[level-membership-for-surgery-category]
Chapter 70A Cirrhosis and portal hypertension
Pathologic aspects
Overview
Cirrhosis is the final stage of a variety of chronic diseases involving the liver. Histopathologically, cirrhosis is characterized by remodeling of the vascular architecture with septal formation and nodular groups of regenerating hepatocytes. Biochemically, the cirrhotic liver functions less efficiently than the normal liver. Decompensated cirrhosis manifests as portal hypertension and liver failure, both of which are associated with numerous clinically important complications (see Chapter 70B, Chapter 71, Chapter 72, Chapter 73, Chapter 74, Chapter 75A, Chapter 75B, Chapter 75C, Chapter 76A, Chapter 76B, Chapter 76C, Chapter 76D, Chapter 76E, Chapter 77 ). In addition, regardless of the underlying etiology, cirrhosis is a major predisposing condition to the development of hepatocellular carcinoma (HCC) (Suriawinata & Xu, 2004). Portal hypertension secondary to cirrhosis develops as a result of the alterations of the hepatic microcirculatory system. Portal hypertension may also occur in the absence of the structural alterations of cirrhosis (noncirrhotic portal hypertension). The conditions associated with this may be idiopathic; all are subdivided into prehepatic, intrahepatic, or posthepatic. This chapter focuses on the pathology of cirrhosis and noncirrhotic portal hypertension and many of the conditions that result in portal hypertension.
Cirrhosis
The true prevalence of cirrhosis in the United States is unknown because many patients with compensated cirrhosis do not exhibit symptoms or signs of liver failure, but cirrhosis is among the top 10 causes of death in the Western world. In autopsy series, cirrhosis is documented in 5% to 10% of cases, but autopsy subjects may not be representative of the general population (Karsan et al, 2004). Currently, the only available and definitive treatment for established cirrhosis is liver transplantation.
Pathogenesis and Reversibility
The transformation from normal hepatic architecture to cirrhosis ultimately results from progressive deposition of fibrous tissue in regions of “extinct” parenchyma and vascular remodeling, regardless of the underlying etiology. Fibrogenesis is dynamic, and it is triggered by liver injury and mediated by a complex interplay of cellular necrosis, inflammation, apoptosis, and various cytokines. The cells that contribute to the development of hepatic fibrosis are the activated hepatic stellate cells and portal myofibroblasts. Once activated, these cell types are primed to respond to a wide variety of chemokines and cytokines (Friedman, 2008). In particular, transforming growth factor (TGF)-α seems to be a major fibrogenic mediator that induces these cells to produce collagens and other types of extracellular matrix and degradatory products (Canbay et al, 2004; Friedman, 2008; Pinzani & Marra, 2001; Ramadori & Saile, 2004). Recent work has shown the fibrogenic nature of leptin, the anorectic hormone produced in adipose tissue (LeClercq et al, 2002). During pathologic fibrogenesis, prevention of matrix degradation by metalloproteinases is orchestrated by the release of potent tissue metalloproteinase inhibitors; apoptosis of fibrogenic hepatic stellate cells also is inhibited. Scar formation ultimately is the consequence of the imbalance of collagen synthesis, deposition, and degradation. Another increasingly recognized pathway of fibrogenesis in the liver is that of epithelial-mesenchymal transformation (EMT), in which “injury” results in a cascade of cytokines that lead to epithelial cells developing and expressing mesenchymal markers (Rygiel et al, 2008); the exact role EMT plays in fibrogenesis is being studied.
Established cirrhosis has traditionally been considered irreversible. Accumulating clinical and experimental evidence suggests, however, that reversal or regression of liver fibrosis and cirrhosis may be possible (Fallowfield & Iredale, 2004). A study by Poynard and colleagues (2002) analyzed 3010 patients with chronic hepatitis C virus (HCV) included in four major clinical trials to receive randomized treatment regimens with interferon or pegylated interferon, with or without additional ribavirin. Reversal of cirrhosis was reported in 75 (49%) of 153 patients with baseline cirrhosis. Pol and associates (2004) examined 64 immunocompetent patients with HCV-related cirrhosis and found that cirrhosis disappeared in five patients (7.8%) on follow-up biopsies at a mean interval of 4.6 years. In addition, three of four patients undergoing dialysis showed reversal of cirrhosis secondary to HCV infection, and resolution of cirrhosis was shown in two patients upon examination of the whole liver explants at the time of transplantation.
More recently, Falize and colleagues (2006) analyzed 36 patients with hereditary hemochromatosis and demonstrated that regression of fibrosis was seen in 69% of those with bridging fibrosis and in 35% of patients with cirrhosis after institution of venosection therapy. Regression of cirrhosis has also been reported in patients with other diseases, such as hepatitis B virus (HBV), alcoholism, autoimmune hepatitis, and primary biliary cirrhosis (Arthur, 2002; Fallowfield & Iredale, 2004; Serpaggi, et al, 2006; Wanless et al, 2000). Spontaneous resolution of fibrosis also has been described in rats treated with carbon tetrachloride (Iredale et al, 1998; Issa et al, 2004). The proposed mechanisms for breakdown and remodeling of liver fibrosis include loss of activated stellate cells via apoptosis, decreased expression of metalloproteinase inhibitors, and increased production and activity of metalloproteinases or collagenases (Arthur, 2002; Benyon & Arthur, 2001; Fallowfield & Iredale, 2004; Gieling, et al, 2008). It is unclear, however, how these molecular and cellular events are initiated and regulated.
Currently, the extent to which cirrhosis is truly reversible is the subject of debate (Arthur, 2002; Chedid, 2000; Desmet & Roskams, 2003; Geller, 2000; Ray, 2000). Many important questions remain to be answered. Even if scar tissue is resorbed, can normal hepatic architecture be completely restored? Were there sampling differences or interpretation errors on biopsies in the studies showing reversibility? It is possibile that some of the reversed cases might have undergone a conversion of micronodular to macronodular cirrhosis, as shown in animal models (Issa et al, 2004), a condition that made the diagnosis of cirrhosis more difficult on a needle core biopsy sample (Desmet & Roskams, 2003)? Regardless, this discussion may have important clinical implications. Many liver diseases progress insidiously, and patients may seek medical attention only at the advanced stage, when treatment choices are limited. If cirrhosis is truly reversible, many patients with advanced liver disease still may benefit from medical treatment for specific etiologies, particularly with the discovery of new drug therapies.
Role of Liver Biopsy
In modern medicine, investigation of patients with chronic liver disease and cirrhosis involves multiple disciplines that include pathology, radiology, chemistry, biochemistry, virology, serology, and molecular biology. Liver biopsy evaluation remains the primary diagnostic tool, despite the drawback of invasiveness (Rockey et al, 2009). Noninvasive screening methods show promise in excluding patients with advanced fibrosis; however, in a recent study evaluating one of these tests, the positive predictive value for advanced fibrosis was still quite low, and the test fared poorly at distinguishing between intermediate stages of fibrosis (Zaman et al, 2007). For cirrhotic patients, liver biopsy can serve several important purposes, such as establishing or confirming diagnoses; assessing the possible underlying causes of disease; analyzing the grade of ongoing necroinflammatory activity; detecting dysplastic lesions or an occult HCC; and providing tissue for chemical, biochemical, molecular, or ultrastructural studies (Brunt, 2000a).
Two broad types of biopsy are used to sample liver tissue: needle biopsy and wedge biopsy. Needle biopsy has proved to be the most useful technique to obtain representative liver tissue for analysis. This procedure can be done percutaneously, transjugularly, or during open surgery or laparoscopy; a cutting needle or the Menghini aspiration needle may be used, although the former generates a better biopsy specimen. If cirrhosis is suspected clinically, a cutting needle is the preferred method of biopsy because an aspiration needle often provides a fragmented specimen that makes histologic evaluation difficult. The size of the needle biopsy specimen is important in avoiding sampling error. Traditionally, it has been recommended that an adequate biopsy specimen should be no smaller than 20 gauge and at least 1.5 cm in length, or it should contain at least five portal tracts (Afdhal & Nunes, 2004; Rockey et al, 2009). It has been suggested more recently, however, that for accurate and reliable grading and staging of chronic viral hepatitis, a biopsy specimen of 2 cm in length or longer that contains at least 11 complete portal tracts is needed (Guido & Rugge, 2004).
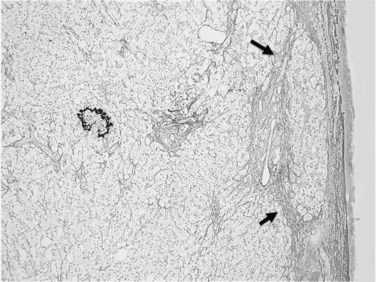
Usually performed during open surgery or laparoscopy, wedge biopsy is discouraged for evaluation of diffuse parenchymal liver diseases because this technique samples primarily the subcapsular liver parenchyma, which may contain misleading fibrous septa extending from the capsule that could be easily confused with cirrhosis or bridging fibrosis (Fig. 70A.1). The same issue can also occur in a subcapsular needle biopsy. In addition, nonspecific necroinflammatory change can occur quickly in the subcapsular region during surgical procedures, which may further confuse histology. A wedge biopsy is more suitable for focal lesions present on or immediately below the capsule. Even during open surgery, a needle biopsy to sample deep liver parenchyma is preferred (Guido & Rugge, 2004).
Morphology
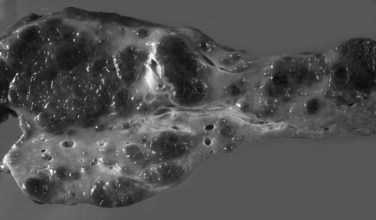
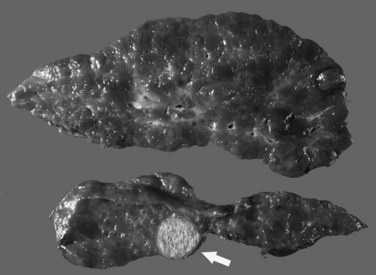
Grossly, the liver with established cirrhosis exhibits a nodular appearance that diffusely involves the entire liver (Fig. 70A.2). Microscopically, the liver parenchyma is divided by variable-sized fibrovascular septa, which contain profiles of ductular structures and diverse inflammatory cell types, into nodules that typically no longer retain a terminal hepatic venule or identifiable portal tracts. The normal portal-central relationship is completely effaced; the exception to this is biliary cirrhosis, in which the terminal hepatic venule may retain its central location. The hepatocytes within the nodules may appear morphologically normal, or they may show evidence of regeneration. The latter may be characterized by thickened cell plates with up to two cells across; anisonucleosis; large cell change with maintenance of nuclear/cytoplasmic (N/C) ratio; or small, crowded cells with markedly increased N/C ratios. The reticulin stain is useful to show cord thickening. The increased ductular profiles (ductular proliferation) are referred to as a ductular reaction (Roskams et al, 2004). Some cases may retain lymphoid aggregates in the septa, and some may have interface hepatitis.
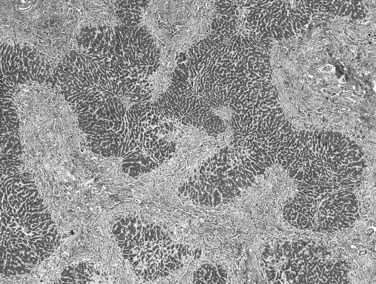
Biliary cirrhosis is caused by disorders such as primary biliary cirrhosis, primary or secondary sclerosing cholangitis, and biliary atresia. This type of cirrhosis exhibits unique morphologic features characterized by a highly irregular “jigsaw puzzle” or “geographic” nodular pattern (Fig. 70A.3). As noted, the terminal hepatic venule may be retained, and loss or effacement of native bile ducts by either lymphoid aggregates or scar tissue may be evident. Ductular reaction (proliferation) may be more pronounced than that seen in other types of cirrhosis. A characteristic clue to biliary cirrhosis is the constellation of findings in periseptal hepatocytes referred to as cholate stasis. These findings include periseptal hepatocyte ballooning, Mallory-Denk bodies (Mallory’s hyaline), and copper deposition. In addition, foamy cell aggregates may be seen in the nodules. Some investigators have attributed large cell change to chronic cholestasis. Another subtle histologic clue to biliary cirrhosis is the presence of nodular regenerative hyperplasia in the regenerative nodules.
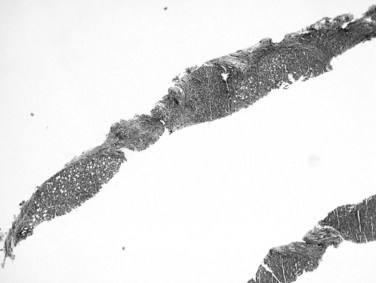
Recognition of cirrhosis is usually straightforward when an adequate biopsy specimen is examined, even on hematoxylin and eosin–stained sections. It may be helpful to use Masson trichrome stain to highlight the dense perisinusoidal fibrosis of alcoholic hepatitis and cirrhosis, particularly for fragmented needle biopsy specimens (Fig. 70A.4). Fragmentation of the specimen noted at the time of biopsy should raise the suspicion of cirrhosis. On histologic sections, a thin layer of collagen, better appreciated on trichrome and reticulin stains, tends to adhere to the surface of detached nodules. Cirrhosis also may be difficult to diagnose on needle biopsy specimens, when a macronodule is sampled, or when cirrhosis is incomplete (incomplete septal cirrhosis). In the last case, the complete spectrum of morphologic features of cirrhosis are not exhibited but vascular relationships are markedly altered, and ectatic, eccentrically located portal veins may be noted. In addition, the septa are thin, and some may be seen to extend into the parenchyma and end blindly. This type of “cirrhosis” is not uncommon in HBV-related liver disease.
Cancer Risk
The most common cancer that arises in cirrhotic livers is HCC (Fig. 70A.5; see Chapter 80), although cholangiocarcinoma is being increasingly recognized in cirrhosis (see Chapter 50A; Shaib, et al, 2007). Although these cancers may occur in cirrhosis of any etiology, particular risks include HBV and HCV infection, alcoholic liver disease (ALD), and hemochromatosis. Grossly, HCC may be multifocal, or it may grow as a well-circumscribed mass; it may show evidence of necrosis, or it may possess a different color and texture from background cirrhotic nodules. It also may bulge from the cut surface on sectioning. Most cases of HCC arising in cirrhotic livers are found in nodules larger than 1 cm (Bruix et al, 2001). Studies have shown, however, that nodules smaller than 1 cm can be malignant in the setting of cirrhosis (Caturelli et al, 2004). Premalignant lesions, or dysplastic nodules, may be indistinguishable from HCC by gross examination.
Cholangiocarcinoma (CCa) in cirrhosis may or may not be distinguishable grossly from HCC. The diagnosis rests with the microscopic appearance of atypical glandular structures, commonly in desmoplastic stroma. CCa also develops in longstanding primary sclerosing cholangitis and is seen more often than HCC in noncirrhotic livers (see Chapter 50A). The tumor may infiltrate the liver diffusely, or it may occur as a mass lesion that may be intrahepatic or extrahepatic. The cirrhotic liver is considered to be resistant to metastases from extrahepatic tumors.
Not all large nodules detected in cirrhotic livers are HCC. A macroregenerative nodule or large regenerative nodule usually measures 0.5 to 1.5 cm and is rarely 5 cm or more in diameter (International Working Party, 1995). It is seen more commonly in macronodular cirrhosis and may be distinct from surrounding cirrhotic nodules on gross examination. Histologically, a macroregenerative nodule may contain portal structures or short fibrovascular septa. The hepatocytes within the nodule are similar to the hepatocytes in smaller cirrhotic nodules but almost always exhibit hyperplastic change, evidenced by thickened plates. The clonal nature of the macroregenerative nodule has been shown, and its malignant potential is an area of ongoing discussion (Bailey & Brunt, 2002).
A dysplastic nodule is a premalignant lesion that usually measures more than 0.5 cm (International Working Party, 1995). Evolution to HCC within months or a few years has been well documented (Hytiroglou, 2004). A high-grade dysplastic nodule exhibits more clear-cut architectural or cytologic atypia, such as bulging or maplike clonal growth; pseudoglandular formation; unpaired arteries; and small cell change characterized by increased cell density, high nuclear/cytoplasmic cell ratio, and nuclear hyperchromasia. These morphologic changes are insufficient, however, for the diagnosis of HCC because a dysplastic nodule does not invade the surrounding stroma or blood vessels and it maintains cell plates no more than three cells wide (Roncalli, 2004).
Distinguishing a high-grade dysplastic nodule from well-differentiated HCC can be extremely difficult or impossible from a needle biopsy specimen, although it may be less ambiguous when an explant is examined (Kojiro, 2004). Identification of ductular reaction at the periphery aids in positive identification of cirrhosis or dysplastic nodule (Park et al, 2007; Lennerz et al, 2009). Clinical management of patients with cirrhosis and dysplasia in biopsy specimens is challenging.
Assessment of Underlying Etiology
Cirrhosis is best classified by its underlying etiology (Box 70A.1), which can be determined by clinical history and laboratory investigation in many but not all cases. Morphologic examination may help establish the diagnosis or guide the clinical investigation. In the following discussion, the morphologic features characteristic of many chronic liver diseases that commonly cause cirrhosis are illustrated. At end-stage liver disease, however, the histopathologic findings may be nonspecific and indiscriminate even to experienced hepatopathologists. Many cases of “cryptogenic” cirrhosis represent burned-out processes, for which no identifying clinical or morphologic features remain. These cases are frequently due to ALD, autoimmune hepatitis, and nonalcoholic fatty liver disease (NAFLD). The contribution of as yet unnamed viral agents is not known but is recognized.
Alcoholic Liver Disease
Excessive alcohol consumption is the leading cause of liver disease in the Western world, which encompasses a clinicopathologic spectrum that includes fatty liver, alcoholic hepatitis, and alcoholic cirrhosis (Haber et al, 2003; Menon et al, 2001; O’Connor & Schottenfeld, 1998; Tome & Lucey, 2004). Alcoholic cirrhosis, or Laënnec cirrhosis, is classically micronodular and may retain some of the features of alcoholic hepatitis at the initial stage. The liver may appear pale or yellow, enlarged, and greasy on gross examination. Histologically, lesions predominate in the perivenular region (zone 3) of the acinus and include various combinations of steatosis (fatty change), ballooning, Mallory-Denk bodies, and satellitosis. Steatosis may be predominantly macrovesicular, defined by the presence of large fat droplets in the cytoplasm of hepatocytes displacing the nuclei. Microvesicular steatosis also can be seen, characterized by fine fat droplets surrounding the central nuclei. Alcoholic hepatitis is characterized by an inflammatory infiltrate rich in neutrophils, most frequently distributed in the lobules adjacent to hepatocytes containing Mallory-Denk bodies, and dense, perisinusoidal fibrosis (Fig. 70A.6). Lymphocytes and histiocytes also may be present, sometimes in the form of lipogranulomas, and megamitochondria also may be evident. In the portal and periportal regions, ductular reaction with numerous neutrophils may occur. Cholangiolitis and canalicular bile plugs are worrisome lesions for concomitant pancreatitis.
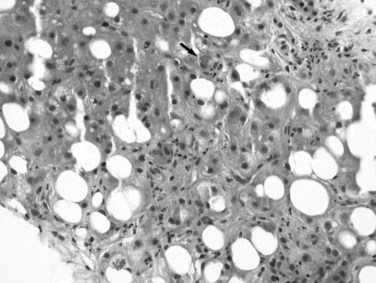
The patterns of fibrosis in alcoholic hepatitis are characteristic. Fibrosis usually involves the terminal hepatic venules, leading to the thickening of the wall; luminal occlusion may be seen with necrosis of adjacent hepatocytes and Mallory-Denk bodies within remaining hepatocytes, a lesion referred to as central hyaline necrosis. Subendothelial fibrosis is a venoocclusive lesion of alcoholism that may be noted even in end-stage cirrhotic livers. Fibrosis may extend into the lobules in perisinusoidal spaces as delicate or dense strands, giving rise to a distinctive “chicken wire” pericellular (perisinusoidal) distribution (Brunt, 2002). Trichrome stain is particularly useful in detecting this unique form of fibrosis. With time, the liver may be replaced by micronodular cirrhosis; often, the septa of ALD are quite broad as manifestations of the microvascular obliteration.
Nonalcoholic Fatty Liver Disease
NAFLD is increasingly recognized as a significant form of potentially progressive liver disease. Prevalence studies estimate that approximately 20% to 25% of the U.S. population is affected by fatty liver; of these individuals, 15% to 20% are at risk for progression to cirrhosis (Angulo, 2002). The lesions noted in liver tissue may resemble many of the features of alcohol-induced liver damage in individuals who are not heavy drinkers (Brunt, 2004; Salt, 2004; te Sligte et al, 2004; Zafrani, 2004). The disease is etiologically attributed primarily to insulin resistance and is considered the hepatic manifestation of the metabolic syndrome, a constellation of obesity, hypertension, diabetes, and hyperlipidemia (Marchesini et al, 2001). Studies have also correlated the lesions of nonalcoholic steatohepatitis (NASH) with features of metabolic syndrome.
Most, but not all, of the histopathologic features described for alcoholic hepatitis can be found in NASH. These mainly include steatosis, which is predominantly macrovesicular and commonly of zone 3 (centrilobular) distribution; hepatocyte ballooning; lobular inflammation, which is usually mild and includes neutrophils; small lipogranulomas; and zone 3 perisinusoidal fibrosis (Fig. 70A.7). Hepatocytes also may exhibit prominent glycogenated nuclei in NASH, particularly when associated with diabetes. Portal inflammation may be present in some cases but is usually mild and less intense than lobular inflammation. The presence of heavy portal inflammation, particularly when accompanied by lymphoid aggregates or abundant plasma cells, should raise the suspicion of overlapping disease, such as chronic viral hepatitis or autoimmune hepatitis (Brunt et al, 2003). However, extensive portal chronic inflammation in the absence of an overlapping disease has been shown to correlate with progressive NASH and more severe metabolic features (Brunt et al, 2009). In addition, Mallory-Denk bodies may or may not be seen in NASH; if present, they may be poorly formed. In contrast to ALD, in which Mallory-Denk bodies may be present in apoptotic hepatocytes, in NASH they are restricted to ballooned hepatocytes. Recently, an immunohistochemical stain to detect loss of K8/18 in ballooning, as well as presence of clumped K8/18 in Mallory-Denk bodies, has been introduced (Lackner et al, 2008). If numerous Mallory-Denk bodies are present in the background of steatohepatitis, an alcoholic origin is more likely (Brunt, 2002, 2004). Likewise, marked cholestasis is highly unlikely in NASH but can occur in ALD. The broad septa of ALD are rare in NAFLD as well.
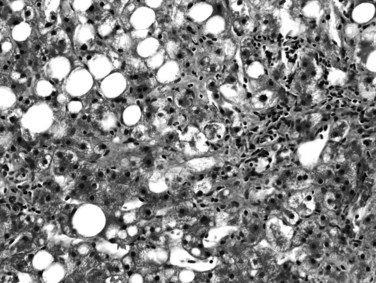
Fibrosis in NASH begins in the pericentral perisinusoidal spaces; with time, portal and periportal fibrosis develop, and eventually bridging fibrosis and cirrhosis may occur. This typically does not obliterate the terminal hepatic venules, a feature dissimilar to alcoholic hepatitis. A grading and staging system has been proposed for NASH primarily based on the extent and severity of the constellation of steatosis, ballooning change, the grade of lobular and portal inflammation, and the stage of fibrosis and remodeling (Brunt, 2004; Brunt et al, 1999). Similar to the widespread use of grading and staging in other forms of chronic liver disease, this proposal was made in recognition of the unique features of steatohepatitis to facilitate further reproducible evaluation for clinical and laboratory investigation. A revision of this system has been introduced as a method of semiquantification for use in clinical trials (Kleiner et al, 2005).
Chronic Hepatitis C
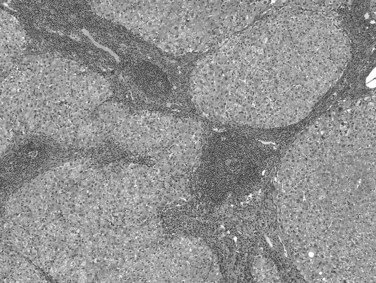
A high proportion of patients infected with HCV develop cirrhosis (Poynard et al, 2003), especially patients with a long duration of infection, concurrent alcohol consumption, coinfection with HBV or human immunodeficiency virus, nonresponse to antiviral therapy, and those of male gender (de Torres & Poynard, 2003; McCaughan & George, 2004). HCV-related cirrhosis is virtually always macronodular or mixed macronodular and micronodular. The fibrovascular septa vary in width and usually are infiltrated by mononuclear cells, predominantly lymphocytes, but also include plasma cells and eosinophils. Lymphoid aggregates, often with well-formed germinal centers, are characteristic although not pathognomonic (Fig. 70A.8). A mild degree of bile duct damage (the Poulsen lesion) also may be seen in some cases, and interface hepatitis may also be present. The lobular inflammation is typically spotty and mild, with or without acidophilic bodies. Subendothelial inflammation in portal veins, identical to endotheliitis, also may occur in HCV infection, but no reliable antibody for immunohistochemical detection of HCV proteins has been found.
Steatosis is a common histologic finding in chronic HCV and may be a manifestation of host phenotype (increased body weight, presence of the metabolic syndrome) or viral cytopathic effect, as in genotypes 3 and 1. Although a small subset of the patients may have concurrent steatohepatitis of either nonalcoholic or alcoholic origin (Brunt et al, 2003; Ramalho, 2003), viral proteins have been shown to promote fat accumulation in hepatocytes directly (Lai, 2002; Lonardo et al, 2004; Ramalho, 2003). Accumulating evidence indicates that steatosis contributes to fibrosis progression, particularly in patients infected with genotype 3 HCV (Brunt & Tiniakos, 2002; Ramalho, 2003; Rubbia-Brandt et al, 2004a).
Liver biopsy plays a central role in assessing the grade of necroinflammation and the portal-based progression of fibrosis and in evaluating the effect of antiviral therapy. Numerous grading and staging systems for chronic hepatitis have been developed; these systems share the assessment of portal and lobular inflammation and necrosis for grade and portal-based fibrosis for stage (Brunt, 2000b).
Chronic Hepatitis B
Chronic hepatitis secondary to HBV infection is another common cause of cirrhosis (Fattovich, 2003; Ganem & Prince, 2004; Lai et al, 2003). Compared with hepatitis caused by HCV, HBV hepatitis may exhibit more severe portal and lobular necroinflammation, particularly when there is an acute exacerbation. Confluent or multiacinar bridging necrosis with collapse of the lobular framework may be seen. A relatively specific finding in chronic HBV hepatitis is the presence of “ground-glass” inclusions within hepatocytes, which are uniform, pale, or eosinophilic cytoplasmic alterations resulting from enriched smooth endoplasmic reticulum filled with hepatitis B surface antigen (Fig. 70A.9). However, changes resembling ground-glass inclusions have been observed in a variety of conditions (Wisell et al, 2006). A definitive diagnosis can be established by histochemical stains (orcein, Victoria blue) or with immunohistochemical detection of hepatitis B surface antigen in the cytoplasm and hepatitis B core antigen in the nucleus.
Coinfection or superinfection with hepatitis D virus (HDV) in HBV patients usually causes more severe liver damage and accelerates the development of cirrhosis (Farci, 2003). Immunohistochemical detection of HDV intranuclear antigen is helpful in establishing the diagnosis.
Autoimmune Hepatitis
The diagnosis of autoimmune hepatitis relies on a constellation of clinical, laboratory, and histopathologic findings, none of which alone is considered specific (Alvarez et al, 1999; McFarlane, 2002a, 2002b; Vergani & Mieli-Vergani, 2003). Histopathologic examination of liver tissue serves important roles in confirming the clinical concern and excluding diseases secondary to other etiologies (Carpenter & Czaja, 2002). Classic autoimmune hepatitis exhibits a dense portal, septal, and lobular mononuclear cell infiltrate, with marked periportal or periseptal interface hepatitis enriched in plasma cells. The presence of numerous plasma cells in portal inflammation and within lobular foci of necrosis and the formation of hepatitic “rosettes” are characteristic (Fig. 70A.10). In severe cases, confluent or bridging necrosis may be seen, sometimes accompanied by pseudoacinar formation. Predominantly centrilobular necrosis with relatively mild portal inflammation also has been described in autoimmune hepatitis (Misdraji et al, 2004). Fibrosis usually develops rapidly in untreated patients. At the cirrhotic stage, the liver parenchyma is divided by broad, fibrous bands into variable-sized nodules, similar to those caused by alcoholic or chronic viral hepatitis; plasma cells and rosettes may become less prominent. Burned-out autoimmune hepatitis may be a cause of “cryptogenic” cirrhosis, and morphologic features do not separate different types of autoimmune hepatitis, classified by either autoantibody profiles or immunogenetic markers (McFarlane, 2002a).
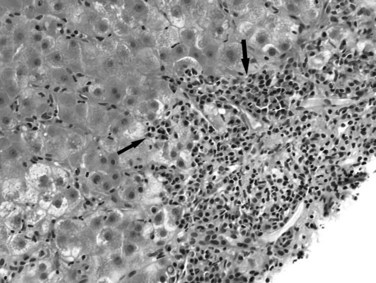
Primary Biliary Cirrhosis
Primary biliary cirrhosis (PBC) is an autoimmune disorder that leads to progressive destruction of intrahepatic bile ducts (Leuschner, 2003; Selmi et al, 2004). In the early stage, the disease is characterized by mixed chronic portal inflammation with lymphocytes, plasma cells, eosinophils, and the pathognomonic florid duct lesion, characterized by lymphocytic or granulomatous bile duct infiltration (Fig. 70A.11). Ductular reaction and interface hepatitis may be seen in more advanced cases. In the setting of positive serum antimitochondrial antibody (AMA), the florid duct lesion is essentially diagnostic. The affected duct may rupture, or the duct epithelium may exhibit degenerative change. With disease progression, ductopenia becomes evident. The native ducts may be absent or obscured by lymphoid aggregates or collections of foamy macrophages. Cholate stasis, as previously described, may be an initial clue to the diagnosis. Features of autoimmune hepatitis also may occur in AMA-positive primary biliary cirrhosis, referred to as overlap syndrome (Dienes et al, 2002; Durazzo et al, 2003); however, it must be kept in mind that significant lobular and interface activity can be seen in patients with PBC alone.
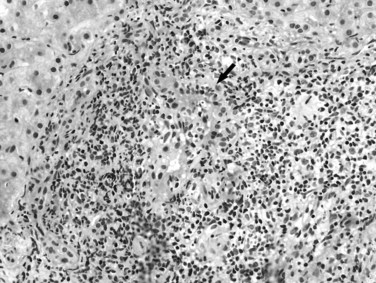
The term autoimmune cholangiopathy—also known as antinuclear antibody–positive, AMA-negative PBC—has been used to describe the disease seen in a small subset of patients who present with typical clinical and histopathologic findings of PBC but are seronegative for AMA. These patients may have other autoantibodies, such as antinuclear antibody and anti–smooth muscle antibody, as seen in autoimmune hepatitis. Accumulating evidence suggests that autoimmune cholangiopathy and PBC are likely to be a single disease with variations in the type and concentration of autoantibodies (Vierling, 2004).
Primary Sclerosing Cholangitis
Primary sclerosing cholangitis (PSC) is another autoimmune inflammatory and fibrosing process that segmentally affects the extrahepatic and intrahepatic biliary tree, leading to biliary stricture and cirrhosis (Cullen & Chapman, 2003; Mendes & Lindor, 2004; Rodriguez & Bass, 2003). It is strongly associated with inflammatory bowel disease, including both Crohn disease and ulcerative colitis (Broomé et al, 2006), and with the development of cholangiocarcinoma.
The definitive diagnosis of PSC rests on characteristic cholangiographic findings. Histopathologic examination of a liver biopsy specimen may or may not reveal diagnostic features because of the patchy nature of the disease. Mild portal lymphocytic infiltration and mild bile ductular reaction may be the only observations. The relatively specific finding of concentric periductal fibrosis with an “onionskin” appearance indicates chronic biliary obstruction of any cause and thus is not pathognomonic of PSC. The duct epithelium usually exhibits atrophic and degenerative changes, and a characteristic fibro-obliterative scar eventually replaces the bile duct (Fig. 70A.12). As in other forms of biliary cirrhosis, inhomogeneity of cirrhosis and histologic evidence of cholate stasis are recognized features in PSC. Careful evaluation of explant livers for occult cholangiocarcinoma with sampling of the entire hilar tissue is recommended.
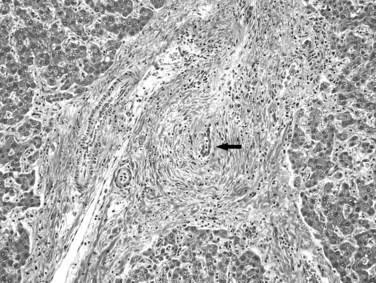
Hereditary Hemochromatosis
Hereditary hemochromatosis (HH) is an autosomal recessive iron-overload disorder primarily associated with mutation of the HFE gene (Limdi & Crampton, 2004; Pietrangelo, 2004). Iron deposition initially occurs in zone 1 (periportal) hepatocytes with a decreasing gradient toward the centrilobular area. The iron granules are concentrated along the border of the canaliculi; this is best shown by Perls’ Prussian blue stain (Fig. 70A.13). With progression, untreated HH results in iron deposition throughout the entire lobule, and iron granules are seen not only in hepatocytes but also in Kupffer cells, biliary epithelial cells, and portal macrophages. Kupffer cell clusters, or siderotic nodules, are common, and usually little to no significant portal and/or lobular inflammation is evident. This pattern of iron deposition is not restricted to HH; the diagnosis rests with genetic testing.
Secondary iron overload in the liver is a common finding in a variety of conditions, including cirrhosis of nonbiliary origins (Limdi & Crampton, 2004). Iron deposition secondary to transfusion can be heavy, but initially, it principally involves Kupffer cells in the sinusoids. Significant amounts of stainable iron may be detected in chronic HCV and HBV hepatitis, alcoholic steatohepatitis and NASH, and cirrhosis unrelated to HH or the above-mentioned chronic liver diseases. Iron deposition in these conditions is usually mild and rarely exceeds 2+ when a semiquantitative histologic score of 1 to 4 is used. Iron granules may be found in hepatocytes, Kupffer cells, and endothelial cells. If a higher score is appreciated in hepatocytes (e.g., 3+ or 4+, which is more commonly associated with HH), age-adjusted chemical quantitation of iron concentration in dry hepatic tissue, the hepatic iron index, or genetic testing for HFE mutations should be performed. An hepatic iron index greater than 2 was developed to distinguish HH from iron overload secondary to alcoholic siderosis, but this is not considered a diagnostic test for HH (Bacon, 2001). Patients with chronic viral hepatitis or steatohepatitis who also have an increased liver iron content have a higher chance of harboring HFE mutations. It has been suggested that these patients are less likely to respond to therapy and are more prone to develop fibrosis and cirrhosis (Bonkovsky et al, 2003). Other genetic markers have been developed to correlate with the more unusual cases of non-HFE iron overload (Pietrangelo, 2004).
Wilson Disease
Wilson disease is an autosomal recessive inherited metabolic disorder of copper metabolism. Any young patient with unexplained chronic or fulminant liver disease should be investigated for Wilson disease (Ferenci, 2004; Gitlin, 2003). Morphologic features in the liver vary widely at different stages of the disease and lack specificity. At the precirrhotic stage, lymphocytic portal inflammation with interface hepatitis that mimics chronic viral hepatitis or autoimmune hepatitis may be evident. Hepatocytes may show variable degrees of steatosis, necrosis, anisocytosis, and anisonucleosis. In later stages, atypical lipofuscin, canalicular cholestasis, and Kupffer cell iron accumulation may be noted. Periportal hepatocytes may contain glycogenated nuclei and Mallory-Denk bodies, and cirrhosis is usually micronodular.
Copper and copper binding protein accumulation may be shown on tissue sections by histochemical techniques, such as rhodanine, rubeanic acid, orcein, or Victoria blue stains (Fig. 70A.14) and is most likely present in cirrhosis. Copper deposition also may be seen in other chronic cholestatic liver diseases, such as PBC or PSC, usually in zone 1 or in periseptal hepatocytes. Alternatively, the absence of stainable copper in a noncirrhotic liver does not exclude the diagnosis of Wilson disease. Copper quantitation in hepatic tissue is the diagnostic test, which can be performed reliably on FFPE tissue. A value of greater than 250 µg/g dry hepatic tissue has been used as a cutoff value. Currently, genetic testing has not yet become practical for clinical diagnosis.
α1-Antitrypsin Deficiency
Liver injury in α1-antitrypsin deficiency, an autosomal recessive inherited metabolic disorder, results from retention of mutant α1-antitrypsin molecules in hepatocytes (Carrell & Lomas, 2002; Perlmutter, 2000). The histopathologic hallmark of the disease is the presence of eosinophilic globules of varying sizes in zone 1 hepatocytes in patients older than infants. These globules are best shown by PAS-d stain (Fig. 70A.15), but immunohistochemical stain may confirm the diagnosis. The globules can be seen in homozygous (PiZZ) and heterozygous (PiMZ) phenotypes, which may be determined by serum protein electrophoresis, but homozygous patients seem to be more likely to develop liver disease and cirrhosis. Other morphologic findings in the liver are relatively nonspecific, which may or may not include hepatic inflammatory infiltration. When chronic hepatitis is seen in α1-antitrypsin deficiency, other causes, such as HCV or alcohol, should be excluded (Ishak, 2002). Cirrhosis developing from α1-antitrypsin deficiency can be micronodular, macronodular, biliary, or mixed in pattern. Dysplasia and HCC may occur in cirrhotic livers, just as it may in other cases of chronic hepatitis.
Cryptogenic Cirrhosis
In approximately 10% to 15% of patients, no clinically or pathologically identifiable cause of cirrhosis is identified. However, accumulating evidence suggests that a large proportion of cryptogenic cirrhosis may represent burned-out NAFLD. This suggestion is based on reported biopsy series of diagnosed NASH followed by subsequent cryptogenic cirrhosis with complete loss of the features of active steatohepatitis. In addition, several authors have shown that many of these patients have type 2 diabetes, obesity, or both compared with patients with cirrhosis of other etiologies (Ayata et al, 2002; Caldwell et al, 1999; Ong et al, 2001; Poonawala et al, 2000; Sakugawa et al, 2003; Sanjeevi et al, 2003). Development of posttransplant NAFLD also is frequent in this group of patients (Ong et al, 2001; Sanjeevi et al, 2003), but whether this represents actual recurrence of NASH or de novo NASH is an area of discussion (Czaja, 1997).
An autoimmune etiology also has been proposed based on clinical and histopathologic findings (Ayata et al, 2002; Kaymakoglu et al, 1998), but autoantibodies may no longer be detectable in these cases (Carpenter & Czaja, 2002). PSC and alcoholic cirrhosis may present as cryptogenic cirrhosis, and one case report documented prior biopsy-proven Budd-Chiari syndrome presenting as cryptogenic cirrhosis 34 years later (Havlioglu et al, 2003). Finally, the possibility of an as yet unknown viral infection or metabolic condition cannot be excluded in cryptogenic cirrhosis. Evaluation of patients with unexplained cirrhosis should include careful review of all prior liver biopsy specimens, especially those from several years prior.
Noncirrhotic Portal Hypertension
An increase in the pressure of the portal venous system can be seen in a heterogeneous group of prehepatic, intrahepatic, and posthepatic conditions in the absence of cirrhosis (Box 70A.2). The etiopathogenetic mechanisms leading to portal hypertension in these conditions, the clinical presentation, and the prognoses vary widely (Molina & Reddy, 2001; Okuda, 2002). Portal hypertension in Budd-Chiari syndrome (see Chapter 77) is secondary to posthepatic vein obstruction and may have an acute or subacute clinical course with liver failure. Portal hypertension associated with infiltrative amyloid or hematologic disorders—such as leukemia, mastocytosis, and Gaucher disease—is believed to occur primarily at the intrahepatic sinusoidal level. Precirrhotic alcoholic hepatitis also may cause portal hypertension because of hepatocyte swelling, sinusoidal fibrosis, and central sclerosis, which may be more insidious clinically. Liver biopsy functions to confirm the absence of cirrhosis and helps establish or suggest the diagnosis (Roskams et al, 2003). The histopathologic features of several selected entities associated with noncirrhotic portal hypertension are discussed briefly.
Box 70A.2
Causes of Noncirrhotic Portal Hypertension
Modified from Geller SA, Petrovic LM, 2004: Biopsy Interpretation of the Liver. Philadelphia, Lippincott Williams & Wilkins.
Nodular Regenerative Hyperplasia
Portal hypertension has been reported in about half of patients with nodular regenerative hyperplasia (Al-Mukhaizeem et al, 2004). It is associated with a wide range of conditions, mainly including hematologic disorders, connective tissue diseases, and medications. The pathogenesis is unclear, but it may involve portal venous thrombosis leading to a microcirculatory disturbance in the liver that causes localized, nonlethal ischemia and compensatory hepatocyte hyperplasia. The liver may be normal in size or enlarged when associated with a hematologic disease. On cut surface, the liver is diffusely nodular in appearance, with nodules ranging from 0.1 to 1 cm in diameter. Microscopically, the nodular appearance is best appreciated with reticulin stain (Fig. 70A.16), which highlights regenerative hepatocytes within the nodules and atrophic hepatocytes at the edges of the nodules. Nodular regenerative hyperplasia differs from cirrhosis in that fibrosis, if present, is minimal, and the portal tract architecture is usually unaltered. These characteristic features may be shown more easily on a wedge biopsy specimen but may be difficult to appreciate on a needle biopsy specimen.
Venous Outflow Obstruction
Obstruction of hepatic venous outflow increases sinusoidal pressure and results in subsequent portal hypertension. Etiologically, it ranges from congestive heart failure, narrowing or occlusion of large hepatic veins (Budd-Chiari syndrome; see Chapter 77), to obliteration of the terminal or sublobular hepatic veins. Liver injury caused by congestive heart failure is characterized by zone 3 sinusoidal dilation and congestion and, when severe or acute, extravasation of red blood cells under the space of Disse into the hepatic cords. Hepatocellular necrosis is usually uncommon but may become evident if accompanied by systemic hypotension and hypoperfusion. The portal tracts are typically unremarkable and devoid of significant inflammatory cell infiltration. In longstanding cases, zone 3 hepatocytes exhibit atrophic change with markedly attenuated cell plates. Lipofuscin pigment and sinusoidal lining cell iron may accrue, and perivenular fibrosis and bridging fibrosis also may develop and rarely may result in septa formation, reverse lobulation, and cardiac cirrhosis.
Budd-Chiari syndrome results from obstruction at any level of the hepatic venous system between the liver and the inferior vena cava or the right atrium. It involves a variety of thrombotic and nonthrombotic causes, among which hypercoagulable states secondary to myeloproliferative disorders are the most common (Menon et al, 2004). The histopathologic features of Budd-Chiari syndrome are similar to those of congestive heart failure, but acute onset also may give rise to a hemorrhagic appearance at zone 3 with extravasation of red blood cells under the space of Disse, to replace hepatocytes within the cords, and more significant hepatocyte necrosis. If unrelieved, Budd-Chiari syndrome results in cord atrophy, replacement by fibrosis, and eventual cirrhosis.
In contrast to Budd-Chiari syndrome, venoocclusive disease rarely results in cirrhosis and has variable clinical manifestations that range from elevation of liver function tests to life-threatening ascites and liver failure. This process affects the distal sinusoids, the intrahepatic portion of the hepatic venous system, and the terminal hepatic and sublobular veins. It has been stressed that the injury in this process is to the sinusoidal lining cells and surrounding hepatocytes. DeLeve and colleagues (2002) recommended a nomenclature change to sinusoidal obstruction syndrome. It is common after bone marrow or hematopoietic stem cell transplantation, is more likely to occur in HCV-infected subjects, and is associated with chemotherapeutic agents and hepatic radiation (Coppell et al, 2003; Kumar et al, 2003; Wadleigh et al, 2003).
Oxaliplatin-based chemotherapy is increasingly recognized as a cause of marked damage to the sinusoids, with the subsequent risks of nodular regenerative hyperplasia, perisinusoidal and outflow vein fibrosis, and liver failure (Rubbia-Brandt et al, 2004b). Patients usually present within 30 days after chemotherapy; however, late-onset venoocclusive disease has been reported (Carreras et al, 2007). The lesion is characterized by subendothelial edema, zone 3 sinusoidal dilation, congestion, hemorrhage, and perivenular hepatocyte necrosis, followed by collagen deposition in the subendothelial space and sinusoids. A reticulin stain is useful in highlighting the obstruction of sinusoids and terminal hepatic veins by collagen. As the lesion progresses, the terminal hepatic veins become thickened and narrowed and are eventually obliterated by fibrosis (Fig. 70A.17). Bridging fibrosis, cirrhosis, or nodular regenerative hyperplasia may ensue in more advanced cases.
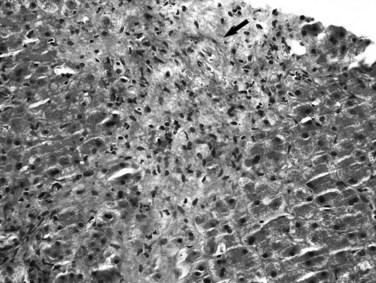
Sarcoidosis
The histopathologic hallmark of hepatic sarcoidosis is the presence of multiple granulomas composed of well-defined, rounded collections of epithelioid histiocytes rimmed by variable amounts of lymphocytes, multinucleated giant cells, plasma cells, and eosinophils (Ishak, 1998). The giant cells sometimes contain Schaumann and asteroid bodies. Small areas of central fibrinoid necrosis may be noted, but the caseating necrosis seen in tuberculosis is never present. The granulomas typically are traversed by reticulin fibers and surrounded in a concentric fashion by collagen. They are noted scattered throughout the liver but tend to be more frequent in the portal and periportal regions (Fig. 70A.18). They may coalesce to become confluent, masslike lesions. Portal hypertension may develop in some patients as a result of extensive fibrosis or cirrhosis, which may or may not be related directly to fibrous conversion and hyalinization of the granulomas. Portal hypertension also occurs in the absence of marked fibrosis, possibly as a result of compression of portal vein branches and sinusoids by granulomatous inflammation (Blich & Edoute, 2004; Valla & Benhamou, 2000). Nodular regenerative hyperplasia and Budd-Chiari syndrome also have been proposed as potential mechanisms.
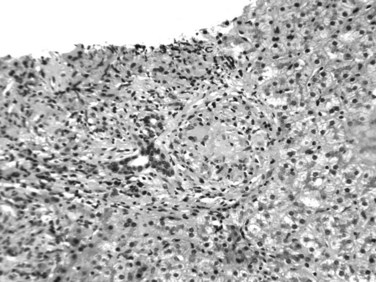
FIGURE 70A.18 Noncaseating, nonnecrotizing epithelioid granulomas in a patient with hepatic sarcoidosis.
Not all granulomas in the liver are due to sarcoid. Noncaseating granulomas can be identified in the liver in a wide variety of disorders, and the pathologist may not always be able to differentiate them (Gaya et al, 2003; Valla & Benhamou, 2000). Common examples include PBC, HCV hepatitis, autoimmune hepatitis, mycobacterial or fungal infections, schistosomiasis, drug reaction, Hodgkin lymphoma, and idiopathic granulomatous hepatitis. A diagnosis of hepatic sarcoidosis requires correlation with clinical, radiologic, and laboratory findings and cannot be rendered solely based on histopathologic examination.
Schistosomiasis
In endemic areas, infestation by Schistosoma japonicum or S. mansoni is a frequent cause of portal hypertension (Bica et al, 2000; Okuda, 2002). The mechanism involves ova deposition in the portal venules, which incites a granulomatous inflammatory response and extensive, so-called clay pipe stem fibrosis, leading to hemodynamic disturbance. Definitive diagnosis can be made by showing the presence of schistosomal ova.
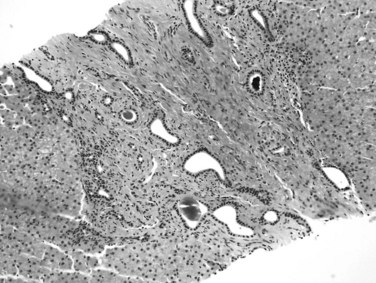
Congenital Hepatic Fibrosis
Congenital hepatic fibrosis is a developmental disorder of the ductal plate predominantly seen in children and only rarely seen in adults. It is inherited in an autosomal recessive or, less commonly, autosomal dominant fashion and may be part of the spectrum of polycystic kidney and liver disease (Kamath & Piccoli, 2003). The affected patient may initially come to medical attention with portal hypertension, and the liver is usually enlarged and of firm consistency. Microscopically, the portal tracts are expanded by fibrous tissue and may show portal-to-portal bridging fibrous bands that do not have the characteristic features of septa described above. An increased number of aberrant duct profiles are distributed at the periphery of the portal tracts, which are believed to represent remnants of incompletely remodeled ductal plates (Fig. 70A.19). Inspissated bile may be noted in the lumina, and the portal vein branches may be hypoplastic or decreased in number, but the branches of hepatic artery may be hypertrophic and abnormally numerous (Desmet, 1992), suggesting arteriovenous anastomosis.
Drugs and Toxins
Chronic liver injury attributable to drugs or toxins may cause extensive fibrosis and cirrhosis, leading to portal hypertension. Examples include methotrexate toxicity, long-term exposure to arsenic or vinyl chloride, and chronic hypervitaminosis A. Drugs and toxins also induce noncirrhotic portal hypertension via different mechanisms, such as venoocclusive disease, Budd-Chiari syndrome, and nodular regenerative hyperplasia. Herbal medicines have been increasingly recognized as hepatotoxic agents in recent years (Stedman, 2002). A classic example found in a variety of herbal medicines is pyrrolizidine alkaloids, which can cause venoocclusive disease and portal hypertension.
Idiopathic Portal Hypertension
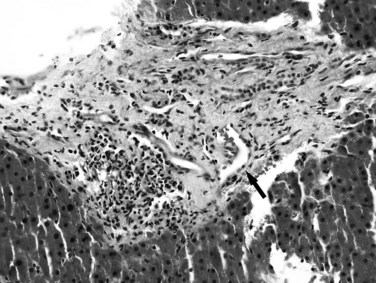
As the name implies, idiopathic portal hypertension is a disorder of unknown etiology, characterized by splenomegaly, anemia, and longstanding portal hypertension in the absence of cirrhosis (Nakanuma et al, 2001; Okudaira et al, 2002). It is primarily a disorder of middle-aged adults and generally has a better prognosis than cirrhotic portal hypertension because liver function is usually preserved. It is also called hepatoportal sclerosis and noncirrhotic portal fibrosis in the literature (Dhiman et al, 2002). Subtle differences may exist among these terms, however, which may reflect different underlying etiologies (Sarin & Kapoor, 2002).
Although the etiology is obscure, idiopathic portal hypertension is frequently associated with autoimmune disorders; immunologic disturbance is thought to be involved in pathogenesis. Bacterial infection leading to repeated stimulation also has been proposed as a candidate mechanism, but this remains speculative. In addition, some authors (Hillaire et al, 2002; Nakanuma et al, 2001; Sarin & Kapoor, 2002) suggest that prothrombotic disorders and thromboembolism play a role in etiopathogenesis, but this hypothesis has not been accepted by others (Okudaira et al, 2002).
Because idiopathic portal hypertension is essentially a diagnosis of exclusion, morphologic examination of liver tissue is imperative to rule out the presence of cirrhosis or other known etiology. The pathologic changes in idiopathic portal hypertension are believed to represent the effects of longstanding portal venous insufficiency, which may or may not be related to the initiating factors (Nakanuma et al, 2001). They are subtle, heterogenous, and nonpathognomonic and may be missed on a needle biopsy specimen. Macroscopically, the liver may have a reduced weight, and the surface may be irregularly undulant or finely wrinkled, owing to subcapsular parenchymal atrophy (Krasinskas et al, 2005). The cut surface may show portal and perivascular fibrosis, dilation and wall thickening of the veins, and unusual distribution and approximation of the portal and outflow vascular structures. Microscopically, the normal relationship between the portal and central areas is distorted. The portal tracts are either abnormally approximated to each other or widely separated. The terminal hepatic vein may be eccentrically located in the lobule adjacent to a portal tract, and sometimes multiple ectatic tributaries (angiomatous lesions) are seen in a single lobule. Conspicuous fibrosis usually is present in the portal tracts, which may extend into the periportal areas and the lobules in a pericellular fashion. The portal veins may show loss of the normal muscle coat or marked wall thickening, and luminal narrowing or obliteration may also be evident (Fig. 70A.20).
Afdhal NH, Nunes D. Evaluation of liver fibrosis: a concise review. Am J Gastroenterol. 2004;99:1160-1174.
Al-Mukhaizeem KA, et al. Nodular regenerative hyperplasia of the liver: an under-recognized cause of portal hypertension in hematological disorders. Am J Hematol. 2004;75:225-230.
Alvarez F, et al. International Autoimmune Hepatitis Group report: review of criteria for diagnosis of autoimmune hepatitis. J Hepatol. 1999;31:929-938.
Angulo P. Nonalcoholic fatty liver disease. N Engl J Med. 2002;346:1221-1231.
Arthur MJ. Reversibility of liver fibrosis and cirrhosis following treatment for hepatitis C. Gastroenterology. 2002;122:1525-1528.
Ayata G, et al. Cryptogenic cirrhosis: clinicopathologic findings at and after liver transplantation. Hum Pathol. 2002;33:1098-1104.
Bacon BR. Hemochromatosis: diagnosis and management. Gastroenterology. 2001;120:718-725.
Bailey MA, Brunt EM. Hepatocellular carcinoma: predisposing conditions and precursor lesions. Gastroenterol Clin North Am. 2002;31:641-662.
Benyon RC, Arthur MJ. Extracellular matrix degradation and the role of hepatic stellate cells. Semin Liver Dis. 2001;21:373-384.
Bica I, et al. Hepatic schistosomiasis. Infect Dis Clin North Am. 2000;14:583-604.
Blich M, Edoute Y. Clinical manifestations of sarcoid liver disease. J Gastroenterol Hepatol. 2004;19:732-737.
Bonkovsky HL, et al. Iron as a co-morbid factor in nonhemochromatotic liver disease. Alcohol. 2003;30:137-144.
Broomé U, Bergquist A. Primary sclerosing cholangitis, inflammatory bowel disease, and colon cancer. Semin Liver Dis. 2006;26:31-41.
Bruix J, et al. Clinical management of hepatocellular carcinoma: conclusions of the Barcelona-2000 EASL conference. J Hepatol. 2001;35:421-430.
Brunt EM. Liver biopsy interpretation for the gastroenterologist. Curr Gastroenterol Rep. 2000;2:27-32.
Brunt EM. Grading and staging the histopathological lesions of chronic hepatitis: the Knodell histology activity index and beyond. Hepatology. 2000;31:241-246.
Brunt EM. Alcoholic and nonalcoholic steatohepatitis. Clin Liver Dis. 2002;6:399-420.
Brunt EM. Nonalcoholic steatohepatitis. Semin Liver Dis. 2004;24:3-20.
Brunt EM, Tiniakos DG. Steatosis, steatohepatitis: review of effects on chronic hepatitis C. Curr Hepatitis Rep. 2002;1:38-44.
Brunt EM, et al. Nonalcoholic steatohepatitis: a proposal for grading and staging the histological lesions. Am J Gastroenterol. 1999;94:2467-2474.
Brunt EM, et al. Concurrence of histologic features of steatohepatitis with other forms of chronic liver disease. Mod Pathol. 2003;16:49-56.
Brunt EM, et al. Portal chronic inflammation in nonalcoholic fatty liver disease (NAFLD): a histologic marker of advanced NAFLD—clinicopathologic correlations from the Nonalcoholic Steatohepatitis Clinical Research Network. Hepatology. 2009;49:809-820.
Caldwell SH, et al. Cryptogenic cirrhosis: clinical characterization and risk factors for underlying disease. Hepatology. 1999;29:664-669.
Canbay A, et al. Apoptosis: the nexus of liver injury and fibrosis. Hepatology. 2004;39:273-278.
Carpenter HA, Czaja AJ. The role of histologic evaluation in the diagnosis and management of autoimmune hepatitis and its variants. Clin Liver Dis. 2002;6:397-417.
Carrell RW, Lomas DA. Alpha1-antitrypsin deficiency: a model for conformational diseases. N Engl J Med. 2002;346:45-53.
Carreras E, et al. Veno-occlusive disease of the liver after high-dose cytoreductive therapy with busulfan and melphalan for autologous blood stem cell transplantation in multiple myeloma patients. Biol Blood Marrow Transplant. 2007;13:1448-1454.
Caturelli E, et al. Ultrasound-guided fine needle biopsy of early hepatocellular carcinoma complicating liver cirrhosis: a multicentre study. Gut. 2004;53:1356-1362.
Chedid A. Regression of human cirrhosis. Arch Pathol Lab Med. 2000;124:1591.
Coppell JA, et al. Veno-occlusive disease: cytokines, genetics, and haemostasis. Blood Rev. 2003;17:63-70.
Cullen S, Chapman R. Primary sclerosing cholangitis. Autoimmun Rev. 2003;2:305-312.
Czaja AJ. Recurrence of nonalcoholic steatohepatitis after liver transplantation. Liver Transpl Surg. 1997;3:185-186.
DeLeve LD, et al. Toxic injury to hepatic sinusoids: sinusoidal obstruction syndrome (veno-occlusive disease). Semin Liver Dis. 2002;22:27-42.
Desmet VJ. What is congenital hepatic fibrosis? Histopathology. 1992;20:465-477.
Desmet VJ, Roskams T. Reversal of cirrhosis: evidence-based medicine? Gastroenterology. 2003;125:629-630.
de Torres M, Poynard T. Risk factors for liver fibrosis progression in patients with chronic hepatitis C. Ann Hepatol. 2003;2:5-11.
Dhiman RK, et al. Non-cirrhotic portal fibrosis (idiopathic portal hypertension): experience with 151 patients and a review of the literature. J Gastroenterol Hepatol. 2002;17:6-16.
Dienes HP, et al. Autoimmune hepatitis and overlap syndromes. Clin Liver Dis. 2002;6:349-362.
Durazzo M, et al. Overlap syndromes of autoimmune hepatitis: what is known so far. Dig Dis Sci. 2003;48:423-430.
Falize, et al. Reversibility of hepatic fibrosis in treated genetic hemachromatosis: a study of 36 cases. Hepatology. 2006;44:472-477.
Fallowfield JA, Iredale JP. Reversal of liver fibrosis and cirrhosis: an emerging reality. Scott Med J. 2004;49:3-6.
Farci P. Delta hepatitis: an update. J Hepatol. 2003;39:S212-S219.
Fattovich G. Natural history and prognosis of hepatitis B. Semin Liver Dis. 2003;23:47-58.
Ferenci P. Diagnosis and current therapy of Wilson’s disease. Aliment Pharmacol Ther. 2004;19:157-165.
Friedman SL. Mechanisms of hepatic fibrogenesis. Gastroenterology. 2008;134:1655-1669.
Ganem D, Prince AM. Hepatitis B virus infection: natural history and clinical consequences. N Engl J Med. 2004;350:1118-1129.
Gaya DR, et al. Hepatic granulomas: a 10-year single centre experience. J Clin Pathol. 2003;56:850-853.
Geller SA. Coming or going? What is cirrhosis? Arch Pathol Lab Med. 2000;124:1587-1588.
Gieling RG, et al. Fibrosis and cirrhosis reversibility: molecular mechanisms. Clin Liver Dis. 2008;12:915-937.
Gitlin JD. Wilson disease. Gastroenterology. 2003;125:1868-1877.
Guido M, Rugge M. Liver biopsy sampling in chronic viral hepatitis. Semin Liver Dis. 2004;24:89-97.
Haber PS, et al. Pathogenesis and management of alcoholic hepatitis. J Gastroenterol Hepatol. 2003;18:1332-1344.
Havlioglu N, et al. Budd–Chiari syndrome and hepatocellular carcinoma: a case report and review of the literature. Am J Gastroenterol. 2003;98:201-204.
Hillaire S, et al. Idiopathic non-cirrhotic intrahepatic portal hypertension in the West: a re-evaluation in 28 patients. Gut. 2002;51:275-280.
Hytiroglou P. Morphological changes of early human hepatocarcinogenesis. Semin Liver Dis. 2004;24:65-75.
International Working Party. Terminology of nodular hepatocellular lesions. Hepatology. 1995;22:983-993.
Iredale JP, et al. Mechanisms of spontaneous resolution of rat liver fibrosis: hepatic stellate cell apoptosis and reduced hepatic expression of metalloproteinase inhibitors. J Clin Invest. 1998;102:538-549.
Ishak KG. Sarcoidosis of the liver and bile ducts. Mayo Clin Proc. 1998;73:467-472.
Ishak KG. Inherited metabolic diseases of the liver. Clin Liver Dis. 2002;6:455-479.
Issa R, et al. Spontaneous recovery from micronodular cirrhosis: evidence for incomplete resolution associated with matrix cross-linking. Gastroenterology. 2004;126:1795-1808.
Kamath BM, Piccoli DA. Heritable disorders of the bile ducts. Gastroenterol Clin North Am. 2003;32:857-875.
Karsan HA, et al. Primary prevention of cirrhosis: public health strategies that can make a difference. Postgrad Med. 2004;115:25-30.
Kaymakoglu S, et al. Is severe cryptogenic chronic hepatitis similar to autoimmune hepatitis? J Hepatol. 1998;28:78-83.
Kleiner DM, et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology. 2005;41:1313-1321.
Kojiro M. Focus on dysplastic nodules and early hepatocellular carcinoma: an Eastern point of view. Liver Transpl. 2004;10:S3-S8.
Krasinskas AM, et al. Liver transplantation for severe intrahepatic noncirrhotic portal hypertension. Liver Transpl. 2005;11:627-634.
Kumar S, et al. Hepatic veno-occlusive disease (sinusoidal obstruction syndrome) after hematopoietic stem cell transplantation. Mayo Clin Proc. 2003;78:589-598.
Lackner C, et al. Ballooned hepatocytes in steatohepatitis: the value of keratin immunohistochemistry for diagnosis. J Hepatol. 2008;48:821-828.
Lai CL, et al. Viral hepatitis B. Lancet. 2003;362:2089-2094.
Lai MM. Hepatitis C virus proteins: direct link to hepatic oxidative stress, steatosis, carcinogenesis and more. Gastroenterology. 2002;122:568-571.
LeClercq IA, et al. Leptin is essential for the hepatic fibrogenic response to chronic liver injury. J Hepatol. 2002;37:206-213.
Lennerz JKM, et al. Perinodular K19 patterns are generic to advanced chronic liver disease, a study of 176 hepatocellular nodules (HCN) in explanted livers. Mod Pathol. 2009;22:314A.
Leuschner U. Primary biliary cirrhosis: presentation and diagnosis. Clin Liver Dis. 2003;7:741-758.
Limdi JK, Crampton JR. Hereditary haemochromatosis. QJM. 2004;97:315-324.
Lonardo A, et al. Steatosis and hepatitis C virus: mechanisms and significance for hepatic and extrahepatic disease. Gastroenterology. 2004;126:586-597.
Marchesini G, et al. Nonalcoholic fatty liver disease: a feature of the metabolic syndrome. Diabetes. 2001;50:1844-1850.
McCaughan GW, George J. Fibrosis progression in chronic hepatitis C virus infection. Gut. 2004;53:318-321.
McFarlane IG. Autoimmune hepatitis: diagnostic criteria, subclassifications, and clinical features. Clin Liver Dis. 2002;6:317-333.
McFarlane IG. Definition and classification of autoimmune hepatitis. Semin Liver Dis. 2002;22:317-324.
Mendes FD, Lindor KD. Primary sclerosing cholangitis. Clin Liver Dis. 2004;8:195-211.
Menon KV, et al. Pathogenesis, diagnosis, and treatment of alcoholic liver disease. Mayo Clin Proc. 2001;76:1021-1029.
Menon KV, et al. The Budd–Chiari syndrome. N Engl J Med. 2004;350:578-585.
Misdraji J, et al. Autoimmune hepatitis with centrilobular necrosis. Am J Surg Pathol. 2004;28:471-478.
Molina E, Reddy KR. Noncirrhotic portal hypertension. Clin Liver Dis. 2001;5:769-787.
Nakanuma Y, et al. Pathology and pathogenesis of idiopathic portal hypertension with an emphasis on the liver. Pathol Res Pract. 2001;197:65-76.
O’Connor PG, Schottenfeld RS. Patients with alcohol problems. N Engl J Med. 1998;338:592-602.
Okuda K. Non-cirrhotic portal hypertension versus idiopathic portal hypertension. J Gastroenterol Hepatol. 2002;17:S204-S213.
Okudaira M, et al. Idiopathic portal hypertension and its pathology. Semin Liver Dis. 2002;22:59-72.
Ong J, et al. Cryptogenic cirrhosis and posttransplantation nonalcoholic fatty liver disease. Liver Transpl. 2001;7:797-801.
Park YN, et al. Ductular reaction is helpful in defining early stromal invasion, small hepatocellular carcinomas, and dysplastic nodules. Cancer. 2007;109:915-923.
Perlmutter DH. Liver injury in alpha 1-antitrypsin deficiency. Clin Liver Dis. 2000;4:387-408.
Pietrangelo A. Hereditary hemochromatosis: a new look at an old disease. N Engl J Med. 2004;350:2383-2397.
Pinzani M, Marra F. Cytokine receptors and signaling in hepatic stellate cells. Semin Liver Dis. 2001;21:397-416.
Pol S, et al. Reversibility of hepatitis C virus–related cirrhosis. Hum Pathol. 2004;35:107-112.
Poonawala A, et al. Prevalence of obesity and diabetes in patients with cryptogenic cirrhosis: a case-control study. Hepatology. 2000;32:689-692.
Poynard T, et al. Impact of pegylated interferon alfa-2b and ribavirin on liver fibrosis in patients with chronic hepatitis C. Gastroenterology. 2002;122:1303-1313.
Poynard T, et al. Viral hepatitis C. Lancet. 2003;362:2095-2100.
Ramadori G, Saile B. Portal tract fibrogenesis in the liver. Lab Invest. 2004;84:153-159.
Ramalho F. Hepatitis C virus infection and liver steatosis. Antiviral Res. 2003;60:125-127.
Ray MB. Regression of cirrhosis: a timely topic. Arch Pathol Lab Med. 2000;124:1589-1590.
Rockey DC, et al. Liver biopsy. Hepatology. 2009;49:1017-1044.
Rodriguez HJ, Bass NM. Primary sclerosing cholangitis. Semin Gastrointest Dis. 2003;14:189-198.
Roncalli M. Hepatocellular nodules in cirrhosis: focus on diagnostic criteria on liver biopsy: a Western experience. Liver Transpl. 2004;10:S9-S15.
Roskams TA, et al. Histopathology of portal hypertension: a practical guideline. Histopathology. 2003;42:2-13.
Roskams TA, et al. Nomenclature of the finer branches of the biliary tree: canals, ductules, and ductular reactions in human livers. Hepatology. 2004;39:1739-1745.
Rubbia-Brandt L, et al. Steatosis affects chronic hepatitis C progression in a genotype specific way. Gut. 2004;53:406-412.
Rubbia-Brandt L, et al. Severe hepatic sinusoidal obstruction associated with oxaliplatin-based chemotherapy in patients with metastatic colorectal cancer. Ann Oncol. 2004;15:460-466.
Rygiel KA, et al. Epithelial–mesenchymal transition contributes to portal tract fibrogenesis during human chronic liver disease. Lab Invest. 2008;88:112-123.
Sakugawa H, et al. Clinical characteristics of patients with cryptogenic liver cirrhosis in Okinawa, Japan. Hepatogastroenterology. 2003;50:2005-2008.
Salt WBII. Nonalcoholic fatty liver disease (NAFLD): a comprehensive review. J Insur Med. 2004;36:27-41.
Sanjeevi A, et al. Outcomes of liver transplantation for cryptogenic cirrhosis: a single-center study of 71 patients. Transplant Proc. 2003;35:2977-2980.
Sarin SK, Kapoor D. Non-cirrhotic portal fibrosis: current concepts and management. J Gastroenterol Hepatol. 2002;17:526-534.
Selmi C, et al. Epidemiology and pathogenesis of primary biliary cirrhosis. J Clin Gastroenterol. 2004;38:264-271.
Serpaggi J, et al. Direct and indirect evidence for the reversibility of cirrhosis. Hum Pathol. 2006;37:1519-1526.
Shaib YH, et al. Risk factors for intrahepatic and extrahepatic cholangiocarcinoma: a hospital-based case-control study. Am J Gastroenterol. 2007;102:1016-1021.
Stedman C. Herbal hepatotoxicity. Semin Liver Dis. 2002;22:195-206.
Suriawinata A, Xu R. An update on the molecular genetics of hepatocellular carcinoma. Semin Liver Dis. 2004;24:77-88.
te Sligte K, et al. Nonalcoholic steatohepatitis: review of a growing medical problem. Eur J Intern Med. 2004;15:10-21.
Tome S, Lucey MR. Current management of alcoholic liver disease. Aliment Pharmacol Ther. 2004;19:707-714.
Valla DC, Benhamou JP. Hepatic granulomas and hepatic sarcoidosis. Clin Liver Dis. 2000;4:269-285.
Vergani D, Mieli-Vergani G. Autoimmune hepatitis. Autoimmun Rev. 2003;2:241-247.
Vierling JM. Primary biliary cirrhosis and autoimmune cholangiopathy. Clin Liver Dis. 2004;8:177-194.
Wadleigh M, et al. Hepatic veno-occlusive disease: pathogenesis, diagnosis and treatment. Curr Opin Hematol. 2003;10:451-462.
Wanless IR, et al. Regression of human cirrhosis: morphologic features and the genesis of incomplete septal cirrhosis. Arch Pathol Lab Med. 2000;124:1599-1607.
Wisell J, et al. Glycogen pseudoground glass change in hepatocytes. Am J Surg Pathol. 2006;30:1085-1090.
Zafrani ES. Nonalcoholic fatty liver disease: an emerging pathological spectrum. Virchows Arch. 2004;444:3-12.
Zaman, et al. Assessment of FIBROSPECT II to detect hepatic fibrosis in chronic hepatitis patients. Am J Med. 2007;120:280.
[/level-membership-for-surgery-category][not-level-membership-for-surgery-category]
Chapter 70A Cirrhosis and portal hypertension
Pathologic aspects
Overview
Cirrhosis is the final stage of a variety of chronic diseases involving the liver. Histopathologically, cirrhosis is characterized by remodeling of the vascular architecture with septal formation and nodular groups of regenerating hepatocytes. Biochemically, the cirrhotic liver functions less efficiently than the normal liver. Decompensated cirrhosis manifests as portal hypertension and liver failure, both of which are associated with numerous clinically important complications (see Chapter 70B, Chapter 71, Chapter 72, Chapter 73, Chapter 74, Chapter 75A, Chapter 75B, Chapter 75C, Chapter 76A, Chapter 76B, Chapter 76C, Chapter 76D, Chapter 76E, Chapter 77 ). In addition, regardless of the underlying etiology, cirrhosis is a major predisposing condition to the development of hepatocellular carcinoma (HCC) (Suriawinata & Xu, 2004). Portal hypertension secondary to cirrhosis develops as a result of the alterations of the hepatic microcirculatory system. Portal hypertension may also occur in the absence of the structural alterations of cirrhosis (noncirrhotic portal hypertension). The conditions associated with this may be idiopathic; all are subdivided into prehepatic, intrahepatic, or posthepatic. This chapter focuses on the pathology of cirrhosis and noncirrhotic portal hypertension and many of the conditions that result in portal hypertension.
Cirrhosis
The true prevalence of cirrhosis in the United States is unknown because many patients with compensated cirrhosis do not exhibit symptoms or signs of liver failure, but cirrhosis is among the top 10 causes of death in the Western world. In autopsy series, cirrhosis is documented in 5% to 10% of cases, but autopsy subjects may not be representative of the general population (Karsan et al, 2004). Currently, the only available and definitive treatment for established cirrhosis is liver transplantation.
Pathogenesis and Reversibility
The transformation from normal hepatic architecture to cirrhosis ultimately results from progressive deposition of fibrous tissue in regions of “extinct” parenchyma and vascular remodeling, regardless of the underlying etiology. Fibrogenesis is dynamic, and it is triggered by liver injury and mediated by a complex interplay of cellular necrosis, inflammation, apoptosis, and various cytokines. The cells that contribute to the development of hepatic fibrosis are the activated hepatic stellate cells and portal myofibroblasts. Once activated, these cell types are primed to respond to a wide variety of chemokines and cytokines (Friedman, 2008). In particular, transforming growth factor (TGF)-α seems to be a major fibrogenic mediator that induces these cells to produce collagens and other types of extracellular matrix and degradatory products (Canbay et al, 2004; Friedman, 2008; Pinzani & Marra, 2001; Ramadori & Saile, 2004). Recent work has shown the fibrogenic nature of leptin, the anorectic hormone produced in adipose tissue (LeClercq et al, 2002). During pathologic fibrogenesis, prevention of matrix degradation by metalloproteinases is orchestrated by the release of potent tissue metalloproteinase inhibitors; apoptosis of fibrogenic hepatic stellate cells also is inhibited. Scar formation ultimately is the consequence of the imbalance of collagen synthesis, deposition, and degradation. Another increasingly recognized pathway of fibrogenesis in the liver is that of epithelial-mesenchymal transformation (EMT), in which “injury” results in a cascade of cytokines that lead to epithelial cells developing and expressing mesenchymal markers (Rygiel et al, 2008); the exact role EMT plays in fibrogenesis is being studied.
Established cirrhosis has traditionally been considered irreversible. Accumulating clinical and experimental evidence suggests, however, that reversal or regression of liver fibrosis and cirrhosis may be possible (Fallowfield & Iredale, 2004). A study by Poynard and colleagues (2002) analyzed 3010 patients with chronic hepatitis C virus (HCV) included in four major clinical trials to receive randomized treatment regimens with interferon or pegylated interferon, with or without additional ribavirin. Reversal of cirrhosis was reported in 75 (49%) of 153 patients with baseline cirrhosis. Pol and associates (2004) examined 64 immunocompetent patients with HCV-related cirrhosis and found that cirrhosis disappeared in five patients (7.8%) on follow-up biopsies at a mean interval of 4.6 years. In addition, three of four patients undergoing dialysis showed reversal of cirrhosis secondary to HCV infection, and resolution of cirrhosis was shown in two patients upon examination of the whole liver explants at the time of transplantation.
More recently, Falize and colleagues (2006) analyzed 36 patients with hereditary hemochromatosis and demonstrated that regression of fibrosis was seen in 69% of those with bridging fibrosis and in 35% of patients with cirrhosis after institution of venosection therapy. Regression of cirrhosis has also been reported in patients with other diseases, such as hepatitis B virus (HBV), alcoholism, autoimmune hepatitis, and primary biliary cirrhosis (Arthur, 2002; Fallowfield & Iredale, 2004; Serpaggi, et al, 2006; Wanless et al, 2000). Spontaneous resolution of fibrosis also has been described in rats treated with carbon tetrachloride (Iredale et al, 1998; Issa et al, 2004). The proposed mechanisms for breakdown and remodeling of liver fibrosis include loss of activated stellate cells via apoptosis, decreased expression of metalloproteinase inhibitors, and increased production and activity of metalloproteinases or collagenases (Arthur, 2002; Benyon & Arthur, 2001; Fallowfield & Iredale, 2004; Gieling, et al, 2008). It is unclear, however, how these molecular and cellular events are initiated and regulated.
Currently, the extent to which cirrhosis is truly reversible is the subject of debate (Arthur, 2002; Chedid, 2000; Desmet & Roskams, 2003; Geller, 2000; Ray, 2000). Many important questions remain to be answered. Even if scar tissue is resorbed, can normal hepatic architecture be completely restored? Were there sampling differences or interpretation errors on biopsies in the studies showing reversibility? It is possibile that some of the reversed cases might have undergone a conversion of micronodular to macronodular cirrhosis, as shown in animal models (Issa et al, 2004), a condition that made the diagnosis of cirrhosis more difficult on a needle core biopsy sample (Desmet & Roskams, 2003)? Regardless, this discussion may have important clinical implications. Many liver diseases progress insidiously, and patients may seek medical attention only at the advanced stage, when treatment choices are limited. If cirrhosis is truly reversible, many patients with advanced liver disease still may benefit from medical treatment for specific etiologies, particularly with the discovery of new drug therapies.
Role of Liver Biopsy
In modern medicine, investigation of patients with chronic liver disease and cirrhosis involves multiple disciplines that include pathology, radiology, chemistry, biochemistry, virology, serology, and molecular biology. Liver biopsy evaluation remains the primary diagnostic tool, despite the drawback of invasiveness (Rockey et al, 2009). Noninvasive screening methods show promise in excluding patients with advanced fibrosis; however, in a recent study evaluating one of these tests, the positive predictive value for advanced fibrosis was still quite low, and the test fared poorly at distinguishing between intermediate stages of fibrosis (Zaman et al, 2007). For cirrhotic patients, liver biopsy can serve several important purposes, such as establishing or confirming diagnoses; assessing the possible underlying causes of disease; analyzing the grade of ongoing necroinflammatory activity; detecting dysplastic lesions or an occult HCC; and providing tissue for chemical, biochemical, molecular, or ultrastructural studies (Brunt, 2000a).
Two broad types of biopsy are used to sample liver tissue: needle biopsy and wedge biopsy. Needle biopsy has proved to be the most useful technique to obtain representative liver tissue for analysis. This procedure can be done percutaneously, transjugularly, or during open surgery or laparoscopy; a cutting needle or the Menghini aspiration needle may be used, although the former generates a better biopsy specimen. If cirrhosis is suspected clinically, a cutting needle is the preferred method of biopsy because an aspiration needle often provides a fragmented specimen that makes histologic evaluation difficult. The size of the needle biopsy specimen is important in avoiding sampling error. Traditionally, it has been recommended that an adequate biopsy specimen should be no smaller than 20 gauge and at least 1.5 cm in length, or it should contain at least five portal tracts (Afdhal & Nunes, 2004; Rockey et al, 2009). It has been suggested more recently, however, that for accurate and reliable grading and staging of chronic viral hepatitis, a biopsy specimen of 2 cm in length or longer that contains at least 11 complete portal tracts is needed (Guido & Rugge, 2004).
Usually performed during open surgery or laparoscopy, wedge biopsy is discouraged for evaluation of diffuse parenchymal liver diseases because this technique samples primarily the subcapsular liver parenchyma, which may contain misleading fibrous septa extending from the capsule that could be easily confused with cirrhosis or bridging fibrosis (Fig. 70A.1). The same issue can also occur in a subcapsular needle biopsy. In addition, nonspecific necroinflammatory change can occur quickly in the subcapsular region during surgical procedures, which may further confuse histology. A wedge biopsy is more suitable for focal lesions present on or immediately below the capsule. Even during open surgery, a needle biopsy to sample deep liver parenchyma is preferred (Guido & Rugge, 2004).
Morphology
Grossly, the liver with established cirrhosis exhibits a nodular appearance that diffusely involves the entire liver (Fig. 70A.2). Microscopically, the liver parenchyma is divided by variable-sized fibrovascular septa, which contain profiles of ductular structures and diverse inflammatory cell types, into nodules that typically no longer retain a terminal hepatic venule or identifiable portal tracts. The normal portal-central relationship is completely effaced; the exception to this is biliary cirrhosis, in which the terminal hepatic venule may retain its central location. The hepatocytes within the nodules may appear morphologically normal, or they may show evidence of regeneration. The latter may be characterized by thickened cell plates with up to two cells across; anisonucleosis; large cell change with maintenance of nuclear/cytoplasmic (N/C) ratio; or small, crowded cells with markedly increased N/C ratios. The reticulin stain is useful to show cord thickening. The increased ductular profiles (ductular proliferation) are referred to as a ductular reaction (Roskams et al, 2004). Some cases may retain lymphoid aggregates in the septa, and some may have interface hepatitis.
Biliary cirrhosis is caused by disorders such as primary biliary cirrhosis, primary or secondary sclerosing cholangitis, and biliary atresia. This type of cirrhosis exhibits unique morphologic features characterized by a highly irregular “jigsaw puzzle” or “geographic” nodular pattern (Fig. 70A.3). As noted, the terminal hepatic venule may be retained, and loss or effacement of native bile ducts by either lymphoid aggregates or scar tissue may be evident. Ductular reaction (proliferation) may be more pronounced than that seen in other types of cirrhosis. A characteristic clue to biliary cirrhosis is the constellation of findings in periseptal hepatocytes referred to as cholate stasis. These findings include periseptal hepatocyte ballooning, Mallory-Denk bodies (Mallory’s hyaline), and copper deposition. In addition, foamy cell aggregates may be seen in the nodules. Some investigators have attributed large cell change to chronic cholestasis. Another subtle histologic clue to biliary cirrhosis is the presence of nodular regenerative hyperplasia in the regenerative nodules.
Recognition of cirrhosis is usually straightforward when an adequate biopsy specimen is examined, even on hematoxylin and eosin–stained sections. It may be helpful to use Masson trichrome stain to highlight the dense perisinusoidal fibrosis of alcoholic hepatitis and cirrhosis, particularly for fragmented needle biopsy specimens (Fig. 70A.4). Fragmentation of the specimen noted at the time of biopsy should raise the suspicion of cirrhosis. On histologic sections, a thin layer of collagen, better appreciated on trichrome and reticulin stains, tends to adhere to the surface of detached nodules. Cirrhosis also may be difficult to diagnose on needle biopsy specimens, when a macronodule is sampled, or when cirrhosis is incomplete (incomplete septal cirrhosis). In the last case, the complete spectrum of morphologic features of cirrhosis are not exhibited but vascular relationships are markedly altered, and ectatic, eccentrically located portal veins may be noted. In addition, the septa are thin, and some may be seen to extend into the parenchyma and end blindly. This type of “cirrhosis” is not uncommon in HBV-related liver disease.
Cancer Risk
The most common cancer that arises in cirrhotic livers is HCC (Fig. 70A.5; see Chapter 80), although cholangiocarcinoma is being increasingly recognized in cirrhosis (see Chapter 50A; Shaib, et al, 2007). Although these cancers may occur in cirrhosis of any etiology, particular risks include HBV and HCV infection, alcoholic liver disease (ALD), and hemochromatosis. Grossly, HCC may be multifocal, or it may grow as a well-circumscribed mass; it may show evidence of necrosis, or it may possess a different color and texture from background cirrhotic nodules. It also may bulge from the cut surface on sectioning. Most cases of HCC arising in cirrhotic livers are found in nodules larger than 1 cm (Bruix et al, 2001). Studies have shown, however, that nodules smaller than 1 cm can be malignant in the setting of cirrhosis (Caturelli et al, 2004). Premalignant lesions, or dysplastic nodules, may be indistinguishable from HCC by gross examination.
Cholangiocarcinoma (CCa) in cirrhosis may or may not be distinguishable grossly from HCC. The diagnosis rests with the microscopic appearance of atypical glandular structures, commonly in desmoplastic stroma. CCa also develops in longstanding primary sclerosing cholangitis and is seen more often than HCC in noncirrhotic livers (see Chapter 50A). The tumor may infiltrate the liver diffusely, or it may occur as a mass lesion that may be intrahepatic or extrahepatic. The cirrhotic liver is considered to be resistant to metastases from extrahepatic tumors.
Not all large nodules detected in cirrhotic livers are HCC. A macroregenerative nodule or large regenerative nodule usually measures 0.5 to 1.5 cm and is rarely 5 cm or more in diameter (International Working Party, 1995). It is seen more commonly in macronodular cirrhosis and may be distinct from surrounding cirrhotic nodules on gross examination. Histologically, a macroregenerative nodule may contain portal structures or short fibrovascular septa. The hepatocytes within the nodule are similar to the hepatocytes in smaller cirrhotic nodules but almost always exhibit hyperplastic change, evidenced by thickened plates. The clonal nature of the macroregenerative nodule has been shown, and its malignant potential is an area of ongoing discussion (Bailey & Brunt, 2002).
A dysplastic nodule is a premalignant lesion that usually measures more than 0.5 cm (International Working Party, 1995). Evolution to HCC within months or a few years has been well documented (Hytiroglou, 2004). A high-grade dysplastic nodule exhibits more clear-cut architectural or cytologic atypia, such as bulging or maplike clonal growth; pseudoglandular formation; unpaired arteries; and small cell change characterized by increased cell density, high nuclear/cytoplasmic cell ratio, and nuclear hyperchromasia. These morphologic changes are insufficient, however, for the diagnosis of HCC because a dysplastic nodule does not invade the surrounding stroma or blood vessels and it maintains cell plates no more than three cells wide (Roncalli, 2004).
Distinguishing a high-grade dysplastic nodule from well-differentiated HCC can be extremely difficult or impossible from a needle biopsy specimen, although it may be less ambiguous when an explant is examined (Kojiro, 2004). Identification of ductular reaction at the periphery aids in positive identification of cirrhosis or dysplastic nodule (Park et al, 2007; Lennerz et al, 2009). Clinical management of patients with cirrhosis and dysplasia in biopsy specimens is challenging.
Assessment of Underlying Etiology
Cirrhosis is best classified by its underlying etiology (Box 70A.1), which can be determined by clinical history and laboratory investigation in many but not all cases. Morphologic examination may help establish the diagnosis or guide the clinical investigation. In the following discussion, the morphologic features characteristic of many chronic liver diseases that commonly cause cirrhosis are illustrated. At end-stage liver disease, however, the histopathologic findings may be nonspecific and indiscriminate even to experienced hepatopathologists. Many cases of “cryptogenic” cirrhosis represent burned-out processes, for which no identifying clinical or morphologic features remain. These cases are frequently due to ALD, autoimmune hepatitis, and nonalcoholic fatty liver disease (NAFLD). The contribution of as yet unnamed viral agents is not known but is recognized.
Alcoholic Liver Disease
Excessive alcohol consumption is the leading cause of liver disease in the Western world, which encompasses a clinicopathologic spectrum that includes fatty liver, alcoholic hepatitis, and alcoholic cirrhosis (Haber et al, 2003; Menon et al, 2001; O’Connor & Schottenfeld, 1998; Tome & Lucey, 2004). Alcoholic cirrhosis, or Laënnec cirrhosis, is classically micronodular and may retain some of the features of alcoholic hepatitis at the initial stage. The liver may appear pale or yellow, enlarged, and greasy on gross examination. Histologically, lesions predominate in the perivenular region (zone 3) of the acinus and include various combinations of steatosis (fatty change), ballooning, Mallory-Denk bodies, and satellitosis. Steatosis may be predominantly macrovesicular, defined by the presence of large fat droplets in the cytoplasm of hepatocytes displacing the nuclei. Microvesicular steatosis also can be seen, characterized by fine fat droplets surrounding the central nuclei. Alcoholic hepatitis is characterized by an inflammatory infiltrate rich in neutrophils, most frequently distributed in the lobules adjacent to hepatocytes containing Mallory-Denk bodies, and dense, perisinusoidal fibrosis (Fig. 70A.6). Lymphocytes and histiocytes also may be present, sometimes in the form of lipogranulomas, and megamitochondria also may be evident. In the portal and periportal regions, ductular reaction with numerous neutrophils may occur. Cholangiolitis and canalicular bile plugs are worrisome lesions for concomitant pancreatitis.
The patterns of fibrosis in alcoholic hepatitis are characteristic. Fibrosis usually involves the terminal hepatic venules, leading to the thickening of the wall; luminal occlusion may be seen with necrosis of adjacent hepatocytes and Mallory-Denk bodies within remaining hepatocytes, a lesion referred to as central hyaline necrosis. Subendothelial fibrosis is a venoocclusive lesion of alcoholism that may be noted even in end-stage cirrhotic livers. Fibrosis may extend into the lobules in perisinusoidal spaces as delicate or dense strands, giving rise to a distinctive “chicken wire” pericellular (perisinusoidal) distribution (Brunt, 2002). Trichrome stain is particularly useful in detecting this unique form of fibrosis. With time, the liver may be replaced by micronodular cirrhosis; often, the septa of ALD are quite broad as manifestations of the microvascular obliteration.
Nonalcoholic Fatty Liver Disease
NAFLD is increasingly recognized as a significant form of potentially progressive liver disease. Prevalence studies estimate that approximately 20% to 25% of the U.S. population is affected by fatty liver; of these individuals, 15% to 20% are at risk for progression to cirrhosis (Angulo, 2002). The lesions noted in liver tissue may resemble many of the features of alcohol-induced liver damage in individuals who are not heavy drinkers (Brunt, 2004; Salt, 2004; te Sligte et al, 2004; Zafrani, 2004). The disease is etiologically attributed primarily to insulin resistance and is considered the hepatic manifestation of the metabolic syndrome, a constellation of obesity, hypertension, diabetes, and hyperlipidemia (Marchesini et al, 2001). Studies have also correlated the lesions of nonalcoholic steatohepatitis (NASH) with features of metabolic syndrome.
[/not-level-membership-for-surgery-category]
