[level-membership-for-pulmolory-and-respiratory-category]
Clinical Assessment of Acid-Base and Oxygenation Status
After reading this chapter, you will be able to:
• Differentiate between arterial blood gas classification and interpretation
• Apply a systematic arterial blood gas classification method
• Differentiate between oxygenation and ventilation defects
• Describe the basis for compensatory activity in all acid-base disturbances
• Explain how the anion gap computation can help the clinician differentiate the causes of metabolic acidosis
• Explain why acute changes in PaCO2 affect plasma [HCO3−]
• Explain how acid-base disturbances affect plasma [K+] and [Cl−]
• Explain why the standard bicarbonate and base excess measurements more accurately reflect purely metabolic acid-base disturbances than plasma [HCO3−]
• Differentiate between the traditional Henderson-Hasselbalch approach and Stewart’s strong ion approach to acid-base physiology
• Distinguish between pulmonary and cardiovascular factors that affect tissue oxygenation
• Classify the causes and severity of oxygenation defects
• Interpret various pulmonary and cardiovascular tissue oxygenation indicators
Classification Versus Interpretation
Chapter 10 discusses the concept of respiratory and metabolic acid-base disturbances and the compensatory responses they elicit. This chapter introduces a systematic method for the classification, or categorization, of arterial blood gases in terms of acid-base balance and oxygenation status. Classification is the first essential step in the development of a rational therapeutic basis for correcting acid-base and oxygenation problems. The classification process focuses attention on the general problem areas and provides a starting point for interpretation, or the in-depth exploration and comprehension of the underlying disorder.
Classification of Acid-Base Disturbances
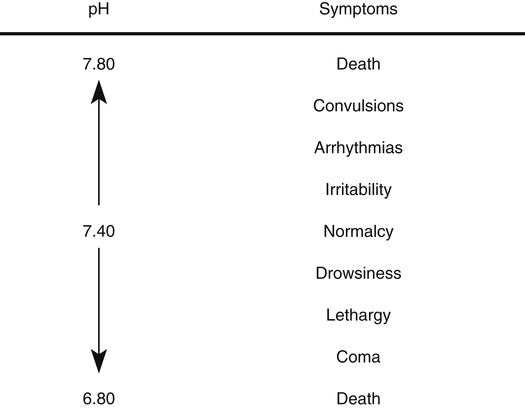
The relevant arterial blood gas components in evaluating acid-base status are pH, PCO2, and [HCO3−]. Arterial pH reflects hydrogen ion activity of extracellular fluid (plasma), which generally correlates with intracellular fluid pH. Chapter 10 emphasizes the importance of regulating the pH from a cellular enzyme standpoint. Abnormal pH values also affect the central nervous system, causing the clinical manifestations shown in Figure 13-1. Generally, a low arterial pH (acidemia) depresses neuronal excitability, whereas a high pH (alkalemia) has the opposite effect (greatly increased excitability).1 An extremely low pH (pH <7.00) causes a coma, whereas an extremely high pH (pH >7.80) causes convulsions and tetany (a state of sustained muscle spasm). Low arterial pH values (pH <7.10) also predispose patients to ventricular arrhythmias and reduce heart muscle contractility.2
Systematic Classification
The most consistent, reliable results are achieved when an orderly, systematic, unvarying approach is used to analyze acid-base problems. If exactly the same step-by-step approach is used for each problem, confusion and premature conclusions can be avoided. A systematic approach also helps identify inconsistencies and errors in blood gas data. Box 13-1 outlines four steps in acid-base classification. The order of steps 1 through 3 is not as important as following the same sequence for each situation.
Step 3: Analyze Nonrespiratory (Metabolic) Involvement ([HCO3−])
Plasma [HCO3−] is not as specific and accurate in indicating metabolic acid-base disturbances as PaCO2 is in indicating respiratory disturbances. The reason is that plasma bicarbonate responds slightly to pure respiratory (i.e., PCO2) changes. The carbon dioxide hydration reaction explains this phenomenon. As explained in Chapter 10, an acute increase in PaCO2 of 10 mm Hg increases [HCO3−] by about 1 mEq/L. Thus, the plasma [HCO3−] must be evaluated with caution; if it falls outside of the normal range but the abnormality can be explained by the effect of the carbon dioxide hydration reaction, the [HCO3−] level is not responsible for any existing acid-base disorder.
Step 4: Assess for Compensation
It is assumed that compensation for a primary acid-base disorder is never truly complete in the sense of restoring arterial pH all the way to 7.40.3 In a compensated disorder, the pH is on the acid or alkaline side of the normal range, depending on whether the primary causative disorder created an acidosis or an alkalosis. That is, if the pH is on the acid side of normal (<7.40 but at least ≥7.35), the main cause of the original acid-base imbalance is the component (PaCO2 or HCO3−) that, by itself, would cause an acidosis. For example, if the pH is 7.36, PaCO2 is 80 mm Hg, and HCO3− is 44 mEq/L, compensation is present because the pH is in the normal range, although it is on the acid side of normal. The primary cause of the original acid-base disturbance (before compensatory activity started) must be the factor that would produce acidemia (i.e., the increased PaCO2 of 80 mm Hg). Thus, this set of blood gases would be classified as compensated (chronic) respiratory acidosis. The primary disturbance is of respiratory origin, and the increased [HCO3−] is a secondary metabolic compensatory response. The reason the pH is on the acid side of the normal range is that the body generally does not overcompensate; it does not increase the [HCO3−] so much that it converts the previously acidotic environment to one with a pH on the alkaline side of normal. Conversely, if the pH is 7.15, PaCO2 is 80 mm Hg, and HCO3− is 26 mEq/L, no compensation has occurred; that is, HCO3− is still normal, not elevated as would be expected if compensatory action were underway.
Compensatory activity does not correct the primary acid-base disturbance; the primary defect is still present. Compensatory activity merely works to restore the pH to the normal range. Table 13-1 summarizes acid-base and ventilatory classification. Table 13-2 classifies the degree of compensation for acid-base disturbances.
TABLE 13-1
Acid-Base and Ventilatory Classification
| Component | Classification | Ranges |
| pH (arterial) | Normal status | 7.35-7.45 |
| Acidemia | <7.35 | |
| Alkalemia | >7.45 | |
| PaCO2 (mm Hg) | Normal ventilatory statusRespiratory acidosis (hypoventilation)Respiratory alkalosis (hyperventilation) | 35-45>45<35 |
| [HCO3−] (mEq/L) | Normal metabolic statusMetabolic acidosisMetabolic alkalosis | 22-26<22>26 |
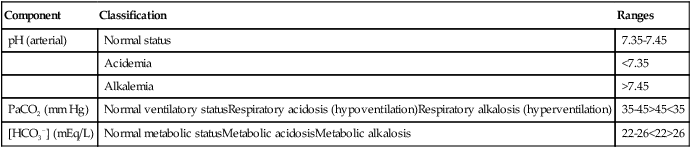
TABLE 13-2
Degrees of Acid-Base Compensation
| Compensating (Noncausative) Component | pH | Classification |
| Within normal range | Abnormal | Noncompensated (acute) |
| Out of normal range in the expected direction | Abnormal | Partially compensated |
| Out of normal range in the expected direction | Normal | Compensated (chronic) |
Respiratory Acidosis (Inadequate Ventilation)
< ?xml:namespace prefix = "mml" />
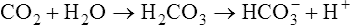
Therefore, hypercapnia is synonymous with respiratory acidosis.
Causes
cause respiratory acidosis. Chronic obstructive pulmonary disease (COPD) is the most frequent cause of respiratory acidosis, mostly because the airways resistance of a patient with severe COPD is so high that the patient cannot sustain the ventilatory work required to maintain a normal PaCO2.3 Central nervous system depression (drug-induced), extreme obesity (impaired diaphragmatic movement), and neuromuscular disorders (spinal cord lesions, paralytic neuromuscular diseases) are other causes of hypoventilation and respiratory acidosis. Hypercapnia may occur in different ways. A person may have an absolute decrease in ventilation because of drug-induced central nervous system depression, or a patient with COPD and a limited ventilatory reserve may sustain a normal PaCO2 at rest but may not accommodate the increased carbon dioxide production associated with increased physical activity.
Uncompensated hypercapnia implies the presence of acute ventilatory failure; the resulting respiratory acidosis is manifested by a low arterial pH, increased PaCO2, and normal or slightly high [HCO3−]. In this situation, a slightly increased [HCO3−] is not a sign that the kidneys have started compensatory activity; it merely reflects the effect of the carbon dioxide hydration reaction on [HCO3−] (see Chapter 10).
Compensation
Renal (kidney) compensation for respiratory acidosis begins as soon as PaCO2 increases. The kidney reclaims HCO3− from the renal tubular filtrate, returning it to the blood. The arterial pH is brought into the normal range because the [HCO3−]-dissolved carbon dioxide ratio is restored near its normal 20:1 range (see Chapter 10). However, this process cannot keep pace with an acutely increasing PaCO2. Full compensation may take several days.
Clinical Manifestations
Patients with neuromuscular weakness or mechanical breathing difficulties usually breathe shallowly and rapidly and are short of breath; they are often anxious and in obvious distress. Drug-induced central nervous system depression produces slow, shallow breathing and possibly apnea. Acute respiratory acidosis produces more serious physiological consequences than chronic respiratory acidosis. Rapidly increasing PaCO2 causes cerebral vasodilation and increased intracranial pressure (ICP), possibly leading to retinal venous distention and retinal hemorrhages; in addition, the patient may develop myoclonus (spasmodic muscle jerks), asterixis (a hand-flapping tremor in which the patient cannot keep the wrists flexed with the arms extended), and mental confusion.3 An abrupt onset of hypercapnia in which PaCO2 rises beyond 70 mm Hg can lead to coma, although patients with chronic hypercapnia can tolerate much higher PaCO2 values.3 Hypercapnia increases the cardiac output and dilates peripheral vessels, often resulting in a bounding pulse and warm, flushed skin.
Correction
needed to restore ventilation. However, if hypoventilation is chronic and compensation has restored arterial pH to the normal range, corrective action aimed at reducing the PaCO2 is inappropriate and possibly harmful. In this situation, a rapidly decreasing PaCO2 induces a sudden alkalosis because of renal compensation and elevated blood HCO3− levels.
Respiratory Alkalosis (Alveolar Hyperventilation)
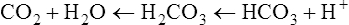
Therefore, hypocapnia is synonymous with respiratory alkalosis.
Causes
Hyperventilation causes hypocapnia. Any process in which ventilatory elimination of carbon dioxide exceeds its production causes respiratory alkalosis. Causes can be divided into three categories: (1) hypoxia, (2) pulmonary diseases, and (3) central nervous system diseases. Probably the most common cause of hyperventilation in patients with pulmonary disease is arterial hypoxemia, mediated through the peripheral chemoreceptors.3 Hypoxia-induced respiratory alkalosis can be caused by high altitude or pulmonary diseases characterized by intrapulmonary shunting (e.g., pneumonia, pulmonary edema). Other pulmonary diseases associated with hyperventilation and respiratory alkalosis include interstitial fibrosis, pulmonary embolism, and acute asthma. J-receptor stimulation in the lung parenchyma may be involved in interstitial diseases, pneumonia, and pulmonary edema. The feeling of dyspnea from high airways resistance and resulting anxiety is a probable mechanism for respiratory alkalosis in acute asthma. General anxiety, fever, stimulating drugs, pain, and injuries of the central nervous system are other possible causes of hyperventilation.
Clinical Manifestations
cramping of the hands or feet (carpopedal spasm), and possibly tetanic convulsions (a fusion of many muscle spasms producing a sustained contraction without relaxation). The low PaCO2 level may constrict cerebral vessels enough to impair cerebral circulation, causing light-headedness, dizziness, and syncope (fainting). If respiratory alkalosis is anxiety-induced, the patient may show signs of panic and express feelings of impending doom. Vision may become impaired (tunnel vision), and speaking may become difficult.3
Compensation
The kidneys compensate for respiratory alkalosis by excreting HCO3− in the urine (bicarbonate diuresis). This activity brings the arterial pH down toward the normal range because the [HCO3−]-dissolved carbon dioxide ratio is restored near its normal 20:1 range. However, renal compensation rarely causes [HCO3−] to fall below 18 mEq/L.3 Renal compensation is a relatively slow process; it may take days until the compensation process is finally completed.
Metabolic (Nonrespiratory) Acidosis
Anion Gap
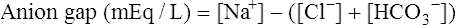
The “anion gap” can be thought of as a measure of the concentrations of other anions in the plasma besides Cl− and HCO3−. Figure 13-2, A, shows that normal concentrations of the routinely measured electrolytes in the plasma are about 140 mEq/L for Na+, 105 mEq/L for Cl−, and 24 mEq/L for HCO3−, producing an apparent anion gap of 11 mEq/L (140 − [105 + 24] = 11). The normal range for the anion gap is 8 to 16 mEq/L.3 Because [Na+] exceeds ([Cl−] + [HCO3−]), the unmeasured anions must outnumber the unmeasured cations (see Figure 13-2, A). Therefore, the anion gap also can be thought of as the difference between the unmeasured anions and unmeasured cations:
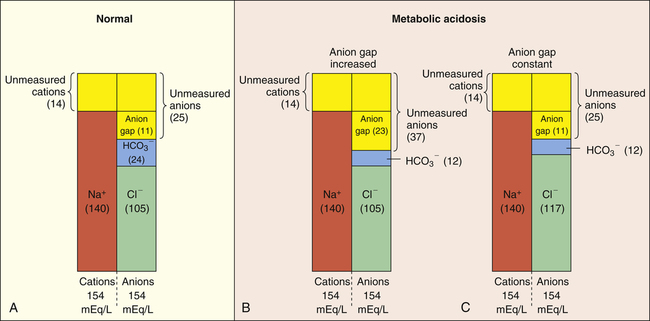
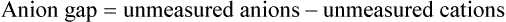
If total anion and cation concentrations each equal 154 mEq/L (see Figure 13-2, A), there are normally 14 mEq/L of unmeasured cations and 25 mEq/L of unmeasured anions. Figure 13-2, A, shows that the anion gap is 11 mEq/L:

Metabolic acidosis caused by an accumulation of fixed acids in the body increases the anion gap (>16 mEq/L). The hydrogen ions of these fixed acids react with HCO3−, lowering its concentration in the plasma. This leads to an increased number of unmeasured anions; that is, when HCO3− buffers the fixed acid’s H+, the anion portion of the fixed acid remains in the plasma, increasing the unmeasured anion concentration (see Figure 13-2, B). Therefore, a high anion gap indicates the fixed acid concentration has increased in the body. The patient’s clinical history and current status help clarify which type of acid this may be. For example, in severe shock, tissue hypoxia may generate lactic acid. A history of diabetes is consistent with ketoacid formation. A history of kidney failure is consistent with uremic acidosis.
Metabolic acidosis caused by HCO3− loss from the body does not produce an increased anion gap because HCO3− loss is accompanied by Cl− gain, keeping the anion gap within normal limits (see Figure 13-2, C). The law of electroneutrality helps explain the reciprocal nature of [HCO3−] and [Cl−] in this situation. With a constant cation concentration, the loss of HCO3− means another anion must be gained to maintain electroneutrality. In this situation, the kidney increases its reabsorption of the most abundant anion in the tubular filtrate, Cl−. (This disturbance in electroneutrality does not occur when fixed acids accumulate and are buffered by HCO3− because no anions are actually lost from the body in that situation.) The type of metabolic acidosis in which HCO3− is lost from the body is sometimes called hyperchloremic acidosis because of the characteristic increase in plasma [Cl−] that occurs. Examples of conditions in which HCO3− is lost or Cl− is gained include severe diarrhea and pancreatic fistulas. A fistula is an abnormal tubelike channel connecting one body cavity to another (in this situation, abnormally connecting the pancreas to the intestinal tract). Pancreatic juice is fairly alkaline, as are all intestinal fluids below the stomach.2 Thus much bicarbonate can be lost during prolonged diarrhea. Box 13-2 summarizes causes of anion gap and non–anion gap metabolic acidosis.
Clinical Manifestations
Metabolic acidosis generally causes a great increase in minute ventilation. Patients may complain of dyspnea and, in extreme acidosis, may descend into a stupor and coma.3 Hyperpnea (increased tidal volume depth) is a common finding on physical examination. In severe metabolic acidosis (e.g., diabetic ketoacidosis), an extremely deep, gasping type of breathing develops, called Kussmaul’s respiration.
Arterial pH values less than 7.00 to 7.10 predispose the heart to potentially fatal ventricular arrhythmias and can reduce not only heart muscle contractility but also the heart’s ability to increase its contractile force in response to the body’s secretion of catecholamines, such as epinephrine.3 Neurological symptoms range from lethargy to coma in severe metabolic acidosis (see Figure 13-1).
Correction
The initial goal in treating severe acidemia is to raise the arterial pH above 7.2, a level at which serious cardiac arrhythmias are less likely, contractility is restored, and responsiveness to catecholamines improves.2 If increased ventilation maintains the pH at or above this level, immediate corrective action is usually not indicated. Treatment should be focused on correction of the underlying metabolic disorder. For example, in diabetic acidosis, insulin is given; in shock-induced lactic acidosis, measures are taken to improve blood pressure and cardiac output.
Historically, sodium bicarbonate (NaHCO3) was routinely infused during cardiac arrest to combat the lactic acidosis of tissue hypoxia; this practice has generally been discontinued. The 2010 American Heart Association (AHA) Guidelines for Cardiopulmonary Resuscitation and Emergency Cardiovascular Care recommend against the routine use of NaHCO3 in cardiac arrest because of the lack of evidence showing improved survival, and because of the negative side effects of NaHCO3, which include intracellular acidosis, cardiac arrhythmias, and inhibited hemoglobin release of oxygen.4 The AHA maintains that rapid return of spontaneous circulation (ROSC) through effective cardiopulmonary resuscitation is the mainstay of restoring acid-base balance during cardiac arrest. Rapid correction of arterial pH to values greater than 7.20 by NaHCO3 infusion can be harmful for the following reasons: when HCO3− buffers H+, the hydration reaction moves to the left, rapidly producing carbon dioxide, which quickly diffuses across the blood-brain barrier into the CSF. Because blood HCO3− cannot follow carbon dioxide across the blood-brain barrier, the CSF rapidly becomes more acidic, possibly aggravating the neurological symptoms (coma) of metabolic acidosis.2 The decreased CSF pH increases central chemoreceptor stimulation, causing the successfully resuscitated patient to hyperventilate, which coupled with the prior NaHCO3 infusion may rapidly swing the previously acidotic arterial pH into an alkalotic range. In the same way, carbon dioxide produced by the buffering action of NaHCO3 diffuses rapidly across all cell membranes, including the cell membranes of the heart; this may precipitate cardiac arrhythmias.3 Intracellular pH generally tends to decrease with the rapid infusion of NaHCO3 because carbon dioxide diffuses across cell membranes into the cell much more rapidly than bicarbonate ions.3 The 2010 AHA Guidelines list special circumstances in which NaHCO3 infusion may be beneficial during cardiac arrest such as in patients with preexisting metabolic acidosis, and in patients with severe hyperkalemia (high plasma potassium concentration).
Metabolic (Nonrespiratory) Alkalosis
Causes
Metabolic alkalosis can occur in two general ways: (1) loss of fixed acids or (2) gain of blood buffer base. Both processes produce an increased plasma [HCO3−]. It is easy to understand why plasma [HCO3−] increases if it is ingested or administered intravenously. It is not as apparent why [HCO3−] increases when fixed acids are lost from the body. For an explanation of this phenomenon, consider a situation in which vomiting or nasogastric suction removes gastric hydrochloric acid (HCl) from the body (Figure 13-3). In response to HCl loss, H+ diffuses out of the gastric cell, accompanied by Cl− from the blood. The loss of H+ forces the carbon dioxide hydration reaction in the gastric cell to the right, which generates HCO3−; the HCO3− ion diffuses into the blood in exchange for Cl−. Thus, the plasma gains a bicarbonate ion for each chloride ion (or hydrogen ion) lost (see Figure 13-3).5
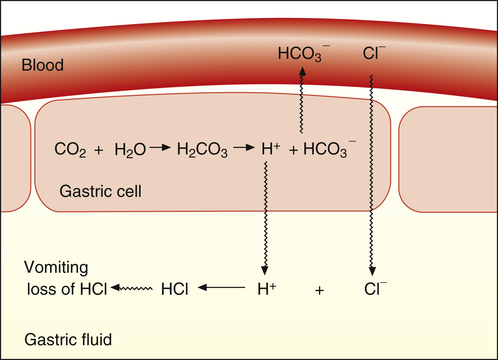
Box 13-3 summarizes the causes of metabolic alkalosis. Increased HCO3− ingestion rarely causes metabolic alkalosis because the normal kidney can rapidly excrete large amounts of HCO3− in the urine.2 Metabolic alkalosis is most often caused by the loss of gastric acid through protracted vomiting or continuous nasogastric suctioning.3 It is also caused by hypochloremia (low plasma [Cl−]) and hypokalemia (low plasma [K+]). Metabolic alkalosis is quite common in acutely ill patients and probably the most complicated acid-base imbalance to treat because it involves fluid and electrolyte imbalances. Many causes of metabolic alkalosis are iatrogenic, resulting from the use of diuretics, low-salt diets, and nasogastric drainage.
The mechanisms by which volume depletion, hypokalemia, and hypochloremia produce alkalosis are discussed in Chapter 22. Briefly, the kidney’s strong stimulus to reabsorb Na+ from the renal tubule filtrate is predominant over its need to maintain chloride, potassium, or acid-base balance. A low plasma [Cl−] (hypochloremia) produces a low tubular filtrate Cl− concentration; this presents a special problem because chloride ions must accompany sodium ions when they are reabsorbed from the tubular filtrate. If chloride ions are scarce (as may be induced by diuretic therapy, low-salt diets, or gastric HCl loss), the kidney reabsorbs Na+ by secreting abnormally large amounts of H+ and K+ into the filtrate. In other words, these ions are secreted in exchange for Na+ to maintain electroneutrality of the filtrate. This process leads to alkalosis (H+ loss) and hypokalemia
(K+ loss). Volume depletion (hypovolemia) induced by diuretics exaggerates alkalosis and hypokalemia because hypovolemia profoundly increases the kidney’s Na+ reabsorption stimulus.
Compensation
occurs because alkalemia eventually leads to CSF alkalosis, decreasing the stimulation of central chemoreceptors. During hypoventilation, the increase in PaCO2 increases the plasma carbonic acid concentration, offsetting the metabolically generated alkalosis. On average, the PaCO2 increases about 0.7 mm Hg for every 1-mEq/L increase in plasma [HCO3−].6
Traditionally, the belief was that the hypoxemia induced by hypoventilation limited respiratory compensation for metabolic alkalosis. In other words, as PACO2 increases during hypoventilation, PAO2 decreases, leading to hypoxemia, which would stimulate peripheral chemoreceptors and counteract compensatory hypoventilation. However, more recent evidence contradicts this theory; alkalosis blunts the sensitivity of the carotid bodies to hypoxemia (see Chapter 11). Alkalemic patients with PaO2 values less than 50 mm Hg may hypoventilate to PaCO2 values much greater than 60 mm Hg.6
Underlying pulmonary disease may also limit compensatory hypoventilation in metabolic alkalosis. Pulmonary edema, fibrosis, vascular congestion, and inflammatory processes such as pneumonia stimulate intrapulmonary irritant receptors in airway walls and J-receptors in the alveolar interstitium. Such stimulation causes tachypnea and hyperventilation, which may limit compensatory hypoventilation and carbon dioxide retention.2,6 Other factors that tend to limit compensatory hypoventilation for metabolic alkalosis include anxiety, pain, infection, and fever.
Symptoms
Symptoms directly related to metabolic alkalemia are most likely related to causative factors (e.g., volume depletion [weakness and dizziness on standing] and hypokalemia [muscle weakness and high urine output]). As with respiratory alkalosis, patients may exhibit hyperactive reflexes, muscle cramping, and possibly tetanic convulsions.3 Similar to acidosis, severe alkalosis may also induce cardiac arrhythmias. Alkalosis, whether of respiratory or metabolic origin, promotes the movement of K+ into the cells, creating a hypokalemia that also can contribute to cardiac arrhythmias.
Correction
Corrective action is aimed at the underlying causes of metabolic alkalosis (e.g., restoring normal fluid volume, potassium, and chloride levels). After these fluids and electrolytes are restored to normal levels, the kidney can reabsorb Na+ normally (i.e., Cl− can be reabsorbed with Na+). The kidney does not need to reabsorb HCO3− with Na+ as it did when Cl− was scarce; this allows excess HCO3− to be excreted in the urine. Restoring plasma K+ levels causes K+ to diffuse into the red blood cells in exchange for H+, which moves out of the cell into the plasma. This reciprocal shift of K+ and H+ helps bring the plasma pH back toward normal.2
In extremely severe metabolic alkalosis (pH >7.55), acidifying agents such as dilute HCl or ammonium chloride may be infused intravenously.3 (The liver metabolizes NH4Cl, producing HCl.)
Metabolic Acid-Base Disturbance Indicators
Expected pH Relationships
Table 13-3 illustrates acute PaCO2 and pH relationships. If PaCO2 changes suddenly as a result of an acute ventilatory change, the subsequent change in pH must be caused by the change in PaCO2 (i.e., a respiratory change). The expected pH for a given acute change in PaCO2 can be precisely predicted using the Henderson-Hasselbalch equation. Table 13-3 illustrates this relationship. If PCO2 acutely increases by 20 mm Hg, the pH should decrease by (20 × 0.006), or 0.12 units, producing an expected pH of 7.28 (i.e., [7.40 − 0.12] = 7.28). This calculated expected pH differs from the actual measured blood pH only if a metabolic acid-base disturbance is present. For example, if the actual measured pH is more acidic than the calculated expected pH, nonrespiratory metabolic factors are causing the acidosis. Likewise, if the actual measured pH is more alkalotic than the expected pH, the alkalosis must be of metabolic origin. The following pH formulas can be derived from Table 13-3 and used to determine whether a measured abnormal pH is strictly due to the abnormal PCO2 or whether the abnormal pH is partly due to nonrespiratory (metabolic) factors:
TABLE 13-3
Acute Arterial Carbon Dioxide Pressure–pH Relationship
| PaCO2 Change | pH Change |
| Decrease | Increase |
| 1 mm Hg | 0.01 |
| 10 mm Hg | 0.10 |
| Increase | Decrease |
| 1 mm Hg | 0.006 |
| 10 mm Hg | 0.06 |
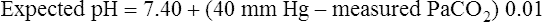
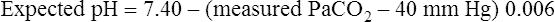
Stewart’s Strong Ion Approach to Acid-Base Balance
In the strong ion approach, substances that affect acid-base balance in body fluids are classified into three groups, based on their degree of dissociation in aqueous solution: strong ions, weak ions, and nonelectrolytes. Strong ions (e.g., Na+ and Cl−) are always fully dissociated, existing only as ions in aqueous solutions. (The term “strong” means strongly dissociated.) In other words, the number of strong ions in body fluids can never be changed by conversion back to the parent compound (e.g., NaCl or KCl), as occurs with weak ions. Weak ions are produced from compounds that only partially (weakly) dissociate in solution, including volatile CO2-related carbonic acid ions (HCO3− and H+) and nonvolatile acid ions such as phosphates and proteins.8 Weak acids dissociate until they reach equilibrium with their component ions, each in accordance with its unique equilibrium constant (see Chapter 10). The nonvolatile weak acids include proteins (chiefly albumin) and inorganic phosphates. Nonelectrolytes are substances that never dissociate in solution but contribute to the solution’s osmotic pressure; thus they affect the movement of ions and water across biological membranes that separate body fluids.
Independent Variables That Affect [H+]: [SID], [ATOT], and [CO2]
Stewart distinguished between independent and dependent variables involved in [H+] regulation. He showed through a series of complex equations that the [H+] of a solution is a dependent variable determined solely by three independent variables:9 (1) strong ion difference [SID]; (2) total concentration of nonvolatile weak acids [ATOT]; and (3) dissolved [CO2], which is a function of PCO2. The [SID] is the summative difference between the concentrations of all strong cations (positively charged ions) and all strong anions (negatively charged ions) in body fluids; for clinical purposes, [SID] = ([Na+] + [K+] + [Ca++] + [Mg++]) − ([Cl−] + [unidentified strong anions]).8,9 Normal body fluids contain more strong cations than strong anions8; the [SID] thus represents a net positive charge that must be balanced by weak anions in solution, as explained in the following paragraphs. The kidneys are responsible for maintaining a normal [SID] of about 40 to 42 mEq/L, primarily by excreting or reabsorbing Na+, K+, and Cl− ions.
Stewart’s theory of acid-base assessment rests on three basic assumptions about physiological solutions:3 (1) All solutions must obey the powerful principle of electrical neutrality; that is, the sum of all strong and weak cations must equal the sum of all strong and weak anions. (2) All weak nonvolatile acids in body fluids attain equilibrium with their dissociated ions according to their equilibrium constants (law of mass action). (3) The total amount of a substance in solution must remain constant unless it is added, removed, generated, or destroyed (conservation of mass).
The principle of electrical neutrality demands that the normal [SID] of body fluids, which represents a net positive charge, must be equal to the net negative charge of all weak anions in solution, that is, anions contributed by volatile and nonvolatile weak acids.8 This principle means that normally the [SID] is equal to [HCO3−] + [A−], or, stated differently, the [SID] is equal to the sum of the bicarbonate [HCO3−] and nonbicarbonate [A−] buffers, or the total buffer base (see Chapter 10). The number of weak anions supplied by volatile acid (i.e., HCO3− originating from H2CO3) is determined by the independent variable, PCO2, which is controlled by ventilation. The number of weak anions supplied by all nonvolatile weak acids is determined by the dissociation constant (temperature dependent) of the independent variable [ATOT]. The total amount of nonvolatile weak acid present ([ATOT]) at any given time is constant, such that [ATOT] = [H+] + [A−], where the universal symbols H+ and A− represent dissociated weak acid molecules. All weak acids in the plasma can be treated as a single acid because they are in equilibrium with the same solution; the dissociation of this combined weak acid group is represented by HA 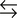 H+ + A−, which has an apparent dissociation constant that determines the number of weak anions contributed by [ATOT] in balancing the positive [SID].
H+ + A−, which has an apparent dissociation constant that determines the number of weak anions contributed by [ATOT] in balancing the positive [SID].
At any given time, PaCO2 is determined by ventilation, and the dissociation of [ATOT] is determined by its apparent dissociation constant. With these two independent variables predetermined, a change in the [SID] generates powerful electrochemical forces that cause H2O molecules to dissociate or reassociate in a way that maintains electrical neutrality. In this way, changes in the [SID] affect the solution’s [H+]; a decrease in the [SID] (a decrease in net positive charges) increases H2O dissociation, liberating more H+ to maintain electrical neutrality, whereas an increase in the [SID] (an increase in net positive charges) has the opposite effect.8 (This is consistent with a decrease or increase in total buffer base.)
Determinants of Metabolic Acid-Base Balance: [SID] and [ATOT]
In Stewart’s system, metabolic (nonrespiratory) acid-base disturbances can be caused only by changes in [SID] and nonvolatile weak acid concentration [ATOT], not by changes in [HCO3−]. Stewart asserts that [HCO3−] and [H+] are dependent variables, that is, dependent on the [SID], [ATOT], and PCO2. In this scheme, the kidneys manipulate the [SID] to change the plasma [H+] by excreting or reabsorbing Na+, K+, and Cl−. The kidneys place priority on keeping [Na+] constant to maintain intravascular volume. [K+], [Ca++], and [Mg++] do not vary enough to affect the [SID] significantly; the kidneys change the [SID] primarily by excreting or reabsorbing Cl− ions.8 With a constant [Na+], an increase in plasma [Cl−] reduces the [SID], causing hyperchloremic acidosis, whereas a decrease in plasma [Cl−] causes hypochloremic alkalosis; these changes cause equal magnitude losses and gains in [HCO3−]. Thus, in responding to metabolic acid-base disturbances, the kidney does not manipulate the HCO3− ion; instead it regulates the [SID], primarily by controlling [Cl−].
This perspective is a departure from the Henderson-Hasselbalch concept of acid-base balance in which [HCO3−] is treated as though it varies independently of dissolved [CO2], independently influencing pH or [H+]. However, in reality, [HCO3−] and [CO2] cannot vary independently of each other as the CO2 hydration reaction clearly shows; instead, both [HCO3−] and [H+] depend on [CO2]:9
Clinical Practicality of Strong Ion Approach
The complex nature of the equations involved in Stewart’s strong ion approach make this method clinically unwieldy; knowledge of protein and phosphate concentrations is required for accurate acid-base assessment. PCO2 and pH can be easily and directly measured in the clinical setting, and [HCO3−] can be calculated with sufficient precision using the Henderson-Hasselbalch equation; there is no need to calculate [HCO3−] and pH from the concentrations of electrolytes and weak acids, which are often unknown.3 Although the strong ion approach is a more conceptually correct approach, it is not sufficiently superior to the Henderson-Hasselbalch approach in the clinical context to merit its universal adoption. It is reasonable and clinically appropriate to explain metabolic acid-base physiology in terms of [H+] and [HCO3−] from the Henderson-Hasselbalch perspective.3,8,10 Hence, the traditional approach is retained in this book.
Assessment and Treatment of Hypoxia
Tissue oxygenation cannot be adequately evaluated by examining only the PaO2 and SaO2 (see Chapter 8). The focus in evaluating oxygenation must always be on oxygen delivery to the tissues. This process involves the breathing of an adequate concentration of oxygen (FIO2), the efficient transfer of oxygen from the alveolus to arterial blood (PAO2-PaO2 relationship), the maintenance of an adequate blood oxygen-carrying capacity (hemoglobin concentration), and adequate oxygen transport to all body tissues (blood flow or cardiac output). The clinician must consider all of these factors to assess oxygenation status adequately.
Classifying Tissue Hypoxia
Traditionally, the causes of tissue hypoxia have been classified arbitrarily into four categories: hypoxic hypoxia, anemic hypoxia, stagnant (hypoperfusion) hypoxia, and histotoxic hypoxia; the acronym HASH helps one recall these categories. Table 13-4 summarizes the characteristics of these four different types of hypoxia. One can make appropriate, effective treatment decisions only if one understands the root causes of hypoxia.
TABLE 13-4
| Category | Mean PAO2 | PaO2 | CaO2 | C O2 O2 |
Oxygen Delivery | Helped by Higher FIO2? |
| Hypoxic hypoxia | ||||||
| Hypoventilation | Low | Low | Low | Low | Low | Yes |
| High altitude | Low | Low | Low | Low | Low | Yes |
| Absolute shunt | Low | Low | Low | Low | Low | Limited∗ |
 / / mismatch mismatch |
Low | Low | Low | Low | Low | Yes |
| Anemic hypoxia | ||||||
| Low hemoglobin | Normal | Normal | Low | Low | Low | No∗ |
| Carbon monoxide poisoning | Normal | Normal | Low | Low | Low | Yes |
| Stagnant (hypoperfusion) | ||||||
| Shock (heart failure) | Normal | Normal | Normal | Low | Low | No∗ |
| Histotoxic hypoxia | ||||||
| Cyanide poisoning | Normal | Normal | Normal | High | Normal | No |
| Diffusion defect | Normal | Low | Low | Low | Low | Yes |
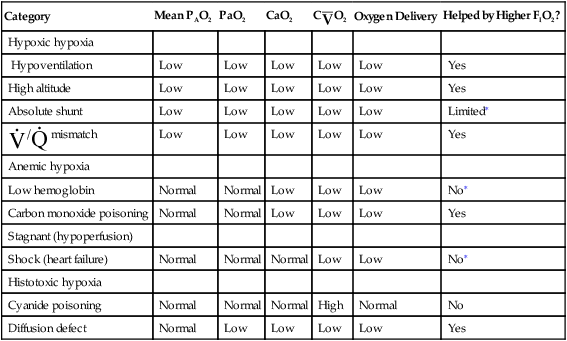
∗Although its effectiveness is limited, oxygen therapy is generally administered in these clinical conditions. The small gains in CaO2 may be critical in severely hypoxic patients, especially patients with myocardial hypoxia and cardiac arrest.
Hypoxic Hypoxia (Decreased PAO2)
Hypoxic hypoxia refers to mechanisms that decrease the alveolar PO2 (see Table 13-4). These mechanisms (hypoventilation, shunt, 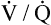 mismatch, and high-altitude breathing) were discussed in detail in Chapter 12. In shunt, the PO2 of collapsed or otherwise airless alveoli is effectively 0 mm Hg, reducing the overall average PAO2. Likewise, in
mismatch, and high-altitude breathing) were discussed in detail in Chapter 12. In shunt, the PO2 of collapsed or otherwise airless alveoli is effectively 0 mm Hg, reducing the overall average PAO2. Likewise, in 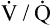 mismatch, PAO2 of underventilated lung regions is low, reducing the mean PAO2. Alveolar hypoxia produces low PaO2 values, leading to low arterial oxygen content and reduced oxygen delivery to the tissues. As discussed in Chapter 12, the effectiveness of oxygen therapy is limited in shunt because the added oxygen cannot reach collapsed alveoli. Treatment in this situation must be aimed at reopening and ventilating collapsed alveoli.
mismatch, PAO2 of underventilated lung regions is low, reducing the mean PAO2. Alveolar hypoxia produces low PaO2 values, leading to low arterial oxygen content and reduced oxygen delivery to the tissues. As discussed in Chapter 12, the effectiveness of oxygen therapy is limited in shunt because the added oxygen cannot reach collapsed alveoli. Treatment in this situation must be aimed at reopening and ventilating collapsed alveoli.
Anemic Hypoxia (Decreased Hemoglobin Concentration)
Carbon monoxide poisoning occurs in the setting of smoke inhalation from house fires, automobile exhaust, or poorly ventilated charcoal fires or gas furnaces. Oxygen therapy cannot add much oxygen to the blood because the available functional hemoglobin is already almost 100% saturated with room air breathing. Nevertheless, oxygen breathing hastens the displacement of carbon monoxide from the hemoglobin molecule, and the administration of 100% inspired oxygen is extremely important in treating carbon monoxide poisoning. The ideal treatment of this condition involves hyperbaric oxygen therapy, in which the individual’s body is placed in an oxygen-enriched environment pressurized above atmospheric pressure (i.e., a hyperbaric oxygen chamber). In this way, PaO2 and the amount of oxygen dissolved in the plasma can be raised far above values achievable while breathing 100% oxygen at atmospheric pressure; this significantly increases the total amount of oxygen the blood can carry. In addition, the extremely high plasma oxygen pressure facilitates the release of carbon monoxide by the hemoglobin molecule, shortening the half-life of carbon monoxide. Severe anemia caused by blood loss or decreased red blood cell production may be treated by blood transfusion (see Chapter 8).
Histotoxic Hypoxia (Blocked Oxidative Metabolism)
Histotoxic hypoxia refers to poisoning of the cellular oxygen use mechanisms. Cyanide poisoning is the typical example of histotoxic hypoxia. Cyanide combines with and inactivates cellular enzymes necessary for oxidative metabolism. Cyanide poisoning often occurs with carbon monoxide poisoning in the setting of smoke inhalation from house fires.11 High blood lactic acid levels with high mixed venous oxygen content are evidence that cellular oxygen consumption has been blocked and should raise the suspicion of cyanide poisoning. Treatment includes administration of 100% oxygen and inhaled amyl nitrite and intravenous sodium nitrite, which convert hemoglobin to methemoglobin; methemoglobin is an effective scavenger of free cyanide molecules.11
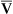
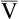
Diffusion Defects
Conditions that increase the diffusion pathway distance across the alveolar capillary membrane include pulmonary edema and processes leading to interstitial alveolar fibrosis. Normally, capillary blood and alveolar gas PO2 values equilibrate in the first one third of the time required to travel through the alveolar capillary. Thickened membranes require longer equilibrium times, and during exercise, equilibrium may not occur, leading to arterial hypoxemia (see Chapter 7). The contribution of diffusion defects to arterial hypoxemia when the patient is at rest is uncertain; the major mechanism is probably uneven ventilation-perfusion relationships caused by regional differences in lung compliance secondary to nonuniform distribution of lung disease.7 For this reason, some physiologists classify diffusion defects in the hypoxic hypoxia category. Increased FIO2 is helpful in treating diffusion defects because the increased diffusion gradient greatly enhances oxygen transfer across the alveolar capillary membrane into the blood.
Physiological Effects of Hypoxia
The body’s compensatory response to hypoxemia is increased cardiac output, improved tissue perfusion (as a result of capillary recruitment), and increased red blood cell production.1 Increased cardiac output and perfusion are immediate responses, whereas increased red blood cell production is a response to chronic hypoxemia. Increased cardiac output increases oxygen delivery to the tissues. Even if the blood is hypoxemic, circulating it more rapidly supplies the tissues with greater amounts of oxygen per minute.
Clinical Signs and Symptoms
If hemoglobin concentration and cardiac output are normal, PaO2 must acutely decrease to less than 50 to 60 mm Hg before clinical manifestations appear.7 At this PaO2, most people experience mild nausea, light-headedness, and dizziness. These symptoms do not necessarily occur in slowly progressing, chronic hypoxemia of equal severity. In chronic hypoxemia, the body compensates by producing more erythrocytes, increasing the CaO2.
In acute hypoxemia in the range of 50 to 60 mm Hg, peripheral chemoreceptor stimulation increases minute ventilation, although dyspnea is generally not perceived. PaO2 values from 35 to 50 mm Hg cause mental confusion; PaO2 less than 35 mm Hg causes decreased renal blood flow and cardiac conduction disturbances. This level of hypoxemia causes maximal minute ventilation and severe lethargy. Loss of consciousness and respiratory center depression occur at PaO2 values less than 25 mm Hg.12 In the presence of anemia or cardiac insufficiency, these manifestations become apparent at higher PaO2 values. Box 13-4 summarizes clinical signs and symptoms of acute hypoxemia from mild to severe conditions. Tissues with high oxygen consumption requirements, such as brain, heart, and kidney cells, are the most susceptible to hypoxia. A total lack of oxygen causes irreversible brain cell injury or death in minutes.
Normal and Abnormal Oxygenation Values
Normal PaO2 Values
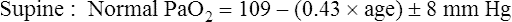
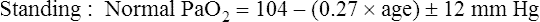
Normal PaO2 at High Altitude
Altitude also affects normal, expected PaO2. High altitude decreases barometric pressure and therefore it decreases inspired PO2. Up to an altitude of 10,000 feet above sea level, barometric pressure decreases about 24 mm Hg per 1000 feet of elevation.1 For example, in Denver, Colorado, where the elevation is 5280 feet, barometric pressure is about 633 mm Hg. At 10,000 feet elevation, barometric pressure is only 523 mm Hg.

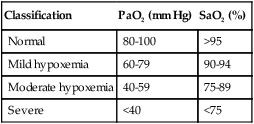
Severity of Arterial Hypoxemia
Table 13-5 arbitrarily classifies the severity of arterial hypoxemia. Traditionally, only PaO2 is classified in evaluating oxygenation. (Corresponding SaO2 values are included in Table 13-5.) Both CaO2 and cardiac output (i.e. oxygen delivery) are essential in evaluating oxygenation. (These factors are discussed later in this chapter.) Table 13-5 assumes a normal cardiac output and blood hemoglobin content.
TABLE 13-5
Classifying Severity of Hypoxemia∗
| Classification | PaO2 (mm Hg) | SaO2 (%) |
| Normal | 80-100 | >95 |
| Mild hypoxemia | 60-79 | 90-94 |
| Moderate hypoxemia | 40-59 | 75-89 |
| Severe | <40 | <75 |
Evaluating Pulmonary Oxygenation Defects
FIO2-PaO2 Relationship
CLINICAL FOCUS 13-10 Normal PaO2 for Denver Residents
At sea level, PAO2 is about 100 mm Hg while a person breathes room air, as the following equation shows:∗
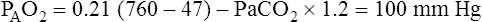
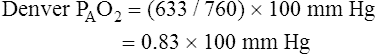
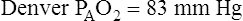
increase in inspired oxygen concentration. Beginning with a normal sea-level, room air PaO2 of about 100 mm Hg, FIO2 of 0.30 should produce a minimum PaO2 value of 150 mm Hg, FIO2 of 0.40 should produce a PaO2 of at least 200 mm Hg, and FIO2 of 1.0 should produce a PaO2 of at least 500 mm Hg. If a patient breathing oxygen has a PaO2 less than the value predicted by these calculations, the patient will be hypoxemic while breathing room air.
Indicators of Oxygen Transfer Efficiency
P(A-a)O2, PaO2/PAO2, and PaO2/FIO2 are indicators of the lung’s efficiency in transferring oxygen from alveolar gas to capillary blood. The advantages and disadvantages of these indicators are reviewed in Chapter 12. All of these indicators are sensitive to the degree of intrapulmonary shunt present, although PaO2/PAO2 is the most stable and reliable in this regard. Calculating the intrapulmonary shunt fraction using the classic shunt equation is the most accurate way to quantify the degree of shunt present (see Chapter 12).
Through observation of the PaO2 response to increased FIO2, shunt can be differentiated from  A/
A/ C mismatch as the major cause of hypoxemia (see Chapter 12). If PaO2 is low but oxygenation efficiency indicators and shunt fraction are normal, overall hypoventilation may be the cause of hypoxemia; this would be confirmed by an increased PaCO2, reciprocally related to PaO2.
C mismatch as the major cause of hypoxemia (see Chapter 12). If PaO2 is low but oxygenation efficiency indicators and shunt fraction are normal, overall hypoventilation may be the cause of hypoxemia; this would be confirmed by an increased PaCO2, reciprocally related to PaO2.
Evaluating Oxygenation Defects of Cardiovascular Origin
Oxygen Delivery to the Tissues
Arterial blood gases alone reveal nothing about the cardiovascular component of oxygenation. CaO2 and cardiac output must be evaluated to estimate the most critical aspect of oxygenation status: oxygen delivery (O2DEL) to the tissues. Oxygen transport, or O2DEL, is equal to cardiac output (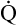 T) multiplied by CaO2 (see Chapter 8), as the following equation shows:
T) multiplied by CaO2 (see Chapter 8), as the following equation shows:
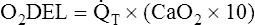
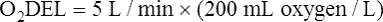
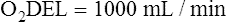
Resting oxygen consumption is about 250 mL per minute. The tissues normally use about one fourth of the oxygen delivered to them (i.e., oxygen-extraction ratio is about 0.25) (see Chapter 8).
If cardiac output decreases to abnormally low levels, blood circulates more slowly through tissue capillary beds. However, tissue cells continue to extract oxygen from the blood at the same rate, regardless of blood flow. Slower circulation means blood has longer contact time with oxygen-consuming tissue cells. Therefore, more oxygen diffuses out of each milliliter of blood into the tissues. When blood leaves the tissue capillary beds and enters the venous system, its oxygen content is lower than normal (i.e., C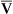 O2 is less than normal) (see Chapter 8). A low cardiac output increases C(a-
O2 is less than normal) (see Chapter 8). A low cardiac output increases C(a-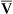 )O2 (arterial venous oxygen-content difference).
)O2 (arterial venous oxygen-content difference).
The Fick cardiac output equation (see Chapter 8) illustrates the relationships among tissue oxygen consumption (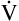 O2), C(a-
O2), C(a-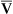 )O2, and
)O2, and 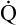 T. This is shown as follows:
T. This is shown as follows:
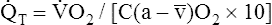
This equation shows that a reduction in C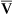 O2 implies a decreased cardiac output.
O2 implies a decreased cardiac output.
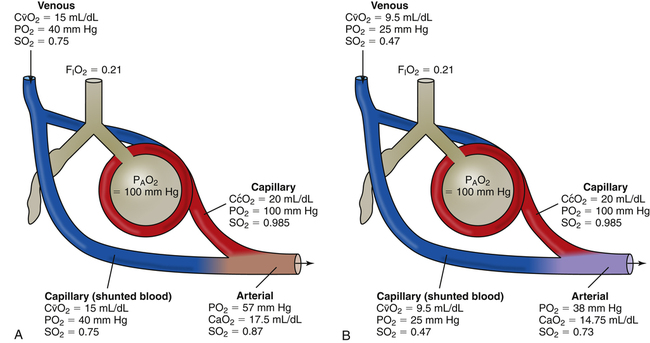
Effect of Cardiac Output on PaO2
Changes in cardiac output do not significantly affect PaO2 or CaO2 of individuals with a normal intrapulmonary shunt fraction (3% to 5%). However, as mentioned in Chapter 12, a decrease in cardiac output can significantly lower CaO2 and PaO2 in persons with increased shunt fractions because it lowers C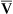 O2 and P
O2 and P O2. Likewise, an increase in cardiac output can mask the hypoxemic effects of shunt by increasing C
O2. Likewise, an increase in cardiac output can mask the hypoxemic effects of shunt by increasing C O2 and P
O2 and P O2.
O2.
The foregoing becomes clear when one considers the fact that arterial blood is a mixture of blood draining both normally oxygenated alveoli and airless alveoli (shunt units) (Figure 13-4, A and B). Shunted blood is actually unoxygenated venous blood. This blood mixes directly with the oxygenated blood that leaves ventilated alveoli. Low cardiac output reduces the mixed venous oxygen content and lowers the oxygen content of the shunted blood (see Figure 13-4, B). In this way, a reduced cardiac output decreases CaO2 and PaO2 as the more profoundly hypoxemic shunted blood mixes with arterial blood.
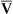
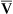
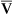
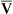
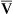
CONCEPT QUESTION 13-12
Why does an increase in cardiac output increase C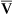 O2, with all other factors remaining constant?
O2, with all other factors remaining constant?
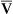
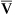
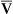
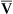
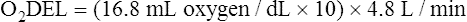
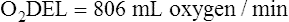
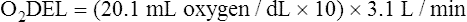
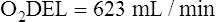
[/level-membership-for-pulmolory-and-respiratory-category][not-level-membership-for-pulmolory-and-respiratory-category]
Clinical Assessment of Acid-Base and Oxygenation Status
After reading this chapter, you will be able to:
• Differentiate between arterial blood gas classification and interpretation
• Apply a systematic arterial blood gas classification method
• Differentiate between oxygenation and ventilation defects
• Describe the basis for compensatory activity in all acid-base disturbances
• Explain how the anion gap computation can help the clinician differentiate the causes of metabolic acidosis
• Explain why acute changes in PaCO2 affect plasma [HCO3−]
• Explain how acid-base disturbances affect plasma [K+] and [Cl−]
• Explain why the standard bicarbonate and base excess measurements more accurately reflect purely metabolic acid-base disturbances than plasma [HCO3−]
• Differentiate between the traditional Henderson-Hasselbalch approach and Stewart’s strong ion approach to acid-base physiology
• Distinguish between pulmonary and cardiovascular factors that affect tissue oxygenation
• Classify the causes and severity of oxygenation defects
• Interpret various pulmonary and cardiovascular tissue oxygenation indicators
Classification Versus Interpretation
Chapter 10 discusses the concept of respiratory and metabolic acid-base disturbances and the compensatory responses they elicit. This chapter introduces a systematic method for the classification, or categorization, of arterial blood gases in terms of acid-base balance and oxygenation status. Classification is the first essential step in the development of a rational therapeutic basis for correcting acid-base and oxygenation problems. The classification process focuses attention on the general problem areas and provides a starting point for interpretation, or the in-depth exploration and comprehension of the underlying disorder.
Classification of Acid-Base Disturbances
The relevant arterial blood gas components in evaluating acid-base status are pH, PCO2, and [HCO3−]. Arterial pH reflects hydrogen ion activity of extracellular fluid (plasma), which generally correlates with intracellular fluid pH. Chapter 10 emphasizes the importance of regulating the pH from a cellular enzyme standpoint. Abnormal pH values also affect the central nervous system, causing the clinical manifestations shown in Figure 13-1. Generally, a low arterial pH (acidemia) depresses neuronal excitability, whereas a high pH (alkalemia) has the opposite effect (greatly increased excitability).1 An extremely low pH (pH <7.00) causes a coma, whereas an extremely high pH (pH >7.80) causes convulsions and tetany (a state of sustained muscle spasm). Low arterial pH values (pH <7.10) also predispose patients to ventricular arrhythmias and reduce heart muscle contractility.2
Systematic Classification
The most consistent, reliable results are achieved when an orderly, systematic, unvarying approach is used to analyze acid-base problems. If exactly the same step-by-step approach is used for each problem, confusion and premature conclusions can be avoided. A systematic approach also helps identify inconsistencies and errors in blood gas data. Box 13-1 outlines four steps in acid-base classification. The order of steps 1 through 3 is not as important as following the same sequence for each situation.
Step 3: Analyze Nonrespiratory (Metabolic) Involvement ([HCO3−])
Plasma [HCO3−] is not as specific and accurate in indicating metabolic acid-base disturbances as PaCO2 is in indicating respiratory disturbances. The reason is that plasma bicarbonate responds slightly to pure respiratory (i.e., PCO2) changes. The carbon dioxide hydration reaction explains this phenomenon. As explained in Chapter 10, an acute increase in PaCO2 of 10 mm Hg increases [HCO3−] by about 1 mEq/L. Thus, the plasma [HCO3−] must be evaluated with caution; if it falls outside of the normal range but the abnormality can be explained by the effect of the carbon dioxide hydration reaction, the [HCO3−] level is not responsible for any existing acid-base disorder.
Step 4: Assess for Compensation
It is assumed that compensation for a primary acid-base disorder is never truly complete in the sense of restoring arterial pH all the way to 7.40.3 In a compensated disorder, the pH is on the acid or alkaline side of the normal range, depending on whether the primary causative disorder created an acidosis or an alkalosis. That is, if the pH is on the acid side of normal (<7.40 but at least ≥7.35), the main cause of the original acid-base imbalance is the component (PaCO2 or HCO3−) that, by itself, would cause an acidosis. For example, if the pH is 7.36, PaCO2 is 80 mm Hg, and HCO3− is 44 mEq/L, compensation is present because the pH is in the normal range, although it is on the acid side of normal. The primary cause of the original acid-base disturbance (before compensatory activity started) must be the factor that would produce acidemia (i.e., the increased PaCO2 of 80 mm Hg). Thus, this set of blood gases would be classified as compensated (chronic) respiratory acidosis. The primary disturbance is of respiratory origin, and the increased [HCO3−] is a secondary metabolic compensatory response. The reason the pH is on the acid side of the normal range is that the body generally does not overcompensate; it does not increase the [HCO3−] so much that it converts the previously acidotic environment to one with a pH on the alkaline side of normal. Conversely, if the pH is 7.15, PaCO2 is 80 mm Hg, and HCO3− is 26 mEq/L, no compensation has occurred; that is, HCO3− is still normal, not elevated as would be expected if compensatory action were underway.
Compensatory activity does not correct the primary acid-base disturbance; the primary defect is still present. Compensatory activity merely works to restore the pH to the normal range. Table 13-1 summarizes acid-base and ventilatory classification. Table 13-2 classifies the degree of compensation for acid-base disturbances.
TABLE 13-1
Acid-Base and Ventilatory Classification
| Component | Classification | Ranges |
| pH (arterial) | Normal status | 7.35-7.45 |
| Acidemia | <7.35 | |
| Alkalemia | >7.45 | |
| PaCO2 (mm Hg) | Normal ventilatory statusRespiratory acidosis (hypoventilation)Respiratory alkalosis (hyperventilation) | 35-45>45<35 |
| [HCO3−] (mEq/L) | Normal metabolic statusMetabolic acidosisMetabolic alkalosis | 22-26<22>26 |
TABLE 13-2
Degrees of Acid-Base Compensation
| Compensating (Noncausative) Component | pH | Classification |
| Within normal range | Abnormal | Noncompensated (acute) |
| Out of normal range in the expected direction | Abnormal | Partially compensated |
| Out of normal range in the expected direction | Normal | Compensated (chronic) |
Respiratory Acidosis (Inadequate Ventilation)
< ?xml:namespace prefix = "mml" />
Therefore, hypercapnia is synonymous with respiratory acidosis.
Causes
cause respiratory acidosis. Chronic obstructive pulmonary disease (COPD) is the most frequent cause of respiratory acidosis, mostly because the airways resistance of a patient with severe COPD is so high that the patient cannot sustain the ventilatory work required to maintain a normal PaCO2.3 Central nervous system depression (drug-induced), extreme obesity (impaired diaphragmatic movement), and neuromuscular disorders (spinal cord lesions, paralytic neuromuscular diseases) are other causes of hypoventilation and respiratory acidosis. Hypercapnia may occur in different ways. A person may have an absolute decrease in ventilation because of drug-induced central nervous system depression, or a patient with COPD and a limited ventilatory reserve may sustain a normal PaCO2 at rest but may not accommodate the increased carbon dioxide production associated with increased physical activity.
Uncompensated hypercapnia implies the presence of acute ventilatory failure; the resulting respiratory acidosis is manifested by a low arterial pH, increased PaCO2, and normal or slightly high [HCO3−]. In this situation, a slightly increased [HCO3−] is not a sign that the kidneys have started compensatory activity; it merely reflects the effect of the carbon dioxide hydration reaction on [HCO3−] (see Chapter 10).
Compensation
Renal (kidney) compensation for respiratory acidosis begins as soon as PaCO2 increases. The kidney reclaims HCO3− from the renal tubular filtrate, returning it to the blood. The arterial pH is brought into the normal range because the [HCO3−]-dissolved carbon dioxide ratio is restored near its normal 20:1 range (see Chapter 10). However, this process cannot keep pace with an acutely increasing PaCO2. Full compensation may take several days.
Clinical Manifestations
Patients with neuromuscular weakness or mechanical breathing difficulties usually breathe shallowly and rapidly and are short of breath; they are often anxious and in obvious distress. Drug-induced central nervous system depression produces slow, shallow breathing and possibly apnea. Acute respiratory acidosis produces more serious physiological consequences than chronic respiratory acidosis. Rapidly increasing PaCO2 causes cerebral vasodilation and increased intracranial pressure (ICP), possibly leading to retinal venous distention and retinal hemorrhages; in addition, the patient may develop myoclonus (spasmodic muscle jerks), asterixis (a hand-flapping tremor in which the patient cannot keep the wrists flexed with the arms extended), and mental confusion.3 An abrupt onset of hypercapnia in which PaCO2 rises beyond 70 mm Hg can lead to coma, although patients with chronic hypercapnia can tolerate much higher PaCO2 values.3 Hypercapnia increases the cardiac output and dilates peripheral vessels, often resulting in a bounding pulse and warm, flushed skin.
Correction
needed to restore ventilation. However, if hypoventilation is chronic and compensation has restored arterial pH to the normal range, corrective action aimed at reducing the PaCO2 is inappropriate and possibly harmful. In this situation, a rapidly decreasing PaCO2 induces a sudden alkalosis because of renal compensation and elevated blood HCO3− levels.
Respiratory Alkalosis (Alveolar Hyperventilation)
Therefore, hypocapnia is synonymous with respiratory alkalosis.
Causes
Hyperventilation causes hypocapnia. Any process in which ventilatory elimination of carbon dioxide exceeds its production causes respiratory alkalosis. Causes can be divided into three categories: (1) hypoxia, (2) pulmonary diseases, and (3) central nervous system diseases. Probably the most common cause of hyperventilation in patients with pulmonary disease is arterial hypoxemia, mediated through the peripheral chemoreceptors.3 Hypoxia-induced respiratory alkalosis can be caused by high altitude or pulmonary diseases characterized by intrapulmonary shunting (e.g., pneumonia, pulmonary edema). Other pulmonary diseases associated with hyperventilation and respiratory alkalosis include interstitial fibrosis, pulmonary embolism, and acute asthma. J-receptor stimulation in the lung parenchyma may be involved in interstitial diseases, pneumonia, and pulmonary edema. The feeling of dyspnea from high airways resistance and resulting anxiety is a probable mechanism for respiratory alkalosis in acute asthma. General anxiety, fever, stimulating drugs, pain, and injuries of the central nervous system are other possible causes of hyperventilation.
Clinical Manifestations
cramping of the hands or feet (carpopedal spasm), and possibly tetanic convulsions (a fusion of many muscle spasms producing a sustained contraction without relaxation). The low PaCO2 level may constrict cerebral vessels enough to impair cerebral circulation, causing light-headedness, dizziness, and syncope (fainting). If respiratory alkalosis is anxiety-induced, the patient may show signs of panic and express feelings of impending doom. Vision may become impaired (tunnel vision), and speaking may become difficult.3
Compensation
The kidneys compensate for respiratory alkalosis by excreting HCO3− in the urine (bicarbonate diuresis). This activity brings the arterial pH down toward the normal range because the [HCO3−]-dissolved carbon dioxide ratio is restored near its normal 20:1 range. However, renal compensation rarely causes [HCO3−] to fall below 18 mEq/L.3 Renal compensation is a relatively slow process; it may take days until the compensation process is finally completed.
Metabolic (Nonrespiratory) Acidosis
Anion Gap
The “anion gap” can be thought of as a measure of the concentrations of other anions in the plasma besides Cl− and HCO3−. Figure 13-2, A
[/not-level-membership-for-pulmolory-and-respiratory-category]
