CHAPTER 103 Chronic Pain
The Basic Science
However, chronic pain, pain that persists long after tissue healing due to injury or in tandem with disease, serves no useful biologic function.1 Chronic pain leads to heightened anxiety and diminished social functioning.2 The economic costs, in terms of both health care costs and lost productivity, are staggering and expected to rise even further as the population ages.3–6 Thus there is a vital need to understand the mechanism of chronic pain and to develop mechanism-based analgesic therapies.
Chronic Spinal Cord Injury Pain
Chronic pain following spinal cord injury (SCI) is estimated to occur in 65% to 81% of SCI patients, wherein about one third of patients rate it as severe.2,7 The addition of intractable pain to deficits in voluntary motor functions and autonomic dysfunction following SCI severely diminishes patient social and psychologic well-being.8 In order to improve patient quality of life, pain relief is an important consideration along with other SCI-related complications in an overall treatment and rehabilitation strategy.
Spinal Cord Injury Pain Classification
The International Association for the Study of Pain (IASP) has proposed a taxonomy that attempts to group SCI pain states to assist consistent clinical diagnosis and aid in designing effective pain management strategies (Table 103–1).9 In addition, classification also aids researchers in designing appropriate experiments that will uncover mechanisms underlying SCI pain and hopefully uncover new analgesic targets.
TABLE 103-1 Proposed IASP Classification of Pain Following Spinal Cord Injury
| Broad Type (Tier 1) | Broad System (Tier 2) | Specific structures/Pathology (Tier 3) |
|---|---|---|
| Nociceptive | Musculoskeletal | Bone, joint, muscle trauma, or inflammation Mechanical instability |
| Muscle spasm | ||
| Secondary overuse syndromes | ||
| Visceral | Renal calculus, bowel, sphincter dysfunction, etc. | |
| Dysreflexic headache | ||
| Neuropathic | Above level | Compressive mononeuropathies |
| Complex regional pain syndromes | ||
| At level | Nerve root compression (including cauda equina) | |
| Syringomyelia | ||
| Spinal cord trauma/ischemia (central dysesthesia syndrome, etc.) | ||
| Below level | Spinal cord trauma/ischemia (central dysesthesia syndrome, etc.) |
Reprinted with permission from Siddall PJ: Management of neuropathic pain following spinal cord injury: now and in the future. Spinal Cord 47:352-359, 2009.
Table 103–1 first divides pain into two general “types,” nociceptive and neuropathic, and further divides these pain types by possible underlying pathologies. The classification of the various pains (musculoskeletal, visceral, and neuropathic pain based on spinal lesion level) does not suggest definitive mechanisms, and the construct validity of the divisions has yet to be confirmed in clinical studies. However, such an uncomplicated taxonomy is a positive first step in systematically addressing the cause of SCI pain and developing useful therapies.
The first type of SCI pain, nociceptive pain, arises from stimulation of either somatic or visceral primary afferent nociceptors. Musculoskeletal pain has been characterized as dull aching pain that worsens with movement and eases with rest.7 In addition, this particular pain is localized to musculoskeletal structures. Sources of musculoskeletal pain include injury to muscles or ligaments related to the initial injury, overuse of the shoulders and arms of a wheelchair-bound patient, and vertebral instability and osteoporosis due to SCI.10 Given the initial challenges of novel self-propulsion and injury-associated pain, the onset of musculoskeletal pain is comparatively rapid, within weeks of the injury, and is the most commonly reported SCI-associated pain.11 Chronic musculoskeletal pain has been noted long after the injury (at least 5 years) and could be associated with the long-term skeletal changes in posture due to injury.7 Visceral pain has been described as spontaneous, dull, poorly localized, or cramping apparently originating from deep visceral structures.7 The occurrence of pain may or may not be coupled with visceral pathology and, unlike musculoskeletal pain, the onset of visceral pain may be months or years following SCI.7,12 Although the incidence of chronic visceral SCI pain is low, the pain is described as either severe or excruciating.7
The second type of SCI pain is neuropathic pain, which results from trauma or disease to the nervous system. Neuropathic pain has been described as unevoked pain characterized as sharp, burning, shooting, stabbing, and electric, occurring continuously or as paroxysms. In addition to spontaneous pain, there may also be exaggerated painfulness evoked by non-noxious stimulation (e.g., allodynia). Spontaneous and evoked neuropathic pain may occur at the level of injury due to a combination of damaged segmental spinal nerve roots or disinhibited spinal dorsal horn nociceptive neurons. About 40% of SCI patients experience at-level neuropathic pain.7 In contrast to at-level pain, below-level pain appears to have a delayed onset post-SCI and this could be due to dysfunction of brain regions postsynaptic to the spinothalamic tract.7 Below-level pain is as severe and persistent as at-level neuropathic SCI pain.7 Despite a lack of cutaneous thermal detection (either cold or heat) below the lesion, below-level neuropathic pain occurs in about one third of SCI patients. Interestingly, at-level and below-level cutaneous hypersensitivity correlates with the presence of below-level spontaneous pain, which suggests a common mechanism underlying these symptoms.13,14 A positive correlation between spontaneous pain and cutaneous mechanical hypersensitivity of the painful area has been reported for other neuropathic pain states.15 Such a correlation suggests that it may be possible to use defined stimuli to quantify spontaneous pain, beyond a subjective patient report, and objectively compare and contrast the efficacies of pain treatments.
General Mechanism
A general outline of the normal pain “pathway” will be presented. Detailed neuroanatomic and neurochemical schemes have been elaborated elsewhere.16 Noxious cutaneous stimuli (either thermal or mechanical) stimulate myelinated or unmyelinated small diameter primary afferents, conducting the “noxious signal” to the spinal cord superficial dorsal horn (or in the face, to the brainstem trigeminal sensory nucleus). Within the dorsal horn, excitatory neurotransmitters released from primary afferent central terminals stimulate postsynaptic dorsal horn neurons. A number of neuroactive substances (e.g., adenosine 5′-triphosphate [ATP]), excitatory amino acids, and neuropeptides activate their respective receptors on the postsynaptic neuron. By contrast, non-nociceptive, large-diameter, primary afferents terminate in deep dorsal horn and also in the brainstem dorsal column nuclei.
Axons of dorsal horn nociceptive neurons ascend to the brain via a number of tracts. Axons of nociceptive neurons decussate in the spinal cord and ascend via the contralateral ventral funiculus (spinothalamic tract) and terminate in the ventroposterior lateral thalamus. The pain signal may be dispersed from the thalamic nucleus to various brain areas with diverse functions such as the sensory cortex, hypothalamus, limbic lobes, and motor nuclei. Although there are a number of indirect pathways between the spinal dorsal horn and brain, direct projections of dorsal horn neurons to a number of brain areas have also been reported.17 By virtue of the numerous direct and indirect connections between nociceptive neurons to higher brain areas, pain evokes multiple physiologic and psychologic responses. Given human genetic diversity, it is apparent that the pain experience, as well as potential treatment strategies, differs between individuals and groups.18,19
In the normally functioning pain system, the excitatory component is counterbalanced by endogenous inhibitory components such that the initial pain sensation is not permanently propagated or does not evoke an exaggerated response. Primary afferents not only synapse with dorsal horn nociceptive neurons but also inhibitory interneurons, which, in turn, synapse with spinal nociceptive neurons, thus moderating pain transmission.20 Also, axons of nociceptive spinal neurons that terminate in the brainstem activate serotonergic, catecholaminergic, and GABAergic (gamma-aminobutyrate) neurons, which in turn send axons down to the dorsal horn. Because terminals are found presynaptic to primary afferents and spinal nociceptive neurons, activation of these brainstem neurons leads to diminished pain perception. Direct application by intrathecal injection of inhibitory neurotransmitters and opioid neuropeptides reduce spinal nociceptive neuron responses to noxious peripheral stimulation and are antinociceptive in rats.21 Analgesia is also observed in humans, following intrathecal injection of similar substances (α-adrenergic, GABAergic, and opioid receptor agonists).22 These findings a considerable parallel between the human and rat spinal dorsal horn neurochemistry, which, furthermore, points out the utility of preclinical models in evaluating drugs for possible clinical use.
In contrast to the normal state, experimental evidence suggests that decreased inhibition, increased excitation, or a combination of both initiates and perpetuates a chronic pain state because of the pathology of the nervous system. For example, acute lumbar intrathecal injection of an antagonist to either the GABAA or GABAB receptor subtype or the glycine receptor leads to a transient yet robust hind paw hypersensitivity and vocalization in rats. This indicates that a tonic inhibition is present in the normal state, which “dampens” responses to peripheral stimulation. Similarly, intrathecal injection of an excitatory glutamate receptor agonist (e.g., N-methyl-D-aspartate) induces a long-lasting hind paw hypersensitivity.23 The sustained excitation of nociceptive neurons may lead to increased intracellular cation levels, upregulation of second messenger systems, and gene expression.16 These intracellular processes then lead to persistent hyperactivity and increased responsiveness to peripheral stimulation. Furthermore, such abnormal activity may be found throughout the pain neuraxis. Preclinical pain models have demonstrated considerable changes to normal neuroanatomy and neurochemistry following painful peripheral nerve injury or inflammation. For example, the central terminals of non-nociceptive primary afferents extend to spinal laminae normally receiving nociceptors, and these same afferents now express neuropeptides typically found in nociceptors.24,25 Changes in brain activity response to peripheral stimulation following an injury, including an SCI, have been observed.26
On the basis of findings in preclinical pain models, to attenuate clinical chronic pain states, it would be reasonable to increase inhibition (e.g., intrathecal GABAB receptor agonist baclofen) or decrease excitation (e.g., intrathecal [N-methyl-D-aspartate] NMDA receptor antagonist ketamine) at the level of the spinal cord, the first site of interaction between the peripheral and central nervous systems (see later).22,27 Even though such acute measures may prove efficacious, they are temporary. It is likely that a number of regions within the nervous system may express abnormal excitation and that these changes have been made permanent via genetic and structural mechanisms. Implantation of cells that continuously release endogenous analgesic substances and gene therapy may provide long-term pain relief.28–32
Most experimental pain studies have focused on neural function for obvious reasons. However, glial cells (e.g., microglia, astrocytes) vastly outnumber neurons in the central nervous system (CNS). Accumulating evidence indicates that glia have a key role in maintaining the neuropathic SCI pain state.33 In the normal state, glia appear to maintain the homeostasis of the extracellular milieu. Because glia express receptors and ion channels, similar to neurons, they may respond to neuroactive substances. Following exposure to these substances, activated glia may release, in turn, a number of neuroactive substances and proinflammatory cytokines. The glial response following injury has been intensely characterized because modulating the response is believed to be crucial to promoting motor and sensory recovery.34 Injury of the thoracic spinal cord leads to dramatic increases in microglia and astrocytes not only at the site of injury but at the level of the lumbar enlargement, several segments away.35,36 Interestingly, similar increases in spinal glial activity in the lumbar spinal cord are observed in models of painful peripheral neuropathies.37 Treatments designed to decrease glial function following SCI in order to improve motor function may also have a secondary effect of reducing SCI-induced pain. Such treatment studies should include sensory outcomes if the patient has SCI pain.
Neuropathic Spinal Cord Injury Animal Models
The majority of preclinical SCI pain experiments have been done in rodents, not only because of the convenient availability of near homogenous subjects but also because general clinical aspects of the histopathology following an SCI can be replicated in rats.38 In addition, numerous analgesic treatments initially screened in peripheral-injury chronic pain models have gone on to demonstrate clinical efficacy. Thus animal models of SCI pain may also be useful in both elucidating SCI pain mechanisms and developing novel clinical treatments.
Considerations
A degree of controversy exists surrounding the clinical relevance of rodent models of pain.39 Such controversy plagues other fields of neurologic research as well.40 The main clinical diagnosis of pain is based on the patient’s verbal report, which would include pain severity, duration, and frequency. By contrast, in chronic pain models, a specific response to a given stimulus is interpreted as pain by experimenters. An exaggerated change in response to either non-noxious or noxious (hyperalgesia) cutaneous stimuli is interpreted to mean a pathologic alteration in the underlying pain mechanism. However, such changes in sensory perception may not always be reported by patients, who primarily present with unevoked spontaneous pain.
An additional controversy that arises concerning animal models is deciding which would be best to use for preclinical research such that the information obtained could be readily translated into clinical practice. In terms of evaluating new therapeutics, predictive validity and reliability are crucial.41 A model with predictive validity allows one to accurately foresee the effect of a treatment in humans.
If the animal displays behaviors that are analogous to clinical symptoms, the model is said to have face validity. In pain research, however, the outcome measures in the preclinical and clinical situations may not be identical. Most preclinical analgesic drug studies measure changes in response to stimulation, whereas few clinical drug trials have solely used quantitative sensory testing. Despite the stark difference in outcome measures, the concordance between the animal and clinical results is between 61% and 88%.42 In general, the high concordance suggests that the presence of cutaneous sensitivity (e.g., withdrawal threshold) is predictive of spontaneous pain (e.g., pain rating on a visual analog scale), but the wide range also indicates that some models may have better predictive value over others.
In an attempt to bridge the divide between clinical and preclinical outcome measures, basic scientists are working to quantify spontaneous pain-related behavior and changes in mood associated with chronic pain in animals.7,39 As mentioned earlier, changes in affect may impair SCI rehabilitation, further degrading overall well-being. Some of the well-defined experimental methods used in other behavioral research fields such as psychiatry and drug addiction have been adopted to detect and quantify spontaneous pain-related behavior, with varying degrees of success.43–45 Given the current concordance between preclinical and clinical results and the difficulty of quantifying affect in animals, it is not entirely clear how much more translational or clinical value will be gained with the addition of nonspontaneous measures to current testing procedures.40
If a model has face validity, the assumption that follows is that it also has construct validity. Although it is desirable to mimic the clinical pathology in the rat, this may not always be possible. Demonstrating construct validity requires knowledge of the clinical mechanism, which is often lacking and extremely difficult to obtain. Also, as Geyer points out, the theoretical basis of neurologic disorders is constantly evolving.40 Thus construct validity should not be the sole determinant to judge the usefulness of a model.
Finally, reliability refers to the “consistency and stability” of both the experimental procedure and the symptoms resulting from it.40 On the one hand, this quality is highly desirable in basic science research because a highly predictable outcome following a manipulation reduces the chance of error and the need to use large numbers of subjects. On the other hand, clinical SCI is not homogeneous, and although many SCI patients suffer from chronic pain, there are those who do not.
Excitotoxin Injection
A series of microinjections of the nonsubtype selective glutamate receptor agonist quisqualate is placed into the deep dorsal horn of the thoraco-lumbar spinal cord.46 The lesion leads to a dramatic necrosis of the deep dorsal horn but spares the superficial dorsal horn, thus preserving ipsilateral sensory input. Furthermore, syrinx formation is observed in these rats. Significant bilateral hind paw hypersensitivity to both thermal and mechanical stimuli are observed as early as 8 days following microinjection. Also, spontaneous “grooming” behavior, excessive hind paw scratching directed within the injected dermatome, begins about 2 days postmicroinjection and may progressively expand and worsen over time. The grooming behavior is suggestive of at-level unevoked pain. Both the grooming behavior and cutaneous hypersensitivity persist for weeks following injection.
The excitotoxic SCI model is unique in that the spinal cord lesion is generally restricted to the dorsal horn. Because of this selective destruction, there is little or no hind paw motor dysfunction. Several novel treatments have been tried in this rat model that attenuated pain-related symptoms including grooming.28,47 The predictive validity of this model is not known because extensive evaluation of clinically available analgesic drugs has yet to be done.
Acute Trauma to the Spinal Cord
Following lateral hemisection of spinal cord at level T13, rats develop bilateral hind paw hypersensitivity to heat (decreased withdrawal latency) and mechanical (decreased withdrawal threshold) stimuli beginning 10 days after injury.48 Loss of ipsilateral hind limb function is also observed, but functional recovery begins about 7 days after injury. Despite mild-to-moderate hind paw dysfunction, as demonstrated by Basso-Beattie-Bresnahan (BBB49) locomotor rating scores, robust ipsilateral hind paw responses can be evoked in hemisected rats in order to evaluate therapeutics. In addition, a bilateral forepaw (above-level) hypersensitivity is observed beginning about 2 weeks after injury. A few clinically relevant analgesic drugs have been tested in these rats, along with novel treatments.30,50 Systemic and intrathecal (catheter terminating at the spinal level T13) injection of morphine ameliorated hind paw mechanical hypersensitivity.51 Intrathecal baclofen increased hind paw withdrawal thresholds in injured rats.52 By contrast, intrathecal injection of the selective serotonin reuptake inhibitor (SSRI) fluvoxamine attenuated forepaw mechanical hypersensitivity to a greater degree than hind paw hypersensitivity.53 The differing effect suggests a differential neuropharmacology between above- and below-level neuropathic pains. Hemisection of the spinal cord could be useful in comparing neurochemical and anatomic changes rostral or caudal to the lesion with the uninjured side. The lesion is relatively easy to generate and fairly consistent from rat to rat, which in part explains why this is the most commonly used SCI injury model.54 However, the relevance of hemisection to clinical injury is not entirely clear because there are few human cases of spinal hemisection injuries.
Most spinal cord injuries are due to acute extradural impact and compressive forces.38 These are traditionally modeled in animals using contusive impact devices in which the severity of the injury can be varied, resulting in locomotor dysfunction in a force-dependent manner.55 In these animals, the effect of various intervention strategies can be examined.54
Even with standardized procedures and devices, it is still difficult to uncover a correlation between the degree of tissue damage and symptoms of neuropathic pain. Following a “moderate” contusion of the thoracic dorsal spinal cord, the forepaws develop hypersensitivity to heat and mechanical stimuli, but the sensitivity of the hind paws to these stimuli are variable.56,57 A “moderate” contusion also yields long-lasting at-level hypersensitivity, whereas fewer rats with a “severe” injury displayed such hypersensitivity.58 Hind limb functionality scores in moderately injured rats were considerably decreased but recovered such that scores were eventually the same as uninjured rats. Cutaneous hypersensitivity persisted, however, long after functional recovery.58 Other studies have demonstrated significant at-level and below-level hypersensitivity with severe injury.59,60 Clinical studies have yet to find a correlation among the extent of spinal cord injury, specific loss of certain tracts, or gray matter areas and pain.61
Similar to the excitotoxic SCI model, there has been little pharmacologic validation of either the hemisection or contusion model with clinical analgesic drugs, except for the anticonvulsant gabapentin.62 The positive effect of an SSRI on above-level SCI pain in the hemisection suggests that this class of drug may be clinically useful. However, there is a lack of positive clinical evidence supporting the use of an SSRI in SCI pain.27 In fact, the use of SSRI and tricyclic antidepressants (TCAs) such as amitriptyline may enhance SCI-induced spasticity and TCAs may also be contraindicated because of side effects such as urinary retention and constipation.27,63 To increase confidence that novel analgesic interventions tested in these models will succeed, more pharmacologic validation studies are necessary.
Compression Injury
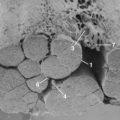
To mimic “circumferential compression,” a combination of impact and compression at multiple levels rather than a single point of the spinal cord, a modified aneurism clip is used, whereby one jaw is slid under the anterior thoracic spinal cord, the other jaw is over the posterior cord, and the clip is closed for a given length of time (e.g., 1 minute).64 Another important distinction of this model compared with others is the prolonged injury duration. A modified version of the “circumferential compression” model was used to evaluate several clinical analgesic drugs. As with other models, significant hind paw hypersensitivity to thermal and mechanical stimuli developed and hind limb locomotor function diminished; these effects were noted as early as 1 week after injury.65,66 On the basis of a limited number of drugs, it appears that this model can distinguish analgesic from nonanalgesic drugs. For example, a sedating and anxiolytic dose of diazepam in SCI rats does not ameliorate hind paw hypersensitivity (Table 103–2). The antiarrhythmic sodium channel blocker mexiletine was not analgesic in clinical SCI pain and did not demonstrate efficacy in compression-injured rats (Table 103–3). By contrast, in an ischemic model of neuropathic SCI pain, mexiletine ameliorated mechanical hypersensitivity.67
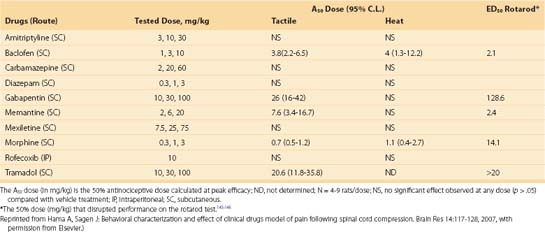
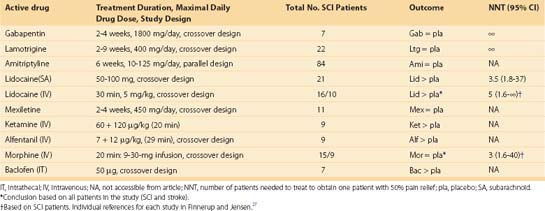
A number of drugs were significantly antinociceptive, however, including systemic baclofen, morphine, and the anticonvulsant gabapentin. Recent data from the rat spinal compression model suggests that memantine, tramadol, a cannabinoid receptor agonist, and the N-type voltage-gated calcium channel blocker ziconotide may be effective for neuropathic SCI pain (compare Table 103–2 and Table 103–3).66,68 Clinical trials with appropriate controls are necessary to confirm the clinical utility of these drugs and establish predictive validity of the model.69 The pharmacologic data suggest a selective responsiveness to antinociceptive agents rather than generalized responses to ataxic or sedating drugs. The data also suggest differences between models (beyond the type of injury) in the mechanisms that maintain neuropathic SCI pain. Further drug efficacy comparisons between models are necessary to support this contention. In general, the diverse animal drug efficacy data imply that some drugs may be useful within specific populations of SCI patients.
Interestingly, drugs that have been shown to be efficacious for peripheral neuropathic pain such as amitriptyline and the anticonvulsant carbamazepine do not show efficacy in this model.27,70 This suggests significant mechanistic differences between peripheral and central neuropathic pain and implies that distinct treatment approaches may be necessary depending on the injury, regardless of similarities in symptoms (e.g., “allodynia”).
Photochemical Ischemia
Ischemia in the CNS may lead to neuropathic pain.2 Hao and colleagues71 developed a neuropathic SCI pain model that did not involve mechanical destruction of CNS tissue. Following intravenous injection of erythrosin B, laser irradiation of the thoracic vertebrae causes coagulation of the spinal blood vessels, which is limited to immediate spinal segments. A short-term (several days) mechanical hypersensitivity develops within the thoracic dermatome in which the ischemia occurred.71 With greater ischemic damage, thoracic hypersensitivity re-emerges several weeks after the initial hypersensitivity and persists for months. Furthermore, some of these animals (40%) mutilate (“autotomy”) their hind limbs, suggesting the presence of below-level spontaneous pain or dysesthesia.72
Several clinical drugs have been evaluated in these rats.67,72–75 The thoracic hypersensitivity was opioid sensitive and also suppressed with the antiarrhythmic drugs tocainide and mexiletine, the sedative pentobarbital, and gabapentin. However, clonidine and baclofen did not significantly affect hypersensitivity. By contrast, in clinical SCI pain, intrathecal clonidine and baclofen demonstrated efficacy, whereas (oral) mexiletine did not (Table 103–3).27,76,77 It is possible that at-level pain in these rats and the clinical neuropathic SCI pain are entirely different in terms of mechanism. The experimental approaches greatly differ as well: At-level pain was not specifically evaluated in the clinical trials, and there is a lack of below-level pain in this model. Other data using this model suggest that the NMDA receptor antagonist dextromethorphan could be useful in the treatment of SCI pain, as it appears to be for some types of peripheral neuropathic pain.78 A drug with similar NMDA receptor channel blocking properties, ketamine, has been shown to relieve neuropathic SCI pain.79 The outcome of clinical studies using dextromethorphan will test the predictive value of the ischemia SCI model.
What Is the Mechanism of Neuropathic Spinal Cord Injury Pain in Humans?
Using microelectrodes, spontaneous activity in dorsal horn neurons was recorded both rostrally and caudally to the injury in SCI pain patients.80,81 Similar activity in spinal neurons was also found in SCI rats.82–84 In addition, dorsal horn neurons in rats displayed exaggerated excitation (central sensitization) to distal cutaneous stimulation, but analogous neurons have yet to be described in humans. Changes in CNS neural function have been observed beyond the spinal cord in both rats and humans.85–87 The cause of the abnormal neural function is not known. One possibility is that, as observed in rats, lesions to dorsal gray matter lead to neuropathic SCI pain. On the basis of limited analyses of spinal lesions using magnetic resonance imaging, a significant distinction between pain patients and pain-free patients and the extent of tract or gray matter damage was not observed.61 Perhaps the temporal change in abnormal neural function parallels that of pain following SCI. A clinical time course study may not be technically feasible to address this issue, however.
At the cellular level, it is possible that changes in excitatory or inhibitory neurotransmission may underlie abnormal activity of, for example, spinal neurons. Changes in neuropeptide content in the spinal dorsal horn following SCI in animals have been reported. An excitatory neuropeptide found in primary afferent nociceptors, calcitonin gene-related peptide (CGRP), was increased in the deep dorsal horn of lumbar spinal cord, far removed from the thoracic injury site.88,89 Both the spinal compression and hemisection models have significant below-level hypersensitivity to noxious and innocuous cutaneous stimuli, which are transiently ameliorated with a CGRP receptor antagonist.50 The elevated CGRP content in deep dorsal horn could in part underlie both increased neuronal activity and chronic pain. The CGRP content in the deep dorsal horn from SCI patients is also increased, although pain was not measured in these patients.90 In the case of humans, then, the consequence of the change in neuropeptide content on sensation is not clear. The amelioration of neuropathic SCI pain with NMDA receptor antagonists such as ketamine and dextromethorphan indirectly suggests that glutamate receptor activity has a key role in maintaining pain. There is no direct evidence to demonstrate significant loss of GABAergic neurons, for example, in the spinal cord of neuropathic SCI pain patients. However, the indirect pharmacologic evidence such as analgesic effects with baclofen suggests that a loss of inhibition could be as important as the increase in excitation. Noninvasive magnetic resonance spectroscopy could be used to evaluate changes in inhibitory amino acids.86,91
Few clinical studies have evaluated the role of glia in clinical neuropathic SCI pain. Spinal cord tissue taken acutely following injury (15 to 60 days) exhibited marked microglia/macrophage infiltration proximal and distal to the injury.92 A correlation between cellular activation and pain cannot be determined because the pain status of the patients was not determined. Although clinical data correlating possible dysfunction of brain nuclei with SCI pain are starting to emerge, postmortem tissue is currently the only way to evaluate in detail the neurochemical and anatomic processes following SCI.85
Currently, treatments for SCI pain are based more or less on trial and error rather empirically. However, with increasing sophistication of medical imaging techniques combined with receptor selective radioligands, it may be possible to observe and quantify the effect of SCI on neural areas that are pain related and assess the effect of treatment on these areas.93 Drugs that are analgesic could be characterized by not only receptor-binding function but also activity at defined pain-related areas.
Non-Neural Molecular Targets for Spinal Cord Injury Pain
Although there are numerous interactions between glia and neurons following injury, research on chronic pain—SCI pain in particular—has been generally slanted toward neural events.94,95 As a result, the development of novel analgesic drugs has focused on neural dysfunction. These include ziconotide and cannabinoids, both of which, coincidentally, are naturally based products.69,96 Given that glia also express membrane proteins similar to those found on neurons, it is likely that drugs targeted to neurons may also affect glial function. In this section, a few of the cellular and molecular processes, with respect to glia, underlying the development of neuropathic SCI pain are summarized.
In addition to the sensitization of nociceptors and spinal dorsal horn neurons, an early and permanent “gliopathy” contributes to maintenance of neuropathic SCI pain. A gliopathy has been defined as the “dysfunctional and maladaptive” glia response to injury.33 Like the response observed in neurons following injury, the glia response is persistent and elevated. Gliopathy has also been reported in other neuropathic pain models.97 Activated glia secrete proinflammatory cytokines, nociceptor sensitizers, and other substances that directly activate nociceptors and alter ion channel expression.98,99 Glial NF-κB, a protein complex transcriptional factor with a key role in the cellular response to infection and stress, is presumed to be crucial in the intracellular process that underlies the long-term response to substances released from injured spinal cord. Activated NF-κB stimulates cytokine gene transcription. Knockout of glial NF-κB in mice reduced peripheral tissue injured–induced pain.100 Nonsteroidal anti-inflammatory drugs, glucocorticoids, and immunosuppressants inhibit the activation of NF-κB.101 It is possible that early postinjury blocking of the NF-κB signaling cascade may attenuate the severity of later neuropathic SCI pain. It should be noted that NF-κB is also crucial in tissue healing. Thus an optimal, probably narrow, level of activation of NF-κB is necessary for recovery. In terms of SCI, the optimal level that will lead to both functional motor and sensory recovery remains to be defined.
Lipid Signaling
A number of proteins that have been identified by proteomic and genomic analyses in SCI can be functionally categorized in several groups such as cell-cycling, chemokines, cellular metabolism, lipid and protein degradation, neuronal survival, and regeneration.102–105 In addition to cellular membrane formation, lipids also have important roles in extracellular and intracellular signaling. One such class of lipids is the lysophospholipids, lipids involved in cell proliferation and chemotaxis.
In addition to cytokines, NF-κB also induces transcriptional upregulation of ectonucleotide pyrophosphatase/phosphodiesterase 2 (ENPP2/autotoxin), the enzyme that produces lysophosphatidic acid (LPA).106 Activation of the peripheral LPA1 receptor leads to demyelination and vascular remodeling, processes that in part mediate the initiation of nerve injury–induced pain.107–109 In the spinal cord, LPA induces proliferation of astrocytes and also inhibits glutamate uptake by reactive astrocytes, thereby exacerbating glutamate neurotoxicity.110 Because these pathologies are also observed following SCI and LPA activation appears to have a key role in initiating nerve injury–induced neuropathic pain, LPA may also contribute to the onset of neuropathic SCI pain. Antagonists to either the enzyme or the receptor may be useful in reducing the tissue pathology caused by LPA and perhaps ameliorating SCI pain. FTY720-phosphate (Fingolimod), an immunosuppressant derived from the fungal product myriocin, has been demonstrated to have antitumor effects and is a potential treatment for multiple sclerosis.111 It is an inhibitor of ENPP2 and a sphingosine-1-phosphate (S1P) receptor agonist. S1P activation leads to similar events observed with LPA including the increased synthesis of prostaglandins, but FTY720-phosphate reduces pain following peripheral nerve injury.111 A number of S1P receptor subtypes exists, so the effect of FTY720-phosphate could be mediated through one of these or via an entirely novel mechanism.
Glial Inhibitors
A number of intracellular or membrane targets modulate the glial response to injury.
In spinal nociceptive neurons, glutamate excitotoxicity results in the upregulation of microglia-specific activator cysteine-cysteine chemokine ligand 21 (CCL21). This ligand is transported preferentially down axons and released.112,113 The protein binds specifically to CXCR3 receptors expressed on microglia and not those found on neurons and astrocytes. The tetracycline derivative minocycline decreases microglia activation and expression of CCL21.114 The presence of the microglia activator CCL21 is not restricted to the site of SCI but may be found rostrally in brain regions such as the thalamus.114 Thus it is important to note here that an apparently discrete CNS lesion may lead to glia activation throughout the pain neuraxis, and systemic rather than localized treatment may be required to effectively treat neuropathic SCI pain.
On exposure to growth-inducing proinflammatory cytokines and nucleotides (e.g., ATP), glia begin to proliferate or re-enter the cell cycle in a cyclin D1–dependent manner.115,116 Cyclin-dependent kinases (CDKs) and cyclins assist the transition through various steps of the cell cycle. Postmitotic cells such as oligodendrocytes and neurons respond to these proliferation signals by undergoing apoptosis. CDK inhibitors that are under investigation as anticancer drugs such as olomoucine, roscovitine, and flavopiridol decrease spinal lesion volume following SCI.117,118 These compounds also inhibited TNF-α-mediated microglial proliferation. Alvocidib, a flavopiridol derivative, reduced inflammation-induced peripheral tissue destruction.119 The capsaicin-sensitive cation channel TRPV1 is found on peripheral nociceptors (as well as glia). Cyclin-dependent kinase 5 (CDK5) mediates TRPV1 phosphorylation, which leads to sensitized nociceptors. TNF-α, abundantly found in injured tissues, also sensitizes TRPV1 by a CDK5-mediated pathway.120–123 Roscovitine, another nucleotide analogue, prevents activation of CDK2 and CDK5 and has been shown to be effective in attenuating inflammatory pain.124,125 At higher concentrations, roscovitine could inhibit the activity of CDK5 inducible signal transduction pathway kinases such as ERK1 (extracellular signal-regulated kinase 1) and ERK2.126 The ERKs have been implicated in dorsal horn neural hyperexcitability and glial activation.95 Thus prevention of injury-induced reinitiation of the glial cell cycle may be useful in not only limiting tissue damage but sensory dysfunction caused by overexpression of cytokines and other inflammatory mediators.
Propentofylline, a xanthine derivative with neuroprotective properties, has been shown to reduce neuropathic SCI pain via numerous possible mechanisms.36,127 Propentofylline is both an adenosine reuptake inhibitor and phosphodiesterase (PDE) inhibitor.128,129 Treatment of SCI rats with propentofylline leads to decreased glia activation and increased GABAergic function as indicated by increased protein expression of the enzyme that produces GABA in neurons, which coincided with decreased pain.127 Similarly, ibudilast, another nonselective PDE inhibitor, suppressed glial cell activation and pain-related behavior following nerve injury.130 Phosphodiesterase inhibitors including sildenafil could be a novel class of analgesics for SCI pain.
Finally, a membrane protein not involved in membrane electrical conduction may have potential as an analgesic target. The aquaporins are integral membrane proteins that are permeable to water and found in various mammalian cells.131 Aquaporin 4 (AQP4) is highly expressed in brain and has been found exclusively in astrocytes.132 Following SCI, these channels were found to be upregulated, which may in part explain the presence of numerous fluid-filled cavities (syrinx) within the injured spinal cord.105,133 An SCI-induced increase in AQP1, found in neurons and ependymal cells, may also be involved in syrinx formation. The carbonic anhydrase inhibitor acetazolamide, a clinically used diuretic, may inhibit AQP4 function, reducing tissue damage and possibly SCI pain.134,135
The degree to which glial function can be attenuated to obtain pain relief is currently unknown. This problem also arises in blocking neurotransmission for pain relief—total block leads to anesthesia and adverse neurologic side effects. Likewise, complete block of glial function or completely destroying proteins associated with glial scarring following SCI leads to injury exacerbation.136,137 Also, time to treatment would be an important consideration. Although immediate blockade of gliosis with cytokine neutralizing antibodies or genetic manipulation significantly improved functional recovery after experimental SCI, few patients can be treated immediately following SCI.138–140 In addition, neuropathic SCI pain emerges months or years following injury, making universal early treatment in all SCI cases questionable.7 Further studies in terms of treatment timing and better understanding of the effects of novel glial inhibitors on eventual functional outcome are necessary.
Summary
A key test of the usefulness of a preclinical model is whether it can predict the clinical efficacy of a given treatment. Researchers believe that their models may be generalized to a given clinical situation, but this may lead to clinical failure on the basis of incorrect assumptions. Many of the neuropathic SCI pain mechanisms observed in rodents have yet to be demonstrated in humans. There should also be confidence that the animal end points and clinical end points are similar or the same. On the basis of a review of the preclinical and clinical literature, such confidence is lacking. Thus if animal models are to shoulder the burden of producing novel treatments for neuropathic SCI pain, the validity of these models should be carefully ascertained. Failures and successes should be openly presented.141,142
Acknowledgment
Key Points
1 Falci S, Best L, Bayles R, et al. Dorsal root entry zone microcoagulation for spinal cord injury-related central pain: operative intramedullary electrophysiological guidance and clinical outcome. J Neurosurg. 2002;97(2 Suppl):193-200.
2 Finnerup NB, Jensen TS. Spinal cord injury pain—mechanisms and treatment. Eur J Neurol. 2004;11:73-82.
3 Gwak YS, Crown ED, Unabia GC, et al. Propentofylline attenuates allodynia, glial activation and modulates GABAergic tone after spinal cord injury in the rat. Pain. 2008;138:410-422.
4 Hulsebosch CE, Hains BC, Crown ED, et al. Mechanisms of chronic central neuropathic pain after spinal cord injury. Brain Res Rev. 2009;60:202-213.
Introduces the concept of “gliopathy” and its contribution to maintenance of neuropathic SCI pain.
5 Lenz FA, Tasker RR, Dostrovsky JO, et al. Abnormal single-unit activity recorded in the somatosensory thalamus of a quadriplegic patient with central pain. Pain. 1987;31(2):225-236.
6 Nesic O, Lee J, Unabia GC, et al. Aquaporin 1—a novel player in spinal cord injury. J Neurochem. 2008;105(3):628-640.
1 Merskey H, Bogduk N. Classification of Chronic Pain: Descriptions of Chronic Pain Syndromes and Definitions of Pain Terms, 2nd ed. Seattle: IASP Press; 1994:pp 240.
2 Bonica JJ. Management of Pain, 2nd ed. Philadelphia, PA: Lippincott Williams & Wilkins; 1990:pp 2120.
3 Straus BN. Chronic pain of spinal origin: the costs of intervention. Spine. 2002;27:2614-2619. discussion 2620
4 Mapel DW, Shainline M, Paez K, et al. Hospital, pharmacy, and outpatient costs for osteoarthritis and chronic back pain. J Rheumatol. 2004;31:573-583.
5 Allaire SH, Prashker MJ, Meenan RF. The costs of rheumatoid arthritis. Pharmacoeconomics. 1994;6:513-522.
6 Pentland W, McColl MA, Rosenthal C. The effect of aging and duration of disability on long term health outcomes following spinal cord injury. Paraplegia. 1995;33:367-373.
7 Siddall PJ, McClelland JM, Rutkowski SB, et al. A longitudinal study of the prevalence and characteristics of pain in the first 5 years following spinal cord injury. Pain. 2003;103:249-257.
8 Widerstrom-Noga EG, Felipe-Cuervo E, Yezierski RP. Chronic pain after spinal injury: interference with sleep and daily activities. Arch Phys Med Rehabil. 2001;82:1571-1577.
9 Siddall PJ, Yezierski RP, Loeser JD. Pain following spinal cord injury: clinical features, prevalence, and taxonomy. International Association for the Study of Pain. 2000;3:1-10.
10 Middleton JW, Leong G, Mann L. Management of spinal cord injury in general practice—part 2. Aust Fam Physician. 2008;37:331-332. 335-338
11 Siddall PJ, Taylor DA, McClelland JM, et al. Pain report and the relationship of pain to physical factors in the first 6 months following spinal cord injury. Pain. 1999;81:187-197.
12 Kogos SCJr, Richards JS, Banos JH, et al. Visceral pain and life quality in persons with spinal cord Injury: a brief report. J Spinal Cord Med. 2005;28:333-337.
13 Finnerup NB, Johannesen IL, Fuglsang-Frederiksen A, et al. Sensory function in spinal cord injury patients with and without central pain. Brain. 2003;126(Pt 1):57-70.
14 Widerstrom-Noga E. Chronic pain and nonpainful sensations after spinal cord injury: is there a relation? Clin J Pain. 2003;19:39-47.
15 Attal N, Fermanian C, Fermanian J, et al. Neuropathic pain: are there distinct subtypes depending on the aetiology or anatomical lesion? Pain. 2008;138:343-353.
16 Millan MJ. The induction of pain: an integrative review. Prog Neurobiol. 1999;57:1-164.
17 Cliffer KD, Burstein R, Giesler GJJr. Distributions of spinothalamic, spinohypothalamic, and spinotelencephalic fibers revealed by anterograde transport of PHA-L in rats. J Neurosci. 1991;11:852-868.
18 Campbell CM, France CR, Robinson ME, et al. Ethnic differences in the nociceptive flexion reflex (NFR). Pain. 2008;134:91-96.
19 Tan EC, Lim Y, Teo YY, et al. Ethnic differences in pain perception and patient-controlled analgesia usage for postoperative pain. J Pain. 2008;9:849-855.
20 Millan MJ. Descending control of pain. Prog Neurobiol. 2002;66:355-474.
21 Yaksh TL, Hua XY, Kalcheva I, et al. The spinal biology in humans and animals of pain states generated by persistent small afferent input. Proc Natl Acad Sci U S A. 1999;96:7680-7686.
22 Bennett G, Serafini M, Burchiel K, et al. Evidence-based review of the literature on intrathecal delivery of pain medication. J Pain Symptom Manage. 2000;20:S12-S36.
23 Yaksh TL. Behavioral and autonomic correlates of the tactile evoked allodynia produced by spinal glycine inhibition: effects of modulatory receptor systems and excitatory amino acid antagonists. Pain. 1989;37:111-123.
24 Neumann S, Doubell TP, Leslie T, et al. Inflammatory pain hypersensitivity mediated by phenotypic switch in myelinated primary sensory neurons. Nature. 1996;384:360-364.
25 Shortland P, Woolf CJ. Chronic peripheral nerve section results in a rearrangement of the central axonal arborizations of axotomized A beta primary afferent neurons in the rat spinal cord. J Comp Neurol. 1993;330:65-82.
26 Endo T, Spenger C, Hao J, et al. Functional MRI of the brain detects neuropathic pain in experimental spinal cord injury. Pain. 2008;138:292-300.
27 Finnerup NB, Jensen TS. Spinal cord injury pain–mechanisms and treatment. Eur J Neurol. 2004;11:73-82.
28 Brewer KL, Yezierski RP. Effects of adrenal medullary transplants on pain-related behaviors following excitotoxic spinal cord injury. Brain Res. 1998;798:83-92.
29 Eaton MJ, Wolfe SQ, Martinez M, et al. Subarachnoid transplant of a human neuronal cell line attenuates chronic allodynia and hyperalgesia after excitotoxic spinal cord injury in the rat. J Pain. 2007;8:33-50.
30 Hains BC, Johnson KM, Eaton MJ, et al. Serotonergic neural precursor cell grafts attenuate bilateral hyperexcitability of dorsal horn neurons after spinal hemisection in rat. Neuroscience. 2003;116:1097-1110.
31 Gajavelli S, Castellanos DA, Furmanski O, et al. Sustained analgesic peptide secretion and cell labeling using a novel genetic modification. Cell Transplant. 2008;17:445-455.
32 Wolfe D, Mata M, Fink DJ. A human trial of HSV-mediated gene transfer for the treatment of chronic pain. Gene Ther. 2009;16:455-460.
33 Hulsebosch CE, Hains BC, Crown ED, et al. Mechanisms of chronic central neuropathic pain after spinal cord injury. Brain Res Rev. 2009;60:202-213.
34 Deumens R, Koopmans GC, Joosten EA. Regeneration of descending axon tracts after spinal cord injury. Prog Neurobiol. 2005;77:57-89.
35 Detloff MR, Fisher LC, McGaughy V, et al. Remote activation of microglia and pro-inflammatory cytokines predict the onset and severity of below-level neuropathic pain after spinal cord injury in rats. Exp Neurol. 2008;212:337-347.
36 Gwak YS, Hulsebosch CE. Remote Astrocytic and Microglial Activation Modulate Neuronal Hyperexcitability and Below-Level Neuropathic Pain after Spinal Injury in Rat. Neuroscience. 2009;161:895-903.
37 Hansson E. Could chronic pain and spread of pain sensation be induced and maintained by glial activation? Acta Physiol (Oxf). 2006;187:321-327.
38 Bunge RP, Puckett WR, Becerra JL, et al. Observations on the pathology of human spinal cord injury. A review and classification of 22 new cases with details from a case of chronic cord compression with extensive focal demyelination. Adv Neurol. 1993;59:75-89.
39 Blackburn-Munro G. Pain-like behaviours in animals—how human are they? Trends Pharmacol Sci. 2004;25:299-305.
40 Geyer MA, Markou A. Animal models of psychiatric disorders. In: Bloom FE, Kupfer DJ, editors. Psychopharamacology: The Fourth Generation of Progress. New York: Raven Press; 1995:787-798.
41 Geyer MA, Markou A. The role of preclincal models in the development of psychotropic drugs. In: Davis KL, Charney D, Coyle JT, Nemeroff C, editors. Neuropsychopharmacology: The Fifth Generation of Progress. Philadelphia: Lippincott Williams & Wilkins; 2002:445-455.
42 Kontinen VK, Meert TF. Predictive validity of neuropathic pain models in pharmacological studies with a behavioral outcome in the rat: a systematic review. In: Dostrovsky JO, Carr DB, Koltzenburg M, editors. Proceedings of the 10th World Congress on Pain, Vol. 24. Seattle: IASP Press; 2003:489-498.
43 Hasnie FS, Breuer J, Parker S, et al. Further characterization of a rat model of varicella zoster virus-associated pain: Relationship between mechanical hypersensitivity and anxiety-related behavior, and the influence of analgesic drugs. Neuroscience. 2007;144:1495-1508.
44 Kontinen VK, Kauppila T, Paananen S, et al. Behavioural measures of depression and anxiety in rats with spinal nerve ligation-induced neuropathy. Pain. 1999;80:341-346.
45 Mills CD, Grady JJ, Hulsebosch CE. Changes in exploratory behavior as a measure of chronic central pain following spinal cord injury. J Neurotrauma. 2001;18:1091-1105.
46 Yezierski RP, Liu S, Ruenes GL, et al. Excitotoxic spinal cord injury: behavioral and morphological characteristics of a central pain model. Pain. 1998;75:141-155.
47 Yu CG, Fairbanks CA, Wilcox GL, et al. Effects of agmatine, interleukin-10, and cyclosporin on spontaneous pain behavior after excitotoxic spinal cord injury in rats. J Pain. 2003;4:129-140.
48 Christensen MD, Everhart AW, Pickelman JT, et al. Mechanical and thermal allodynia in chronic central pain following spinal cord injury. Pain. 1996;68:97-107.
49 Basso DM, Beattie MS, Bresnahan JC. A sensitive and reliable locomotor rating scale for open field testing in rats. J Neurotrauma. 1995;12:1-21.
50 Bennett AD, Chastain KM, Hulsebosch CE. Alleviation of mechanical and thermal allodynia by CGRP(8-37) in a rodent model of chronic central pain. Pain. 2000;86:163-175.
51 Kim J, Jung JI, Na HS, et al. Effects of morphine on mechanical allodynia in a rat model of central neuropathic pain. Neuroreport. 2003;14:1017-1020.
52 Gwak YS, Tan HY, Nam TS, et al. Activation of spinal GABA receptors attenuates chronic central neuropathic pain after spinal cord injury. J Neurotrauma. 2006;23:1111-1124.
53 Hains BC, Everhart AW, Fullwood SD, et al. Changes in serotonin, serotonin transporter expression and serotonin denervation supersensitivity: involvement in chronic central pain after spinal hemisection in the rat. Exp Neurol. 2002;175:347-362.
54 Rosenzweig ES, McDonald JW. Rodent models for treatment of spinal cord injury: research trends and progress toward useful repair. Curr Opin Neurol. 2004;17:121-131.
55 Stokes BT, Jakeman LB. Experimental modelling of human spinal cord injury: a model that crosses the species barrier and mimics the spectrum of human cytopathology. Spinal Cord. 2002;40:101-109.
56 Hains BC, Yucra JA, Hulsebosch CE. Reduction of pathological and behavioral deficits following spinal cord contusion injury with the selective cyclooxygenase-2 inhibitor NS-398. J Neurotrauma. 2001;18:409-423.
57 Mills CD, Hains BC, Johnson KM, et al. Strain and model differences in behavioral outcomes after spinal cord injury in rat. J Neurotrauma. 2001;18:743-756.
58 Siddall P, Xu CL, Cousins M. Allodynia following traumatic spinal cord injury in the rat. Neuroreport. 1995;6:1241-1244.
59 Berrocal YA, Pearse DD, Andrade CM, et al. Increased spinal c-Fos expression with noxious and non-noxious peripheral stimulation after severe spinal contusion. Neurosci Lett. 2007;413:58-62.
60 Hubscher CH, Johnson RD. Chronic spinal cord injury induced changes in the responses of thalamic neurons. Exp Neurol. 2006;197:177-188.
61 Finnerup NB, Sorensen L, Biering-Sorensen F, et al. Segmental hypersensitivity and spinothalamic function in spinal cord injury pain. Exp Neurol. 2007;207:139-149.
62 Hulsebosch CE, Xu GY, Perez-Polo JR, et al. Rodent model of chronic central pain after spinal cord contusion injury and effects of gabapentin. J Neurotrauma. 2000;17:1205-1217.
63 Stolp-Smith KA, Wainberg MC. Antidepressant exacerbation of spasticity. Arch Phys Med Rehabil. 1999;80:339-342.
64 Robins S, Fehlings M. Models of experimental spinal cord injury: translational relevance and impact. Drug Discovery Today: Disease Models. 2008;5:5-11.
65 Hama A, Sagen J. Behavioral characterization and effect of clinical drugs in a rat model of pain following spinal cord compression. Brain Res. 2007;1185:117-128.
66 Hama A, Sagen J. Antinociceptive effects of the marine snail peptides conantokin-G and conotoxin MVIIA alone and in combination in rat models of pain. Neuropharmacology. 2009;56:556-563.
67 Xu XJ, Hao JX, Seiger A, et al. Systemic mexiletine relieves chronic allodynialike symptoms in rats with ischemic spinal cord injury. Anesth Analg. 1992;74:649-652.
68 Hama A, Sagen J. Antinociceptive effect of cannabinoid agonist WIN 55,212-2 in rats with a spinal cord injury. Exp Neurol. 2007;204:454-457.
69 Saulino M. Successful reduction of neuropathic pain associated with spinal cord injury via of a combination of intrathecal hydromorphone and ziconotide: a case report. Spinal Cord. 2007;45:749-752.
70 Finnerup NB, Otto M, McQuay HJ, et al. Algorithm for neuropathic pain treatment: an evidence based proposal. Pain. 2005;118:289-305.
71 Hao JX, Xu XJ, Aldskogius H, et al. Allodynia-like effects in rat after ischaemic spinal cord injury photochemically induced by laser irradiation. Pain. 1991;45:175-185.
72 Hao JX, Xu XJ. Treatment of a chronic allodynia-like response in spinally injured rats: effects of systemically administered excitatory amino acid receptor antagonists. Pain. 1996;66:279-285.
73 Hao JX, Xu XJ. Animal models of spinal cord injury pain and their implications for pharmacological treatments. J Rehabil Med. 2003;41(Suppl):81-84.
74 Hao JX, Xu XJ, Urban L, et al. Repeated administration of systemic gabapentin alleviates allodynia-like behaviors in spinally injured rats. Neurosci Lett. 2000;280:211-214.
75 Kouya PF, Hao JX, Xu XJ. Buprenorphine alleviates neuropathic pain-like behaviors in rats after spinal cord and peripheral nerve injury. Eur J Pharmacol. 2002;450:49-53.
76 Glynn CJ, Jamous MA, Teddy PJ, et al. Role of spinal noradrenergic system in transmission of pain in patients with spinal cord injury. Lancet. 1986;2:1249-1250.
77 Taira T, Kawamura H, Tanikawa T, et al. A new approach to control central deafferentation pain: spinal intrathecal baclofen. Stereotact Funct Neurosurg. 1995;65:101-105.
78 Sang CN, Booher S, Gilron I, et al. Dextromethorphan and memantine in painful diabetic neuropathy and postherpetic neuralgia: efficacy and dose-response trials. Anesthesiology. 2002;96:1053-1061.
79 Eide PK, Stubhaug A, Stenehjem AE. Central dysesthesia pain after traumatic spinal cord injury is dependent on N-methyl-D-aspartate receptor activation. Neurosurgery. 1995;37:1080-1087.
80 Falci S, Best L, Bayles R, et al. Dorsal root entry zone microcoagulation for spinal cord injury-related central pain: operative intramedullary electrophysiological guidance and clinical outcome. J Neurosurg. 2002;97(2 Suppl):193-200.
81 Loeser JD, Ward AAJr, White LEJr. Chronic deafferentation of human spinal cord neurons. J Neurosurg. 1968;29:48-50.
82 Drew GM, Siddall PJ, Duggan AW. Mechanical allodynia following contusion injury of the rat spinal cord is associated with loss of GABAergic inhibition in the dorsal horn. Pain. 2004;109:379-388.
83 Wang J, Kawamata M, Namiki A. Changes in properties of spinal dorsal horn neurons and their sensitivity to morphine after spinal cord injury in the rat. Anesthesiology. 2005;102:152-164.
84 Yezierski RP, Park SH. The mechanosensitivity of spinal sensory neurons following intraspinal injections of quisqualic acid in the rat. Neurosci Lett. 1993;157:115-119.
85 Wrigley PJ, Press SR, Gustin SM, et al. Neuropathic pain and primary somatosensory cortex reorganization following spinal cord injury. Pain. 2009;141:52-59.
86 Pattany PM, Yezierski RP, Widerstrom-Noga EG, et al. Proton magnetic resonance spectroscopy of the thalamus in patients with chronic neuropathic pain after spinal cord injury. AJNR Am J Neuroradiol. 2002;23:901-905.
87 Lenz FA, Tasker RR, Dostrovsky JO, et al. Abnormal single-unit activity recorded in the somatosensory thalamus of a quadriplegic patient with central pain. Pain. 1987;31:225-236.
88 Christensen MD, Hulsebosch CE. Spinal cord injury and anti-NGF treatment results in changes in CGRP density and distribution in the dorsal horn in the rat. Exp Neurol. 1997;147:463-475.
89 Weaver LC, Verghese P, Bruce JC, et al. Autonomic dysreflexia and primary afferent sprouting after clip-compression injury of the rat spinal cord. J Neurotrauma. 2001;18:1107-1119.
90 Ackery AD, Norenberg MD, Krassioukov A. Calcitonin gene-related peptide immunoreactivity in chronic human spinal cord injury. Spinal Cord. 2007;45:678-686.
91 Grachev ID, Fredrickson BE, Apkarian AV. Brain chemistry reflects dual states of pain and anxiety in chronic low back pain. J Neural Transm. 2002;109:1309-1334.
92 Chang HT. Subacute human spinal cord contusion: few lymphocytes and many macrophages. Spinal Cord. 2007;45:174-182.
93 Borsook D, Becerra L. Phenotyping central nervous system circuitry in chronic pain using functional MRI: considerations and potential implications in the clinic. Curr Pain Headache Rep. 2007;11:201-207.
94 Darian-Smith C. Synaptic plasticity, neurogenesis, and functional recovery after spinal cord injury. Neuroscientist. 2009;15:149-165.
95 Ji RR, Suter MR. p38 MAPK, microglial signaling, and neuropathic pain. Mol Pain. 2007;3:33.
96 Baastrup C, Finnerup NB. Pharmacological management of neuropathic pain following spinal cord injury. CNS Drugs. 2008;22:455-475.
97 Watkins LR, Hutchinson MR, Ledeboer A, et al. Norman Cousins Lecture. Glia as the “bad guys”: implications for improving clinical pain control and the clinical utility of opioids. Brain Behav Immun. 2007;21:131-146.
98 Binshtok AM, Wang H, Zimmermann K, et al. Nociceptors are interleukin-1beta sensors. J Neurosci. 2008;28:14062-14073.
99 Cramer SW, Baggott C, Cain J, et al. The role of cation-dependent chloride transporters in neuropathic pain following spinal cord injury. Mol Pain. 2008;4:36.
100 Fu ES, Zhang YP, Sagen J, et al. Transgenic glial nuclear factor-kappa B inhibition decreases formalin pain in mice. Neuroreport. 2007;18:713-717.
101 Yamamoto Y, Gaynor RB. Therapeutic potential of inhibition of the NF-kappaB pathway in the treatment of inflammation and cancer. J Clin Invest. 2001;107:135-142.
102 Kang SK, So HH, Moon YS, et al. Proteomic analysis of injured spinal cord tissue proteins using 2-DE and MALDI-TOF MS. Proteomics. 2006;6:2797-2812.
103 Ding Q, Wu Z, Guo Y, et al. Proteome analysis of up-regulated proteins in the rat spinal cord induced by transection injury. Proteomics. 2006;6:505-518.
104 Velardo MJ, Burger C, Williams PR, et al. Patterns of gene expression reveal a temporally orchestrated wound healing response in the injured spinal cord. J Neurosci. 2004;24:8562-8576.
105 Nesic O, Lee J, Johnson KM, et al. Transcriptional profiling of spinal cord injury-induced central neuropathic pain. J Neurochem. 2005;95:998-1014.
106 Li S, Zhang J. Lipopolysaccharide induces autotaxin expression in human monocytic THP-1 cells. Biochem Biophys Res Commun. 2009;378:264-268.
107 Inoue M, Ma L, Aoki J, et al. Autotaxin, a synthetic enzyme of lysophosphatidic acid (LPA), mediates the induction of nerve-injured neuropathic pain. Mol Pain. 2008;4:6.
108 Inoue M, Rashid MH, Fujita R, et al. Initiation of neuropathic pain requires lysophosphatidic acid receptor signaling. Nat Med. 2004;10:712-718.
109 Ueda H. Peripheral mechanisms of neuropathic pain—involvement of lysophosphatidic acid receptor-mediated demyelination. Mol Pain. 2008;4:11.
110 Shano S, Moriyama R, Chun J, et al. Lysophosphatidic acid stimulates astrocyte proliferation through LPA1. Neurochem Int. 2008;52:216-220.
111 Coste O, Pierre S, Marian C, et al. Antinociceptive activity of the S1P-receptor agonist FTY720. J Cell Mol Med. 2008;12:995-1004.
112 de Jong EK, Dijkstra IM, Hensens M, et al. Vesicle-mediated transport and release of CCL21 in endangered neurons: a possible explanation for microglia activation remote from a primary lesion. J Neurosci. 2005;25:7548-7557.
113 de Jong EK, Vinet J, Stanulovic VS, et al. Expression, transport, and axonal sorting of neuronal CCL21 in large dense-core vesicles. Faseb J. 2008;22:4136-4145.
114 Zhao P, Waxman SG, Hains BC. Modulation of thalamic nociceptive processing after spinal cord injury through remote activation of thalamic microglia by cysteine cysteine chemokine ligand 21. J Neurosci. 2007;27:8893-8902.
115 Neary JT, Kang Y, Shi YF. Cell cycle regulation of astrocytes by extracellular nucleotides and fibroblast growth factor-2. Purinergic Signal. 2005;1:329-336.
116 Sawynok J, Liu XJ. Adenosine in the spinal cord and periphery: release and regulation of pain. Prog Neurobiol. 2003;69:313-340.
117 Byrnes KR, Stoica BA, Fricke S, et al. Cell cycle activation contributes to post-mitotic cell death and secondary damage after spinal cord injury. Brain. 2007;130(Pt 11):2977-2992.
118 Diaz-Padilla I, Siu LL, Duran I. Cyclin-dependent kinase inhibitors as potential targeted anticancer agents. Invest New Drugs. 2009.
119 Sekine C, Sugihara T, Miyake S, et al. Successful treatment of animal models of rheumatoid arthritis with small-molecule cyclin-dependent kinase inhibitors. J Immunol. 2008;180:1954-1961.
120 Utreras E, Futatsugi A, Rudrabhatla P, et al. Tumor necrosis factor-alpha regulates cyclin-dependent kinase 5 activity during pain signaling through transcriptional activation of p35. J Biol Chem. 2009;284:2275-2284.
121 Saikkonen B, Pareek TK, Agarwal N, et al. Conditional deletion of cyclin-dependent kinase 5 in primary sensory neurons leads to atypical skin lesions. Cell Cycle. 2008;7:750-753.
122 Pareek TK, Kulkarni AB. Cdk5: a new player in pain signaling. Cell Cycle. 2006;5:585-588.
123 Pareek TK, Keller J, Kesavapany S, et al. Cyclin-dependent kinase 5 modulates nociceptive signaling through direct phosphorylation of transient receptor potential vanilloid 1. Proc Natl Acad Sci U S A. 2007;104:660-665.
124 Yang YR, He Y, Zhang Y, et al. Activation of cyclin-dependent kinase 5 (Cdk5) in primary sensory and dorsal horn neurons by peripheral inflammation contributes to heat hyperalgesia. Pain. 2007;127:109-120.
125 Rossi AG, Sawatzky DA, Walker A, et al. Cyclin-dependent kinase inhibitors enhance the resolution of inflammation by promoting inflammatory cell apoptosis. Nat Med. 2006;12:1056-1064.
126 Mizushima T, Obata K, Katsura H, et al. Intensity-dependent activation of extracellular signal-regulated protein kinase 5 in sensory neurons contributes to pain hypersensitivity. J Pharmacol Exp Ther. 2007;321:28-34.
127 Gwak YS, Crown ED, Unabia GC, et al. Propentofylline attenuates allodynia, glial activation and modulates GABAergic tone after spinal cord injury in the rat. Pain. 2008;138:410-422.
128 Numagami Y, Marro PJ, Mishra OP, et al. Effect of propentofylline on free radical generation during cerebral hypoxia in the newborn piglet. Neuroscience. 1998;84:1127-1133.
129 Kehlen A, Lauterbach R, Santos AN, et al. IL-1 beta- and IL-4-induced down-regulation of autotaxin mRNA and PC-1 in fibroblast-like synoviocytes of patients with rheumatoid arthritis (RA). Clin Exp Immunol. 2001;123:147-154.
130 Ledeboer A, Hutchinson MR, Watkins LR, et al. Ibudilast (AV-411). A new class therapeutic candidate for neuropathic pain and opioid withdrawal syndromes. Expert Opin Investig Drugs. 2007;16:935-950.
131 Agre P, Preston GM, Smith BL, et al. Aquaporin CHIP: the archetypal molecular water channel. Am J Physiol. 1993;265(4 Pt 2):F463-F476.
132 Jessica Chen M, Sepramaniam S, Armugam A, et al. Water and ion channels: crucial in the initiation and progression of apoptosis in central nervous system? Curr Neuropharmacol. 2008;6:102-116.
133 Nesic O, Lee J, Unabia GC, et al. Aquaporin 1—a novel player in spinal cord injury. J Neurochem. 2008;105:628-640.
134 Warner JS, Wamil AW, McLean MJ. Acetazolamide for the treatment of chronic paroxysmal hemicrania. Headache. 1994;34:597-599.
135 Woehlck HJ, Otterson M, Yun H, et al. Acetazolamide reduces referred postoperative pain after laparoscopic surgery with carbon dioxide insufflation. Anesthesiology. 2003;99:924-928.
136 Faulkner JR, Herrmann JE, Woo MJ, et al. Reactive astrocytes protect tissue and preserve function after spinal cord injury. J Neurosci. 2004;24:2143-2155.
137 Rolls A, Shechter R, Schwartz M. The bright side of the glial scar in CNS repair. Nat Rev Neurosci. 2009;10:235-241.
138 Okada S, Nakamura M, Mikami Y, et al. Blockade of interleukin-6 receptor suppresses reactive astrogliosis and ameliorates functional recovery in experimental spinal cord injury. J Neurosci Res. 2004;76:265-276.
139 Marchand F, Tsantoulas C, Singh D, et al. Effects of Etanercept and Minocycline in a rat model of spinal cord injury. Eur J Pain. 2009;13:673-681.
140 Peng XM, Zhou ZG, Glorioso JC, et al. Tumor necrosis factor-alpha contributes to below-level neuropathic pain after spinal cord injury. Ann Neurol. 2006;59:843-851.
141 Diguet E, Gross CE, Tison F, et al. Rise and fall of minocycline in neuroprotection: need to promote publication of negative results. Exp Neurol. 2004;189:1-4.
142 Gordon PH, Moore DH, Miller RG, et al. Efficacy of minocycline in patients with amyotrophic lateral sclerosis: a phase III randomised trial. Lancet Neurol. 2007;6:1045-1053.
143 Erichsen HK, Hao JX, Xu XJ, et al. Comparative actions of the opioid analgesics morphine, methadone and codeine in rat models of peripheral and central neuropathic pain. Pain. 2005;116:347-358.
144 Guneli E, Karabay Yavasoglu NU, Apaydin S, et al. Analysis of the antinociceptive effect of systemic administration of tramadol and dexmedetomidine combination on rat models of acute and neuropathic pain. Pharmacol Biochem Behav. 2007;88:9-17.
145 Medvedev IO, Malyshkin AA, Belozertseva IV, et al. Effects of low-affinity NMDA receptor channel blockers in two rat models of chronic pain. Neuropharmacology. 2004;47:175-183.
146 Patel S, Naeem S, Kesingland A, et al. The effects of GABA(B) agonists and gabapentin on mechanical hyperalgesia in models of neuropathic and inflammatory pain in the rat. Pain. 2001;90:217-226.