distinguish various subtleties in tactile sensation and lack of feedback control of movement.
Lhermitte’s Symptom
This is the occurrence of tingling and “electric current-like” sensation in the arms, down the back, into the legs, or in all the three areas, associated with forward flexion of the neck.14,94,95 This symptom occurs in 3% of patients at the onset of the disease and probably occurs in 30% to 40% of patients overall at some time during the clinical course. It is most frequently associated with MS, although it is not specific to this disease. Symptoms are usually precipitated by forward flexion of the neck but can also be brought on by forward flexion of the spine or even other minor movements. This symptom may sometimes occur spontaneously. In unusual circumstances, Lhermitte’s symptom can be disabling in the absence of other significant neurological deficits, usually because of its frequency, duration, or intensity. Such disabling symptoms usually respond somewhat to gabapentin, carbamazepine, or benzodiazepine therapy.
Paroxysmal Symptoms
Other paroxysmal symptoms in MS include ataxias, vertigo, painful and disturbing paresthesias, dysesthesias, monocular blurring or blindness, pain, and weakness. Paroxysmal motor phenomena include not only weakness but also spasticity and spasms, tonic seizures, akinesia, diplopia, dysarthria, and chorea in addition to focal and generalized seizures.96,97
The best known of these paroxysmal disturbances is the painful tonic spasm. This is usually manifested by tonic flexion or extension of a limb with a writhing movement reminiscent of paroxysmal athetosis.98 The muscular tension in the limb is so high that it becomes quite painful. A particularly difficult situation can be due to the paroxysmal loss of neurological function, as in paroxysmal monoparesis or hemiparesis or paroxysmal blindness. Paroxysmal itching has also been described.99
Pain in Multiple Sclerosis
There are two major categories of pain (Table 76-5): paroxysmal (such as trigeminal neuralgia) and chronic.14,100 Less than one half of the patients have musculoskeletal pain of various types. It makes sense that patients with weak muscles and poor support of the spine would have such pains (e.g., low backache, neckache, joint pains). Pain can be seen at the onset of MS.101
Pain of trigeminal neuralgia has been well discussed in MS. The trigeminal neuralgia features in MS are essentially the same as in idiopathic trigeminal neuralgia.100 Interestingly, trigeminal neuralgia in MS is more frequently bilateral (32%) than idiopathic trigeminal neuralgia (4%).14
Weakness and Spasticity
Weakness in MS is usually pyramidal in distribution but can be remarkably focal.14 For example, marked interosseous weakness can occur with preservation of thenar muscle strength. The weakness is usually more distal than proximal. Foot drop may be seen in the presence of normal proximal strength.
Spasticity is one of the more frequent features encountered in MS. As many as 90% of patients will develop some signs of spasticity during the clinical course. In some patients with severely weak legs, spasticity is actually useful for walking or standing. Weak legs may be able to support considerable weight because of involuntary contraction of the antigravity muscles; attempts to treat spasticity may be counterproductive in such patients. Occasional patients have a syndrome of severe increase in muscle tone with a paradoxical reduction in deep tendon reflexes. This is due to a combination of extrapyramidal and pyramidal lesions.
Cerebellar Features in Multiple Sclerosis
As mentioned earlier, cerebellar involvement may produce one of the most disabling forms of the disease.14 Cerebellar intention tremor is usually related to lesions in the cerebellar outflow pathways. Cerebellar manifestations include intention tremor, titubation, gait ataxia, and dysarthria. Action tremor may be superimposed on intention tremor. The intention tremor may be severe enough to appear even with an intention to move, thus manifesting practically at rest. The characteristic tremor in MS is the oscillating tremor at 5 to 7Hz perpendicular to the direction of the movement. Besides this, clumsiness, dyssynergia, and dysmetria are present.
Amyotrophy (Muscle Wasting) in Multiple Sclerosis
Amyotrophy is considered an uncommon clinical finding in MS.103 However, autopsy studies have shown that it is quite common. The three most common causes described are:
Gray matter plaques, now being increasingly recognized in MS, may also explain myoatrophy.
Peripheral Neuropathy
This is very uncommon, and many researchers have described subtle electrophysiological findings suggesting peripheral neuropathy in MS patients.106–109 Chance occurrences of clinical peripheral neuropathy and acute and chronic inflammatory demyelinating polyradiculoneuropathy have been described.
Autonomic Disturbances in Multiple Sclerosis
Nearly 3% of patients of MS present with isolated bladder symptoms.110
Forty-three percent of MS patients have been shown to have constipation.111 Fecal incontinence occurred at least once in 51% of patients and once per week or more frequently in about 25% of patients. The overall prevalence of bowel dysfunction was 68%. In a small number of patients, malabsorption,112 incontinence, and frequent diarrheal episodes113 are also reported.
Sexual dysfunction is another major concern in MS and can also be a presenting symptom.114–118 There is a high degree of correlation between sexual dysfunction and urinary dysfunction in MS. The reported prevalence is 80% in males and 33% in females.119 Male sexual dysfunction in MS is usually organic, but psychosocial factors are important, too. Female patients may have decreased arousal, decreased libido, lesser frequency of orgasm, fatigue, and decreased sensation.120 Males complain of erectile dysfunction, decreased sensation, fatigue, decreased libido, and orgasmic dysfunction. The prognosis of neurogenic erectile dysfunction is poor. Other treatable causes should always be excluded.
Nonsphincteric Autonomic Problems
These can be quite troublesome in some patients. Postural hypotension is common. A cyanotic hue or bluish-red mottling in paretic limbs, especially legs, with some edema may also be commonly seen. Other less common symptoms in this category include hypothermia, paroxysmal atrial fibrillation,121–123 orthostatic hypotension,124 exercise-induced tachycardia, and breathlessness.125 These symptoms are believed to be contributed by lesions in the ascending autonomic pathways in the brainstem or spinal cord.126 Yokota and colleagues127 reported abnormal sympathetic skin responses in 75% of MS patients studied.
Chronic hypothermia is sometimes seen.128 Paty and Ebers14 described six cases, some of which were severe enough to produce obtundation or coma. It may be precipitated by urinary or other infection. Patients usually respond to warming measures and antibiotics. Hypothalamic MRI lesions may be seen. Electrolyte disturbances and syndrome of inappropriate antidiuretic hormone secretion have also been reported.
As MS can affect white matter anywhere in the brain and spine, many unusual presentations are possible and have been reported in the literature (Table 76-6).
TABLE 76-6 Unusual (but Not Rare) Presentations of Multiple Sclerosis
Natural Progression
Relapsing-Remitting Multiple Sclerosis
In the RRMS category, the mean age at symptomatic onset is 29 years, with the majority of patients in the age group between 20 and 39 years. This compares well with reports from other researchers such as the Lyon, France, group, where the mean age at onset was 31 years (SD, 10 years; range, 5 to 67 years).129,130 In a study at the Mayo Clinic, the median age at onset was reported to be 37.2 years (range, 16.7 to 65.3 years) for men, 35.4 years (range, 17.3 to 59.6 years) for women, and 36.2 years (range, 16.7 to 65.3 years) overall.131 In approximately 10% to 15% of our patients, onset is before the age of 20 (mostly in their late teens, although pediatric MS is more often recognized), whereas about 5% of patients have MS onset after the age of 49 years.
A selective gender predilection is commonly seen in MS, as it is often reported in other autoimmune conditions. The male-to-female ratio is 1:2, and there is a much more conspicuous female preponderance among younger RRMS patients compared with persons with older progressive MS cases. In a study from the Mayo Clinic, newly diagnosed MS cases over a period of 15 years from 1985 through 2000 included 38 men and 94 women, with the male-to-female ratio being 1:2.4.131 Another study from Lyon, France, reported a male-to-female ratio of 1:1.7.129,130
The attack rate in the early years of MS is an important predictor of long-term outcome. In London, Ontario studies, the mean annual attack rate in the first year after onset was 1.5 in the total population. However, attack rates vary greatly in different studies, particularly when patients are specifically selected (e.g., for participation in clinical trials) or represent selected subgroups (e.g., hospitalized, and so on). Several studies serve to illustrate such variation. Of course, the relapse rates reported differ for treated and untreated patients. Table 76-7 compares the reported relapse rates in some relevant trials.
TABLE 76-7 Baseline Annual Relapse Rates Reported in Different Multiple Sclerosis Studies
| Year | Senior Author | Annual Relapse Rate Reported |
|---|---|---|
| 1971 | Gudmundsson157 | 0.14 |
| 1982 | Patzold7 | 1.1 |
| 1989 | Goodkin132 | 0.64 |
| 1994 | Durelli133 | 0.94 |
| 1995 | Johnson134 | 2.90 |
| 1996 | Jacobs135 | 1.20 |
| 2003 | Russo136 | 0.87 |
| 2004 | Kalanie137 | 1.0 |
Interestingly, although there may be variation when surveying untreated populations as well, figures tend to be more closely grouped. However, it is typical for there to be a remarkable drop in the relapse rate after the second year. In 1952, McAlpine12 described attack rates of 1.23 in the first year and 0.42 in the second year. Now, early studies from our London, Ontario geographically based cohort demonstrated attack rates of 1.57 in the first year and 0.35 in the second year for the natural history cohort.138 Most studies report a reduced attack rate with time. It is therefore difficult to assess the true worth and efficacy of disease-modifying agents like interferons and other drugs with respect to reduction in attack rates.
The subsequent disease course is less unpredictable for populations compared with any given individual patient. Relapses occur varying frequency in various patients. The frequency of relapses varies even in a given patient at different times. However, there is a clear tendency for the frequency of relapses to be greater in the initial years, and the recovery more complete.13,138–141
With the passage of time, relapses may leave behind more neurological and functional deficits. The contribution of attacks to progression is unclear. After this early relapsing phase, most patients enter the progressive phase of the disease. After the first decade from onset, over 50% of patients whose disease was initially relapsing-remitting enter a progressive phase of MS. Approximately a cumulative 90% develop progressive disease after 25 years of follow-up.142 It has been commented that if followed up long enough, eventually nearly all RRMS patients will be found to have developed the progressive form of the disease. Our personal observations have shown a mean duration of conversion from the time of the first attack to the onset of the progressive form of the disease is approximately 11 years, although it has been reported to be up to 19 years.143,144
PROGRESSIVE MULTIPLE SCLEROSIS
Clinically, PPMS begins at a relatively later age compared with RRMS. Its onset peaks in the late fourth decade in contrast to early fourth decade in RRMS. The female preponderance in RRMS is not as prominent as in PPMS, with a female-to-male ratio of 1.3:1. Progression appears to worsen at a faster pace, but this may be apparent only in the early years. The progression rates in both groups are similar once progression begins.34,129
Relapses in the preprogressive phase of SPMS do not seem to significantly impact the later speed or severity of the progressive phase. Of course, the preexisting neurodeficits (relapse remnants) may exaggerate the disability on initial assessment. Worth noticing, though, is the fact that in individuals with one single attack predating the progressive phase of MS, the severity of the inaugural attack may be negatively associated with the onset of secondary progression (involvement of more than three neurological systems due to attack is associated with earlier onset of progression).34,145
Progression does not seem to begin specifically at the sites affected by earlier relapses. The site of the original attack has been suspected of becoming a locus minoris resistentiae where progression begins.146 However, most researchers agree that there appears to be no demonstrable relationship between the site of the initial clinically evident expression of disease and the location of the progressive deficit. In our samples, patients with optic neuritis, brainstem, and spinal sensory MS onset are all characterized by an overwhelming predominance of distal central motor dysfunction at outset of the progressive phase of the disease. Progression is seldom joined by overlapping relapses in its march, but when relapses occur, they tend to behave like RRMS relapses, with typical temporal profile and partial or complete recovery.
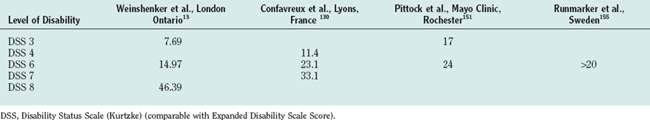
Over the years, disability accumulates and patients reach for a cane. This happens on an average of about 8 to 12 years after the beginning of the progressive phase. At this stage, the progression seems to relatively stabilize for some time, but this appearance of stability probably represents the limitations of the present disability scale used.14,139 Table 76-8 shows the median time in years from onset of MS to reach selected levels of disability.
The next hallmark of progression is wheelchair dependence, which usually comes approximately 10 and 20 years from the onset of progression.139 Around this period, many things can happen simultaneously. The patient may develop many medical conditions that usually accompany this age group. Lack of mobility, stress of disability, need for catheterization, and resultant infections increase a patient’s morbidity for life-threatening consequences. Falls, aspirations due to bulbar weakness, depression, memory retrieval deficits, and emotional lability occur more frequently. Coexisting morbidity for strokes and cardiac, pulmonary, or renal disease may further complicate the scene.
DISABILITY IN MULTIPLE SCLEROSIS
The EDSS has been designed to objectively evaluate MS patients.147,148 This is necessary when longitudinally evaluating patients in clinical trials for comparable outcome measures. The EDSS is based on assessment of clinical deficits in various functional systems of the central nervous system, rating them according to severity, rating the ambulation capacity and in its absence the use of upper limbs, and scoring the total not by addition but by overall review of the individual system scores and deficits in ambulation and effective use of hands. The functional systems included are visual, sensory, pyramidal, cerebellar, brainstem, bowel-bladder, cerebral, and others. EDSS has 20 scoring points from 0 to 10; each point after 1 is divided in two.
The key milestones in EDSS are as follows:
The EDSS is not linear in its function. Also, with lack of comparability of neurodeficits in various neurological systems, the scale has to depend mainly on the disability in ambulation as a comparable criterion. After the score of 5, the scale mainly depends on ambulation, with progression in other systems being sidelined.139
In RRMS, the initial deficit, as mentioned, is quite variable due to diverse neurological system involvements of different degrees; hence, the score of 3 on the EDSS grossly represents a disability level signifying moderate compromise in at least one functional system, with or without mild deficits in others. This level forms a good comparable level for natural history evaluation. The mean duration taken by the patients to reach this level is discrepant in RRMS and PPMS, quite understandably, given the late onset and probably faster early progression of PPMS.14,139,149
The mean time for an RRMS patient to reach an EDSS score of 3 is 6 to 8 years, whereas that for a PPMS patient is 1 to 2 years. This observation may differ depending on whether the patient was observed retrospectively or prospectively.14,139,149,150
The next level generally used for comparison is EDSS score 6, and the score is reached by most RRMS patients in 9 to 15 years, whereas most PPMS patients reach this score in 3 to 5 years.139
Most RRMS patients achieve wheelchair dependency or an EDSS score of 8 in their progressive (SPMS) phase, and the time required for this varies from 18 to 30 years. PPMS patients reach this score in an average 20 years from onset of progression.139
The Mayo Clinic group reported recently that the median time from diagnosis to EDSS score of 3 and 6 was 17 and 24 years, respectively. Only 25% of patients with RRMS were expected to reach an EDSS score of 3 within 20 years based on Kaplan-Meier plots of time. The median time expected for SPMS patients to reach EDSS scores of 3, 6, and 8 was about 3, 10, and 38 years, respectively. The median time from diagnosis to EDSS scores of 6 and 8 for patients with PPMS was 3 and 25 years, respectively.151 In a study from Lyon, France, it was reported that the median time from onset of MS to the assignment of a score of 4, 6, and 7 was 8.4 years (range, 7.8 to 9.6 years), 20.1 years (range, 18.1 to 22.5 years), and 29.9 years (range, 25.1 to 34.5 years), respectively. Runmarker and associates155 reported comparable results (see Table 76-8).
These observations are analogous to the data published by our group, with minor variations. The median interval from onset of disease to reach each of these scores was significantly longer in females than in males and in patients with a younger age at onset of MS. The interval was also longer in those with an initial relapsing remitting course of MS, in those with complete recovery from the first relapse, and those with longer first interattack intervals. Interestingly, the median intervals to reach these target scores were significantly longer for cases with isolated optic neuritis at onset compared with those with isolated long tract dysfunction.127
A series of studies drew a very useful conclusion: Approximately 50% of patients with MS are still able to ambulate independently after 15 years of disease.137
PREDICTORS OF PROGNOSIS, LONG-TERM OUTCOME
Given the fact that in their later course both RRMS and PPMS have similar paces of progression and the disabilities are comparable, many predictors have been evaluated in the early course138,139,145,152 (Table 76-9).
| Better | Worse |
|---|---|
| Female | Male |
| Onset: RRMS; optic neuritis, sensory; complete recovery | Onset: polysymptomatic, motor, incomplete recovery |
| Younger age at onset | Late onset |
| Longer interattack intervals in early course | More frequent attacks in early course |
| Long time to DSS 3 | Short time to DSS 3 |
DSS, Disability Status Scale (Kurtzke) (comparable with Expanded Disability Scale Score).
A shorter time of achievement of an EDSS score of 3 from the onset denotes faster early accumulation of disability. This has been associated in many studies with an earlier achievement of further milestones of disability (i.e., EDSS scores of 6 and 8) compared with those who had slower early accumulation of disability.
MRI is increasingly changing the way we evaluate MS patients in recent years, and new techniques are widening our understanding of MS in multiple dimensions. The typical MRI lesions seen with demyelination aid in documenting time and space dissemination so vital in the diagnosis of RRMS. Although correlations are so far relatively and disappointingly poor, measures such as T2 hypointensities in gray matter, white matter atrophy, residual brain volume, spinal cord atrophy, early lesion load, and magnetization transfer ratio analysis of normal appearing brain tissue have all been directly associated with increased disability in MS.153
Comorbidity with other diseases definitely negatively affects long-term outcome.
MORTALITY IN MULTIPLE SCLEROSIS
Mortality in MS is not significantly different from the age-matched general population in the early, low disability years of MS. As the disease ages, MS-specific complications set in. The cause of death in about 50% patients of MS is due to some complication of MS, most commonly, pneumonia and urosepsis. In the other one half of patients, the common causes of death are the same as in the general population and include acute myocardial infarction, stroke, and malignancy. A significant minority of MS patients commit suicide, and this could be due to increased incidences of depression and disability.139
A greater propensity of MS patients to succumb to depression, fatigue, and other medical conditions, combined with multisystem neurological deficits, may be responsible for a minor shortening (≈5 years) of total life span compared with the normal population. There does not seem to be any difference in the causes of mortality when RRMS (and subsequent SPMS) patients are compared with PPMS patients154 (Table 76-10).
TABLE 76-10 Primary Causes of Death in Multiple Sclerosis Patients Listed on 312 Death Certificates in the London, Ontario, Natural History Cohort152
| Cause of Death | No. of Deaths (N = 312) |
|---|---|
| Pneumonia | 99 |
| Multiple sclerosis | 43 |
| Cancer | 43 |
| Heart disease | 28 |
| Septicemia | 19 |
| Respiratory failure | 15 |
| Stroke | 15 |
| Cardiac arrest | 13 |
| Suicide | 6 |
| Pulmonary embolism | 4 |
| Aspiration | 3 |
| Cachexia | 3 |
| Gastrointestinal bleeding | 3 |
| Accident | 2 |
| Dehydration | 2 |
| Respiratory arrest | 2 |
| Other* (1 case each) | 12 |
* Other causes of death (1 case each): acute renal failure, assisted suicide, encephalopathy, bowel infarction, cardiac arrhythmia, cardiac failure, chronic bronchitis, chronic renal failure, pyelonephritis, intestinal obstruction, head injury, pulmonary edema.
Cook SD, editor. Handbook of Multiple Sclerosis, 3rd ed., New York: Marcel Dekker, 2001.
Filippi M, Comi G. Primary Progressive Multiple Sclerosis. Milan, Italy: Top Neurosci Springer, 2002.
Glaser JS. Neuro-ophthalmology, 3rd ed. Philadelphia: Lippincott Williams & Wilkins, 1999.
Paty DW, Ebers GC. Multiple Sclerosis. Contemporary Neurology Series (50). Philadelphia: FA Davis, 1997.
Scheinberg L, Raine CS. Multiple sclerosis: experimental and clinical aspects. Ann N Y Acad Sci. 1984:436.
1 McAlpine D, Lumsden CE, Acheson ED. Multiple Sclerosis—A Reappraisal. Edinburgh: Livingstone, 1965.
2 Lublin FD, Reingold SC. Defining the clinical course of multiple sclerosis: results of an international survey. National Multiple Sclerosis Society (USA) Advisory Committee on Clinical Trials of New Agents in Multiple Sclerosis. Neurology. 1996;46:907-911.
3 Kremenchutzky M, Cottrell D, Rice G, et al. The natural history of multiple sclerosis: a geographically based study. VII. Progressive-relapsing and relapsing-progressive multiple sclerosis: a re-evaluation. Brain. 1999;122:1941-1949.
4 Weinshenker BG. Progressive forms of multiple sclerosis: classification streamlined or consensus overturned? Lancet. 2000;355:162-163.
5 Andersson PB, Waubant E, Gee L, et al. Multiple sclerosis that is progressive from the time of onset: clinical characteristics and progression of disability. Arch Neurol. 1999;56:1138-1142.
6 Schumacher GA, Beebe GW, Kibler RF, et al. Problems of experimental trials of therapy in multiple sclerosis: report by the panel on the evaluation of experimental trials of therapy in multiple sclerosis. Ann N Y Acad Sci. 1965;122:552-568.
7 Patzold U, Pocklington PR. Course of multiple sclerosis. First results of a prospective study out of 102MS patients from 1976–1980. Acta Neurol Scand. 1982;65:248-266.
8 Goodkin DE, Hertsgaard D, Rudick RA. Exacerbation rates and adherence to disease type in a prospectively followed-up population with multiple sclerosis. Implication for clinical trials. Arch Neurol. 1989;46:1107-1112.
9 Fog T, Linnemann F. The course of multiple sclerosis in 73 cases with computer designed curves. Acta Neurol Scand. 1970;47(Suppl):3-175.
10 Leibowitz U, Alter M. Clinical factors associated with increased disability in multiple sclerosis. Acta Neurol Scand. 1970;46:53-70.
11 Panelius M. Studies on epidemiological, clinical and etiological aspects of multiple sclerosis. Acta Neurol Scand. 1969;45(suppl 39):1-82.
12 McAlpine D, Compston N. Some aspects of the natural history of disseminated sclerosis. Q J Med. 1952;21:135-167.
13 Weinshenker BG, Ebers GC. The natural history of multiple sclerosis. Can J Neurol Sci. 1987;14:255-261.
14 Paty DW, Ebers GC. Multiple Sclerosis. Contemporary Neurology Series (50). Philadelphia: FA Davis, 1997.
15 Bamford CR, Sibley WA, Thies C. Seasonal variation of multiple sclerosis exacerbations in Arizona. Neurology. 1983;33:697-701.
16 Birk K, Ford C, Smeltzer S, et al. The clinical course of multiple sclerosis during pregnancy and the puerperium. Arch. Neurol. 1990;47:738-742.
17 Birk K, Rudick RA. Pregnancy and multiple sclerosis. Arch Neurol. 1986;43:719-726.
18 Frith JA, McLeod JG. Pregnancy and multiple sclerosis. J Neurol Neurosurg Psychiatry. 1988;51:495-498.
19 Nelson LM, Franklin GM, Jones MC, the Multiple Sclerosis Study Group. Risk of multiple sclerosis exacerbation during pregnancy and breastfeeding. JAMA. 1988;359:3441-3443.
20 Schapira K, Poskanzer DC, Newell DJ, et al. Marriage, pregnancy, and multiple sclerosis. Brain. 1966;89:419-428.
21 Sadovnick AD, Eisen K, Hashimoto SA, et al. Pregnancy and multiple sclerosis: a prospective study. Arch Neurol. 1994;51:1120-1124.
22 Miller H, Cendrowski W, Schapira K. Multiple sclerosis and vaccination. BMJ. 1967;2:210-213.
23 Andersen E, Isager H, Hyllested K. Risk factors in multiple sclerosis: tuberculin reactivity, age at measles infection, tonsillectomy and appendectomy. Acta Neurol Scand. 1981;63:131-135.
24 Martyn CN, Colquhoun I. Radiological evidence of sinus infection in patients with multiple sclerosis. J Neurol Neurosurg Psychiatry. 1991;54:925-926.
25 Sibley WA. Therapeutic Claims in Multiple Sclerosis. New York: Demos Publications, 1988.
26 Sibley WA, Bamford CR, Clark K. Clinical viral infections and multiple sclerosis. Lancet. 1985;1:1313-1315.
27 Bamford CR, Sibley WA, Thies C, et al. Trauma as an etiologic and aggravating factor in multiple sclerosis. Neurology. 1981;31:1229-1234.
28 Sibley WA, Bamford CR, Clark K, et al. A prospective study of physical trauma and multiple sclerosis. J Neurol Neurosurg Psychiatry. 1991;54:584-589.
29 Warren S, Greenhill S, Warren KG. Emotional stress and the development of multiple sclerosis: case control evidence of a relationship. J Chronic Dis. 1982;35:821-831.
30 Warren S, Warren KG, Cockerill R. Emotional stress and coping in multiple sclerosis. Exacerbations. J Psychosom Res. 1991;35:37-47.
31 Buljevac D, Hop WCJ, Reedeker W, et al. Self reported stressful life events and exacerbations in multiple sclerosis: prospective study. BMJ. 2003;327:646-653.
32 Mohr DC, Hart SL, Julian L, et al. Association between stressful life events and exacerbation in multiple sclerosis: a meta-analysis. BMJ. 2004;328:731.
33 Rice GPA, Ebers GC. Management of multiple sclerosis in women. Female Patient. 1995;20:19-32.
34 Kremenchutzky M, Rice GPA, Baskerville J, et al: The natural history of multiple sclerosis: a geographically based study. IX. Observations on the progressive phase of the disease (unpublished article).
35 Redekop K, Paty D: PhD thesis. University of British Columbia, Vancouver, 1995.
36 Lennon VA, Wingerchuk DM, Kryzer TJ, et al. A serum autoantibody marker of neuromyelitis optica: distinction from multiple sclerosis. Lancet. 2004;364:2106-2112.
37 Banwell B. Pediatric multiple sclerosis. Curr Neurol Neurosci Rep. 2004;4:245-252.
38 Klonoff H, Clark C, Oger JJF, et al. Neuropsychological performance in patients with mild multiple sclerosis. J Nerv Ment Dis. 1991;179:127-131.
39 Rao SM. Neuropsychology of multiple sclerosis: a critical review. J Clin Exp Neuropsychol. 1986;8:503-542.
40 Ron MA, Feinstein A. Multiple sclerosis and the mind [Editorial]. J Neurol Neurosurg Psychiatry. 1992;55:1-3.
41 Pozzilli C, Bastianello S, Padovani A, et al. Anterior corpus callosum atrophy and verbal fluency in multiple sclerosis. Cortex. 1991;27:441-445.
42 Peyser JM, Edwards KR, Poser CM, et al. Cognitive function in patients with multiple sclerosis. Arch Neurol. 1980;137:577-579.
43 Carroll M, Gates R, Roldan F. Memory impairment in multiple sclerosis. Neuropsychologia. 1984;22:297-302.
44 Boyle EA, Clark CM, Klonoff H, et al. Empirical support for psychological? profiles observed in multiple sclerosis. Arch Neurol. 1991;48:1150-1154.
45 Litvan I, Grafman J, Vendrell P. Multiple memory deficits in patients with multiple sclerosis: exploring the working memory system. Arch Neurol. 1988;45:607-610.
46 Litvan I, Grafman J, Vendrell P, et al. Slowed information processing in multiple sclerosis. Arch Neurol. 1988;45:281-285.
47 Mahler ME. Behavioral manifestations associated with multiple sclerosis. Psychiatr Clin North Am. 1992;15:425-438.
48 Peyser JM, Rao SM, LaRocca NG, et al. Guidelines for neuropsychological research in multiple sclerosis. Arch Neurol. 1990;47:94-97.
49 Rao SM, Leo GJ, Ellington L, et al. Cognitive dysfunction in multiple sclerosis. 2. Impact impact on employment and social functioning. Neurology. 1991;41:692-696.
50 Ruchkin DS, Grafman J, Krauss GL, et al. Event-related brain potential evidence for a verbal working memory deficit in multiple sclerosis. Brain. 1994;117(pt 2):289-305.
51 Joffe RT, Lippert GP, Gray TA, et al. Mood disorder and multiple sclerosis. Arch Neurol. 1987;44:376-378.
52 Remick RA, Sadovnick AD, Allen JM, et al: Mood disorders and multiple sclerosis [Abstract]. Canadian Psychiatric Association annual meeting, Ottawa, 1994.
53 Schiffer RB, Rudick RA, Herndon RM. Psychological aspects of multiple sclerosis. N Y State J Med. 1983;83:312-316.
54 Clark CM, Fleming JA, Li DKB, et al. Sleep disturbance, depression, and lesion site in patients with multiple sclerosis. Arch Neurol. 1992;49:641-643.
55 Ferini-Strambi L, Fillipi M, Martinelli V, et al. Nocturnal sleep in multiple sclerosis: correlations with clinical and brain magnetic resonance imaging findings. J Neurol Sci. 1994;125:194-197.
56 Poirier G, Montplaisir J, Dumont M, et al. Clinical and sleep laboratory study of narcoleptic symptoms in multiple sclerosis. Neurology. 1987;37:693-695.
57 Fisk JD, Pontefract A, Rivto PG, et al. The impact of fatigue on patients with multiple sclerosis. Can J Neurol Sci. 1994;21:9-14.
58 Sandroni P, Walker C, Starr A. “Fatigue” in multiple sclerosis: motor pathway conduction and event related potentials. Arch Neurol. 1992;49:517-524.
59 Krupp LB. Fatigue in multiple sclerosis: definition, pathophysiology and treatment. CNS Drugs. 2003;17:225-234.
60 Truyen L, Barkhof F, Frequin STF, et al: A case-control study of epilepsy in multiple sclerosis using magnetic resonance imaging: implications for treatment trials with 4-aminopyridine. 10th Congress of the European Committee for Treatment and Research in multiple sclerosis, Book of Abstracts, Athens, Greece, 1994, p 38.
61 Rolak LA, Brown S. Headaches and multiple sclerosis: a clinical study and review of the literature. J Neurol. 1990;237:300-302.
62 Paty DW, Ebers GC. Multiple Sclerosis. Contemporary Neurology Series (50). Philadelphia: FA Davis, 1997. Chapter 5.
63 McDonald WI. Pain around the eye: inflammatory and neoplastic causes. Trans Ophthalmol Soc UK. 1980;pt 2:260-262.
64 Bamford CR, Smith MS, Sibley WA. Horner’s syndrome, an unusual manifestation of multiple sclerosis. Can J Neurol Sci. 1980;7:65-66.
65 Nikoskelainen E, Riekkinen P. Optic neuritis: a sign of multiple sclerosis or other diseases of the central nervous system. Acta Neurol Scand. 1974;50:690-718.
66 Compston DAS, Batchelor JR, Earl CJ, et al. Factors influencing the risk of multiple sclerosis developing in patients with optic neuritis. Brain. 1978;101:495-511.
67 Landy PJ, Innis M, Boyle R. Factors likely to affect the development of multiple sclerosis in patients presenting with optic neuritis in a tropical and subtropical area. Clin Exp Neurol Proc Assoc Neurol. 1979;16:175-182.
68 Kinnunen E. The incidence of optic neuritis and its prognosis for multiple sclerosis. Acta Neurol Scand. 1983;68:371-377.
69 Hely MA, McManis PG, Doran TJ, et al. Acute optic neuritis: a prospective study of the risk factors for multiple sclerosis. J Neurol Neurosurg Psychiatry. 1986;49:1125-1130.
70 Francis DA, Compston DAS, Batchelor JR, et al. A reassessment of the risk of multiple sclerosis developing in patients with optic neuritis after extended follow-up. J Neurol Neurosurg Psychiatry. 1987;50:758-765.
71 Riikonen R, Ketonen L, Sipponen J. Magnetic resonance imaging, evoked responses and cerebrospinal fluid findings in a follow-up study of children with optic neuritis. Acta Neurol Scand. 1988;77:44-49.
72 Sanders EACM, Van Lith GHM. Optic neuritis, confirmed by visual evoked response, and the risk of multiple sclerosis: a prospective survey. J Neurol Neurosurg Psychiatry. 1989;52:799-810.
73 Sandberg-Wollheim M, Bynke H, Conqvist S, et al. A long-term prospective study of optic neuritis: evaluation of risk factors. Ann Neurol. 1990;27:386-393.
74 Scholl GB, Song HS, Wray SH. Uhthoff’s symptom in optic neuritis: relationship to magnetic resonance imaging and development of multiple sclerosis. Ann Neurol. 1991;30:180-184.
75 Rolak LA, Beck RW, Paty DW, et al. Cerebrospinal fluid in acute optic neuritis: experience of the optic neuritis treatment trial. Neurology. 1996;46:368-372.
76 Morrissey SP, Miller DH, Kendall BE, et al. The significance of brain magnetic resonance imaging abnormalities at presentation with clinically isolated syndromes suggestive of multiple sclerosis. Brain. 1993;116:135-146.
77 Optic Neuritis Study Group. High- and low-risk profiles for the development of multiple sclerosis within 10 years after optic neuritis: experience of the optic neuritis treatment trial. Arch Ophthalmol. 2003;121:944-949.
78 Sharpe JA, Sanders MD. Atrophy of myelinated nerve fibers in the retina in optic neuritis. Br J Ophthalmol. 1975;59:229-232.
79 Slamovitis TL, Rosen CE, Cheng KP, et al. Visual recovery in patients with optic neuritis and visual loss to no light perception. Am J Ophthalmol. 1991;111:209-214.
80 Gasperini C, Pozzilli C, Bernardi S. Irreversible total blindness in multiple sclerosis [Letter]. Ital J Neurol Sci. 1991;12:329.
81 Uhthoff W. Untersuchungen uber bei multiplen Herdsklerose vorkommenden Augenstorugen. Arch Fur Psychiatr Nervenkrankheiten. 1890;21:55-116. 303–410.
82 Lepore FE. The origin of pain in optic neuritis. Determinants of pain in 101 eyes with optic neuritis. Arch Neurol. 1991;48:748-749.
83 Pulfrich C. Die Stereoskopie im dienste der isochromen und heterochromen photometrie. Naturwissenschaften. 1922;10:533-564.
84 Ansari KA. Olfaction in multiple sclerosis, with a note on the discrepancy between optic and olfactory involvement. Eur Neurol. 1976;14:138-145.
85 Christ T, Pillunat LE, Stodtmeister R. The ‘flicker test’ according to Aulhorn in the diagnosis of acute optic neuritis. Ophthalmologica. 1986;192:220-227.
86 Savitsky N, Rangell L. The ocular findings in multiple sclerosis. Res Publ Assoc Res Nerv Ment Dis. 1950;28:403-413.
87 Wybar KC. The ocular manifestations of disseminated sclerosis. Proc R Soc Med. 1952;45:315-320.
88 Marshall D. Ocular manifestation of multiple sclerosis and relationship to retrobulbar neuritis. Trans Am Ophthalmol Soc. 1950;48:487-525.
89 Brown P. Pathophysiology of spasticity [Editorial and Review]. J Neurol Neurosurg Psychiatry. 1994;57:773-777.
90 D’Costa DF, Vania AK, Millac PA. Multiple sclerosis associated with trismus. Postgrad Med J. 1990;66:853-854.
91 Noffsinger D, Olen WO, Carhart R, et al. Auditory and vestibular aberrations in multiple sclerosis. Acta Otolaryngol Suppl. 1972;303:1-63.
92 Howard RS, Wiles CM, Hirsch NP, et al. Respiratory involvement in multiple sclerosis. Brain. 1992;115:479-494.
93 Oppenheim H. Textbook of Nervous Diseases for Physicians and Students. Edinburgh: O Schulze and Co, 1991.
94 Babinski J, Dubois R. Douleurs a forme de decharge electrique, consecutive aux tramatismes de la nuque. Presse Med. 1998;26:64.
95 L’hermitte J, Bollak, Nicholas M. Les douleurs a type de decharge electrique consecutives a la flexion cephalique dans la sclerose en plaques. Rev Neurol. 1924;42:56-62.
96 Hess DC, Sethi KD. Elipepsia partialis continua in multiple sclerosis. Int J Neurosci. 1990;50:109-111.
97 Hong LS, Wasserstein PH, Adornato BT. Tonic spasms in multiple sclerosis. Anatomic basis and treatment. West J Med. 1991;154:723-726.
98 Shibasaki H, Kuroiwa Y. Painful tonic seizure in multiple sclerosis. Arch Neurol. 1974;30:47-51.
99 Osterman PO. Paroxysmal itching in multiple sclerosis. Int J Dermatol. 1979;18:626-627.
100 Moulin DE. Pain in multiple sclerosis. Neurol Clin. 1989;7:321-331.
101 Portenoy RK, Yang K, Thorton D. Chronic intractable pain: an atypical presentation of multiple sclerosis. J Neurol. 1988;325:226-228.
102 Rushton JG, Olafson RA. Trigeminal neuralgia associated with multiple sclerosis. Arch Neurol. 1965;13:383-384.
103 Fisher M, Long RR, Drachman DA. Hand muscle atrophy in multiple sclerosis. Arch Neurol. 1983;40:811-815.
104 Masur H, Oberwittler C. MS and syringomyelia [Letter to the Editor]. Neurology. 1992;42:464-592.
105 Ransohoff RM, Whitman GJ, Weinstein MA. Noncommunication syringomyelia in multiple sclerosis: detection by magnetic resonance imaging. Neurology. 1990;40:718-721.
106 Miglietta O, Lowenthal M. A study of peripheral nerve involvement in fifty-four patients with multiple sclerosis. Arch Phys Med Rehabil. 1961;42:573-578.
107 Zee PC, Cohen BA, Walczak T, et al. Peripheral nervous system involvement in multiple sclerosis. Neurology. 1991;41:457-460.
108 Eisen AA, Odusote K. Central and peripheral conduction times in multiple sclerosis. Electroencephalogr Clin Neurophysiol. 1980;48:253-265.
109 Waxman SG. Peripheral nerve abnormalities in multiple sclerosis [Editorial]. Muscle Nerve. 1993;16:1-5.
110 Bemelmans BL, Hommes OR, Van Kerrebroeck PE, et al. Evidence for early lower urinary tract dysfunction in clinically silent multiple sclerosis. J Urol. 1991;145:1219-1224.
111 Hinds JP, Eidelman BH, Wald A. Prevalence of bowel dysfunction on multiple sclerosis: a population survey. Gastroenterology. 1990;98:1538-1542.
112 Fantelli FJ, Mitsumoto H, Sebek BA. Multiple sclerosis and malabsorption. Lancet. 1978;1:1039-1040.
113 Sorensen M, Lorentzen M, Petersen J, et al. Anorectal dysfunction in patients with urologic disturbance due to multiple sclerosis. Dis Colon Rectum. 1991;34:136-139.
114 Lilius HG, Valtonen EJ, Wirkstrom J. Sexual problems in patients suffering from multiple sclerosis. J Chronic Dis. 1976;29:643-647.
115 Stenager E, Stenager EN, Jensen K. Sexual aspects of multiple sclerosis. Semin Neurol. 1992;12:120-124.
116 Szasz G, Paty DW, Lawton-Speert S, et al. A sexual functioning scale in multiple sclerosis. Acta Neurol Scand. 1984;70(Suppl 101):37-43.
117 Szasz G, Paty DW, Maurice WL. Sexual dysfunctions in multiple sclerosis. Ann NY Acad Sci. 1984;436:443-452.
118 Vas CJ. Sexual Impotence and some autonomic disturbances in men with multiple sclerosis. Acta Neurol Scand. 1969;45:166-182.
119 Ivers RR, Goldstein NP. Multiple sclerosis: a current appraisal of symptoms and signs. Proc Mayo Clin. 1963;38:457.
120 Valleroy ML, Kraft GH. Sexual dysfunction in multiple sclerosis. Arch Phys Med Rehab. 1984;65:125-128.
121 Pentland B, Ewing DJ. Cardiovascular reflexes in multiple sclerosis. Eur Neurol. 1987;26:46-50.
122 Senaratne MPJ, Carroll D, Warren KG, et al. Evidence for cardiovascular autonomic nerve dysfunction in multiple sclerosis. J Neurol Neurosurg Psychiatry. 1984;47:947-952.
123 Schroth WS, Tenner SM, Rappaport BA, et al. Multiple sclerosis as a cause of atrial fibrillation and electrocardiographic changes. Arch Neurol. 1992;49:422-424.
124 Anema JR, Heijenbrok MW, Faes TJ, et al. Cardiovascular autonomic function in multiple sclerosis. J Neurol Sci. 1991;104:129-134.
125 Cartlidge NE. Autonomic function in multiple sclerosis. Brain. 1972;95:661-664.
126 Vita G, Fazio MC, Milone S, et al. Cardiovascular autonomic dysfunction in multiple sclerosis is likely related to brainstem lesions. J Neurol Sci. 1993;120:82-86.
127 Yokota T, Matsunaga T, Okiyama R, et al. Sympathetic skin response in patients with multiple sclerosis compared with patients with spinal cord transection and normal controls. Brain. 1991;114:1381-1394.
128 Sullivan F, Hutchinson M, Bahandeka S, et al. Chronic hypothermia in multiple sclerosis. J Neurol Neurosurg Psychiatry. 1987;50:813-815.
129 Confavreux C, Vukusic S, Adeleine P. Early clinical predictors and progression of irreversible disability in multiple sclerosis: an amnesic process. Brain. 2003;126:770-782.
130 Confavreux C, Aimard G, Devic M. Course and prognosis of multiple sclerosis assessed by the computerized data processing of 349 patients. Brain. 1980;103:281-300.
131 Mayr WT, Pittock SJ, McClelland RL, et al. Incidence and prevalence of multiple sclerosis in Olmsted County, Minnesota, 1985–2000. Neurology. 2003;61:1373-1377.
132 Goodkin DE, Hertsgaard D, Rudick RA. Exacerbation rates and adherence to disease type in a prospectively followed-up population with multiple sclerosis. Implications for clinical trials. Arch Neurol. 1989;46:1107-1112.
133 Durelli L, Bongioanni MR, Cavallo R, et al. Chronic systemic high-dose recombinant interferon alfa-2a reduces exacerbation rate, MRI signs of disease activity, and lymphocyte interferon gamma production in relapsing-remitting multiple sclerosis. Neurology. 1994;44:406-413.
134 Johnson KP, Brooks BR, Cohen JA, et al. Copolymer 1 reduces relapse rate and improves disability in relapsing-remitting multiple sclerosis: results of a phase III multicenter, double-blind placebo-controlled trial. The Copolymer 1 Multiple Sclerosis Study Group. Neurology. 1995;45:1245-1247.
135 Jacobs LD, Cookfair DL, Rudick RA, et al. Intramuscular interferon beta-1a for disease progression in relapsing multiple sclerosis. The Multiple Sclerosis Collaborative Research Group (MSCRG). Ann Neurol. 1996;39:285-294.
136 Russo P, Paolillo A, Caprino L, et al. Effectiveness of interferon beta treatment in relapsing remitting multiple sclerosis: an Italian cohort study. J Eval Clin Pract. 2004;10:511-518.
137 Kalanie H, Gharagozle K, Hemmatie A, et al. Interferon beta 1a and intravenous immunoglobulin treatment for multiple sclerosis in Iran. Eur Neurol. 2004;52:202-206.
138 Weinshenker BG, Bass B, Rice GP, et al. The natural history of multiple sclerosis: a geographically based study. II. Predictive value of the early clinical course. Brain. 1989;112:1419-1428.
139 Ebers GC, Paty DW. Natural history studies and applications to clinical trials. In: Paty DW, Ebers GC, editors. Multiple Sclerosis. Philadelphia: FA Davis; 1998:192-228.
140 Ebers GC, Paty DW. Natural history studies and applications to clinical trials. In: Paty DW, Ebers GC, editors. Multiple Sclerosis. Philadelphia: FA Davis; 1998:192-228.
141 Poser C. The course of multiple sclerosis. Arch Neurol. 1985;42:1035.
142 Weinshenker BG, Bass B, Rice GP, et al. The natural history of multiple sclerosis: a geographically based study. I. Clinical course and disability. Brain. 1989;112:133-146.
143 Vukusic S, Confavreux C. Prognostic factors for progression of disability in the secondary progressive phase of multiple sclerosis. J Neurol Sci. 2003;206:135-137.
144 Minderhoud JM, Van der Hooven JH, Prange AJ. Course and prognosis of chronic progressive multiple sclerosis: results of an epidemiological study. Acta Neurol Scand. 1988;78:10-15.
145 Poser S, Poser W, Schlaf G, et al. Prognostic indicators in multiple sclerosis. Acta Neurol Scand. 1986;74:387-392.
146 Fog T. Topographic distribution of plaques in the spinal cord in multiple sclerosis. Arch Neurol. 1950;63:382-414.
147 Kurtzke JF. International symposium on MS—Goteberg 1972. Ann NY Acad Sci. 1974;Suppl 58:14.
148 Kurtzke JF. On the evaluation of disability in multiple sclerosis. Neurology. 1961;11:686-694.
149 Kurtzke JF. Clinical manifestations of multiple sclerosis. In: Vinken PJ, Bruyn GW, editors. Handbook of Clinical Neurology. Amsterdam: Elsevier North-Holland; 1970:161-216.
150 Rice GP, Kremenchutzky M, Cottrell D, et al. Observations from the natural history cohort of London, Ontario. In: Filippi M, Comi G, editors. Primary Progressive Multiple Sclerosis Series—Topics in Neuroscience. Milan, Italy: Springer-Verlag, 2001. Chapter 2.
151 Pittock SJ, Mayr WT, McClelland RL, et al. Rodriguez: Disability profile of multiple sclerosis did not change over 10 years in a population based prevalence cohort. Neurology. 2004;62:601-606.
152 Weinshenker BG, Rice GPA, Noseworthy JH, et al. The natural history of multiple sclerosis: a geographically based study. III. Multivariate analysis of predictive factors and models of outcome. Brain. 1991;114:1045-1056.
153 Traboulsee A, Dehmeshki J, Peters KR, et al. Disability in multiple sclerosis is related to normal appearing brain tissue MTR histogram abnormalities. Mult Scler. 2003;9:566-573.
154 Kremenchutzky M, Sim D, Baskerville J, et al. A study of the causes of death in multiple sclerosis patients. Neurology. 2000;54(Suppl 3A):350.
155 Runmarker B, Andersen O. Prognostic factors in a multiple sclerosis incidence cohort with twenty-five years of follow-up. Brain. 1993;116:117-134.
156 Rodriguez M, Siva A, Cross S, O’Brien P, Kurland L. Optic neuritis: A population based study in Olmsted county, Minnesota. Neurology. 1994;44(Suppl. 2):A374.
157 Gudmundsson KR. Clinical Studies of MS in Iceland. Acta Neurol Scand. 1971;47(Suppl. 48):1-78.






