[level-membership-for-otolaryngology-category]
CHAPTER 139 Chronic Otitis Media, Mastoiditis, and Petrositis
Some cases of acute otitis media (AOM) result in persistent otitis media with effusion (OME), which is recognized as the leading cause of childhood hearing loss. The exact cause of OME is unclear; however, more recent data suggest that reflux of gastric contents may be associated with OME in children.1–4 Although eustachian tube dysfunction alone may lead to effusion of the middle ear, there is mounting evidence that most cases of OME occur as a sequelae of AOM, or at least share the same etiologic factors. Specific causes can be identified in many cases of adult-onset otitis media, such as paranasal sinus disease, nasopharyngeal carcinomas and tumors, and postradiation sequelae.
Effects on Mastoid Pneumatization
It has been observed that patients with a history of chronic OME have more sclerotic mastoids with decreased pneumatization compared with healthy subjects. There are two theories to explain this observation: (1) the hereditary theory, which states that children with hypoaeration of the mastoid are prone to OME, and (2) the environmental theory, which states that chronic OME results in hypopneumatization of the mastoid.5 Although measurable correlations between mastoid hypocellularity and OME,6,7 and between the length of the mastoid process or the degree of pneumatization and an abnormal eardrum8 have been proven, a cause-and-effect relationship is unclear. Available evidence generally supports the concept that chronic inflammation in early childhood may lead to new bone formation within the middle ear and mastoid and, subsequently, decreased size of mastoid air cells. Shatz and Sadé9 measured the distance from the lateral sinus to the external auditory canal and found it to be significantly smaller in patients with sclerotic mastoids; they believed that this finding supported the hereditary theory because it was unlikely that otitis would change the position of the lateral sinus. A chronically inflamed mastoid in a young child may not develop normally, however.
Hasebe and colleagues10 carried out a study to establish which type of cholesteatoma is controllable by conservative treatment from the viewpoint of mastoid ventilation. Their clinical observations suggest that progressiveness of cholesteatoma is related to the ventilatory conditions in the mastoid, rather than eustachian tube function, and that conservative treatment may be effective if ears with cholesteatoma have aeration in the mastoid.
Middle Ear Atelectasis and Adhesive Otitis Media
Middle ear atelectasis (Fig. 139-1) is thought to result mainly from long-standing eustachian tube dysfunction. One of the main functions of the eustachian tube is ventilation of the middle ear and mastoid. Opening of the eustachian tube allows exchanging of gases and equalization between the environment and middle ear. The middle ear gases also are exchanged with the middle ear mucosa. Bilateral diffusion between the middle ear cavity and the blood may be an important factor in middle ear atelectasis because the gas composition of the middle ear basically resembles that of venous blood.11
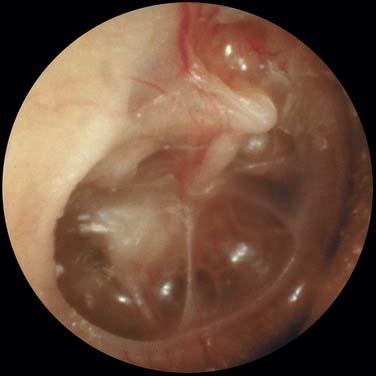
If the atelectasis develops, the tympanic membrane becomes retracted onto the promontory and the ossicles of the middle ear. In atelectatic ears, the middle ear space is partially or completely obliterated, but the tympanic membrane is not adherent to the medial wall of the middle ear, and the mucosal lining of the middle ear is intact. In contrast, adhesive otitis media exists when the middle ear space is totally obliterated, and the tympanic membrane is adherent to the ossicles and promontory; mucosal surfaces are not present. Retraction of the tympanic membrane may lead to erosion of the long process of the incus and the stapes suprastructure (Fig. 139-2).
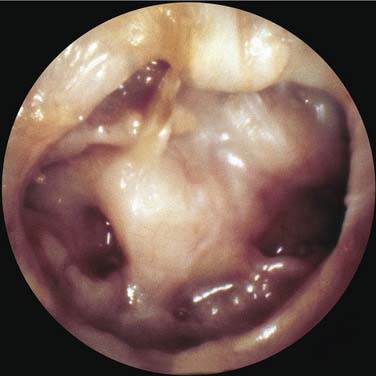
Not all patients with chronic OME develop atelectasis; in most patients with OME, retraction of the tympanic membrane is limited. In patients with bilateral OME, 1.5% of untreated ears and 2% of ears treated with tubes developed severe atelectasis.12 It may be that repeated bouts of AOM lead to weakening and thinning of the membrane, which allows atelectasis. Sadé and Berco13 showed destruction of the collagen-containing fibrous layer of the tympanic membrane in some ears with recurrent infection. Collagen destruction within the tympanic membrane may lead to another complication of OME—tympanosclerosis. Sadé and Berco13 and Tos and Poulsen14 described four stages of tympanic membrane retraction: stage I, retracted tympanic membrane; stage II, retraction with contact onto the incus; stage III, middle ear atelectasis; and stage IV, adhesive otitis media (Fig. 139-3).
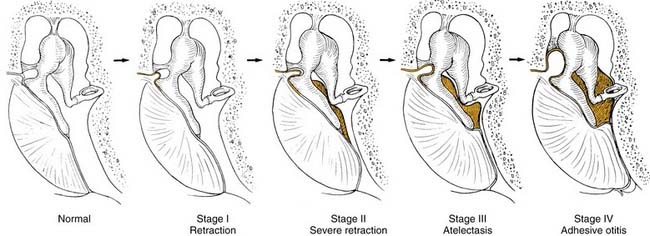
Figure 139-3. Four stages of middle ear atelectasis.
(Adapted from Sadé J, Berco E. Atelectasis and secretory otitis media. Ann Otol Rhinol Laryngol. 1976;85(Suppl 25):66.)
Middle ear atelectasis may be reversible with ventilating tubes. Sadé15 showed that ventilating tubes improved the state of atelectatic ears. Graham and Knight16 reported three cases in which atelectatic tympanic membranes were restored to their normal position by administration of nitrous oxide during anesthesia and insertion of a ventilating tube.
Atelectasis and adhesive otitis media usually coexist with OME, although OME may resolve in these ears, allowing aeration of the attic and mastoid, but leaving a collapsed middle ear. In extreme cases, when hearing loss or ossicular erosion occurs, a myringoplasty for the reinforcement of atelectatic tympanic membrane may be indicated.6,17 Cholesteatomas may originate from deep retraction pockets in which desquamated keratin debris would not be cleared into the ear canal.18,19 These retraction pockets may occur in the pars tensa or pars flaccida of atelectatic ears, and should be considered precursors to cholesteatomas (see discussion of cholesteatoma). Nonpneumatized mastoids may have a limited ability to buffer pressure changes and can manifest as an atelectasis, a retraction pocket, or a cholesteatoma.20
Chronic Otitis Media with Cholesteatoma
Aural cholesteatomas are epidermal inclusion cysts of the middle ear or mastoid (in the case of a retraction pocket cholesteatoma, the “cyst” opens into the external auditory canal). They contain the desquamated debris (principally keratin) from their keratinizing, squamous epithelial lining. Cruveilhier21 first described aural cholesteatoma as a “pearly tumor” of the temporal bone. The term cholesteatoma, coined by the German physiologist Müller22 in 1838, is a misnomer because this entity does not contain cholesterol; the white-yellow keratin flakes found within cholesteatomas grossly resemble cholesterol crystals. Cholesteatomas of the temporal bone may be congenital or acquired. Acquired cholesteatomas are the consequence of OME, AOM, or both. An understanding of the pathogenesis and pathophysiology of aural cholesteatoma is particularly important because the destructive nature of this entity is responsible for much of the morbidity associated with chronic otitis media. The propensity of cholesteatomas to erode bone and the lack of effective, nonsurgical management add importance to the understanding of this disease.
Diagnosis
The diagnosis of aural cholesteatoma is made on otoscopic examination, including endoscopic and microscopic evaluation or surgical exploration. Special imaging procedures, such as high-resolution computed tomography (CT) and magnetic resonance imaging (MRI), may suggest the presence of cholesteatomas within the temporal bone, and may be used to complement the clinical examination. High-resolution CT is useful for operative planning and is recommended for all revision mastoid operations. The symptoms of cholesteatoma vary; some cholesteatomas are asymptomatic, whereas others become infected and rapidly cause bone destruction. Some patients seen by the physician show slowly progressive conductive hearing loss, and frequently patients with cholesteatomas consult a physician because of chronic otitis with purulent otorrhea. The otorrhea from an infected cholesteatoma often is malodorous because of the frequent infection with anaerobic bacteria.23
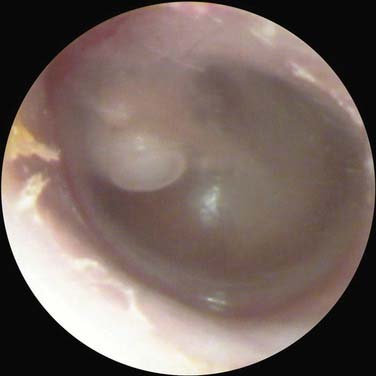
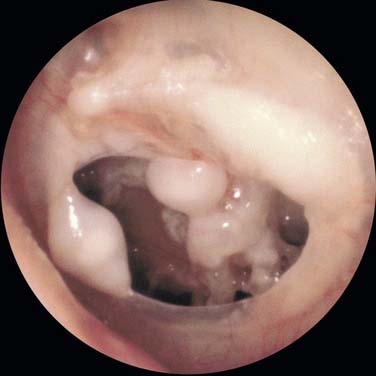
The otoscopic appearance of an aural cholesteatoma also varies. A typical attic retraction cholesteatoma (Fig. 139-4) appears as a defect of variable size adjacent to the posterosuperior portion of the tympanic membrane. The center of the defect contains keratin debris (primary acquired cholesteatoma). In other patients, keratinizing epithelium has migrated through a perforation into the middle ear (secondary acquired cholesteatoma) (Fig. 139-5). Cholesteatomas sometimes appear behind or within an intact tympanic membrane—so-called congenital cholesteatoma (Fig. 139-6). These cholesteatomas have been considered to be congenital, but more recent experimental evidence raises the possibility that they may originate during an inflammatory process.24 An infected cholesteatoma sometimes manifests as an “aural polyp.” These “polyps” actually are granulation tissue at the junction between an eroding cholesteatoma and bone. The presence of an aural polyp in a chronically infected ear should be considered to be a cholesteatoma until proved otherwise. Occasionally, a cholesteatoma cannot be seen otoscopically, but is discovered during tympanomastoid surgery.
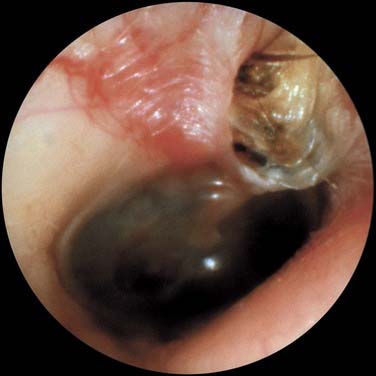
Figure 139-4. Primary acquired cholesteatoma in the region of the pars flaccida with scutum erosion.
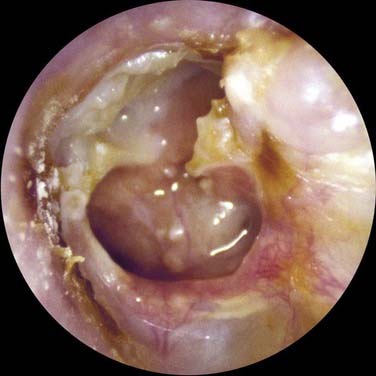
Figure 139-5. Cholesteatoma developing at the margin of perforation (secondary acquired cholesteatoma) with secondary infection.
The exact prevalence of cholesteatoma is unknown. In 1978, there were 4.2 hospital discharges per 100,000 population with cholesteatoma.25 In addition, there were 13.8 hospital discharges per 100,000 population with chronic otitis media without cholesteatoma. Studies by Harker23 documented an annual incidence of 6 cholesteatomas per 100,000 population. Tos26 found an annual incidence of 3 cholesteatomas in children and 12.6 cholesteatomas in adults per 100,000 population. In the human temporal bones with chronic otitis media, cholesteatoma was observed in 36% of ears with perforations and in 4% of ears without the perforated tympanic membranes.27
Pathogenesis
Congenital cholesteatomas, by definition, originate from areas of keratinizing epithelium within the middle ear cleft. Michaels28 showed that a small area in the anterior tympanum in the developing fetus often contains a small area of keratinizing epithelium. He found epidermoid formation in 37 of 68 temporal bones of fetuses at 10 to 33 weeks’ gestation. Congenital cholesteatomas may originate in this region. Although congenital cholesteatomas generally are seen as pearl-like masses behind an intact membrane, Koltai and coworkers29 presented a convincing case that some congenital cholesteatomas advance to perforate and become chronically infected, taking on the appearance of an acquired cholesteatoma. Potsic and colleagues,30 in a review of 172 series of congenital cholesteatomas, developed a useful staging system: stage I, limited to one quadrant; stage II, involving multiple quadrants without ossicular involvement; stage III, ossicular involvement without mastoid extension; and stage IV, mastoid involvement. They showed a correlation between stage and risk of residual disease; stage IV carries a 67% risk of residual cholesteatoma.
The pathogenesis of acquired cholesteatoma has been debated for more than a century. There are four basic theories of the pathogenesis of acquired aural cholesteatoma: (1) invagination of the tympanic membrane (retraction pocket cholesteatoma), (2) basal cell hyperplasia, (3) epithelial ingrowth through a perforation (the migration theory), and (4) squamous metaplasia of middle ear epithelium (Fig. 139-7). Additionally, Sudhoff and Tos19 proposed a combination of the invagination and basal cell theories as an explanation for retraction pocket cholesteatoma formation.
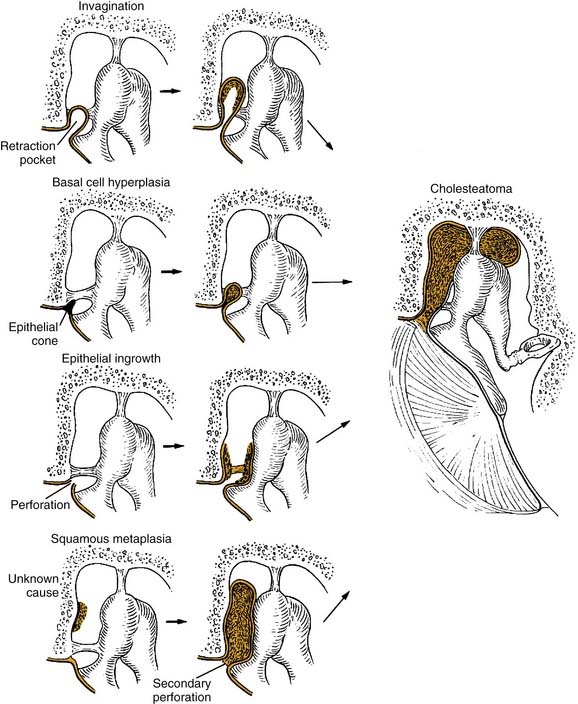
Invagination Theory
The invagination theory5 of the genesis of cholesteatoma generally is regarded as one of the primary mechanisms of the formation of attic cholesteatomas. Retraction pockets of the pars flaccida deepen because of negative middle ear pressure and possibly repeated inflammation (see Fig. 139-2). As the retraction pocket deepens, desquamated keratin cannot be cleared from the recess, and a cholesteatoma results. The origin of such retraction pocket cholesteatomas is thought to be eustachian tube dysfunction (or OME) with resultant negative middle ear pressure (ex vacuo theory). Usually, the pars flaccida, being less fibrous and less resistant to displacement, is the source of the cholesteatoma.
The result of this type of cholesteatoma (so-called primary acquired cholesteatoma) is an apparent defect in the posterosuperior quadrant of the tympanic membrane and erosion of the adjacent canal wall.31 Although these defects have the appearance of a marginal perforation, it is not a perforation, but rather an invagination. Sadé20 showed that epithelial migration patterns within attic retraction pockets are altered. This failure of epithelial migration may allow the accumulation of keratin within a retraction pocket, with subsequent enlargement merely from the accumulation of keratin within a relatively closed space. This theory has been supported by the experimental creation of retraction pockets by use of the eustachian tube obstruction,32 and external auditory canal ligation.33
Ruah and colleagues31 suggested that inflammation of middle ear and persistent mesenchyme lead to a greater inflammatory reaction in pars flaccida and posterosuperior quadrant of the tympanic membrane of the human temporal bones with serous and purulent otitis media in children. These findings support this theory of primary acquired cholesteatoma in children. Retraction pockets are considered as precursors to cholesteatoma. Bacteria can infect the keratin matrix, forming biofilms leading to chronic persistent infection. The presence of bacterial biofilms in the cholesteatoma matrix may lead to epithelial proliferation and invasion of the cholesteatoma.34,35
Basal Cell Hyperplasia Theory
Another possible mechanism for the histogenesis of cholesteatoma was suggested by Lange in the 1920s.49 In this theory, he proposed that epithelial cells (prickle cells) of the pars flaccida could invade the subepithelial tissue by means of proliferating columns of epithelial cells. Nearly 40 years later, Ruedi50 supported this hypothesis with clinical and experimental evidence. For epithelium to invade into the lamina propria, the basal lamina (basement membrane) should be altered. Basal lamina disruptions now have been documented in human51,52 and animal53 cholesteatomas.
Huang and colleagues54 and Masaki and coworkers55 provided experimental support of this theory by showing that epithelial ingrowth from the tympanic membrane can be induced by instillation of propylene glycol into the middle ear of chinchillas. These basal lamina breaks allow the invasion of epithelial cones into the subepithelial connective tissue and the formation of microcholesteatomas. This mechanism may explain some types of human cholesteatomas, even those occurring behind an intact tympanic membrane.56 According to this theory, microcholesteatomas may enlarge and then perforate secondarily through the tympanic membrane, leaving the typical appearance of an attic cholesteatoma. This sequence of events has not been documented, although the alternations in the differentiation of keratinocytes and basal cell layer of cholesteatoma matrix have been observed in several studies.
Abnormal distribution of epidermal differentiation markers, such as filaggrin and involucrin,57 c-jun and p53 proteins,58 and increased epidermal growth factor receptor,59,60 has been shown in middle ear cholesteatoma matrix. Increased levels of proteins cytokeratin 13 and cytokeratin 16, which are markers for differentiation and hyperproliferation, were also found.61 Kim and Chole39 showed the increased expression of cytokeratin 13 and cytokeratin 16 in the area of the peripheral area of the pars tensa of induced cholesteatoma by ear canal ligation and in the peripheral and central area of pars tensa of induced cholesteatoma by eustachian tube obstruction. Sakamoto and associates62 found ErbB-2 protein to be overexpressed, and cell proliferation and apoptosis of keratinocytes accelerated. Caspases play a key role in apoptosis; Miyao and coworkers63 suggested that caspase-8, which is activated by the induction of tumor necrosis factor-α, leads to activation of caspase-3, which activates apoptotic nucleases in cholesteatoma tissue.
Human toll-like receptors are crucial in the induction and activation of innate immunity in the course of an infection. Expression of toll-like receptor–3 on the epithelium and some cells within the perimatrix and the presence of T cells may suggest that apart from innate immune responses, mechanisms of adaptive immunity also operate in cholesteatoma.64 Data from Parisier and colleagues65 suggested that fibroblasts in the subepithelium of cholesteatomas showed an invasive phenotype, whereas fibroblasts from postauricular and ear canal skin appeared either weakly invasive or not invasive. In a similar study, Chole and colleagues66 found that normal fibroblasts and fibroblasts from induced cholesteatomas did not exhibit the invasive phenotype characteristics of true neoplastic cells.
Other lines of evidence support the basal cell hyperplasia/migration theory. Increased expression of human intercellular adhesion molecule-1 and intercellular adhesion molecule-2 has been shown, suggesting a role in cell migration into tissue.40 The presence of heat shock proteins 60 and 70 suggested proliferation and active differentiation of basal keratinocytes in cholesteatomas.58 There are some reports that immune response is involved in the hyperproliferative state of cholesteatoma epithelium.52,67,68 Langerhans cells may initiate immune reaction and promote proliferation of keratinizing epithelium via an interleukin (IL)-1α and transforming growth factor (TGF)-β mechanism.42,67,69
Epithelial Invasion Theory
The epithelial invasion theory36 states that keratinizing squamous epithelium from the surface of the tympanic membrane invades or migrates into the middle ear from a perforation in the tympanic membrane. This theory is supported by clinical observation and experimental evidence. Weiss37 showed that epithelial cells could migrate along a surface by a process that he called contact guidance, and that, when they encounter another epithelial surface, they stop migrating, for which he used the term contact inhibition. van Blitterswijk and Grote38 reported that cytokeratin 10, which was seen in the meatal epidermis and migrating epithelium, was preferentially expressed in the cholesteatoma matrix, rather than in the middle ear mucosa. This finding suggests an epidermal origin of cholesteatoma. Kim and Chole39 showed increased cytokeratin 10 expression in the peripheral area of the pars tensa of induced cholesteatoma by ear canal ligation and in the peripheral and the central area of the pars tensa of induced cholesteatoma by eustachian tube obstruction. The findings of this study also support the basal cell hyperplasia hypotheses for the pathogenesis of aural cholesteatoma, with regard to hyperproliferation, migration, and an altered differentiation of keratinocytes.39 High levels of fibronectin and tenascin and focal disruptions of the basement membrane, were reported in the middle ear cholesteatoma, supporting the concept of the invasion theory.40–43
This theory also is supported by investigations in animal cholesteatoma models and human temporal bones. Jackson and Lim44 gave histologic and ultrastructural evidence that keratinizing epithelium can migrate into the cat bulla by contact guidance. It is likely that in some tympanic membrane perforations, inflammation damages the inner mucosal lining of the tympanic membrane, allowing the outer keratinizing epithelium to migrate inward and generate a cholesteatoma. Hueb and associates45 showed similar evidence in chinchillas. Palva and colleagues46 showed histologic evidence for this theory in human temporal bones. Cholesteatomas originating after temporal bone fractures may result from this mechanism; fractures within the ear canal may allow ingrowth of keratinizing epithelium by contact guidance.47 7,12-Dimethylbenz[a]anthracene, a chemical carcinogen, could induce the advancing of the keratinizing squamous epithelium into or under the mucosal layer, and ingrowth and spreading over the middle ear cavity and eustachian tube in the rat ear.48
Squamous Metaplasia Theory
Wendt70 theorized that the simple squamous or cuboidal epithelium of the middle ear cleft could undergo a metaplastic transformation into keratinizing epithelium. Sadé15,71 supported this theory, noting that epithelial cells are pluripotent and can be stimulated by inflammation to become keratinizing. According to this theory, an area of keratinizing epithelium within the middle ear would enlarge because of accumulated debris and contact with the tympanic membrane. With intercurrent infection and inflammation, the cholesteatoma would lead to lysis of the tympanic membrane and perforation, resulting in the typical appearance of an attic cholesteatoma. This theory is supported by the demonstration that biopsy specimens from the middle ear of children with OME sometimes contain islands of keratinizing epithelium.15
Some experimental evidence supports the contention that middle ear mucosa can become metaplastic and keratinize. Chole and Frush72 showed that extreme vitamin A deficiency leads to the formation of keratinizing epithelium within the middle ear and eustachian tube of rats. None of their experimental animals had developed cholesteatomas. Consequently, there is no direct evidence that cholesteatomas arise by squamous metaplasia of the middle ear mucosa.
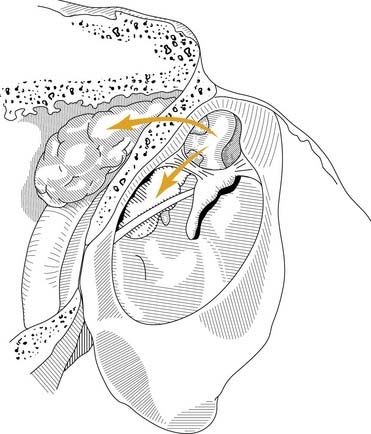
From a clinical perspective, it seems that each of these pathogenic mechanisms accounts for a proportion of acquired cholesteatomas. Regardless of the pathogenesis of aural cholesteatomas, they all share certain properties. Cholesteatomas are prone to recurrent infection, and they characteristically erode the bone of the ossicles and the otic capsule. Aural cholesteatomas originating from the vicinity of the tympanic membrane exhibit typical growth patterns into the temporal bone. Because most acquired cholesteatomas originate by invagination of the pars flaccida, their growth is limited by the mucosal folds and suspensory ligaments of the ossicles. The pars flaccida may invaginate into the lateralmost portion of the epitympanum (Prussak’s space) and then into the recesses of the epitympanum posteriorly, lateral to the body of the incus, inferiorly into the middle ear by way of the pouch of von Tröltsch (Fig. 139-8), or anteriorly into the protympanum (Fig. 139-9).73–75
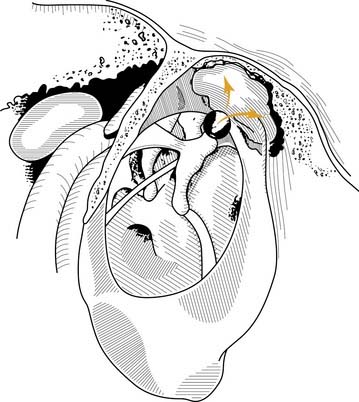
Complications
The expansion of cholesteatoma may result in bone erosion of the ossicles, otic capsule, fallopian canal, tegmen tympani, and tegmen mastoideum. These complications may cause intracranial complications (Box 139-1). The erosion of ossicles, most commonly in the incus, may result in conductive hearing loss. The severity of hearing loss is related to the status of the ossicles and the position of the cholesteatoma sac. Erosion of the otic capsule occurs most commonly in the lateral semicircular canal and rarely in the cochlea. A labyrinthine fistula has been reported in 10% of cholesteatomas in adults and children,76 which may result in sensorineural hearing loss and vertigo. Sensorineural hearing loss may result from the secondary suppurative labyrinthitis or from the cochlear hair cell loss adjacent to cholesteatoma.66 Facial nerve paralysis may occur acutely as a result of infection or slowly as a result of expansion of the cholesteatoma. Erosion of the tegmen tympani or the tegmen mastoideum may lead to development of a brain hernia or cerebrospinal fluid leakage.77
Because cholesteatomas contain keratin debris enclosed in a tissue space, they are subject to recurrent infection. The bacteria found in infected cholesteatomas differ from bacteria found in AOM or OME. Significant anaerobic bacteria are present. The most common aerobic bacterium is Pseudomonas aeruginosa, and the most common anaerobic microorganism is Bacteroides sp (Table 139-1).78
Table 139-1 Bacteriology of Infected Cholesteatomas in 30 Children and Adults
| Bacteria | No. of Cases |
|---|---|
| Aerobes | |
| Pseudomonas aeruginosa | 11 |
| Pseudomonas fluorescens | 2 |
| Streptococcus sp | 8 |
| Proteus sp | 4 |
| Escherichia coli | 4 |
| Klebsiella, Enterobacter, Serratia | 4 |
| Alcaligenes, Achromobacter | 3 |
| Staphylococcus epidermidis | 2 |
| Staphylococcus aureus | 1 |
| CBC group F | |
| Anaerobes | |
| Bacteroides sp | 13 |
| Peptococcus, Peptostreptococcus | 11 |
| Propionibacterium acnes | 8 |
| Fusobacterium sp | 4 |
| Bifidobacterium sp | 3 |
| Clostridium sp | 3 |
| Eubacterium sp | 2 |
Modified from Harker LA, Koontz FP. The bacteriology of cholesteatomas. In: McCabe BF, Sadé J, Abramson M, eds. Cholesteatoma: First International Conference. New York: Aesculapius Publishers; 1977.
Management
Congenital and acquired cholesteatomas can be eradicated from the temporal bone only by surgical resection. The goals of surgery are to eradicate disease and manage complications and, secondarily, to reconstruct the middle ear. The decision whether to perform surgery depends on the nature and extent of disease, the existence of complications, mastoid pneumatization, eustachian tube function, hearing status of both ears, the reliability of the patient, and the experience and skill of the surgeon.77,79 Surgical approaches include atticotomy, simple mastoidectomy, canal wall-up or canal wall-down procedures, radical mastoidectomy, modified radical mastoidectomy, and Bondy procedure.
The open (canal wall-down) and the closed (intact canal with facial recess) procedures have advantages and disadvantages (Table 139-2). The reported results of both procedures vary. Residual disease and recurrent disease occur in 11% to 27% and 5% to 13% of patients undergoing the closed procedure, whereas residual or recurrent disease occurs in 2% to 10% of patients undergoing the open procedure.80 In the cases of labyrinthine fistula, facial nerve paralysis, and intracranial complications, surgery should be performed as soon as possible.
Table 139-2 Advantages and Disadvantages of Canal Wall-Up and Canal Wall-Down Procedures in Chronic Otitis Media with Cholesteatoma
| Advantages | Disadvantages |
|---|---|
| Canal Wall-Up | |
| Physiologic position of tympanic membrane | Residual and recurrent cholesteatoma may occur |
| Enough middle ear space | Incomplete exteriorization of facial recess |
| No mastoid cavity problem | Second-stage operation often required |
| Canal Wall-Down | |
| Residual cholesteatoma easily found on follow-up evaluation | Mastoid cavity problem often |
| Recurrent cholesteatoma rare | Middle ear shallow and difficult to reconstruct |
| Total exteriorization of facial recess | Position of pinna may be altered; second-stage operation sometimes required |
Modified from Harker LA, Koontz FP. The bacteriology of cholesteatomas. In: McCabe BF, Sadé J, Abramson M, eds. Cholesteatoma: First International Conference. New York: Aesculapius Publishers; 1977.
In some patients, a cholesteatoma can be débrided of entrapped keratin by direct removal or by irrigation. In some cases, surgical intervention is impossible or not advisable; the patient may not be medically able to withstand surgery, or the risks of surgery may not outweigh the benefits in some patients with only-hearing ears. Irrigation with 1 :1 : 1 distilled white vinegar, distilled water, and 70% isopropyl alcohol may keep some cholesteatomas stable if their opening into the ear canal is sufficiently large (Box 139-2). If surgery is required for a cholesteatoma in an only-hearing ear, careful preoperative evaluation and operative planning, perioperative use of antibiotics and steroids, ossicular reconstruction, and postoperative care should be considered.81
Chronic Otitis Media without Cholesteatoma
Acute or recurrent infection of the middle ear may result in a permanent perforation of the tympanic membrane. Ears with chronic perforations without cholesteatoma may be chronically or intermittently infected. Three times as many operations were performed in the United States for this disease as were performed for cholesteatomas.25 Paparella and Kim81a reported that of 375 primary tympanomastoid operations for chronic mastoiditis, two thirds were performed in ears with granulation tissue and without cholesteatoma.
Diagnosis
Tympanic membrane perforation (Fig. 139-10) may result from AOM, chronic otitis media, or trauma (injury or surgery). In some instances, a dry, simple perforation results from a single episode of AOM (i.e., necrotizing otitis media). Perforation of the tympanic membrane, especially involving the tympanic anulus, may allow ingrowth of the keratinizing epithelium of the ear canal or tympanic membrane, leading to cholesteatoma. An ear with a simple perforation may become infected because of contamination from the ear canal or because of a smoldering infection in the mastoid. A simple perforation commonly is seen as a low-frequency conductive hearing loss clinically. This finding is supported by experimental perforation in rats.82 The tympanic membrane velocity was found to be decreased in the low frequency in a small perforation and in the high and low frequencies in a large perforation.83
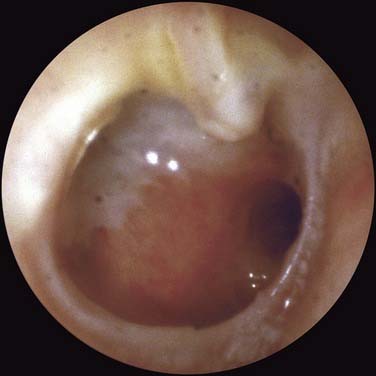
Pathogenesis
Chronic otomastoiditis without cholesteatoma is marked by the presence of irreversible inflammatory changes within the middle ear and mastoid. The factors that allow acute infections within the middle ear and mastoid to develop into chronic infections are unclear. da Costa and colleagues27 found granulation tissue in 96%, ossicular changes in 96%, tympanosclerosis in 43%, cholesteatoma in 36%, and cholesterol granuloma in 21% of the human temporal bones of patients with chronic otitis media with a perforated tympanic membrane (Table 139-3). Aeration of the middle ear, antrum, and mastoid depends on the free movement of gases from the eustachian tube into the mastoid air cells. In the human temporal bone, gases must travel around the ossicles in the epitympanic space to get into the antrum (Fig. 139-11).
Table 139-3 Pathologic Findings in the Temporal Bones with Chronic Otitis Media
| Pathologic Finding | Perforated Tympanic Membrane (n = 116) (%) | Nonperforated Tympanic Membrane (n = 28) (%) |
|---|---|---|
| Granulation tissue | 113 (97.4) | 27 (96.4) |
| Ossicular changes | 105 (90.5) | 27 (96.4) |
| Tympanosclerosis | 23 (19.8) | 12 (42.9) |
| Cholesterol granuloma | 14 (12.1) | 6 (21.4) |
| Cholesteatoma | 5 (4.3) | 10 (35.7) |
Modified from da Costa SS, Paparella MM, Schachern PA, et al. Temporal bone histopathology in chronically infected ears with intact and perforated tympanic membranes. Laryngoscope. 1992;102:1229.
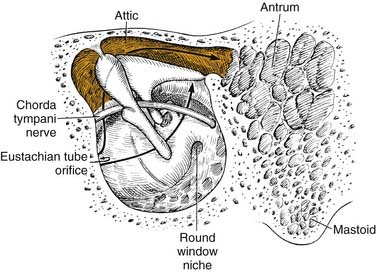
Proctor74 showed that the middle ear is separated from the antrum not only by the ossicles, but also by mucosal folds. He found that there were only two constant openings: (1) between the tendon of the tensor tympani muscle and the stapes, and (2) between the short process of the incus and the stapedial tendon. Edema and inflammation with granulation tissue may block these communicating openings, preventing drainage of the antrum and mastoid. Chronic obstruction of the attic and antrum with infection leads to “irreversible” changes in the mucosa and bone of the antrum and mastoid. Granulation tissue within the temporal bone can lead to bone erosion; 4 of 123 temporal bones in this series had active bone erosion. Thomsen and colleagues84 found that bone erosion in patients with chronic otitis media was more prevalent when cholesteatoma was present, but it still occurred in the absence of cholesteatoma. Moriyama and associates85 found osteoclastic bone resorption in rats with chronic otitis media.
Chronic otitis media may occur in patients who have indwelling tympanostomy tubes. Otorrhea may be a complication of tympanostomy tube insertion; it has been reported to occur in 9% to 34% of children undergoing this procedure.86 Chronic otorrhea that is resistant to therapy has been reported to occur in 5.5% of children with tubes.87 It is unclear whether such chronic infection is a result of the indwelling tube or of the drainage of an already smoldering infection. Giebink and coworkers88 found the risk of otorrhea on the second postoperative day was significantly increased by the presence of a bacterial pathogen in the ear canal or in the middle ear effusion, and by the presence of inflamed middle ear mucosa at tympanostomy tube insertion. The bacteria that are found in such cases are bacteria found in chronic otitis media generally: P. aeruginosa and Staphylococcus aureus. Erkan and colleagues89 reported the culture results of aspiration of exudate in 183 patients with chronic otitis media. Aerobes were found in 39% of the samples, anaerobes were found in 11%, and aerobes and anaerobes were found in 50%. P. aeruginosa, S. aureus, and Klebsiella pneumoniae were commonly found in aerobes, and Bacteroides sp were most commonly isolated in anaerobes. The colonization of tympanostomy tubes in the form of microbial biofilms may lead to persistence and recurrence of infection.90
Management
Most infected perforations can be managed conservatively with topical antibiotics. Antibiotic otic suspensions with or without steroids usually are effective. The antibiotics should be chosen to eradicate the most common pathogens, P. aeruginosa and S. aureus. In cases with recurrent or chronic infections, cultures should be used to adjust antibiotics. Many topical otic antibiotic preparations contain potentially ototoxic substances, including aminoglycoside antibiotics and propylene glycol. Studies of these substances applied to the middle ear have shown ototoxicity in rodents91 and primates.92 Although there are reports suggesting that sensorineural hearing loss may occur after topical use of these preparations,93,94 no conclusive evidence is available proving ototoxicity of commercially available otic preparations in the human middle ear.
Otorrhea occurs after insertion of tympanostomy tubes in approximately 10% of ears. The incidence of immediate postoperative otorrhea usually can be decreased by the use of topical antibiotics at the time of tube insertion.95,96 The use of Silastic tympanostomy tubes impregnated with silver oxide was shown to decrease the incidence of otorrhea significantly in a randomized clinical trial.86 If infection persists, intravenous antibiotic therapy directed at specifically identified organisms should be considered. Because chronically infected tympanostomy tubes may have persistent bacterial biofilms, removal of the tympanostomy tube may be necessary in some refractory cases.90,97
Bone Erosion in Cholesteatoma and Chronic Otitis Media
In Virchow’s description of the pathology of aural cholesteatoma in 1854, he noted that “the cholesteatoma extended through the bone to the external auditory canal, sometimes, also, in the cranial cavity.”98 Since that time, clinicians and investigators have studied the pathophysiology of bone resorption from this disease. Although much progress has been made in the understanding of the resorptive process, the actual sequence of events and their relative importance are not completely understood.
In the first half of the 20th century, it was believed that the bone resorption seen adjacent to cholesteatomas was a result of pressure necrosis,99 although clinical observations led to the abandonment of the pressure necrosis theory. It was thought to be highly unlikely that cholesteatomas could exert pressure exceeding capillary perfusion pressure (approximately 25 mm Hg), and this was later confirmed by Orisek and Chole100 using direct measurements of the pressure exerted by experimental cholesteatomas (1.3 to 11.9 mm Hg). Lautenschlager101 first suggested that cholesteatomas may elaborate enzymes that directly erode bone, and some investigators showed that cultured human cholesteatoma could degrade guinea pig collagen.63,102 Direct extracellular dissolution of bone was never shown, however.
It is now clear that inflammatory processes in the temporal bone induce the development and activation of osteoclasts, the only cell capable of resorbing bone. Several early human temporal bone studies revealed osteoclastic bone resorption adjacent to cholesteatoma matrix.103,104 Because of the relative scarcity of osteoclasts seen in surgical specimens, however, numerous investigators hypothesized that mononuclear histiocytes were the predominant cell associated with erosive cholesteatomas.105–107 Bernstein and colleagues108,109 noted that early studies were based on biopsy material taken during tympanomastoid surgery, and surgery often is preceded by attempts to control active inflammation; biopsy material may represent the reparative phase instead of the resorptive phase. Chole110 showed ultrastructural evidence in human and experimental cholesteatomas that bone resorption is primarily a result of the action of multinucleated osteoclasts on bone (Figs. 139-12 and 139-13). Although many mononuclear cells (histiocytes and fibroblasts) were present in the vicinity of active bone resorption, only multinucleated osteoclasts were seen to disrupt the lamina limitans of bone and cause resorption lacunae. It is established that multinucleated osteoclasts caused bone resorption in patients with cholesteatoma and chronic otitis media.110–114
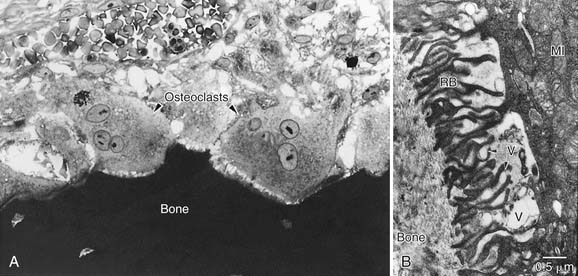
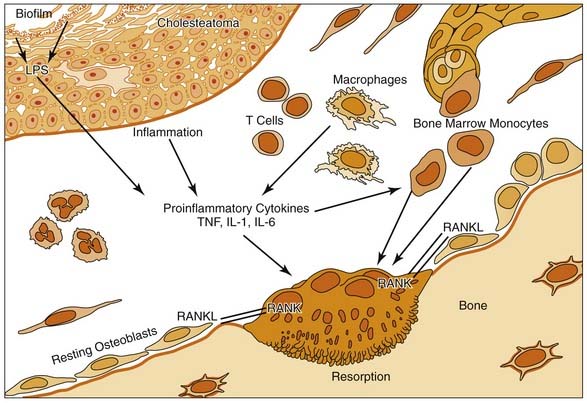
For bone resorption to occur, enzymatic removal of the organic and inorganic components must occur. These enzymes likely are elaborated or activated by the resorbing cells (osteoclasts) in their immediate microenvironment. These enzymes include acid phosphatase,54,110,115,116 collagenase,54,116 and acid proteases.117 In studies by Blair and colleagues,117 a cathepsin-like proteolytic enzyme with maximal activity at pH 4.0 was shown to be active in the microenvironment of the ruffled border of the osteoclast.
Numerous factors may lead to localized osteoclast recruitment and activation in regions of inflammatory osteolysis. It has been shown that bone remodeling and bone loss are controlled by a balance between the receptor activator of NF-κB (RANK) and its ligand, RANKL.113 The RANKL receptor, or RANK, has been identified on dendritic cells, chondrocytes, osteoclast precursors, and mature osteoclasts.113 Hamzei and coworkers118 indicated an increased number of osteoclast precursor cells in the perimatrix of cholesteatoma tissue concomitant with an enhanced expression of RANKL, osteoprotegerin, and macrophage colony-stimulating factor. The cholesteatoma-induced inflammatory process is associated with the release of RANKL from stromal cells and activated T cells, which triggers osteoclastogenesis.113,118
The localized control of osteoclastogenesis is complex, and signaling pathways may vary during the natural course of an inflammatory process. The type of bone undergoing resorption may also modify the process. The endochondral bone of the otic capsule seems to be more resistant to erosion than the intramembranous bone of the middle ear and mastoid.119
Some investigators have noted that the keratin debris within a cholesteatoma sometimes extrudes into the subepithelium adjacent to bone eliciting an inflammatory and osteoclastic response. Yuasa and colleagues120 showed that the pH of keratin debris within cholesteatomas was acidic, which they believed led to demineralization of the hydroxyapatite of bone. Some investigators have shown that the keratin itself may induce an inflammatory reaction (foreign body granuloma), which leads to cellular bone resorption.85,121,122
The extracellular matrix exhibits alterations in cholesteatoma, and many studies have evaluated the role of enzymes in cholesteatoma-induced bone resorption. By immunocytochemistry and zymography, the expression of matrix metalloproteinases (MMP-1, MMP-2, MMP-3, and MMP-9) was found to be strictly confined to the basal and suprabasal cell layers of the cholesteatoma epithelium.123–126 The neutrophil collagenase showed a more disseminated expression in the epithelium and the granulation tissue. The tissue inhibitor of metalloproteinases, TIMP-1, could be detected only in very limited areas of the granulation tissue in a quite randomized manner.126 Because of their destructive capacity, matrix metalloproteinases are normally tightly controlled. A derailment of these regulatory mechanisms in favor of proteolysis was postulated to play a role in the invasion of cholesteatomas into the temporal bone.125,126
Collagenase plays an important role in the mechanism of local invasion by aural cholesteatoma.116,127 Neutral collagenase may stimulate osteoclastic resorption by degrading the osteoid surface of bone, allowing osteoclastic activity.128 Neutral collagenase has not been found within osteoclasts,129 but has been localized in the vicinity of resorbing bone.116 In the collagenase cascade, procollagenase is activated to collagenase by plasmin. Plasminogen is activated to plasmin by plasminogen activator. Plasminogen activator is thought to play a role in keratinocyte proliferation, migration, and differentiation.130 Plasminogen activator can be divided into two types: tissue-type plasminogen activator and urokinase-type plasminogen activator. Tissue-type plasminogen activator is known to activate osteoclasts in vitro.131 Nakamura and associates132 showed both types of plasminogen activator in two of six tissue extracts of human cholesteatoma.
Ohsaki and colleagues133 showed that bone adjacent to cholesteatomas is demineralized. Although their study suggested that demineralization by acidic cholesteatomas may explain bony destruction, intimate contact of the cholesteatoma matrix with bone does not seem to be necessary for bone resorption. Bone resorption has been shown to occur in patients with chronic otitis media with or without cholesteatoma.84 Macri and Chole134 showed that an implanted silicone barrier between a cholesteatoma and underlying bone did not prevent osteoclastic bone resorption in experimental cholesteatoma. It is likely that indirect effects (e.g., pressure or inflammation) may activate the cellular events of bone resorption.135 More recent studies have shown that pressure with85,136 or without32,137 inflammation is sufficient to induce bone resorption in experimental animals. The histologic appearance of pressure-induced bone resorption is similar to that seen in cholesteatoma, showing osteoclasts and granulation tissue.111 It has been postulated that the physical effects (pressure) of cholesteatomas may lead to transient electrical potentials138 and the recruitment of monocytes into the subepithelial space. These monocytes may activate the downstream cellular events leading to bone resorption.
Activated monocytes can produce arachidonic acid metabolites such as prostaglandins and leukotrienes, which are potent inflammatory mediators. Prostaglandin E2 (PGE2)139 has been shown to stimulate bone remodeling similar to the kallikrein-kinin system.51 Prostaglandins, produced from the cyclooxygenase pathway of arachidonic acid metabolism, can be inhibited by aspirin, indomethacin, and ibuprofen. Indomethacin has been shown to impede bone resorption by inhibiting the production of PGE2 in vitro.140 This study also showed that in vivo indomethacin can inhibit pressure-induced bone resorption in an animal model. Ibuprofen, another prostaglandin inhibitor, has also been shown to inhibit bone resorption in a dose-dependent manner.141 Leukotrienes are 5-lipoxygenase metabolites of arachidonic acid. Although the role of leukotrienes in cholesteatoma is unclear, the peptidoleukotrienes (LTC4, LTD4, and LTE4) stimulate isolated osteoclasts to accumulate tartrate-resistant acid phosphatase and resorb bone.142
Although the mechanism is unknown, osteoclast activity can be inhibited by bisphosphonates, also known as antiosteolytic agents such as 1-hydroxyethylidene-1,1-bisphosphonate (HEBP), risedronate, and zoledronate. These drugs inhibit the localized recruitment and activation of osteoclasts in vivo and in vitro.143,144
Proinflammatory cytokines play an important role in middle ear infections and cholesteatoma. They are released by macrophages, T lymphocytes, monocytes, and many other cells at the site of infection. IL-1 was identified in cholesteatoma matrix,8 and was shown to stimulate fibroblasts and macrophages to produce PGE2 and collagenase.105 Cultured cholesteatoma specimens produced IL-1α and IL-1β, and both isoforms have been detected within cholesteatoma tissue.41,145,146 IL-1α and IL-1β are potent inducers of bone resorption and may act by increasing PGE2. Human recombinant IL-1 receptor antagonist blocked bone resorption in mouse calvaria as a result of IL-1β, but not IL-1α.147
Concomitantly, a parallel process of osteoblastic bone deposition invariably accompanies osteoclastic bone resorption. Factors known to induce new bone formation such as transforming growth factor (TGF)-β and bone morphogenetic protein-2 have been identified in cholesteatomas.105,148,149 Cytokines such as TGF-β1 and TGF-β2 may potentially slow the proliferation and tissue destruction associated with human cholesteatoma.150 TGF-β is involved in matrix formation and stimulates numerous matrix proteins, such as collagen, laminin, and fibronectin. TGF-β may also initiate bone formation by recruitment and proliferation of osteoblast precursor, whereas bone morphogenetic protein-2 seems to be important in inducing differentiation of pluripotent progenitor cells.
Fujioka and Huang151 identified platelet-derived growth factor in human cholesteatoma tissue, which stimulates monocytes to form multinucleated osteoclast-like cells. Additionally, Bujia and coworkers60 and Sudhoff and colleagues59 showed abnormal expression of epidermal growth factor in human cholesteatoma, which is a potent stimulator of cell proliferation and differentiation. Production of parathyroid hormone–related protein was detected in cholesteatoma cell cultures and may increase incidence of bone resorption.152
Vascularization within the perimatrix of cholesteatoma showed a fivefold increase compared with middle ear mucosa and a twofold increase compared with skin.152 Sudhoff and colleagues153 found a close relationship between the density of capillaries, degree of inflammation, and expression of different angiogenic factors. Because proliferating tissues such as middle ear cholesteatoma require an enhanced blood supply, angiogenesis seems to be necessary for the expansion of cholesteatoma matrix within the middle ear cavity.
The role of other cells in the pathophysiology of bone erosion in patients with chronic otitis media is unclear. Gantz154 suggested that Langerhans cells within the matrix of cholesteatomas might initiate an immunologic response to the presence of antigens (keratin and bacterial debris); these cells may induce the cellular events outlined previously. These cells may also promote proliferation of keratinized epithelial cells by means of IL-1α in vitro.92 Aberg and associates155 found no increase in Langerhans cells compared with ear canal epithelium, however, suggesting that these cells may not have a primary role in the development of cholesteatoma. Mast cells are seen in cholesteatoma matrix, but their role is unknown.156
The important role of infection in the pathogenesis of cholesteatoma is highlighted by the more recent identification of bacterial biofilms in otitis media157 and cholesteatoma.35,158 Biofilms are bacterial communities enclosed in a self-produced matrix adherent to a surface.147 Many bacterial species relevant to otologic infections are known to form biofilms, including P. aeruginosa, H. influenzae, Streptococcus pneumoniae, and S. aureus.34 Bacteria within biofilms are more resistant to antibiotics and are likely responsible for the chronicity and recurrence of these infections. The presence of antibiotic-resistant bacterial biofilms in cholesteatomas may also explain their aggressiveness.35 Bacterial biofilms within cholesteatomas may elaborate lipopolysaccharide and other bacterial products that stimulate osteoclastogenesis. Zhuang and colleagues159 showed that lipopolysaccharide derived from P. aeruginosa can induce osteoclast development in vitro and potently stimulates in vivo bone resorption through a toll-like receptor–4–dependent mechanism.159
Sensorineural Hearing Loss
The destructive effects of an expanding cholesteatoma within the middle ear or mastoid and concomitant chronic infection are not limited to the bony structures of the temporal bone. Paparella and coworkers160 observed sensorineural hearing loss in patients with chronic otitis media, and Chole and Chiu161 observed loss of cochlear hair cell stereocilia in animals with experimental cholesteatomas with or without infection. In a study of age-matched, staged cholesteatomas, McGinn and Chole162 showed loss of cochlear hair cells in areas subjacent to areas of bone erosion, suggesting that ototoxic substances may traverse the bony wall of the cochlea directly. Meyerhoff and associates163 found that 17.9% of temporal bones with chronic otitis media had histologic evidence of labyrinthitis.
Vartiainen and Karjalainen164 compared 874 chronically infected ears with and without cholesteatoma with 609 control ears, and found significantly worse bone conduction in the infected group; ears with cholesteatomas were generally worse than ears without cholesteatomas. Fria and coworkers165 found that there was a tendency for bone conduction thresholds to be elevated at 2 kHz and 4 kHz in the ears with chronic OME compared with bone conduction thresholds in children with or without OME. In animal experiments, the inner ear is particularly sensitive to injury by middle ear infection. Morizono and associates166 found that otitis media in chinchillas resulted in significant increase in tone-burst elicited compound action potentials after the otitis had cleared, indicating a sensory hearing loss.
In contrast, after correcting for the artificial elevation of bone conduction thresholds from conductive hearing loss, also known as the Carhart effect, Browning and Gatehouse167 found no difference in the bone conduction thresholds between 395 ears with chronic otitis media and 920 control ears. These results were supported by MacAndie and O’Reilly,168 who pointed out that the presence of cholesteatoma or ossicular erosion, or both, was not associated with a significantly increased risk of sensorineural hearing loss. Rahko and colleagues169 studied bone conduction thresholds in 359 children with a history of recurrent AOM and found no correlation between the number of bouts of infection and permanent sensorineural hearing loss.
Tympanosclerosis
Diagnosis
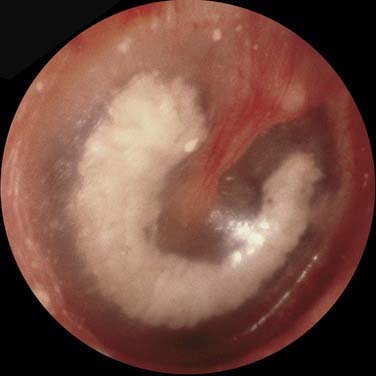
Tympanosclerosis is thought to be a complication of otitis media in which acellular hyalin and calcified deposits accumulate within the tympanic membrane and the submucosa of the middle ear. In most patients, these plaques are clinically insignificant and cause little or no hearing impairment. Tympanosclerotic plaques within the tympanic membrane appear as a semicircular crescent or horseshoe-shaped white plaque within the tympanic membrane (Fig. 139-14).
Pathogenesis
Tympanosclerosis is a consequence of resolved otitis media or trauma. Hussl and Mueller170 found tympanosclerosis to be a frequent sequela of chronic OME; they found tympanosclerosis in 19.7% of drumheads 6 to 8 years after the insertion of ventilating tubes for OME. They also noted that middle ear tympanosclerosis often was seen after recurrent bouts of AOM. Tos and Stangerup171 found a significant increase in tympanosclerosis in ears in which grommets were placed (59%) compared with the contralateral ears, in which only myringotomy was performed (13%). Daly172 reported the weighted average incidence of tympanosclerosis is 10% in children 4 to 15 years old, with an average follow-up period of 4 years. The incidence of tympanosclerosis in chronic otitis media has been reported to range from 9% to 38%. Kinney173 found that 20% of 1495 patients undergoing surgery for chronic otitis media or its sequelae had tympanosclerosis. Mangat and coworkers174 found that 23.6% of 1274 patients treated with tympanostomy tubes had tympanosclerosis.
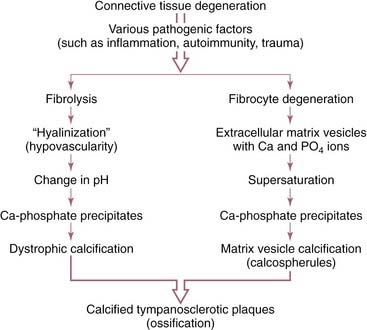
Tympanosclerosis appears histologically as an acellular hyalinization of the subepithelial connective tissue of the tympanic membrane and middle ear. In most instances, calcification is present. Osteoneogenesis also can occur within these lesions. The bone deposition and ossicular fixation occur most frequently in the attic associated with the heads of the malleus and incus. When plaques occur within the tympanic membrane, they are limited to the lamina propria. Hussl and Lim175 found these plaques to be a degenerative process resulting in calcification in connective tissue of the middle ear. They hypothesized that OME or AOM led to a destructive process within connective tissue, which led to degeneration of collagen and subsequently dystrophic calcification and tympanosclerosis.
The degeneration of collagen may be a direct result of inflammation or infection within the middle ear (e.g., by bacterial proteinases and collagenases). Wielinga and colleagues176 showed that eustachian tube obstruction alone, without infection, caused tympanosclerosis in rats; they hypothesized that deformation alone was sufficient to cause the plaques to form. Another possible cause of tympanosclerosis is an autoimmune process occurring within the tympanic membrane. Schiff and associates61 prepared antisera to guinea pig lamina propria and passively immunized guinea pigs. When the tympanic membranes of these animals were traumatized, tympanosclerotic plaques developed. Chole and Henry177 found that LP/J inbred mice spontaneously developed middle ear lesions that resembled tympanosclerosis and may be immunologically mediated.178 Hussl and Lim175 proposed two possible mechanisms for the formation of tympanosclerotic plaques, beginning with collagen degeneration (Fig. 139-15). Russell and Giles179 found that the process of tympanosclerosis started in the submucosal connective tissue layer and progressed to involve all connective tissue sublayers in an animal model. The extent of calcium deposition and fibrosis across the membrane was related to the duration of OME.
Management
Tympanosclerosis within the middle ear (Fig. 139-16) is histologically similar to tympanosclerosis occurring within the tympanic membrane, but it often leads to conductive hearing loss caused by ossicular fixation. Although some authors have stated that tympanosclerosis tends to recur after surgical removal, others have reported stable hearing results in these patients. Smyth and colleagues180 reported excellent hearing results in 79% of tympanosclerotic ears in which ossicular reconstruction (stapedectomy and total ossicular reconstruction) was performed in two stages, although Gormley181 found that only 7% of his cases had an air-bone gap of less than 21 dB on long-term follow-up evaluation, questioning the advisability of stapedectomy in ears with tympanosclerosis. In the earlier series182 in which one-stage procedures were performed, 21% of 57 cases resulted in cochlear losses. Tympanoplasty and ossicular reconstruction can be performed in ears with tympanosclerosis, but the risks of cochlear damage seem to be greater than in other middle ear diseases because of the extensive dissection that is required in tympanosclerotic ears and the coexistence of labyrinthine erosion.
Petrositis
Infection of the mastoid and middle ear may be complicated by the spread of infection within the temporal bone into the petrous apex. Petrous apicitis is an extension of infection from the mastoid air cell tract into a pneumatized anterior or posterior petrous apex. In the preantibiotic era, otitis media often was complicated by a spread into the petrous apex and further intracranial complications. The classic symptoms of petrous apicitis include deep facial pain, otitis media, and ipsilateral abducens nerve paralysis. This triad, called Gradenigo’s syndrome,183 is rare, although suppurative processes in the petrous apex occur in patients with AOM and chronic otitis media, but most often manifest as chronic infection with otorrhea and sometimes deep pain after adequate surgery. More recently, otologic diseases have been observed as complications in patients with acquired immunodeficiency syndrome (AIDS). Chandrasekhar and colleagues184 reported six of seven specimens had petromastoiditis in the temporal bones of AIDS patients.
Historical Note
The history of petrous apicitis and its management has been reviewed more recently.185 A patient with petrous apicitis and Gradenigo’s triad was first described by Goris.186 In Gradenigo’s review of previously published cases of 57 patients with petrositis, 24 actually had the pure triad; others had multiple complications. In the early part of the 20th century, there was a controversy as to whether petrous apicitis can develop in diploic (marrow-filled) or pneumatic (air-filled) petrous apices.187 It is generally believed that petrous apicitis occurs in patients who have pneumatized petrous apices. In the 1930s, Almour188 and Kopetzky and Almour189 described surgical approaches for petrous apicitis in which fistulous tracts were followed into the petrous apex. Ramandier in 1933,190 and soon thereafter Lempert191 described the now classic operation for exenteration of the anterior petrous apex. The histopathology of petrous apicitis was described by Lindsay.192 An additional surgical approach for suppuration of the petrous apex was described in a case report by Hendershot and Wood,193 in which they drained an osteomyelitis of the petrous apex through the middle cranial fossa.
Anatomy
The petrous apex is a truncated pyramid, which is the portion of the temporal bone medial to the inner ear labyrinth (Fig. 139-17). The petrous apex is the most surgically inaccessible portion of the temporal bone.194 The apex may be arbitrarily bisected by a coronal plane through the internal auditory canal (see Fig. 139-17). This plane divides the apex into an anterior portion, the peritubal area, and a posterior portion, the perilabyrinthine area. The posterior petrous apex, which is pneumatized in 33% of patients, is just medial to the semicircular canals. The anterior apex, which is pneumatized in 10% of patients, is anterior and medial to the cochlea. The carotid artery traverses the anterior petrous apex.
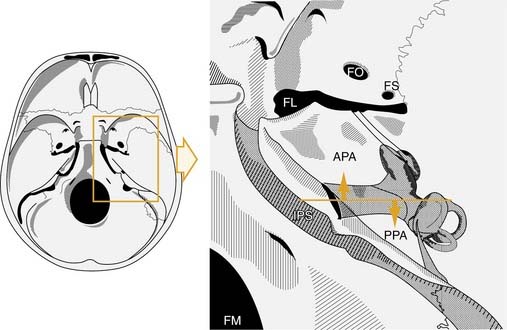
(Modified from Chole RA. Petrous apicitis: surgical anatomy. Ann Otol Rhinol Laryngol. 1985;94:251.)
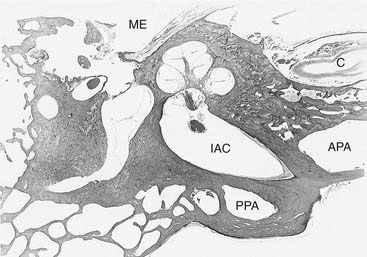
The petrous apex may be pneumatic (air cell filled), diploic (marrow filled), or sclerotic (solid bone). Direct extension of infection from the mastoid and middle ear through pneumatized air cell tracts into the petrous apex is thought to be the etiology of petrous apicitis. It has been estimated that 30% of posterior petrous apices are pneumatized, and in a study of 84 normal human temporal bones, 9% of anterior petrous apices were found to be pneumatized (Fig. 139-18).195
The anatomic relationship at the petrous tip may explain some of the symptoms of petrous apicitis. An undetected and poorly drained infected air cell of the petrous apex must trail through small air cell tracts into the middle ear and mastoid. These cell tracts consist of the infralabyrinthine air cell tract, the retrofacial tract, and the peritubal air cells superior to the eustachian tube. If the bony cortex of the anterior petrous apex is involved by the extension of infection, the infection may cause an epidural abscess in the region or damage nearby cranial nerves. On the superior aspect of the petrous tip lies the trigeminal or gasserian ganglion. Damage or irritation to the ganglion may explain the deep facial pain that some patients with apicitis experience. Extending from the tip of the petrous apex to the clinoid is the petroclinoid ligament. The abducens nerve travels below the petroclinoid ligament in a small canal called Dorello’s canal.196 Entrapment or inflammation in the ear of Dorello’s canal is thought to account for the presence of abducens paralysis in some patients with petrous apicitis.
Diagnosis
Symptoms of petrositis usually are subtle. Typically, a patient who has had previous mastoid surgery complains of persistent infection and deep facial pain. In a series of 22 patients during 20 years, 16 patients (72.7%) had otalgia, and 13 patients (59.1%) had deep facial pain and headache; trigeminal nerve paralysis (68.2%) most frequently involved the cranial nerve; 6 (27.3%) had facial nerve paralysis, 4 (18.2%) had abducens nerve paralysis, and 2 had coma (Table 139-4).197 Patients with suppuration may manifest various symptoms, none of which are pathognomonic for the syndrome. In patients with long-standing chronic otomastoiditis, deep pain, and persistent infection, the diagnosis of petrositis should be considered. In a more recent series of patients with petrous apicitis, the predominant organism was P. aeruginosa. The physical findings of petrositis usually include those of chronic otitis media with chronic otorrhea. In some patients, the infection can be limited to the anterior petrous apex, and the middle ear is normal.197 Many patients have involvement of cranial nerves V, VI, and VII.
Table 139-4 Symptoms Found in 22 Patients with Petrous Apicitis from 1976-1995
| Symptom | No. Patients (%) |
|---|---|
| Deep pain and headache | 13 (59) |
| Otalgia | 16 (72) |
| Otorrhea | 13 (59) |
| Fever | 5 (22) |
| Coma | 2 (9) |
| Cranial nerve paralysis | |
| V | 15 (68.2) |
| VI | 4 (18.2) |
| VII | 6 (27.3) |
| VIII | 9 (40.9) |
| IX | 1 (4.5) |
| X | 1 (4.5) |
From Chole RA, Gadre AK. Petrous apicitis—symptomatology, pathology, and management [Abstract]. Skull Base Surgery Symposium, Sacramento, CA, 1995.
Diagnostic Tests
When the diagnosis of petrous apicitis is suspected on clinical grounds, the most appropriate diagnostic procedure is CT. High-resolution CT usually shows details of the petrous apex and provides important details about potential surgical routes. A pneumatized petrous apex on the uninvolved side can sometimes be contrasted with a fluid-filled or sclerotic petrous apex on the involved side, although Roland and colleagues198 have shown that asymmetry of the petrous apex is not diagnostic for apicitis because asymmetric pneumatization of the apex can occur in healthy subjects. If CT scan indicates a potential apicitis, MRI may add information about the nature of the fluid or tissue within the apex (Fig. 139-19). A gallium bone scan may provide additional information, showing increased uptake on the side of the apicitis. A combination of MRI and CT is necessary to evaluate normal anatomic variations and to be capable of differentiating diagnosis.199
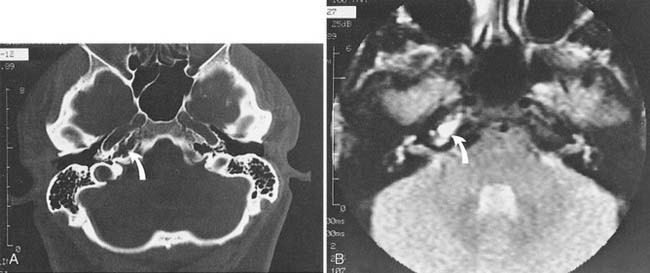
Management
The management of petrous apicitis is directed toward control of the infection. If topical and systemic antibiotic management is inadequate to control the suppuration, various surgical approaches are available. Surgical therapy aims to achieve drainage of the petrous apex through the mastoid and middle ear into the petrous apex. These air cells have been well defined anatomically194; they include the subarcuate and sinodural angle cells toward the posterior petrous apex and the peritubal, retrofacial, infralabyrinthine, and infracochlear tracts toward the anterior petrous apex. The anterior apex may be widely exposed through the glenoid fossa with the approach of Ramandier190 and Lempert.191 If adequate air cells cannot be identified through the middle ear and mastoid, the middle cranial fossa approach can be used to enter the roof of the anterior petrous apex.193 Brackmann and Toh138 found the translabyrinthine approach useful in nonhearing ears. In hearing individuals, anatomy permitting, the transcanal infracochlear approach with stenting was the preferred approach for drainage of the petrous apex.
Brackmann DE, Toh EH. Surgical management of petrous apex cholesterol granulomas. Otol Neurotol. 2002;23:529-533.
Chole RA. Cellular and subcellular events of bone resorption in human and experimental cholesteatoma: the role of osteoclasts. Laryngoscope. 1984;94:76-95.
Chole RA, Donald PJ. Petrous apicitis: clinical considerations. Ann Otol Rhinol Laryngol. 1983;92(6 Pt 1):544-551.
Chole RA, Faddis BT. Evidence for microbial biofilms in cholesteatomas. Arch Otolaryngol Head Neck Surg. 2002;128:1129-1133.
Costerton JW, Stewart PS, Greenberg EP. Bacterial biofilms: a common cause of persistent infections. Science. 1999;284:1318-1322.
Ehrlich GD, Veeh R, Wang X, et al. Mucosal biofilm formation on middle-ear mucosa in the chinchilla model of otitis media. JAMA. 2002;287:1710-1715.
Giebink GS, Daly K, Buran DJ, et al. Predictors for postoperative otorrhea following tympanostomy tube insertion. Arch Otolaryngol Head Neck Surg. 1992;118:491-494.
Hildmann H, Sudhoff H. Middle Ear Surgery. New York: Springer Verlag; 2006.
Hussl B, Lim DJ. Histopathology of tympanosclerosis. In: Lim DJ, Bluestone CD, Klein JO, editors. Recent Advances in Otitis Media with Effusion. St. Louis: Mosby, 1984.
Lempert J. Complete apicectomy (mastoidotympano-apicectomy): new technique for complete apical exenteration of apical carotid portion of petrous pyramid. Arch Otolaryngol Head Neck Surg. 1937;25:144.
Lieu JE, Muthappan PG, Uppaluri R. Association of reflux with otitis media in children. Otolaryngol Head Neck Surg. 2005;133:357-361.
McKennan KX, Chole RA. Post-traumatic cholesteatoma. Laryngoscope. 1989;99(8 Pt 1):779-782.
Michaels L. An epidermoid formation in the developing middle ear: possible source of cholesteatoma. J Otolaryngol. 1986;15:169-174.
Sudhoff H, Linthicum FHJr. Cholesteatoma behind an intact tympanic membrane: histopathologic evidence for a tympanic membrane origin. Otol Neurotol. 2001;22:444-446.
Sudhoff H, Tos M. Pathogenesis of attic cholesteatoma: clinical and immunohistochemical support for combination of retraction theory and proliferation theory. Am J Otol. 2000;21:786-792.
1. Poelmans J, Tack J, Feenstra L. Prospective study on the incidence of chronic ear complaints related to gastroesophageal reflux and on the outcome of antireflux therapy. Ann Otol Rhinol Laryngol. 2002;111:933-938.
2. Tasker A, Dettmar PW, Panetti M, et al. Reflux of gastric juice and glue ear in children. Lancet. 2002;359:493.
3. Tasker A, Dettmar PW, Panetti M, et al. Is gastric reflux a cause of otitis media with effusion in children? Laryngoscope. 2002;112:1930-1934.
4. Lieu JE, Muthappan PG, Uppaluri R. Association of reflux with otitis media in children. Otolaryngol Head Neck Surg. 2005;133:357-361.
5. Wittmaack K. Wie entsteht ein genuines Cholesteatom? Arch Otorhinolaryngol. 1933;137:306.
6. Donaldson JD. Mini-myringoplasty in the treatment of tympanic atelectasis. J Otolaryngol. 1986;15:21-24.
7. Tos M, Stangerup SE, Andreassen UK. Size of the mastoid air cells and otitis media. Ann Otol Rhinol Laryngol. 1985;94(4 Pt 1):386-392.
8. Turgut S, Tos M. Correlation between temporal bone pneumatization, location of lateral sinus and length of the mastoid process. J Laryngol Otol. 1992;106:485-489.
9. Shatz A, Sadé J. Correlation between mastoid pneumatization and position of the lateral sinus. Ann Otol Rhinol Laryngol. 1990;99(2 Pt 1):142-145.
10. Hasebe S, Takahashi H, Honjo I, et al. Mastoid condition and clinical course of cholesteatoma. ORL J Otorhinolaryngol Relat Spec. 2001;63:160-164.
11. Sadé J, Luntz M, Levy D. Middle ear gas composition and middle ear aeration. Ann Otol Rhinol Laryngol. 1995;104:369-373.
12. Maw AR, Bawden R. Tympanic membrane atrophy, scarring, atelectasis and attic retraction in persistent, untreated otitis media with effusion and following ventilation tube insertion. Int J Pediatr Otorhinolaryngol. 1994;30:189-204.
13. Sadé J, Berco E. Atelectasis and secretory otitis media. Ann Otol Rhinol Laryngol. 1976;85(2 Suppl 25 Pt 2):66-72.
14. Tos M, Poulsen G. Attic retractions following secretory otitis. Acta Otolaryngol. 1980;89(5-6):479-486.
15. Sadé J. The atelectatic ear. In: Sade J, editor. Secretory Otitis Media and Its Sequelae. New York: Churchill Livingstone, 1979.
16. Graham MD, Knight PR. Atelectatic tympanic membrane reversal by nitrous oxide supplemented general anesthesia and polyethylene ventilation tube insertion: a preliminary report. Laryngoscope. 1981;91(9 Pt 1):1469-1471.
17. Paparella MM, Jung TT. Experience with tympanoplasty for atelectatic ears. Laryngoscope. 1981;91(9 Pt 1):1472-1477.
18. Ars B. Tympanic membrane: retraction pocket. Acta Otorhinolaryngol Belg. 1995;49:163-171.
19. Sudhoff H, Tos M. Pathogenesis of attic cholesteatoma: clinical and immunohistochemical support for combination of retraction theory and proliferation theory. Am J Otol. 2000;21:786-792.
20. Sadé J. Hyperectasis: the hyperinflated tympanic membrane: the middle ear as an actively controlled system. Otol Neurotol. 2001;22:133-139.
21. Cruveilhier LJB. Anatomie Pathologique du Corpus Hamani, Vol 1. Paris: JB Balliere. 1829.
22. Müller J. Über den Feineren Bau und die Formen der Krankhaften Geschwülste. Berlin: Reimer G; 1838.
23. Harker LA. Cholesteatoma: an incidence study. In: McCabe BF, Sadé J, Abramson M, editors. Cholesteatoma: First International Conference. Birmingham, AL: Aesculapius Publishing, 1977.
24. Liang J, Michaels L, Wright A. Immunohistochemical characterization of the epidermoid formation in the middle ear. Laryngoscope. 2003;113:1007-1014.
25. Ruben RJ. The disease in society: evaluation of chronic otitis media in general and cholesteatoma in particular. In: Sade J, editor. Cholesteatoma and Mastoid Surgery. Amsterdam: Kugler Publishing, 1982.
26. Tos M. Incidence, etiology and pathogenesis of cholesteatoma in children. Adv Otorhinolaryngol. 1988;40:110-117.
27. da Costa SS, Paparella MM, Schachern PA, et al. Temporal bone histopathology in chronically infected ears with intact and perforated tympanic membranes. Laryngoscope. 1992;102:1229-1236.
28. Michaels L. An epidermoid formation in the developing middle ear: possible source of cholesteatoma. J Otolaryngol. 1986;15:169-174.
29. Koltai PJ, Nelson M, Castellon RJ, et al. The natural history of congenital cholesteatoma. Arch Otolaryngol Head Neck Surg. 2002;128:804-809.
30. Potsic WP, Korman SB, Samadi DS, et al. Congenital cholesteatoma: 20 years’ experience at The Children’s Hospital of Philadelphia. Otolaryngol Head Neck Surg. 2002;126:409-414.
31. Ruah CB, Schachern PA, Paparella MM, et al. Mechanisms of retraction pocket formation in the pediatric tympanic membrane. Arch Otolaryngol Head Neck Surg. 1992;118:1298-1305.
32. Wolfman DE, Chole RA. Osteoclast stimulation by positive middle-ear air pressure. Arch Otolaryngol Head Neck Surg. 1986;112:1037-1042.
33. McGinn MD, Chole RA, Henry KR. Cholesteatoma induction: consequences of external auditory canal ligation in gerbils, cats, hamsters, guinea pigs, mice and rats. Acta Otolaryngol. 1984;97(3-4):297-304.
34. Post JC, Hiller NL, Nistico L, et al. The role of biofilms in otolaryngologic infections: update 2007. Curr Opin Otolaryngol Head Neck Surg. 2007;15:347-351.
35. Chole RA, Faddis BT. Evidence for microbial biofilms in cholesteatomas. Arch Otolaryngol Head Neck Surg. 2002;128:1129-1133.
36. Haberman J. Zur entstehung des cholesteatoms des mittlohrs. Arch Ohrenheilk. 1889;27:42.
37. Weiss P. Cell contact. Int Rev Cytol. 1958;7:391.
38. van Blitterswijk CA, Grote JJ. Cytokeratin expression in cholesteatoma matrix, meatal epidermis and middle ear epithelium: a preliminary report. Acta Otolaryngol. 1988;105(5-6):529-532.
39. Kim HJ, Chole RA. Experimental models of aural cholesteatomas in Mongolian gerbils. Ann Otol Rhinol Laryngol. 1998;107:129-134.
40. Bujia J, Holly A, Kim C, et al. Expression of human intercellular adhesion molecules in middle ear cholesteatoma. Am J Otolaryngol. 1994;15:271-275.
41. Bujia J, Kim C, Boyle D, et al. Quantitative analysis of interleukin-1-alpha gene expression in middle ear cholesteatoma. Laryngoscope. 1996;106(2 Pt 1):217-220.
42. Chao WY, Jin YT, Huang CC. Langerhans cells in human middle ear cholesteatomas. Eur Arch Otorhinolaryngol. 1992;249:380-384.
43. Chao WY, Yuan QG, Huang CC. Localization of fibronectin in human middle ear cholesteatoma. Arch Otorhinolaryngol. 1988;245:160-165.
44. Jackson DG, Lim DJ. Fine morphology of the advancing front of cholesteatome—experimental and human. Acta Otolaryngol. 1978;86(1-2):71-88.
45. Hueb MM, Goycoolea MV, Muchow D, et al. In search of missing links in otology, III: development of a new animal model for cholesteatoma. Laryngoscope. 1993;103:774-784.
46. Palva T, Karma P, Makinen J. The invasion theory. In: Sade J, editor. Cholesteatoma and Mastoid Surgery. Amsterdam: Kugler Publishing, 1982.
47. McKennan KX, Chole RA. Post-traumatic cholesteatoma. Laryngoscope. 1989;99(8 Pt 1):779-782.
48. Schmidt SH, Hellstrom S. Experimental cholesteatoma in the rat. Acta Otolaryngol. 1994;114:430-434.
49. Lange W. Über bei entstehung der mittlohrcholesteatome. Z Hals Nas Ohrenheilk. 1925;11:250.
50. Ruedi L. Cholesteatoma formation in the middle ear in animal experiments. Acta Otolaryngol. 1959;50(3-4):233-240.
51. Lim DJ, Birck HG, Saunders WH. Aural cholesteatoma epidermidization: fine morphological study. In: McCabe BD, Sadé J, Abramson M, editors. Cholesteatoma: First International Conference. Birmingham, AL: Aescalapius Publishing, 1977.
52. Sudhoff H, Bujia J, Borkowshi G, et al. Basement membrane in middle ear cholesteatoma: immunohistochemical and ultrastructural observations. Ann Otol Rhinol Laryngol. 1996;105:804-810.
53. Chole RA, Tinling SP. Basal lamina breaks in the histogenesis of cholesteatoma. Laryngoscope. 1985;95:270-275.
54. Huang CC, Shi GS, Yi ZX. Experimental induction of middle ear cholesteatoma in rats. Am J Otolaryngol. 1988;9:165-172.
55. Masaki M, Wright CG, Lee DH, et al. Experimental cholesteatoma: epidermal ingrowth through tympanic membrane following middle ear application of propylene glycol. Acta Otolaryngol. 1989;108(1-2):113-121.
56. Sudhoff H, Linthicum FHJr. Cholesteatoma behind an intact tympanic membrane: histopathologic evidence for a tympanic membrane origin. Otol Neurotol. 2001;22:444-446.
57. Stammberger M, Bujia J, Kastenbauer E. Alteration of epidermal differentiation in middle ear cholesteatoma. Am J Otol. 1995;16:527-531.
58. Shinoda H, Huang CC. Expressions of c-jun and p53 proteins in human middle ear cholesteatoma: relationship to keratinocyte proliferation, differentiation, and programmed cell death. Laryngoscope. 1995;105:1232-1237.
59. Sudhoff H, Bujia J, Holly A, et al. Functional characterization of middle ear mucosa residues in cholesteatoma samples. Am J Otol. 1994;15:217-221.
60. Bujia J, Holly A, Schilling V, et al. Aberrant expression of epidermal growth factor receptor in aural cholesteatoma. Laryngoscope. 1993;103:326-329.
61. Schiff M, Poliquin JF, Catanzaro A, et al. Tympanosclerosis: s theory of pathogenesis. Ann Otol Rhinol Laryngol Suppl. 1980;89(4 Pt 2):1-16.
62. Sakamoto T, Kondo K, Yamasoba T, et al. Overexpression of ErbB-2 protein in human middle ear cholesteatomas. Laryngoscope. 2004;114:1988-1991.
63. Miyao M, Shinoda H, Takahashi S. Caspase-3, caspase-8, and nuclear factor-kappaB expression in human cholesteatoma. Otol Neurotol. 2006;27:8-13.
64. Szczepanski M, Szyfter W, Jenek R, et al. Toll-like receptors 2, 3 and 4 (TLR-2, TLR-3 and TLR-4) are expressed in the microenvironment of human acquired cholesteatoma. Eur Arch Otorhinolaryngol. 2006;263:603-607.
65. Parisier SC, Agresti CJ, Schwartz GK, et al. Alteration in cholesteatoma fibroblasts: induction of neoplastic-like phenotype. Am J Otol. 1993;14:126-130.
66. Chole RA, Faddis BT, Chamberlain S, et al. Invasiveness of fibroblasts from experimental cholesteatomas. Otol Neurotol. 2001;22:15-17.
67. Huisman MA, de Heer E, Ten Dijke P, et al. Transforming growth factor beta and wound healing in human cholesteatoma. Laryngoscope. 2008;118:94-98.
68. Sudhoff H, Bujia J, Fisseler-Eckhoff A, et al. Expression of a cell-cycle-associated nuclear antigen (MIB 1) in cholesteatoma and auditory meatal skin. Laryngoscope. 1995;105:1227-1231.
69. Kamide Y, Sasaki H, Abramson M, et al. Effects of epidermal Langerhans cell’s conditioned medium on keratinocytes: a role of Langerhans cells in cholesteatoma. Am J Otolaryngol. 1991;12:307-315.
70. Wendt H. Dequamative Entzundung des Mittelohrs (Cholesteatom des Felsenbeins). Arch Heilk. 1873;14:428.
71. Sadé J. Cellular differentiation of the middle ear lining. Ann Otol Rhinol Laryngol. 1971;80:376-383.
72. Chole RA, Frush DP. Quantitative studies of eustachian tube epithelium during experimental vitamin A deprivation and reversal. In: Sadé J, editor. Cholesteatoma and Mastoid Surgery. Amsterdam: Krugler Publishing, 1982.
73. Jackler RK. The surgical anatomy of cholesteatoma. Otolaryngol Clin North Am. 1989;22:883-896.
74. Proctor B. The development of the middle ear spaces and their surgical significance. J Laryngol Otol. 1964;78:631-648.
75. Von Tröltsch A. The Diseases of the Ear: Their Diagnosis and Treatment. New York: William Wood & Co; 1864.
76. Parisier SC, Edelstein DR, Han JC, et al. Management of labyrinthine fistulas caused by cholesteatoma. Otolaryngol Head Neck Surg. 1991;104:110-115.
77. Hildmann H, Sudhoff H, Jahnke K. Grundzüge einer differenzierten cholesteatom chirurgie. Laryngorhinootologie. 2000;79(Suppl 2):S73.
78. Harker LA, Koontz FP. The bacteriology of cholesteatomas. In: McCabe BF, Sadé J, Abramson M, editors. Cholesteatoma: First International Conference. Birmingham, AL: Aesculapius Publishing, 1977.
79. Hildmann H, Sudhoff H, editors. Middle Ear Surgery. New York: Springer Verlag, 2006.
80. Karmarkar S, Bhatia S, Saleh E, et al. Cholesteatoma surgery: the individualized technique. Ann Otol Rhinol Laryngol. 1995;104:591-595.
81. Glasscock ME3rd, Johnson GD, Poe DS. Surgical management of cholesteatoma in an only hearing ear. Otolaryngol Head Neck Surg. 1990;102:246-250.
81a. Paparella MM, Kim CS. Mastoidectomy update. Laryngoscope. 1977;87:1977-1988.
82. Bigelow DC, Swanson PB, Saunders JC. The effect of tympanic membrane perforation size on umbo velocity in the rat. Laryngoscope. 1996;106(1 Pt 1):71-76.
83. Voss SE, Rosowski JJ, Merchant SN, et al. Middle-ear function with tympanic-membrane perforations, I: measurements and mechanisms. J Acoust Soc Am. 2001;110(3 Pt 1):1432-1444.
84. Thomsen J, Bretlau P, Balslev Joorgensen M. Bone resorption in chronic otitis media: the role of cholesteatoma, a must or an adjunct? Clin Otolaryngol Allied Sci. 1981;6:179-186.
85. Moriyama H, Huang CC, Kato M, et al. Effects of pressure on bone resorption in the middle ear of rats. Ann Otol Rhinol Laryngol. 1985;94(1 Pt 1):60-64.
86. Chole RA, Hubbell RN. Antimicrobial activity of Silastic tympanostomy tubes impregnated with silver oxide: a double-blind randomized multicenter trial. Arch Otolaryngol Head Neck Surg. 1995;121:562-565.
87. McLelland CA. Incidence of complications from use of tympanostomy tubes. Arch Otolaryngol. 1980;106:97-99.
88. Giebink GS, Daly K, Buran DJ, et al. Predictors for postoperative otorrhea following tympanostomy tube insertion. Arch Otolaryngol Head Neck Surg. 1992;118:491-494.
89. Erkan M, Aslan T, Sevuk E, et al. Bacteriology of chronic suppurative otitis media. Ann Otol Rhinol Laryngol. 1994;103:771-774.
90. Bothwell MR, Smith AL, Phillips T. Recalcitrant otorrhea due to Pseudomonas biofilm. Otolaryngol Head Neck Surg. 2003;129:599-601.
91. Wright CG, Meyerhoff WL. Ototoxicity of otic drops applied to the middle ear in the chinchilla. Am J Otolaryngol. 1984;5:166-176.
92. Wright CG, Halama AR, Meyerhoff WL. Ototoxicity of an ototopical preparation in a primate. Am J Otol. 1987;8:56-60.
93. Murphy KW. Deafness after topical neomycin. BMJ. 1970;2:114.
94. Dumas G, Bessard G, Gavend M, et al. Risque de surdite par instillations de goutes auriculaires contenant des aminosides. Therapie. 1980;35:357.
95. Baker RS, Chole RA. A randomized clinical trial of topical gentamicin after tympanostomy tube placement. Arch Otolaryngol Head Neck Surg. 1988;114:755-757.
96. Garcia P, Gates GA, Schechtman KB. Does topical antibiotic prophylaxis reduce post-tympanostomy tube otorrhea? Ann Otol Rhinol Laryngol. 1994;103:54-58.
97. Gates GA, Avery C, Prihoda TJ, et al. Delayed onset post-tympanotomy otorrhea. Otolaryngol Head Neck Surg. 1988;98:111-115.
98. Virchow R. Uber Perlgeschwulste. Virchows Arch Pathol Anat. 1854;8:371.
99. Walsh TE, Covell WP, Ogura JH. The effect of cholesteatosis on bone. Ann Otol Rhinol Laryngol. 1951;60:1100-1113.
100. Orisek BS, Chole RA. Pressures exerted by experimental cholesteatomas. Arch Otolaryngol Head Neck Surg. 1987;113:386-391.
101. Lautenschlager A. Entstehung, Wachstums und Heilungsbedingungen, des Cholesteatoms. Klin Wochenschr. 1927;6:2101.
102. Abramson M. Collagenolytic activity in middle ear cholesteatoma. Ann Otol Rhinol Laryngol. 1969;78:112-124.
103. Pollack FJ. Pathology of chronic otitis media. Arch Otolaryngol Head Neck Surg. 1959;70:421.
104. Grippaudo M. Histopathological studies of the ossicles in chronic otitis media. J Laryngol Otol. 1958;72:177-189.
105. Ahn JM, Huang CC, Abramson M. Third place—Resident Basic Science Award 1990. Interleukin 1 causing bone destruction in middle ear cholesteatoma. Otolaryngol Head Neck Surg. 1990;103:527-536.
106. Holtrop ME, Cox KA, Glowacki J. Cells of the mononuclear phagocytic system resorb implanted bone matrix: a histologic and ultrastructural study. Calcif Tissue Int. 1982;34:488-494.
107. Thomsen J, Jorgensen MD, Bretlan P, et al. Bone resorption in chronic otitis media: histological and ultrastructural study, II: cholesteatoma. J Laryngol Otol. 1974;88:983.
108. Bernstein JM. Biological mediators of inflammation im middle ear effusions. Ann Otol Rhinol Laryngol. 1976;85(2 Suppl 25 Pt 2):90-96.
109. Bernstein JM, Hausmann M, Wright J. Middle ear disease: release of soluble factors stimulating bone resorption. In: McCabe BF, Sadé J, editors. Cholesteatoma: First International Conference. Birmingham, AL: Aesculapius Publishing, 1977.
110. Chole RA. Cellular and subcellular events of bone resorption in human and experimental cholesteatoma: the role of osteoclasts. Laryngoscope. 1984;94:76-95.
111. Chole RA. Osteoclasts in chronic otitis media, cholesteatoma, and otosclerosis. Ann Otol Rhinol Laryngol. 1988;97(6 Pt 1):661-666.
112. Chole RA, Henry KR, McGinn MD. Cholesteatoma: spontaneous occurrence in the Mongolian gerbil Meriones unguiculatis. Am J Otol. 1981;2:204-210.
113. Jung JY, Chole RA. Bone resorption in chronic otitis media: the role of the osteoclast. ORL J Otorhinolaryngol Relat Spec. 2002;64:95-107.
114. Uno Y, Saito R. Bone resorption in human cholesteatoma: morphological study with scanning electron microscopy. Ann Otol Rhinol Laryngol. 1995;104:463-468.
115. Abramson M, Huang CC. Localization of collagenase in human middle ear cholesteatoma. Laryngoscope. 1977;87(5 Pt 1):771-791.
116. Moriyama H, Honda Y, Huang CC, et al. Bone resorption in cholesteatoma: epithelial-mesenchymal cell interaction and collagenase production. Laryngoscope. 1987;97(7 Pt 1):854-859.
117. Blair HC, Kahn AJ, Crouch EC, et al. Isolated osteoclasts resorb the organic and inorganic components of bone. J Cell Biol. 1986;102:1164-1172.
118. Hamzei M, Ventriglia G, Hagnia M, et al. Osteoclast stimulating and differentiating factors in human cholesteatoma. Laryngoscope. 2003;113:436-442.
119. Chole RA. Differential osteoclast activation in endochondral and intramembranous bone. Ann Otol Rhinol Laryngol. 1993;102(8 Pt 1):616-619.
120. Yuasa R, Ise I, Kaneko T. pH of debris in cholesteatoma mastric. Otologia (Fukuoka). 1978;24(Suppl 1):163.
121. Iino Y, Toriyama M, Ohmi S, et al. Activation of peritoneal macrophages with human cholesteatoma debris and alpha-keratin. Acta Otolaryngol. 1990;109(5-6):444-449.
122. Chole RA, Hughes RM, Faddis BT. Keratin particle-induced osteolysis: a mouse model of inflammatory bone remodeling related to cholesteatoma. J Assoc Res Otolaryngol. 2001;2:65-71.
123. Banerjee AR, James R, Narula AA. Matrix metalloproteinase-2 and matrix metalloproteinase-9 in cholesteatoma and deep meatal skin. Clin Otolaryngol Allied Sci. 1998;23:345-347.
124. Banerjee AR, James R, Narula AA, et al. Matrix metalloproteinase-1 in cholesteatoma, middle ear granulations and deep meatal skin: a comparative analysis. Clin Otolaryngol Allied Sci. 1998;23:515-519.
125. Schonermark M, Mester B, Kempf HG, et al. Expression of matrix-metalloproteinases and their inhibitors in human cholesteatomas. Acta Otolaryngol. 1996;116:451-456.
126. Stark T, Sudhoff H, Fisseler-Eckhoff A, et al. Alterations of basement membrane in middle ear cholesteatoma [Abstract]. In: Sanna M, editor. Proceedings of the Fifth International Conference on Cholesteatoma and Mastoid Surgery. Roma: IC Edizioni Internationali; 1997:238.
127. Abramson M, Moriyama H, Huang CC. Pathogenic factors in bone resorption in cholesteatoma. Acta Otolaryngol. 1984;97(5-6):437-442.
128. Kahn AJ, Partridge NC. New concepts in bone remodeling: an expanding role for the osteoblast. Am J Otolaryngol. 1987;8:258-264.
129. Sakamoto S, Sakamoto M. Biochemical and immunohistochemical studies on collagenase in resorbing bone in tissue culture: a novel hypothesis for the mechanism of bone resorption. J Periodontal Res. 1982;17:523-526.
130. Lazarus GS, Jensen PJ. Plasminogen activators in epithelial biology. Semin Thromb Hemost. 1991;17:210-216.
131. Hoekman K, Lowik CW, vd Ruit M, et al. Regulation of the production of plasminogen activators by bone resorption enhancing and inhibiting factors in three types of osteoblast-like cells. Bone Miner. 1991;14:189-204.
132. Nakamura M, Yamashiro Y, Nakahodo K, et al. Plasminogen activators in tissue extract of aural cholesteatoma. Laryngoscope. 1995;105(3 Pt 1):305-310.
133. Ohsaki K, Yamashita S, Fujita A, et al. Mechanism of bone destruction due to middle ear cholesteatoma as revealed by laser-Raman spectrometry. Am J Otolaryngol. 1988;9:117-126.
134. Macri JR, Chole RA. Bone erosion in experimental cholesteatoma—the effects of implanted barriers. Otolaryngol Head Neck Surg. 1985;93:3-17.
135. Binderman I, Shimshoni Z, Somjen D. Biochemical pathways involved in the translation of physical stimulus into biological message. Calcif Tissue Int. 1984;36(Suppl 1):S82-S85.
136. Huang CC, Yi ZX, Yuan QG, et al. A morphometric study of the effects of pressure on bone resorption in the middle ear of rats. Am J Otol. 1990;11:39-43.
137. Chole RA, McGinn MD, Tinling SP. Pressure-induced bone resorption in the middle ear. Ann Otol Rhinol Laryngol. 1985;94(2 Pt 1):165-170.
138. Brackmann DE, Toh EH. Surgical management of petrous apex cholesterol granulomas. Otol Neurotol. 2002;23:529-533.
139. Moriyama H, Huang CC, Abramson M, et al. Bone resorption factors in chronic otitis media. Otolaryngol Head Neck Surg. 1984;92:322-328.
140. Adachi K, Chole RA, Yee J. Indomethacin inhibition of middle ear bone resorption. Arch Otolaryngol Head Neck Surg. 1991;117:267-269.
141. Jungkeit MC, Chole RA. Ibuprofen inhibits localized bone resorption in the middle ear. Calcif Tissue Int. 1991;48:267-271.
142. Gallwitz WE, Mundy GR, Lee CH, et al. 5-Lipoxygenase metabolites of arachidonic acid stimulate isolated osteoclasts to resorb calcified matrices. J Biol Chem. 1993;268:10087-10094.
143. Adachi K, Chole RA. Inhibition of osteoclast recruitment at a local site by 1-hydroxyethylidene-1,1-bisphosphonate (HEBP). Ann Otol Rhinol Laryngol. 1990;99(9 Pt 1):738-741.
144. Richardson AC, Tinling SP, Chole RA. Risedronate activity in the fetal and neonatal mouse. Otolaryngol Head Neck Surg. 1993;109:623-633.
145. Schilling V, Negri B, Bujia J, et al. Possible role of interleukin 1 alpha and interleukin 1 beta in the pathogenesis of cholesteatoma of the middle ear. Am J Otol. 1992;13:350-355.
146. Kakiuchi H, Kinoshita K, Katoh Y, et al. Interleukin-1 of cholesteatomatous keratinocytes. Ann Otol Rhinol Laryngol Suppl. 1992;157:32-38.
147. Costerton JW, Stewart PS, Greenberg EP. Bacterial biofilms: a common cause of persistent infections. Science. 1999;284:1318-1322.
148. Bonewald LF, Dallas SL. Role of active and latent transforming growth factor beta in bone formation. J Cell Biochem. 1994;55:350-357.
149. Mundy GR. Cytokines and growth factors in the regulation of bone remodeling. J Bone Miner Res. 1993;8(Suppl 2):S505-S510.
150. Marenda SA, Aufdemorte TB. Localization of cytokines in cholesteatoma tissue. Otolaryngol Head Neck Surg. 1995;112:359-368.
151. Fujioka O, Huang CC. Platelet-derived growth factor in middle ear cholesteatoma. Eur Arch Otorhinolaryngol. 1994;251:199-204.
152. Cheshire IM, Blight A, Ratcliffe WA, et al. Production of parathyroid-hormone-related protein by cholesteatoma cells in culture. Lancet. 1991;338:1041-1043.
153. Sudhoff H, Dazert S, Gonzales AM, et al. Angiogenesis and angiogenic growth factors in middle ear cholesteatoma. Am J Otol. 2000;21:793-798.
154. Gantz BJ. Epidermal Langerhans cells in cholesteatoma. Ann Otol Rhinol Laryngol. 1984;93(2 Pt 1):150-156.
155. Aberg B, Jontell M, Edstrom S. Analysis of class II antigen expressing cells in cholesteatoma epithelium. Acta Otolaryngol. 1988;106(3-4):186-191.
156. Berger G, Hawke M, Ekem JK. Bone resorption in chronic otitis media: The role of mast cells. Acta Otolaryngol. 1985;100(1-2):72-80.
157. Ehrlich GD, Veeh R, Wang X, et al. Mucosal biofilm formation on middle-ear mucosa in the chinchilla model of otitis media. JAMA. 2002;287:1710-1715.
158. Wang EW, Jung JY, Pashia ME, et al. Otopathogenic Pseudomonas aeruginosa strains as competent biofilm formers. Arch Otolaryngol Head Neck Surg. 2005;131:983-989.
159. Zhuang L, Jung JY, Wang EW, et al. Pseudomonas aeruginosa lipopolysaccharide induces osteoclastogenesis through a toll-like receptor 4 mediated pathway in vitro and in vivo. Laryngoscope. 2007;117:841-847.
160. Paparella MM, Brady DR, Hoel R. Sensori-neural hearing loss in chronic otitis media and mastoiditis. Trans Am Acad Ophthalmol Otolaryngol. 1970;74:108-115.
161. Chole RA, Chiu M. Cochlear hair cell loss in ears with cholesteatomas: scanning electron microscopy study. Ann Otol Rhinol Laryngol. 1988;97:78-82.
162. McGinn MD, Chole RA. Cochlear bone erosion: effects on cochlear hair cells: a scanning electron microscopy study. Ann Otol Rhinol Laryngol. 1991;100:1015-1019.
163. Meyerhoff WL, Kim CS, Paparella MM. Pathology of chronic otitis media. Ann Otol Rhinol Laryngol. 1978;87(6 Pt 1):749-760.
164. Vartiainen E, Karjalainen S. Factors influencing sensorineural hearing loss in chronic otitis media. Am J Otolaryngol. 1987;8:13-15.
165. Fria TJ, Saad MM, Doyle WJ, et al. The effect of otitis media with effusion (“secretory” otitis media) on hearing sensitivity in children. In: Lim DJ, Bluestone CD, Klein JO, editors. Recent Advances in Otitis Media with Effusion. St. Louis: Mosby, 1984.
166. Morizono T, Giebink GS, Paparella MM, et al. Sensorineural hearing loss in an animal model of purulent otitis media. In: Lim DJ, Bluestone CD, Klein JO, editors. Recent Advances in Otitis Media with Effusion. St. Louis: Mosby, 1984.
167. Browning GG, Gatehouse S. Hearing in chronic suppurative otitis media. Ann Otol Rhinol Laryngol. 1989;98(4 Pt 1):245-250.
168. MacAndie C, O’Reilly BF. Sensorineural hearing loss in chronic otitis media. Clin Otolaryngol Allied Sci. 1999;24:220-222.
169. Rahko T, Karma P, Sipila M. Sensorineural hearing loss and acute otitis media in children. Acta Otolaryngol. 1989;108(1-2):107-112.
170. Hussl B, Mueller K. Physiology and Pathophysiology of Eustachian Tube and Middle Ear. New York: Thieme-Stratton; 1980.
171. Tos M, Stangerup SE. Hearing loss in tympanosclerosis caused by grommets. Arch Otolaryngol Head Neck Surg. 1989;115:931-935.
172. Daly K. Risk factors for otitis media sequelae and chronicity. Ann Otol Rhinol Laryngol Suppl. 1994;163:39-42.
173. Kinney SE. Postinflammatory ossicular fixation in tympanoplasty. Laryngoscope. 1978;88:821-838.
174. Mangat KS, Morrison GA, Ganniwalla TM. T-tubes: a retrospective review of 1274 insertions over a 4-year period. Int J Pediatr Otorhinolaryngol. 1993;25(1-3):119-125.
175. Hussl B, Lim DJ. Histopathology of tympanosclerosis. In: Lim DJ, Bluestone CD, Klein JO, editors. Recent Advances in Otitis Media with Effusion. St. Louis: Mosby, 1984.
176. Wielinga EW, Kuijpers W, Tonnaer EL, et al. An experimental model for tympanosclerosis: a preliminary report. Acta Otolaryngol. 1988;105(5-6):537-542.
177. Chole RA, Henry KR. Otosclerotic lesions in the inbred LP/J mouse. Science. 1983;221:881-882.
178. Brodie HA, Chole RA. The possible role of immunologic injury in the dysplastic bony lesion in LP/J mice. Am J Otolaryngol. 1987;8:342-350.
179. Russell JD, Giles JJ. Tympanosclerosis in the rat tympanic membrane: an experimental study. Laryngoscope. 2002;112:1663-1666.
180. Smyth GD, Patterson CC, Hall S. Tympanostomy tubes: do they significantly benefit the patient? Otolaryngol Head Neck Surg. 1982;90:783-786.
181. Gormley PK. Stapedectomy in tympanosclerosis: a report of 67 cases. Am J Otol. 1987;8:123-130.
182. Smyth GD. Tympanosclerosis. J Laryngol Otol. 1972;86:9-14.
183. Gradenigo G. Über die Paralyses des Nervus Abducens bei Otitis. Archiv Ohrenheilkunde. 1907;774:149.
184. Chandrasekhar SS, Siverls V, Sekhar HK. Histopathologic and ultrastructural changes in the temporal bones of HIV-infected human adults. Am J Otol. 1992;13:207-214.
185. Chole RA, Donald PJ. Petrous apicitis: clinical considerations. Ann Otol Rhinol Laryngol. 1983;92(6 Pt 1):544-551.
186. Goris A. Un cas de chirurgie pour comp d’otite mayenne chronique. Annales des maladies di l’oreille et du larynx. 1903;29:64.
187. Diamant M. Otitis and pneumatization of the mastoid bone. Acta Otolaryngol. 1940;41(Suppl):1.
188. Almour R. Surgical therapy for the release of suppurations of the petrous pyramid. Laryngoscope. 1931;41:405.
189. Kopetzky SJ, Almour R. Suppuration of the petrous pyramid: symptomatology, pathology and surgical treatment. Ann Otol Rhinol Laryngol. 1931;40:396.
190. Ramandier J. Exploration de la pointe du rocher par la voie du canal carotidien. Ann d’Otolaryngol. 1933;4:422.
191. Lempert J. Complete apicectomy (mastoidotympano-apicectomy): new technic for complete apical exenteration of apical carotid portion of petrous pyramid. Arch Otolaryngol Head Neck Surg. 1937;25:144.
192. Lindsay JR. Suppuration in the petrous pyramid. Ann Otol Rhinol Laryngol. 1938;47:3.
193. Hendershot EL, Wood JW. The middle fossa approach in the treatment of petrositis. Arch Otolaryngol. 1973;98:426-427.
194. Chole RA. Petrous apicitis: surgical anatomy. Ann Otol Rhinol Laryngol. 1985;94:251-257.
195. Hentona H, Ohkubo J, Tsutsumi T, et al. [Pneumatization of the petrous apex]. Nippon Jibiinkoka Gakkai Kaiho. 1994;97:450-456.
196. Dorello P. Über die Ursache der transitorischen Abduzenslahmung. Int Zentralblatt fur Ohrenheil. 1906.
197. Chole RA, Gadre AK. Petrous apicitis: symptomatology, pathology and management [Abstract]. Skull Base Surgery Symposium, Sacramento, 1995.
198. Roland PS, Meyerhoff WL, Judge LO, et al. Asymmetric pneumatization of the petrous apex. Otolaryngol Head Neck Surg. 1990;103:80-88.
199. Mosnier I, Cyna-Gorse F, Grayeli AB, et al. Management of cholesterol granulomas of the petrous apex based on clinical and radiologic evaluation. Otol Neurotol. 2002;23:522-528.
[/level-membership-for-otolaryngology-category][not-level-membership-for-otolaryngology-category]
CHAPTER 139 Chronic Otitis Media, Mastoiditis, and Petrositis
Some cases of acute otitis media (AOM) result in persistent otitis media with effusion (OME), which is recognized as the leading cause of childhood hearing loss. The exact cause of OME is unclear; however, more recent data suggest that reflux of gastric contents may be associated with OME in children.1–4 Although eustachian tube dysfunction alone may lead to effusion of the middle ear, there is mounting evidence that most cases of OME occur as a sequelae of AOM, or at least share the same etiologic factors. Specific causes can be identified in many cases of adult-onset otitis media, such as paranasal sinus disease, nasopharyngeal carcinomas and tumors, and postradiation sequelae.
Effects on Mastoid Pneumatization
It has been observed that patients with a history of chronic OME have more sclerotic mastoids with decreased pneumatization compared with healthy subjects. There are two theories to explain this observation: (1) the hereditary theory, which states that children with hypoaeration of the mastoid are prone to OME, and (2) the environmental theory, which states that chronic OME results in hypopneumatization of the mastoid.5 Although measurable correlations between mastoid hypocellularity and OME,6,7 and between the length of the mastoid process or the degree of pneumatization and an abnormal eardrum8 have been proven, a cause-and-effect relationship is unclear. Available evidence generally supports the concept that chronic inflammation in early childhood may lead to new bone formation within the middle ear and mastoid and, subsequently, decreased size of mastoid air cells. Shatz and Sadé9 measured the distance from the lateral sinus to the external auditory canal and found it to be significantly smaller in patients with sclerotic mastoids; they believed that this finding supported the hereditary theory because it was unlikely that otitis would change the position of the lateral sinus. A chronically inflamed mastoid in a young child may not develop normally, however.
Hasebe and colleagues10 carried out a study to establish which type of cholesteatoma is controllable by conservative treatment from the viewpoint of mastoid ventilation. Their clinical observations suggest that progressiveness of cholesteatoma is related to the ventilatory conditions in the mastoid, rather than eustachian tube function, and that conservative treatment may be effective if ears with cholesteatoma have aeration in the mastoid.
Middle Ear Atelectasis and Adhesive Otitis Media
Middle ear atelectasis (Fig. 139-1) is thought to result mainly from long-standing eustachian tube dysfunction. One of the main functions of the eustachian tube is ventilation of the middle ear and mastoid. Opening of the eustachian tube allows exchanging of gases and equalization between the environment and middle ear. The middle ear gases also are exchanged with the middle ear mucosa. Bilateral diffusion between the middle ear cavity and the blood may be an important factor in middle ear atelectasis because the gas composition of the middle ear basically resembles that of venous blood.11
If the atelectasis develops, the tympanic membrane becomes retracted onto the promontory and the ossicles of the middle ear. In atelectatic ears, the middle ear space is partially or completely obliterated, but the tympanic membrane is not adherent to the medial wall of the middle ear, and the mucosal lining of the middle ear is intact. In contrast, adhesive otitis media exists when the middle ear space is totally obliterated, and the tympanic membrane is adherent to the ossicles and promontory; mucosal surfaces are not present. Retraction of the tympanic membrane may lead to erosion of the long process of the incus and the stapes suprastructure (Fig. 139-2).
Not all patients with chronic OME develop atelectasis; in most patients with OME, retraction of the tympanic membrane is limited. In patients with bilateral OME, 1.5% of untreated ears and 2% of ears treated with tubes developed severe atelectasis.12 It may be that repeated bouts of AOM lead to weakening and thinning of the membrane, which allows atelectasis. Sadé and Berco13 showed destruction of the collagen-containing fibrous layer of the tympanic membrane in some ears with recurrent infection. Collagen destruction within the tympanic membrane may lead to another complication of OME—tympanosclerosis. Sadé and Berco13 and Tos and Poulsen14 described four stages of tympanic membrane retraction: stage I, retracted tympanic membrane; stage II, retraction with contact onto the incus; stage III, middle ear atelectasis; and stage IV, adhesive otitis media (Fig. 139-3).
Figure 139-3. Four stages of middle ear atelectasis.
(Adapted from Sadé J, Berco E. Atelectasis and secretory otitis media. Ann Otol Rhinol Laryngol. 1976;85(Suppl 25):66.)
Middle ear atelectasis may be reversible with ventilating tubes. Sadé15 showed that ventilating tubes improved the state of atelectatic ears. Graham and Knight16 reported three cases in which atelectatic tympanic membranes were restored to their normal position by administration of nitrous oxide during anesthesia and insertion of a ventilating tube.
Atelectasis and adhesive otitis media usually coexist with OME, although OME may resolve in these ears, allowing aeration of the attic and mastoid, but leaving a collapsed middle ear. In extreme cases, when hearing loss or ossicular erosion occurs, a myringoplasty for the reinforcement of atelectatic tympanic membrane may be indicated.6,17 Cholesteatomas may originate from deep retraction pockets in which desquamated keratin debris would not be cleared into the ear canal.18,19 These retraction pockets may occur in the pars tensa or pars flaccida of atelectatic ears, and should be considered precursors to cholesteatomas (see discussion of cholesteatoma). Nonpneumatized mastoids may have a limited ability to buffer pressure changes and can manifest as an atelectasis, a retraction pocket, or a cholesteatoma.20
Chronic Otitis Media with Cholesteatoma
Aural cholesteatomas are epidermal inclusion cysts of the middle ear or mastoid (in the case of a retraction pocket cholesteatoma, the “cyst” opens into the external auditory canal). They contain the desquamated debris (principally keratin) from their keratinizing, squamous epithelial lining. Cruveilhier21 first described aural cholesteatoma as a “pearly tumor” of the temporal bone. The term cholesteatoma, coined by the German physiologist Müller22 in 1838, is a misnomer because this entity does not contain cholesterol; the white-yellow keratin flakes found within cholesteatomas grossly resemble cholesterol crystals. Cholesteatomas of the temporal bone may be congenital or acquired. Acquired cholesteatomas are the consequence of OME, AOM, or both. An understanding of the pathogenesis and pathophysiology of aural cholesteatoma is particularly important because the destructive nature of this entity is responsible for much of the morbidity associated with chronic otitis media. The propensity of cholesteatomas to erode bone and the lack of effective, nonsurgical management add importance to the understanding of this disease.
Diagnosis
The diagnosis of aural cholesteatoma is made on otoscopic examination, including endoscopic and microscopic evaluation or surgical exploration. Special imaging procedures, such as high-resolution computed tomography (CT) and magnetic resonance imaging (MRI), may suggest the presence of cholesteatomas within the temporal bone, and may be used to complement the clinical examination. High-resolution CT is useful for operative planning and is recommended for all revision mastoid operations. The symptoms of cholesteatoma vary; some cholesteatomas are asymptomatic, whereas others become infected and rapidly cause bone destruction. Some patients seen by the physician show slowly progressive conductive hearing loss, and frequently patients with cholesteatomas consult a physician because of chronic otitis with purulent otorrhea. The otorrhea from an infected cholesteatoma often is malodorous because of the frequent infection with anaerobic bacteria.23
The otoscopic appearance of an aural cholesteatoma also varies. A typical attic retraction cholesteatoma (Fig. 139-4) appears as a defect of variable size adjacent to the posterosuperior portion of the tympanic membrane. The center of the defect contains keratin debris (primary acquired cholesteatoma). In other patients, keratinizing epithelium has migrated through a perforation into the middle ear (secondary acquired cholesteatoma) (Fig. 139-5). Cholesteatomas sometimes appear behind or within an intact tympanic membrane—so-called congenital cholesteatoma (Fig. 139-6). These cholesteatomas have been considered to be congenital, but more recent experimental evidence raises the possibility that they may originate during an inflammatory process.24 An infected cholesteatoma sometimes manifests as an “aural polyp.” These “polyps” actually are granulation tissue at the junction between an eroding cholesteatoma and bone. The presence of an aural polyp in a chronically infected ear should be considered to be a cholesteatoma until proved otherwise. Occasionally, a cholesteatoma cannot be seen otoscopically, but is discovered during tympanomastoid surgery.
Figure 139-4. Primary acquired cholesteatoma in the region of the pars flaccida with scutum erosion.
Figure 139-5. Cholesteatoma developing at the margin of perforation (secondary acquired cholesteatoma) with secondary infection.
The exact prevalence of cholesteatoma is unknown. In 1978, there were 4.2 hospital discharges per 100,000 population with cholesteatoma.25 In addition, there were 13.8 hospital discharges per 100,000 population with chronic otitis media without cholesteatoma. Studies by Harker23 documented an annual incidence of 6 cholesteatomas per 100,000 population. Tos26 found an annual incidence of 3 cholesteatomas in children and 12.6 cholesteatomas in adults per 100,000 population. In the human temporal bones with chronic otitis media, cholesteatoma was observed in 36% of ears with perforations and in 4% of ears without the perforated tympanic membranes.27
Pathogenesis
Congenital cholesteatomas, by definition, originate from areas of keratinizing epithelium within the middle ear cleft. Michaels28 showed that a small area in the anterior tympanum in the developing fetus often contains a small area of keratinizing epithelium. He found epidermoid formation in 37 of 68 temporal bones of fetuses at 10 to 33 weeks’ gestation. Congenital cholesteatomas may originate in this region. Although congenital cholesteatomas generally are seen as pearl-like masses behind an intact membrane, Koltai and coworkers29 presented a convincing case that some congenital cholesteatomas advance to perforate and become chronically infected, taking on the appearance of an acquired cholesteatoma. Potsic and colleagues,30 in a review of 172 series of congenital cholesteatomas, developed a useful staging system: stage I, limited to one quadrant; stage II, involving multiple quadrants without ossicular involvement; stage III, ossicular involvement without mastoid extension; and stage IV, mastoid involvement. They showed a correlation between stage and risk of residual disease; stage IV carries a 67% risk of residual cholesteatoma.
The pathogenesis of acquired cholesteatoma has been debated for more than a century. There are four basic theories of the pathogenesis of acquired aural cholesteatoma: (1) invagination of the tympanic membrane (retraction pocket cholesteatoma), (2) basal cell hyperplasia, (3) epithelial ingrowth through a perforation (the migration theory), and (4) squamous metaplasia of middle ear epithelium (Fig. 139-7). Additionally, Sudhoff and Tos19 proposed a combination of the invagination and basal cell theories as an explanation for retraction pocket cholesteatoma formation.
Invagination Theory
The invagination theory5 of the genesis of cholesteatoma generally is regarded as one of the primary mechanisms of the formation of attic cholesteatomas. Retraction pockets of the pars flaccida deepen because of negative middle ear pressure and possibly repeated inflammation (see Fig. 139-2). As the retraction pocket deepens, desquamated keratin cannot be cleared from the recess, and a cholesteatoma results. The origin of such retraction pocket cholesteatomas is thought to be eustachian tube dysfunction (or OME) with resultant negative middle ear pressure (ex vacuo theory). Usually, the pars flaccida, being less fibrous and less resistant to displacement, is the source of the cholesteatoma.
The result of this type of cholesteatoma (so-called primary acquired cholesteatoma) is an apparent defect in the posterosuperior quadrant of the tympanic membrane and erosion of the adjacent canal wall.31 Although these defects have the appearance of a marginal perforation, it is not a perforation, but rather an invagination. Sadé20 showed that epithelial migration patterns within attic retraction pockets are altered. This failure of epithelial migration may allow the accumulation of keratin within a retraction pocket, with subsequent enlargement merely from the accumulation of keratin within a relatively closed space. This theory has been supported by the experimental creation of retraction pockets by use of the eustachian tube obstruction,32 and external auditory canal ligation.33
Ruah and colleagues31 suggested that inflammation of middle ear and persistent mesenchyme lead to a greater inflammatory reaction in pars flaccida and posterosuperior quadrant of the tympanic membrane of the human temporal bones with serous and purulent otitis media in children. These findings support this theory of primary acquired cholesteatoma in children. Retraction pockets are considered as precursors to cholesteatoma. Bacteria can infect the keratin matrix, forming biofilms leading to chronic persistent infection. The presence of bacterial biofilms in the cholesteatoma matrix may lead to epithelial proliferation and invasion of the cholesteatoma.34,35
Basal Cell Hyperplasia Theory
Another possible mechanism for the histogenesis of cholesteatoma was suggested by Lange in the 1920s.49 In this theory, he proposed that epithelial cells (prickle cells) of the pars flaccida could invade the subepithelial tissue by means of proliferating columns of epithelial cells. Nearly 40 years later, Ruedi50 supported this hypothesis with clinical and experimental evidence. For epithelium to invade into the lamina propria, the basal lamina (basement membrane) should be altered. Basal lamina disruptions now have been documented in human51,52 and animal53 cholesteatomas.
Huang and colleagues54 and Masaki and coworkers55 provided experimental support of this theory by showing that epithelial ingrowth from the tympanic membrane can be induced by instillation of propylene glycol into the middle ear of chinchillas. These basal lamina breaks allow the invasion of epithelial cones into the subepithelial connective tissue and the formation of microcholesteatomas. This mechanism may explain some types of human cholesteatomas, even those occurring behind an intact tympanic membrane.56 According to this theory, microcholesteatomas may enlarge and then perforate secondarily through the tympanic membrane, leaving the typical appearance of an attic cholesteatoma. This sequence of events has not been documented, although the alternations in the differentiation of keratinocytes and basal cell layer of cholesteatoma matrix have been observed in several studies.
Abnormal distribution of epidermal differentiation markers, such as filaggrin and involucrin,57 c-jun and p53 proteins,58 and increased epidermal growth factor receptor,59,60 has been shown in middle ear cholesteatoma matrix. Increased levels of proteins cytokeratin 13 and cytokeratin 16, which are markers for differentiation and hyperproliferation, were also found.61 Kim and Chole39 showed the increased expression of cytokeratin 13 and cytokeratin 16 in the area of the peripheral area of the pars tensa of induced cholesteatoma by ear canal ligation and in the peripheral and central area of pars tensa of induced cholesteatoma by eustachian tube obstruction. Sakamoto and associates62 found ErbB-2 protein to be overexpressed, and cell proliferation and apoptosis of keratinocytes accelerated. Caspases play a key role in apoptosis; Miyao and coworkers63 suggested that caspase-8, which is activated by the induction of tumor necrosis factor-α, leads to activation of caspase-3, which activates apoptotic nucleases in cholesteatoma tissue.
Human toll-like receptors are crucial in the induction and activation of innate immunity in the course of an infection. Expression of toll-like receptor–3 on the epithelium and some cells within the perimatrix and the presence of T cells may suggest that apart from innate immune responses, mechanisms of adaptive immunity also operate in cholesteatoma.64 Data from Parisier and colleagues65 suggested that fibroblasts in the subepithelium of cholesteatomas showed an invasive phenotype, whereas fibroblasts from postauricular and ear canal skin appeared either weakly invasive or not invasive. In a similar study, Chole and colleagues66 found that normal fibroblasts and fibroblasts from induced cholesteatomas did not exhibit the invasive phenotype characteristics of true neoplastic cells.
Other lines of evidence support the basal cell hyperplasia/migration theory. Increased expression of human intercellular adhesion molecule-1 and intercellular adhesion molecule-2 has been shown, suggesting a role in cell migration into tissue.40 The presence of heat shock proteins 60 and 70 suggested proliferation and active differentiation of basal keratinocytes in cholesteatomas.58 There are some reports that immune response is involved in the hyperproliferative state of cholesteatoma epithelium.52,67,68 Langerhans cells may initiate immune reaction and promote proliferation of keratinizing epithelium via an interleukin (IL)-1α and transforming growth factor (TGF)-β mechanism.42,67,69
Epithelial Invasion Theory
The epithelial invasion theory36 states that keratinizing squamous epithelium from the surface of the tympanic membrane invades or migrates into the middle ear from a perforation in the tympanic membrane. This theory is supported by clinical observation and experimental evidence. Weiss37 showed that epithelial cells could migrate along a surface by a process that he called contact guidance, and that, when they encounter another epithelial surface, they stop migrating, for which he used the term contact inhibition. van Blitterswijk and Grote38 reported that cytokeratin 10, which was seen in the meatal epidermis and migrating epithelium, was preferentially expressed in the cholesteatoma matrix, rather than in the middle ear mucosa. This finding suggests an epidermal origin of cholesteatoma. Kim and Chole39 showed increased cytokeratin 10 expression in the peripheral area of the pars tensa of induced cholesteatoma by ear canal ligation and in the peripheral and the central area of the pars tensa of induced cholesteatoma by eustachian tube obstruction. The findings of this study also support the basal cell hyperplasia hypotheses for the pathogenesis of aural cholesteatoma, with regard to hyperproliferation, migration, and an altered differentiation of keratinocytes.39 High levels of fibronectin and tenascin and focal disruptions of the basement membrane, were reported in the middle ear cholesteatoma, supporting the concept of the invasion theory.40–43
This theory also is supported by investigations in animal cholesteatoma models and human temporal bones. Jackson and Lim44 gave histologic and ultrastructural evidence that keratinizing epithelium can migrate into the cat bulla by contact guidance. It is likely that in some tympanic membrane perforations, inflammation damages the inner mucosal lining of the tympanic membrane, allowing the outer keratinizing epithelium to migrate inward and generate a cholesteatoma. Hueb and associates45 showed similar evidence in chinchillas. Palva and colleagues46 showed histologic evidence for this theory in human temporal bones. Cholesteatomas originating after temporal bone fractures may result from this mechanism; fractures within the ear canal may allow ingrowth of keratinizing epithelium by contact guidance.47 7,12-Dimethylbenz[a]anthracene, a chemical carcinogen, could induce the advancing of the keratinizing squamous epithelium into or under the mucosal layer, and ingrowth and spreading over the middle ear cavity and eustachian tube in the rat ear.48
Squamous Metaplasia Theory
Wendt70 theorized that the simple squamous or cuboidal epithelium of the middle ear cleft could undergo a metaplastic transformation into keratinizing epithelium. Sadé15,71 supported this theory, noting that epithelial cells are pluripotent and can be stimulated by inflammation to become keratinizing. According to this theory, an area of keratinizing epithelium within the middle ear would enlarge because of accumulated debris and contact with the tympanic membrane. With intercurrent infection and inflammation, the cholesteatoma would lead to lysis of the tympanic membrane and perforation, resulting in the typical appearance of an attic cholesteatoma. This theory is supported by the demonstration that biopsy specimens from the middle ear of children with OME sometimes contain islands of keratinizing epithelium.15
Some experimental evidence supports the contention that middle ear mucosa can become metaplastic and keratinize. Chole and Frush72 showed that extreme vitamin A deficiency leads to the formation of keratinizing epithelium within the middle ear and eustachian tube of rats. None of their experimental animals had developed cholesteatomas. Consequently, there is no direct evidence that cholesteatomas arise by squamous metaplasia of the middle ear mucosa.
From a clinical perspective, it seems that each of these pathogenic mechanisms accounts for a proportion of acquired cholesteatomas. Regardless of the pathogenesis of aural cholesteatomas, they all share certain properties. Cholesteatomas are prone to recurrent infection, and they characteristically erode the bone of the ossicles and the otic capsule. Aural cholesteatomas originating from the vicinity of the tympanic membrane exhibit typical growth patterns into the temporal bone. Because most acquired cholesteatomas originate by invagination of the pars flaccida, their growth is limited by the mucosal folds and suspensory ligaments of the ossicles. The pars flaccida may invaginate into the lateralmost portion of the epitympanum (Prussak’s space) and then into the recesses of the epitympanum posteriorly, lateral to the body of the incus, inferiorly into the middle ear by way of the pouch of von Tröltsch (Fig. 139-8), or anteriorly into the protympanum (Fig. 139-9).73–75
Complications
The expansion of cholesteatoma may result in bone erosion of the ossicles, otic capsule, fallopian canal, tegmen tympani, and tegmen mastoideum. These complications may cause intracranial complications (Box 139-1). The erosion of ossicles, most commonly in the incus, may result in conductive hearing loss. The severity of hearing loss is related to the status of the ossicles and the position of the cholesteatoma sac. Erosion of the otic capsule occurs most commonly in the lateral semicircular canal and rarely in the cochlea. A labyrinthine fistula has been reported in 10% of cholesteatomas in adults and children,76 which may result in sensorineural hearing loss and vertigo. Sensorineural hearing loss may result from the secondary suppurative labyrinthitis or from the cochlear hair cell loss adjacent to cholesteatoma.66 Facial nerve paralysis may occur acutely as a result of infection or slowly as a result of expansion of the cholesteatoma. Erosion of the tegmen tympani or the tegmen mastoideum may lead to development of a brain hernia or cerebrospinal fluid leakage.77
Because cholesteatomas contain keratin debris enclosed in a tissue space, they are subject to recurrent infection. The bacteria found in infected cholesteatomas differ from bacteria found in AOM or OME. Significant anaerobic bacteria are present. The most common aerobic bacterium is Pseudomonas aeruginosa, and the most common anaerobic microorganism is Bacteroides sp (Table 139-1).78
Table 139-1 Bacteriology of Infected Cholesteatomas in 30 Children and Adults
| Bacteria | No. of Cases |
|---|---|
| Aerobes | |
| Pseudomonas aeruginosa | 11 |
| Pseudomonas fluorescens | 2 |
| Streptococcus sp | 8 |
| Proteus sp | 4 |
| Escherichia coli | 4 |
| Klebsiella, Enterobacter, Serratia | 4 |
| Alcaligenes, Achromobacter | 3 |
| Staphylococcus epidermidis | 2 |
| Staphylococcus aureus | 1 |
| CBC group F | |
| Anaerobes | |
| Bacteroides sp | 13 |
| Peptococcus, Peptostreptococcus | 11 |
| Propionibacterium acnes | 8 |
| Fusobacterium sp | 4 |
| Bifidobacterium sp | 3 |
| Clostridium sp | 3 |
| Eubacterium sp | 2 |
Modified from Harker LA, Koontz FP. The bacteriology of cholesteatomas. In: McCabe BF, Sadé J, Abramson M, eds. Cholesteatoma: First International Conference. New York: Aesculapius Publishers; 1977.
Management
Congenital and acquired cholesteatomas can be eradicated from the temporal bone only by surgical resection. The goals of surgery are to eradicate disease and manage complications and, secondarily, to reconstruct the middle ear. The decision whether to perform surgery depends on the nature and extent of disease, the existence of complications, mastoid pneumatization, eustachian tube function, hearing status of both ears, the reliability of the patient, and the experience and skill of the surgeon.77,79 Surgical approaches include atticotomy, simple mastoidectomy, canal wall-up or canal wall-down procedures, radical mastoidectomy, modified radical mastoidectomy, and Bondy procedure.
The open (canal wall-down) and the closed (intact canal with facial recess) procedures have advantages and disadvantages (Table 139-2). The reported results of both procedures vary. Residual disease and recurrent disease occur in 11% to 27% and 5% to 13% of patients undergoing the closed procedure, whereas residual or recurrent disease occurs in 2% to 10% of patients undergoing the open procedure.80 In the cases of labyrinthine fistula, facial nerve paralysis, and intracranial complications, surgery should be performed as soon as possible.
Table 139-2 Advantages and Disadvantages of Canal Wall-Up and Canal Wall-Down Procedures in Chronic Otitis Media with Cholesteatoma
| Advantages | Disadvantages |
|---|---|
| Canal Wall-Up | |
| Physiologic position of tympanic membrane | Residual and recurrent cholesteatoma may occur |
| Enough middle ear space | Incomplete exteriorization of facial recess |
| No mastoid cavity problem | Second-stage operation often required |
| Canal Wall-Down | |
| Residual cholesteatoma easily found on follow-up evaluation | Mastoid cavity problem often |
| Recurrent cholesteatoma rare | Middle ear shallow and difficult to reconstruct |
| Total exteriorization of facial recess | Position of pinna may be altered; second-stage operation sometimes required |
Modified from Harker LA, Koontz FP. The bacteriology of cholesteatomas. In: McCabe BF, Sadé J, Abramson M, eds. Cholesteatoma: First International Conference. New York: Aesculapius Publishers; 1977.
In some patients, a cholesteatoma can be débrided of entrapped keratin by direct removal or by irrigation. In some cases, surgical intervention is impossible or not advisable; the patient may not be medically able to withstand surgery, or the risks of surgery may not outweigh the benefits in some patients with only-hearing ears. Irrigation with 1 :1 : 1 distilled white vinegar, distilled water, and 70% isopropyl alcohol may keep some cholesteatomas stable if their opening into the ear canal is sufficiently large (Box 139-2). If surgery is required for a cholesteatoma in an only-hearing ear, careful preoperative evaluation and operative planning, perioperative use of antibiotics and steroids, ossicular reconstruction, and postoperative care should be considered.81
Chronic Otitis Media without Cholesteatoma
Acute or recurrent infection of the middle ear may result in a permanent perforation of the tympanic membrane. Ears with chronic perforations without cholesteatoma may be chronically or intermittently infected. Three times as many operations were performed in the United States for this disease as were performed for cholesteatomas.25 Paparella and Kim81a reported that of 375 primary tympanomastoid operations for chronic mastoiditis, two thirds were performed in ears with granulation tissue and without cholesteatoma.
Diagnosis
Tympanic membrane perforation (Fig. 139-10) may result from AOM, chronic otitis media, or trauma (injury or surgery). In some instances, a dry, simple perforation results from a single episode of AOM (i.e., necrotizing otitis media). Perforation of the tympanic membrane, especially involving the tympanic anulus, may allow ingrowth of the keratinizing epithelium of the ear canal or tympanic membrane, leading to cholesteatoma. An ear with a simple perforation may become infected because of contamination from the ear canal or because of a smoldering infection in the mastoid. A simple perforation commonly is seen as a low-frequency conductive hearing loss clinically. This finding is supported by experimental perforation in rats.82 The tympanic membrane velocity was found to be decreased in the low frequency in a small perforation and in the high and low frequencies in a large perforation.83
Pathogenesis
Chronic otomastoiditis without cholesteatoma is marked by the presence of irreversible inflammatory changes within the middle ear and mastoid. The factors that allow acute infections within the middle ear and mastoid to develop into chronic infections are unclear. da Costa and colleagues27 found granulation tissue in 96%, ossicular changes in 96%, tympanosclerosis in 43%, cholesteatoma in 36%, and cholesterol granuloma in 21% of the human temporal bones of patients with chronic otitis media with a perforated tympanic membrane (Table 139-3
[/not-level-membership-for-otolaryngology-category]
