Chapter 41 Chronic Obstructive Pulmonary Disease
Epidemiology, Pathophysiology, and Clinical Evaluation
Definitions and Diagnostic Considerations
The diagnosis of COPD should be considered in any person with the following:
Pathology
Although the clinical and physiologic presentation of chronic asthma may be indistinguishable from COPD, the pathologic changes are distinct from those in most cases of COPD, largely because of cigarette smoking. The histologic features of COPD in the 15% to 20% of COPD patients who are nonsmokers have not yet been studied in detail. Although complex, the pathology of COPD can be simplified by considering separate disease sites in which pathologic changes occur in smokers to produce a clinical pattern of largely fixed airflow limitation (Box 41-1). The clinicopathologic picture is complicated because chronic bronchitis, bronchiolitis, and emphysema may exist in an individual patient, resulting in the clinical and pathophysiologic heterogeneity seen in patients with COPD.
Box 41-1 Chronic Obstructive Pulmonary Disease (COPD)
Pathologic Changes
Proximal Airways (cartilaginous airways >2 mm in diameter)
Macrophages and CD8 T lymphocytes
Few neutrophils and eosinophils (neutrophils increase with progressive disease)
Submucosal bronchial gland enlargement and goblet cell metaplasia (results in excessive mucus production or chronic bronchitis)
Cellular infiltrates (neutrophils and lymphocytes) of bronchial glands
Airway epithelial squamous metaplasia, ciliary dysfunction, increased smooth muscle and connective tissue
Peripheral Airways (noncartilaginous airways <2 mm in diameter)
Macrophages and T lymphocytes (CD8+ > CD4+)
Few neutrophils or eosinophils
Pathologic extension of goblet cells and squamous metaplasia into peripheral airways
Luminal and inflammatory exudates
B lymphocytes, lymphoid follicles, and fibroblasts
Peribronchial fibrosis and airway narrowing with progressive disease
Lung Parenchyma (respiratory bronchioles and alveoli)
Macrophages and CD8+ T lymphocytes
Alveolar wall destruction caused by loss of epithelial and endothelial cells
Development of emphysema (abnormal enlargement of air spaces distal to terminal bronchioles)
Microscopic emphysematous changes
Chronic Bronchitis
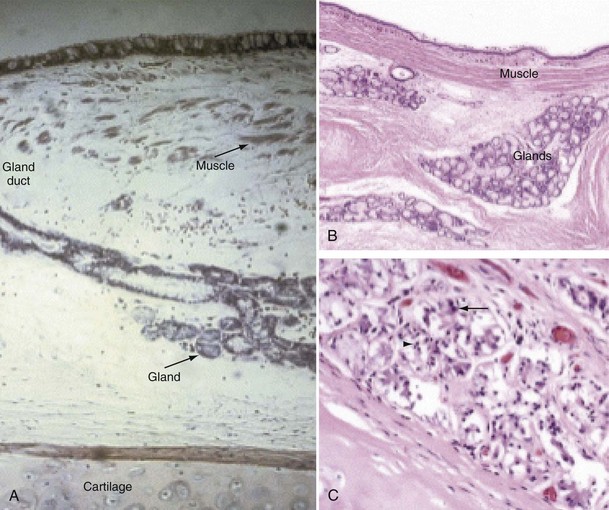
Mucus is produced by mucous glands present in the larger airways and by goblet cells in the airway epithelium. Chronic bronchitis is characterized by hypertrophy of the mucous glands (Figure 41-1). Goblet cells that occur predominantly in the surface epithelium of the larger airways increase in number and change in distribution, extending more peripherally. Bronchial biopsy studies confirm findings in resected lungs and show bronchial wall inflammation in chronic bronchitis. Activated T lymphocytes are prominent in the proximal airway walls, with a predominance of the CD8 suppressor T lymphocyte subset, rather than the CD4 subset, as seen in asthma. Macrophages are also prominent. Sputum volume correlates with the degree of inflammation in the airway wall. Neutrophils are present, particularly in the bronchial mucus-secreting glands (Figure 41-1), and become more prominent as the disease progresses. In stable chronic bronchitis, the high percentage of intraluminal neutrophils is associated with the presence of neutrophil chemotactic factors, including interleukin-8 (IL-8) and leukotriene B4 (LTB4). Elastase released from these cells is a potent stimulant for the secretion of mucus. Macrophages and CD8+ T cells also accumulate in the mucous glands.
Small Airways Disease and Bronchiolitis
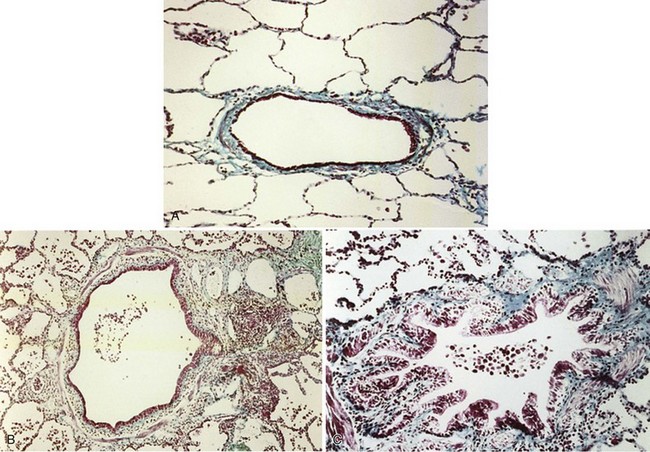
The smaller bronchioles (<2 mm in internal diameter) normally contribute relatively little to the total airway resistance, because there are so many airways of this size in parallel. Considerable narrowing of these airways can occur before pulmonary function becomes impaired and symptoms develop. Small airways inflammation is one of the earliest changes in asymptomatic cigarette smokers. The inflammatory cell profiles in the small airways are similar to those in larger airways, including the predominance of CD8+ lymphocytes, increase in CD8/CD4 ratio, and increased macrophages. Mucosal ulceration, goblet cell hyperplasia, and squamous cell metaplasia may be present, as well as mesenchymal cell accumulation and fibrosis. With progression of the condition, structural remodeling may occur, characterized by increased collagen content and scar tissue formation that narrows the airways and produces fixed airway obstruction (Figure 41-2).
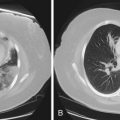
Emphysema
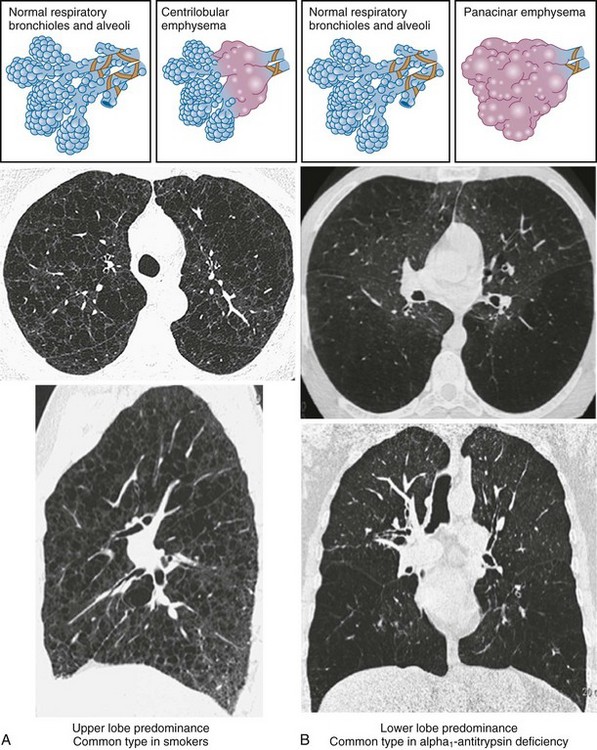
• Centrilobular (or centriacinar) emphysema, in which large air spaces are initially clustered around the terminal bronchiole (Figure 41-3, A).
• Panlobular (or panacinar) emphysema, where the large air spaces are distributed throughout the acinar unit (Figure 41-3, B).
Etiology
Risk Factors
Cigarette Smoking
In adulthood the effect of smoking on FEV1 decline is well known. In general there is a significant dose-response effect, with smokers having lower lung function the more and the longer they smoke. There is, however, considerable variation. Most longitudinal studies indicate that the decline in FEV1 in smokers ranges from 45 to 90 mL per year, in contrast to the normal 30 mL/yr (Figure 41-4). However, values vary considerably among individuals, and some experience significantly greater decline, at least temporarily, which may explain why COPD may seem to surface over a short period in the fifth and sixth decades of life. Some nonsmokers have impaired lung function, and 15% to 20% of COPD patients are lifelong nonsmokers. Conversely, some heavy smokers are able to maintain normal lung function, although the frequently quoted “15% to 20%” of smokers who are thought to develop clinically significant COPD is probably an underestimate. About 35% of smokers with normal lung function initially developed COPD during a 25-year follow-up in the Copenhagen City Heart Study.
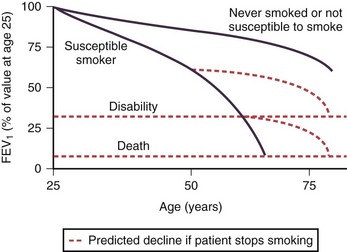
Pipe and cigar smokers have significantly greater morbidity and mortality from COPD than nonsmokers, although the risk is less than that from cigarettes. There is a trend to an increased relative risk of chronic airflow limitation from passive smoking, but the effect is not powerful enough to demonstrate clinical significance. Epidemiologic studies have associated cessation of smoking with a decrease in the prevalence of respiratory symptoms and improvement in the subsequent decline in FEV1 (Figure 41-4). The first effect on lung function after smoking cessation is a small increase of 50 to 100 mL in FEV1. There is some debate on whether decline in FEV1 after smoking cessation completely normalizes, although in general those who quit smoking continue to have an FEV1 decline slightly larger than in those who never smoked.
Host Factors
Genetic Factors
Chronic obstructive pulmonary disease is a prime example of a condition of gene-environment interaction. The observation of a familial association for an increased risk of airflow limitation in smoking siblings of subjects with severe COPD suggests a genetic component to this disease. Genetic linkage analysis has identified several sites in the genome that may contain susceptibility genes, such as chromosome 2q. Genetic association studies show that a number of candidate genes are associated with the development of COPD or with rapid decline in FEV1. However, the associations are not consistent in different populations (Box 41-2).
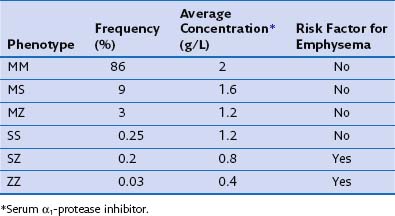
The most consistent association with COPD is alpha1-antitrypsin (α1-proteinase inhibitor) deficiency. Alpha1-antitrypsin is a glycoprotein that is the major inhibitor of serine proteases, including neutrophil elastase. More than 75 biochemical variants of α1-antitrypsin have been described relating to their electrophoretic properties, giving rise to the phase inhibitor (Pi) nomenclature (Table 41-1). The most common allele in all populations is PiM, and the most common genotype is PiMM, which occurs in 93% of the alleles in subjects of Northern European descent. PiMZ and PiMS are the next two most common genotypes and are associated with α1-proteinase inhibitor levels of 15% to 75% of the mean levels of PiMM subjects. Similar levels occur in the much less common PiSS type. The most important other type is PiSZ, in which basal levels are 35% to 50% of normal values. The threshold point for increased risk of emphysema is a level of about 80 mg/dL, which is about 30% of normal.
Natural History and Prognosis
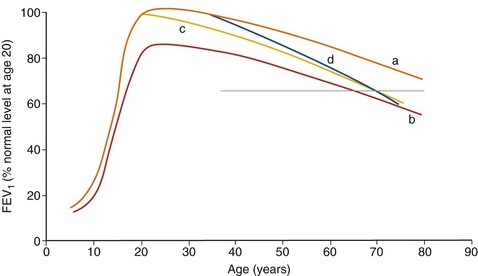
The airway obstruction in susceptible smokers develops slowly because of an accelerated rate of decline in FEV1 that continues for years. As noted previously, impaired lung function development during childhood and adolescence as a result of recurrent infections or exposure to tobacco smoke may lead to lower maximally attained lung function in adulthood. This failure in lung growth, often combined with a shortened plateau phase in teenage smokers, increases the risk of COPD (Figure 41-5). In never-smokers the FEV1 declines at a rate of 20 to 30 mL/yr (see Figure 41-4). Smokers as a population have a faster rate of decline, and reported changes in FEV1 in patients with COPD exceed 50 mL/yr. However, decline in COPD patients varies considerably. The initial level of FEV1 is related to the annual rate of decline in FEV1, and individuals in the highest or lowest FEV1 percentiles remain in the same percentiles over subsequent years. This suggests that susceptible cigarette smokers can be identified in early middle age by a reduction in FEV1.
Pathogenesis
Central to the pathogenesis of COPD is an enhanced inflammatory response to inhaled particles or gases. The following pathogenic processes are involved in this inflammatory response (Figure 41-6):
• Increased air space inflammation
• Increased protease burden and decreased antiprotease function
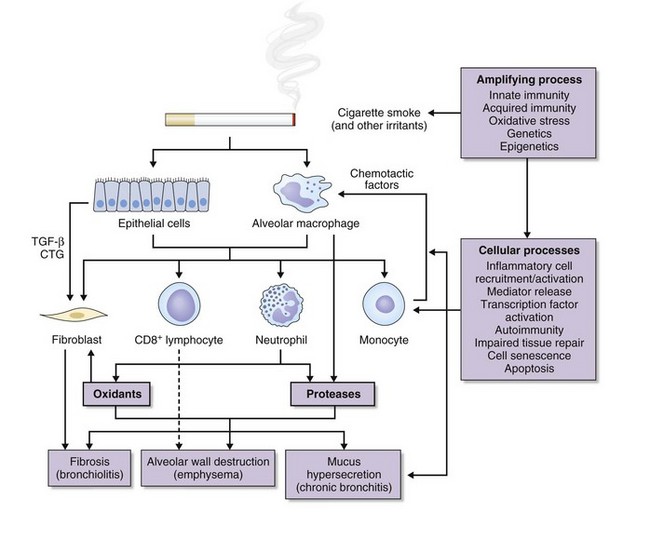
Air Space Inflammation
The innate inflammatory immune system provides primary protection against the continuing insult from inhalation of toxic gases and particles. The first line of defense consists of the mucociliary clearance apparatus and macrophages that clear foreign material from the lower respiratory tract; both are impaired in COPD. The second line of defense of the innate immune system is the exudation of plasma and circulating cells into both large and small conducting airways and the alveoli. This process is controlled by an array of proinflammatory chemokines and cytokines (Box 41-3). COPD is characterized by increased neutrophils, macrophages, T lymphocytes (CD8 > CD4), and dendritic cells in various parts of the lungs (see Box 41-2). Generally, the extent of inflammation is related to degree of airflow limitation. These inflammatory cells are capable of releasing a variety of cytokines and mediators that participate in the disease process. This inflammatory cell pattern is greatly different from that found in asthma.
Both central airways and peripheral airways are inflamed in smokers with COPD. Smokers with chronic bronchitis have greater inflammation in bronchial glands. Recent studies characterizing the inflammation show increased infiltration of mast cells, macrophages, and neutrophils in smokers with chronic bronchitis (see Box 41-1). An increase in T lymphocytes, mainly in the CD8+ subset, occurs, in contrast to the predominance of the CD4 T cell subset in asthma. CD8 lymphocytes may have a role in apoptosis and destruction of alveolar wall epithelial cells, through the release of perforins and tumor necrosis factor alpha (TNF-α). Excessive recruitment of CD8 T lymphocytes may occur in response to repeated viral infections, damaging the lungs in susceptible smokers.
Proteinase/Antiproteinase Imbalance
Important for understanding the pathogenesis of COPD, an association was seen between α1-antitrypsin deficiency and development of early-onset emphysema. Alpha1-antitrypsin is a potent inhibitor of serine proteases and has greatest affinity for the enzyme neutrophil elastase. It is synthesized in the liver and increases from its usual plasma concentration as part of the acute-phase response. The activity of this protein is critically dependent on the methionine-serine sequence at its active site. Table 41-1 provides the average α1-antitrypsin plasma levels for the more common phenotypes.
This simplified protease/antiprotease theory is complicated by the presence of other antiproteases (e.g., antileukoprotease) and other proteases (e.g., metalloproteases) released from macrophages (Table 41-2).
| Proteinases | Antiproteinases |
|---|---|
| Serine proteinases | α1-Antitrypsin |
| Neutrophil elastase | α1-Antitrypsin |
| Cathepsin G | Secretory leukoprotease inhibitor |
| Proteinase 3 | Elafin |
| Cysteine proteinases | Cystatins |
| Cathepsins: B, K, L, S | |
| Matrix metalloproteinases (MMP-8, MMP-9, MMP-12) | Tissue inhibitor of MMPs (TIMP-1 to TIMP-4) |
COPD, chronic obstructive pulmonary disease.
Other Mechanisms
Autoimmunity, apoptosis, and cell senescence also may be involved in the pathogenesis of emphysema (Figure 41-6). Studies show that apoptosis occurs in emphysematous lungs, predominantly involving endothelial cells in the alveolar walls and resulting from a decrease in vascular endothelial growth factor (VEGF) or VEGF signaling, also shown to occur in association with emphysema in human lungs. These data led to the concept of an “alveolar maintenance program” required for the structural preservation of the lungs. Cigarette smoke is thought to cause disruption of this maintenance program, resulting in emphysema. The lung destruction or tissue destruction of emphysema is therefore caused by the mutual interaction of alveolar cell apoptosis, oxidative stress, and protease/antiprotease imbalance.
Pathophysiology
Airflow Limitation and Hyperinflation
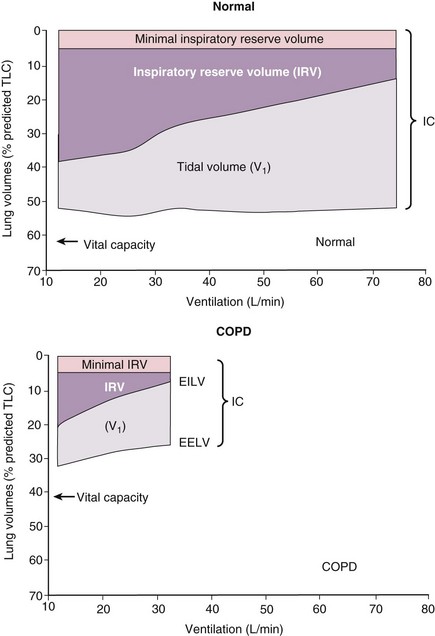
The main site of airflow limitation in COPD occurs in the small conducting airways (<2 mm in diameter) and results from inflammation, narrowing (airway remodeling), and inflammatory exudates in the small airways, features that correlate with the reduction in FEV1. Other factors contributing to the airflow limitation include loss of the lung elastic recoil (caused by destruction of alveolar walls) and destruction of alveolar support (from alveolar attachments). The resultant airway obstruction causes progressive trapping of the air during expiration, resulting in hyperinflation at rest and dynamic hyperinflation during exercise. Lung hyperinflation reduces the inspiratory capacity, and thus functional residual capacity (FRC) increases, particularly during exercise (Figure 41-7). These features are thought to occur early in the course of the disease and result in the breathlessness and limited exercise capacity typical of COPD. Bronchodilators reduce air trapping and thus decrease lung volumes, improving symptoms and exercise capacity. Tests of overall lung mechanics (e.g., FEV1, airway resistance) are usually abnormal in patients with COPD when breathlessness develops.
The resistance to airflow depends on the length and diameter of the airways and the physical properties of the respirable gas. At a constant airway diameter, airflow on inhalation is proportional to the difference between atmospheric gas pressure and alveolar pressure. During exhalation, airflow depends on the difference between alveolar and atmospheric pressures. Throughout inhalation and during the initial portion of exhalation, this relationship is constant. However, at a certain point during exhalation, flow cannot increase despite further increases in alveolar pressure. This is a result of dynamic compression of the airways, which limits flow, as illustrated by the flow-volume loop (Figure 41-8). During exhalation from TLC, flow increases to a point beyond which additional expiratory effort has no effect. During tidal breathing, expiratory flow is well below that attainable during maximum expiration. In COPD, however, the flow-volume loop is different. The major site of the fixed airway narrowing in COPD is in peripheral airways of diameter less than 2 mm. Loss of lung elastic recoil pressure is also an important mechanism of airway obstruction, resulting from a reduction in the distending force applied to the intrathoracic airways. Dynamic expiratory compression of the airways is enhanced by loss of lung recoil and by atrophic changes in the airways and loss of support from the surrounding alveolar walls, allowing flow limitation at lower driving pressure and flow.
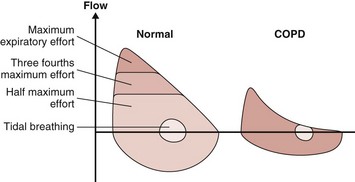
In addition to a decrease in peak expiratory flow, the later expiratory portion of the flow-volume curve is concave relative to the volume axis in patients with COPD. In severe disease the flow generated during tidal breathing may actually reach the maximum possible flow (Figure 41-8). Such patients, in response to the increased metabolic demands of exercise, for example, are unable to increase ventilation. Increased respiratory rate results in gas trapping from incomplete alveolar emptying, so-called dynamic overinflation. This increased lung volume increases the elastic recoil and is associated with an increase in the end-expiratory alveolar pressure. The result is an increase in the work of breathing because pleural pressure must drop below alveolar pressure before inspiration of air can occur.
Pulmonary Hypertension
There are few data on the prevalence of pulmonary hypertension resulting from COPD, because large studies of right-sided heart catheterization or Doppler echocardiography have not been undertaken. An estimate of the prevalence of pulmonary hypertension in COPD can be obtained from calculating the number of subjects with significant hypoxemia who require long-term oxygen therapy. Significant hypoxemia (PaO2 <55 mm Hg, FEV1 <50% of predicted) occurs in 0.3% of the UK population 45 or older. Extrapolation from this figure suggests that in England and Wales, 60,000 patients may be at risk of pulmonary hypertension and eligible for long-term oxygen therapy. Extrapolating these figures to the United States, 300,000 COPD patients are at risk of pulmonary hypertension. Box 41-4 lists the factors leading to pulmonary hypertension in patients with COPD.
Systemic Effects and Comorbidities
Clinical Features
Symptoms
Breathlessness is usually first noticed on climbing hills or stairs, or hurrying on level ground, which later becomes progressive and persistent. It usually heralds at least moderate impairment of expiratory flow. Patients may adapt their breathing pattern and their behavior to minimize the sensation of breathlessness. The perception of breathlessness varies greatly among patients with the same impairment of ventilatory capacity. However, when the FEV1 has fallen to 35% or less of the predicted value, breathlessness is usually present on minimal exertion. Severe breathlessness is often affected by changes in environmental temperature or occupational exposure to dust and fumes. Some patients have severe orthopnea, relieved by leaning forward, whereas others find greatest ease when lying flat. The impact of breathlessness can be assessed on the UK Medical Research Council (MRC) Dyspnea Scale (Table 41-3).
Table 41-3 Modified MRC Dyspnea Scale for Assessing Breathlessness
| Grade | Degree of Breathlessness Related to Activities |
|---|---|
| 1 | Not troubled by breathlessness, except on strenuous exercise. |
| 2 | Short of breath when hurrying or walking up a slight hill. |
| 3 | Walks slower than contemporaries on level ground because of breathlessness, or has to stop for “breather” when walking at own pace. |
| 4 | Stops for breath after walking about 100 m or after a few minutes on level ground. |
| 5 | Too breathless to leave the house, or breathless when dressing or undressing. |
Data from UK Medical Research Council.
Other Aspects of the History
• History of other medical conditions
• Symptoms of anxiety and depression
• Frequency of exacerbations; number of courses of corticosteroids and antibiotics in preceding year
• Occupational and environmental exposure to dust, chemicals, gases, and fumes
• Previous respiratory problems (chronic asthma or tuberculosis)
• Number of days missed from work
• Current smoking status and number of pack-years (1 pack-year is defined as 20 cigarettes [1 pack] smoked per day for 1 year)
Several conditions should be considered in the differential diagnosis in patients with COPD. Asthma can be particularly difficult; scales (e.g., the modified Borg scale) can be used to assess the degree of breathlessness (Table 41-4).
| Scale | Severity Experienced by Patient |
|---|---|
| 0 | Nothing at all |
| 0.5 | Very, very slight (just noticeable) |
| 1 | Very slight |
| 2 | Slight (light) |
| 3 | Moderate |
| 4 | Somewhat severe |
| 5 | Severe (heavy) |
| 6 | Very severe |
| Very, very severe (almost maximal) | |
| Maximal |
Patient Assessment
Physiologic Assessment
Spirometry
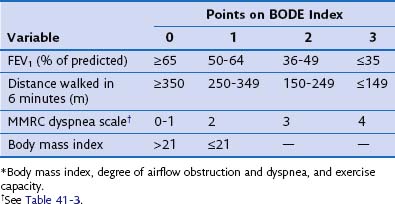
The FEV1 as a percentage of the predicted value can be used to assess the severity of disease (Table 41-5). FEV1 does not fully capture the impact of COPD on the patient’s functional capabilities, however, and thus other measurements in addition to spirometry are required to assess the effect of COPD on functional ability. Breathlessness can be gauged by the MRC scale (see Table 41-3). Exercise capacity can be objectively measured by a reduction in self-paced walking distance (e.g., 6-minute walking distance [6MWD]), which is a strong predictor of health status impairment and prognosis. A combination of these variables to give a more detailed indication of disease severity has been proposed as the BODE index, a composite score of body mass index, airways obstruction, dyspnea, and exercise that appears to be a better predictor of subsequent survival than any of the individual components (Table 41-6).
| Stage | Characteristics |
|---|---|
| I: Mild | FEV1/FVC <0.7 |
| FEV1 ≥80% predicted | |
| II: Moderate | FEV1/FVC <0.7 |
| 50% ≤ FEV1 <80% predicted | |
| III: Severe | FEV1/FVC <0.7 |
| 30% ≤ FEV1 <50% predicted | |
| IV: Very severe | FEV1/FVC <0.7 |
| FEV1 <30% predicted or FEV1 <50% predicted plus chronic respiratory failure |
Other Tests
Respiratory Muscle Function Test
The usual tests of respiratory muscle function in COPD are maximum inspiratory and expiratory mouth pressures. These tests can be useful in cases where breathlessness or hypercapnia is not fully explained by other lung function testing or when peripheral muscle weakness is suspected (see Chapter 42).
Imaging
Plain Chest Radiography
All patients with suspected COPD should have a posteroanterior (PA) chest radiograph performed at diagnosis. COPD does not produce any specific features on plain chest radiography unless features of emphysema are present. There may be no abnormalities, however, even in patients with severe disability. A chest x-ray film is used to discount other causes of respiratory symptoms and to identify complications with COPD, such as bulla formation. The most reliable radiographic signs of emphysema can be divided into those caused by overinflation, by vascular changes, and by bullae (see Computed Tomography). The following radiologic features are indicative of overinflation:
• Low, flattened diaphragm; the border of the diaphragm in the midclavicular line on the PA film is at or below the anterior end of the seventh rib, and it is flattened if the perpendicular height from a line drawn between the costal and cardiophrenic angles to the border of the diaphragm is less than 1.5 cm.
• Increased retrosternal air space, visible on the lateral film at a point 3 cm below the manubrium; it is present when the horizontal distance from the posterior surface of the aorta to the sternum exceeds 4.5 cm.
• Obtuse costophrenic angle on the PA or lateral chest radiograph.
• Inferior margin of the retrosternal air space 3 cm or less from the anterior aspect of the diaphragm.
Computed Tomography
• Areas of low attenuation without obvious margins or walls
Areas of low attenuation correlate best with areas of macroscopic emphysema. Visual inspection of the CT scan can be used to locate macroscopic emphysema, although assessing the extent is insensitive and subject to high intraobserver and interobserver variability. The CT scan can be used to assess different types of emphysema; centrilobular emphysema produces patchy areas of low attenuation prominent in the upper zones, whereas those of panlobular emphysema are diffuse throughout the lung zones (see Figure 41-3).
Agusti A. Systemic effects of chronic obstructive pulmonary disease: what we know and what we don’t know (but should). Proc Am Thorac Soc. 2007;4:522–525.
Calverley PMA, MacNee W, Pride NB, Rennard SI. Chronic obstructive pulmonary disease, ed 2, London: Chapman & Hall, 2003.
Celli BR, MacNee W. Standards for the diagnosis and treatment of patients with COPD. Eur Respir J. 2004;23:841–845.
Cosio MG, Saetta M, Agusti A. Immunologic aspects of chronic obstructive pulmonary disease. N Engl J Med. 2009;360:2445–2454.
Global Initiative for Chronic Obstructive Pulmonary Disease Workshop Report. Medical Communications Resources. www.goldcopd.com, 2008.
Hogg JC, Senior RM. Chronic obstructive pulmonary disease. II. Pathology and biochemistry of emphysema. Thorax. 2002;57:830–834.
Hogg JC, Timens W. The pathology of chronic obstructive pulmonary disease. Ann Rev Pathol. 2009;4:435–459.
Hogg JC, Chu F, Utokaparch S, et al. The nature of small-airway obstruction in chronic obstructive pulmonary disease. N Engl J Med. 2004;350:2645–2653.
MacNee W. Pathogenesis of chronic obstructive pulmonary disease. Proc Am Thorac Soc. 2005;2:258–266.
MacNee W, Tuder R. New paradigms in the pathogenesis of chronic obstructive pulmonary disease. 1. Proc Am Thorac Soc. 2009;6:527–531.
MacNee W, Maclay J, McAllister D. Cardiovascular injury and repair in chronic obstructive pulmonary disease. Proc Am Thorac Soc. 2008;5:824–833.
2006 Management of chronic obstructive pulmonary disease. Eur Respir Monogr. 2006;38. www.ersnet.org.
Mannino DM, Watt G, Hole D, et al. The natural history of chronic obstructive pulmonary disease. Eur Resp J. 2006;27:627–643.
Peinado VI, Pizarro S, Barbera JA. Pulmonary vascular involvement in COPD. Chest. 2008;134:808–814.
Rabinovich RA, MacNee W. Chronic obstructive pulmonary disease and its comorbidities. Br J Hosp Med. 2011;72:137–145.
Vestbo J, Hogg JC. Convergence of the epidemiology and pathology of COPD. Thorax. 2006;61:86–88.
Voelkel NF, MacNee W. Chronic obstructive lung disease, ed 2, Hamilton, Ontario: BC Decker, 2008.
Wouters EF. Local and systemic inflammation in chronic obstructive pulmonary disease. Proc Am Thorac Soc. 2005;2:26–33.