[level-membership-for-hematology-oncology-and-palliative-medicine-category]22
Chronic myeloid leukaemia
Chronic myeloid leukaemia (CML) is a clonal myeloproliferative disorder which results from an acquired genetic change in a pluripotential stem cell. The disease is characterised by a gross overproduction of neutrophils and their precursors (Fig 22.1). It is unusual in having three clinical phases: a relatively benign ‘chronic phase’ is followed by an ominous ‘accelerated phase’ and, finally, an almost invariably fatal acute leukaemic phase termed ‘blast crisis’.

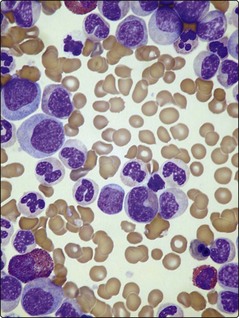
Fig 22.1 Blood sample (right) from a patient with CML.
Note the greatly increased white cell component (‘buffy coat’) compared with the normal sample.
Pathogenesis
The hallmark of CML cells is the presence of a Philadelphia (Ph) chromosome – the t(9;22)(q34;q11) chromosomal translocation. Over 95% of classical CML cases are Ph positive. The Ph translocation causes the fusion of the ABL proto-oncogene from chromosome 9 to the interrupted end of the breakpoint cluster region (BCR) of chromosome 22 (Fig 22.2). The chimeric BCR-ABL gene created on the Ph chromosome (22q−) encodes a protein with considerably greater tyrosine kinase activity than the normal counterpart. In chronic phase CML, cells in the progenitor pool have increased proliferation due to over-expression of BCR-ABL. The mechanism by which the BCR-ABL oncogene affects stem cell kinetics is not well understood. It presumably deregulates signalling pathways involved in proliferation, apoptosis, cellular adhesion and genomic stability. Progression to blast crisis with production of leukaemic stem cells requires complex additional events including increased proliferation and self-renewal capacity avoidance of cell death, a block in differentiation and bypassing of normal immune responses.
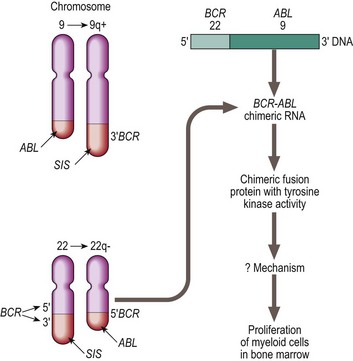
Fig 22.2 The Philadelphia chromosome.
Chromosomal and molecular abnormalities in chronic myeloid leukaemia. In a translocation between chromosomes 9 and 22 (t(9;22) ) the oncogene ABL on chromosome 9 is moved to the breakpoint cluster region (BCR) of chromosome 22. The resulting BCR-ABL hybrid gene encodes a protein with high tyrosine kinase activity.
Diagnosis and monitoring
The major laboratory abnormality in CP-CML is an elevated white cell count; this often exceeds 100 × 109/L. The blood film shows an increase in morphologically normal myeloid cells at all stages of differentiation but with greatest numbers of myelocytes and neutrophils (Fig 22.3). There is usually an absolute basophilia. Thrombocytosis and nucleated red cells may be present.
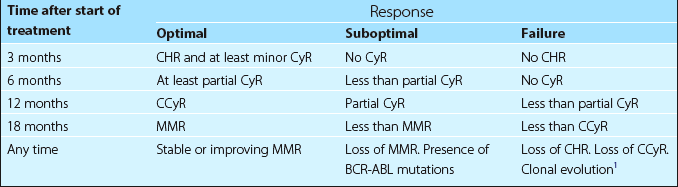
The accelerated phase is characterised by an increase in the number of immature cells in the peripheral blood and in blast crisis the blood appearance is dominated by the presence of myeloblasts (65% of cases) or lymphoblasts (35%). The most widely used staging system, devised by Sokal, is based on patient age, spleen size, blood blast cell count and platelet count. Monitoring of the response to treatment is now central to management. This involves examination of the peripheral blood, bone marrow metaphase cytogenetics and measurement of BCR-ABL transcripts using real-time quantitative polymerase chain reaction (RQ-PCR). The results allow the patient’s response to be defined at key time points (see Table 22.1). Where there is failure or a suboptimal response, alternative therapy is considered.
Treatment
Chronic phase
Drug therapy. Hydroxycarbamide can also be used to rapidly reduce an initial high white cell count. Tyrosine kinase inhibitors (TKIs) are the treatment of choice. Imatinib at a dose of 400 mg daily orally is normally used as first-line. Patients achieving the milestones shown in Table 22.1 have an excellent prognosis, with long-term survival rates exceeding 90% and a very low risk of transformation to accelerated phase or blast crisis. For patients who are unable to tolerate imatinib or who become resistant, so-called second generation TKIs such as dasatinib and nilotinib can be effective. Both these drugs are more potent than imatinib and are likely to be increasingly used as initial treatment. Third generation agents (e.g. bosutinib) are under investigation.
Advanced disease






[/level-membership-for-hematology-oncology-and-palliative-medicine-category][not-level-membership-for-hematology-oncology-and-palliative-medicine-category]22
Chronic myeloid leukaemia
Chronic myeloid leukaemia (CML) is a clonal myeloproliferative disorder which results from an acquired genetic change in a pluripotential stem cell. The disease is characterised by a gross overproduction of neutrophils and their precursors (Fig 22.1). It is unusual in having three clinical phases: a relatively benign ‘chronic phase’ is followed by an ominous ‘accelerated phase’ and, finally, an almost invariably fatal acute leukaemic phase termed ‘blast crisis’.
Fig 22.1 Blood sample (right) from a patient with CML.
Note the greatly increased white cell component (‘buffy coat’) compared with the normal sample.
Pathogenesis
The hallmark of CML cells is the presence of a Philadelphia (Ph) chromosome – the t(9;22)(q34;q11) chromosomal translocation. Over 95% of classical CML cases are Ph positive. The Ph translocation causes the fusion of the ABL proto-oncogene from chromosome 9 to the interrupted end of the breakpoint cluster region (BCR) of chromosome 22 (Fig 22.2). The chimeric BCR-ABL gene created on the Ph chromosome (22q−) encodes a protein with considerably greater tyrosine kinase activity than the normal counterpart. In chronic phase CML, cells in the progenitor pool have increased proliferation due to over-expression of BCR-ABL. The mechanism by which the BCR-ABL oncogene affects stem cell kinetics is not well understood. It presumably deregulates signalling pathways involved in proliferation, apoptosis, cellular adhesion and genomic stability. Progression to blast crisis with production of leukaemic stem cells requires complex additional events including increased proliferation and self-renewal capacity avoidance of cell death, a block in differentiation and bypassing of normal immune responses.
Fig 22.2 The Philadelphia chromosome.
Chromosomal and molecular abnormalities in chronic myeloid leukaemia. In a translocation between chromosomes 9 and 22 (t(9;22) ) the oncogene ABL on chromosome 9 is moved to the breakpoint cluster region (BCR) of chromosome 22. The resulting BCR-ABL hybrid gene encodes a protein with high tyrosine kinase activity.
