7 Chronic Diffuse Lung Diseases
Diffuse or “interstitial” lung diseases (ILDs) include a spectrum of primarily non-neoplastic inflammatory conditions that share the common property of diffuse involvement of the lung parenchyma. The term “ILD” has become so thoroughly entrenched across multiple medical disciplines that it seems practical to continue its usage, although we would emphasize that many of the diseases discussed in this chapter also involve the alveolar spaces and terminal bronchioles to a variable extent and would therefore not be considered entirely “interstitial” by the anatomic purist.1
This chapter focuses on the subacute and chronic forms of ILD (acute ILDs are discussed in Chapter 5), which includes diseases that typically evolve over weeks, months, or years. Patients with ILD share a number of clinical and radiologic manifestations, including: (1) shortness of breath (dyspnea), (2) diffuse abnormalities in lung mechanics and gas transfer (pulmonary function), and (3) diffuse abnormalities on chest radiographs and computed tomography (CT) scans of the chest.2
An overview of ILD from the pathologist’s perspective is presented in Box 7-1. In this chapter we will restrict our focus to a limited number of predominantly inflammatory diseases that come to biopsy relatively frequently (Box 7-2). Our emphasis is on the histopathologic patterns of these diseases as observed through the microscope. These patterns help narrow the differential diagnosis and often allow for a definitive diagnosis when coupled with clinical and radiologic data.
Box 7-1 Overview of the Chronic Diffuse Interstitial Lung Diseases
COP, cryptogenic organizing pneumonia; DIP, desquamative interstitial pneumonia; LIP, lymphoid interstitial pneumonia; NSIP, nonspecific interstitial pneumonia; RBILD, respiratory bronchiolitis interstitial lung disease; UIP, usual interstitial pneumonia.
Box 7-2 Chronic Interstitial Lung Diseases Presented in This Chapter
COP, cryptogenic organizing pneumonia; DIP, desquamative interstitial pneumonia; LIP, lymphoid interstitial pneumonia; NSIP, nonspecific interstitial pneumonia; RBILD, respiratory bronchiolitis interstitial lung disease; UIP, usual interstitial pneumonia.
Unlike neoplasms, which may have distinctive or even unique morphologic features, ILDs are distinguished from one another by (1) location involved (anatomic compartment or structure), (2) distribution (focal or diffuse), and (3) cellular composition (e.g., acute, chronic, histiocytic) of the inflammatory reaction. Additional identification criteria include the mechanism by which repair is taking place (organizing or not) and the stage of the reparative process (acute: fibroblastic proliferation; subacute: fibroblasts accompanied by matrix and epithelial regeneration; chronic: dense fibrosis and structural remodeling).2
Transbronchial and surgical wedge biopsy interpretation in the ILD patient is complicated by several factors.3 First, these diseases involve the interstitium, but they are frequently attended by reactive changes in the surrounding alveolar spaces and associated terminal airways. Such reactive changes can be quite impressive and commonly distract the observer from recognizing the interstitial nature of the process. Second, the inherent variability and natural history of inflammatory diseases pose problems, wherein early phases of a disease may differ in appearance from later phases, and the intensity of a reaction may vary from individual to individual. Third, more than one inflammatory disease can involve the lung simultaneously, adding further complexity to the morphologic picture. Finally, and perhaps of greatest importance, these predominantly medical diseases cannot be diagnosed accurately without some clinical and radiologic correlation.4
One of the most important chronic lung diseases in pulmonary medicine today is known clinically as idiopathic pulmonary fibrosis (IPF). This most devastating of chronic ILDs is often the diagnosis of exclusion from a clinical perspective and one against which all other chronic lung diseases are judged. The reason for this is that IPF is a disease that progresses despite therapy and rivals many cancers in mortality rate, with death often occurring within 3 years of the diagnosis.5 As emphasized in the 2002 joint consensus statement of the American Thoracic Society (ATS) and the European Respiratory Society (ERS), the pathologic manifestation of IPF in the lung is usual interstitial pneumonia (UIP).6 UIP was a term introduced by Liebow in reference to one of five forms of idiopathic interstitial pneumonia (IIP).7 In Liebow’s words, UIP represents “chronic lung fibrosis of the common or usual type,”—a seemingly broad category of chronic lung disease.
Idiopathic Interstitial Pneumonias
Liebow’s initial classification of IIPs is presented for historical purposes in Box 7-3. In the years following the introduction of this classification scheme, new information led to the modification or elimination of certain of these IIPs and the addition of others not previously included8,9 (Box 7-4). Since Liebow’s time, it has been established that desquamative interstitial pneumonia (DIP), initially thought to be an early manifestation of UIP,10 is in fact a smoking-related disease in a majority of cases, most often affecting adults.11,12 Subsequent investigation also showed that giant cell interstitial pneumonia (GIP) was actually a manifestation of cobalt exposure, as a pneumoconiosis in “hard metal disease” (see Chapter 8).13 Finally, it became apparent that many early cases of lymphoid interstitial pneumonia (LIP) evolved into lymphoproliferative disease and probably did not constitute “inflammatory” disease in the true sense of the word.bb0080 bb0085
Box 7-3 Historical Classification of Idiopathic Interstitial Pneumonias
Data from Liebow A, Carrington C: The interstitial pneumonias. In: Simon M, Potchen EJ, LeMay M, eds. Frontiers of Pulmonary Radiology: Pathophysiologic, Roentgenographic and Radioisotopic Considerations. Orlando, FL: Grune & Stratton; 1969:109–142.
Box 7-4 Initial Revised Classification of Idiopathic Interstitial Pneumonias
Reprinted with permission from Katzenstein A, Askin F, eds. Surgical Pathology of Non-Neoplastic Lung Disease, 2nd ed. Philadelphia: WB Saunders; 1990:49, Table 3-1.
Based on this evolution in our understanding, a modification to Liebow’s original classification of the IIPs was proposed by Katzenstein.8,9 This new schema included the major categories of UIP and DIP but coupled DIP with “respiratory bronchiolitis–associated interstitial lung disease” (RBILD) and acknowledged the strong relationship of these diseases to cigarette smoking. Katzenstein also proposed a new category of acute interstitial pneumonia (AIP)15 as an entity separate from UIP, a distinction that Liebow did not make in his initial classification. Finally, Katzenstein created a new category to encompass a group of inflammatory diseases that differed in appearance from UIP, DIP, or AIP. The term nonspecific interstitial pneumonia (NSIP) was proposed for this “new” pattern.16 In our experience, a majority of IIPs can be classified using this scheme. We would add idiopathic (cryptogenic) organizing pneumonia, previously known as “idiopathic bronchiolitis obliterans organizing pneumonia,”17 to this classification, as has been recommended by the 2002 International Workshop on the classification of IIPs.6 That workshop retained LIP in the classification (Table 7-1), acknowledging the presence of some morphologic overlap with NSIP once a lymphoproliferative disease has been excluded by all available means. Importantly, the consensus panel determined that NSIP should be included in the IIPs as a provisional category until additional data accrue.6
Table 7-1 International Consensus Classification of Idiopathic Interstitial Pneumonias (2002)
| Histopathologic Pattern | Clinical-Radiologic-Pathologic Diagnosis |
|---|---|
| Usual interstitial pneumonia | Idiopathic pulmonary fibrosis/cryptogenic fibrosing alveolitis |
| Nonspecific interstitial pneumonia | Nonspecific interstitial pneumonia (“provisional”) |
| Respiratory bronchiolitis | Respiratory bronchiolitis interstitial lung disease |
| Desquamative interstitial pneumonia | Desquamative interstitial pneumonia |
| Organizing pneumonia | Cryptogenic organizing pneumonia |
| Diffuse alveolar damage | Acute interstitial pneumonia |
| Lymphoid interstitial pneumonia | Lymphoid interstitial pneumonia |
Reprinted with permission from American Thoracic Society/European Respiratory Society international multidisciplinary consensus classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med. 2002;165(2):277–304, Table 2.
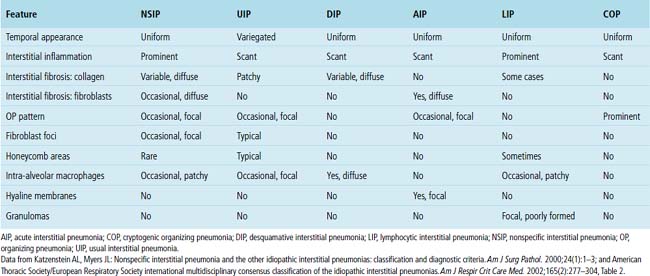
IIPs are classically defined as diffuse pulmonary diseases that involve two or more lobes of the lung; in most patients, such diseases are bilateral in distribution.18 Some localized lesions (such as infection, atelectasis, or tumor) may mimic IIP in a biopsy specimen. It is safe to say that if a disease process is confined to the biopsy area sampled, it is unlikely to be an IIP. AIP is an acute form of IIP (discussed in detail in Chapter 5). A comparison of the histopathologic findings in each of the IIPs is presented in Table 7-2. The importance of accurately diagnosing these IIPs lies mainly with differences in prognosis. UIP is a uniformly fatal disease for which the median survival period historically is less than 3 years in its classic presentation (CT with honeycombing), competing with many cancers in this respect.
Usual Interstitial Pneumonia
Pulmonary pathologists have debated for years what is, and what is not, UIP. To some, UIP is a relatively nonspecific pattern of chronic lung injury with fibrosis and “honeycomb” remodeling (see further on). Today it is recognized that not all lung diseases with fibrosis behave similarly and, in particular, do not run the aggressive course expected for clinical IPF. The most honest answer may be that clinical and radiologic IPF has UIP-type pathologic changes, but that a UIP pattern of parenchymal fibrosis with remodeling may be seen in biopsy specimens and may not necessarily correlate with clinical and radiologic IPF. Fortunately, not all lung diseases that produce scarring fit the pattern now defined as UIP. Asbestosis,19–21 chronic hypersensitivity pneumonitis,22–24 systemic collagen vascular diseases (CVDs),25–28 and even some chronic toxic drug reactions29 can all produce lung fibrosis. Unfortunately, in the 30 years following Liebow’s introduction of UIP as an “idiopathic” interstitial disease, pathologists often used the designation of UIP in a variety of nonidiopathic settings (e.g., “UIP from asbestosis” or “UIP from rheumatoid arthritis”). If UIP is defined as simply any form of lung fibrosis, then applying UIP as a synonym for fibrosis is perfectly reasonable. On the other hand, if UIP is a distinctive pathologic entity that corresponds to an idiopathic clinical disease (IPF), then UIP should have identifiable features that afford it status as a unique disease process. That there is a continued misconception of UIP among pathologists is underscored by feedback from our clinical colleagues, who note that many diagnoses of UIP provided by the pathology laboratory do not correspond to clinical IPF in their patients’ presentation, response to therapy, or observed outcome.
Clinical Presentation
The incidence of UIP varies by gender, with males predominating. The disease may have a prevalence in the United States as high as 43 per 100,000, using broad criteria30; roughly two thirds of patients are older than 60 years of age at diagnosis.5,31 For this reason, caution should be exercised when considering a diagnosis of UIP in patients who are younger than 50 years of age, and preferably expert consultation should be sought in this setting. Symptoms typically progress insidiously for months to years before diagnosis. The onset of a nonproductive cough and slowly progressive dyspnea are characteristic. Dry inspiratory crackles (so-called Velcro crackles) are detected at the lung bases on chest auscultation in more than 80% of patients at presentation.5 Clubbing of the digits is seen in 25% to 50% of patients at presentation. Fever is rare, and its presence should suggest an alternate diagnosis, as should a significantly elevated erythrocyte sedimentation rate (greater than 100 mm/hour). Serologic studies such as antinuclear antibody (ANA) or rheumatoid factor (RF) assays may reveal mildly elevated titers, but when significant elevation is present, a systemic connective tissue disease should be strongly considered. Also, in patients presenting with clinical features of UIP or IPF in whom a defined CVD develops later, reclassification of their disease may be necessary.
Radiologic Findings
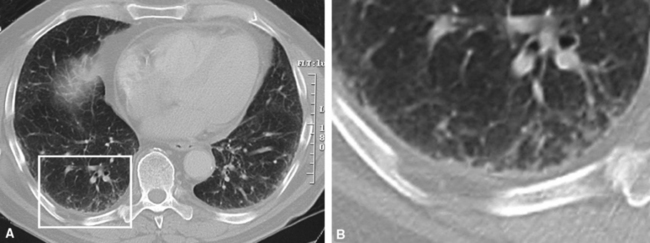
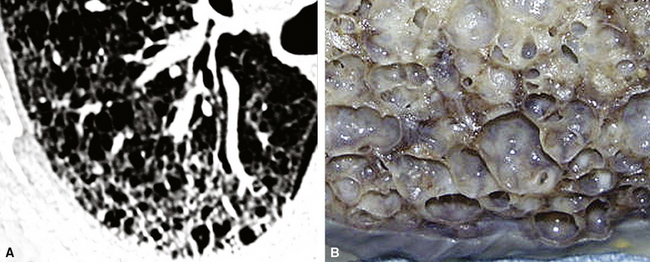
On chest radiographs, peripheral reticular opacities involving the lung bases are a characteristic finding.32 When present, these usually are bilateral and often asymmetrical. Lung volumes are typically decreased at presentation except in cases with severe upper lobe (centriacinar) emphysema.33 Unfortunately, a normal chest radiograph does not exclude the diagnosis.34 Confluent alveolar opacities are rare and, if present, suggest an alternate diagnosis or a comorbid process. CT scans, preferably of the high-resolution type (i.e., with scan sections ≤ 1 mm), commonly show patchy, predominantly peripheral (subpleural) reticular abnormalities involving the lung bases bilaterally.35 Some asymmetry is expected between right and left lungs, and characteristic “skip” areas are present, with coarse pleural-based reticulation alternating with adjacent better-preserved lung (so-called “radiologic heterogeneity”). The earliest findings may be quite subtle, consisting of delicate, peripherally accentuated pleural-based reticular opacities in the lower lung zones (Fig. 7-1). Ground-glass opacities are not typical and, if present, should be limited in extent.36–38 Subpleural cysts—ranging from a few millimeters to a centimeter or more in diameter (“radiologic honeycombing”)—increase in prominence as the disease advances (Fig. 7-2). In areas of more severe involvement, traction bronchiectasis often is evident. Diagnostic accuracy for IPF on high-resolution CT scan by trained observers is in the range of 90% when typical findings are present (high specificity); however, approximately one third of cases of UIP will be missed when relying on high-resolution CT diagnosis alone (low sensitivity).24,39
Histopathologic Findings
UIP cannot be diagnosed with the bronchoscopic or transbronchial biopsy specimen. Surgically derived wedge lung biopsies (3–5 cm in length by 2–3 cm in breadth), obtained from video-assisted thoracoscopic surgery (VATS) or open thoracotomy, are the appropriate samples for diagnosis (see Chapter 2 for additional details on the lung biopsy). Occasionally, UIP will be evident in lobectomy and pneumonectomy specimens obtained for other diseases. UIP is a process that involves the periphery of the lung lobule; these areas are not sampled adequately in even the most ambitious transbronchial biopsy scenario (where many large fragments of alveolar parenchyma may be present, but are mainly derived from the central portion of the lung lobules). More than one biopsy site should be sampled, and preferably a biopsy sample should be obtained from all lobes in the hemithorax chosen for surgical intervention. If only two areas can be sampled, mid-lung and lower lung are preferable to upper and mid-lung, and samples from the lower lobe should be taken above the most advanced areas of fibrosis.
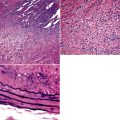
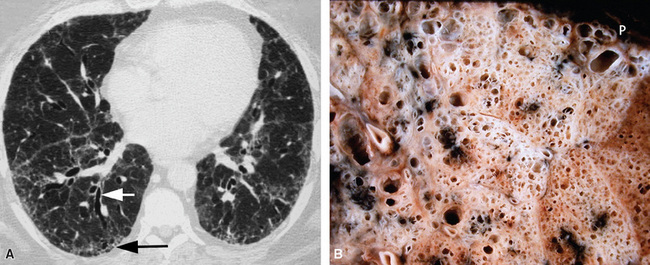
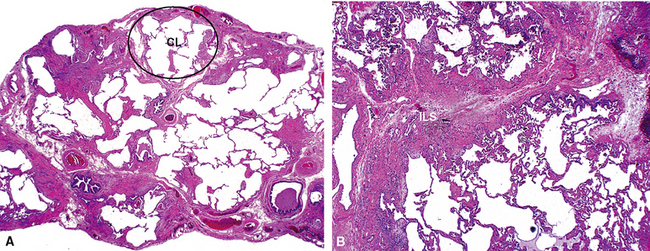
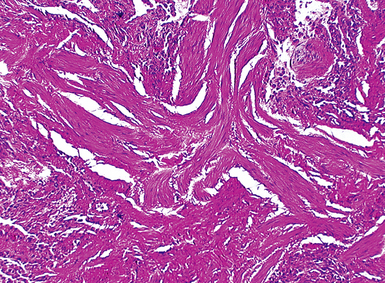
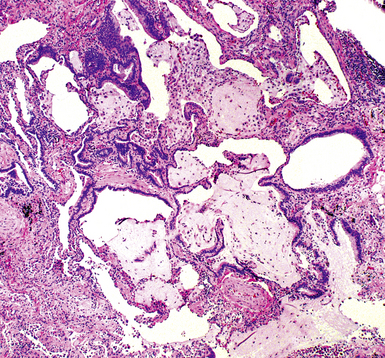
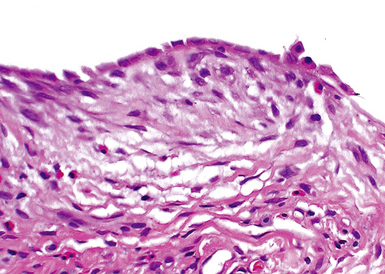
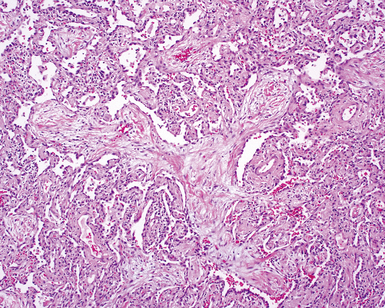
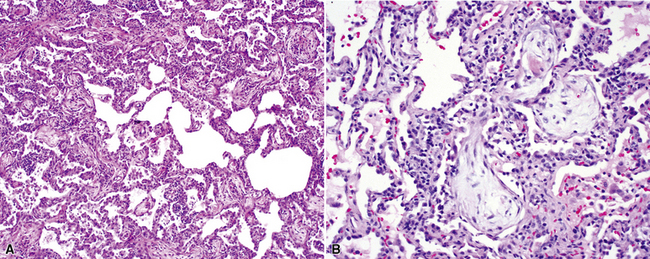
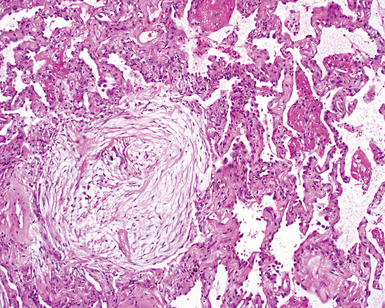
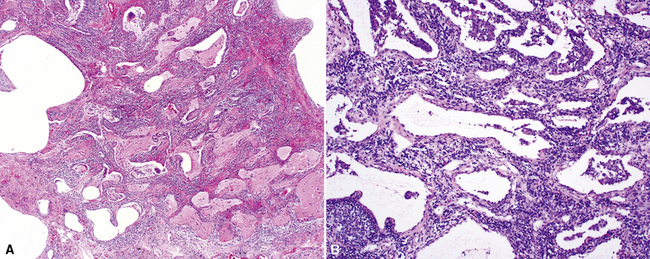
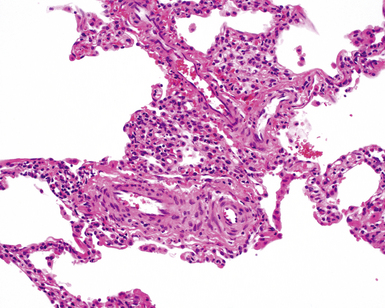
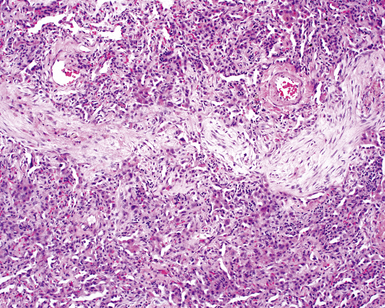
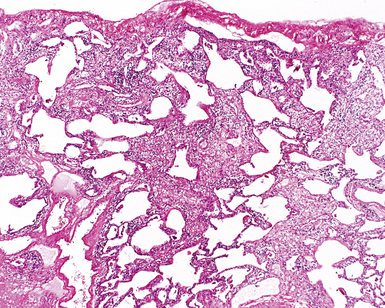
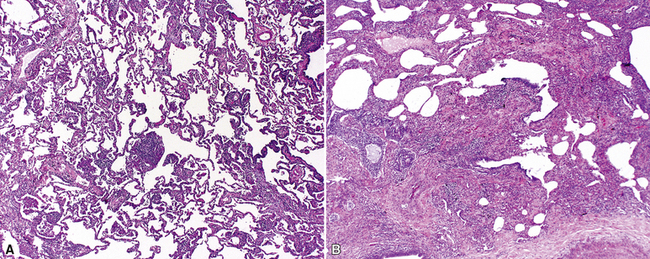
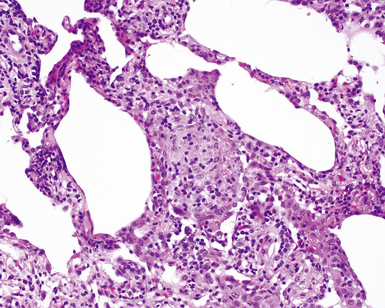
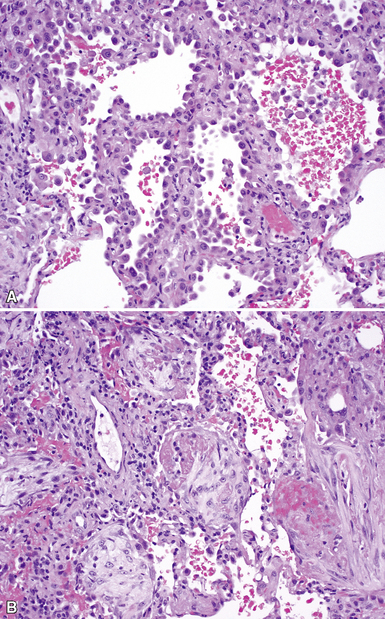
The characteristic histopathologic findings of UIP have been referred to as being “temporally heterogeneous” or having a “patchwork quilt” appearance,32,40–43 concepts and terms that are often misunderstood by surgical pathologists and pulmonologists. An expanded description of “temporal heterogeneity” is that of transitions in the biopsy from dense scar (the “past”) to normal lung (the “future”—lung tissue yet to be involved). At the juncture of these, transitions occur through patches of active lung injury referred to as “fibroblast” or “fibroblastic” foci (Fig. 7-3). The remodeled lung is present mainly beneath the pleura and at the periphery of the secondary lobule, adjacent to interlobular septa (Fig. 7-4). When UIP is recognizable as a distinct pathologic entity, the pleural fibrosis contains smooth muscle proliferation in disorganized fascicles (Fig. 7-5) and foci of microscopic honeycombing are evident, even when the overall process appears to be mild or early in its evolution (Fig. 7-6). Microscopic honeycombing probably represents one of the early manifestations of the gross honeycomb cysts seen in the end-stage of UIP. As used by radiologists, the term honeycombing refers to an array of much larger cysts (in the range of 0.5–3 cm or larger) as a localized manifestation of advanced lung remodeling (Fig. 7-7). Microscopic honeycomb cysts are considerably smaller (in the range of 1–3 mm) and typically are present subpleurally (Fig. 7-8). The cysts are lined by columnar ciliated epithelium and typically are filled with mucus, with variable amounts of acute inflammation and inflammatory debris (Fig. 7-9). When dense chronic inflammation is present in UIP, it is seen around these localized inflammatory lesions.
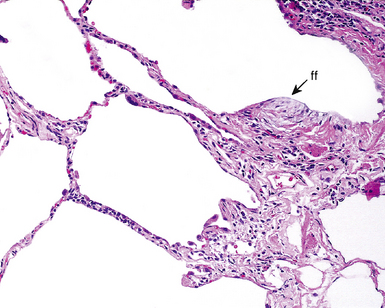
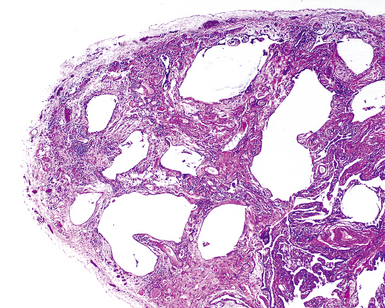
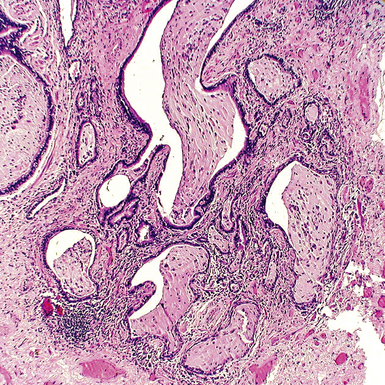
Between the two temporal extremes of “old” peripheral fibrosis and uninvolved lung present centrally in the lobule is the presumed active zone of injury in UIP, evidenced by a crescent-shaped bulge of immature fibroblasts (technically, myofibroblasts) and ground substance (see Fig. 7-3). This lesion is known as the fibroblastic focus and typically is not extensive in the biopsy. Fibroblast foci have been shown to be continuous linear structures in three-dimensional reconstruction.44 Some investigators have postulated that the increased number of these foci in a given UIP patient’s biopsy is associated with a worse prognosis, and that a relative lack of fibroblastic foci may be an explanation for the better prognosis observed for patients with UIP-like lung fibrosis related to systemic CVDs.45
UIP is not an overtly inflammatory condition, in the absence of so-called acute exacerbation (see further on). This is not to imply that fibrosis occurs “mysteriously” in the disease. Some form of injury is occurring in UIP, but it seems to be subtle and probably is directed at the alveolar epithelium and its underlying basement membrane (epithelial-mesenchymal transitions). The fibroblastic foci of UIP appear immediately beneath reactive-appearing alveolar lining epithelium, where they obscure the epithelial basement membrane and bulge into the adjacent air space (Fig. 7-10), as though they were aborted “Masson polyps” of the type seen in organizing pneumonia (see later under “Cryptogenic Organizing Pneumonia”). Further evidence of an injury repair phenotype for UIP/IPF is the consistent presence of reactive type II cell proliferation overlying fibroblastic foci. Conceptually, the subtle inflammatory disease of UIP burns like a smoldering fire through the lung, leaving fibrosis, smooth muscle proliferation, microscopic honeycombing, and fibrosis in its path.
Acute Exacerbation
In his writings, Liebow conceived of UIP as a chronic lung disease resulting from repeated subclinical episodes of “diffuse alveolar damage” (DAD).7 In support of this hypothesis, episodic deterioration is typical in patients with IPF.5 In some patients with IPF, however, clinical deterioration is abrupt and overwhelming. Many of these acute deteriorations are of unidentifiable cause and have been referred to as “acute exacerbations of IPF.” Acute exacerbations have been the subject of considerable laboratory investigation, but the mechanism of their occurrence remains unknown. We do know that when such episodes are biopsied, the most consistent pathologic finding is that of DAD.46 Acute exacerbations of IPF can manifest as other patterns of acute lung injury, such as organizing pneumonia, and despite the implication of the term, the “acute” exacerbation tends to evolve over several weeks, rather than a few days.47 The mixed histopathologic changes can be confusing to the surgical pathologist examining the lung biopsy (and to the radiologist) because the background older fibrosis with microscopic honeycombing of UIP is often overshadowed by diffuse acute lung injury (Fig. 7-11).
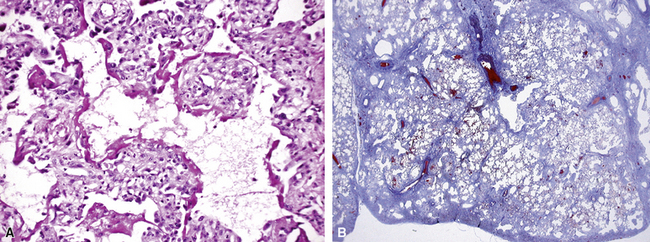
The three patients described by Kondoh and coworkers all showed some degree of improvement in the short term after high-dose corticosteroid therapy, but no consistently effective therapy has emerged.46 Several investigators have proposed that acute exacerbations may be the common terminal episode in many patients with IPF, even though respiratory failure has always been presumed to be of slower evolution.48 Based on all available data, including data from the placebo arms of several large randomized, double-blind, placebo-controlled trials in patients with IPF, an estimated 10% to 15% of patients with UIP experience overwhelming acute exacerbation during the course of their disease, and this is often the fatal event for those affected.47,49
Differential Diagnosis
The differential diagnosis for the UIP pattern includes a number of diseases that produce lung fibrosis. When this is a diffuse bilateral process, the main entities in the differential diagnosis are listed in Box 7-5. There are cases showing coexistence of histopathologic patterns of NSIP and UIP in the same patient in multiple lobe biopsies.50 Such cases can be considered “discordant” UIP.51 The clinical course of discordant UIP is still more like that of “non-discordant” UIP, however, with possibly longer survival.52
Clinical Course
The most common causes of death among patients with IPF are listed in Box 7-6. As defined clinically, IPF patients have a median survival time of less than 3 years.53 At present, no effective therapy has been established for IPF, but newer therapies are on the horizon using human recombinant cytokines as agents antagonistic to the effects of potentially “responsible” molecules.
Box 7-6 Cause of Death in 543 Patients with Idiopathic Pulmonary Fibrosis*
Data from Panos RJ, Mortenson RL, Niccoli SA, King TE Jr: Clinical deterioration in patients with idiopathic pulmonary fibrosis: causes and assessment. Am J Med. 1990;88(4):396–404.
A number of therapeutic approaches have been attempted in clinical trials. These include use of human recombinant interferon γ-1β, the antifibrotic compound pirfenidone, the antioxidant N-acetylcysteine, and several endothelin receptor antagonists (e.g., bosentan, ambrisentan). To date, no trial has revealed a successful cure for the disease. However, perfenidone has emerged as a candidate for slowing functional loss in IPF patients and is approved for the treatment of IPF in Japan.54,55
Essential Requirements for Accurate Diagnosis
For pulmonary physicians, a pathologic diagnosis of UIP implies clinical IPF; accordingly, UIP should never be diagnosed in the absence of clinical and radiologic correlation. The gravity of the prognosis and the lack of current available therapy strongly support this notion.5 If the pathologic findings are compelling for a UIP pattern, it is reasonable to use a descriptive diagnosis such as that presented in Box 7-7. This approach provides an opportunity for further correlation by clinical colleagues and radiologists in solidifying the diagnosis.
Data from Leslie K, Colby T, Swensen S: Anatomic distribution and histopathologic patterns in interstitial lung disease. In: Schwarz M, King TJ, eds. Interstitial Lung Disease. Hamilton, ON: BC Decker; 2002:31–50; and American Thoracic Society/European Respiratory Society international multidisciplinary consensus classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med. 2002;165(2):277–304.
Familial Idiopathic Pulmonary Fibrosis
There is a small subset of patients with IPF who have a history of unexplained lung disease in first-degree relatives. This form of pulmonary fibrosis has been referred to as familial IPF or familial interstitial pneumonia (although in most studies of familial interstitial pneumonia, fibrosis seems to be the dominant pattern of disease). A compelling body of evidence suggests that IPF is a genetic disorder,56,57 and its familial occurrence is not surprising. Steele and colleagues examined the population of persons with familial interstitial pneumonia from 111 candidate families and found that more than 80% of these individuals had clinical IPF, followed by NSIP.58 Genetic analysis was performed in search of the mechanism underlying familial IPF, and telomerase germ line mutations were identified in 8%.59 The role of telomerase mutations was hypothesized to be a function of excess telomere shortening over time, resulting in cellular dysfunction and premature cell death.
Nonspecific Interstitial Pneumonia
For many years after Liebow’s classification of IIPs was widely adopted, a number of diffuse inflammatory lung diseases were identified that did not fit well within this classification scheme. Various terms were applied to such diffuse lung diseases, including “chronic cellular” and “unclassifiable” interstitial pneumonia.60 In 1994 the term nonspecific interstitial pneumonia was proposed by Katzenstein and Fiorelli, based on data from 64 patients who presented with diffuse lung disease and a chief complaint of dyspnea, usually present for several months before evaluation.16 Radiologic studies showed bilateral interstitial infiltrates with variable consolidation. Importantly, the 64 patients in this study had a significantly better prognosis than that observed for patients with UIP.
Katzenstein and Fiorelli recognized that the constellation of histopathologic patterns seen in NSIP did not represent one disease and, in follow-up investigations, found that these patients often had hypersensitivity, resolving infection, or systemic CVD, among other occurrences. Nagai and coworkers studied a group of patients with cellular interstitial pneumonia and rigorously excluded possible etiologies. The reported survival rate in this “idiopathic NSIP” was 90% at 5 years.61 Thus, when used in the true idiopathic context, the designation “NSIP” may actually be useful if it consistently implies an interstitial chronic inflammatory disease of unknown etiology, with an expected good response to therapy and excellent survival rate. If, on the other hand, the term is applied indiscriminately as a substitute for any histopathologically unrecognized ILD, clinical behavior will be impossible to predict, thereby significantly reducing the benefit of lung biopsy.
Clinical Presentation
Some general statements can be made regarding the clinical presentation in NSIP, recognizing that most of the available data have been derived from studies in which a heterogeneous group of disorders were represented. Patients with NSIP histopathology in lung biopsies (that is, an NSIP pattern) tend to be younger than patients with UIP61–63; the NSIP pattern might also appear in children.16 As with many of the chronic diffuse lung diseases, symptoms develop gradually. Shortness of breath, cough, fatigue, and weight loss are the most common complaints. Fever and digital clubbing have been reported but are uncommon.16,62
Radiologic Findings
As in UIP, most of the chest x-ray abnormalities in NSIP are confined to the lower lung zones and tend to be bilateral and symmetrical.64 Less than 40% of the lung volume is typically involved. Patchy parenchymal (alveolar) opacification is a commonly reported abnormality,64 but reticular (interstitial) changes have also been identified.16 High-resolution CT findings are variable and nonspecific.65 The most common findings are a reticular pattern and traction bronchiectasis, followed by lobar volume loss and ground-glass attenuation. As uncommon features, subpleural sparing, irregular linear opacities, patchy honeycombing, and nodular opacities can be seen.61,62 As might be anticipated, some of the findings described for NSIP overlap with those in other ILDs, such as hypersensitivity pneumonitis and COP. In the stage before honeycomb cysts are visible, even UIP can be indistinguishable from NSIP.
Histopathologic Findings
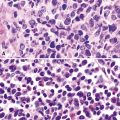
Katzenstein and Fiorelli emphasized that the histopathologic pattern in NSIP was temporally uniform (Fig. 7-12), in contrast with the UIP pattern, in which variable zones of established (dense) fibrosis, more active fibroplasia, and normal lung all coexist in the same biopsy specimen (i.e., temporal heterogeneity). As initially defined, the inflammatory process in NSIP is diffuse and uniform, mainly involving the alveolar walls (Fig. 7-13) and variably affecting the bronchovascular sheaths (Fig. 7-14) and pleura16 (Fig. 7-15). In some patients, infiltrates are predominantly peribronchial, whereas in others, germinal centers may be seen along with chronic pleuritis. When air space organization (the organizing pneumonia pattern) is present, it is not uniformly distributed (Fig. 7-16) as might occur in organizing infectious pneumonia.16 When fibrosis occurs in NSIP, it is usually mild and preserves lung structure (Fig. 7-17). Peribronchiolar metaplasia of variable extent may be seen, but microscopic honeycombing is characteristically absent.16,66
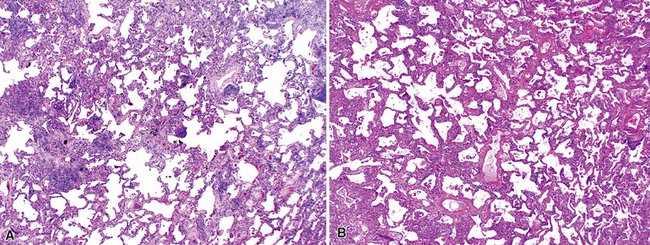
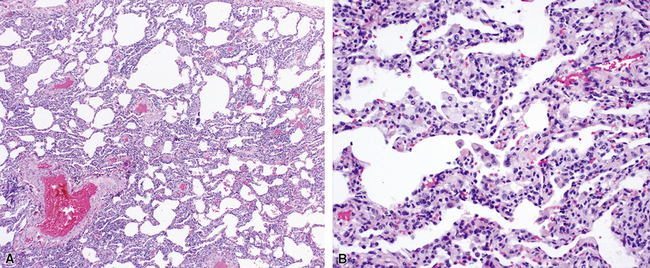
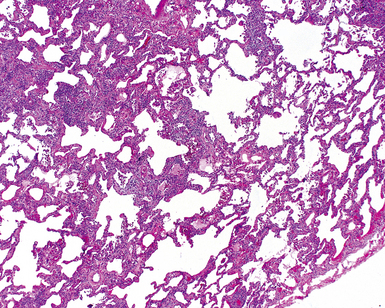
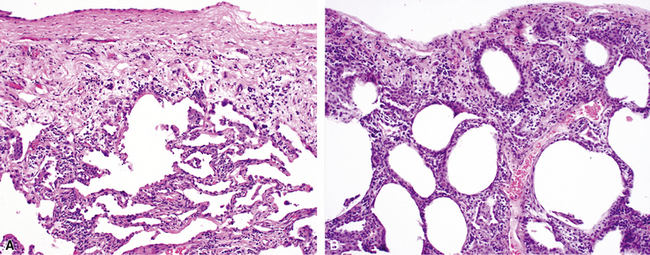
There has been debate as to whether NSIP is a new “interstitial lung disease” or simply a wastebasket category of diseases with some overlapping features. An American Thoracic Society project concluded that idiopathic NSIP is likely a distinct clinical entity with characteristic radiologic and pathologic features.66 Caution is advised in using this term for any lung disease with interstitial inflammation, just as it is imprudent to diagnose all fibrosing lung diseases as UIP.
Differential Diagnosis
The main entities in the differential diagnosis of the NSIP pattern include hypersensitivity pneumonitis, systemic CVDs manifesting in the lung, resolving infection, and low-grade lymphoproliferative disease masquerading as LIP (see later on). Cellular NSIP and LIP may be difficult to distinguish from one another on histopathologic grounds, so they might be considered synonymous from the pathologist’s perspective, once lymphoproliferative disease has been rigorously excluded. Kinder and associates hypothesized that a majority of NSIP cases fall into the category of undifferentiated connective tissue disease manifesting in the lung. Because of significant overlap between NSIP and ILD in CVD, careful follow-up with serologic testing is recommended.67 In clinical practice, in view of the limited arsenal of available therapies for ILD, managing NSIP as a systemic autoimmune disorder with immunosuppressive strategies (even though it may not be initially diagnosable by a rheumatologist) often proves to be the best course of action for the patient.
Clinical Course
The overall survival rate for patients with NSIP is estimated to be in the range of 82.3% at 5 years and 73.2% at 10 years.66 The purely “cellular” form of NSIP seems to be a disease with a good prognosis, compared with UIP and AIP.
When significant fibrous remodeling with microscopic honeycombing is permitted in the diagnosis of NSIP, 5- and 10-year survival rates change significantly for the worse.61,68 This observation suggests that fibrotic forms of NSIP may be within the spectrum of other fibrosing lung diseases, such as UIP of IPF, and certain systemic connective tissue diseases that manifest in the lung with fibrosis.
Cryptogenic Organizing Pneumonia
Air space organization is an extremely common manifestation of lung injury and can be seen after a wide variety of insults, from organizing lung infarction to bacterial pneumonia (Box 7-8). For this reason, the organizing pneumonia pattern in the lung biopsy is the least specific and perhaps the most misunderstood.
Box 7-8 Causes of the Organizing Pneumonia Pattern
Modified from Leslie K, Colby T, Swensen S: Anatomic distribution and histopathologic patterns in interstitial lung disease. In: Schwarz M, King TJ, eds. Interstitial Lung Disease. Hamilton, ON: BC Decker; 2002:31–50.
It is well known that lung repair following a wide spectrum of injuries frequently evolves through a phase of air space organization. When organization is diffuse, involving the entire surgical biopsy, organizing pneumonia (or “diffuse air space organization”) is an appropriate designation. When no etiology can be identified for an organizing pneumonia pattern, the clinical diagnosis of cryptogenic organizing pneumonia (COP) has been proposed (referred to previously as “idiopathic bronchiolitis obliterans organizing pneumonia”).6,17,69
The term bronchiolitis obliterans organizing pneumonia (BOOP), as an idiopathic disease, was first proposed by Davison and coworkers,69 and later used by Epler and associates,17 to define a specific clinical disease course in a group of patients in whom lung biopsies showed variable amounts of air space organization (organizing pneumonia pattern) of unexplained etiology. The importance of recognizing the pattern of organizing pneumonia in the clinical context defined relates to therapy and prognosis. Patients with clinical COP respond well to systemic corticosteroid administration, and pulmonologists expect a good prognosis when this diagnosis is implied histopathologically. When “BOOP” is used in a pathology report as a descriptive term for the occurrence of organizing pneumonia in a biopsy, the clinician may misinterpret this to mean “idiopathic BOOP” is the correct diagnosis. For example, the “BOOP” pattern may be seen in a disease with abundant background lung fibrosis. In this setting, the prognosis is best considered to be guarded.70
Clinical Presentation
As described by Epler and coworkers for the original “idiopathic BOOP,” the patient typically presents several weeks after an episode of clinical symptoms suggesting upper respiratory tract infection.17 The mean age at onset is 55 years, and a majority of patients are nonsmokers.71,72 Slowly worsening symptoms of cough (sometimes productive) and dyspnea are typically present, often leading to surgical lung biopsy within 3 months of disease onset. Weight loss, night sweats, chills, intermittent fever, and myalgias are common. Mild to moderate restrictive pulmonary function studies are identified in a majority of patients.72–74 Hemoptysis and wheezing typically are absent. Often there is a marked increase in the erythrocyte sedimentation rate (ESR). Digital clubbing is not a feature of the disease.
Radiologic Findings
Chest radiography and CT show a number of abnormalities, none of which are specific for one disease. Patchy air space consolidation (loss of visible structure underlying opacification) is the most consistent finding and is present in 90% of cases.75,76 Air bronchograms can be seen in areas of consolidation. Ground-glass attenuation accompanies consolidation in more than one half of the patients. The disease involves the lower lung zones more often than the upper lung zones.77 Small nodular opacities can be seen in 10% to 50% of patients.78
In a small percentage of patients, large nodules may be seen78; rarely, reticulonodular infiltrates occur.74 It is speculated that this latter finding identifies a subset of COP that may not respond to therapy. Opacities may be recurrent and/or migratory.79,80 Lung volumes are normal in most patients, and pleural effusions rarely occur.75,76,81
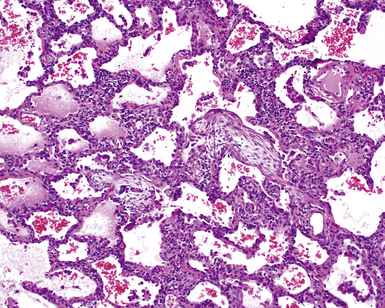
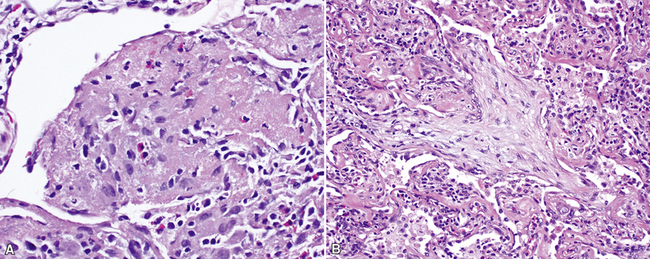
Histopathologic Findings
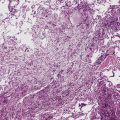
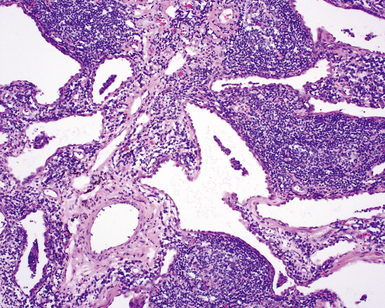
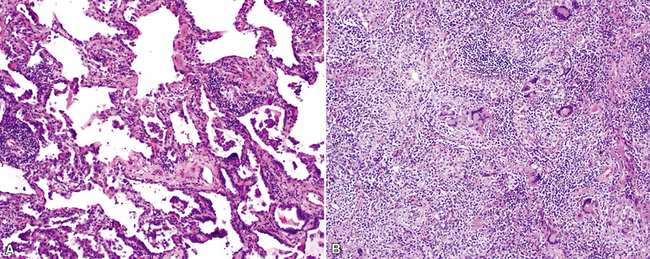
The organizing pneumonia pattern is characterized by variably dense air space aggregates of fibroblasts in ground substance (immature collagen matrix) (Fig. 7-18). This alveolar filling process can be seen to extend into or from terminal bronchioles (Fig. 7-19). Typically, the lung architecture is preserved in COP, and lymphocytes, plasma cells, and histiocytes are present in variable numbers within the interstitium17,82,83 (Fig. 7-20). Fibrin may be seen focally in association with air space organization (Fig. 7-21). Alveolar macrophage accumulation may be present, attesting to some degree of airway obstruction. 17,82,83 When air space organization is confluent and diffuse in the biopsy, COP is less likely to be the accurate diagnosis. Interstitial fibrosis and honeycomb lung remodeling are not components of the cryptogenic (idiopathic) form of organizing pneumonia. 17,82,83
Treatment and Prognosis
The expected response to systemic corticosteroid administration therapy is excellent.17,79,84 Because relapses may occur if therapy is stopped abruptly, patients with COP generally require extended corticosteroid tapering, sometimes over a year or more.17,79,84
Differential Diagnosis
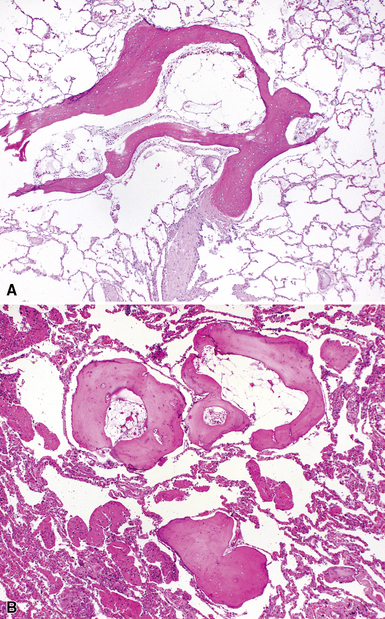
As mentioned previously, the differential diagnosis for the organizing pneumonia histopathologic pattern is too broad in scope to be of clinical use. In general, it is fair to say that the presence of the organizing pneumonia pattern is much more commonly associated with slowly organizing infection, systemic connective tissue diseases, hypersensitivity pneumonitis, and idiosyncratic reaction to drug or medication, rather than a “cryptogenic” disease. Rarely, air space organization may ossify and produced so-called “racemose” or “dendriform” ossification (Fig. 7-22).
Respiratory Bronchiolitis–Associated Interstitial Lung Disease
Respiratory bronchiolitis (RB) is a histopathologic lesion of the small airways that is common in cigarette smokers.85 In some smokers, an exuberant form of RB occurs as a clinical and radiologic manifestation of diffuse “interstitial” lung disease. This ILD manifestation of RB has been referred to as respiratory bronchiolitis–associated interstitial lung disease (RBILD).86 RB, RBILD, and DIP have been proposed as existing along a continuum in smokers,87 with RB on the asymptomatic end of a spectrum that culminates in DIP on the other. Whether RB, RBILD, and DIP are truly manifestations of a single disease process remains to be proved. Certainly, all three have some histopathologic elements in common, but the two main clinical manifestations in the spectrum (RBILD and DIP) also differ in a number of ways clinically and radiologically.
Clinical Presentation
Patients with RBILD are typically a decade younger than those with DIP and present in early midlife, with a mean age of 36 years in two studies.86,88 A relationship between smoking pack-years and onset of disease suggests a dose-related effect, with a threshold in the vicinity of 30 pack-years. There tends to be a gender predilection toward men,87,89 but men and women were equally affected in one study.88 Mild breathlessness and cough are the most common initial complaints.86,88 Clubbing of the digits is unusual in RBILD.88,90,91 Pulmonary function abnormalities parallel the mild clinical symptoms and may show evidence of both obstruction and restriction, with mild reduction in the diffusing capacity.89
Radiologic Findings
The chest x-ray appearance of RBILD reflects the presence of disease centered on the airways, mainly with thickening of airway walls.87 Ground-glass opacity is seen in more than 50% of chest radiographs in RBILD. On CT scans, ground-glass opacities and centrilobular nodules are typical findings, often best seen at the periphery of the upper lung zones.87
Histopathologic Findings
RB is a common reactive process in the lungs of cigarette smokers; its presence alone does not imply the diffuse lung disease manifestation.91 Moreover, even when the histopathologic changes are diffuse and distinctive in the biopsy specimen, clinical correlation is required for accurate diagnosis. For example, a patient with a lung mass, resected and found to be a bronchogenic carcinoma, may have extensive RB in surrounding lung parenchyma. In the absence of a clinically and radiologically defined ILD, a diagnosis of RBILD would be inappropriate.
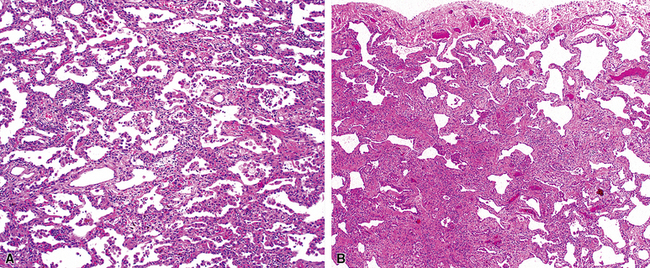
The essential morphologic constituents of RB are (1) scant inflammation around the terminal airways (Fig. 7-23), (2) metaplastic bronchiolar epithelium extending out from terminal airways to involve alveolar ducts (Fig. 7-24), and (3) variable numbers of lightly pigmented, dusty brown air space macrophages within bronchiolar lumens and in immediate surrounding alveoli (Fig. 7-25). Scant peribronchiolar fibrosis may be present and may extend to involve contiguous alveolar walls (Fig. 7-26). When bronchiolocentric scarring is prominent, an alternative diagnosis, such as chronic hypersensitivity pneumonitis, should be considered. Dense collagenous thickening of the alveolar septa without inflammation can be seen in RBILD. Such fibrosis does not appear to progress to honeycomb fibrosis.92 Presence of fibroblastic foci or destruction of the lung architecture should always raise the possibility of an alternative diagnosis, especially undersampled UIP.
Differential Diagnosis
RB may be confused with bronchiolitis of some other etiology. When bronchiolar metaplasia is a prominent component, distinction from other small-airway disease, such as idiopathic constrictive bronchiolitis, may be difficult. Patients with idiopathic constrictive bronchiolitis in surgical biopsies tend to have more severe pulmonary function abnormalities than patients with RB or RBILD (see Chapter 8 for a discussion of small airways disease).
Clinical Course
RBILD generally carries an excellent prognosis. However, symptomatic or physiologic improvement occurs in a limited number of patients. Smoking cessation, with or without immunosuppressive therapy, has been recommended, but a recent report demonstrated benefit in only a small subset of patients.93
Desquamative Interstitial Pneumonia
Leibow7 proposed the term desquamative interstitial pneumonia for a diffuse lung disease that occurred in patients who were typically 10 years or more younger than patients who developed UIP. The disease often presented in mid-adulthood, and most patients were cigarette smokers.94 Liebow also believed that the “desquamated” cells that filled the air spaces in DIP were epithelial cells. It has now been established that the air space cells of Liebow’s DIP are actually macrophages, and that DIP is not a credible precursor lesion for UIP, as was proposed by a number of authorities.
Clinical Presentation
As currently defined, DIP is a very rare smoking-related lung disease. Patients with DIP are typically older than those with RBILD87 and roughly a decade younger than those with UIP.91,94 Most patients with DIP are cigarette smokers; men are more frequently affected than women. Like UIP, the clinical presentation is dominated by insidious onset of dyspnea and dry cough over several weeks or months.91,95 Digital clubbing is present in 50% of patients with DIP, a finding in sharp contrast with RBILD. The symptoms of DIP are usually more pronounced and more severe than those of RBILD,87 supported by pulmonary function testing showing mild restriction and moderate reduction in diffusing capacity.95
Radiologic Findings
The chest radiograph may be normal in 3% to 22% of patients. When abnormalities are present, patchy areas of ground-glass opacification predominate. The lung bases and periphery are most commonly affected.87,96,97 On CT scans, ground-glass opacification is universally present, mostly in a bibasilar distribution.96 Linear and reticular opacities may accompany ground-glass opacities at the bases but tend to be quite limited in extent. Focal areas of peripheral honeycombing may be identified in as many as one third of patients,96 but when prominent and associated with more pronounced reticular abnormalities, an alternate diagnosis should be considered (probably UIP).
Histopathologic Findings
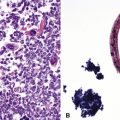
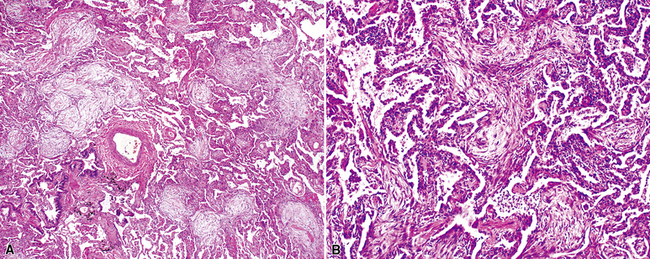
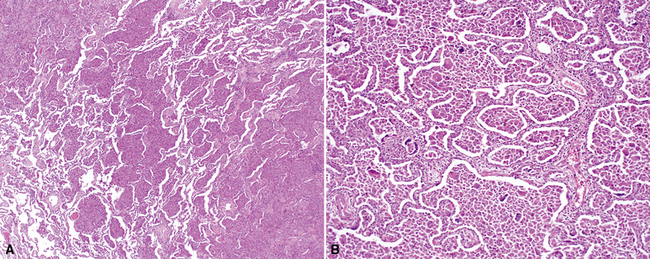
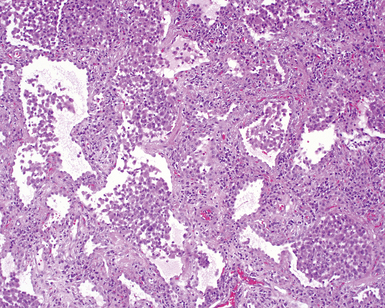
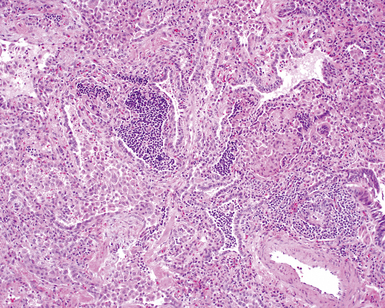
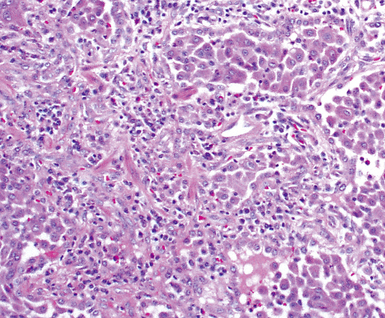
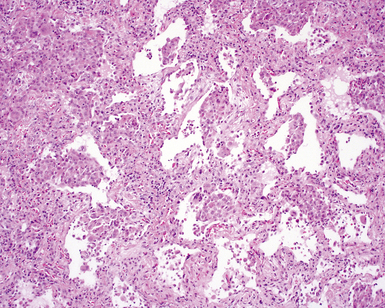
On scanning magnification, the surgical lung biopsy in DIP has an eosinophilic appearance due to the presence of eosinophilic macrophages uniformly filling air spaces11,88 (Fig. 7-27). Mild interstitial thickening by fibrous tissue is the rule and is uniform in appearance (Fig. 7-28). When chronic inflammation is evident at scanning magnification, it is centrilobular and associated with respiratory bronchioles (Fig. 7-29). Scant numbers of plasma cells and rare eosinophils may be seen within slightly thickened alveolar walls at high magnification (Fig. 7-30).
Lymphoid Interstitial Pneumonia
LIP was originally conceived as a chronic cellular interstitial pneumonia with distinctive histopathologic features, quite different in cellular composition and form from Liebow’s other IIPs (e.g., UIP, BIP, DIP, GIP).7 LIP became controversial because many of the cases originally classified as LIP by Liebow (and his contemporaries) evolved into (or were actually indolent forms of) low-grade lymphoproliferative disease involving the lung.98–100 It is now generally acknowledged that the accrual of dense lymphoid tissue in the lung carries strong implications for lymphoproliferative disease, especially small B cell lymphomas of extranodal marginal zone type (so-called lymphomas of the mucosa-associated lymphoid tissue [MALT]) and polymorphous lymphoproliferative disorders associated with viral infection, including EBV or HTLV-1.98–103 “Lymphoid interstitial pneumonia,” as currently defined, is included as an entity in this chapter because a recent international consensus panel chose to keep LIP as a form of IIP, partly for historical reasons. The panel participants acknowledged that many pulmonary pathologists might classify the described histopathologic findings of “idiopathic LIP” as a cellular form of NSIP.
More recently, Cha and colleagues104 described a series of non-lymphoma LIP cases in which 9 of 15 patients were found to have a CVD, mainly Sjögren syndrome. In that series, three patients with idiopathic LIP were identified. All three survived longer than 10 years, and their disease did not progress to lymphoma or leukemia, despite the fact that one of them had a monoclonal gammopathy.104
Clinical Presentation
The clinical manifestations of the idiopathic form of the LIP pattern are not well studied but seem to be similar to those associated with definable systemic conditions, such as CVD. Women are more commonly affected than men; patients are typically between 40 and 50 years of age.105 Interestingly, all of the “idiopathic LIP” patients described by Cha and colleagues were men, whereas most of secondary LIP patients were women.104 Slowly progressive breathlessness is a common feature, with or without nonproductive cough; the disease may evolve over months or years.
In the classic description of LIP, systemic signs and symptoms such as weight loss, pleuritic pain, arthralgias, adenopathy, and fever were reported, depending on whether an associated systemic condition was present.105–107 Findings may include bibasilar crackles, cyanosis, and clubbing. Immunoglobulin abnormalities in serum are present in some patients.108 More commonly, the LIP pattern is associated with a systemic condition that dominates the clinical presentation and clinical course (e.g., Sjögren syndrome, pernicious anemia, hypogammaglobulinemia).
Radiologic Findings
The published radiologic features of idiopathic LIP seem to describe more than one pattern of disease.109 Bibasilar reticular opacities along with ground-glass attenuation and thickening of interlobular septa are frequently observed abnormalities.104,109–111 There may be mixed alveolar and interstitial infiltrates and thin-walled cysts, honeycombing, and changes suggesting pulmonary hypertension late in the disease.112,113 Nodular patterns can also occur.114 Pleural effusion is rare and, if present, should increase concern for low-grade malignant lymphoma.
A distinctive cystic disease has also been referred to by radiologists as “lymphocytic interstitial pneumonia” (or simply, LIP); on biopsy, however, the process has no significant interstitial inflammatory infiltrates or fibrosis, exhibiting only thin-walled, dilated airways with scant associated bronchiolitis.115 An association with Sjögren syndrome has been documented.116
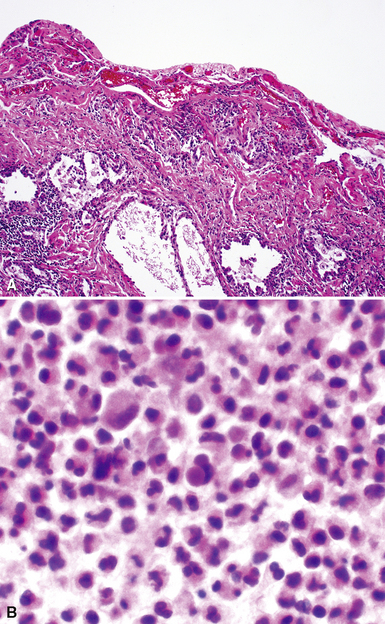
Histopathologic Findings
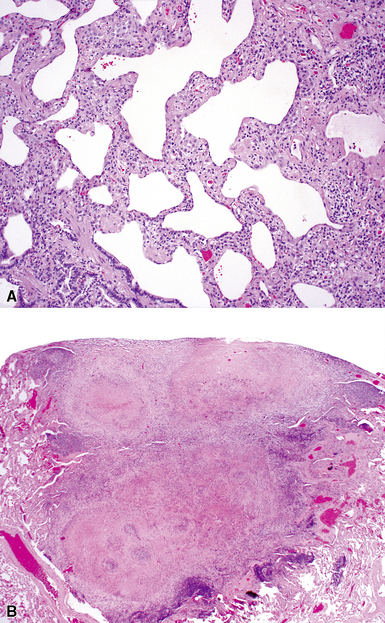
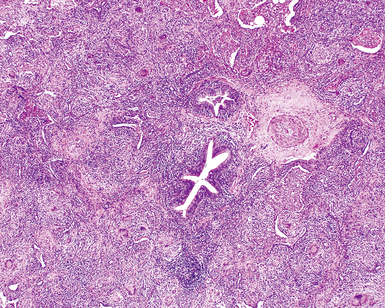
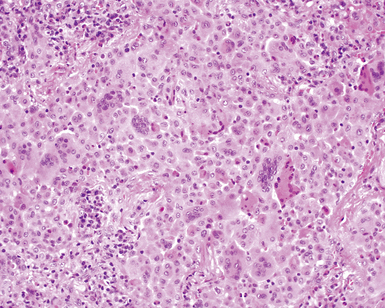
The histopathologic pattern in LIP is characterized by the presence of a dense and diffuse alveolar septal infiltrate made up of lymphocytes, plasma cells, and histiocytes (Fig. 7-31). This definition helps exclude diseases with less intense cellular interstitial infiltrates, such as certain hypersensitivity reactions and systemic connective tissue diseases. Multinucleated giant cells or small, ill-defined granulomas in the interstitium have been described in the idiopathic form of LIP, but microscopic honeycomb remodeling, with some interstitial fibrosis (Fig. 7-32), can also be a part of idiopathic LIP. To a variable extent, germinal centers may be present along airways and lymphatic routes (Fig. 7-33). When these are prominent and interstitial lymphocytic infiltration is less remarkable, diffuse lymphoid hyperplasia has been used as a preferable term. In this situation, lymphoproliferative disorders, such as multicentric Castleman disease, idiopathic plasmacytic lymphadenopathy with hyperimmunoglobulinemia, or even MALT lymphoma, are the main considerations.117 When the idiopathic form of the LIP pattern is identified, immunophenotyping and gene rearrangement studies typically show an absence of clonality.118 When nodular lymphoid hyperplasia is prominent around bronchioles, typically accompanied by an interstitial infiltrate, Sjögren syndrome should be rigorously investigated as a potential etiology.
Differential Diagnosis
The LIP pattern is most consistently seen when systemic CVDs manifest in the lung.105,119 The LIP pattern may also be seen in the setting of bone marrow transplantation120 and has frequently been observed in both children and adults who have congenital or acquired immunodeficiency syndromes121 and in the setting of adult HIV infection including vertical transmission from mother to child.122–124
Much of what has been written about the histopathology of LIP is similar to that written about the histopathologic patterns of NSIP. If LIP and cellular NSIP can be distinguished from each other microscopically, it is usually on the basis of the sheer density of the lymphoid infiltrate in LIP, accompanied by fibrosis and some degree of remodeling (the latter would be unexpected for the cellular form of NSIP). Naturally, in this setting, gene rearrangement studies are important in distinguishing idiopathic LIP from low-grade lymphoproliferative disease (see Chapter 15 for further discussion). Once the pattern is established, it is useful to suggest the potential systemic conditions that may be associated with this pattern (Box 7-9) in a “comment” section of the surgical pathology report.
Box 7-9 Systemic Conditions Associated with the Lymphoid Interstitial Pneumonia Histopathologic Pattern
Modified from Leslie K, Colby T, Swensen S: Anatomic distribution and histopathologic patterns in interstitial lung disease. In: Schwarz M, King TJ, eds. Interstitial Lung Disease. Hamilton, ON: BC Decker; 2002:31–50.
Clinical Course
The clinical outcome and response to therapy for patients with the LIP pattern is largely dependant on whether systemic disease is present. In the idiopathic form, an accurate prognosis has not been forthcoming, although in the series reported by Cha and coworkers, three patients survived more than 10 years.104 In symptomatic patients, corticosteroid administration may result in significant benefit,105 lending further support to LIP’s being an immunologic disease, rather than a neoplastic one, in most instances. When honeycomb cysts, clubbing, or cor pulmonale are present, the prognosis is less favorable, with as many as one third of patients succumbing to the disease.105,114 Infection is a common complication, especially when LIP is associated with dysproteinemia.105,106,125
Chronic Manifestations of Systemic Collagen Vascular Disease
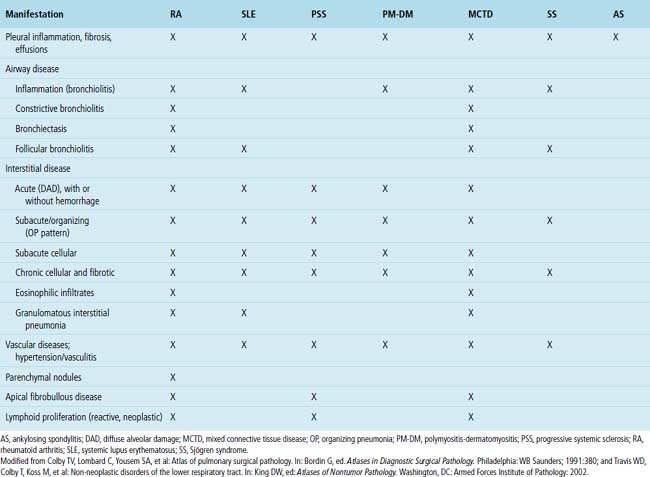
Systemic CVDs play an extremely important role in the etiology of ILDs. Knowledge of rheumatic ILD is derived mainly from retrospective studies, typically including small numbers of patients. Because of differences in patient populations reported and in the rheumatic disease severity (and duration) at the time of study, many important questions remain concerning the frequency, pathogenesis, natural history, clinical relevance, and prognosis of ILD occurring in the rheumatic diseases. It is estimated that ILD in CVD is responsible for 1600 deaths annually in the United States, accounting for roughly 25% of all ILD deaths and 2% of all deaths from respiratory causes.126 Not suprisingly, most interstitial pneumonia patterns raise CVD as a consideration in the differential diagnosis. On the other hand, certain CVDs are associated with reasonably reproducible findings in the lung.2Table 7-3 summarizes the different patterns of inflammatory lung disease that have been described as lung manifestations of the known connective tissue diseases. The five rheumatic diseases that are more commonly associated with ILD are (1) rheumatoid arthritis (RA), (2) progressive systemic sclerosis (PSS), (3) systemic lupus erythematosus (SLE), (4) polymyositis-dermatomyositis (PM-DM), and (5) Sjögren syndrome. The estimated frequency of lung involvement and the patterns produced are presented in Table 7-4. This section is restricted to the more chronic manifestations of these diseases. Acute lung manifestations of the rheumatic diseases are described in Chapter 5.
Rheumatoid Arthritis
RA is a chronic systemic disease that produces symmetrical arthritis and occurs more commonly in women than in men. ILD was not recognized as a manifestation of RA until 1948,138 possibly because lung manifestations of the disease are difficult to recognize on purely clinical grounds. Today, with the use of pulmonary function testing, bronchoalveolar lavage, and CT imaging, significant lung disease is identified in 14% of patients who meet the American College of Rheumatology (formerly the American Rheumatism Association) criteria for RA; subclinical disease is seen in as many as 44%.139 Interestingly, men are three times more likely to develop ILD with RA than are women.25 Clinically significant ILD in RA is associated with increased morbidity and mortality.25
Clinical Presentation
Diffuse lung disease in RA typically is noted in patients with diagnosed RA, but rarely, ILD may precede articular manifestations.140,141 The clinical presentation is dominated by shortness of breath and cough. Adults are more commonly affected than children,142 and despite a higher incidence of RA in women, men with long-standing rheumatoid disease and subcutaneous nodules seem to develop lung manifestations more often.140 Physical examination may reveal bibasilar inspiratory crackles, digital clubbing, and evidence of cor pulmonale, the last due to pulmonary hypertension arising as a result of hypoxic vasoconstriction.25,140 When fibrosis and honeycomb remodeling accompany diffuse lung disease in RA, UIP enters into the differential diagnosis. Affected patients often are younger than those with idiopathic UIP. Cigarette smoking has been reported to be an independent predictor of lung disease in persons with RA.143
Radiologic Findings
Several radiologic manifestations are described in RA, including reticular opacities with or without honeycombing, airway-associated abnormalities (bronchiectasis, nodules, centrilobular branching lines), and parenchymal opacities.144 When ground-glass infiltrates and reticular opacities are present, there is a predilection for involving the bases and lung periphery. High-resolution CT findings include ground-glass attenuation with mixed alveolar and interstitial infiltrates. As lung disease advances, dense reticular and nodular opacities appear, and honeycomb lung may be seen in late stages of the disease.144,145
Histopathologic Findings
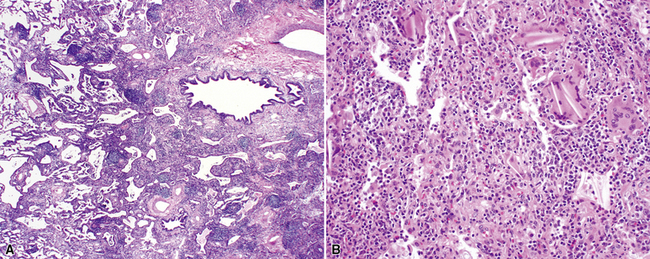
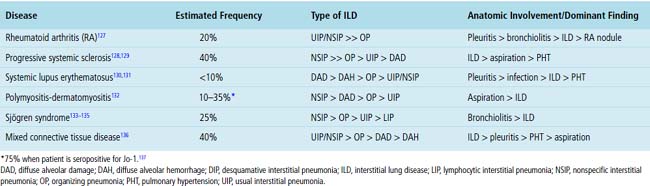
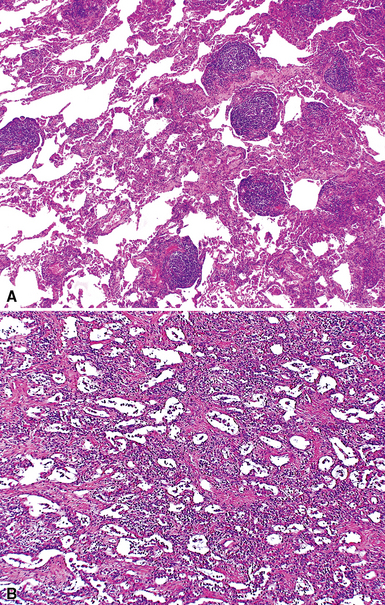
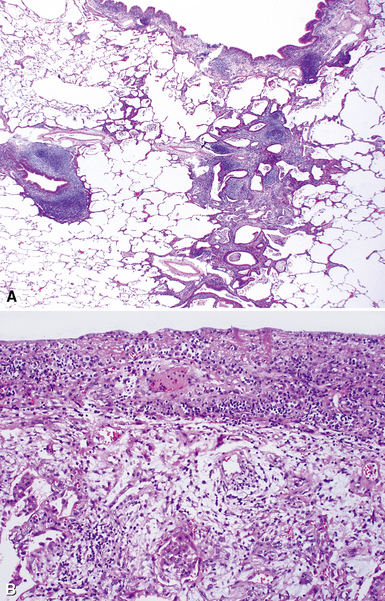
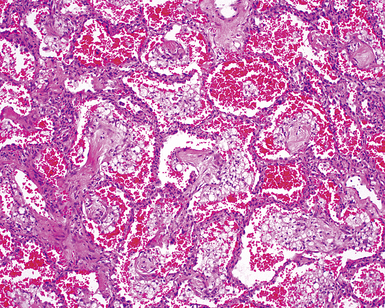
Despite the seemingly nonspecific nature of the histopathologic manifestations of RA, a few key elements emerge on review of many well-documented cases of RA-associated ILD. Chronic inflammation, in the form of lymphocyte aggregates and germinal centers, is typical, although not unique (Fig. 7-34) Most of the lymphoid aggregations are present around the terminal airways (“follicular bronchiolitis” when lymphoid germinal centers are prominent) (Fig. 7-35), but lymphoid follicles may also present in the pleura. In fact, the presence of chronic pleuritis should always raise RA as a consideration in the differential diagnosis. Areas of subacute lung injury, attended by reactive type II cells and air space organization (Fig. 7-36), can be seen with cellular interstitial pneumonia (Fig. 7-37) and variable interstitial fibrosis (Fig. 7-38). This combination of subacute and chronic inflammatory reactions haphazardly involving the same lung biopsy, including the pleura, should raise the possibility of RA lung disease. When fibrosis is prominent, it is often difficult to classify as UIP or NSIP. That may be one of the reasons for the variable reported incidence of these two patterns of fibrosis in the disease. Fibroblastic foci are usually less prominent, and normal lung may be absent. Vasculitis (including capillaritis) and even pulmonary hemorrhage have been described as manifestations of RA lung. When silicosis occurs with RA, the resulting disease is referred to as Caplan syndrome. Rheumatoid nodules can occur in the lung and pleura and must be distinguished from granulomatous infection or Wegener granulomatosis. Intrapulmonary lymph nodes may become prominent in RA and typically show reactive lymphoid hyperplasia when subjected to biopsy.
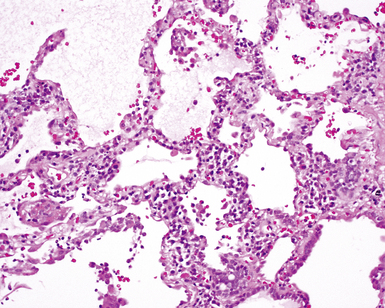
Clinical Course
As with other connective tissue diseases, therapeutic strategies in RA have focused on immunosuppression.146 Although the reported survival significance of RA-ILD varies, a majority of published papers indicate better survival rates for patients with RA-ILD than for those with UIP/IPF.147 Needless to say, the development of pulmonary fibrosis with a UIP pattern has a significant negative impact on survival.141,143,148,149
Progressive Systemic Sclerosis
PSS is a relatively rare systemic autoimmune disease, with cutaneous manifestations (dermal sclerosis) frequently accompanied by Raynaud phenomenon. Pulmonary involvement (mainly ILD) occurs more commonly in patients with PSS than in those with any other connective tissue disease,128,150 with lung disease ranking fourth in frequency (after skin, peripheral vascular, and esophageal manifestations) in the disease, but is the primary cause of death in PSS.151 As in RA, lung involvement in PSS is associated with increased morbidity and mortality.151
Clinical Presentation
Chronic exertional dyspnea is the most common presentation, followed in frequency by chronic cough. Bibasilar inspiratory crackles are present in two thirds of patients.128,152 Digital clubbing may be present but is uncommon. Pulmonary fibrosis and cor pulmonale may develop eventually.153 As in other CVDs, lung involvement can precede the development of diagnostic systemic manifestations.128,154
Radiologic Findings
Bibasilar interstitial infiltrates with relative sparing of the upper lung zones are typical radiologic features.155,156 Loss of lung volume, honeycomb cysts, and findings consistent with pulmonary hypertension may also be seen. Mixed reticular and nodular infiltrates are common.155,156 Pleural effusion and pleural thickening may occur as minor findings.
Histopathologic Findings
The pulmonary manifestations of PSS can be quite characteristic. The interstitial fibrosis of PSS is paucicellular and diffuse, with preservation of underlying lung architecture (Fig. 7-39). This distinctive “collagenization” of the lung interstitium has been confused with the pattern of lung fibrosis seen in idiopathic UIP.153 The lack of so-called “temporal heterogeneity” (see the earlier section, “Usual Interstitial Pneumonia”) is a useful finding and helps exclude UIP (of IPF) from the differential diagnosis. Pulmonary hypertensive changes (Fig. 7-40) may be present and merit careful attention because this is a major cause of death in patients with scleroderma and lung disease.157 Because patients with PSS can also develop esophageal motility problems, subclinical chronic aspiration should be carefully excluded as a comorbid disease process.158,159
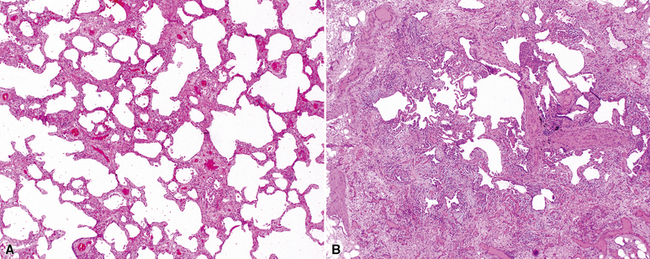
Clinical Course
The mean survival time for patients with scleroderma is 12 years from the time of diagnosis; pulmonary disease has emerged as the major cause of death.160 Pulmonary function status at presentation is a reasonable predictor of survival; high-dose immunosuppressive therapy, typically in combination with a cytotoxic agent, seems to benefit those patients with severe manifestations.161
Systemic Lupus Erythematosus
SLE is a chronic systemic autoimmune disorder characterized by arthropathy, mucocutaneous manifestations, renal disease, and serositis.162 The lung may be the major site of involvement in SLE, ranging from acute lupus pneumonitis on one end of the spectrum to fibrotic forms of NSIP on the other.130,163,164 Acute lung injury and pulmonary hemorrhage are more commonly associated with SLE than with other systemic connective tissue diseases,130,165 but hemoptysis occurs only in little more than one half of the affected patients.166,167
Clinical Presentation
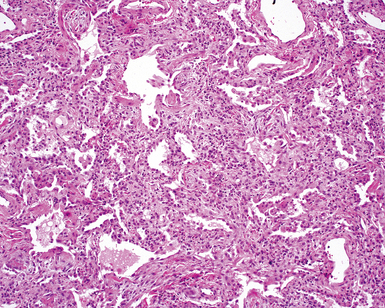
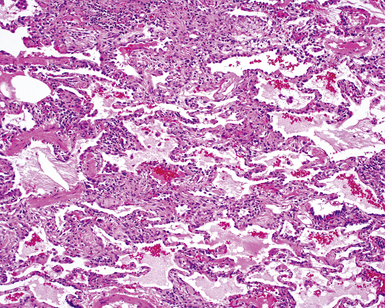
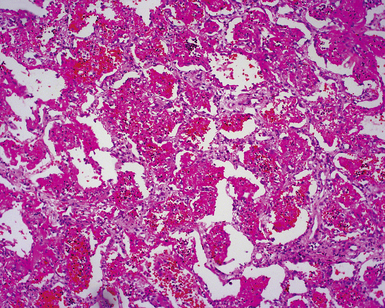
SLE rivals PSS as the leader in pleuropulmonary manifestations in CVD.131,163,168,169 The spectrum of lung disease in SLE is quite broad130,164,165,170 and includes pleuritis (Fig. 7-41), acute lupus pneumonitis (Fig. 7-42), NSIP with fibrosis (Fig. 7-43), and diffuse alveolar hemorrhage (Fig. 7-44). Constrictive small-airway disease, pulmonary arterial hypertension, and pulmonary embolism can also occur as rare manifestations. Patients with lung disease often have high serum ANA or RF titers.
Radiologic Findings
The radiologic findings are similar to those in other connective tissue diseases: variable ground-glass attenuation, pleural thickening, pleural and pericardial effusions, and linear parenchymal opacities.130,171–174 Acute pneumonitis can produce more extensive changes, but, rarely, chest radiography and high-resolution CT may show normal findings.175
Histopathologic Findings
Two general categories of pulmonary disease are described in SLE. The first is acute lupus pneumonitis (ALP). ALP is characterized by alveolitis with variable interstitial inflammation and edema (Fig. 7-45). Siderophages and capillaritis occur to a variable degree (Fig. 7-46). Pleuritis is commonly present. The second category of disease includes cellular interstitial pneumonia (lymphocytes and plasma cells) with variable interstitial fibrosis (Fig. 7-47). This latter NSIP pattern is associated with a better prognosis than that for ALP. When pulmonary hemorrhage occurs, the prognosis may be adversely affected. One dramatic but fortunately rare complication of SLE is the occurrence of lung infarction related to the lupus anticoagulant.176–178 Whenever lung infarction is encountered in a young, otherwise healthy patient, this possibility should be considered (even if the patient does not have evident SLE).
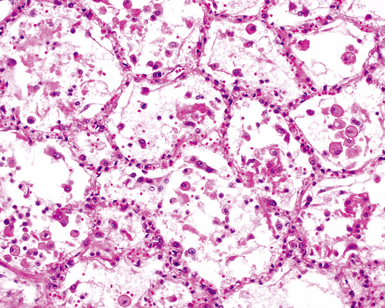
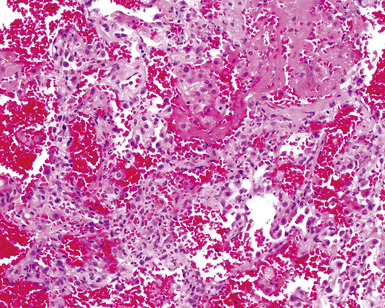
Clinical Course
Systemic corticosteroid therapy may be effective in SLE-associated lung disease, although sometimes the addition of a cytotoxic agent (e.g., cyclophosphamide, azathioprine) may be required.179 Acute lupus pneumonitis carries a reported high mortality rate in some series.168 More chronic forms of diffuse lung disease in SLE have a relatively good prognosis and response to therapy.180
Polymyositis-Dermatomyositis
Polymyositis and dermatomyositis (PM-DM) are inflammatory disorders of the skeletal muscle and dermis. Five groups of primary or secondary disease are recognized, including childhood forms and overlap syndromes.181,182 Pulmonary complications in PM-DM occur less commonly than in other systemic connective tissue diseases, but in a percentage of patients, the pulmonary manifestations can be quite dramatic.183
Clinical Presentation
Although most patients with PM-DM develop lung manifestations after the clinical diagnosis has been established, occasionally lung disease can precede the clinical and serologic diagnosis by months or even years.184 Onset of pulmonary symptoms may occur at any age, with a mean occurrence in the sixth decade.150,184,185 Women are more commonly affected than men. Digital clubbing is rare. In contrast with most other systemic connective tissue diseases (with the exception of SLE), patients with PM-DM can present with acute lung disease, typically manifesting as rapidly progressive DAD.183,185 Importantly, both acute aspiration pneumonitis secondary to underlying respiratory muscle weakness and bronchopneumonia occurring in the setting of immunosuppressive therapy are more common in PM-DM than is chronic diffuse lung disease.186,187
Radiologic Findings
As with other CVDs manifesting in the lung, radiologic abnormalities in PM-DM predominantly affect the lung bases.185 Ikezoe and colleagues reviewed the high-resolution CT findings in 23 of 25 patients with PM-DM who had high-resolution CT abnormalities.188 These researchers identified ground-glass opacities in 92%, linear opacities in 92%, irregular interfaces in 88%, air space consolidation in 52%, parenchymal micronodules in 28%, and honeycombing in 16% of the cases. The most dramatic radiologic finding associated with PM-DM is the rapid onset of air space consolidation associated with the development of DAD.184,185
Histopathologic Findings
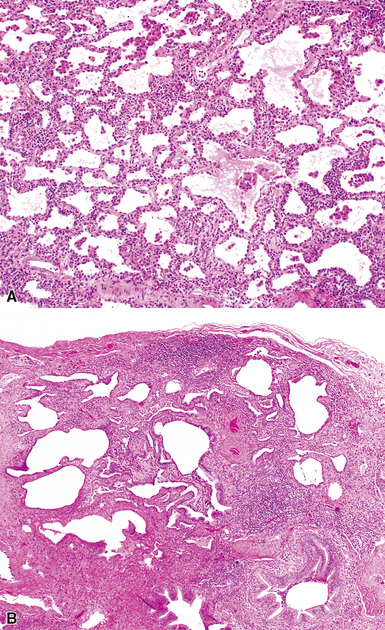
The most frequent lung manifestation of PM-DM is a cellular interstitial pneumonia (Fig. 7-48) with some fibrosis,184,185 indistinguishable from nonspecific interstitial pneumonia (see the later section, “Idiopathic Interstitial Pneumonias”). The next most common pattern is DAD (Fig. 7-49). The fibrosis associated with PM-DM is distinguishable from that of idiopathic UIP based on a relative lack of peripheral accentuation (Fig. 7-50) and absence of the typical transitions from older lung fibrosis to normal lung through fibroblastic foci. Pleuritis, inflammatory small-airway disease, and pulmonary hypertension are unusual findings; their occurrence should suggest a manifestation of a different connective tissue disease.
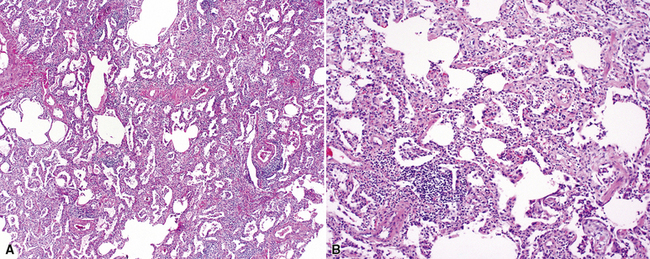
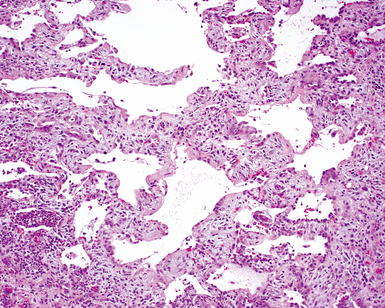
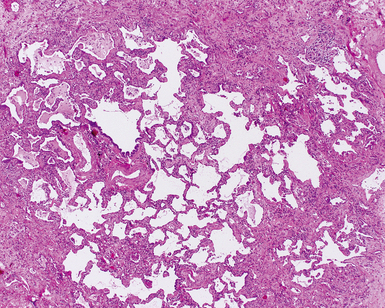
Sjögren Syndrome
Sjögren syndrome is an immune-mediated exocrinopathy characterized by lymphocytic infiltration of the salivary glands, with resulting dry mouth and dry eyes.189 Lung involvement is common and is similar to that in other CVDs manifesting in the lung.107,190,191 Sjögren syndrome can occur as a primary connective tissue disease or as a complication associated with other connective tissue diseases (occurrence rates for the two forms are approximately equal).189 In both primary and secondary forms, the consistent pathologic manifestation is that of lymphoid accumulation in a bronchiolocentric distribution and the NSIP/LIP pattern of cellular interstitial pneumonia.
Clinical Presentation
Women are more commonly affected with lung disease in Sjögren syndrome than men; the most frequent presenting complaint is cough and dyspnea.107,190 Positive results on rheumatoid factor and ANA assays are expected findings, as well as positive reactions to extractable nuclear antigens (anti-SSA, anti-SSB).192 These latter serologic tests are specific for the primary form of the disease.193
Radiologic Findings
Mixed alveolar and interstitial infiltrates are characteristic in Sjögren syndrome; they usually have a finely reticular or nodular pattern.191,194 The occurrence of pleural effusion or hilar/mediastinal adenopathy in patients with Sjögren syndrome should raise concern for lymphoma.195
Histopathologic Findings
The histopathologic spectrum of pulmonary Sjögren syndrome includes bronchiolitis (Fig. 7-51) with or without air space organization, follicular lymphoid hyperplasia along airways (Fig. 7-52), diffuse NSIP or LIP pattern interstitial inflammation (Fig. 7-53), and rarely, interstitial fibrosis (Fig. 7-54), the last raising concern for an inflammatory version of UIP. Small, non-necrotizing granulomas, resembling those of hypersensitivity pneumonitis, are frequently identified in the interstitium (Fig. 7-55).196 In some cases, more prominent granulomatous inflammation can be seen, especially in association with the LIP pattern of cellular infiltration. Patients with Sjögren syndrome are at risk of developing lymphoid hyperplasia and lymphoproliferative diseases; this predilection should be kept in mind in evaluating the lung biopsy in this setting.
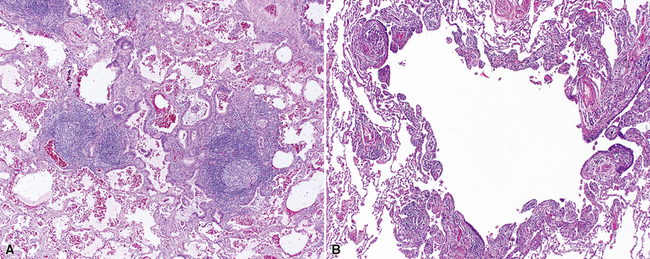
Diffuse Eosinophilic Lung Disease (Pulmonary Eosinophilia)
Several conditions have been described in which the lungs become infiltrated by eosinophils.197,198 An etiologic and clinical classification of the “eosinophil-rich lung diseases” is presented in Box 7-10. Asthma and hypersensitivity play important roles in a number of these. Eosinophilic lung diseases are discussed together here, but in practice, only acute onset of diffuse lung injury accompanied by extravascular eosinophils is relatively predictable in terms of clinical behavior and responsiveness to therapy (i.e., corticosteroids). When pulmonary eosinophilia presents as a chronic condition, the behavior seems to be less predictable. The term chronic eosinophilic pneumonia has been applied in such cases, even though the histopathologic findings in biopsy specimens may not reflect this longer evolution with observable fibrosis or structural remodeling.
Box 7-10 Etiologic and Clinical Classification of Eosinophilic Pneumonia
Modified from Travis WD, Colby T, Koss M, et al: Non-neoplastic disorders of the lower respiratory tract. In: King DW, ed: Atlases of Nontumor Pathology. Washington, DC: Armed Forces Institute of Pathology: 2002:161, Table 3-31.
Clinical Features
Patients with the “chronic” form of eosinophilic pneumonia often exhibit a typical clinical syndrome and radiographic appearance.199 The condition frequently affects middle-aged women, and asthma is present in approximately one fourth of the cases. Nasal symptoms occur in roughly one third of affected individuals. The disease typically presents with severe systemic symptoms, including fever, sweats, weight loss, cough, and dyspnea. Peripheral blood eosinophilia can often be identified, but this may be transient or absent altogether.
Radiologic Findings
Chronic eosinophilic pneumonia presents the most consistent radiologic pattern, with bilateral, poorly defined, subpleural air space consolidation on chest radiographs, most commonly distributed at the lung apices and in the axillary region. One unifying concept for the diffuse forms of eosinophilic lung disease is the “migratory” infiltrate. These infiltrates may disappear spontaneously and recur in the same position or elsewhere. In the most extreme cases, the infiltrates are densest in the periphery of the lung and spare the central region. This phenomenon has been referred to as the photographic negative of pulmonary edema.199 CT scans detect the peripheral location of infiltrates even when this distribution is not apparent on the chest radiograph.200 Once the characteristic presentation is recognized, corticosteroid administration can lead to dramatic improvement in patients with some forms of the disease and may even be used as a diagnostic test. Atypical presentations occur, so surgical wedge biopsy may be required to establish the diagnosis.
Histopathologic Findings
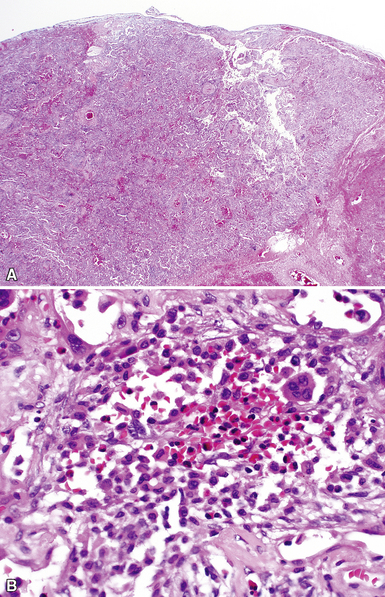
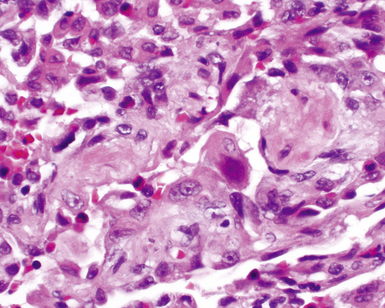
The histopathologic features of chronic pulmonary eosinophilia are similar to those of the acute form; it has been said that the distinction must rely on the clinical course rather than on the constellation of morphologic findings.1 Alveolar spaces are filled with eosinophils and plump eosinophilic macrophages (Fig. 7-56), and there is an associated mild interstitial pneumonia. Type II hyperplasia is characteristic (Fig. 7-57), and fibrin often is present in the air spaces (Fig. 7-58). Angiitis of small vessels may be seen, and patchy air space and alveolar duct organization may be present. A vaguely granulomatous accumulation of dense macrophages may be seen within the alveolar spaces (Fig. 7-59), sometimes accompanied by multinucleate giant cells whose nuclei and cytoplasm closely resemble those of adjacent macrophages (Fig. 7-60). Chronicity may be suggested by the presence of variable interstitial fibrosis on histopathologic examination, but as mentioned earlier, this is not a prerequisite for the diagnosis.
Differential Diagnosis
When air space organization is prominent, disorders with the organizing pneumonia pattern must be considered in the differential diagnosis (see Box 7-8). Some investigators have proposed an overlap syndrome between COP and eosinophilic pneumonia with subacute clinical course; it is interesting that both conditions are expected to have favorable responses to systemic corticosteroid administration. When corticosteroids have been administered before biopsy (which is quite common), eosinophils may be absent or inconspicuous in lung sections. In such cases, the differential diagnosis may include granulomatous disease if the dense histiocytic response and multinucleate giant cells dominate the picture. When fibrin is prominent in the setting of pretreatment with corticosteroids, generic acute lung injury (including DAD and acute fibrinous and organizing pneumonia, because both patterns can be seen in acute eosinophilic pneumonia [AEP]; see Chapter 5) may enter the histopathologic differential diagnosis. Some patients with long-standing symptoms may have fibrosis on biopsy; in such cases, a fibrosing lung disease, such as UIP or NSIP, may enter the differential diagnosis, with eosinophilic pneumonia possibly manifesting as a comorbid process (e.g., drug reaction superimposed on NSIP).
Clinical Course
The clinical course is somewhat dependent on the underlying cause of the eosinophilic pneumonia, but in general, most affected individuals will benefit from high-dose corticosteroid therapy (although the speed of recovery may not be as rapid as that seen in acute onset eosinophilic pneumonia). As in all cases of eosinophilic pneumonia, it is always worthwhile to suggest the possibility of Churg-Strauss syndrome (see Chapter 10) because the pulmonary manifestations of that systemic vasculitic disease in the lung are most commonly those of eosinophilic pneumonia.
Drug-Associated Diffuse Lung Disease
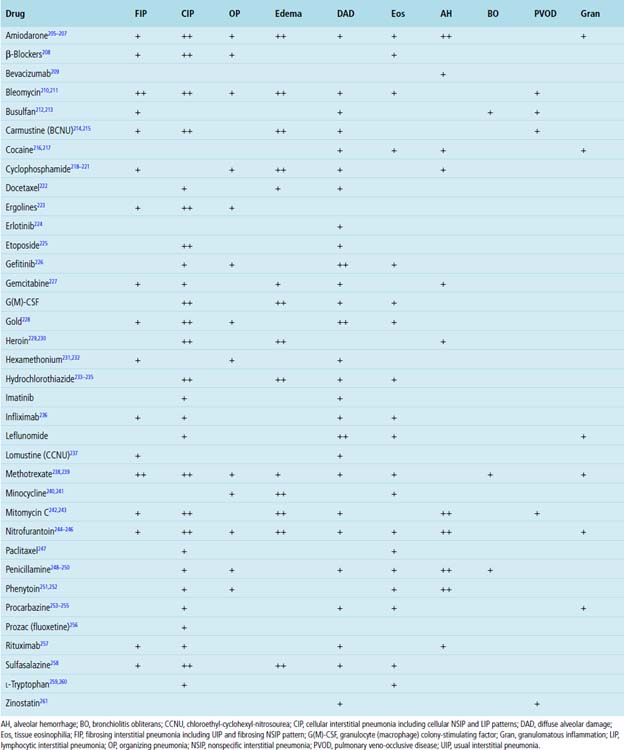
An increasing number of medications have been implicated in chronic diffuse lung disease.201,202 Three distinct forms are recognized: (1) drug-mediated chronic diffuse lung disease, (2) acute lung injury associated with drug administration, and (3) vascular diseases produced by medications.203,204 The latter two conditions are dealt with in Chapters 5 and 10, respectively.
The recognition of drug-induced diffuse lung disease is a major challenge in lung pathology because most of the histopathologic changes identified are nonspecific (Table 7-5) and simulate those seen with other causes of diffuse lung disease.262 Moreover, many affected patients have underlying diseases for which a drug has been administered, and some of these diseases also have pulmonary manifestations. The diagnosis of a drug-mediated diffuse lung disease requires careful exclusion of other causes.262 Unfortunately, a clear onset of pulmonary symptoms with drug administration and abatement of symptoms on cessation of the drug may not be easily discernable. Clinical information regarding specific drug type, dose, and timing of administration relative to onset of symptoms is essential to an accurate diagnosis.
General Histopathologic Findings
Most of the inflammatory changes in the lung related to drug toxicity are nonspecific. More often than not, a mixture of both acute and chronic disease is apparent and can be a clue to the diagnosis of drug-mediated injury.238 In chronic drug toxicity, lung fibrosis may occur, sometimes with honeycomb remodeling. In such cases, UIP may be simulated. Type II cell hyperplasia with or without atypia, cytoplasmic vacuolation in type II cells and macrophages, and tissue eosinophilia can occur in drug reactions. Also, some drugs are associated with the production in the lung of small, poorly formed granulomas, simulating infection, hypersensitivity, or even Sjögren syndrome.201
General Treatment and Prognosis
Most patients with drug-mediated diffuse lung disease have a favorable prognosis when the implicated drug is withdrawn. Certain newer targeted molecular therapies have been associated with a higher mortality rate when diffuse acute injury occurs.224,263 Once fibrosis has occurred, changes are likely to be stable, with little improvement. Systemic corticosteroid therapy may be added when symptoms are severe.
Specific Drugs Associated with Interstitial Lung Disease
Methotrexate
Methotrexate (MTX) lung toxicity is uncommon; when it occurs, it is predominantly a manifestation in women taking the drug.264,265 Because MTX is used in the treatment of a variety of disorders (e.g., RA, some leukemias, some visceral cancers), these tend to be the associated underlying diseases that must be considered in the differential diagnosis for the lung manifestations identified. Interstitial inflammation and fibrosis (Fig. 7-61) are common findings in the surgical lung biopsy.239,266,267Giant cells and small non-necrotizing granulomas (Fig. 7-62) are the only relatively specific markers for MTX, in comparison with other drugs.239 Type II pneumocyte hyperplasia and tissue eosinophilia may be seen (Fig. 7-63). Hyaline membranes are rarely identified.239
Amiodarone
Amiodarone is the drug of choice for the treatment of certain refractory cardiac arrhythmias. Pulmonary toxicity has been reported in 5% to 10% of patients taking this medication. Older patients are more likely to develop lung disease. The clinical onset is characterized by slowly progressive dyspnea and dry cough, occurring within months of initiating therapy. Approximately one third of patients experience an acute febrile illness mimicking infectious pneumonia.268–271 High-resolution CT scans show diffuse infiltrates combined with basal or peripheral high-attenuation opacities and nonspecific infiltrates.272,273 The most common pathologic manifestation is a cellular interstitial pneumonia (Fig. 7-64) associated with prominent intra-alveolar macrophages whose cytoplasm shows fine vacuolation.268,274–276 This vacuolation can also be seen in reactive type II pneumocytes (Fig. 7-65); published reports have described the presence of characteristic lamellar cytoplasmic inclusions ultrastructurally.200 Unfortunately, these cytoplasmic changes are an expected manifestation of the drug, so the mere presence of such changes is not sufficient to warrant a diagnosis of amiodarone toxicity.274 Pleural inflammation and pleural effusion have also been reported.277 Some patients with amiodarone toxicity may develop an organizing pneumonia (OP) pattern (Fig. 7-66), a nodular pattern,278 or even DAD.274,279,280 A majority of patients with amiodarone-related pulmonary toxicity will recover once the drug is discontinued.268,269,274–276
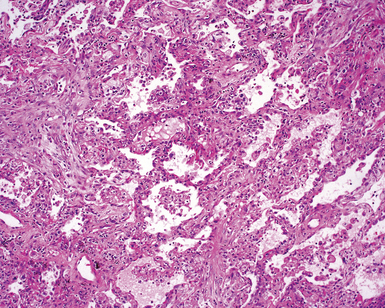
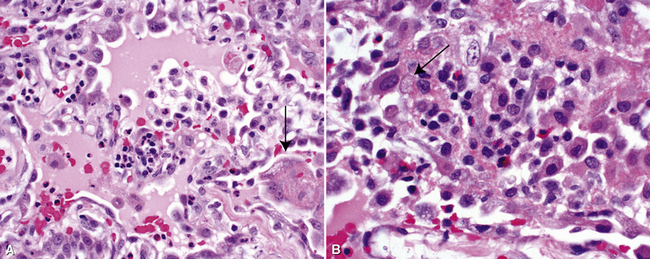
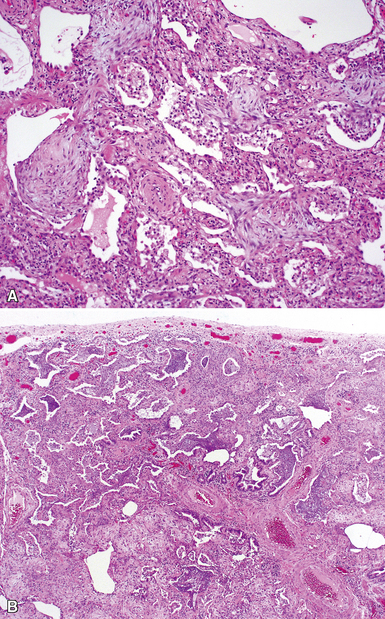
BCNU
BCNU (carmustine) is the treatment of choice for patients with certain brain tumors and is used in some combination chemotherapy regimens. Acute and organizing DAD is the most common manifestationof acute BCNU pulmonary toxicity.281–284 Delayed lung toxicity has been described in survivors of childhood brain tumors who received BCNU, with lung changes appearing 8 to 20 years after cessation of treatment.281,285,286 The histopathologic features of BCNU toxicity can be generally grouped within the NSIP pattern (Fig. 7-67). In children, fibrosis develops in the upper lung zones, with peripheral accentuation.286 In adults, this upper lobe distribution is uncommon.287 Pleural disease can accompany pulmonary abnormalities in some patients.
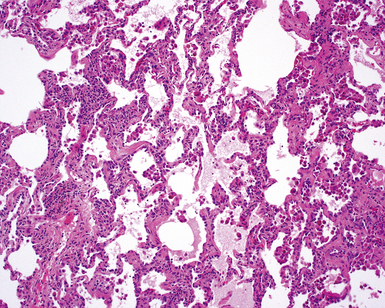
Busulfan
Busulfan is an alkylating agent that has been used in the treatment of chronic myelogenous leukemia. Pulmonary toxicity has been reported to occur in 4% of patients,288–290 most commonly as acute lung injury. Unfortunately, the prognosis for patients with busulfan-induced acute lung disease is poor. Rarely, patients with busulfan toxicity develop chronic diffuse lung disease212 (Fig. 7-68).
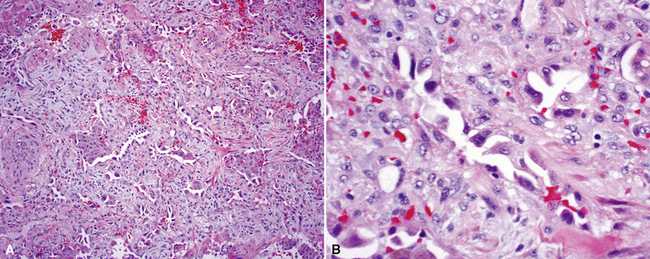
Bleomycin
Bleomycin is a chemotherapeutic agent used in the treatment of lymphomas, epidermoid carcinomas, and malignant testicular tumors. Lung toxicity appears to be dose-related, but irradiation or oxygen therapy may predispose the lung to injury.210,291–293 The typical clinical presentation begins with dry cough and progresses to breathlessness. Chest imaging reveals areas of nodular consolidation or diffuse reticulation.294 In experimental models, the initial site of injury seems to be the venous endothelial cell, followed by necrosis of type I cells with consequent fibroplasia.295–298 In humans, toxicity results in acute lung injury with air space organization in the early phase and fibrosis as a late consequence (Fig. 7-69).
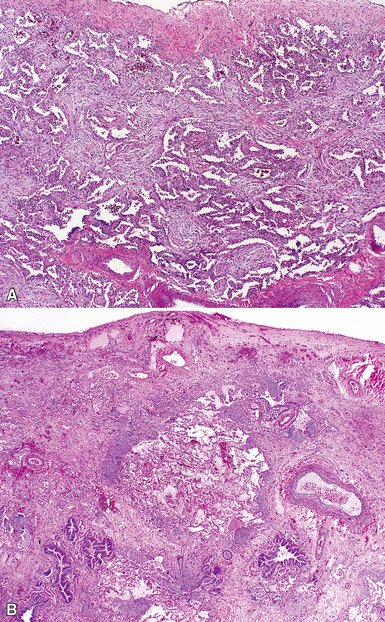
Targeted Molecular Therapies
Progress in the application of newer anti-cancer therapies using antibodies and small molecules directed at molecular signaling pathways has lead to increasing reports of pulmonary toxicity. For example, the epidermal growth factor receptor tyrosine kinase inhibitor (EGFR-TKI) gefitinib is said to produce ILD in 1% of patients treated with this agent (2% to 4% in Japanese patients and 0.3% in U.S. patients).299,300 The most common histopathologic pattern associated with gefitinib use is DAD. The other main EGFR-TKI, erlotinib, is also reported to produce a DAD reaction pattern.224 The common risk factors for toxicity with these agents are smoking history, age, and the presence of pre-existing ILD.263 Similar trends are seen with conventional cytotoxic chemotherapies, such as paclitaxel, docetaxel, and gemcitabine.
Newer additions to the anti-inflammatory drug arsenal, such as the anti-TNF-α inhibitor infliximab, are also associated with adverse reactions, including an apparent risk for certain pulmonary infections, such as tuberculosis and aspergillosis, followed by DAD and pulmonary fibrosis.301
Illicit Drug Abuse
Many of the described pulmonary manifestations of illicit drug use (Box 7-11) are related to infections derived from the use of contaminated needles for intravenous injection or comorbidity related to the use of inhaled substances.302 For a minority of substance abusers, intravenously injected solubilized analgesic tablets is the drug of choice. In current practice, the presence of perivascular talc particles in giant cells usually indicates a residual injury from earlier in the patient’s life, because tablet manufacturers today most often use microcrystalline cellulose compounds as binding and filling agents, rather than talc, as was the practice several decades ago. When intravenous drug use entails injection of crushed analgesic tablets nowadays, microcrystalline cellulose is the commonly identified injurious particle in the lung parenchyma. The physical characteristics and histochemical staining reactions of this material are different than those of talc; these may be useful distinguishing features, on occasion.303
Injected particles generally average 10 to 15 μm in diameter, 303–305 in contrast with inhaled substances, for which particle sizes tend to be smaller, usually less than 5 μm (with red blood cells used for size comparison). Cornstarch is another filler agent; it appears as a spherical structure with a “Maltese cross” pattern under plane-polarized light. Talc appears as irregular, plate-like, crystals that are strongly birefringent (Fig. 7-70) and may be slightly yellow on routine hematoxylin and eosin staining when a single polarizing filter is in place. Microcrystalline cellulose particles appear as elongated crystalline structures (Fig. 7-71) that may stain positively using digested PAS, methenamine silver, and Congo red stains. The reaction in the lung is generally interstitial and perivascular (Fig. 7-72), rather than intra-alveolar. Intravenous foreign material may rarely become encrusted with iron (ferruginated), and sometimes colored tablet coatings (such as blue crospovidone) may be present and quite striking in histopathologic appearance.
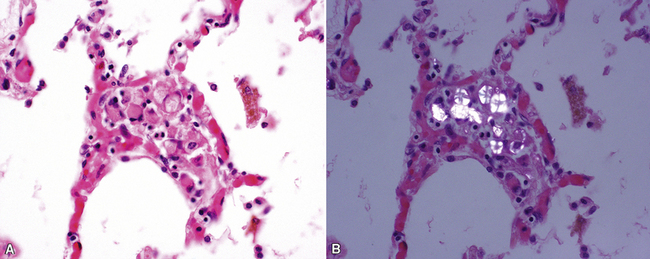
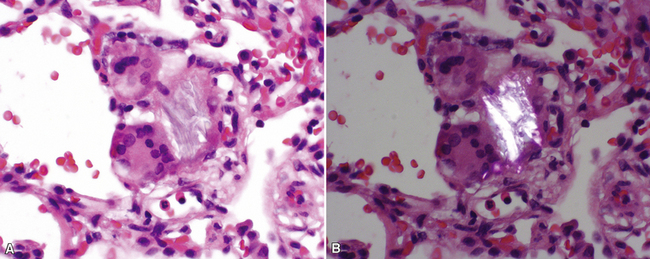
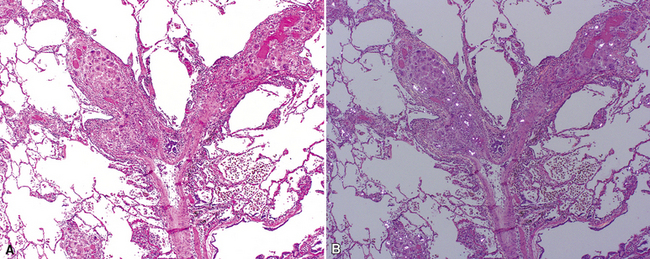
Histopathologic Findings
As the intravenous material becomes lodged in the pulmonary microvasculature, a perivascular interstitial foreign body–type granulomatous reaction is produced (Fig. 7-73) that may be associated with prominent vascular changes (including pulmonary hypertension and thrombotic lesions and others),306 as well as interstitial fibrosis (Fig. 7-74) that may be focal or diffuse and sometimes massive.307 Among the pathologic manifestations presented in Table 7-6, pulmonary hypertension is the most common, whereas emphysema is quite uncommon. Emphysema associated with intravenous drug abuse is typically of the panacinar type (similar to that in α1-antitrypsin deficiency) and has been most frequently associated with methylphenidate (Ritalin) abuse.308 In contrast with smoking-associated emphysema, the radiologic changes seen in so-called “Ritalin lung” are more severe in the lower lobes. Bullae may be present. In one report it was suggested that the presence of basilar pulmonary emphysema should always alert the radiologist to the possibility of intravenous drug abuse.308 The pathogenesis of panlobular emphysema associated with intravenous drug abuse is unknown. Some of the postulated mechanisms include synergism with cigarette smoke, direct toxic effects of the drug, and induced intravascular leukocyte sequestration causing proteolytic pulmonary injury.
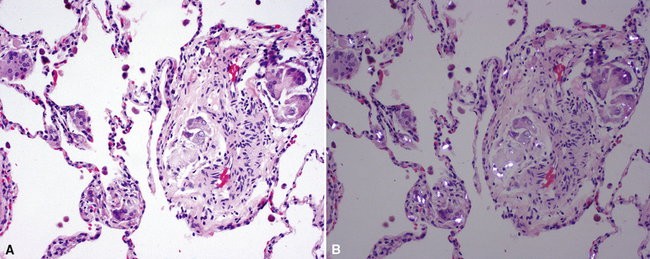
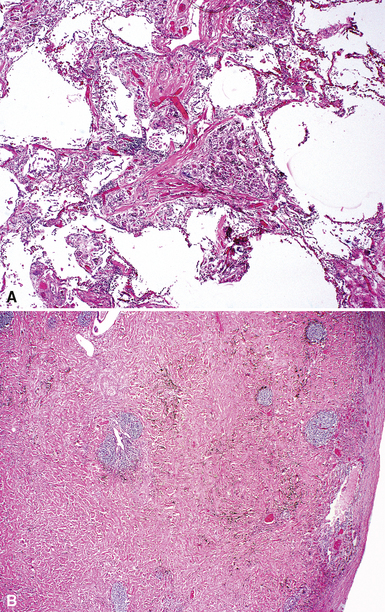
Table 7-6 Pathologic Lesions and Clinical Syndromes Related to Intravenous Drug Use
| Pathologic Lesion | Clinical Syndrome |
|---|---|
| Vascular, perivascular, and interstitial foreign body granulomas with: | |
| Pulmonary arterial hypertension (with or without thrombotic lesions) | Pulmonary hypertension (may cause sudden death) |
| Interstitial fibrosis | Interstitial lung disease |
| Massive fibrosis | Complicated “pneumoconiosis” (tends to be bilateral and involves mid- and upper lung zones) |
| Panacinar emphysema | Emphysema/chronic airflow obstruction |
Because the foreign material remains in the lung, progression of the clinical and pathologic lesions may occur after discontinuation of intravenous drug use. Recurrence of the changes of intravenous drug abuse have been described rarely in transplanted lung tissue, although such recurrence appears to be a consequence of resumed intravenous drug abuse.309
Diffuse Lung Diseases with Granulomas
Granulomas occur in the lung in a variety of infectious and noninfectious diseases310 (Box 7-12). Infectious diseases with granulomas are discussed in Chapter 6. Although non-infectious granulomas can occur diffusely in the lungs in certain drug reactions and in Sjögren syndrome, the relatively specific disease entities of sarcoidosis, berylliosis, and hypersensitivity pneumonitis are discussed here separately because of their distinctive clinical, radiologic, and histopathologic presentations.
Box 7-12 Granulomatous Interstitial Pneumonias
Adapted from Cheung OY, Muhm JR, Helmers RA, et al: Surgical pathology of granulomatous interstitial pneumonia. Ann Diagn Pathol. 2003;7(2):127–138.
Sarcoidosis
Clinical Presentation
Sarcoidosis is a systemic disease of uncertain etiology, with frequent lung manifestations.311 Lung disease is usually mild, accompanied by variable degrees of shortness of breath, chest pain, and cough.311 As many as two thirds of patients are asymptomatic.270,312 Sarcoidosis is predominantly a disease of young adults but has been described in patients of all ages.313–315 Sarcoidosis can have an acute, subacute, or chronic presentation in the lung. In symptomatic patients, restrictive defects and decreased diffusing capacity are commonly described. Serum angiotensin-converting enzyme (ACE) is elevated in 30% to 80% of patients. ACE has also been detected within granulomas, bronchial alveolar lavage fluid, tears, and even cerebrospinal fluid in these patients. Unfortunately, ACE levels can be elevated in a variety of disorders, including infectious granulomatous diseases, lymphoma, hepatitis, and diabetes.270 The Kveim test270 is relatively specific for sarcoidosis, employing an injected antigenic extract derived from human sarcoid granulomas, but the test is not widely used today,311 given the widespread adoption of bronchoscopic biopsy for confirming the diagnosis in patients suspected of having the disease on clinical and radiologic grounds.
Radiologic Findings
The clinical staging of sarcoidosis is based on chest radiographic findings. Five stages of pulmonary sarcoidosis are described, corresponding to the extent of disease. Stage I and stage II disease are most common, with only 15% of patients presenting with parenchymal infiltrates alone.270,311 When sarcoidosis involves the lung parenchyma, the disease is upper lobe predominant.311 CT scans are not usually necessary for the diagnosis but will show reticulonodular opacities along lymphatic routes, with or without alveolar infiltrates.270,316–318 Bullae with honeycombing may be seen in advanced disease, sometimes associated with progressive lung fibrosis.319,320 A peculiar form of sarcoidosis with rapid onset of symptoms and a diffuse alveolar filling pattern on CT scans has been referred to as “alveolar sarcoidosis.”321 The pathologic manifestations of this form of the disease are distinctive only for the large number of small interstitial granulomas present throughout the lung parenchyma.
Histopathologic Findings
The characteristic histopathologic lesion of pulmonary sarcoidosis is the non-necrotizing (immune) granuloma, typically occurring within areas of sclerotic fibrosis (Fig. 7-75). In sarcoidosis, small granulomas have a tendency to coalesce to form larger nodular lesions, all embedded in refractile eosinophilic collagen (Fig. 7-76). A narrow rim of lymphocytic inflammation is typically seen at the periphery of these confluent nodules (Fig. 7-77). Granulomas are distributed along lymphatic routes in the pleura, within the intralobular septa, and along the bronchovascular bundles (Fig. 7-78). Multinucleate giant cells are characteristically present in the disease, often accompanied by a variety of distinctive cytoplasmic inclusions (e.g., Schaumann bodies, asteroid bodies) (Fig. 7-79). A mild inflammatory interstitial infiltrate is said to occur occasionally in pulmonary sarcoidosis, but in practice this is rarely seen. In bronchoscopic or transbronchial biopsies, granulomas are typically seen immediately beneath the airway mucosa (Fig. 7-80).
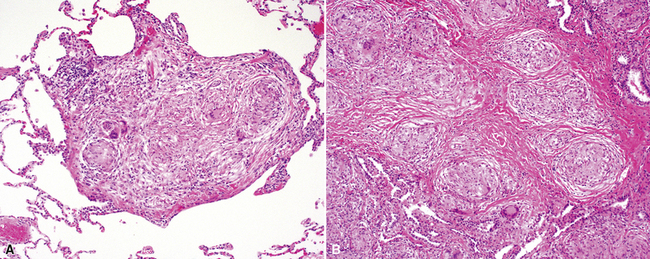
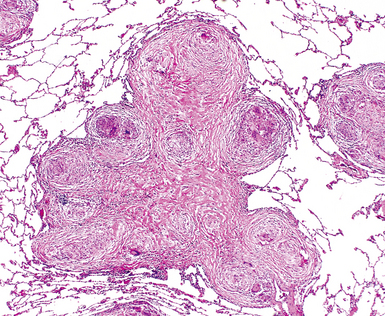
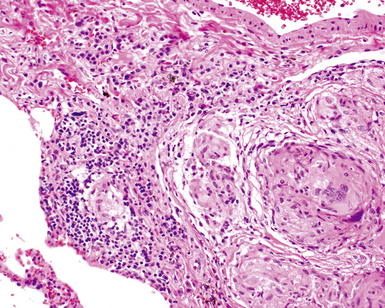
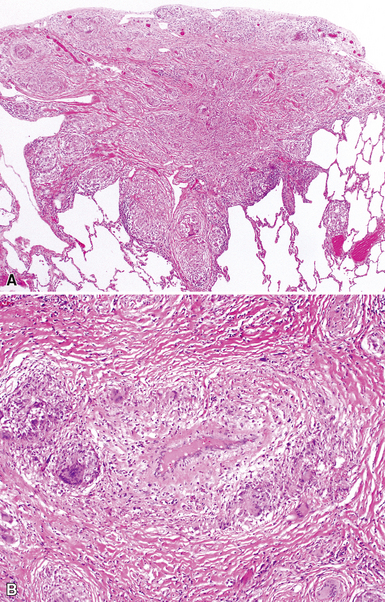
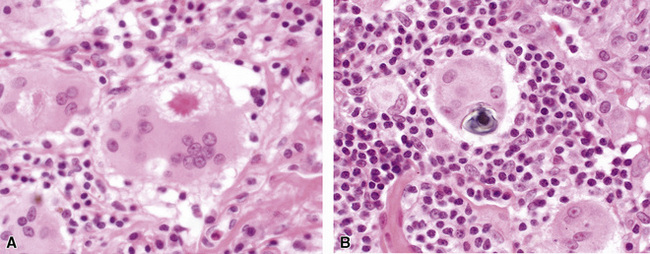
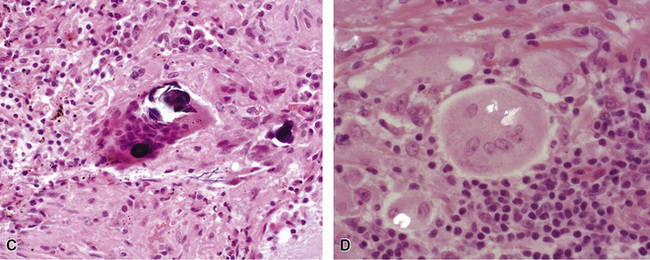
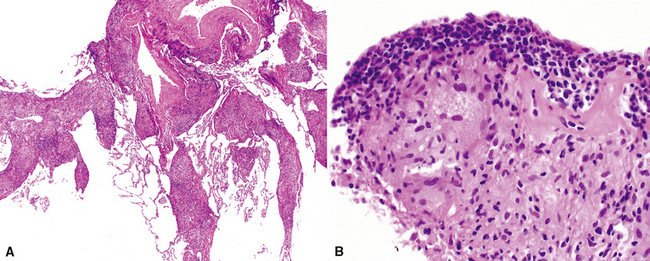
Gilman and coworkers showed that the chance of obtaining a positive result in patients with sarcoidosis increased to 90% when four biopsy specimens were obtained.322 When five to six samples were obtained, the probability rose to 100% for patients with stage II and stage III disease.
Special stains for organisms (acid-fast stains and silver stains) should be routinely used when granulomas are identified in lung biopsies to exclude infection, even in the absence of necrosis. In a retrospective study performed by Hsu and colleagues, positive microbiological cultures were identified in 11% of biopsies in which granulomas were present despite negative special stains of tissue sections.323 In the culture-positive cases, clinical and radiographic findings were judged to be of low or intermediate suspicion for sarcoidosis. As might be expected, necrosis in granulomas was more frequently associated with culture-positive cases.
Chronic Berylliosis
Beryllium, derived from the mineral beryl, is the etiologic agent for berylliosis.324–326 The disease occurs after inhalation of this metal or its salts.327 Exposure to beryllium occurs today mainly as an occupational lung disease, particularly in the computer manufacturing and aerospace engineering fields. However, recent reports indicate that individuals with low-level exposure, such as residents living near facilities that use beryllium, can also develop berylliosis328–330 (for additional discussion, see Chapter 8).
Acute berylliosis occurs after heavy exposure; fortunately, it is rare today as a consequence of strict industrial exposure regulations and aggressive surveillance in this setting. When acute berylliosis occurs, the histopathologic findings are similar to those of DAD. The chronic form of berylliosis is indistinguishable from sarcoidosis on histopathologic grounds.324–326 Like sarcoidosis, chronic berylliosis produces fibrosis of variable severity and has distinct granulomas with giant cells (Fig. 7-81). Chronic berylliosis may produce large, centrally hyalinized nodules (Fig. 7-82), which can mimick resolved lesions of histoplasmosis. When granulomas are less prominent, chronic berylliosis may also simulate hypersensitivity pneumonitis histopathologically (Fig. 7-83).
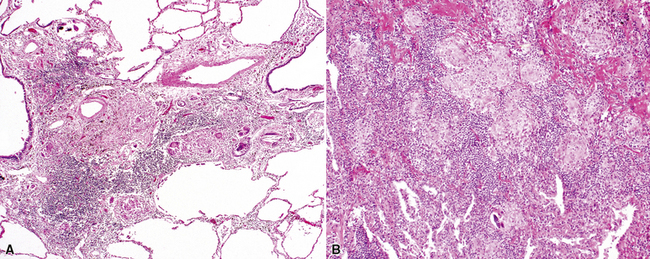
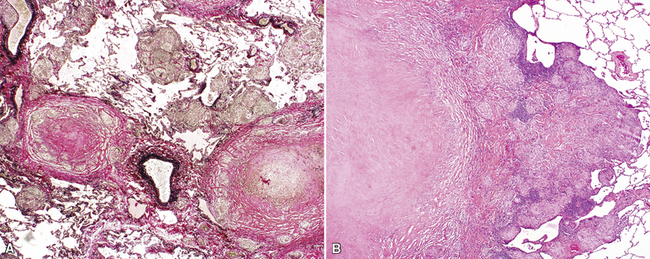
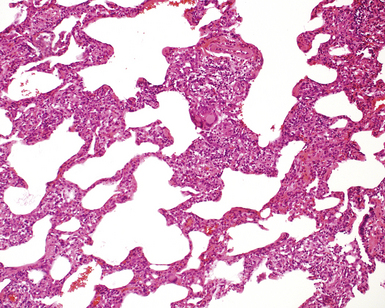
Hypersensitivity Pneumonitis (Extrinsic Allergic Alveolitis)
Environmental antigens (typically, “organic” protein antigens) are known to produce characteristic inflammatory reactions in the lung in certain predisposed individuals.331–334 The classic descriptions of farmer’s lung, resulting from exposure to thermophilic actinomycetes in hay, and bird fancier’s lung, resulting from inhalation from avian antigens, are examples of hypersensitivity pneumonitis. Similar reactions to ingested antigens associated with some medications can also occur, but in general usage, hypersensitivity pneumonitis refers to disease occurring as a result of inhalation exposure. The more common antigens implicated in hypersensitivity pneumonitis are presented in Table 7-7.
Table 7-7 Agents of Hypersensitivity Pneumonitis (Extrinsic Allergic Alveolitis)
| Antigen | Source | Disease |
|---|---|---|
| Thermophilic bacteria | ||
| Micropolyspora faeni | Moldy hay | Farmer’s lung |
| Thermoactinomyces vulgaris | Moldy compost | Mushroom worker’s disease |
| Thermoactinomyces saccharii | Moldy sugar cane | Bagassosis |
| Thermoactinomyces vulgaris | Air conditioners, humidifiers | Air conditioner lung/ humidifier lung |
| Thermoactinomyces candidus | Air conditioners, humidifiers | Air conditioner lung/ humidifier lung |
| Molds | ||
| Cryptostroma corticale | Moldy maple bark | Maple bark stripper’s disease |
| Aspergillus clavatus | Moldy barley | Malt worker’s lung |
| Graphium spp. | Moldy wood dust | Sequoiosis |
| Pullularia spp. | Moldy wood dust | Sequoiosis |
| Trichosporon cutaneum | Home environment | Summer-type hypersensitivity pneumonitis (Japan) |
| Other bacteria | ||
| Bacillus subtilis | Water | Detergent worker’s lung |
| Bacillus cereus | Water | Humidifier lung |
| Bacterial products | Cotton | Byssinosis |
| Amebae | Water | Humidifier lung |
| Insect products | Grain | Wheat weevil disease |
| Chemicals | ||
| Trimellitic anhydride (TMA) | Plastics, rubber manufacturing | Chemical worker’s lung |
| Methylene diisocyanate (MDI) | Plastics, rubber manufacturing | Chemical worker’s lung |
| Toluene diisocyanate (TDI) | Plastics, rubber manufacturing | Chemical worker’s lung |
| Pyromellitic dianhydride (PMDA) | Epoxy resin | Chemical worker’s lung |
Reprinted with permission from Katzenstein A, Askin F, eds. Surgical Pathology of Non-Neoplastic Lung Disease, 2nd ed. Philadelphia: WB Saunders; 1990:139.
Clinical Presentation
Hypersensitivity pneumonitis can occur as acute or subacute and chronic forms. The acute form occurs within hours of inhalational exposure to antigen. Affected individuals experience malaise, dyspnea, dry cough, and, occasionally, fever and chills. With this symptom complex, the differential diagnosis will include viral infection. Recurrence of symptoms and signs follows subsequent exposure episodes.335 Women are more commonly affected than men. Cigarette smoking seems to reduce the risk of developing hypersensitivity, although the mechanism for this protective effect is unknown.336
The chronic form of hypersensitivity pneumonitis probably results from lower levels of antigen exposure over time, presumably accompanied by more subtle symptoms, so that the affected person may not associate the symptom with a specific exposure.335,337 The frequency of disease occurrence in smokers and men is higher in the chronic form than in the acute and subacute forms of the disease. Unfortunately, the chronic form of hypersensitivity pneumonitis may be progressive, eventuating in death from end-stage lung fibrosis.335,337 As in the acute phase, patients experience chronic malaise and varying degrees of breathlessness on exertion, sometimes accompanied by weight loss. Several excellent reviews of hypersensitivity pneumonitis are available.333–335,337,338
Radiologic Findings
In acute hypersensitivity pneumonitis, a diffuse ground-glass appearance is seen on CT scans of the chest, sometimes accompanied by fine nodules.339 In subacute disease, abnormalities tend to be confined to the upper one half or two thirds of the lung and are characterized radiologically as ill-defined centrilobular nodules. In more chronic forms, small nodules, variable ground-glass change, and irregular linear opacities may be seen, most often in the middle lung zones (with relative sparing of the apices and bases) or without a specific zonal predilection.23 The presence of irregular linear opacities correlates with the presence of lung fibrosis.335,338 In very-late-stage chronic hypersensitivity pneumonitis, honeycomb remodeling may be identified radiologically, mimicking UIP.23,24 Interestingly, the CT appearance of subacute hypersensitivity pneumonitis may be indistinguishable from that in low-grade atypical mycobacterial infection occurring in the immunocompetent host as a result of bioaerosol exposure to non-tuberculous mycobacteria340,341 (so-called “hot tub lung”; see Chapter 6).
Histopathologic Findings
The histopathologic features of hypersensitivity pneumonitis are typically those of a chronic inflammatory interstitial pneumonia associated with bronchiolitis, and small, indistinct, non-necrotizing interstitial granulomas331,342,343 (Fig. 7-84). In early reports of farmer’s lung, acute inflammation and vasculitis were also observed.344 The histopathology of hypersensitivity pneumonitis raises a differential diagnosis that includes other cellular interstitial pneumonias, such as NSIP. At low magnification, the surgical lung biopsy shows a moderately dense interstitial infiltrate, composed of plasma cells and small lymphocytes, causing slight widening of the alveolar walls (Fig. 7-85). A bronchiolocentric distribution may be evident, either by the presence of a terminal bronchiole or by some degree of nodularity in the infiltrates at low magnification (Fig. 7-86). The interstitial “granulomas” of hypersensitivity pneumonitis are inconspicuous (Fig. 7-87) and may easily escape notice in many cases. Multinucleate giant cells (Fig. 7-88) may be seen in the interstitium at scanning magnification and constitute a helpful feature in prompting closer examination of the interstitium for epithelioid histiocytes in small aggregates. Necrosis is not a component of the granulomatous reaction in hypersensitivity pneumonitis. Air space organization (Fig. 7-89) with immature fibroblast and matrix (organizing pneumonia pattern) can be seen in as many as 60% of patients with hypersensitivity pneumonitis.342 In general, this is not confluent organization, as seen in organizing pneumonia of infectious etiology. Rather, small, tufted patches of organization can be seen scattered throughout the lung biopsy. Some degree of bronchiolitis is expected in hypersensitivity pneumonitis; this is characterized by aggregates of lymphocytes and plasma cells surrounding terminal airways (Fig. 7-90). A mild perivascular lymphoid accumulation typically is present in hypersensitivity pneumonitis, but prominent germinal centers or dense lymphoplasmacellular infiltration along vascular sheaths (especially veins in the interlobular septa) are not typical and should raise concern for lymphoproliferative disease (which on occasion can simulate the cellular phase of chronic hypersensitivity pneumonitis). In the chronic form, dense fibrosis with microscopic honeycombing can be seen (Fig. 7-91). Peribronchiolar metaplasia along with peribronchiolar fibrosis is frequent. There may be bridging formations between peribronchiolar areas and subpleural scar, analogous to “bridging fibrosis” in liver disease.345 Refractile oxalate crystals in giant cells may be sufficiently prominent to suggest aspiration pneumonia or even pneumoconiosis (Fig. 7-92).
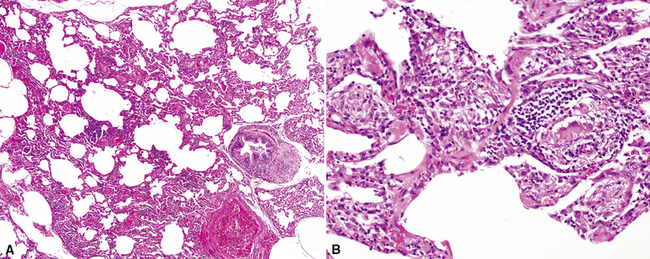
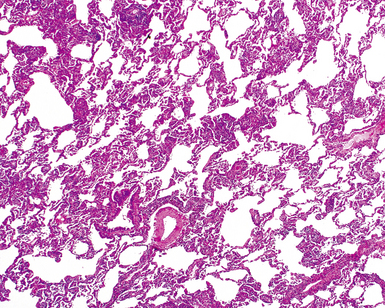
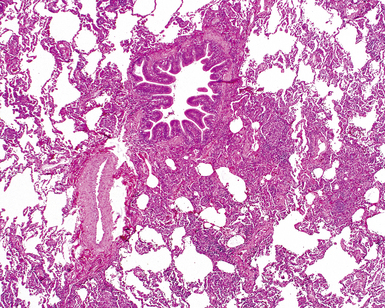
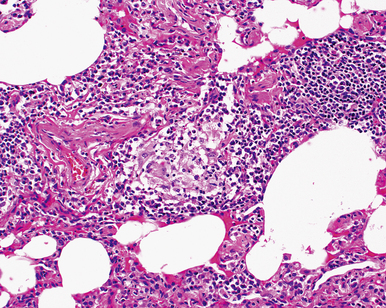
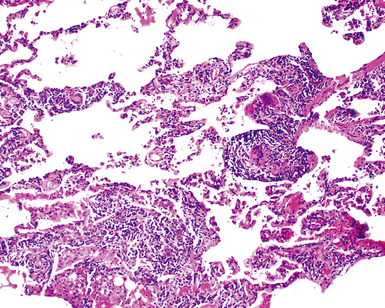
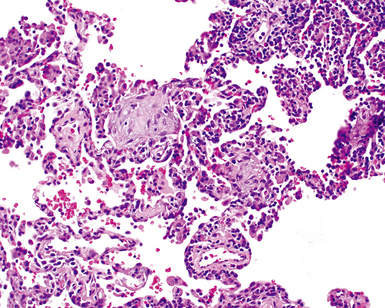
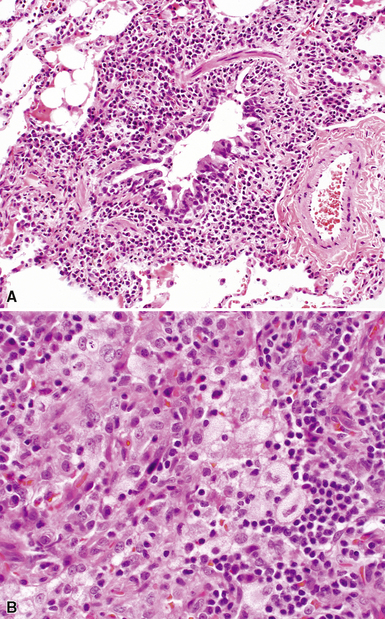
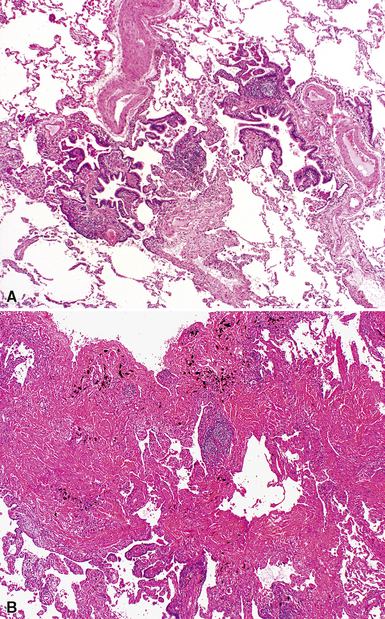
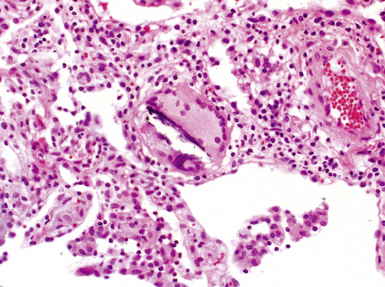
Clinical Course
The clinical course in hypersensitivity pneumonitis varies with the intensity and chronicity of exposure.335,344,346 If an offending antigen cannot be identified in the patient’s environment, the prognosis is guarded since patients may not respond to corticosteroid therapy in the continued presence of the initiating antigen.347–351 Once lung fibrosis ensues, immunosuppressive therapy is of little benefit, especially if continued antigen exposure occurs.351
Miscellaneous Diffuse Lung Diseases
Pulmonary Langerhans Cell Histiocytosis
In addition to smoking-related ILD (i.e., RBILD, as described earlier in the chapter), another important consequence of smoking is pulmonary Langerhans cells histiocytosis (PLCH), a lung disease previously referred to as “pulmonary eosinophilic granuloma” or “pulmonary histiocytosis X.” PLCH is considered to be a reactive proliferative disease of Langerhans cells,352,353 in contrast to extrapulmonary forms of Langerhans cell histiocytosis, which are thought to be neoplasms.354 A compelling association with cigarette smoking has led to the general acceptance of this lung disorder as a smoking-related disease.355,356 The pathobiology of PLCH has been comprehensively reviewed by Vassalo and associates.353 Although PLCH is unrelated to the systemic diseases of Langerhans cells (eosinophilic granuloma, Letterer-Siwi disease, Hand-Schüller-Christian disease), systemic eosinophilic granuloma involving the lung, when it occurs, is said to be indistinguishable from the cellular phase of PLCH on histopathologic grounds.357
Two distinctive histopathologic manifestations of PLCH occur—a cellular form and a fibrotic form. The natural history of PLCH suggests that the early lesions of PLCH are cellular, with many Langerhans cells and tissue eosinophils, whereas older lesions are predominantly fibrotic. Such a natural progression may occur continuously in the same patient, with “younger” cellular lesions admixed with “older” ones, but in most surgical biopsy specimens, one form of the disease seems to predominate.353 Often the fibrotic lesions of the disease are not recognized because of the paucity or absence of Langerhans cells, and the proliferative lesions are typically mistaken for neoplasm.
Clinical Presentation
The true incidence and prevalence of PLCH are unknown. Most patients are between 20 and 50 years of age at the onset of symptoms; women may be more commonly affected than men.353,355,358 Chronic cough and dyspnea are typical presenting complaints. However, a significant percentage of patients are asymptomatic. Rarely, hemoptysis or pneumothorax can occur.
Radiologic Findings
PLCH is a disease of the upper lung zones. Small nodules and cysts are present to a variable degree as seen on plain films of the chest.358–361 The nodules range in size from 0.2 to 1 cm. When fibrosis occurs, it is best seen on high-resolution CT scans and appears as reticular opacities.360,362 When significant fibrosis occurs in PLCH, the disease may simulate IPF.359,360,362
Histopathologic Findings
Proliferative (Cellular) Phase
At scanning magnification, the nodules of PLCH have a stellate appearance and are centered on the small airways (Fig. 7-93). Cysts are formed at the periphery of nodules by traction on surrounding alveolar walls or the central terminal airway, resulting in variably sized spaces, typically lacking distinctive lining cells (Fig. 7-94). Stellate cellular nodules may be as large as 1.5 cm,357 and confluence of nodules affecting adjacent airways may impart a serpentine outline to the lesions (Fig. 7-95). As suggested by a three-dimensional reconstruction study, the lesions of PLCH seem to form a sheath around the small airways exclusively and extend proximally and distally in a continuous fashion.363 In many cases, lightly pigmented brown macrophages (“smoker’s macrophages”) are present in and around the nodules (Fig. 7-96). Eosinophils in variable numbers occupy the next innermost layer of the nodules (Fig. 7-97); this is the location in which aggregated Langerhans cells are most easily found in the thickened interstitium (Fig. 7-98). The Langerhans cells have a pale basophilic nucleus with characteristic sharp nuclear infoldings, imparting a “crumpled tissue paper” nuclear contour (Fig. 7-99). The cytoplasm of the Langerhans cell is granular and mildly eosinophilic, with indistinct margins. In these cellular lesions of PLCH, immunohistochemical stains for S100 protein and CD1a can be used to highlight the presence of Langerhans cells (Fig. 7-100), but in most instances, the morphology of the lesions is sufficiently compelling enough for a definitive diagnosis to be established without the aid of special stains. In some cases, patchy interstitial and air space organization (Fig. 7-101) may be seen, and RB is typically present. Other smoking-related lung changes may be present, adding further complexity to the morphologic picture (e.g., DIP/RBILD, small-airway disease with mucostasis, areas of bronchiolization).
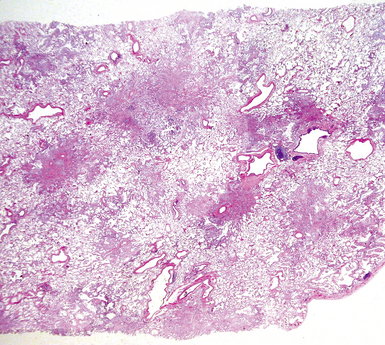
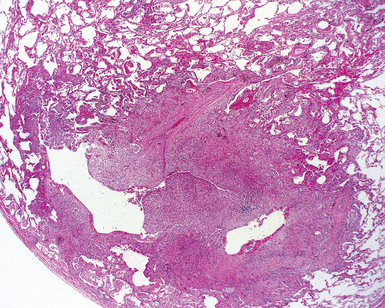
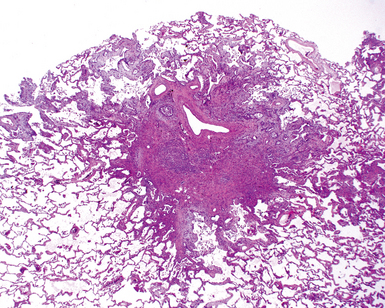
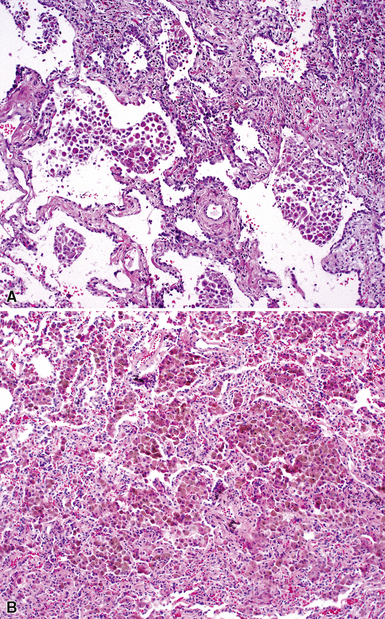
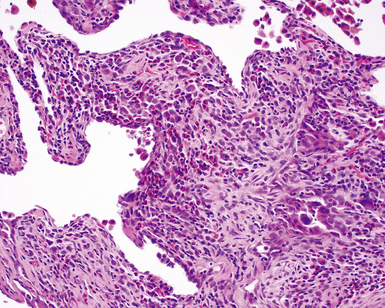
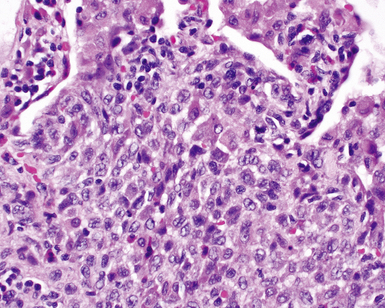
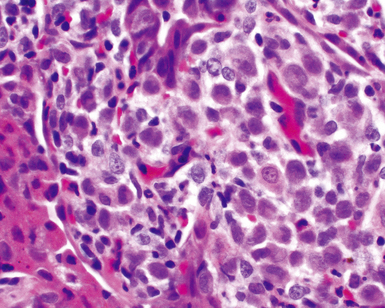
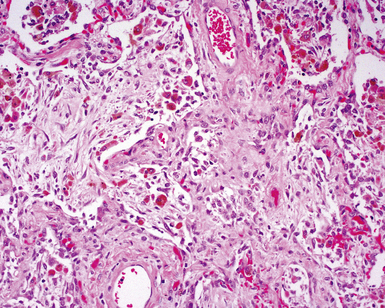
Fibrotic Lesions
With “aging” of the lesions of PLCH, Langerhans cells become progressively depleted and overshadowed by fibrosis (Fig. 7-102). The mechanism for this transformation is unknown. In some patients, only residual stellate parenchymal scars are found, and in these patients, pulmonary function may be significantly compromised (by a process analogous to constrictive bronchiolitis). Lung function and radiologic studies may suggest a diffuse lung disease, but biopsy tissue may exhibit only stellate fibrotic lesions centered on the terminal airways, without an identifiable interstitial inflammatory disease (Fig. 7-103). Another scenario inwhich such scars can be seen is in portions of lung removed for other reasons (e.g., bronchogenic carcinoma). Presumably these “footprints” of previous PLCH are incidental findings and do not necessarily imply active disease elsewhere in the lung.
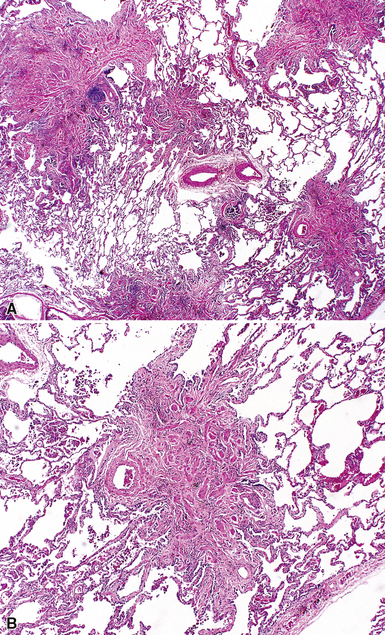
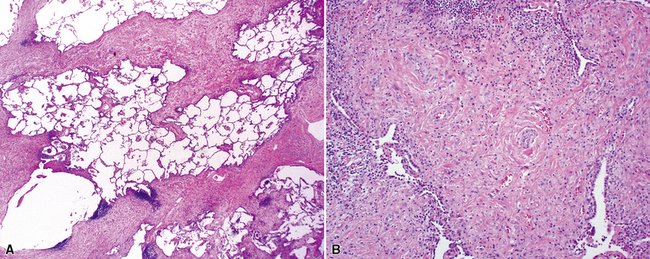
Differential Diagnosis
Nodular infections and neoplasm are common in the differential diagnosis radiologically, the latter especially in the setting of known breast carcinoma in a young woman undergoing screening for metastatic disease to the lungs. Pathologically, the proliferative lesions may be mistaken for neoplasm as well, but careful attention to the layered or zonal composition of the nodules and the distinctive morphology of the Langerhans cells will typically bring the diagnosis of PLCH to the forefront. When cysts are prominent, lymphangioleiomyomatosis (LAM) enters the differential diagnosis (see further on). In this setting, immunohistochemical stains may be useful in establishing the correct diagnosis (S100 protein immunoreactivity in Langerhans cells versus HMB45 and actin in LAM cells). Other nodular lung diseases are occasionally confused with PLCH, including Wegener granulomatosis, Hodgkin disease, and certain metastatic low-grade sarcomas. When fibrotic lesions are dominant, UIP (clinical IPF) and other fibrosing lung disorders may be suggested. Helpful distinguishing features include the stellate appearance of the PLCH lesion, the tendency for lesions to spare the pleura and immediate subpleural lung, and the association of scars with the airways. Rarely, pneumothorax (from any cause) can produce subpleural fibrosis with sheets of tissue eosinophils (so-called “reactive eosinophilic pleuritis”).364,365 Such lesions are not usually confused with PLCH but on occasion may be the only pathologic change identified on biopsy following pneumothorax (sometimes during surgical intervention for the pneumothorax), raising concern of undersampled or “upstream” PLCH. Radiologic correlation may be very helpful in this setting.
Clinical Course
Smoking cessation is the most effective management approach and is the “treatment” of choice.353 Immunosuppression with corticosteroids, with or without the addition of a cytotoxic agent, has met with variable success. In larger reviews, the median survival time is 12 years,353,366 with 5- and 10-year survival rates of 70% and 60%, respectively. The most frequent cause of death is the respiratory complications associated with neoplasms of hematologic, pulmonary, or other organs. Survival rates are worse for patients with poorer respiratory function at diagnosis.353
Erdheim-Chester Disease
Erdheim-Chester disease (ECD) is a rare, nonfamilial, “non–Langerhans cell,” systemic histiocytosis affecting middle-aged adults; it has no gender predilection.367–369 The disease is characterized by xanthogranulomatous infiltration of the long tubular bones, resulting in symmetrical osteosclerosis.367,370–373 Extraskeletal involvement is relatively common, occurring in approximately one half of patients. Of the reported extraskeletal sites, the pituitary area, skin, orbit, pericardium, and retroperitoneum are most often involved.
Clinical Presentation
Pulmonary involvement occurs in approximately one third of patients with ECD and is associated with significant morbidity and mortality.367,374–378 Progressive breathlessness is the typical presentation.
Radiologic Findings
CT scans reveal thickening of the visceral pleura and interlobular septa, fine reticular and centrilobular opacities, and ground-glass attenuation.377
Histopathologic Findings
In surgical wedge lung biopsies, a distinctive interstitial infiltrate is observed consisting of xanthomatous histiocytes, lymphocytes, and scattered Touton-type giant cells376 (Fig. 7-104). Prominent fibrosis is present in a characteristic subpleural and lymphatic distribution (Fig. 7-105). In immunohistochemical studies, the cells of ECD express histiocytic markers (CD68 and factor XIIIa) but typically lack CD1a immunoreactivity. S100 protein immunoreactivity occurs in a subset of patients.
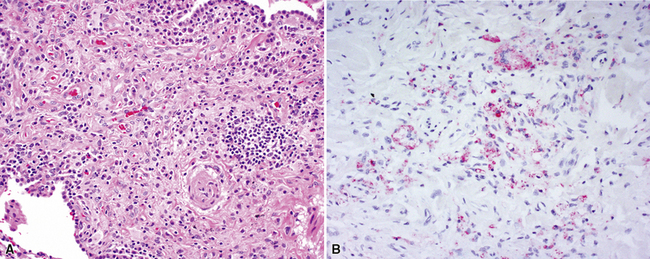
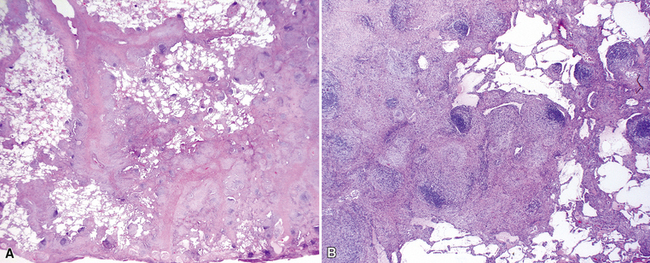
Differential Diagnosis
The differential diagnosis includes advanced lung fibrosis associated with pulmonary Langerhans cell histiocytosis, and the exceedingly rare occurrence of lung manifestations of Rosai-Dorfman disease (sinus histiocytosis with massive lymphadenopathy—see Chapter 18 for discussion). In advanced PLCH, the fibrosis tends to be more confluent and particularly bronchiolocentric, with blunt, stellate-shaped scars. In Rosai-Dorfman disease, the histiocytic nature of the process is often overshadowed by the inflammatory component (Fig. 7-106).
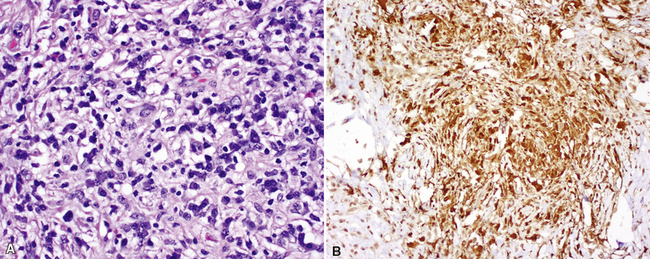
Lymphangioleiomyomatosis
Lymphangioleiomyomatosis (LAM) is a rare chronic lung disease characterized by the presence of cysts accompanied by bundles of distinctive smooth muscle cells.379–384 LAM is a disease that affects women; it has a distinctive relationship to the genetic disorder known as the tuberous sclerosis complex (TSC).385 Recent experimental data have demonstrated loss of heterozygosity on 9p and and 16p and mutation in the tuberous sclerosis complex gene (TSC2), suggesting that LAM is a neoplastic disease.386–388 Nevertheless, debate continues as to whether LAM is a true neoplasm or represents hyperplasia of genetically aberrant cells. Although LAM was considered to be a disease unique to women, rare cases of LAM in males have been reported.389 Likewise, we have seen an additional case in consultation (unpublished data) in a phenotypic male with TSC. LAM is included in this chapter because the disease typically presents as a chronic diffuse lung process clinically and radiologically.
Clinical Presentation
LAM occurs most frequently in women of childbearing age, with a peak incidence in the fourth decade of life.379,383 Two forms of the disease occur, one sporadically and the other in association with the genetic marker for TSC. In both forms of the disease, extrathoracic (especially renal) angiomyolipomas are common.390–395 Of note, the rare male patients reported to have LAM also had TSC.389 LAM is often asymptomatic in its early stage, and many affected individuals are diagnosed by chest radiographs performed for other reasons. The most common clinical complaint is shortness of breath.379,383,384 Pneumothorax occurs, and pleurocentesis may reveal a chylous effusion.
Radiologic Features
Plain films of the chest may be interpreted as normal,396 but with pneumothorax, air or fluid may be present in the pleural space. Small nodules and cysts may be detected and typically are present diffusely in the lungs, in contrast with the upper lobe distribution of lesions in PLCH. CT scans show diagnostic changes, with small nodules and thin-walled cysts seen throughout both lungs.396–401
Histopathologic Features
LAM lesions may be difficult to appreciate at scanning magnification, particularly in cases where smooth muscle lesions are sparse or of small size (Fig. 7-107). In most cases, thin walled cysts are easily visible (Fig. 7-108) and useful for identifying diagnostic smooth muscle bundles at higher magnification (Fig. 7-109). The smooth muscle of LAM is distinctive (Fig. 7-110). The LAM cell is fusiform and plump. The nucleus is larger than that of other smooth muscle cells in the lung (Fig. 7-111) and the nuclear-cytoplasmic ratio is typically higher. Smooth muscle bundles may be small or attenuated at the periphery of cysts (Fig. 7-112) or quite cellular and prominent (Fig. 7-113). In the lung lymphatics or pleural space, unattached aggregations of cells enveloped by lymphatic endothelium may be seen; these have been referred to as LAM cell clusters.402 These unattached cell clusters have been proposed as the mechanism for dissemination of the disease.402
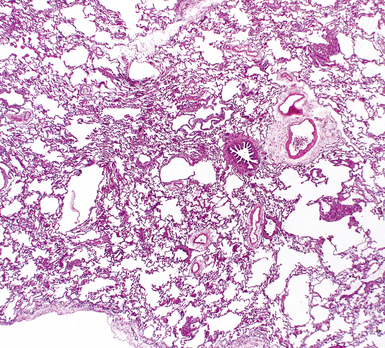
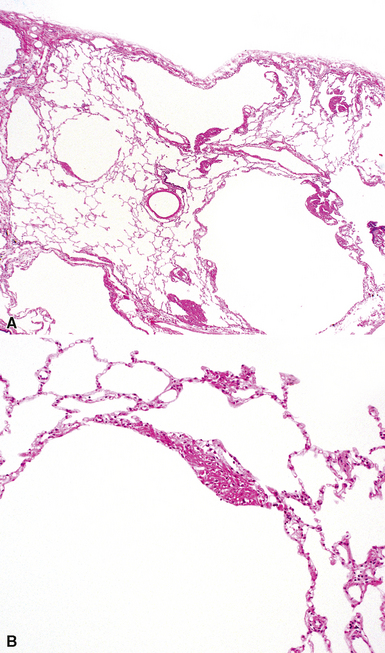
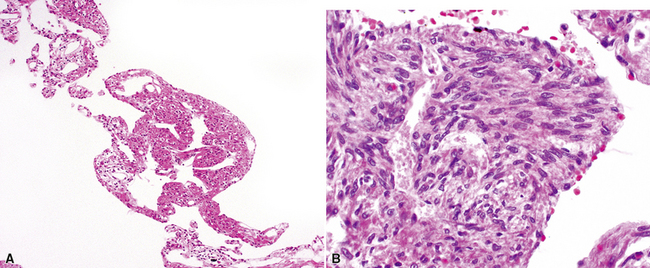
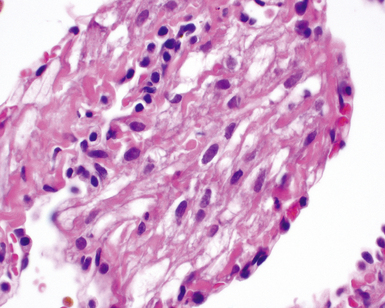
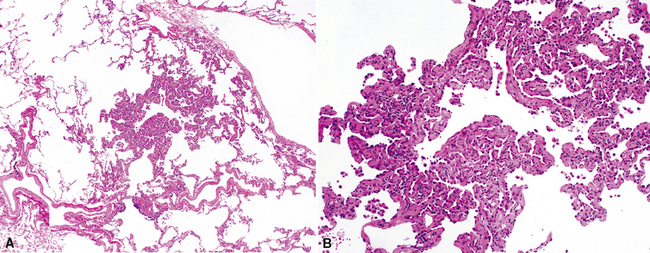
Differential Diagnosis
The differential diagnosis for LAM includes Birt-Hogg-Dubé syndrome,403 alveolar duct smooth muscle hyperplasia, pulmonary Langerhans cell histiocytosis, metastatic low-grade sarcomas in the lung, thin-walled cysts seen in so-called “radiologic LIP” or in Sjögren syndrome, and cystic terminal airways in small-airway disease, especially when these diseases occur in the setting of recurrent pneumothorax in a woman.
Clinical Course
No effective therapy has emerged for LAM. Nevertheless, antihormonal therapy404–407 has been the mainstay of treatment, based on the presence of estrogen and progesterone receptors in the abnormal smooth muscle cells in this disease.270,384,406,408 There is no evidence of improvement with the use of estrogen antagonists.409 Currently several multicenter clinical trials are under way using agents such as the mTOR inhibitor, sirolimus, Rheb inhibitors, selective estrogen antagonists, tyrosine kinase inhibitors, angiogenesis inhibitors, and lymphangiogenesis inhibitors (anti–vascular endothelial growth factor D antibody).409 Lung transplantation may also be considered an effective option.410 Recurrence of LAM in transplanted lung has been reported.411 The median survival period is in the range of 10 years, with an early subset of deaths occurring within the first 5 years of diagnosis.309,412
Hermansky-Pudlak Syndrome
The Hermansky-Pudlak syndrome (HPS) encompasses a group of genetic disorders of autosomal recessive inheritance that share features of oculocutaneous albinism, platelet storage pool deficiency, and variable tissue lipofuchsinosis.413–415 The most common form of HPS arises from a 16–base pair duplication in the HPS1 gene at exon 15 on the long arm of chromosome 10 (10q23).416 This form is referred to as HPS type 1 (HPS-1) and is associated with progressive, lethal pulmonary fibrosis.417 HPS-1 affects between 400 and 500 individuals in northwest Puerto Rico.418,419 Pulmonary fibrosis typically begins in the fourth decade and results in death from respiratory failure within 1 to 6 years of onset.420 A granulomatous colitis may also occur in HPS patients.
Radiologic Findings
Avila and coworkers421 reviewed chest radiographs and CT scans from 67 patients with HPS-1. CT scans with minimal abnormalities had normal corresponding chest radiographs. When abnormalities were present on the radiographs, these consisted of diffuse reticulonodular interstitial infiltrates, perihilar fibrosis, and pleural thickening. High-resolution CT findings included peribronchovascular thickening, ground-glass opacification, and septal thickening. Increasing severity of these changes, as assessed using a fibrosis scoring system, was inversely correlated with forced vital capacity.
Histopathologic Findings
Surgical biopsies and autopsy lungs from five patients with HPS were studied by Nakatani and colleagues.422 These researchers described alveolar septal thickening (Fig. 7-114) associated with prominent clear vacuolated type II pneumocytes (Fig. 7-115), patchy zones of fibrosis with an apparent bronchiolocentric distribution, some evidence of constrictive bronchiolitis, and haphazard microscopic honeycombing without a consistent peripheral lobular or subpleural distribution. Many giant lamellar bodies were present in the macrophages and type II cells on ultrastructural examination, and the phospholipid material in the vacuoles was weakly positive with antibodies directed against surfactant apoprotein by immunohistochemistry.
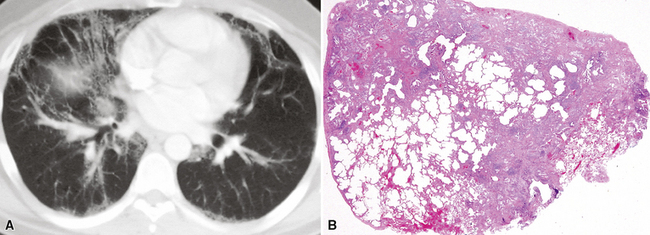
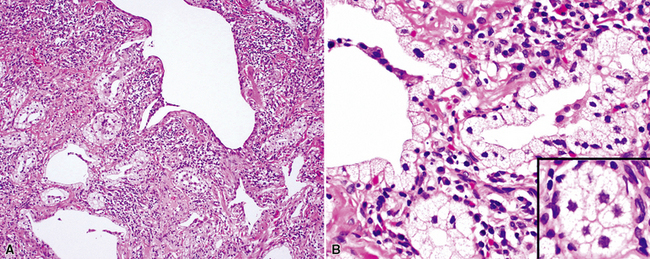
Pulmonary Alveolar Microlithiasis
Pulmonary alveolar microlithiasis (PAM) is a rare but distinctive lung disease of autosomal recessive inheritance, with approximately 300 cases reported in the literature.424–428 A significant number of cases identified are familial (affecting siblings); however, PAM occurring in both a parent and a child is uncommon. The exact incidence of the disease is unknown, but in a 46-year period, the Mayo Clinic identified only eight cases.426 Patients are typically diagnosed in the fourth decade of life, but diagnosis can occur from childhood to 80 years of age.426 There appears to be an increased incidence in individuals of Turkish descent; a Turkish family with six affected members has been described.429 The responsible gene mutation has been found in the SLC34A2 gene, which encodes the sodiumphosphate cotransporter in patients with PAM.430
Clinical Presentation
Most cases are diagnosed in adult life, but the disease can manifest at any age.425–427 It is hypothesized that the condition results from a congenital metabolic disorder, resulting in slowly progressive disease during life. The most common presentation is that of an asymptomatic patient. Some patients may experience various degrees of dyspnea, and death has been reported to occur after accelerated respiratory failure. Affected patients typically exhibit a restrictive pulmonary function defect,425 but this is generally mild when compared with the dramatic appearance of the chest radiograph, especially in young patients. Associated nephrolithiasis and pleural calcification have been reported.431
Radiologic Findings
The chest radiograph is almost immediately diagnostic, with sand-like micronodular opacities present throughout both lungs diffusely. The lower lung zones tend to be more opacified than the upper lobes. Chest radiographs may remain static for many years.425–427 High-resolution CT scans reveal intra-alveolar microcalcifications bilaterally, with increased concentration of microliths along bronchovascular bundles and interlobular fissures and in the subpleural lung parenchyma.432
Histopathologic Findings
On gross examination, the lungs are firm and gritty and rigidly maintain their shape before fixation. On microscopic examination, the air spaces contain innumerable tiny calcified bodies that are concentrically laminated and have radial striations (Fig. 7-116). These microliths are composed of calcium and phosphorus, in concentrations similar to those in bone.427 Magnesium and iron are typically present in small amounts. Microliths may be as large as 1 mm or more in diameter, but usually they are of uniform size, with diameters in the range of 250 μm. Rarely, microlithiasis may involve the alveolar walls, often accompanied by some degree of interstitial thickening and chronic inflammation (Fig. 7-117).
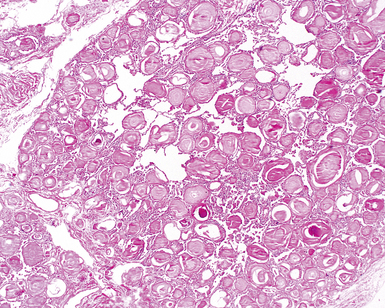
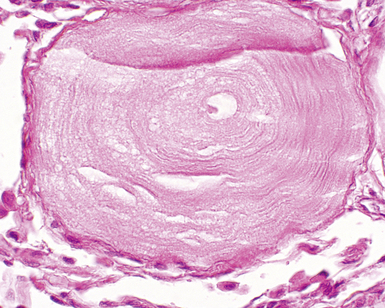
Pulmonary Alveolar Proteinosis
Pulmonary alveolar proteinosis (PAP) is a diffuse lung process characterized by the presence of alveolar spaces filled with amorphous eosinophilic material. Recent data suggest that most patients with primary PAP develop this disorder as part of an autoimmune disease.433,434 In a recent series of PAP patients, 223 of 248 were seropositive for anti–granulocyte-monocyte colony-stimulating factor (anti–GM-CSF) autoantibody.435 PAP is often multilobar, bilateral, and of subacute or chronic onset. Other subacute or chronic ILDs that include an alveolar component are considerations in the differential diagnosis for PAP (e.g., COP, RBILD/DIP, LIP, NSIP).
Clinical Presentation
The disease occurs commonly as a primary idiopathic form, whether congenital or adult onset. However, PAP may also be seen as a secondary phenomenon in the settings of occupational disease (especially dust-related), drug-induced injury, hematologic diseases, and in many settings of immunodeficiency.436–440 The disease is commonly associated with exposure to crystalline material and silica, although other substances have also been implicated.437,441 The idiopathic form is the most common presentation. Most PAP patients are male and range in age from 30 to 50 years. The usual presenting symptom is dyspnea on exertion, followed in frequency by insidious breathlessness, sometimes with cough.435,440–442
Radiologic Findings
Chest radiographs show extensive bilateral air space consolidation, involving mainly the perihilar regions; often the symptoms belie the severity of the radiologic abnormalities. CT demonstrates smooth thickening of lobular septa that is not seen on the chest radiograph. Thickened lobular septa sharply demarcate areas of ground-glass attenuation. These characteristics of alveolar proteinosis on CT are referred to as “crazy paving pattern.”443
Histopathologic Findings
PAP (i.e., alveolar lipoproteinosis) is characterized by an intra-alveolar accumulation of lipid-rich eosinophilic material.436 In primary PAP, it occurs as a result of impaired clearance of surfactant by alveolar macrophages due to the effects of an autoantibody directed against GM-CSF. The gross lung shows firm yellow-white nodules, some as large as 2 cm in diameter. Microscopically, the scanning magnification appearance is distinctive, if not diagnostic. Pink granular material fills the air spaces, sometimes with a rim of retraction that separates the alveolar wall slightly from the exudates (Fig. 7-118). Closer inspection of this material shows embedded clumps of dense globular material and cholesterol clefts (Fig. 7-119). Amorphous solid eosinophilic globules are seen frequently inside the pools of proteinaceous material. The periodic acid/Schiff reagent (PAS) stain may be useful in demonstrating a diastase-resistant, positive reaction in the globular inclusions of PAP. By immunohistochemistry this material is immunoreactive with antibodies directed against surfactant. There may be few other associated changes in the lung biopsy, although patients with long-standing disease may develop some interstitial fibrosis and chronic inflammation. More dramatic inflammatory changes should suggest comorbid disease, such as infection. For example, Nocardia, Aspergillus, or mycobacterial infection can be associated with PAP.435 PAP can be patchy in the lungs and even in biopsy specimens, so a high index of suspicion is required, coupled with good clinical and radiologic data.
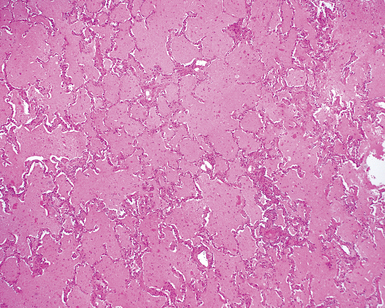
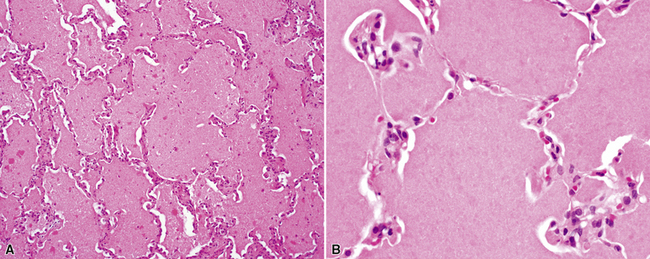
Clinical Course
Despite significant radiologic abnormalities, the disease may be unusually silent, producing few if any clinical manifestations in the absence of superimposed infection. When patients have severe dyspnea and hypoxemia, treatment can be accomplished using one or more sessions of whole-lung lavage, which usually induces remission and excellent long-term survival.444 Based on the recent evidence of a high incidence of anti–GM-CSF antibody in primary PAP patients, clinical trials using aerosolized GM-CSF have been initiated with good results.445,446
Lymphangitic Carcinomatosis
Metastatic carcinoma involving the lung, primarily within lymphatics, is known as pulmonary lymphangitic carcinomatosis (lymphangitis carcinomatosa). The tumor type is most often adenocarcinoma; it accounts for as much as 8% of all cases of metastasis to the lung. The most common sites of origin are breast, lung, and stomach, although primary disease in pancreas, ovary, kidney, and uterine cervix can also spread to the lungs in this manner.447,448 Published reports on this subject are found predominantly in the radiology literature.447–450
Radiologic Findings
On plain chest films, the changes are often subtle and nonspecific. In one study, the correct diagnosis was made in only 20 of 87 cases (23%), and 50% of the films were interpreted as normal.451 Abnormalities include linear opacities, horizontal linear lines abutting on the pleura, mostly the lower lobes (Kerley B lines), subpleural edema, and commonly, hilar and mediastinal lymph node enlargement.449 By contrast, the high-resolution CT findings are highly characteristic and accurately reflect the macroscopic abnormalities in this disease. Because the major lymphatic vessels in the lung are located in the pleura, interlobular septa, and bronchovascular bundles, the abnormalities are found primarily in this distribution, thickening and accentuating these structures. High-resolution CT scanning shows irregular thickening of the bronchovascular bundles and lobular septa, giving them a beaded appearance.448,450 Distinction from sarcoidosis (another “lymphatic” disease) can be made by examining distribution (sarcoidosis is an upper lung zone disease).
Histopathologic Findings
Small aggregates of tumor cells are present within lymphatic channels of the bronchovascular sheath and pleura (Fig. 7-120). Because the lymphatics of the bronchi are involved in this process, lymphangitic carcinoma is one of the few diffuse lung diseases that can be diagnosed definitively by bronchial or transbronchial biopsy452 (Fig. 7-121). Variable amounts of tumor may be present throughout the lung, involving the interstitium of the alveolar walls, the air spaces themselves, and the lumens of small muscular pulmonary arteries. This latter finding (microangiopathic obliterative endarteritis) may be the origin of the edema, inflammation, and interstitial fibrosis that frequently accompany the disease and probably accounts for the clinical and radiologic impression of non-neoplastic diffuse lung disease.447,449
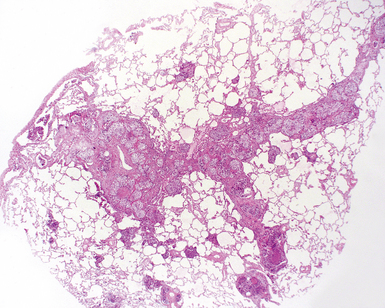
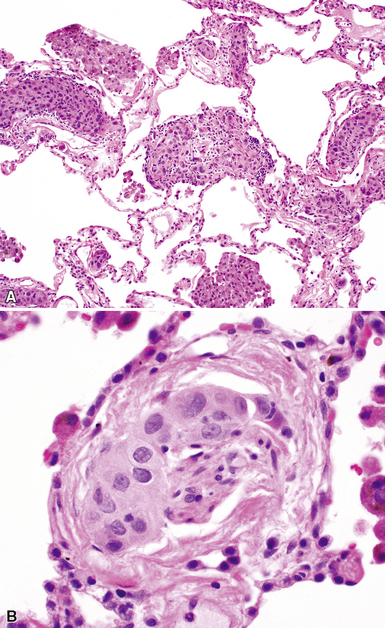
The pathogenesis of lymphangitic carcinoma is unknown. Seeding of the lung lymphatics from retrograde spread of microvascular tumor emboli has been postulated and supported by observations of intravascular tumor present in other organs at autopsy from patients so affected.447,449
Idiopathic Pleuroparenchymal Fibroelastosis
In 2004, Frankel and coworkers described five distinctive cases of an ILD referred to as idiopathic pleuroparenchymal fibroelastosis (IPFE).453 This upper lobe–predominant fibrotic disease slowly progresses to involve the lower lung. On histopathologic examination, elastotic fibrosis, similar to that seen in so-called apical cap, is the dominant feature. The major differences between apical cap and IPFE are that the patients with IPFE are symptomatic and the disease is progressive,eventuating in death within several years. In addition to the original series described by Frankel’s group, only two reports of IPFE have appeared in the English literature to date; however, several case reports from Japan and the United States seem to describe a similar disease.454–457
Radiologic Findings
Bilateral upper lung volume loss with marked apical pleural thickening is the most common finding on plain films. CT images show marked visceral pleural thickening extending into the lung parenchyma with subpleural reticular abnormalities. Traction bronchiectasis is a common finding in areas of fibrosis. Lower lung zones may be hyperinflated (Fig. 7-122A).
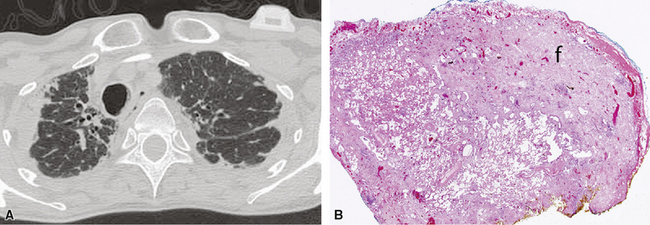
Histopathologic Findings
The histopathology of IPFE overlaps with that of apical cap. Fibrotic areas show dense elastotic scarring without inflammation. Scattered lymphoid aggregations are common, but there is no associated cellular interstitial pneumonia. Margins between normal lung and affected fibrotic areas are sharply defined. Uninvolved lung shows minimal abnormalities, mainly involving the airways (bronchiolar fibrosis or ectasia). Broad pleural adhesions, probably due to the common history of pneumothorax, may be present (see Fig. 7-122B).
1 Leslie K., Colby T. Classification and pathology of diffuse interstitial lung disease. Semin Clin Immunol. 1999;1:7-15.
2 Leslie K.O. My approach to interstitial lung disease using clinical, radiological and histopathologic patterns. J Clin Pathol. 2009;62(5):387-401.
3 Leslie K.O., Gruden J.F., Parish J.M., Scholand M.B. Transbronchial biopsy interpretation in the patient with diffuse parenchymal lung disease. Arch Pathol Lab Med. 2007;131(3):407-423.
4 Elicker B., Pereira C.A., Webb R., Leslie K.O. High-resolution computed tomography patterns of diffuse interstitial lung disease with clinical and pathological correlation. J Bras Pneumol. 2008;34(9):715-744.
5 American Thoracic Society. Idiopathic pulmonary fibrosis: diagnosis and treatment. International consensus statement. American Thoracic Society (ATS), and the European Respiratory Society (ERS). Am J Respir Crit Care Med. 2000;161(2 Pt 1):646-664.
6 American Thoracic Society/European Respiratory Society. International Multidisciplinary Consensus Classification of the Idiopathic Interstitial Pneumonias. Am Rev Respir Crit Care Med. 2002;165(2):227-304.
7 Liebow A.L., Carrington C.B. The interstitial pneumonias. In: Simon M., Potchen E.J., LeMay M., editors. Frontiers of Pulmonary Radiology: Pathophysiologic, Roentgenographic and Radioisotopic Considerations. New York: Grune & Stratton; 1969:102-141.
8 Katzenstein A.L. Idiopathic interstitial pneumonia: classification and diagnosis. Monogr Pathol. 1993;36:1-31.
9 Katzenstein A.L., Myers J.L. Nonspecific interstitial pneumonia and the other idiopathic interstitial pneumonias: classification and diagnostic criteria. Am J Surg Pathol. 2000;24(1):1-3.
10 Crystal R., Fulmer J.D., Roberts W.C., et al. Idiopathic pulmonary fibrosis: clinical histologic, radiographic, physiologic, scintigraphic, cytologic and biochemical aspects. Ann Intern Med. 1976;85:769-788.
11 Carrington C.B., Gaensler E.A., Coutu R.E., et al. Usual and desquamative interstitial pneumonia. Chest. 1976;69(suppl 2):261-263.
12 Aubry M.C., Wright J.L., Myers J.L. The pathology of smoking-related lung diseases. Clin Chest Med. 2000;21(1):11-35. vii
13 Ohori N., Sciurba F.C., Owens G.R., et al. Giant-cell interstitial pneumonia and hard-metal pneumoconiosis. A clinicopathologic study of four cases and review of the literature. Am J Surg Pathol. 1989;13(7):581-587.
14 Lewis C.C., Yang J.Y., Huang X., et al. Disease-specific gene expression profiling in multiple models of lung disease. Am J Respir Crit Care Med. 2008;177(4):376-387.
15 Katzenstein A., Myers J., Mazur M. Acute interstitial pneumonia: a clinicopathologic, ultrastructural, and cell kinetic study. Am J Surg Pathol. 1986;10:256-267.
16 Katzenstein A.L., Fiorelli R.F. Nonspecific interstitial pneumonia/fibrosis. Histologic features and clinical significance. Am J Surg Pathol. 1994;18(2):136-147.
17 Epler G.R., Colby T.V., McLoud T.C., et al. Bronchiolitis obliterans organizing pneumonia. N Engl J Med. 1985;312(3):152-158.
18 Leslie K.O. Pathology of the idiopathic interstitial pneumonias. Exp Lung Res. 2005;31(suppl 1):23-40.
19 Gough J. Differential diagnosis in the pathology of asbestosis. Ann N Y Acad Sci. 1965;132:368-372.
20 Gadek J., Hunninghake G., Schoenberger C., et al. Pulmonary asbestosis and idiopathic pulmonary fibrosis: pathogenetic parallels. Chest. 1981;80(suppl 1):63-64.
21 Yamamoto S. Histopathological features of pulmonary asbestosis with particular emphasis on the comparison with those of usual interstitial pneumonia. Osaka City Med J. 1997;43(2):225-242.
22 Leslie K., King T.E.Jr, Low R. Smooth muscle actin is expressed by air space fibroblast-like cells in idiopathic pulmonary fibrosis and hypersensitivity pneumonitis. Chest. 1991;99(suppl 3):47S-48S.
23 Adler B.D., Padley S.P., Müller N.L., et al. Chronic hypersensitivity pneumonitis: high resolution CT and radiographic features in 16 patients. Radiology. 1992;185:91-95.
24 Lynch D.A., Newell J.D., Logan P.M., et al. Can CT distinguish hypersensitivity pneumonitis from idiopathic pulmonary fibrosis? AJR Am J Roentgenol. 1995;165(4):807-811.
25 Hunninghake G., Fauci A. Pulmonary involvement in the collagen vascular diseases. Am Rev Respir Dis. 1979;119:471-503.
26 Colby T.V. Pathology of the lung in collagen vascular diseases. In: Cannon G., Zimmerman G., editors. The Lung Rheumatic Diseases. New York: Marcel Dekker; 1990:145-178.
27 Leslie K.O., Trahan S., Gruden J. Pulmonary pathology of the rheumatic diseases. Semin Respir Crit Care Med. 2007;28(4):369-378.
28 Lamblin C., Bergoin C., Saelens T., Wallaert B. Interstitial lung diseases in collagen vascular diseases. Eur Respir J Suppl. 2001;32:69s-80s.
29 Rossi S.E., Erasmus J.J., McAdams H.P., et al. Pulmonary drug toxicity: radiologic and pathologic manifestations. Radiographics. 2000;20(5):1245-1259.
30 Raghu G., Weycker D., Edelsberg J., et al. Incidence and prevalence of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2006;174(7):810-816.
31 Coultas D.B. Epidemiology of idiopathic pulmonary fibrosis. Semin Respir Med. 1993;14:181-196.
32 Guerry-Force M., Müller N.L., Wright J.L., et al. A comparison of bronchiolitis obliterans with organizing pneumonia, usual interstitial pneumonia and small airways disease. Am Rev Respir Dis. 1987;135:705-712.
33 Staples C., Müller N.L., Vedal S., et al. Usual interstitial pneumonia: correlations of CT with clinical, functional and radiologic findings. Radiology. 1987;162:377-381.
34 Epler G., McLoud T.C., Gaensler E.A., et al. Normal chest roentgenograms in chronic diffuse infiltrative lung disease. N Engl J Med. 1978;298:934-939.
35 Akira M., Sakatani M., Ueda E. Idiopathic pulmonary fibrosis: progression of honeycombing at thin-section CT. Radiology. 1993;189(3):687-691.
36 Nishimura K., Itoh H. High-resolution computed tomographic features of bronchiolitis obliterans organizing pneumonia. Chest. 1992;102:26S-31S.
37 Lynch D.A. Ground glass attenuation on CT in patients with idiopathic pulmonary fibrosis. Chest. 1996;110(2):312-313.
38 Kazerooni E.A., Martinez F.J., Flint A., et al. Thin-section CT obtained at 10-mm increments versus limited three-level thin-section CT for idiopathic pulmonary fibrosis: correlation with pathologic scoring. AJR Am J Roentgenol. 1997;169(4):977-983.
39 Hunninghake G.W., Zimmerman M.B., Schwartz D.A., et al. Utility of a lung biopsy for the diagnosis of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2001;164(2):193-196.
40 Katzenstein A., Myers J.L., Prophet W.D., et al. Bronchiolitis obliterans and usual interstitial pneumonia. A comparative clinicopathologic study. Am J Surg Pathol. 1986;10:373-376.
41 Lee J.S., Gong G., Song K.S., et al. Usual interstitial pneumonia: relationship between disease activity and the progression of honeycombing at thin-section computed tomography. J Thorac Imaging. 1998;13(3):199-203.
42 Myers J.L. NSIP, UIP, and the ABCs of idiopathic interstitial pneumonias. Eur Respir J. 1998;12(5):1003-1004.
43 Nagao T., Nagai S., Kitaichi M., et al. Usual interstitial pneumonia: idiopathic pulmonary fibrosis versus collagen vascular diseases. Respiration. 2001;68(2):151-159.
44 Cool C.D., Groshong S.D., Rai P.R., et al. Fibroblast foci are not discrete sites of lung injury or repair: the fibroblast reticulum. Am J Respir Crit Care Med. 2006;174(6):654-658.
45 Flaherty K.R., Colby T.V., Travis W.D., et al. Fibroblastic foci in usual interstitial pneumonia: idiopathic versus collagen vascular disease. Am J Respir Crit Care Med. 2003;167(10):1410-1415.
46 Kondoh Y., Taniguchi H., Kawabata Y., et al. Acute exacerbation in idiopathic pulmonary fibrosis. Analysis of clinical and pathologic findings in three cases. Chest. 1993;103(6):1808-1812.
47 Collard H.R., Moore B.B., Flaherty K.R., et al. Acute exacerbations of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2007;176(7):636-643.
48 Panos R.J., Mortenson R.L., Niccoli S.A., King T.E.Jr. Clinical deterioration in patients with idiopathic pulmonary fibrosis: causes and assessment. Am J Med. 1990;88(4):396-404.
49 Kim D.S., Park J.H., Park B.K., et al. Acute exacerbation of idiopathic pulmonary fibrosis: frequency and clinical features. Eur Respir J. 2006;27(1):143-150.
50 Katzenstein A.L., Zisman D.A., Litzky L.A., et al. Usual interstitial pneumonia: histologic study of biopsy and explant specimens. Am J Surg Pathol. 2002;26(12):1567-1577.
51 Flaherty K.R., Travis W.D., Colby T.V., et al. Histopathologic variability in usual and nonspecific interstitial pneumonias. Am J Respir Crit Care Med. 2001;164(9):1722-1727.
52 Noth I., Martinez F.J. Recent advances in idiopathic pulmonary fibrosis. Chest. 2007;132(2):637-650.
53 Bjoraker J.A., Ryu J.H., Edwin M.K., et al. Prognostic significance of histopathologic subsets in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 1998;157(1):199-203.
54 Raghu G., Brown K.K., Costabel U., et al. Treatment of idiopathic pulmonary fibrosis with etanercept: an exploratory, placebo-controlled trial. Am J Respir Crit Care Med. 2008;178(9):948-955.
55 Ask K., Martin G.E., Kolb M., Gauldie J. Targeting genes for treatment in idiopathic pulmonary fibrosis: challenges and opportunities, promises and pitfalls. Proc Am Thorac Soc. 2006;3(4):389-393.
56 Grutters J.C., du Bois R.M. Genetics of fibrosing lung diseases. Eur Respir J. 2005;25(5):915-927.
57 Wang Y., Kuan P.J., Xing C., et al. Genetic defects in surfactant protein A2 are associated with pulmonary fibrosis and lung cancer. Am J Hum Genet. 2009;84(1):52-59.
58 Steele M.P., Speer M.C., Loyd J.E., et al. Clinical and pathologic features of familial interstitial pneumonia. Am J Respir Crit Care Med. 2005;172(9):1146-1152.
59 Armanios M.Y., Chen J.J., Cogan J.D., et al. Telomerase mutations in families with idiopathic pulmonary fibrosis. N Engl J Med. 2007;356(13):1317-1326.
60 Kitaichi M. Pathologic features and the classification of interstitial pneumonia of unknown etiology. Bull Chest Dis Res Inst Kyoto Univ. 1990;23(1–2):1-18.
61 Nagai S., Kitaichi M., Itoh H., et al. Idiopathic nonspecific interstitial pneumonia/fibrosis: Comparison with idiopathic pulmonary fibrosis and BOOP. Eur Respir J. 1998;12(5):1010-1019.
62 Cottin V., Donsbeck A.V., Revel D., et al. Nonspecific interstitial pneumonia. Individualization of a clinicopathologic entity in a series of 12 patients. Am J Respir Crit Care Med. 1998;158(4):1286-1293.
63 Daniil Z.D., Gilchrist F.C., Nicholson A.G., et al. A histologic pattern of nonspecific interstitial pneumonia is associated with a better prognosis than usual interstitial pneumonia in patients with cryptogenic fibrosing alveolitis. Am J Respir Crit Care Med. 1999;160(3):899-905.
64 Park J.S., Lee K.S., Kim J.S., et al. Nonspecific interstitial pneumonia with fibrosis: radiographic and CT findings in seven patients. Radiology. 1995;195(3):645-648.
65 Hartman T.E., Swensen S.J., Hansell D.M., et al. Nonspecific interstitial pneumonia: variable appearance at high-resolution chest CT. Radiology. 2000;217(3):701-705.
66 Travis W.D., Hunninghake G., King T.E.Jr, et al. Idiopathic nonspecific interstitial pneumonia: report of an American Thoracic Society project. Am J Respir Crit Care Med. 2008;177(12):1338-1347.
67 Kinder B.W., Collard H.R., Koth L., et al. Idiopathic nonspecific interstitial pneumonia: lung manifestation of undifferentiated connective tissue disease? Am J Respir Crit Care Med. 2007;176(7):691-697.
68 Travis W.D., Matsui K., Moss J., Ferrans V.J. Idiopathic nonspecific interstitial pneumonia: prognostic significance of cellular and fibrosing patterns: survival comparison with usual interstitial pneumonia and desquamative interstitial pneumonia. Am J Surg Pathol. 2000;24(1):19-33.
69 Davison A.G., Heard B.E., McAllister W.A., Turner-Warwick M.E. Cryptogenic organizing pneumonitis. Q J Med. 1983;52:382-394.
70 Yousem S.A., Lohr R.H., Colby T.V. Idiopathic bronchiolitis obliterans organizing pneumonia/cryptogenic organizing pneumonia with unfavorable outcome: pathologic predictors. Mod Pathol. 1997;10(9):864-871.
71 Izumi T., Nagai S., Nishimura K., et al. BALF cell findings in patients with BOOP, particularly in comparison with UIP. Nihon Kyobu Shikkan Gakkai Zasshi. 1989;27(4):474-480.
72 King T.J., Mortensen R. Cryptogenic organizing pneumonitis. Chest. 1992;102:8S-13S.
73 Müller N.L., Guerry-Force M.L., Staples C.A., et al. Differential diagnosis of bronchiolitis obliterans with organizing pneumonia and usual interstitial pneumonia: clinical, functional, and radiologic findings. Radiology. 1987;162(1 Pt 1):151-156.
74 Cordier J.F., Loire R., Brune J. Idiopathic bronchiolitis obliterans organizing pneumonia: definition of clinical profiles in a series of 16 patients. Chest. 1989;96:999-1004.
75 Müller N.L., Staples C.A., Miller R.R. Bronchiolitis obliterans organizing pneumonia: CT features in 14 patients. AJR Am J Roentgenol. 1990;154(5):983-987.
76 Lee K.S., Kullnig P., Hartman T.E., Müller N.L. Cryptogenic organizing pneumonia. CT findings in 43 patients. AJR Am J Roentgenol. 1994;62:543-546.
77 Preidler K.W., Szolar D.M., Moelleken S., et al. Distribution pattern of computed tomography findings in patients with bronchiolitis obliterans organizing pneumonia. Invest Radiol. 1996;31(5):251-255.
78 Akira M., Yamamoto S., Sakatani S. Bronchiolitis obliterans organizing pneumonia manifesting as multiple large nodules or masses. AJR Am J Roentgenol. 1998;170(2):291-295.
79 Izumi T., Kitaichi M., Nishimura K., Nagai S. Bronchiolitis obliterans organizing pneumonia. Clinical features and differential diagnosis. Chest. 1992;102(3):715-719.
80 King T.E.Jr. BOOP: An important cause of migratory pulmonary infiltrates? Eur Respir J. 1995;8(2):193-195.
81 Bouchardy L.M., Kuhlman J.E., Ball W.C.Jr, et al. CT findings in bronchiolitis obliterans organizing pneumonia (BOOP) with radiographic, clinical, and histologic correlation. J Comput Assist Tomogr. 1993;17:352-357.
82 Colby T.V. Pathologic aspects of bronchiolitis obliterans organizing pneumonia. Chest. 1992;102(suppl 1):38S-43S.
83 Kitaichi M. Bronchiolitis obliterans organizing pneumonia (BOOP). In: Takishima T., editor. Basic and Clinical Aspects of Pulmonary Fibrosis. Boca Raton, FL: CRC Press; 1994:463-488.
84 Cordier J.F. Cryptogenic organizing pneumonitis. Bronchiolitis obliterans organizing pneumonia. Clin Chest Med. 1993;14(4):677-692.
85 Niewoehner D., Kleinerman J., Rice D. Pathologic changes in the the peripheral airways in young cigarette smokers. N Engl J Med. 1974;291:755-758.
86 Myers J., Veal C.F.Jr, Shin M.S., Katzenstein A.L. Respiratory bronchiolitis causing interstitial lung disease. A clinicopathologic study of six cases. Am Rev Respir Dis. 1987;135:880-884.
87 Heyneman L.E., Ward S., Lynch D.A., et al. Respiratory bronchiolitis, respiratory bronchiolitis–associated interstitial lung disease, and desquamative interstitial pneumonia: different entities or part of the spectrum of the same disease process? AJR Am J Roentgenol. 1999;173(6):1617-1622.
88 Yousem S.A., Colby T.V., Gaensler E.A. Respiratory bronchiolitis–associated interstitial lung disease and its relationship to desquamative interstitial pneumonia. Mayo Clin Proc. 1989;64(11):1373-1380.
89 Myers J.L., Veal C.F.Jr, Shin M.S., Katzenstein A.L. Respiratory bronchiolitis causing interstitial lung disease. A clinicopathologic study of six cases. Am Rev Respir Dis. 1987;135(4):880-884.
90 King T.E.Jr. Respiratory bronchiolitis–associated interstitial lung disease. Clin Chest Med. 1993;14(4):693-698.
91 Moon J., du Bois R.M., Colby T.V., et al. Clinical significance of respiratory bronchiolitis on open lung biopsy and its relationship to smoking related interstitial lung disease. Thorax. 1999;54(11):1009-1014.
92 Yousem S.A. Respiratory bronchiolitis–associated interstitial lung disease with fibrosis is a lesion distinct from fibrotic nonspecific interstitial pneumonia: A proposal. Mod Pathol. 2006;19(11):1474-1479.
93 Portnoy J., Veraldi K.L., Schwarz M.I., et al. Respiratory bronchiolitis–interstitial lung disease: long-term outcome. Chest. 2007;131(3):664-671.
94 Carrington C.B., Gaensler E.A., Coutu R.E., et al. Natural history and treated course of usual and desquamative interstitial pneumonia. N Engl J Med. 1978;298(15):801-809.
95 Katzenstein A.L., Myers J.L. Idiopathic pulmonary fibrosis: clinical relevance of pathologic classification. Am J Respir Crit Care Med. 1998;157(4 Pt 1):1301-1315.
96 Hartman T.E., Primack S.L., Swensen S.J., et al. Desquamative interstitial pneumonia: thin-section CT findings in 22 patients. Radiology. 1993;187(3):787-790.
97 Gruden J.F., Webb W.R. CT findings in a proved case of respiratory bronchiolitis. AJR Am J Roentgenol. 1993;161(1):44-46.
98 Nicholson A.G., Wotherspoon A.C., Diss T.C., et al. Pulmonary B-cell non-Hodgkin’s lymphomas. The value of immunohistochemistry and gene analysis in diagnosis. Histopathology. 1995;26(5):395-403.
99 Nicholson A.G., Wotherspoon A.C., Diss T.C., et al. Reactive pulmonary lymphoid disorders. Histopathology. 1995;26(5):405-412.
100 Nicholson A.G., Wotherspoon A.C., Jones A.L., et al. Pulmonary B-cell non-Hodgkin’s lymphoma associated with autoimmune disorders: a clinicopathological review of six cases. Eur Respir J. 1996;9(10):2022-2025.
101 Koss M.N. Pulmonary lymphoproliferative disorders. Monogr Pathol. 1993;18(36):145-194.
102 Herbert A., Walters M.T., Cawley M.I., Godfrey R.C. Lymphocytic interstitial pneumonia identified as lymphoma of mucosa-associated lymphoid tissue. J Pathol. 1985;146(2):129-138.
103 Schuurman H.J., Gooszen H.C., Tan I.W., et al. Low-grade lymphoma of immature T-cell phenotype in a case of lymphocytic interstitial pneumonia and Sjögren’s syndrome. Histopathology. 1987;11(11):1193-1204.
104 Cha S.I., Fessler M.B., Cool C.D., et al. Lymphoid interstitial pneumonia: clinical features, associations and prognosis. Eur Respir J. 2006;28(2):364-369.
105 Strimlan C., Rosenow E.C.3rd, Weiland L.H., Brown L.R. Lymphocytic interstitial pneumonitis. Review of 13 cases. Ann Intern Med. 1978;88:616-621.
106 Liebow A.A., Carrington C.B. Diffuse pulmonary lymphoreticular infiltrations associated with dysproteinemia. Med Clin North Am. 1973;57:809-843.
107 Strimlan C.V., Rosenow E.C.3rd, Divertie M.B., Harrison E.G.Jr. Pulmonary manifestations of Sjögren’s syndrome. Chest. 1976;70(03):354-361.
108 DeCoteau W.E., Tourville D., Ambrus J.L., et al. Lymphoid interstitial pneumonia and autoerythrocyte sensitization syndrome. A case with deposition of immunoglobulins on the alveolar basement membrane. Arch Intern Med. 1974;134(3):519-522.
109 Julsrud P.R., Brown L.R., Li C.Y., et al. Pulmonary processes of mature-appearing lymphocytes: pseudolymphoma, well-differentiated lymphocytic lymphoma, and lymphocytic interstitial pneumonitis. Radiology. 1978;127(2):289-296.
110 Schwarz M.I. A man with dyspnea, productive cough, and chest radiograph showing hyperinflation and a diffuse nodular pattern. Chest. 1998;113(4):1123-1124.
111 Johkoh T., Ichikado K., Akira M., et al. Lymphocytic interstitial pneumonia: follow-up CT findings in 14 patients. J Thorac Imaging. 2000;15(3):162-167.
112 Macfarlane A., Davies D. Diffuse lymphoid interstitial pneumonia. Thorax. 1973;28(6):768-776.
113 Strimlan C., Rosenow E.C.3rd, Weiland L.H., Brown L.R. Lymphocytic interstitial pneumonitis. Review of 13 cases. Ann Intern Med. 1978;88:616-621.
114 Johkoh T., Müller N.L., Pickford H.A., et al. Lymphocytic interstitial pneumonia: thin-section CT findings in 22 patients. Radiology. 1999;212(2):567-572.
115 Silva C.I., Flint J.D., Levy R.D., Müller N.L. Diffuse lung cysts in lymphoid interstitial pneumonia: high-resolution CT and pathologic findings. J Thorac Imaging. 2006;21(3):241-244.
116 Lohrmann C., Uhl M., Warnatz K., et al. High-resolution CT imaging of the lung for patients with primary Sjögren’s syndrome. Eur J Radiol. 2004;52(2):137-143.
117 Torii K., Ogawa K., Kawabata Y., et al. Lymphoid interstitial pneumonia as a pulmonary lesion of idiopathic plasmacytic lymphadenopathy with hyperimmunoglobulinemia. Intern Med. 1994;33(4):237-241.
118 Julsrud P.R., Brown L.R., Li C.Y., et al. Pulmonary processes of mature-appearing lymphocytes: pseudolymphoma, well-differentiated lymphocytic lymphoma, and lymphocytic interstitial pneumonitis. Radiology. 1978;127:289-296.
119 Strimlan C.V. Pulmonary function in patients with primary Sjögren’s syndrome. Ann Rheum Dis. 2001;60(4):429.
120 Palmas A., Tefferi A., Myers J.L., et al. Late-onset noninfectious pulmonary complications after allogeneic bone marrow transplantation. Br J Haematol. 1998;100(4):680-687.
121 Grieco M., Chinoy-Acharya P. Lymphoid interstitial pneumonia associated with the acquired immune deficiency syndrome. Am Rev Respir Dis. 1985;131:952-955.
122 Berdon W.E., Mellins R.B., Abramson S.J., Ruzal-Shapiro C. Pediatric HIV infection in its second decade—the changing pattern of lung involvement. Clinical, plain film, and computed tomographic findings. Radiol Clin North Am. 1993;31(3):453-463.
123 Ikeogu M.O., Wolf B., Mathe S. Pulmonary manifestations in HIV seropositivity and malnutrition in Zimbabwe. Arch Dis Child. 1997;76(2):124-128.
124 Khare M.D., Sharland M. Pulmonary manifestations of pediatric HIV infection. Indian J Pediatr. 1999;66(6):895-904.
125 Church J.A. Lymphoid interstitial pneumonia and rheumatoid arthritis. J Pediatr. 1985;107(3):485.
126 Black L., Katz S. Respiratory Disease: Task Force Report on Problems, Research Approaches, Needs. Washington, DC: Department of Health, Education, and Welfare, National Heart and Lung Institute, 1977.
127 Banks J., Banks C., Cheong B., et al. An epidemiological and clinical investigation of pulmonary function and respiratory symptoms in patients with rheumatoid arthritis. Q J Med. 1992;85:795-806.
128 Cheema G.S., Quismorio F.P.Jr. Interstitial lung disease in systemic sclerosis. Curr Opin Pulm Med. 2001;7(5):283-290.
129 Minai O.A., Dweik R.A., Arroliga A.C. Manifestations of scleroderma pulmonary disease. Clin Chest Med.. 1998;19(4):713-731. viii–ix
130 Cheema G.S., Quismorio F.P.Jr. Interstitial lung disease in systemic lupus erythematosus. Curr Opin Pulm Med. 2000;6(5):424-429.
131 Murin S., Wiedemann H.P., Matthay R.A. Pulmonary manifestations of systemic lupus erythematosus. Clin Chest Med.. 1998;19(4):641-665. viii
132 Dickey B.F., Myers A.R. Pulmonary disease in polymyositis/dermatomyositis. Semin Arthritis Rheum.. 1984;14(1):60-76.
133 Ito I., Nagai S., Kitaichi M., et al. Pulmonary manifestations of primary Sjogren’s syndrome: a clinical, radiologic, and pathologic study. Am J Respir Crit Care Med. 2005;171(6):632-638.
134 Koyama M., Johkoh T., Honda O., et al. Pulmonary involvement in primary Sjögren’s syndrome: spectrum of pulmonary abnormalities and computed tomography findings in 60 patients. J Thorac Imaging.. 2001;16(4):290-296.
135 Sarkar P.K., Patel N., Furie R.A., Talwar A. Pulmonary manifestations of primary Sjögren’s syndrome. Indian J Chest Dis Allied Sci.. 2009;51(2):93-101.
136 Prakash U.B. Respiratory complications in mixed connective tissue disease. Clin Chest Med.. 1998;19(4):733-746. ix
137 Tillie-Leblond I., Wislez M., Valeyre D., et al. Interstitial lung disease and anti-Jo-1 antibodies: difference between acute and gradual onset. Thorax. 2008;63(1):53-59.
138 Ellman P., Ball R.E. “Rheumatoid disease” with joint and pulmonary manifestations. BMJ. 1948;2:816-820.
139 Gabbay E., Tarala R., Will R., et al. Interstitial lung disease in recent onset rheumatoid arthritis. Am J Respir Crit Care Med. 1997;156(2 Pt 1):528-535.
140 Anaya J.M., Diethelm L., Ortiz L.A., et al. Pulmonary involvement in rheumatoid arthritis. Semin Arthritis Rheum. 1995;24(4):242-254.
141 Hakala M. Poor prognosis in patients with rheumatoid arthritis hospitalized for interstitial lung fibrosis. Chest. 1988;93(1):114-118.
142 Athreya B.H., Doughty R.A., Bookspan M., et al. Pulmonary manifestations of juvenile rheumatoid arthritis. A report of eight cases and review. Clin Chest Med. 1980;1(3):361-374.
143 Saag K.G., Kolluri S., Koehnke R.K., et al. Rheumatoid arthritis lung disease. Determinants of radiographic and physiologic abnormalities. Arthritis Rheum. 1996;39(10):1711-1719.
144 Akira M., Sakatani M., Hara H. Thin-section CT findings in rheumatoid arthritis–associated lung disease: CT patterns and their courses. J Comput Assist Tomogr. 1999;23(6):941-948.
145 Remy-Jardin M., Remy J., Cortet B., et al. Lung changes in rheumatoid arthritis: CT findings. Radiology. 1994;193(2):375-382.
146 Hunninghake G.W., Fauci A.S. Pulmonary involvement in the collagen vascular diseases. Am Rev Respir Dis. 1979;119(3):471-503.
147 Hubbard R., Venn A. The impact of coexisting connective tissue disease on survival in patients with fibrosing alveolitis. Rheumatology (Oxford). 2002;41(6):676-679.
148 Laitinen O., Nissilä M., Salorinne Y., Aalto P. Pulmonary involvement in patients with rheumatoid arthritis. Scand J Respir Dis. 1975;56:297-304.
149 Lee H.K., Kim D.S., Yoo B., et al. Histopathologic pattern and clinical features of rheumatoid arthritis–associated interstitial lung disease. Chest. 2005;127(6):2019-2027.
150 Schwarz M.I., Matthay R.A., Sahn S.A., et al. Interstitial lung disease in polymyositis and dermatomyositis: analysis of six cases and review of the literature. Medicine (Baltimore). 1976;55(1):89-104.
151 Khanna D., Brown K.K., Clements P.J., et al. Systemic sclerosis-associated interstitial lung disease-proposed recommendations for future randomized clinical trials. Clin Exp Rheumatol.. 2010;28(2 Suppl 58):S55-S62.
152 Hunninghake G.W., Gadek J.E., Kawanami O., et al. Inflammatory and immune processes in the human lung in health and disease: evaluation by bronchoalveolar lavage. Am J Pathol. 1979;97:149-206.
153 Bouros D., Wells A.U., Nicholson A.G., et al. Histopathologic subsets of fibrosing alveolitis in patients with systemic sclerosis and their relationship to outcome. Am J Respir Crit Care Med. 2002;165(12):1581-1586.
154 Kim D.S., Yoo B., Lee J.S., et al. The major histopathologic pattern of pulmonary fibrosis in scleroderma is nonspecific interstitial pneumonia. Sarcoidosis Vasc Diffuse Lung Dis. 2002;19(2):121-127.
155 Kim E.A., Johkoh T., Lee K.S., et al. Interstitial pneumonia in progressive systemic sclerosis: serial high-resolution CT findings with functional correlation. J Comput Assist Tomogr. 2001;25(5):757-763.
156 Cozzi F., Chiesura Corona M., Rizzi M., et al. Lung fibrosis quantified by HRCT in scleroderma patients with different disease forms and ANA specificities. Reumatismo. 2001;53(1):55-62.
157 Yamane K., Ihn H., Asano Y., et al. Clinical and laboratory features of scleroderma patients with pulmonary hypertension. Rheumatology (Oxford). 2000;39(11):1269-1271.
158 Johnson D.A., Drane W.E., Curran J., et al. Pulmonary disease in progressive systemic sclerosis. A complication of gastroesophageal reflux and occult aspiration? Arch Intern Med. 1989;149(3):589-593.
159 Montesi A., Pesaresi A., Cavalli M.L., et al. Oropharyngeal and esophageal function in scleroderma. Dysphagia. 1991;6(4):219-223.
160 Tashkin D.P., Elashoff R., Clements P.J., et al. Effects of 1-year treatment with cyclophosphamide on outcomes at 2 years in scleroderma lung disease. Am J Respir Crit Care Med. 2007;176(10):1026-1034.
161 McSweeney P.A., Nash R.A., Sullivan K.M., et al. High-dose immunosuppressive therapy for severe systemic sclerosis: initial outcomes. Blood. 2002;100(5):1602-1610.
162 Dubois E., Tufanelli D.L. Clinical manifestations of systemic lupus erythematosus. JAMA. 1964;190:104-111.
163 Eisenberg H., Dubois E.L., Sherwin R.P., Balchum O.J. Diffuse interstitial lung disease in systemic lupus erythematosus. Ann Intern Med. 1973;79:37-45.
164 Gammon R.B., Bridges T.A., al-Nezir H., et al. Bronchiolitis obliterans organizing pneumonia associated with systemic lupus erythematosus. Chest. 1992;102:1171-1174.
165 Myers J.L., Katzenstein A.A. Microangiitis in lupus-induced pulmonary hemorrhage. Am J Clin Pathol. 1986;85(5):552-556.
166 Chang M.Y., Fang J.T., Chen Y.C., Huang C.C. Diffuse alveolar hemorrhage in systemic lupus erythematosus: a single center retrospective study in Taiwan. Ren Fail. 2002;24(6):791-802.
167 Santos-Ocampo A.S., Mandell B.F., Fessler B.J. Alveolar hemorrhage in systemic lupus erythematosus: presentation and management. Chest. 2000;118(4):1083-1090.
168 Matthay R.A., Schwarz M.I., Petty T.L., et al. Pulmonary manifestations of systemic lupus erythematosus: review of twelve cases with acute lupus pneumonitis. Medicine. 1974;54:397-409.
169 Kim J.S., Lee K.S., Koh E.M., et al. Thoracic involvement of systemic lupus erythematosus: clinical, pathologic, and radiologic findings. J Comput Assist Tomogr. 2000;24(1):9-18.
170 Hughson M.D., He Z., Henegar J., McMurray R. Alveolar hemorrhage and renal microangiopathy in systemic lupus erythematosus. Arch Pathol Lab Med. 2001;125(4):475-483.
171 Bankier A.A., Kiener H.P., Wiesmayr M.N., et al. Discrete lung involvement in systemic lupus erythematosus: CT assessment. Radiology. 1995;196(3):835-840.
172 Fenlon H.M., Doran M., Sant S.M., Breatnach E. High-resolution chest CT in systemic lupus erythematosus. AJR Am J Roentgenol. 1996;166(2):301-307.
173 Sant S.M., Doran M., Fenelon H.M., Breatnach E.S. Pleuropulmonary abnormalities in patients with systemic lupus erythematosus: assessment with high resolution computed tomography, chest radiography and pulmonary function tests. Clin Exp Rheumatol. 1997;15(5):507-513.
174 Ooi G.C., Ngan H., Peh W.C., et al. Systemic lupus erythematosus patients with respiratory symptoms: the value of HRCT. Clin Radiol. 1997;52(10):775-781.
175 Susanto I., Peters J.I. Acute lupus pneumonitis with normal chest radiograph. Chest. 1997;111(6):1781-1783.
176 Anderson N.E., Ali M.R. The lupus anticoagulant, pulmonary thromboembolism, and fatal pulmonary hypertension. Ann Rheum Dis. 1984;43(5):760-763.
177 Howe H.S., Boey M.L., Fong K.Y., Feng P.H. Pulmonary haemorrhage, pulmonary infarction, and the lupus anticoagulant. Ann Rheum Dis. 1988;47(10):869-872.
178 Auger W.R., Permpikul P., Moser K.M. Lupus anticoagulant, heparin use, and thrombocytopenia in patients with chronic thromboembolic pulmonary hypertension: a preliminary report. Am J Med. 1995;99(4):392-396.
179 Urman J.D., Rothfield N.F. Corticosteroid treatment in systemic lupus erythematosus. Survival studies. JAMA. 1977;238(21):2272. 1176
180 Rosner S., Ginzler E.M., Diamond H.S., et al. A multicenter study of outcome in systemic lupus erythematosus. II. Causes of death. Arthritis Rheum. 1982;25(6):612-617.
181 Bohan A., Peter J.B., Bowman R.L., Pearson C.M. Computer-assisted analysis of 153 patients with polymyositis and dermatomyositis. Medicine (Baltimore). 1977;56(4):255-286.
182 Bohan A. History and classification of polymyositis and dermatomyositis. Clin Dermatol. 1988;6(2):3-8.
183 Nobutoh T., Kohda M., Doi Y., Ueki H. An autopsy case of dermatomyositis with rapidly progressive diffuse alveolar damage. J Dermatol. 1998;25(1):32-36.
184 Tazelaar H.D., Viggiano R.W., Pickersgill J., Colby T.V. Interstitial lung disease in polymyositis and dermatomyositis. Clinical features and prognosis as correlated with histologic findings. Am Rev Respir Dis. 1990;141(3):727-733.
185 Douglas W.W., Tazelaar H.D., Hartman T.E., et al. Polymyositis-dermatomyositis–associated interstitial lung disease. Am J Respir Crit Care Med. 2001;164(7):1182-1185.
186 Hepper N., Ferguson R.H., Howard F.M. Three types of pulmonary involvement in polymyositis. Med Clin North Am. 1964;48:1031-1042.
187 Benbassat J., Gefel D., Larholt K., et al. Prognostic factors in polymyositis/dermatomyositis. A computer-assisted analysis of ninety-two cases. Arthritis Rheum. 1985;28(3):249-255.
188 Ikezoe J., Johkoh T., Kohno N., et al. High-resolution CT findings of lung disease in patients with polymyositis and dermatomyositis. J Thorac Imaging. 1996;11(4):250-259.
189 Whaley K., Alspaugh M.A. Sjögren’s syndrome. In: Kelley H., Harris E.D.Jr, Ruddy S., Sledge C.B., editors. Textbook of Rheumatology. 2nd ed. Philadelphia: WB Saunders; 1985:956.
190 Baruch H.H., Firooznia H., Sackler J.P., et al. Pulmonary disorders associated with Sjögren’s syndrome. Rev Interam Radiol. 1977;2(2):77-81.
191 Franquet T., Giménez A., Monill J.M., et al. Primary Sjögren’s syndrome and associated lung disease: CT findings in 50 patients. AJR Am J Roentgenol. 1997;169(3):655-658.
192 Deheinzelin D., Capelozzi V.L., Kairalla R.A., et al. Interstitial lung disease in primary Sjögren’s syndrome. Clinical-pathological evaluation and response to treatment. Am J Respir Crit Care Med. 1996;154(3 Pt 1):794-799.
193 Reveille J.D., Wilson R.W., Provost T.T., et al. Primary Sjögren’s syndrome and other autoimmune diseases in families. Prevalence and immunogenetic studies in six kindreds. Ann Intern Med. 1984;101(6):748-756.
194 Meyer C.A., Pina J.S., Taillon D., Godwin J.D. Inspiratory and expiratory high-resolution CT findings in a patient with Sjögren’s syndrome and cystic lung disease. AJR Am J Roentgenol. 1997;168(1):101-103.
195 Hansen L.A., Prakash U.B., Colby T.V. Pulmonary lymphoma in Sjögren’s syndrome. Mayo Clin Proc. 1989;64(8):920-931.
196 Kokosi M., Riemer E.C., Highland K.B. Pulmonary involvement in Sjögren syndrome. Clin Chest Med. 2010;31(3):489-500.
197 Carrington C.B., et al. Chronic eosinophilic pneumonia. N Engl J Med. 1969;280:787-798.
198 Liebow A., Carrington C. The eosinophilic pneumonias. Medicine (Baltimore). 1969;48:251-285.
199 Gaensler E., Carrington C. Peripheral opacities in chronic eosinophilic pneumonia: the photographic negative of pulmonary edema. AJR Am J Roentgenol. 1977;128:1-13.
200 Mayo J., Müller N.L., Road J., et al. Chronic eosinophilic pneumonia: CT findings in six cases. AJR Am J Roentgenol. 1989;153:727-730.
201 Copper J.A.Jr. Drug-induced lung disease. Adv Intern Med. 1997;42:231-268.
202 Camus P.H., Foucher P., Bonniaud P.H., Ask K. Drug-induced infiltrative lung disease. Eur Respir J Suppl. 2001;32:93s-100s.
203 Ozkan M., Dweik R.A., Ahmad M. Drug-induced lung disease. Cleve Clin J Med. 2001;68(9):782-785. 789–795
204 Cuellar M.L. Drug-induced vasculitis. Curr Rheumatol Rep. 2002;4(1):55-59.
205 Ruangchira-Urai R., Colby T.V., Klein J., et al. Nodular amiodarone lung disease. Am J Surg Pathol. 2008;32(11):1654-1660.
206 Camus P., Lombard J.N., Perrichon M., et al. Bronchiolitis obliterans organising pneumonia in patients taking acebutolol or amiodarone. Thorax.. 1989;44(9):711-715.
207 Kennedy J.I., Myers J.L., Plumb V.J., Fulmer J.D. Amiodarone pulmonary toxicity. Clinical, radiologic, and pathologic correlations. Arch Intern Med. 1987;147(1):50-55.
208 Musk A.W., Pollard J.A. Pindolol and pulmonary fibrosis. BMJ. 1979;2(6190):581-582.
209 Sandler A., Gray R., Perry M.C., et al. Paclitaxel-carboplatin alone or with bevacizumab for non-small-cell lung cancer. N Engl J Med.. 2006;355(24):2542-2550.
210 Iacovino J.R., Leitner J., Abbas A.K., et al. Fatal pulmonary reaction from low doses of bleomycin. An idiosyncratic tissue response. JAMA. 1976;235(12):1253-1255.
211 Luna M.A., Bedrossian C.W., Lichtiger B., Salem P.A. Interstitial pneumonitis associated with bleomycin therapy. Am J Clin Pathol.. 1972;58(5):501-510.
212 Feingold M.L., Koss L.G. Effects of long-term administration of busulfan. Report of a patient with generalized nuclear abnormalities, carcinoma of vulva, and pulmonary fibrosis. Arch Intern Med. 1969;124(1):66-71.
213 Littler W.A., Kay J.M., Hasleton P.S., Heath D. Busulphan lung. Thorax. 1969;24(6):639-655.
214 Lombard C.M., Churg A., Winokur S. Pulmonary veno-occlusive disease following therapy for malignant neoplasms. Chest. 1987;92(5):871-876.
215 Mitsudo S.M., Greenwald E.S., Banerji B., Koss L.G. BCNU (1,3-bis-(2-chloroethyl)-1-nitrosurea) lung. Drug-induced pulmonary changes. Cancer. 1984;54(4):751-755.
216 Forrester J.M., Steele A.W., Waldron J.A., Parsons P.E. Crack lung: an acute pulmonary syndrome with a spectrum of clinical and histopathologic findings. Am Rev Respir Dis. 1990;142(2):462-467.
217 Murray R.J., Albin R.J., Mergner W., Criner G.J. Diffuse alveolar hemorrhage temporally related to cocaine smoking. Chest. 1988;93(2):427-429.
218 Abdel Karim F.W., Ayash R.E., Allam C., Salem P.A. Pulmonary fibrosis after prolonged treatment with low-dose cyclophosphamide. A case report. Oncology. 1983;40(3):174-176.
219 Patel A.R., Shah P.C., Rhee H.L., et al. Cyclophosphamide therapy and interstitial pulmonary fibrosis. Cancer. 1976;38(4):1542-1549.
220 Spector J.I., Zimbler H., Ross J.S. Early-onset cyclophosphamide-induced interstitial pneumonitis. JAMA. 1979;242(26):2852-2854.
221 Slavin R.E., Millan J.C., Mullins G.M. Pathology of high dose intermittent cyclophosphamide therapy. Hum Pathol. 1975;6(6):693-709.
222 Read W.L., Mortimer J.E., Picus J. Severe interstitial pneumonitis associated with docetaxel administration. Cancer. 2002;94(3):847-853.
223 Pfitzenmeyer P., Foucher P., Dennewald G., et al. Pleuropulmonary changes induced by ergoline drugs. Eur Respir J. 1996;9(5):1013-1019.
224 Liu V., White D.A., Zakowski M.F., et al. Pulmonary toxicity associated with erlotinib. Chest. 2007;132(3):1042-1044.
225 Gurjal A., An T., Valdivieso M., Kalemkerian G.P. Etoposide-induced pulmonary toxicity. Lung Cancer. 1999;26(2):109-112.
226 Inoue A., Saijo Y., Maemondo M., et al. Severe acute interstitial pneumonia and gefitinib. Lancet. 2003;361(9352):137-139.
227 Briasoulis E., Froudarakis M., Milionis H.J., et al. Chemotherapy-induced noncardiogenic pulmonary edema related to gemcitabine plus docetaxel combination with granulocyte colony-stimulating factor support. Respiration. 2000;67(6):680-683.
228 Tomioka R., King T.E.Jr. Gold-induced pulmonary disease: clinical features, outcome, and differentiation from rheumatoid lung disease. Am J Respir Crit Care Med. 1997;155(3):1011-1020.
229 Warnock M.L., Ghahremani G.G., Rattenborg C., et al. Pulmonary complication of heroin intoxication. Aspiration pneumonia and diffuse bronchiectasis. JAMA. 1972;219(8):1051-1053.
230 Karne S., D’Ambrosio C., Einarsson O., O’Connor P.G. Hypersensitivity pneumonitis induced by intranasal heroin use. Am J Med. 1999;107(4):392-395.
231 Petersen A.G., Dodge M., Helwig F.C. Pulmonary changes associated with hexamethonium therapy. Arch Intern Med. 1959;103:285-288.
232 Perry H.M.Jr, O’Neal R.M., Thomas W.A. Pulmonary disease following chronic chemical ganglionic blockade. Am J Med. 1957;22:37-50.
233 Beaudry C., Laplante L. Severe allergic pneumonitis from hydrochlorothiazide. Ann Intern Med. 1973;78(2):251-253.
234 Kaufman A., Montilla E., Helfgott M., et al. Pneumonitis and hydrochlorothiazide. Ann Intern Med. 1973;79(2):282-283.
235 Dorn M.R., Walker B.K. Noncardiogenic pulmonary edema associated with hydrochlorothiazide therapy. Chest. 1981;79(4):482-483.
236 Tengstrand B., Ernestam S., Engvall I.L., et al. TNF blockade in rheumatoid arthritis can cause severe fibrosing alveolitis. Six case reports. Lakartidningen. 2005;102(49):3788-3790. 3793
237 Cordonnier C., Vernant J.P., Mital P., et al. Pulmonary fibrosis subsequent to high doses of CCNU for chronic myeloid leukemia. Cancer. 1983;51(10):1814-1818.
238 Myers J.L. Pathology of drug-induced lung disease. In: Katzenstein A.L., editor. Katzenstein and Askin’s Surgical Pathology of Non-Neoplastic Lung Disease. Philadelphia: WB Saunders; 1997:81-111.
239 Imokawa S., Colby T.V., Leslie K.O., Helmers R.A. Methotrexate pneumonitis: review of the literature and histopathological findings in nine patients. Eur Respir J. 2000;15(2):373-381.
240 Bentur L., Bar-Kana Y., Livni E., et al. Severe minocycline-induced eosinophilic pneumonia: extrapulmonary manifestations and the use of in vitro immunoassays. Ann Pharmacother. 1997;31(6):733-735.
241 Piperno D., Donné C., Loire R., Cordier J.F. Bronchiolitis obliterans organizing pneumonia associated with minocycline therapy: a possible cause. Eur Respir J. 1995;8(6):1018-1020.
242 Waldhorn R.E., Tsou E., Smith F.P., Kerwin D.M. Pulmonary veno-occlusive disease associated with microangiopathic hemolytic anemia and chemotherapy of gastric adenocarcinoma. Med Pediatr Oncol. 1984;12(6):394-396.
243 Fielding J.W., Crocker J., Stockley R.A., Brookes V.S. Interstitial fibrosis in a patient treated with 5-fluorouracil and mitomycin C. BMJ. 1979;2(6189):551-552.
244 Israel K.S., Brashear R.E., Sharma H.M., et al. Pulmonary fibrosis and nitrofurantoin. Am Rev Respir Dis. 1973;108(2):353-356.
245 Geller M., Dickie H.A., Kass D.A., et al. The histopathology of acute nitrofurantoin-associated pneumonitis. Ann Allergy. 1976;37(4):275-279.
246 Magee F., Wright J.L., Chan N., et al. Two unusual pathological reactions to nitrofurantoin: case reports. Histopathology. 1986;10(7):701-706.
247 Ayoub J.P., North L., Greer J., et al. Pulmonary changes in patients with lymphoma who receive paclitaxel. J Clin Oncol. 1997;15(6):2476.
248 Epler G.R., Snider G.L., Gaensler E.A., et al. Bronchiolitis and bronchitis in connective tissue disease. A possible relationship to the use of penicillamine. JAMA. 1979;242(6):528-532.
249 Sternlieb I., Bennett B., Scheinberg I.H. D-Penicillamine induced Goodpasture’s syndrome in Wilson’s disease. Ann Intern Med. 1975;82(5):673-676.
250 Davies D., Jones J.K. Pulmonary eosinophilia caused by penicillamine. Thorax. 1980;35(12):957-958.
251 Chamberlain D.W., Hyland R.H., Ross D.J. Diphenylhydantoin-induced lymphocytic interstitial pneumonia. Chest. 1986;90(3):458-460.
252 Michael J.R., Rudin M.L. Acute pulmonary disease caused by phenytoin. Ann Intern Med. 1981;95(4):452-454.
253 Dohner V.A., Ward H.P., Standord R.E. Alveolitis during procarbazine, vincristine and cyclophosphamide therapy. Chest. 1972;62(5):636-639.
254 Farney R.J., Morris A.H., Armstrong J.D., Hammer S. Diffuse pulmonary disease after therapy with nitrogen mustard, vincristine, procarbazine, and prednisone. Am Rev Respir Dis. 1977;115(1):135-145.
255 Horton L.W., Chappell A.G., Powell D.E. Diffuse interstitial pulmonary fibrosis complicating Hodgkin’s disease. Br J Dis Chest. 1977;71(1):44-48.
256 Gonzalez-Rothi R.J., Zander D.S., Ros P.R. Fluoxetine hydrochloride (Prozac)-induced pulmonary disease. Chest. 1995;107(6):1763-1765.
257 Burton C., Kaczmarski R., Jan-Mohamed R. Interstitial pneumonitis related to rituximab therapy. N Engl J Med. 2003;348(26):2690-2691. discussion 2690–2691
258 Parry S.D., et al. Sulphasalazine and lung toxicity. Eur Respir J. 2002;19(4):756-764.
259 Travis W.D., Kalafer M.E., Robin H.S., Luibel F.J. Hypersensitivity pneumonitis and pulmonary vasculitis with eosinophilia in a patient taking an L-tryptophan preparation. Ann Intern Med. 1990;112(4):301-303.
260 Catton C.K., Elmer J.C., Whitehouse A.C., et al. Pulmonary involvement in the eosinophilia-myalgia syndrome. Chest. 1991;99(2):327-329.
261 Calvo D.B.3rd, Legha S.S., McKelvey E.M., et al. Zinostatin-related pulmonary toxicity. Cancer Treat Rep. 1981;65(1–2):165-167.
262 Zitnik R.J., Matthay R.A. Drug-induced lung disease. In: Schwartz M.I., King T.E., editors. Interstitial Lung Disease. London: BC Decker; 1998:423-449.
263 Kudoh S., Kato H., Nishiwaki Y., et al. Interstitial lung disease in Japanese patients with lung cancer: a cohort and nested case-control study. Am J Respir Crit Care Med.. 2008;177(12):1348-1357.
264 Sostman H.D., Matthay R.A., Putman C.E., Smith G.J. Methotrexate-induced pneumonitis. Medicine (Baltimore). 1976;55(5):371-388.
265 Goodman T.A., Polisson R.P. Methotrexate: adverse reactions and major toxicities. Rheum Dis Clin North Am. 1994;20(2):513-528.
266 Bedrossian C.W., Miller W.C., Luna M.A. Methotrexate-induced diffuse interstitial pulmonary fibrosis. South Med J. 1979;72(3):313-318.
267 Zisman D.A., McCune W.J., Tino G., Lynch J.P.3rd. Drug-induced pneumonitis: the role of methotrexate. Sarcoidosis Vasc Diffuse Lung Dis. 2001;18(3):243-252.
268 Kennedy J.I., Myers J.L., Plumb V.J., Fulmer J.D. Amiodarone pulmonary toxicity. Clinical, radiologic, and pathologic correlations. Arch Intern Med. 1987;147(1):50-55.
269 Dusman R.E., Stanton M.S., Miles W.M., et al. Clinical features of amiodarone-induced pulmonary toxicity. Circulation. 1990;82(1):51-59.
270 Newman L.S., Rose C.S., Bresnitz E.A., et al. A case control etiologic study of sarcoidosis: environmental and occupational risk factors. Am J Respir Crit Care Med. 2004;170(12):1324-1330.
271 Fraire A.E., Guntupalli K.K., Greenberg S.D., et al. Amiodarone pulmonary toxicity: a multidisciplinary review of current status. South Med J. 1993;86(1):67-77.
272 Nicholson A.A., Hayward C. The value of computed tomography in the diagnosis of amiodarone-induced pulmonary toxicity. Clin Radiol. 1989;40(6):564-567.
273 Kuhlman J.E., Teigen C., Ren H., et al. Amiodarone pulmonary toxicity: CT findings in symptomatic patients. Radiology. 1990;177(1):121-125.
274 Myers J.L., Kennedy J.I., Plumb V.J. Amiodarone lung: Pathologic findings in clinically toxic patients. Hum Pathol. 1987;18(4):349-354.
275 Martin W.J.2nd, Rosenow E.C.3rd. Amiodarone pulmonary toxicity. Recognition and pathogenesis (Part I). Chest. 1988;93(5):1067-1075.
276 Martin W.J.2nd, Rosenow E.C.3rd. Amiodarone pulmonary toxicity. Recognition and pathogenesis (Part 2). Chest. 1988;93(6):1242-1248.
277 Gonzalez-Rothi R.J., Hannan S.E., Hood C.I., Franzini D.A. Amiodarone pulmonary toxicity presenting as bilateral exudative pleural effusions. Chest. 1987;92(1):179-182.
278 Ruangchira-Urai R., Colby T.V., Klein J., et al. Nodular amiodarone lung disease. Am J Surg Pathol.. 2008;32(11):1654-1660.
279 Wood D.L., Osborn M.J., Rooke J., Holmes D.R.Jr. Amiodarone pulmonary toxicity: report of two cases associated with rapidly progressive fatal adult respiratory distress syndrome after pulmonary angiography. Mayo Clin Proc. 1985;60(9):601-603.
280 Van Mieghem W., Coolen L., Malysse I., et al. Amiodarone and the development of ARDS after lung surgery. Chest. 1994;105(6):1642-1645.
281 Durant J.R., Norgard M.J., Murad T.M., et al. Pulmonary toxicity associated with bischloroethylnitrosourea (BCNU). Ann Intern Med. 1979;90(2):191-194.
282 Lieberman A., Ruoff M., Estey E., et al. Irreversible pulmonary toxicity after single course of BCNU. Am J Med Sci. 1980;279(1):53-56.
283 Litam J.P., Dail D.H., Spitzer G., et al. Early pulmonary toxicity after administration of high-dose BCNU. Cancer Treat Rep. 1981;65(1–2):39-44.
284 Cao T.M., Negrin R.S., Stockerl-Goldstein K.E., et al. Pulmonary toxicity syndrome in breast cancer patients undergoing BCNU-containing high-dose chemotherapy and autologous hematopoietic cell transplantation. Biol Blood Marrow Transplant. 2000;6(4):387-394.
285 Holoye P.Y., Jenkins D.E., Greenberg S.D. Pulmonary toxicity in long-term administration of BCNU. Cancer Treat Rep. 1976;60(11):1691-1694.
286 O’Driscoll B.R., Hasleton P.S., Taylor P.M., et al. Active lung fibrosis up to 17 years after chemotherapy with carmustine (BCNU) in childhood. N Engl J Med. 1990;323(6):378-382.
287 Parish J.M., Muhm J.R., Leslie K.O. Upper lobe pulmonary fibrosis associated with high-dose chemotherapy containing BCNU for bone marrow transplantation. Mayo Clin Proc. 2003;78(5):630-634.
288 Littler W., Kay J.M., Hasleton P.S., Heath D. Busulphan lung. Thorax. 1969;24(6):639-655.
289 Cooper J.A.Jr, White D.A., Matthay R.A. Drug-induced pulmonary disease. Part 1: Cytotoxic drugs. Am Rev Respir Dis. 1986;133(2):321-340.
290 Kreisman H., Wolkove N. Pulmonary toxicity of antineoplastic therapy. Semin Oncol. 1992;19(5):508-520.
291 Holoye P.Y., Luna M.A., MacKay B., Bedrossian C.W. Bleomycin hypersensitivity pneumonitis. Ann Intern Med. 1978;8:47-49.
292 Hay J.G., Haslam P.L., Dewar A., et al. Development of acute lung injury after the combination of intravenous bleomycin and exposure to hyperoxia in rats. Thorax. 1987;42(5):374-382.
293 Borzone G., Moreno R., Urrea R., et al. Bleomycin-induced chronic lung damage does not resemble human idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2001;163(7):1648-1653.
294 Lien H.H., Brodahl U., Telhaug R., et al. Pulmonary changes at computed tomography in patients with testicular carcinoma treated with cis-platinum, vinblastine and bleomycin. Acta Radiol Diagn (Stockh). 1985;26(5):507-510.
295 Adler K.B., Callahan L.M., Evans J.N. Cellular alterations in the alveolar wall in bleomycin-induced pulmonary fibrosis in rats. An ultrastructural morphometric study. Am Rev Respir Dis. 1986;133(6):1043-1048.
296 Lazo J.S. Bleomycin. Cancer Chemother Biol Response Modif. 1999;18:39-45.
297 Bowler R.P., Nicks M., Warnick K., Crapo J.D. Role of extracellular superoxide dismutase in bleomycin-induced pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2002;282(4):L719-L726.
298 Taatjes D.J., Leslie K.O., von Turkovich M., et al. Alveolocapillary remodeling in bleomycin-induced rat lung injury: interpretation from lectin-binding studies. Prog Histochem Cytochem. 1991;23(1–4):194-199.
299 Cohen M.H., Williams G.A., Sridhara R., et al. FDA drug approval summary: gefitinib (ZD1839) (Iressa) tablets. Oncologist. 2003;8(4):303-306.
300 Ando M., Okamoto I., Yamamoto N., et al. Predictive factors for interstitial lung disease, antitumor response, and survival in non-small-cell lung cancer patients treated with gefitinib. J Clin Oncol. 2006;24(16):2549-2556.
301 Ostör A.J., Chilvers E.R., Somerville M.F., et al. Pulmonary complications of infliximab therapy in patients with rheumatoid arthritis. J Rheumatol. 2006;33(3):622-628.
302 Stern W., Subbarao K. Pulmonary complications of drug addiction. Semin Roentgenol. 1983;28:183-197.
303 Abraham J., Brambilla C. Particle size for differentiation between inhalation and injection pulmonary talcosis. Environ Res. 1980;21(1):94-96.
304 Abraham J.L. Identification and quantitative analysis of tissue particulate burden. Ann N Y Acad Sci. 1984;428:60-67.
305 Abraham J.L., Burnett B.R., Hunt A. Development and use of a pneumoconiosis database of human pulmonary inorganic particulate burden in over 400 lungs. Scanning Microsc. 1991;5(1):95-104. discussion 105–108
306 Tomashefski J., Hirsch C. The pulmonary vascular lesions of intravenous drug abuse. Hum Pathol. 1980;11:133-145.
307 Crouch E., Churg A. Progressive massive fibrosis of the lung secondary to intravenous injection of talc. A pathologic and mineralogical analysis. Am J Clin Pathol. 1983;80:520-526.
308 Stern E.J., Frank M.S., Schmutz J.F., et al. Panlobular pulmonary emphysema caused by i.v. injection of methylphenidate (Ritalin): findings on chest radiographs and CT scans. AJR Am J Roentgenol. 1994;162:555-560.
309 Schiavina M., Di Scioscio V., Contini P., et al. Pulmonary lymphangioleiomyomatosis in a karyotypically normal man without tuberous sclerosis complex. Am J Respir Crit Care Med. 2007;176(1):96-98.
310 Cheung O.Y., Muhm J.R., Helmers R.A., et al. Surgical pathology of granulomatous interstitial pneumonia. Ann Diagn Pathol. 2003;7(2):127-138.
311 Hunninghake G.W., Costabel U., Ando M., et al. ATS/ERS/WASOG statement on sarcoidosis. American Thoracic Society/European Respiratory Society/World Association of Sarcoidosis and other Granulomatous Disorders. Sarcoidosis Vasc Diffuse Lung Dis. 1999;16(2):149-173.
312 Stirling R.G., Cullinan P., Du Bois R.M.P. Sarcoidosis. In: Schwartz M.I., King T.E., editors. Interstitial Lung Disease. London: BC Decker; 1998:279-322.
313 Milman N., Selroos O. Pulmonary sarcoidosis in the Nordic countries 1950–1982. Epidemiology and clinical picture. Sarcoidosis. 1990;7(1):50-57.
314 Milman N., Selroos O. Pulmonary sarcoidosis in the Nordic countries 1950–1982. II. Course and prognosis. Sarcoidosis. 1990;7(2):113-118.
315 Sartwell P.E., Edwards L.B. Epidemiology of sarcoidosis in the U.S. Navy. Am J Epidemiol. 1974;99(4):250-257.
316 Lynch D.A., Webb W.R., Gamsu G., et al. Computed tomography in pulmonary sarcoidosis. J Comput Assist Tomogr. 1989;13:405-410.
317 Kuhlman J.E., Fishman E.K., Hamper U.M., et al. The computed tomographic spectrum of thoracic sarcoidosis. Radiographics. 1989;9(3):449-466.
318 Müller N.L., Kullnig P., Miller R.R. The CT findings of pulmonary sarcoidosis: analysis of 25 patients. AJR Am J Roentgenol. 1989;152(6):1179-1182.
319 Padley S.P., Padhani A.R., Nicholson A., Hansell D.M. Pulmonary sarcoidosis mimicking cryptogenic fibrosing alveolitis on CT. Clin Radiol. 1996;51(11):807-810.
320 Abehsera M., Valeyre D., Grenier P., et al. Sarcoidosis with pulmonary fibrosis: CT patterns and correlation with pulmonary function. AJR Am J Roentgenol. 2000;174(6):1751-1757.
321 Battesti J.P., Saumon G., Valeyre D., et al. Pulmonary sarcoidosis with an alveolar radiographic pattern. Thorax. 1982;37:448-452.
322 Gilman M.J., Wang K.P. Transbronchial lung biopsy in sarcoidosis. An approach to determine the optimal number of biopsies. Am Rev Respir Dis. 1980;122(5):721-724.
323 Hsu R.M., Connors A.F.Jr, Tomashefski J.F.Jr. Histologic, microbiologic, and clinical correlates of the diagnosis of sarcoidosis by transbronchial biopsy. Arch Pathol Lab Med. 1996;120(4):364-368.
324 Freiman D., Hardy H. Beryllium disease: the relation of pulmonary pathology to clinical course and prognosis based on a study of 130 cases from the U.S. Beryllium Case Registry. Hum Pathol. 1970;1:25-44.
325 Matilla A., Galera H., Pascual E., Carapeto R. Chronic berylliosis. Br J Dis Chest. 1973;67:308-314.
326 Aronchik J.M., Rossman M.D., Miller W.T. Chronic beryllium disease: diagnosis, radiographic findings, and correlation with pulmonary function tests. Radiology. 1987;163:677-678.
327 Kriebel D., Brain J.D., Sprince N.L., Kazemi H. The pulmonary toxicity of beryllium. Am Rev Respir Dis. 1988;137(2):464-473.
328 Kriebel D., Sprince N.L., Eisen E.A., Greaves I.A. Pulmonary function in beryllium workers: assessment of exposure. Br J Ind Med. 1988;45(2):83-92.
329 Kriebel D., Sprince N.L., Eisen E.A., et al. Beryllium exposure and pulmonary function: a cross sectional study of beryllium workers. Br J Ind Med. 1988;45(3):167-173.
330 Maier L.A., Martyny J.W., Liang J., Rossman M.D. Recent chronic beryllium disease in residents surrounding a beryllium facility. Am J Respir Crit Care Med. 2008;177(9):1012-1017.
331 Kawanami O., Basset F., Barrios R., et al. Hypersensitivity pneumonitis in man: light- and electron-microscopic studies of 18 lung biopsies. Am J Pathol. 1983;110:275-289.
332 Salvaggio J., Karr R. Hypersensitivity pneumonitis: state of the art. Chest. 1979;75(suppl 2):270-274.
333 Wild L.G., Lopez M. Hypersensitivity pneumonitis: a comprehensive review. J Investig Allergol Clin Immunol. 2001;11(1):3-15.
334 Bourke S.J., Dalphin J.C., Boyd G., et al. Hypersensitivity pneumonitis: current concepts. Eur Respir J Suppl. 2001;32:81s-92s.
335 Selman M. Hypersensitivity pneumonitis. In: Schwartz M.I., King T.E., editors. Interstitial Lung Disease. London: BC Decker; 1998:393-422.
336 McSharry C., Banham S.W., Boyd G. Effect of cigarette smoking on the antibody response to inhaled antigens and the prevalence of extrinsic allergic alveolitis among pigeon breeders. Clin Allergy. 1985;15:487.
337 Patel A.M., Ryu J.H., Reed C.E. Hypersensitivity pneumonitis: current concepts and future questions. J Allergy Clin Immunol. 2001;108(5):661-670.
338 Glazer C., Rose C., Lynch D. Clinical and radiologic manifestations of hypersensitivity pneumonitis. J Thorac Imaging. 2002;17(4):261-272.
339 Cook P.G., Wells I.P., McGavin C.R. The distribution of pulmonary shadowing in farmer’s lung. Clin Radiol.. 1988;39(1):21-27.
340 Khoor A., Leslie K.O., Tazelaar H.D., et al. Diffuse pulmonary disease caused by nontuberculous mycobacteria in immunocompetent people (hot tub lung). Am J Clin Pathol. 2001;115(5):755-762.
341 Pham R., Vydareny K., Gal A. High-resolution computed tomography appearance of pulmonary Mycobacterium avium complex infection after exposure to hot tub: case of hot-tub lung. J Thorac Imaging. 2003;18(1):48-52.
342 Coleman A., Colby T.V. Histologic diagnosis of extrinsic allergic alveolitis. Am J Surg Pathol. 1988;12(7):514-518.
343 Colby T.V., Coleman A. Histologic differential diagnosis of extrinsic allergic alveolitis. Prog Surg Pathol. 1989;10:11-26.
344 Seal R.M., Hapke E.J., Thomas G.O., et al. The pathology of the acute and chronic stages of farmer’s lung. Thorax. 1968;23(5):469-489.
345 Takemura T., Akashi T., Ohtani Y., et al. Pathology of hypersensitivity pneumonitis. Curr Opin Pulm Med. 2008;14(5):440-454.
346 Barbee R.A., Callies Q., Dickie H.A., Rankin J. The long-term prognosis in farmer’s lung. Am Rev Respir Dis. 1968;97(2):223-231.
347 Kokkarinen J., Tukiainen H., Seppä A., Terho E.O. Hypersensitivity pneumonitis due to native birds in a bird ringer. Chest. 1994;106(4):1269-1271.
348 Kokkarinen J., Tukiainen H., Terho E.O. Mortality due to farmer’s lung in Finland. Chest. 1994;106(2):509-512.
349 Kokkarinen J.I., Tukiainen H.O., Terho E.O. Effect of corticosteroid treatment on the recovery of pulmonary function in farmer’s lung. Am Rev Respir Dis. 1992;145(1):3-5.
350 Sansores R., Salas J., Chapela R., et al. Clubbing in hypersensitivity pneumonitis. Its prevalence and possible prognostic role. Arch Intern Med. 1990;150(9):1849-1851.
351 Pérez-Padilla R., Gaxiola M., Salas J., et al. Bronchiolitis in chronic pigeon breeder’s disease. Morphologic evidence of a spectrum of small airway lesions in hypersensitivity pneumonitis induced by avian antigens. Chest. 1996;110(2):371-377.
352 Brabencova E., Tazi A., Lorenzato M., et al. Langerhans cells in Langerhans cell granulomatosis are not actively proliferating cells. Am J Pathol. 1998;152(5):1143-1149.
353 Vassallo R., Ryu J.H., Colby T.V., et al. Pulmonary Langerhans’-cell histiocytosis. N Engl J Med. 2000;342(26):1969-1978.
354 Willman C.L., Busque L., Griffith B.B., et al. Langerhans’-cell histiocytosis (histiocytosis X)—a clonal proliferative disease. N Engl J Med. 1994;331(3):154-160.
355 Travis W.D., Borok Z., Roum J.H., et al. Pulmonary Langerhans cell granulomatosis (histiocytosis X). A clinicopathologic study of 48 cases. Am J Surg Pathol. 1993;17(10):971-986.
356 Parambil J.G., Myers J.L., Aubry M.C., Ryu J.H. Causes and prognosis of diffuse alveolar damage diagnosed on surgical lung biopsy. Chest. 2007;132(1):50-57.
357 Colby T.V., Lombard C. Histiocytosis X in the lung. Hum Pathol. 1983;14(10):847-856.
358 Niku S., Stark P., Levin D.L., Friedman P.J. Lymphangioleiomyomatosis: clinical, pathologic, and radiologic manifestations. J Thorac Imaging. 2005;20(2):98-102.
359 Lacronique J., Roth C., Battesti J.P., et al. Chest radiological features of pulmonary histiocytosis X: a report based on 50 adult cases. Thorax. 1982;37(2):104-109.
360 Moore A.D., Godwin J.D., Müller N.L., et al. Pulmonary histiocytosis X: comparison of radiographic and CT findings. Radiology. 1989;172(1):249-254.
361 Kulwiec E.L., Lynch D.A., Aguayo S.M., et al. Imaging of pulmonary histiocytosis X. Radiographics. 1992;12(3):515-526.
362 Brauner M.W., Grenier P., Tijani K., et al. Pulmonary Langerhans cell histiocytosis: evolution of lesions on CT scans. Radiology. 1997;204(2):497-502.
363 Kambouchner M., Basset F., Marchal J., et al. Three-dimensional characterization of pathologic lesions in pulmonary Langerhans cell histiocytosis. Am J Respir Crit Care Med. 2002;166(11):1419-1421.
364 McDonnell T.J., Crouch E.C., Gonzalez J.G. Reactive eosinophilic pleuritis. A sequela of pneumothorax in pulmonary eosinophilic granuloma. Am J Clin Pathol. 1989;91(1):107-111.
365 Askin F.B., McCann B.G., Kuhn C. Reactive eosinophilic pleuritis: a lesion to be distinguished from pulmonary eosinophilic granuloma. Arch Pathol Lab Med. 1977;101:187-192.
366 Delobbe A., Durieu J., Duhamel A., Wallaert B. Determinants of survival in pulmonary Langerhans’ cell granulomatosis (histiocytosis X). Groupe d’Etude en Pathologie Interstitielle de la Societe de Pathologie Thoracique du Nord. Eur Respir J. 1996;9(10):2002-2006.
367 Veyssier-Belot C., Cacoub P., Caparros-Lefebvre D., et al. Erdheim-Chester disease. Clinical and radiologic characteristics of 59 cases. Medicine (Baltimore). 1996;75(3):157-169.
368 Cottin V., Nunes H., Brillet P.Y., et al. Combined pulmonary fibrosis and emphysema: a distinct underrecognised entity. Eur Respir J. 2005;26(4):586-593.
369 Bisceglia M., Cammisa M., Suster S., Colby T.V. Erdheim-Chester disease: clinical and pathologic spectrum of four cases from the Arkadi M. Rywlin slide seminars. Adv Anat Pathol. 2003;10(3):160-171.
370 Bancroft L.W., Berquist T.H. Erdheim-Chester disease: radiographic findings in five patients. Skeletal Radiol. 1998;27(3):127-132.
371 Breuil V., Brocq O., Pellegrino C., et al. Erdheim-Chester disease: typical radiological bone features for a rare xanthogranulomatosis. Ann Rheum Dis. 2002;61(3):199-200.
372 Gottlieb R., Chen A. MR findings of Erdheim-Chester disease. J Comput Assist Tomogr. 2002;26(2):257-261.
373 Olmos J.M., Canga A., Velero C., González-Macías J. Imaging of Erdheim-Chester disease. J Bone Miner Res. 2002;17(3):381-383.
374 Kambouchner M., Colby T.V., Domenge C., et al. Erdheim-Chester disease with prominent pulmonary involvement associated with eosinophilic granuloma of mandibular bone. Histopathology. 1997;30(4):353-358.
375 Egan A.J., Boardman L.A., Tazelaar H.D., et al. Erdheim-Chester disease: clinical, radiologic, and histopathologic findings in five patients with interstitial lung disease. Am J Surg Pathol. 1999;23(1):17-26.
376 Rush W.L., Andriko J.A., Galateau-Salle F., et al. Pulmonary pathology of Erdheim-Chester disease. Mod Pathol. 2000;13(7):747-754.
377 Wittenberg K.H., Swensen S.J., Myers J.L. Pulmonary involvement with Erdheim-Chester disease: radiographic and CT findings. AJR Am J Roentgenol. 2000;174(5):1327-1331.
378 Shamburek R.D., Brewer H.B.Jr, Gochuico B.R. Erdheim-Chester disease: a rare multisystem histiocytic disorder associated with interstitial lung disease. Am J Med Sci. 2001;321(1):66-75.
379 Taylor J.R., Ryu J., Colby T.V., Raffin T.A. Lymphangioleiomyomatosis. Clinical course in 32 patients. N Engl J Med. 1990;323(18):1254-1260.
380 Kitaichi M., Nishimura K., Itoh H., Izumi T. Pulmonary lymphangioleiomyomatosis: a report of 46 patients including a clinicopathologic study of prognostic factors. Am J Respir Crit Care Med. 1995;151(2 Pt 1):527-533.
381 Kalassian K.G., Doyle R., Kao P., et al. Lymphangioleiomyomatosis: new insights. Am J Respir Crit Care Med. 1997;155(4):1183-1186.
382 Athavale A., Chhajed P., Singhal P., et al. Pulmonary lymphangioleiomyomatosis. J Assoc Physicians India. 1999;47(6):649-650.
383 Urban T., Lazor R., Lacronique J., et al. Pulmonary lymphangioleiomyomatosis. A study of 69 patients. Groupe d’Etudes et de Recherche sur les Maladies “Orphelines” Pulmonaires (GERM“O”P). Medicine (Baltimore). 1999;78(5):321-337.
384 Chu S.C., Horiba K., Usuki J., et al. Comprehensive evaluation of 35 patients with lymphangioleiomyomatosis. Chest. 1999;115(4):1041-1052.
385 Costello L.C., Hartman T.E., Ryu J.H. High frequency of pulmonary lymphangioleiomyomatosis in women with tuberous sclerosis complex. Mayo Clin Proc. 2000;75(6):591-594.
386 Sato T., Seyama K., Fujii H., et al. Mutation analysis of the TSC1 and TSC2 genes in Japanese patients with pulmonary lymphangioleiomyomatosis. J Hum Genet. 2002;47(1):20-28.
387 Yu J., Astrinidis A., Henske E.P. Chromosome 16 loss of heterozygosity in tuberous sclerosis and sporadic lymphangiomyomatosis. Am J Respir Crit Care Med. 2001;164(8 Pt 1):1537-1540.
388 Carsillo T., Astrinidis A., Henske E.P. Mutations in the tuberous sclerosis complex gene TSC2 are a cause of sporadic pulmonary lymphangioleiomyomatosis. Proc Natl Acad Sci U S A. 2000;97(11):6085-6090.
389 Aubry M.C., Myers J.L., Ryu J.H., et al. Pulmonary lymphangioleiomyomatosis in a man. Am J Respir Crit Care Med. 2000;162(2 Pt 1):749-752.
390 Lack E.E., Dolan M.F., Finisio J., et al. Pulmonary and extrapulmonary lymphangioleiomyomatosis. Report of a case with bilateral renal angiomyolipomas, multifocal lymphangioleiomyomatosis, and a glial polyp of the endocervix. Am J Surg Pathol. 1986;10(9):650-657.
391 Uzzo R.G., Libby D.M., Vaughan E.D.Jr, Levey S.H. Coexisting lymphangioleiomyomatosis and bilateral angiomyolipomas in a patient with tuberous sclerosis. J Urol. 1994;151(6):1612-1615.
392 Bernstein S.M., Newell J.D.Jr, Adamczyk D., et al. How common are renal angiomyolipomas in patients with pulmonary lymphangiomyomatosis? Am J Respir Crit Care Med. 1995;152(6 Pt 1):2138-2143.
393 Maziak D.E., Kesten S., Rappaport D.C., Maurer J. Extrathoracic angiomyolipomas in lymphangioleiomyomatosis. Eur Respir J. 1996;9(3):402-405.
394 Neumann H.P., Schwarzkopf G., Henske E.P. Renal angiomyolipomas, cysts, and cancer in tuberous sclerosis complex. Semin Pediatr Neurol. 1998;5(4):269-275.
395 Tüzel E., Kirkali Z., Mungan U., et al. Giant angiomyolipoma associated with marked pulmonary lesions suggesting lymphangioleiomyomatosis in a patient with tuberous sclerosis. Int Urol Nephrol. 2000;32(2):219-222.
396 Müller N.L., Chiles C., Kullnig P. Pulmonary lymphangiomyomatosis: correlation of CT with radiographic and functional findings. Radiology. 1990;175(2):335-339.
397 Rappaport D.C., Weisbrod G.L., Herman S.J., Chamberlain D.W. Pulmonary lymphangioleiomyomatosis: high-resolution CT findings in four cases. AJR Am J Roentgenol. 1989;152(5):961-964.
398 Templeton P.A., McLoud T.C., Müller N.L., et al. Pulmonary lymphangioleiomyomatosis: CT and pathologic findings. J Comput Assist Tomogr. 1989;13(1):54-57.
399 Sherrier R.H., Chiles C., Roggli V. Pulmonary lymphangioleiomyomatosis: CT findings. AJR Am J Roentgenol. 1989;153(5):937-940.
400 Templeton P.A., McLoud T.C., Müller N.L., et al. Pulmonary lymphangioleiomyomatosis: CT and pathologic findings. J Comput Assist Tomogr. 1989;13:54-57.
401 Kirchner J., Stein A., Viel K., et al. Pulmonary lymphangioleiomyomatosis: high-resolution CT findings. Eur Radiol. 1999;9(1):49-54.
402 Kumasaka T., Seyama K., Mitani K., et al. Lymphangiogenesis-mediated shedding of LAM cell clusters as a mechanism for dissemination in lymphangioleiomyomatosis. Am J Surg Pathol. 2005;29(10):1356-1366.
403 Toro J.R., Wei M.H., Glenn G.M., et al. BHD mutations, clinical and molecular genetic investigations of Birt-Hogg-Dubé syndrome: a new series of 50 families and a review of published reports. J Med Genet.. 2008;45(6):321-331.
404 Shen A., Iseman M.D., Waldron J.A., King T.E. Exacerbation of pulmonary lymphangioleiomyomatosis by exogenous estrogens. Chest. 1987;91(5):782-785.
405 Eliasson A.H., Phillips Y.Y., Tenholder M.F. Treatment of lymphangioleiomyomatosis. A meta-analysis. Chest. 1989;96(6):1352-1355.
406 Ohori N.P., Yousem S.A., Sonmez-Alpan E., Colby T.V. Estrogen and progesterone receptors in lymphangioleiomyomatosis, epithelioid hemangioendothelioma, and sclerosing hemangioma of the lung. Am J Clin Pathol. 1991;96(4):529-535.
407 Rajjoub S., Blatt M.W., Ritterspach J. Response to treatment with progesterone in a patient with pulmonary lymphangioleiomyomatosis. W V Med J. 1995;91(7):322-323.
408 Colley M.H., Geppert E., Franklin W.A. Immunohistochemical detection of steroid receptors in a case of pulmonary lymphangioleiomyomatosis. Am J Surg Pathol. 1989;13(9):803-807.
409 McCormack F.X. Lymphangioleiomyomatosis: a clinical update. Chest. 2008;133(2):507-516.
410 Boehler A., Speich R., Russi E.W., Weder W. Lung transplantation for lymphangioleiomyomatosis. N Engl J Med. 1996;335(17):1275-1280.
411 Bittmann I., Dose T.B., Müller C., et al. Lymphangioleiomyomatosis: recurrence after single lung transplantation. Hum Pathol. 1997;28(12):1420-1423.
412 Gunji Y., Akiyoshi T., Sato T., et al. Mutations of the Birt Hogg Dube gene in patients with multiple lung cysts and recurrent pneumothorax. J Med Genet. 2007;44(9):588-593.
413 Davies B.H., Tuddenham E.G. Familial pulmonary fibrosis associated with oculocutaneous albinism and platelet function defect. A new syndrome. Q J Med. 1976;45(178):219-232.
414 DePinho R.A., Kaplan K.L. The Hermansky-Pudlak syndrome. Report of three cases and review of pathophysiology and management considerations. Medicine (Baltimore). 1985;64(3):192-202.
415 Dimson O., Drolet B.A., Esterly N.B. Hermansky-Pudlak syndrome. Pediatr Dermatol. 1999;16(6):475-477.
416 Huizing M., Gahl W.A. Disorders of vesicles of lysosomal lineage: the Hermansky-Pudlak syndromes. Curr Mol Med. 2002;2(5):451-467.
417 Pierson D.M., Ionescu D., Qing G., et al. Pulmonary fibrosis in Hermansky-Pudlak syndrome. A case report and review. Respiration. 2006;73(3):382-395.
418 Anikster Y., Huizing M., White J., et al. Mutation of a new gene causes a unique form of Hermansky-Pudlak syndrome in a genetic isolate of central Puerto Rico. Nat Genet. 2001;28(4):376-380.
419 Hermos C.R., Huizing M., Kaiser-Kupfer M.I., Gahl W.A. Hermansky-Pudlak syndrome type 1: gene organization, novel mutations, and clinical-molecular review of non–Puerto Rican cases. Hum Mutat. 2002;20(6):482.
420 Okano A., Sato A., Chida K., et al. Pulmonary interstitial pneumonia in association with Hermansky-Pudlak syndrome. Nihon Kyobu Shikkan Gakkai Zasshi. 1991;29(12):1596-1602.
421 Avila N.A., Brantly M., Premkumar A., et al. Hermansky-Pudlak syndrome: radiography and CT of the chest compared with pulmonary function tests and genetic studies. AJR Am J Roentgenol. 2002;179(4):887-892.
422 Nakatani Y., Nakamura N., Sano J., et al. Interstitial pneumonia in Hermansky-Pudlak syndrome: significance of florid foamy swelling/degeneration (giant lamellar body degeneration) of type-2 pneumocytes. Virchows Arch. 2000;437(3):304-313.
423 Gahl W.A., Brantly M., Troendle J., et al. Effect of pirfenidone on the pulmonary fibrosis of Hermansky-Pudlak syndrome. Mol Genet Metab. 2002;76(3):234-242.
424 Sosman M.C., Dodd G.D., Jones W.D., Pillmore G.U. The familial occurrence of pulmonary alveolar microlithiasis. Am J Roentgenol Radium Ther Nucl Med. 1957;77:947-952.
425 Fuleihan F.J., Abboud R.T., Balikian J.P., Nucho C.K. Pulmonary alveolar microlithiasis: lung function in five cases. Thorax. 1969;24:84-90.
426 Prakash U.B., Barham S.S., Rosenow E.C.3rd, et al. Pulmonary alveolar microlithiasis. A review including ultrastructural and pulmonary function studies. Mayo Clin Proc. 1983;58(5):290-300.
427 Edelweiss M., Medeiros L.J., Suster S., Moran C.A. Lymph node involvement by Langerhans cell histiocytosis: a clinicopathologic and immunohistochemical study of 20 cases. Hum Pathol. 2007;38(10):1463-1469.
428 Castellana G., Gentile M., Castellana R., et al. Pulmonary alveolar microlithiasis: clinical features, evolution of the phenotype, and review of the literature. Am J Med Genet. 2002;111(2):220-224.
429 Senyiğit A., Yaramiş A., Gürkan F., et al. Pulmonary alveolar microlithiasis: a rare familial inheritance with report of six cases in a family. Contribution of six new cases to the number of case reports in Turkey. Respiration. 2001;68(2):204-209.
430 Huqun I.S., Miyazawa H,., Ishii K., et al. Mutations in the SLC34A2 gene are associated with pulmonary alveolar microlithiasis. Am J Respir Crit Care Med. 2007;175(3):263-268.
431 Pant K., Shah A., Mathur R.K., et al. Pulmonary alveolar microlithiasis with pleural calcification and nephrolithiasis. Chest. 1990;98:245-246.
432 Chang Y., Yang P.C., Luh K.T., et al. High-resolution computed tomography of pulmonary alveolar microlithiasis. J Formos Med Assoc. 1999;98(6):440-443.
433 Trapnell B.C., Whitsett J.A., Nakata K. Pulmonary alveolar proteinosis. N Engl J Med. 2003;349(26):2527-2539.
434 Kitamura T., Tanaka N., Watanabe J., et al. Idiopathic pulmonary alveolar proteinosis as an autoimmune disease with neutralizing antibody against granulocyte/macrophage colony-stimulating factor. J Exp Med. 1999;190(6):875-880.
435 Inoue Y., Trapnell B.C., Tazawa R., et al. Characteristics of a large cohort of patients with autoimmune pulmonary alveolar proteinosis in Japan. Am J Respir Crit Care Med. 2008;177(7):752-762.
436 Singh G., Katyal S.L., Bedrossian C.W., Rogers R.M. Pulmonary alveolar proteinosis. Staining for surfactant apoprotein in alveolar proteinosis and in conditions simulating it. Chest. 1983;83:82-86.
437 Miller R.R., Churg A.M., Hutcheon M., Lom S. Pulmonary alveolar proteinosis and aluminum dust exposure. Am Rev Respir Dis. 1984;130:312-315.
438 Bedrossian C.W., Luna M.A., Conklin R.H., Miller W.C. Alveolar proteinosis as a consequence of immunosuppression. A hypothesis based on clinical and pathologic observations. Hum Pathol. 1980;11(suppl 5):527-535.
439 Wang Y., Xia J., Wang Q.W. Clinical report on 62 cases of acute dimethyl sulfate intoxication. Am J Ind Med. 1988;13(4):455-462.
440 Seymour J., Presneill J. Pulmonary alveolar proteinosis: progress in the first 44 years. Am J Respir Crit Care Med. 2002;166(2):215-235.
441 Shah P., Hansell D., Lawson P.R., et al. Pulmonary alveolar proteinosis: clinical aspects and current concepts in pathogenesis. Thorax. 2000;55:67-77.
442 Davidson J., MacLeod W. Pulmonary alveolar proteinosis. Br J Dis Chest. 1969;63:13-16.
443 Lynch D.A., Godwin J.D., Safrin S., et al. High-resolution computed tomography in idiopathic pulmonary fibrosis: diagnosis and prognosis. Am J Respir Crit Care Med. 2005;172(4):488-493.
444 Claypool W., Roger R., Matuschak G. Update on the clinical diagnosis, management, and pathogenesis of pulmonary alveolar proteinosis (phospholipidosis). Chest. 1984;85:550-558.
445 Wylam M.E., Ten R., Prakash U.B., et al. Aerosol granulocyte-macrophage colony-stimulating factor for pulmonary alveolar proteinosis. Eur Respir J. 2006;27(3):585-593.
446 Tazawa R., Hamano E., Arai T., et al. Granulocyte-macrophage colony-stimulating factor and lung immunity in pulmonary alveolar proteinosis. Am J Respir Crit Care Med. 2005;171(10):1142-1149.
447 Heitzman E. The Lung: Radiologic-Pathologic Correlations, 2nd ed. St. Louis: Mosby, 1984.
448 Munk P.L., Müller N.L., Miller R.R., Ostrow D.N. Pulmonary lymphangitic carcinomatosis: CT and pathologic findings. Radiology. 1988;166:705-709.
449 Janower M., Blennerhassett J. Lymphangitic spread of metastatic cancer to the lung: a radiologic-pathologic classification. Radiology. 1971;101:267-273.
450 Stein M.G., Mayo J., Müller N., et al. Pulmonary lymphangitic spread of carcinoma: appearance on CT scans. Radiology. 1987;162:371-375.
451 Goldsmith H.S., Bailey H.D., Callahan E.L., Beattie E.J.Jr. Pulmonary lymphangitic metastases from breast carcinoma. Arch Surg. 1967;94:483-488.
452 Wall C.P., Gaensler E.A., Carrington C.B., Hayes J.A. Comparison of transbronchial and open biopsies in chronic infiltrative lung disease. Am Rev Respir Dis. 1981;123:280-285.
453 Frankel S.K., Cool C.D., Lynch D.A., Brown K.K. Idiopathic pleuroparenchymal fibroelastosis: description of a novel clinicopathologic entity. Chest. 2004;126(6):2007-2013.
454 Becker C.D., Gil J., Padilla M.L. Idiopathic pleuroparenchymal fibroelastosis: an unrecognized or misdiagnosed entity? Mod Pathol. 2008;21(6):784-787.
455 Shiota S., Shimizu K., Suzuki M., et al. Seven cases of marked pulmonary fibrosis in the upper lobe. Nihon Kokyuki Gakkai Zasshi. 1999;37(2):87-96.
456 Kamoi H., Okamoto T., Yoshimura N., et al. A case of interstitial pneumonia in the upper lung field histologically diagnosed as nonspecific interstitial pneumonia complicated by bilateral pneumothorax. Nihon Kokyuki Gakkai Zasshi. 2002;40(12):936-940.
457 Krakówka P., Meleniewska-Maciszewska A., Lesiak B., et al. Pulmonary fibrosis of the upper lobe resembling tuberculosis in patients without ankylosing spondylitis. Pneumonol Pol. 1985;53(1):39-47.