Part 3: Genes, the Environment, and Disease
82 |
Principles of Human Genetics |
IMPACT OF GENETICS AND GENOMICS ON MEDICAL PRACTICE
The prevalence of genetic diseases, combined with their potential severity and chronic nature, imposes great human, social, and financial burdens on society. Human genetics refers to the study of individual genes, their role and function in disease, and their mode of inheritance. Genomics refers to an organism’s entire genetic information, the genome, and the function and interaction of DNA within the genome, as well as with environmental or nongenetic factors, such as a person’s lifestyle. With the characterization of the human genome, genomics complements traditional genetics in our efforts to elucidate the etiology and pathogenesis of disease and to improve therapeutic interventions and outcomes. Following impressive advances in genetics, genomics, and health care information technology, the consequences of this wealth of knowledge for the practice of medicine are profound and play an increasingly prominent role in the diagnosis, prevention, and treatment of disease (Chap. 84).
Personalized medicine, the customization of medical decisions to an individual patient, relies heavily on genetic information. For example, a patient’s genetic characteristics (genotype) can be used to optimize drug therapy and predict efficacy, adverse events, and drug dosing of selected medications (pharmacogenetics) (Chap. 5). The mutational profile of a malignancy allows the selection of therapies that target mutated or overexpressed signaling molecules. Although still investigational, genomic risk prediction models for common diseases are beginning to emerge.
Genetics has traditionally been viewed through the window of relatively rare single-gene diseases. These disorders account for ~10% of pediatric admissions and childhood mortality. Historically, genetics has focused predominantly on chromosomal and metabolic disorders, reflecting the long-standing availability of techniques to diagnose these conditions. For example, conditions such as trisomy 21 (Down’s syndrome) or monosomy × (Turner’s syndrome) can be diagnosed using cytogenetics (Chap. 83e). Likewise, many metabolic disorders (e.g., phenylketonuria, familial hypercholesterolemia) are diagnosed using biochemical analyses. The advances in DNA diagnostics have extended the field of genetics to include virtually all medical specialties and have led to the elucidation of the pathogenesis of numerous monogenic disorders. In addition, it is apparent that virtually every medical condition has a genetic component. As is often evident from a patient’s family history, many common disorders such as hypertension, heart disease, asthma, diabetes mellitus, and mental illnesses are significantly influenced by the genetic background. These polygenic or multifactorial (complex) disorders involve the contributions of many different genes, as well as environmental factors that can modify disease risk (Chap. 84). Genome-wide association studies (GWAS) have elucidated numerous disease-associated loci and are providing novel insights into the allelic architecture of complex traits. These studies have been facilitated by the availability of comprehensive catalogues of human single-nucleotide polymorphism (SNP) haplotypes generated through the HapMap Project. The sequencing of whole genomes or exomes (the exons within the genome) is increasingly used in the clinical realm in order to characterize individuals with complex undiagnosed conditions or to characterize the mutational profile of advanced malignancies in order to select better targeted therapies.
Cancer has a genetic basis because it results from acquired somatic mutations in genes controlling growth, apoptosis, and cellular differentiation (Chap. 101e). In addition, the development of many cancers is associated with a hereditary predisposition. Characterization of the genome (and epigenome) in various malignancies has led to fundamental new insights into cancer biology and reveals that the genomic profile of mutations is in many cases more important in determining the appropriate chemotherapy than the organ in which the tumor originates. Hence, comprehensive mutational profiling of malignancies has increasing impact on cancer taxonomy, the choice of targeted therapies, and improved outcomes.
Genetic and genomic approaches have proven invaluable for the detection of infectious pathogens and are used clinically to identify agents that are difficult to culture such as mycobacteria, viruses, and parasites, or to track infectious agents locally or globally. In many cases, molecular genetics has improved the feasibility and accuracy of diagnostic testing and is beginning to open new avenues for therapy, including gene and cellular therapy (Chaps. 90e and 91e). Molecular genetics has also provided the opportunity to characterize the microbiome, a new field that characterizes the population dynamics of bacteria, viruses, and parasites that coexist with humans and other animals (Chap. 86e). Emerging data indicate that the microbiome has significant effects on normal physiology as well as various disease states.
Molecular biology has significantly changed the treatment of human disease. Peptide hormones, growth factors, cytokines, and vaccines can now be produced in large amounts using recombinant DNA technology. Targeted modifications of these peptides provide the practitioner with improved therapeutic tools, as illustrated by genetically modified insulin analogues with more favorable kinetics. Lastly, there is reason to believe that a better understanding of the genetic basis of human disease will also have an increasing impact on disease prevention.
The astounding rate at which new genetic information is being generated creates a major challenge for physicians, health care providers, and basic investigators. Although many functional aspects of the genome remain unknown, there are many clinical situations where sufficient evidence exits for the use of genetic and genomic information to optimize patient care and treatment. Much genetic information resides in databases or is being published in basic science journals. Databases provide easy access to the expanding information about the human genome, genetic disease, and genetic testing (Table 82-1). For example, several thousand monogenic disorders are summarized in a large, continuously evolving compendium, referred to as the Online Mendelian Inheritance in Man (OMIM) catalogue (Table 82-1). The ongoing refinement of bioinformatics is simplifying the analysis and access to this daunting amount of new information.
|
SELECTED DATABASES RELEVANT FOR GENOMICS AND GENETIC DISORDERS |
THE HUMAN GENOME
Structure of the Human Genome • HUMAN GENOME PROJECT The Human Genome Project was initiated in the mid-1980s as an ambitious effort to characterize the entire human genome. Although the prospect of determining the complete sequence of the human genome seemed daunting several years ago, technical advances in DNA sequencing and bioinformatics led to the completion of a draft human sequence in 2000 and the completion of the DNA sequence for the last of the human chromosomes in May 2006. Currently, facilitated by rapidly decreasing costs for comprehensive sequence analyses and improvement of bioinformatics pipelines for data analysis, the sequencing of whole genomes and exomes is used with increasing frequency in the clinical setting. The scope of a whole genome sequence analysis can be illustrated by the following analogy. Human DNA consists of ~3 billion base pairs (bp) of DNA per haploid genome, which is nearly 1000-fold greater than that of the Escherichia coli genome. If the human DNA sequence were printed out, it would correspond to about 120 volumes of Harrison’s Principles of Internal Medicine.
In addition to the human genome, the genomes of numerous organisms have been sequenced completely (~4000) or partially (~10,000) (Genomes Online Database [GOLD]; Table 82-1). They include, among others, eukaryotes such as the mouse (Mus musculus), Saccharomyces cerevisiae, Caenorhabditis elegans, and Drosophila melanogaster; bacteria (e.g., E. coli); and Archaea, viruses, organelles (mitochondria, chloroplasts), and plants (e.g., Arabidopsis thaliana). Genomic information of infectious agents has significant impact for the characterization of infectious outbreaks and epidemics. Other ramifications arising from the availability of genomic data include, among others, (1) the comparison of entire genomes (comparative genomics), (2) the study of large-scale expression of RNAs (functional genomics) and proteins (proteomics) to detect differences between various tissues in health and disease, (3) the characterization of the variation among individuals by establishing catalogues of sequence variations and SNPs (HapMap Project), and (4) the identification of genes that play critical roles in the development of polygenic and multifactorial disorders.
CHROMOSOMES The human genome is divided into 23 different chromosomes, including 22 autosomes (numbered 1–22) and the × and Y sex chromosomes (Fig. 82-1). Adult cells are diploid, meaning they contain two homologous sets of 22 autosomes and a pair of sex chromosomes. Females have two × chromosomes (XX), whereas males have one × and one Y chromosome (XY). As a consequence of meiosis, germ cells (sperm or oocytes) are haploid and contain one set of 22 autosomes and one of the sex chromosomes. At the time of fertilization, the diploid genome is reconstituted by pairing of the homologous chromosomes from the mother and father. With each cell division (mitosis), chromosomes are replicated, paired, segregated, and divided into two daughter cells.
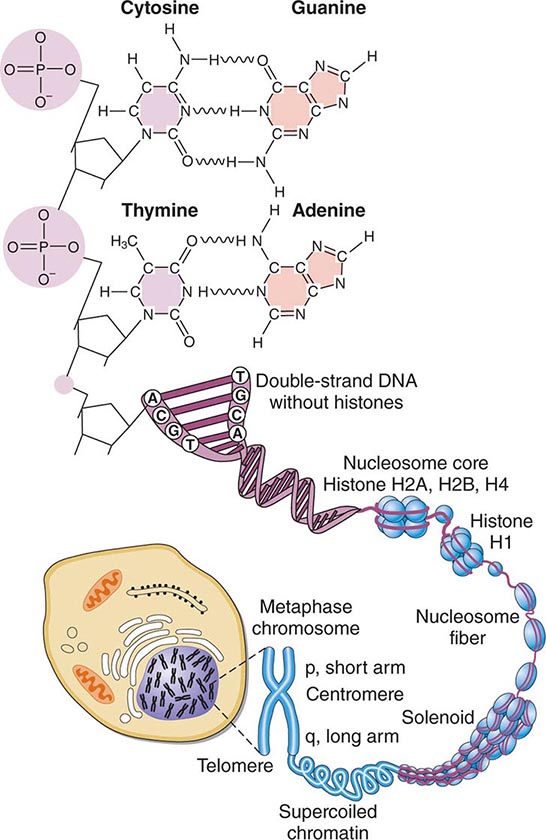
FIGURE 82-1 Structure of chromatin and chromosomes. Chromatin is composed of double-strand DNA that is wrapped around histone and nonhistone proteins forming nucleosomes. The nucleosomes are further organized into solenoid structures. Chromosomes assume their characteristic structure, with short (p) and long (q) arms at the metaphase stage of the cell cycle.
STRUCTURE OF DNA DNA is a double-stranded helix composed of four different bases: adenine (A), thymidine (T), guanine (G), and cytosine (C). Adenine is paired to thymidine, and guanine is paired to cytosine, by hydrogen bond interactions that span the double helix (Fig. 82-1). DNA has several remarkable features that make it ideal for the transmission of genetic information. It is relatively stable, and the double-stranded nature of DNA and its feature of strict base-pair complementarity permit faithful replication during cell division. Complementarity also allows the transmission of genetic information from DNA → RNA → protein (Fig. 82-2). mRNA is encoded by the so-called sense or coding strand of the DNA double helix and is translated into proteins by ribosomes.
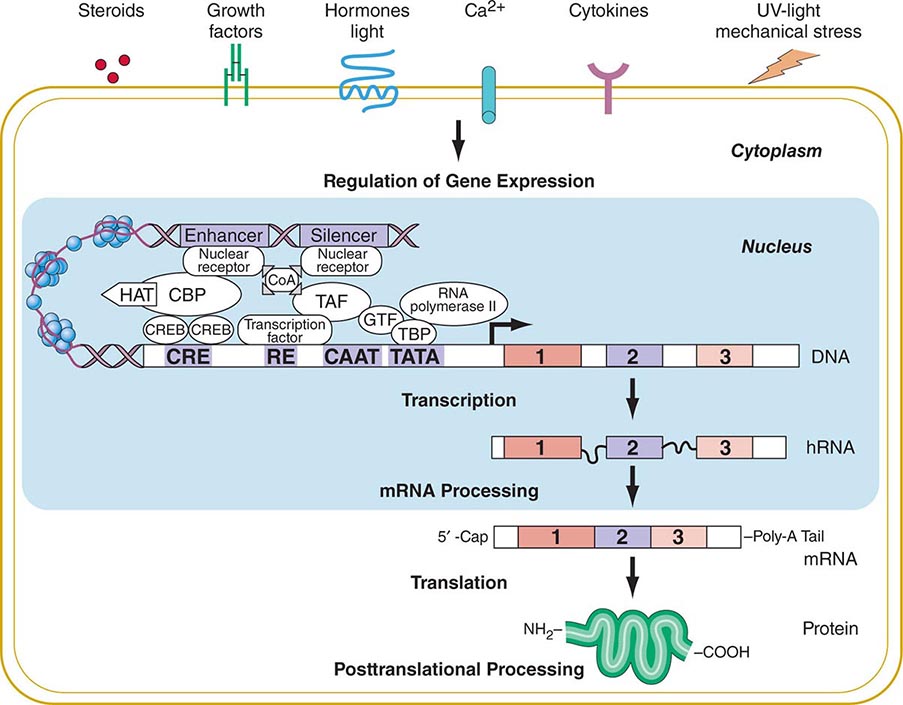
FIGURE 82-2 Flow of genetic information. Multiple extracellular signals activate intracellular signal cascades that result in altered regulation of gene expression through the interaction of transcription factors with regulatory regions of genes. RNA polymerase transcribes DNA into RNA that is processed to mRNA by excision of intronic sequences. The mRNA is translated into a polypeptide chain to form the mature protein after undergoing posttranslational processing. CBP, CREB-binding protein; CoA, co-activator; COOH, carboxyterminus; CRE, cyclic AMP responsive element; CREB, cyclic AMP response element–binding protein; GTF, general transcription factors; HAT, histone acetyl transferase; NH2, aminoterminus; RE, response element; TAF, TBP-associated factors; TATA, TATA box; TBP, TATA-binding protein.
The presence of four different bases provides surprising genetic diversity. In the protein-coding regions of genes, the DNA bases are arranged into codons, a triplet of bases that specifies a particular amino acid. It is possible to arrange the four bases into 64 different triplet codons (43). Each codon specifies 1 of the 20 different amino acids, or a regulatory signal such as initiation and stop of translation. Because there are more codons than amino acids, the genetic code is degenerate; that is, most amino acids can be specified by several different codons. By arranging the codons in different combinations and in various lengths, it is possible to generate the tremendous diversity of primary protein structure.
DNA length is normally measured in units of 1000 bp (kilobases, kb) or 1,000,000 bp (megabases, Mb). Not all DNA encodes genes. In fact, genes account for only ~10–15% of DNA. Much of the remaining DNA consists of sequences, often of highly repetitive nature, the function of which is poorly understood. These repetitive DNA regions, along with nonrepetitive sequences that do not encode genes, serve, in part, a structural role in the packaging of DNA into chromatin (i.e., DNA bound to histone proteins, and chromosomes) and exert regulatory functions (Fig. 82-1).
GENES A gene is a functional unit that is regulated by transcription (see below) and encodes an RNA product, which is most commonly, but not always, translated into a protein that exerts activity within or outside the cell (Fig. 82-3). Historically, genes were identified because they conferred specific traits that are transmitted from one generation to the next. Increasingly, they are characterized based on expression in various tissues (transcriptome). The size of genes is quite broad; some genes are only a few hundred base pairs, whereas others are extraordinarily large (2 Mb). The number of genes greatly underestimates the complexity of genetic expression, because single genes can generate multiple spliced messenger RNA (mRNA) products (isoforms), which are translated into proteins that are subject to complex posttranslational modification such as phosphorylation. Exons refer to the portion of genes that are eventually spliced together to form mRNA. Introns refer to the spacing regions between the exons that are spliced out of precursor RNAs during RNA processing. The gene locus also includes regions that are necessary to control its expression (Fig. 82-2). Current estimates predict 20,687 protein-coding genes in the human genome with an average of about four different coding transcripts per gene. Remarkably, the exome only constitutes 1.14% of the genome. In addition, thousands of noncoding transcripts (RNAs of various length such as microRNAs and long noncoding RNAs), which function, at least in part, as transcriptional and posttranscriptional regulators of gene expression, have been identified. Aberrant expression of microRNAs has been found to play a pathogenic role in numerous diseases.
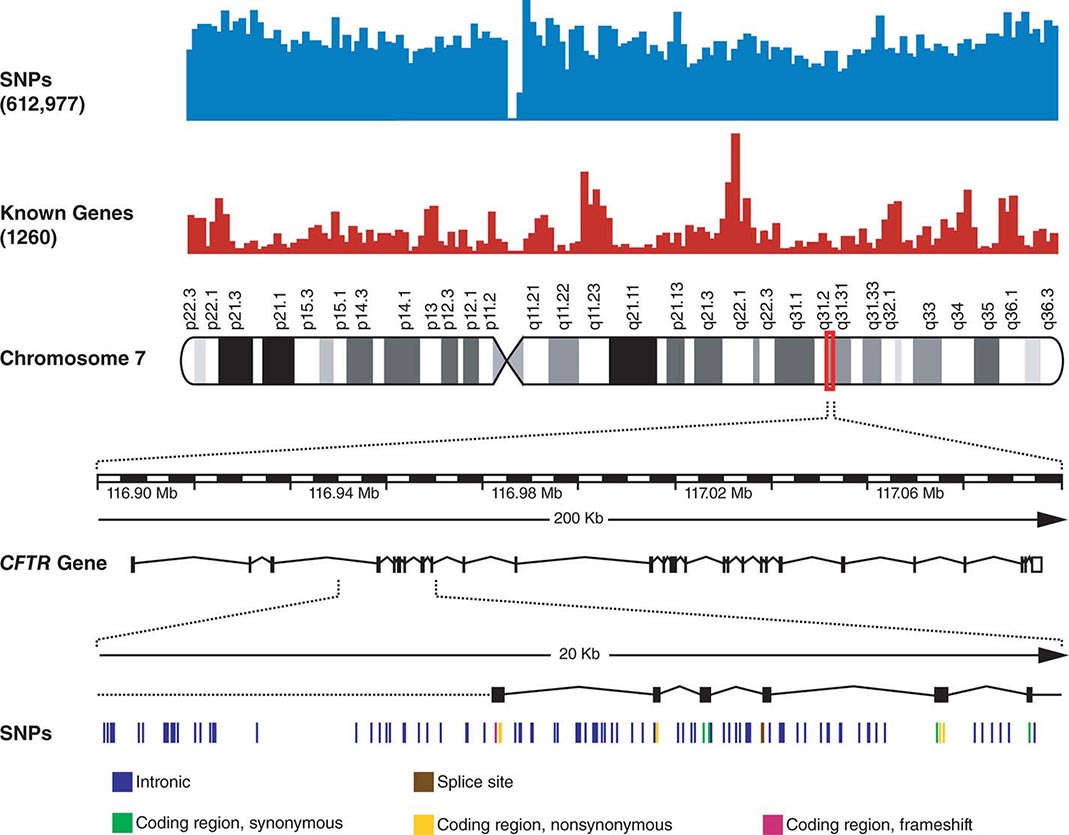
FIGURE 82-3 Chromosome 7 is shown with the density of single-nucleotide polymorphisms (SNPs) and genes above. A 200-kb region in 7q31.2 containing the CFTR gene is shown below. The CFTR gene contains 27 exons. More than 1900 mutations in this gene have been found in patients with cystic fibrosis. A 20-kb region encompassing exons 4–9 is shown further amplified to illustrate the SNPs in this region.
SINGLE-NUCLEOTIDE POLYMORPHISMS An SNP is a variation of a single base pair in the DNA. The identification of the ~10 million SNPs estimated to occur in the human genome has generated a catalogue of common genetic variants that occur in human beings from distinct ethnic backgrounds (Fig. 82-3). SNPs are the most common type of sequence variation and account for ~90% of all sequence variation. They occur on average every 100 to 300 bases and are the major source of genetic heterogeneity. Remarkably, however, the primary DNA sequence of humans has ~99.9% similarity compared to that of any other human. SNPs that are in close proximity are inherited together (e.g., they are linked) and are referred to as haplotypes (Fig. 82-4). The HapMap describes the nature and location of these SNP haplotypes and how they are distributed among individuals within and among populations. The haplotype map information, referred to as HapMap, is greatly facilitating GWAS designed to elucidate the complex interactions among multiple genes and lifestyle factors in multifactorial disorders (see below). Moreover, haplotype analyses are useful to assess variations in responses to medications (pharmacogenomics) and environmental factors, as well as the prediction of disease predisposition.
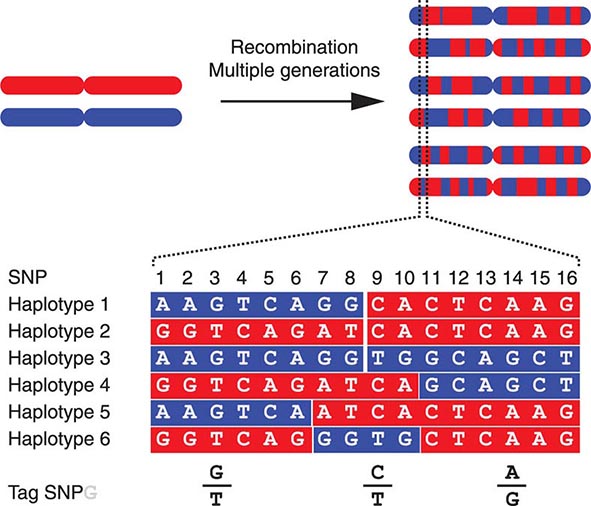
FIGURE 82-4 The origin of haplotypes is due to repeated recombination events occurring in multiple generations. Over time, this leads to distinct haplotypes. These haplotype blocks can often be characterized by genotyping selected Tag single-nucleotide polymorphisms (SNPs), an approach that facilitates performing genome-wide association studies (GWAS).
COPY NUMBER VARIATIONS Copy number variations (CNVs) are relatively large genomic regions (1 kb to several Mb) that have been duplicated or deleted on certain chromosomes (Fig. 82-5). It has been estimated that as many as 1500 CNVs, scattered throughout the genome, are present in an individual. When comparing the genomes of two individuals, approximately 0.4–0.8% of their genomes differ in terms of CNVs. Of note, de novo CNVs have been observed between monozygotic twins, who otherwise have identical genomes. Some CNVs have been associated with susceptibility or resistance to disease, and CNVs can be elevated in cancer cells.
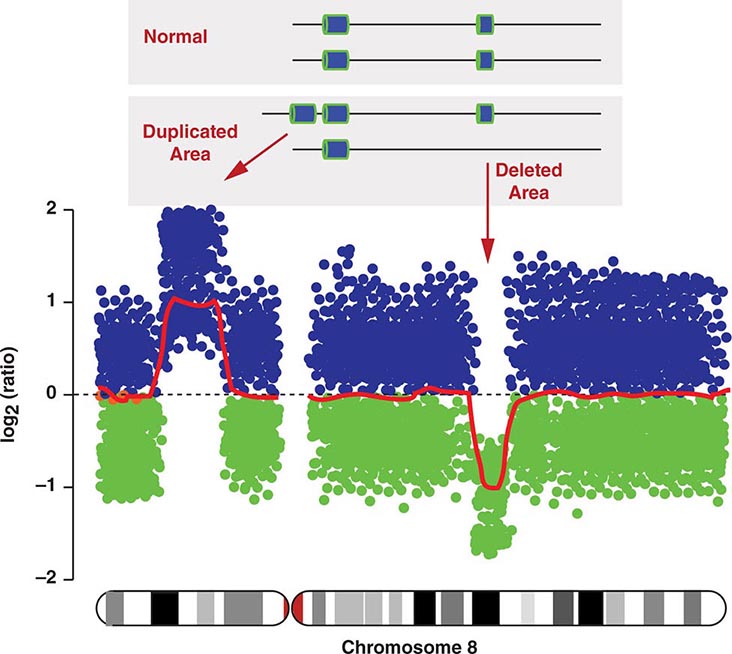
FIGURE 82-5 Copy number variations (CNV) encompass relatively large regions of the genome that have been duplicated or deleted. Chromosome 8 is shown with CNV detected by genomic hybridization. An increase in the signal strength indicates a duplication, a decrease reflects a deletion of the covered chromosomal regions.
Replication of DNA and Mitosis Genetic information in DNA is transmitted to daughter cells under two different circumstances: (1) somatic cells divide by mitosis, allowing the diploid (2n) genome to replicate itself completely in conjunction with cell division; and (2) germ cells (sperm and ova) undergo meiosis, a process that enables the reduction of the diploid (2n) set of chromosomes to the haploid state (1n).
Prior to mitosis, cells exit the resting, or G0 state, and enter the cell cycle (Chap. 101e). After traversing a critical checkpoint in G1, cells undergo DNA synthesis (S phase), during which the DNA in each chromosome is replicated, yielding two pairs of sister chromatids (2n → 4n). The process of DNA synthesis requires stringent fidelity in order to avoid transmitting errors to subsequent generations of cells. Genetic abnormalities of DNA mismatch/repair include xeroderma pigmentosum, Bloom’s syndrome, ataxia telangiectasia, and hereditary nonpolyposis colon cancer (HNPCC), among others. Many of these disorders strongly predispose to neoplasia because of the rapid acquisition of additional mutations (Chap. 101e). After completion of DNA synthesis, cells enter G2 and progress through a second checkpoint before entering mitosis. At this stage, the chromosomes condense and are aligned along the equatorial plate at metaphase. The two identical sister chromatids, held together at the centromere, divide and migrate to opposite poles of the cell. After formation of a nuclear membrane around the two separated sets of chromatids, the cell divides and two daughter cells are formed, thus restoring the diploid (2n) state.
Assortment and Segregation of Genes During Meiosis Meiosis occurs only in germ cells of the gonads. It shares certain features with mitosis but involves two distinct steps of cell division that reduce the chromosome number to the haploid state. In addition, there is active recombination that generates genetic diversity. During the first cell division, two sister chromatids (2n → 4n) are formed for each chromosome pair and there is an exchange of DNA between homologous paternal and maternal chromosomes. This process involves the formation of chiasmata, structures that correspond to the DNA segments that cross over between the maternal and paternal homologues (Fig. 82-6). Usually there is at least one crossover on each chromosomal arm; recombination occurs more frequently in female meiosis than in male meiosis. Subsequently, the chromosomes segregate randomly. Because there are 23 chromosomes, there exist 223 (>8 million) possible combinations of chromosomes. Together with the genetic exchanges that occur during recombination, chromosomal segregation generates tremendous diversity, and each gamete is genetically unique. The process of recombination and the independent segregation of chromosomes provide the foundation for performing linkage analyses, whereby one attempts to correlate the inheritance of certain chromosomal regions (or linked genes) with the presence of a disease or genetic trait (see below).
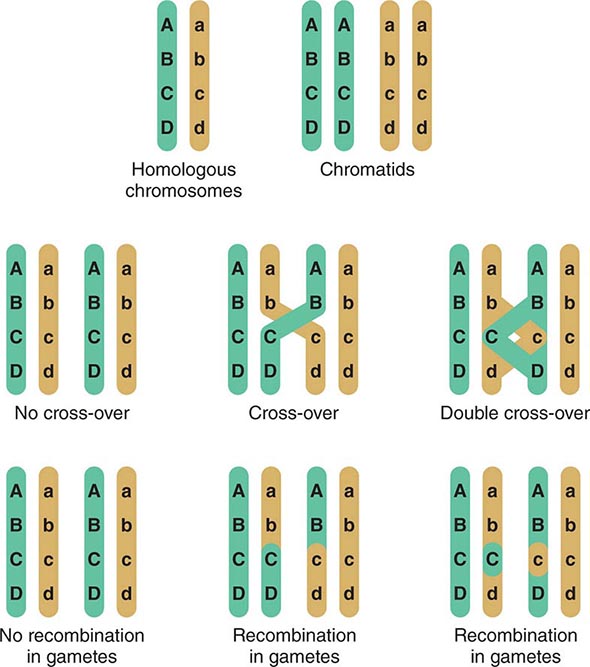
FIGURE 82-6 Crossing-over and genetic recombination. During chiasma formation, either of the two sister chromatids on one chromosome pairs with one of the chromatids of the homologous chromosome. Genetic recombination occurs through crossing-over and results in recombinant and nonrecombinant chromosome segments in the gametes. Together with the random segregation of the maternal and paternal chromosomes, recombination contributes to genetic diversity and forms the basis of the concept of linkage.
After the first meiotic division, which results in two daughter cells (2n), the two chromatids of each chromosome separate during a second meiotic division to yield four gametes with a haploid state (1n). When the egg is fertilized by sperm, the two haploid sets are combined, thereby restoring the diploid state (2n) in the zygote.
REGULATION OF GENE EXPRESSION
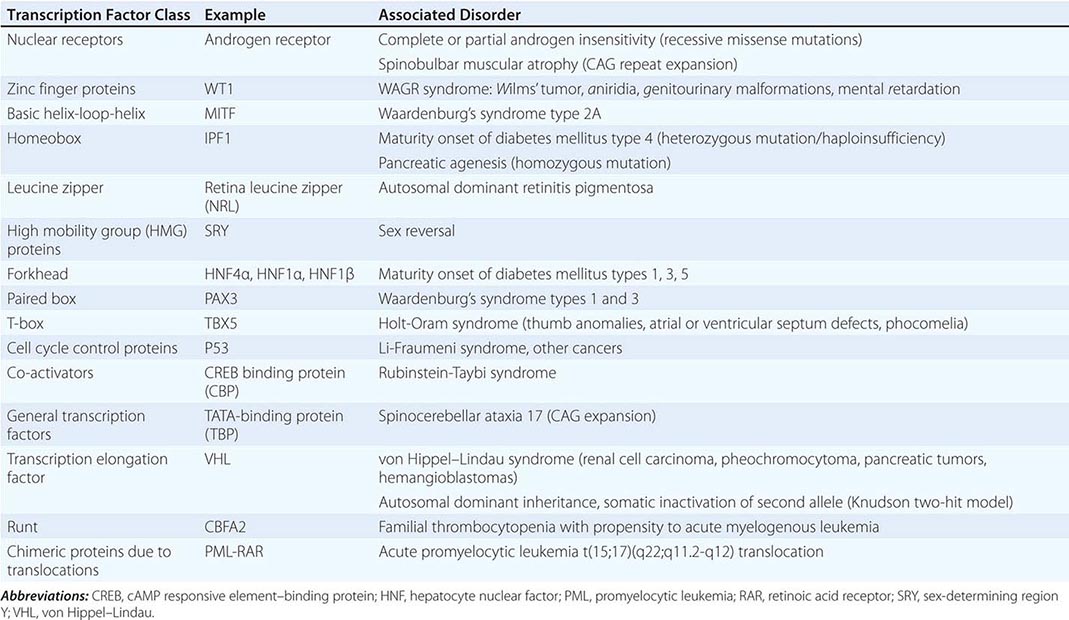
Regulation by Transcription Factors The expression of genes is regulated by DNA-binding proteins that activate or repress transcription. The number of DNA sequences and transcription factors that regulate transcription is much greater than originally anticipated. Most genes contain at least 15–20 discrete regulatory elements within 300 bp of the transcription start site. This densely packed promoter region often contains binding sites for ubiquitous transcription factors such as CAAT box/enhancer binding protein (C/EBP), cyclic AMP response element–binding (CREB) protein, selective promoter factor 1 (Sp-1), or activator protein 1 (AP-1). However, factors involved in cell-specific expression may also bind to these sequences. Key regulatory elements may also reside at a large distance from the proximal promoter. The globin and the immunoglobulin genes, for example, contain locus control regions that are several kilobases away from the structural sequences of the gene. Specific groups of transcription factors that bind to these promoter and enhancer sequences provide a combinatorial code for regulating transcription. In this manner, relatively ubiquitous factors interact with more restricted factors to allow each gene to be expressed and regulated in a unique manner that is dependent on developmental state, cell type, and numerous extracellular stimuli. Regulatory factors also bind within the gene itself, particularly in the intronic regions. The transcription factors that bind to DNA actually represent only the first level of regulatory control. Other proteins—co-activators and co-repressors—interact with the DNA-binding transcription factors to generate large regulatory complexes. These complexes are subject to control by numerous cell-signaling pathways and enzymes, leading to phosphorylation, acetylation, sumoylation, and ubiquitination. Ultimately, the recruited transcription factors interact with, and stabilize, components of the basal transcription complex that assembles at the site of the TATA box and initiator region. This basal transcription factor complex consists of >30 different proteins. Gene transcription occurs when RNA polymerase begins to synthesize RNA from the DNA template. A large number of identified genetic diseases involve transcription factors (Table 82-2).
|
SELECTED EXAMPLES OF DISEASES CAUSED BY MUTATIONS AND REARRANGEMENTS IN TRANSCRIPTION FACTOR CLASSES |
The field of functional genomics is based on the concept that understanding alterations of gene expression under various physiologic and pathologic conditions provides insight into the underlying functional role of the gene. By revealing specific gene expression profiles, this knowledge may be of diagnostic and therapeutic relevance. The large-scale study of expression profiles, which takes advantage of microarray and bead array technologies, is also referred to as transcriptomics because the complement of mRNAs transcribed by the cellular genome is called the transcriptome.
Most studies of gene expression have focused on the regulatory DNA elements of genes that control transcription. However, it should be emphasized that gene expression requires a series of steps, including mRNA processing, protein translation, and posttranslational modifications, all of which are actively regulated (Fig. 82-2).
Epigenetic Regulation of Gene Expression Epigenetics describes mechanisms and phenotypic changes that are not a result of variation in the primary DNA nucleotide sequence, but are caused by secondary modifications of DNA or histones. These modifications include heritable changes such as X-inactivation and imprinting, but they can also result from dynamic posttranslational protein modifications in response to environmental influences such as diet, age, or drugs. The epigenetic modifications result in altered expression of individual genes or chromosomal loci encompassing multiple genes. The term epigenome describes the constellation of covalent modifications of DNA and histones that impact chromatin structure, as well as noncoding transcripts that modulate the transcriptional activity of DNA. Although the primary DNA sequence is usually identical in all cells of an organism, tissue-specific changes in the epigenome contribute to determining the transcriptional signature of a cell (transcriptome) and hence the protein expression profile (proteome).
Mechanistically, DNA and histone modifications can result in the activation or silencing of gene expression (Fig. 82-7). DNA methylation involves the addition of a methyl group to cytosine residues. This is usually restricted to cytosines of CpG dinucleotides, which are abundant throughout the genome. Methylation of these dinucleotides is thought to represent a defense mechanism that minimizes the expression of sequences that have been incorporated into the genome such as retroviral sequences. CpG dinucleotides also exist in so-called CpG islands, stretches of DNA characterized by a high CG content, which are found in the majority of human gene promoters. CpG islands in promoter regions are typically unmethylated, and the lack of methylation facilitates transcription.
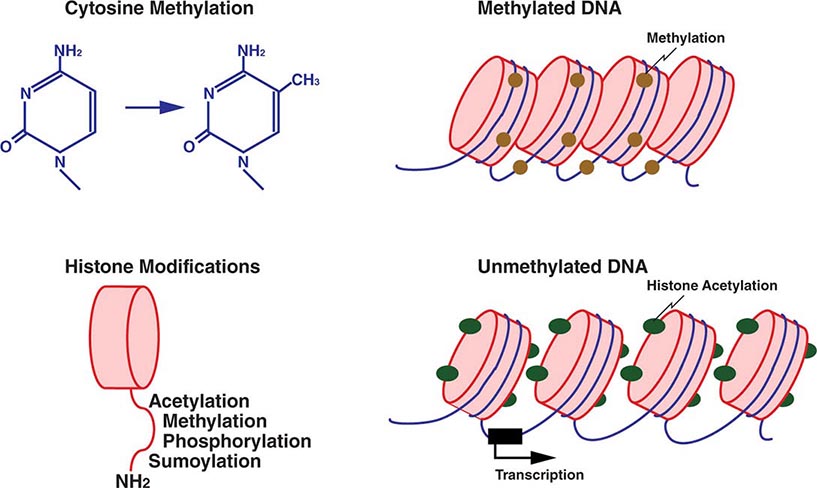
FIGURE 82-7 Epigenetic modifications of DNA and histones. Methylation of cytosine residues is associated with gene silencing. Methylation of certain genomic regions is inherited (imprinting), and it is involved in the silencing of one of the two × chromosomes in females (X-inactivation). Alterations in methylation can also be acquired, e.g., in cancer cells. Covalent posttranslational modifications of histones play an important role in altering DNA accessibility and chromatin structure and hence in regulating transcription. Histones can be reversibly modified in their amino-terminal tails, which protrude from the nucleosome core particle, by acetylation of lysine, phosphorylation of serine, methylation of lysine and arginine residues, and sumoylation. Acetylation of histones by histone acetylases (HATs), e.g., leads to unwinding of chromatin and accessibility to transcription factors. Conversely, deacetylation by histone deacetylases (HDACs) results in a compact chromatin structure and silencing of transcription.
Histone methylation involves the addition of a methyl group to lysine residues in histone proteins (Fig. 82-7). Depending on the specific lysine residue being methylated, this alters chromatin configuration, either making it more open or tightly packed. Acetylation of histone proteins is another well-characterized mechanism that results in an open chromatin configuration, which favors active transcription. Acetylation is generally more dynamic than methylation, and many transcriptional activation complexes have histone acetylase activity, whereas repressor complexes often contain deacetylases and remove acetyl groups from histones. Other histone modifications, whose effects are incompletely characterized, include phosphorylation and sumoylation. Lastly, noncoding RNAs that bind to DNA can have a significant impact on transcriptional activity.
Physiologically, epigenetic mechanisms play an important role in several instances. For example, X-inactivation refers to the relative silencing of one of the two × chromosome copies present in females. The inactivation process is a form of dosage compensation such that females (XX) do not generally express twice as many X-chromosomal gene products as males (XY). In a given cell, the choice of which chromosome is inactivated occurs randomly in humans. But once the maternal or paternal × chromosome is inactivated, it will remain inactive, and this information is transmitted with each cell division. The X-inactive specific transcript (Xist) gene encodes a large noncoding RNA that mediates the silencing of the × chromosome from which it is transcribed by coating it with Xist RNA. The inactive × chromosome is highly methylated and has low levels of histone acetylation.
Epigenetic gene inactivation also occurs on selected chromosomal regions of autosomes, a phenomenon referred to as genomic imprinting. Through this mechanism, a small subset of genes is only expressed in a monoallelic fashion. Imprinting is heritable and leads to the preferential expression of one of the parental alleles, which deviates from the usual biallelic expression seen for the majority of genes. Remarkably, imprinting can be limited to a subset of tissues. Imprinting is mediated through DNA methylation of one of the alleles. The epigenetic marks on imprinted genes are maintained throughout life, but during zygote formation, they are activated or inactivated in a sex-specific manner (imprint reset) (Fig. 82-8), which allows a differential expression pattern in the fertilized egg and the subsequent mitotic divisions. Appropriate expression of imprinted genes is important for normal development and cellular functions. Imprinting defects and uniparental disomy, which is the inheritance of two chromosomes or chromosomal regions from the same parent, are the cause of several developmental disorders such as Beckwith-Wiedemann syndrome, Silver-Russell syndrome, Angelman’s syndrome, and Prader-Willi syndrome (see below). Monoallelic loss-of-function mutations in the GNAS1 gene lead to Albright’s hereditary osteodystrophy (AHO). Paternal transmission of GNAS1 mutations leads to an isolated AHO phenotype (pseudopseudohypoparathyroidism), whereas maternal transmission leads to AHO in combination with hormone resistance to parathyroid hormone, thyrotropin, and gonadotropins (pseudohypoparathyroidism type IA). These phenotypic differences are explained by tissue-specific imprinting of the GNAS1 gene, which is expressed primarily from the maternal allele in the thyroid, gonadotropes, and the proximal renal tubule. In most other tissues, the GNAS1 gene is expressed biallelically. In patients with isolated renal resistance to parathyroid hormone (pseudohypoparathyroidism type IB), defective imprinting of the GNAS1 gene results in decreased Gsα expression in the proximal renal tubules. Rett’s syndrome is an X-linked dominant disorder resulting in developmental regression and stereotypic hand movements in affected girls. It is caused by mutations in the MECP2 gene, which encodes a methyl-binding protein. The ensuing aberrant methylation results in abnormal gene expression in neurons, which are otherwise normally developed.
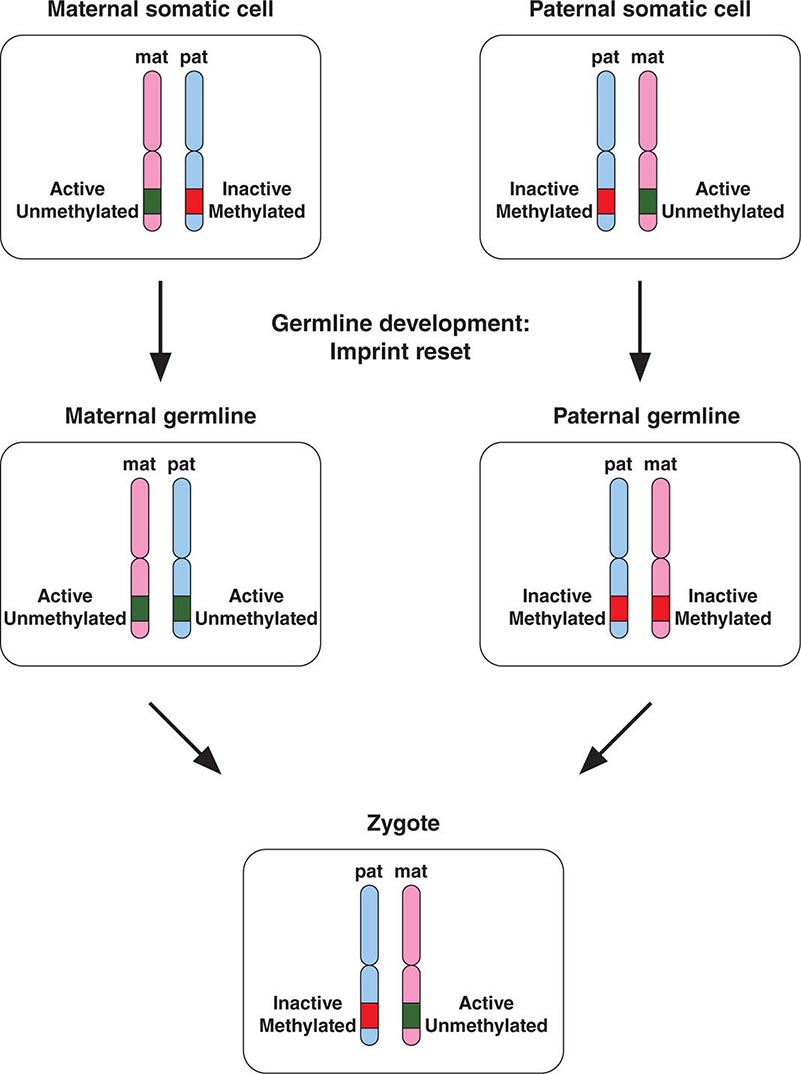
FIGURE 82-8 A few genomic regions are imprinted in a parent-specific fashion. The unmethylated chromosomal regions are actively expressed, whereas the methylated regions are silenced. In the germline, the imprint is reset in a parent-specific fashion: both chromosomes are unmethylated in the maternal (mat) germline and methylated in the paternal (pat) germline. In the zygote, the resulting imprinting pattern is identical with the pattern in the somatic cells of the parents.
Remarkably, epigenetic differences also occur among monozygotic twins. Although twins are epigenetically indistinguishable during the early years of life, older monozygotic twins exhibit differences in the overall content and genomic distribution of DNA methylation and histone acetylation, which would be expected to alter gene expression in various tissues.
In cancer, the epigenome is characterized by simultaneous losses and gains of DNA methylation in different genomic regions, as well as repressive histone modifications. Hyper- and hypomethylation are associated with mutations in genes that control DNA methylation. Hypomethylation is thought to remove normal control mechanisms that prevent expression of repressed DNA regions. It is also associated with genomic instability. Hypermethylation, in contrast, results in the silencing of CpG islands in promoter regions of genes, including tumor-suppressor genes. Epigenetic alterations are considered to be more easily reversible compared to genetic changes, and modification of the epigenome with demethylating agents and histone deacetylases is being explored in clinical trials.
MODELS OF GENETIC DISEASE
Several organisms have been studied extensively as genetic models, including M. musculus (mouse), D. melanogaster (fruit fly), C. elegans (nematode), S. cerevisiae (baker’s yeast), and E. coli (colonic bacterium). The ability to use these evolutionarily distant organisms as genetic models that are relevant to human physiology reflects a surprising conservation of genetic pathways and gene function. Transgenic mouse models have been particularly valuable, because many human and mouse genes exhibit similar structure and function and because manipulation of the mouse genome is relatively straightforward compared to that of other mammalian species. Transgenic strategies in mice can be divided into two main approaches: (1) expression of a gene by random insertion into the genome, and (2) deletion or targeted mutagenesis of a gene by homologous recombination with the native endogenous gene (knock-out, knock-in). Previous versions of this chapter provide more detail about the technical principles underlying the development of genetically modified animals. Several databases provide comprehensive information about natural and transgenic animal models, the associated phenotypes, and integrated genetic, genomic, and biologic data (Table 82-1).
TRANSMISSION OF GENETIC DISEASE
Origins and Types of Mutations A mutation can be defined as any change in the primary nucleotide sequence of DNA regardless of its functional consequences. Some mutations may be lethal, others are less deleterious, and some may confer an evolutionary advantage. Mutations can occur in the germline (sperm or oocytes); these can be transmitted to progeny. Alternatively, mutations can occur during embryogenesis or in somatic tissues. Mutations that occur during development lead to mosaicism, a situation in which tissues are composed of cells with different genetic constitutions. If the germline is mosaic, a mutation can be transmitted to some progeny but not others, which sometimes leads to confusion in assessing the pattern of inheritance. Somatic mutations that do not affect cell survival can sometimes be detected because of variable phenotypic effects in tissues (e.g., pigmented lesions in McCune-Albright syndrome). Other somatic mutations are associated with neoplasia because they confer a growth advantage to cells. Epigenetic events may also influence gene expression or facilitate genetic damage. With the exception of triplet nucleotide repeats, which can expand (see below), mutations are usually stable.
Mutations are structurally diverse—they can involve the entire genome, as in triploidy (one extra set of chromosomes), or gross numerical or structural alterations in chromosomes or individual genes (Chap. 83e). Large deletions may affect a portion of a gene or an entire gene, or, if several genes are involved, they may lead to a contiguous gene syndrome. Unequal crossing-over between homologous genes can result in fusion gene mutations, as illustrated by color blindness. Mutations involving single nucleotides are referred to as point mutations. Substitutions are called transitions if a purine is replaced by another purine base (A ↔ G) or if a pyrimidine is replaced by another pyrimidine (C ↔ T). Changes from a purine to a pyrimidine, or vice versa, are referred to as transversions. If the DNA sequence change occurs in a coding region and alters an amino acid, it is called a missense mutation. Depending on the functional consequences of such a missense mutation, amino acid substitutions in different regions of the protein can lead to distinct phenotypes.
Mutations can occur in all domains of a gene (Fig. 82-9). A point mutation occurring within the coding region leads to an amino acid substitution if the codon is altered (Fig. 82-10). Point mutations that introduce a premature stop codon result in a truncated protein. Large deletions may affect a portion of a gene or an entire gene, whereas small deletions and insertions alter the reading frame if they do not represent a multiple of three bases. These “frameshift” mutations lead to an entirely altered carboxy terminus. Mutations in intronic sequences or in exon junctions may destroy or create splice donor or splice acceptor sites. Mutations may also be found in the regulatory sequences of genes, resulting in reduced or enhanced gene transcription.
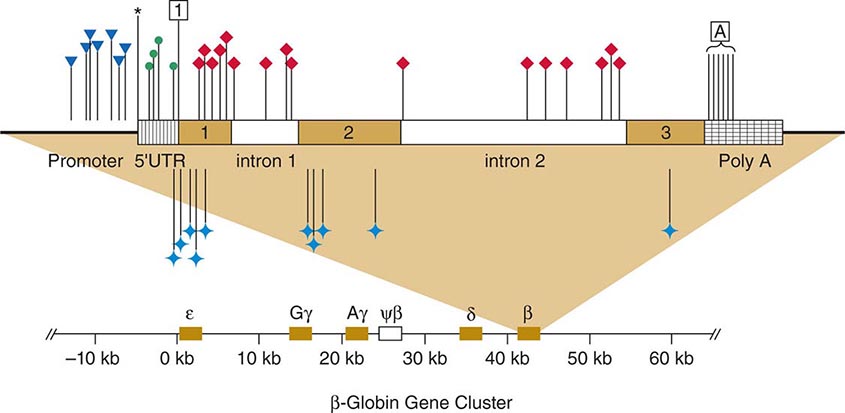
FIGURE 82-9 Point mutations causing β thalassemia as example of allelic heterogeneity. The β-globin gene is located in the globin gene cluster. Point mutations can be located in the promoter, the CAP site, the 5’-untranslated region, the initiation codon, each of the three exons, the introns, or the polyadenylation signal. Many mutations introduce missense or nonsense mutations, whereas others cause defective RNA splicing. Not shown here are deletion mutations of the β-globin gene or larger deletions of the globin locus that can also result in thalassemia. ![]() , promoter mutations; *, CAP site; •, 5’UTR;
, promoter mutations; *, CAP site; •, 5’UTR;![]() , initiation codon; ♦, defective RNA processing;
, initiation codon; ♦, defective RNA processing; ![]() , missense and nonsense mutations;
, missense and nonsense mutations; ![]() , Poly A signal.
, Poly A signal.
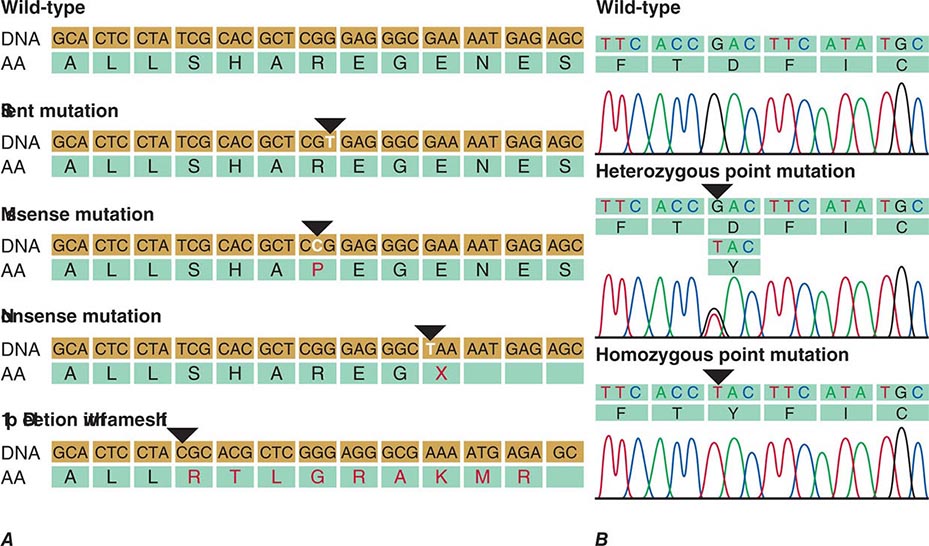
FIGURE 82-10 A. Examples of mutations. The coding strand is shown with the encoded amino acid sequence. B. Chromatograms of sequence analyses after amplification of genomic DNA by polymerase chain reaction.
Certain DNA sequences are particularly susceptible to mutagenesis. Successive pyrimidine residues (e.g., T-T or C-C) are subject to the formation of ultraviolet light–induced photoadducts. If these pyrimidine dimers are not repaired by the nucleotide excision repair pathway, mutations will be introduced after DNA synthesis. The dinucleotide C-G, or CpG, is also a hot spot for a specific type of mutation. In this case, methylation of the cytosine is associated with an enhanced rate of deamination to uracil, which is then replaced with thymine. This C → T transition (or G → A on the opposite strand) accounts for at least one-third of point mutations associated with polymorphisms and mutations. In addition to the fact that certain types of mutations (C → T or G → A) are relatively common, the nature of the genetic code also results in overrepresentation of certain amino acid substitutions.
Polymorphisms are sequence variations that have a frequency of at least 1%. Usually, they do not result in a perceptible phenotype. Often they consist of single base-pair substitutions that do not alter the protein coding sequence because of the degenerate nature of the genetic code (synonymous polymorphism), although it is possible that some might alter mRNA stability, translation, or the amino acid sequence (nonsynonymous polymorphism) (Fig. 82-10). The detection of sequence variants poses a practical problem because it is often unclear whether it creates a mutation with functional consequences or a benign polymorphism. In this situation, the sequence alteration is described as variant of unknown significance (VUS).
MUTATION RATES Mutations represent an important cause of genetic diversity as well as disease. Mutation rates are difficult to determine in humans because many mutations are silent and because testing is often not adequate to detect the phenotypic consequences. Mutation rates vary in different genes but are estimated to occur at a rate of ~10–10/bp per cell division. Germline mutation rates (as opposed to somatic mutations) are relevant in the transmission of genetic disease. Because the population of oocytes is established very early in development, only ~20 cell divisions are required for completed oogenesis, whereas spermatogenesis involves ~30 divisions by the time of puberty and 20 cell divisions each year thereafter. Consequently, the probability of acquiring new point mutations is much greater in the male germline than the female germline, in which rates of aneuploidy are increased (Chap. 83e). Thus, the incidence of new point mutations in spermatogonia increases with paternal age (e.g., achondrodysplasia, Marfan’s syndrome, neurofibromatosis). It is estimated that about 1 in 10 sperm carries a new deleterious mutation. The rates for new mutations are calculated most readily for autosomal dominant and X-linked disorders and are ~10–5–10–6/locus per generation. Because most monogenic diseases are relatively rare, new mutations account for a significant fraction of cases. This is important in the context of genetic counseling, because a new mutation can be transmitted to the affected individual but does not necessarily imply that the parents are at risk to transmit the disease to other children. An exception to this is when the new mutation occurs early in germline development, leading to gonadal mosaicism.
UNEQUAL CROSSING-OVER Normally, DNA recombination in germ cells occurs with remarkable fidelity to maintain the precise junction sites for the exchanged DNA sequences (Fig. 82-6). However, mispairing of homologous sequences leads to unequal crossover, with gene duplication on one of the chromosomes and gene deletion on the other chromosome. A significant fraction of growth hormone (GH) gene deletions, for example, involve unequal crossing-over (Chap. 402). The GH gene is a member of a large gene cluster that includes a GH variant gene as well as several structurally related chorionic somatomammotropin genes and pseudogenes (highly homologous but functionally inactive relatives of a normal gene). Because such gene clusters contain multiple homologous DNA sequences arranged in tandem, they are particularly prone to undergo recombination and, consequently, gene duplication or deletion. On the other hand, duplication of the PMP22 gene because of unequal crossing-over results in increased gene dosage and type IA Charcot-Marie-Tooth disease. Unequal crossing-over resulting in deletion of PMP22 causes a distinct neuropathy called hereditary liability to pressure palsy (Chap. 459).
Glucocorticoid-remediable aldosteronism (GRA) is caused by a gene fusion or rearrangement involving the genes that encode aldosterone synthase (CYP11B2) and steroid 11β-hydroxylase (CYP11B1), normally arranged in tandem on chromosome 8q. These two genes are 95% identical, predisposing to gene duplication and deletion by unequal crossing-over. The rearranged gene product contains the regulatory regions of 11β-hydroxylase fused to the coding sequence of aldosterone synthetase. Consequently, the latter enzyme is expressed in the adrenocorticotropic hormone (ACTH)–dependent zona fasciculata of the adrenal gland, resulting in overproduction of mineralocorticoids and hypertension (Chap. 406).
Gene conversion refers to a nonreciprocal exchange of homologous genetic information. It has been used to explain how an internal portion of a gene is replaced by a homologous segment copied from another allele or locus; these genetic alterations may range from a few nucleotides to a few thousand nucleotides. As a result of gene conversion, it is possible for short DNA segments of two chromosomes to be identical, even though these sequences are distinct in the parents. A practical consequence of this phenomenon is that nucleotide substitutions can occur during gene conversion between related genes, often altering the function of the gene. In disease states, gene conversion often involves intergenic exchange of DNA between a gene and a related pseudogene. For example, the 21-hydroxylase gene (CYP21A2) is adjacent to a nonfunctional pseudogene (CYP21A1P). Many of the nucleotide substitutions that are found in the CYP21A2 gene in patients with congenital adrenal hyperplasia correspond to sequences that are present in the CYP21A1P pseudogene, suggesting gene conversion as one cause of mutagenesis. In addition, mitotic gene conversion has been suggested as a mechanism to explain revertant mosaicism in which an inherited mutation is “corrected” in certain cells. For example, patients with autosomal recessive generalized atrophic benign epidermolysis bullosa have acquired reverse mutations in one of the two mutated COL17A1 alleles, leading to clinically unaffected patches of skin.
INSERTIONS AND DELETIONS Although many instances of insertions and deletions occur as a consequence of unequal crossing-over, there is also evidence for internal duplication, inversion, or deletion of DNA sequences. The fact that certain deletions or insertions appear to occur repeatedly as independent events indicates that specific regions within the DNA sequence predispose to these errors. For example, certain regions of the DMD gene, which encodes dystrophin, appear to be hot spots for deletions and result in muscular dystrophy (Chap. 462e). Some regions within the human genome are rearrangement hot spots and lead to CNVs.
ERRORS IN DNA REPAIR Because mutations caused by defects in DNA repair accumulate as somatic cells divide, these types of mutations are particularly important in the context of neoplastic disorders (Chap. 102e). Several genetic disorders involving DNA repair enzymes underscore their importance. Patients with xeroderma pigmentosum have defects in DNA damage recognition or in the nucleotide excision and repair pathway (Chap. 105). Exposed skin is dry and pigmented and is extraordinarily sensitive to the mutagenic effects of ultraviolet irradiation. More than 10 different genes have been shown to cause the different forms of xeroderma pigmentosum. This finding is consistent with the earlier classification of this disease into different complementation groups in which normal function is rescued by the fusion of cells derived from two different forms of xeroderma pigmentosum.
Ataxia telangiectasia causes large telangiectatic lesions of the face, cerebellar ataxia, immunologic defects, and hypersensitivity to ionizing radiation (Chap. 450). The discovery of the ataxia telangiectasia mutated (ATM) gene reveals that it is homologous to genes involved in DNA repair and control of cell cycle checkpoints. Mutations in the ATM gene give rise to defects in meiosis as well as increasing susceptibility to damage from ionizing radiation. Fanconi’s anemia is also associated with an increased risk of multiple acquired genetic abnormalities. It is characterized by diverse congenital anomalies and a strong predisposition to develop aplastic anemia and acute myelogenous leukemia (Chap. 132). Cells from these patients are susceptible to chromosomal breaks caused by a defect in genetic recombination. At least 13 different complementation groups have been identified, and the loci and genes associated with Fanconi’s anemia have been cloned. HNPCC (Lynch’s syndrome) is characterized by autosomal dominant transmission of colon cancer, young age (<50 years) of presentation, predisposition to lesions in the proximal large bowel, and associated malignancies such as uterine cancer and ovarian cancer. HNPCC is predominantly caused by mutations in one of several different mismatch repair (MMR) genes including MutS homologue 2 (MSH2), MutL homologue 1 and 6 (MLH1, MLH6), MSH6, PMS1, and PMS2 (Chap. 110). These proteins are involved in the detection of nucleotide mismatches and in the recognition of slipped-strand trinucleotide repeats. Germline mutations in these genes lead to microsatellite instability and a high mutation rate in colon cancer. Genetic screening tests for this disorder are now being used for families considered to be at risk (Chap. 84). Recognition of HNPCC allows early screening with colonoscopy and the implementation of prevention strategies using nonsteroidal anti-inflammatory drugs.
UNSTABLE DNA SEQUENCES Trinucleotide repeats may be unstable and expand beyond a critical number. Mechanistically, the expansion is thought to be caused by unequal recombination and slipped mispairing. A premutation represents a small increase in trinucleotide copy number. In subsequent generations, the expanded repeat may increase further in length and result in an increasingly severe phenotype, a process called dynamic mutation (see below for discussion of anticipation). Trinucleotide expansion was first recognized as a cause of the fragile × syndrome, one of the most common causes of intellectual disability. Other disorders arising from a similar mechanism include Huntington’s disease (Chap. 448), X-linked spinobulbar muscular atrophy (Chap. 452), and myotonic dystrophy (Chap. 462e). Malignant cells are also characterized by genetic instability, indicating a breakdown in mechanisms that regulate DNA repair and the cell cycle.
Functional Consequences of Mutations Functionally, mutations can be broadly classified as gain-of-function and loss-of-function mutations. Gain-of-function mutations are typically dominant (e.g., they result in phenotypic alterations when a single allele is affected). Inactivating mutations are usually recessive, and an affected individual is homozygous or compound heterozygous (e.g., carrying two different mutant alleles of the same gene) for the disease-causing mutations. Alternatively, mutation in a single allele can result in haploinsufficiency, a situation in which one normal allele is not sufficient to maintain a normal phenotype. Haploinsufficiency is a commonly observed mechanism in diseases associated with mutations in transcription factors (Table 82-2). Remarkably, the clinical features among patients with an identical mutation in a transcription factor often vary significantly. One mechanism underlying this variability consists in the influence of modifying genes. Haploinsufficiency can also affect the expression of rate-limiting enzymes. For example, haploinsufficiency in enzymes involved in heme synthesis can cause porphyrias (Chap. 430).
An increase in dosage of a gene product may also result in disease, as illustrated by the duplication of the DAX1 gene in dosage-sensitive sex reversal (Chap. 410). Mutation in a single allele can also result in loss of function due to a dominant-negative effect. In this case, the mutated allele interferes with the function of the normal gene product by one of several different mechanisms: (1) a mutant protein may interfere with the function of a multimeric protein complex, as illustrated by mutations in type 1 collagen (COL1A1, COL1A2) genes in osteogenesis imperfecta (Chap. 427); (2) a mutant protein may occupy binding sites on proteins or promoter response elements, as illustrated by thyroid hormone resistance, a disorder in which inactivated thyroid hormone receptor β binds to target genes and functions as an antagonist of normal receptors (Chap. 405); or (3) a mutant protein can be cytotoxic as in α1 antitrypsin deficiency (Chap. 314) or autosomal dominant neurohypophyseal diabetes insipidus (Chap. 404), in which the abnormally folded proteins are trapped within the endoplasmic reticulum and ultimately cause cellular damage.
Genotype and Phnotype • ALLELES, GENOTYPES, AND HAPLOTYPES An observed trait is referred to as a phenotype; the genetic information defining the phenotype is called the genotype. Alternative forms of a gene or a genetic marker are referred to as alleles. Alleles may be polymorphic variants of nucleic acids that have no apparent effect on gene expression or function. In other instances, these variants may have subtle effects on gene expression, thereby conferring adaptive advantages associated with genetic diversity. On the other hand, allelic variants may reflect mutations that clearly alter the function of a gene product. The common Glu6Val (E6V) sickle cell mutation in the β-globin gene and the ΔF508 deletion of phenylalanine (F) in the CFTR gene are examples of allelic variants of these genes that result in disease. Because each individual has two copies of each chromosome (one inherited from the mother and one inherited from the father), he or she can have only two alleles at a given locus. However, there can be many different alleles in the population. The normal or common allele is usually referred to as wild type. When alleles at a given locus are identical, the individual is homozygous. Inheriting identical copies of a mutant allele occurs in many autosomal recessive disorders, particularly in circumstances of consanguinity or isolated populations. If the alleles are different on the maternal and the paternal copy of the gene, the individual is heterozygous at this locus (Fig. 82-10). If two different mutant alleles are inherited at a given locus, the individual is said to be a compound heterozygote. Hemizygous is used to describe males with a mutation in an × chromosomal gene or a female with a loss of one × chromosomal locus.
Genotypes describe the specific alleles at a particular locus. For example, there are three common alleles (E2, E3, E4) of the apolipoprotein E (APOE) gene. The genotype of an individual can therefore be described as APOE3/4 or APOE4/4 or any other variant. These designations indicate which alleles are present on the two chromosomes in the APOE gene at locus 19q13.2. In other cases, the genotype might be assigned arbitrary numbers (e.g., 1/2) or letters (e.g., B/b) to distinguish different alleles.
A haplotype refers to a group of alleles that are closely linked together at a genomic locus (Fig. 82-4). Haplotypes are useful for tracking the transmission of genomic segments within families and for detecting evidence of genetic recombination, if the crossover event occurs between the alleles (Fig. 82-6). As an example, various alleles at the histocompatibility locus antigen (HLA) on chromosome 6p are used to establish haplotypes associated with certain disease states. For example, 21-hydroxylase deficiency, complement deficiency, and hemochromatosis are each associated with specific HLA haplotypes. It is now recognized that these genes lie in close proximity to the HLA locus, which explains why HLA associations were identified even before the disease genes were cloned and localized. In other cases, specific HLA associations with diseases such as ankylosing spondylitis (HLA-B27) or type 1 diabetes mellitus (HLA-DR4) reflect the role of specific HLA allelic variants in susceptibility to these autoimmune diseases. The characterization of common SNP haplotypes in numerous populations from different parts of the world through the HapMap Project is providing a novel tool for association studies designed to detect genes involved in the pathogenesis of complex disorders (Table 82-1). The presence or absence of certain haplotypes may also become relevant for the customized choice of medical therapies (pharmacogenomics) or for preventive strategies.
Genotype-phenotype correlation describes the association of a specific mutation and the resulting phenotype. The phenotype may differ depending on the location or type of the mutation in some genes. For example, in von Hippel–Lindau disease, an autosomal dominant multisystem disease that can include renal cell carcinoma, hemangioblastomas, and pheochromocytomas, among others, the phenotype varies greatly and the identification of the specific mutation can be clinically useful in order to predict the phenotypic spectrum.
ALLELIC HETEROGENEITY Allelic heterogeneity refers to the fact that different mutations in the same genetic locus can cause an identical or similar phenotype. For example, many different mutations of the β-globin locus can cause β thalassemia (Table 82-3) (Fig. 82-9). In essence, allelic heterogeneity reflects the fact that many different mutations are capable of altering protein structure and function. For this reason, maps of inactivating mutations in genes usually show a near-random distribution. Exceptions include (1) a founder effect, in which a particular mutation that does not affect reproductive capacity can be traced to a single individual; (2) “hot spots” for mutations, in which the nature of the DNA sequence predisposes to a recurring mutation; and (3) localization of mutations to certain domains that are particularly critical for protein function. Allelic heterogeneity creates a practical problem for genetic testing because one must often examine the entire genetic locus for mutations, because these can differ in each patient. For example, there are currently 1963 reported mutations in the CFTR gene (Fig. 82-3). Mutational analysis may initially focus on a panel of mutations that are particularly frequent (often taking the ethnic background of the patient into account), but a negative result does not exclude the presence of a mutation elsewhere in the gene. One should also be aware that mutational analyses generally focus on the coding region of a gene without considering regulatory and intronic regions. Because disease-causing mutations may be located outside the coding regions, negative results need to be interpreted with caution. The advent of more comprehensive sequencing technologies greatly facilitates concomitant mutational analyses of several genes after targeted enrichment, or even mutational analysis of the whole exome or genome. However, comprehensive sequencing can result in significant diagnostic challenges because the detection of a sequence alteration alone is not always sufficient to establish that it has a causal role.
|
SELECTED EXAMPLES OF LOCUS HETEROGENEITY AND PHENOTYPIC HETEROGENEITY |
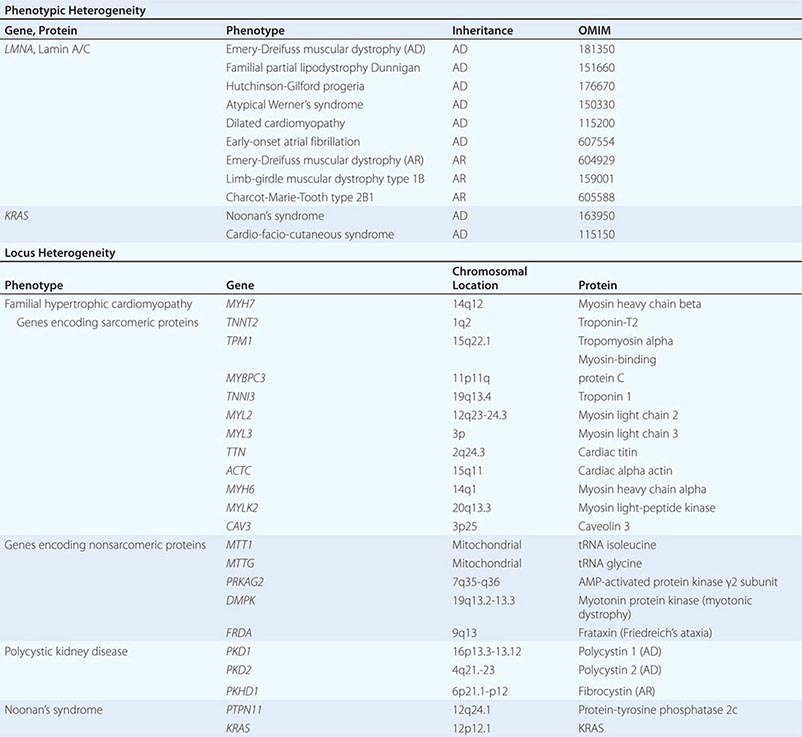
PHENOTYPIC HETEROGENEITY Phenotypic heterogeneity occurs when more than one phenotype is caused by allelic mutations (e.g., different mutations in the same gene) (Table 82-3). For example, laminopathies are monogenic multisystem disorders that result from mutations in the LMNA gene, which encodes the nuclear lamins A and C. Twelve autosomal dominant and four autosomal recessive disorders are caused by mutations in the LMNA gene. They include several forms of lipodystrophies, Emery-Dreifuss muscular dystrophy, progeria syndromes, a form of neuronal Charcot-Marie-Tooth disease (type 2B1), and a group of overlapping syndromes. Remarkably, hierarchical cluster analysis has revealed that the phenotypes vary depending on the position of the mutation (genotype-phenotype correlation). Similarly, identical mutations in the FGFR2 gene can result in very distinct phenotypes: Crouzon’s syndrome (craniofacial synostosis) or Pfeiffer’s syndrome (acrocephalopolysyndactyly).
LOCUS OR NONALLELIC HETEROGENEITY AND PHENOCOPIES Nonallelic or locus heterogeneity refers to the situation in which a similar disease phenotype results from mutations at different genetic loci (Table 82-3). This often occurs when more than one gene product produces different subunits of an interacting complex or when different genes are involved in the same genetic cascade or physiologic pathway. For example, osteogenesis imperfecta can arise from mutations in two different procollagen genes (COL1A1 or COL1A2) that are located on different chromosomes, and at least eight other genes (Chap. 427). The effects of inactivating mutations in these two genes are similar because the protein products comprise different subunits of the helical collagen fiber. Similarly, muscular dystrophy syndromes can be caused by mutations in various genes, consistent with the fact that it can be transmitted in an X-linked (Duchenne or Becker), autosomal dominant (limb-girdle muscular dystrophy type 1), or autosomal recessive (limb-girdle muscular dystrophy type 2) manner (Chap. 462e). Mutations in the X-linked DMD gene, which encodes dystrophin, are the most common cause of muscular dystrophy. This feature reflects the large size of the gene as well as the fact that the phenotype is expressed in hemizygous males because they have only a single copy of the × chromosome. Dystrophin is associated with a large protein complex linked to the membrane-associated cytoskeleton in muscle. Mutations in several different components of this protein complex can also cause muscular dystrophy syndromes. Although the phenotypic features of some of these disorders are distinct, the phenotypic spectrum caused by mutations in different genes overlaps, thereby leading to nonallelic heterogeneity. It should be noted that mutations in dystrophin also cause allelic heterogeneity. For example, mutations in the DMD gene can cause either Duchenne’s or the less severe Becker’s muscular dystrophy, depending on the severity of the protein defect.
Recognition of nonallelic heterogeneity is important for several reasons: (1) the ability to identify disease loci in linkage studies is reduced by including patients with similar phenotypes but different genetic disorders; (2) genetic testing is more complex because several different genes need to be considered along with the possibility of different mutations in each of the candidate genes; and (3) novel information is gained about how genes or proteins interact, providing unique insights into molecular physiology.
Phenocopies refer to circumstances in which nongenetic conditions mimic a genetic disorder. For example, features of toxin- or drug-induced neurologic syndromes can resemble those seen in Huntington’s disease, and vascular causes of dementia share phenotypic features with familial forms of Alzheimer’s dementia (Chap. 448). As in nonallelic heterogeneity, the presence of phenocopies has the potential to confound linkage studies and genetic testing. Patient history and subtle differences in phenotype can often provide clues that distinguish these disorders from related genetic conditions.
VARIABLE EXPRESSIVITY AND INCOMPLETE PENETRANCE The same genetic mutation may be associated with a phenotypic spectrum in different affected individuals, thereby illustrating the phenomenon of variable expressivity. This may include different manifestations of a disorder variably involving different organs (e.g., multiple endocrine neoplasia [MEN]), the severity of the disorder (e.g., cystic fibrosis), or the age of disease onset (e.g., Alzheimer’s dementia). MEN 1 illustrates several of these features. In this autosomal dominant tumor syndrome, affected individuals carry an inactivating germline mutation that is inherited in an autosomal dominant fashion. After somatic inactivation of the alternate allele, they can develop tumors of the parathyroid gland, endocrine pancreas, and the pituitary gland (Chap. 408). However, the pattern of tumors in the different glands, the age at which tumors develop, and the types of hormones produced vary among affected individuals, even within a given family. In this example, the phenotypic variability arises, in part, because of the requirement for a second somatic mutation in the normal copy of the MEN1 gene, as well as the large array of different cell types that are susceptible to the effects of MEN1 gene mutations. In part, variable expression reflects the influence of modifier genes, or genetic background, on the effects of a particular mutation. Even in identical twins, in whom the genetic constitution is essentially the same, one can occasionally see variable expression of a genetic disease.
Interactions with the environment can also influence the course of a disease. For example, the manifestations and severity of hemochromatosis can be influenced by iron intake (Chap. 428), and the course of phenylketonuria is affected by exposure to phenylalanine in the diet (Chap. 434e). Other metabolic disorders, such as hyperlipidemias and porphyria, also fall into this category. Many mechanisms, including genetic effects and environmental influences, can therefore lead to variable expressivity. In genetic counseling, it is particularly important to recognize this variability, because one cannot always predict the course of disease, even when the mutation is known.
Penetrance refers to the proportion of individuals with a mutant genotype that express the phenotype. If all carriers of a mutant express the phenotype, penetrance is complete, whereas it is said to be incomplete or reduced if some individuals do not exhibit features of the phenotype. Dominant conditions with incomplete penetrance are characterized by skipping of generations with unaffected carriers transmitting the mutant gene. For example, hypertrophic obstructive cardiomyopathy (HCM) caused by mutations in the myosin-binding protein C gene is a dominant disorder with clinical features in only a subset of patients who carry the mutation (Chap. 283). Patients who have the mutation but no evidence of the disease can still transmit the disorder to subsequent generations. In many conditions with postnatal onset, the proportion of gene carriers who are affected varies with age. Thus, when describing penetrance, one has to specify age. For example, for disorders such as Huntington’s disease or familial amyotrophic lateral sclerosis, which present later in life, the rate of penetrance is influenced by the age at which the clinical assessment is performed. Imprinting can also modify the penetrance of a disease. For example, in patients with Albright’s hereditary osteodystrophy, mutations in the Gsα subunit (GNAS1 gene) are expressed clinically only in individuals who inherit the mutation from their mother (Chap. 424).
SEX-INFLUENCED PHENOTYPES Certain mutations affect males and females quite differently. In some instances, this is because the gene resides on the × or Y sex chromosomes (X-linked disorders and Y-linked disorders). As a result, the phenotype of mutated X-linked genes will be expressed fully in males but variably in heterozygous females, depending on the degree of X-inactivation and the function of the gene. For example, most heterozygous female carriers of factor VIII deficiency (hemophilia A) are asymptomatic because sufficient factor VIII is produced to prevent a defect in coagulation (Chap. 141). On the other hand, some females heterozygous for the X-linked lipid storage defect caused by α-galactosidase A deficiency (Fabry’s disease) experience mild manifestations of painful neuropathy, as well as other features of the disease (Chap. 432e). Because only males have a Y chromosome, mutations in genes such as SRY, which causes male-to-female sex reversal, or DAZ (deleted in azoospermia), which causes abnormalities of spermatogenesis, are unique to males (Chap. 410).
Other diseases are expressed in a sex-limited manner because of the differential function of the gene product in males and females. Activating mutations in the luteinizing hormone receptor cause dominant male-limited precocious puberty in boys (Chap. 411). The phenotype is unique to males because activation of the receptor induces testosterone production in the testis, whereas it is functionally silent in the immature ovary. Biallelic inactivating mutations of the follicle-stimulating hormone (FSH) receptor cause primary ovarian failure in females because the follicles do not develop in the absence of FSH action. In contrast, affected males have a more subtle phenotype, because testosterone production is preserved (allowing sexual maturation) and spermatogenesis is only partially impaired (Chap. 411). In congenital adrenal hyperplasia, most commonly caused by 21-hydroxylase deficiency, cortisol production is impaired and ACTH stimulation of the adrenal gland leads to increased production of androgenic precursors (Chap. 406). In females, the increased androgen level causes ambiguous genitalia, which can be recognized at the time of birth. In males, the diagnosis may be made on the basis of adrenal insufficiency at birth, because the increased adrenal androgen level does not alter sexual differentiation, or later in childhood, because of the development of precocious puberty. Hemochromatosis is more common in males than in females, presumably because of differences in dietary iron intake and losses associated with menstruation and pregnancy in females (Chap. 428).
Chromosomal Disorders Chromosomal or cytogenetic disorders are caused by numerical or structural aberrations in chromosomes. For a detailed discussion of disorders of chromosome number and structure, see Chap. 83e. Deviations in chromosome number are common causes of abortions, developmental disorders, and malformations. Contiguous gene syndromes (e.g., large deletions affecting several genes) have been useful for identifying the location of new disease-causing genes. Because of the variable size of gene deletions in different patients, a systematic comparison of phenotypes and locations of deletion breakpoints allows positions of particular genes to be mapped within the critical genomic region.
Monogenic Mendelian Disorders Monogenic human diseases are frequently referred to as Mendelian disorders because they obey the principles of genetic transmission originally set forth in Gregor Mendel’s classic work. The continuously updated OMIM catalogue lists several thousand of these disorders and provides information about the clinical phenotype, molecular basis, allelic variants, and pertinent animal models (Table 82-1). The mode of inheritance for a given phenotypic trait or disease is determined by pedigree analysis. All affected and unaffected individuals in the family are recorded in a pedigree using standard symbols (Fig. 82-11). The principles of allelic segregation, and the transmission of alleles from parents to children, are illustrated in Fig. 82-12. One dominant (A) allele and one recessive (a) allele can display three Mendelian modes of inheritance: autosomal dominant, autosomal recessive, and X-linked. About 65% of human monogenic disorders are autosomal dominant, 25% are autosomal recessive, and 5% are X-linked. Genetic testing is now available for many of these disorders and plays an increasingly important role in clinical medicine (Chap. 84).
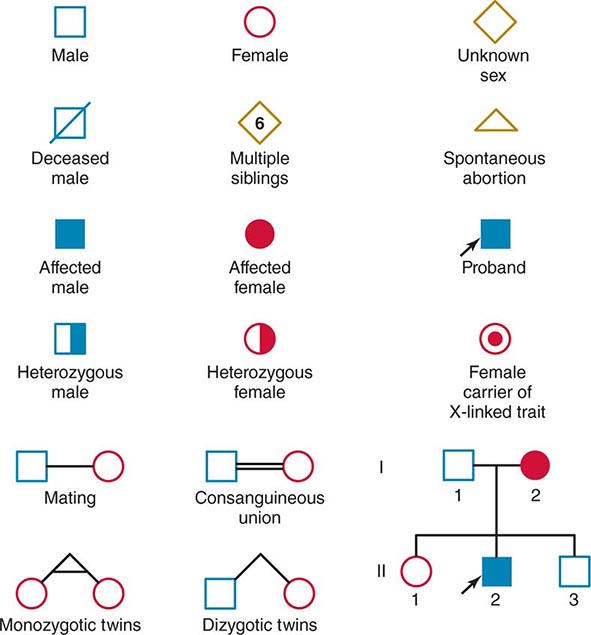
FIGURE 82-11 Standard pedigree symbols.
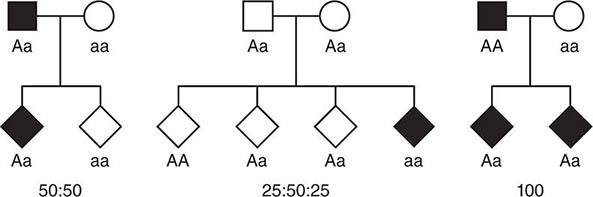
FIGURE 82-12 Segregation of alleles. Segregation of genotypes in the offspring of parents with one dominant (A) and one recessive (a) allele. The distribution of the parental alleles to their offspring depends on the combination present in the parents. Filled symbols = affected individuals.
AUTOSOMAL DOMINANT DISORDERS These disorders assume particular relevance because mutations in a single allele are sufficient to cause the disease. In contrast to recessive disorders, in which disease pathogenesis is relatively straightforward because there is loss of gene function, dominant disorders can be caused by various disease mechanisms, many of which are unique to the function of the genetic pathway involved.
In autosomal dominant disorders, individuals are affected in successive generations; the disease does not occur in the offspring of unaffected individuals. Males and females are affected with equal frequency because the defective gene resides on one of the 22 autosomes (Fig. 82-13A). Autosomal dominant mutations alter one of the two alleles at a given locus. Because the alleles segregate randomly at meiosis, the probability that an offspring will be affected is 50%. Unless there is a new germline mutation, an affected individual has an affected parent. Children with a normal genotype do not transmit the disorder. Due to differences in penetrance or expressivity (see above), the clinical manifestations of autosomal dominant disorders may be variable. Because of these variations, it is sometimes challenging to determine the pattern of inheritance.
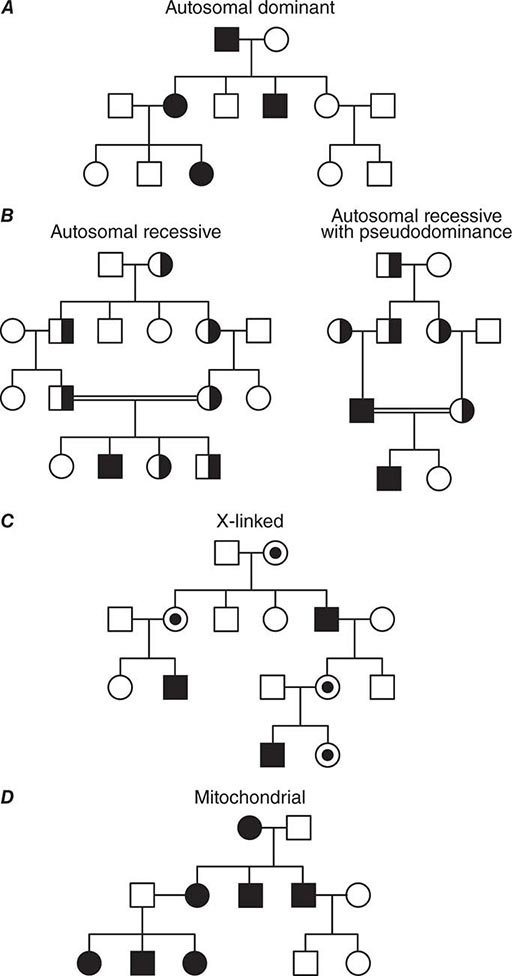
FIGURE 82-13 (A) Dominant, (B) recessive, (C) X-linked, and (D) mitochondrial (matrilinear) inheritance.
It should be recognized, however, that some individuals acquire a mutated gene from an unaffected parent. De novo germline mutations occur more frequently during later cell divisions in gametogenesis, which explains why siblings are rarely affected. As noted before, new germline mutations occur more frequently in fathers of advanced age. For example, the average age of fathers with new germline mutations that cause Marfan’s syndrome is ~37 years, whereas fathers who transmit the disease by inheritance have an average age of ~30 years.
AUTOSOMAL RECESSIVE DISORDERS In recessive disorders, the mutated alleles result in a complete or partial loss of function. They frequently involve enzymes in metabolic pathways, receptors, or proteins in signaling cascades. In an autosomal recessive disease, the affected individual, who can be of either sex, is a homozygote or compound heterozygote for a single-gene defect. With a few important exceptions, autosomal recessive diseases are rare and often occur in the context of parental consanguinity. The relatively high frequency of certain recessive disorders such as sickle cell anemia, cystic fibrosis, and thalassemia, is partially explained by a selective biologic advantage for the heterozygous state (see below). Although heterozygous carriers of a defective allele are usually clinically normal, they may display subtle differences in phenotype that only become apparent with more precise testing or in the context of certain environmental influences. In sickle cell anemia, for example, heterozygotes are normally asymptomatic. However, in situations of dehydration or diminished oxygen pressure, sickle cell crises can also occur in heterozygotes (Chap. 127).
In most instances, an affected individual is the offspring of heterozygous parents. In this situation, there is a 25% chance that the offspring will have a normal genotype, a 50% probability of a heterozygous state, and a 25% risk of homozygosity for the recessive alleles (Figs. 82-10, 82-13B). In the case of one unaffected heterozygous and one affected homozygous parent, the probability of disease increases to 50% for each child. In this instance, the pedigree analysis mimics an autosomal dominant mode of inheritance (pseudodominance). In contrast to autosomal dominant disorders, new mutations in recessive alleles are rarely manifest because they usually result in an asymptomatic carrier state.
X-LINKED DISORDERS Males have only one × chromosome; consequently, a daughter always inherits her father’s × chromosome in addition to one of her mother’s two × chromosomes. A son inherits the Y chromosome from his father and one maternal × chromosome. Thus, the characteristic features of X-linked inheritance are (1) the absence of father-to-son transmission, and (2) the fact that all daughters of an affected male are obligate carriers of the mutant allele (Fig. 82-13 C). The risk of developing disease due to a mutant X-chromosomal gene differs in the two sexes. Because males have only one × chromosome, they are hemizygous for the mutant allele; thus, they are more likely to develop the mutant phenotype, regardless of whether the mutation is dominant or recessive. A female may be either heterozygous or homozygous for the mutant allele, which may be dominant or recessive. The terms X-linked dominant or X-linked recessive are therefore only applicable to expression of the mutant phenotype in women. In addition, the expression of X-chromosomal genes is influenced by × chromosome inactivation.
Y-LINKED DISORDERS The Y chromosome has a relatively small number of genes. One such gene, the sex-region determining Y factor (SRY), which encodes the testis-determining factor (TDF), is crucial for normal male development. Normally there is infrequent exchange of sequences on the Y chromosome with the × chromosome. The SRY region is adjacent to the pseudoautosomal region, a chromosomal segment on the × and Y chromosomes with a high degree of homology. A crossing-over event occasionally involves the SRY region with the distal tip of the × chromosome during meiosis in the male. Translocations can result in XY females with the Y chromosome lacking the SRY gene or XX males harboring the SRY gene on one of the × chromosomes (Chap. 410). Point mutations in the SRY gene may also result in individuals with an XY genotype and an incomplete female phenotype. Most of these mutations occur de novo. Men with oligospermia/azoospermia frequently have microdeletions on the long arm of the Y chromosome that involve one or more of the azoospermia factor (AZF) genes.
Exceptions to Simple Mendelian Inheritance Patterns • MITOCHONDRIAL DISORDERS Mendelian inheritance refers to the transmission of genes encoded by DNA contained in the nuclear chromosomes. In addition, each mitochondrion contains several copies of a small circular chromosome (Chap. 85e). The mitochondrial DNA (mtDNA) is ~16.5 kb and encodes transfer and ribosomal RNAs and 13 core proteins that are components of the respiratory chain involved in oxidative phosphorylation and ATP generation. The mitochondrial genome does not recombine and is inherited through the maternal line because sperm does not contribute significant cytoplasmic components to the zygote. A noncoding region of the mitochondrial chromosome, referred to as D-loop, is highly polymorphic. This property, together with the absence of mtDNA recombination, makes it a valuable tool for studies tracing human migration and evolution, and it is also used for specific forensic applications.
Inherited mitochondrial disorders are transmitted in a matrilineal fashion; all children from an affected mother will inherit the disease, but it will not be transmitted from an affected father to his children (Fig. 82-13D). Alterations in the mtDNA that involves enzymes required for oxidative phosphorylation lead to reduction of ATP supply, generation of free radicals, and induction of apoptosis. Several syndromic disorders arising from mutations in the mitochondrial genome are known in humans and they affect both protein-coding and tRNA genes (Chap. 85e). The broad clinical spectrum often involves (cardio) myopathies and encephalopathies because of the high dependence of these tissues on oxidative phosphorylation. The age of onset and the clinical course are highly variable because of the unusual mechanisms of mtDNA transmission, which replicates independently from nuclear DNA. During cell replication, the proportion of wild-type and mutant mitochondria can drift among different cells and tissues. The resulting heterogeneity in the proportion of mitochondria with and without a mutation is referred to as heteroplasmia and underlies the phenotypic variability that is characteristic of mitochondrial diseases.
Acquired somatic mutations in mitochondria are thought to be involved in several age-dependent degenerative disorders affecting predominantly muscle and the peripheral and central nervous system (e.g., Alzheimer’s and Parkinson’s diseases). Establishing that an mtDNA alteration is causal for a clinical phenotype is challenging because of the high degree of polymorphism in mtDNA and the phenotypic variability characteristic of these disorders. Certain pharmacologic treatments may have an impact on mitochondria and/or their function. For example, treatment with the antiretroviral compound azidothymidine (AZT) causes an acquired mitochondrial myopathy through depletion of muscular mtDNA.
MOSAICISM Mosaicism refers to the presence of two or more genetically distinct cell lines in the tissues of an individual. It results from a mutation that occurs during embryonic, fetal, or extrauterine development. The developmental stage at which the mutation arises will determine whether germ cells and/or somatic cells are involved. Chromosomal mosaicism results from nondisjunction at an early embryonic mitotic division, leading to the persistence of more than one cell line, as exemplified by some patients with Turner’s syndrome (Chap. 410). Somatic mosaicism is characterized by a patchy distribution of genetically altered somatic cells. The McCune-Albright syndrome, for example, is caused by activating mutations in the stimulatory G protein α (Gsα) that occur early in development (Chap. 424). The clinical phenotype varies depending on the tissue distribution of the mutation; manifestations include ovarian cysts that secrete sex steroids and cause precocious puberty, polyostotic fibrous dysplasia, café-au-lait skin pigmentation, growth hormone–secreting pituitary adenomas, and hypersecreting autonomous thyroid nodules (Chap. 412).
X-INACTIVATION, IMPRINTING, AND UNIPARENTAL DISOMY According to traditional Mendelian principles, the parental origin of a mutant gene is irrelevant for the expression of the phenotype. There are, however, important exceptions to this rule. X-inactivation prevents the expression of most genes on one of the two × chromosomes in every cell of a female. Gene inactivation through genomic imprinting occurs on selected chromosomal regions of autosomes and leads to inheritable preferential expression of one of the parental alleles. It is of pathophysiologic importance in disorders where the transmission of disease is dependent on the sex of the transmitting parent and, thus, plays an important role in the expression of certain genetic disorders. Two classic examples are the Prader-Willi syndrome and Angelman’s syndrome (Chap. 83e). Prader-Willi syndrome is characterized by diminished fetal activity, obesity, hypotonia, mental retardation, short stature, and hypogonadotropic hypogonadism. Deletions of the paternal copy of the Prader-Willi locus located on the short arm of chromosome 15 result in a contiguous gene syndrome involving missing paternal copies of the necdin and SNRPN genes, among others. In contrast, patients with Angelman’s syndrome, characterized by mental retardation, seizures, ataxia, and hypotonia, have deletions involving the maternal copy of this region on chromosome 15. These two syndromes may also result from uniparental disomy. In this case, the syndromes are not caused by deletions on chromosome 15 but by the inheritance of either two maternal chromosomes (Prader-Willi syndrome) or two paternal chromosomes (Angelman’s syndrome). Lastly, the two distinct phenotypes can also be caused by an imprinting defect that impairs the resetting of the imprint during zygote development (defect in the father leads to Prader-Willi syndrome; defect in the mother leads to Angelman’s syndrome).
Imprinting and the related phenomenon of allelic exclusion may be more common than currently documented, because it is difficult to examine levels of mRNA expression from the maternal and paternal alleles in specific tissues or in individual cells. Genomic imprinting, or uniparental disomy, is involved in the pathogenesis of several other disorders and malignancies (Chap. 83e). For example, hydatidiform moles contain a normal number of diploid chromosomes, but they are all of paternal origin. The opposite situation occurs in ovarian teratomata, with 46 chromosomes of maternal origin. Expression of the imprinted gene for insulin-like growth factor II (IGF-II) is involved in the pathogenesis of the cancer-predisposing Beckwith-Wiedemann syndrome (BWS) (Chap. 101e). These children show somatic overgrowth with organomegalies and hemihypertrophy, and they have an increased risk of embryonal malignancies such as Wilms’ tumor. Normally, only the paternally derived copy of the IGF-II gene is active and the maternal copy is inactive. Imprinting of the IGF-II gene is regulated by H19, which encodes an RNA transcript that is not translated into protein. Disruption or lack of H19 methylation leads to a relaxation of IGF-II imprinting and expression of both alleles. Alterations of the epigenome through gain and loss of DNA methylation, as well as altered histone modifications, play an important role in the pathogenesis of malignancies.
SOMATIC MUTATIONS Cancer can be considered a genetic disease at the cellular level (Chap. 101e). Cancers are monoclonal in origin, indicating that they have arisen from a single precursor cell with one or several mutations in genes controlling growth (proliferation or apoptosis) and/or differentiation. These acquired somatic mutations are restricted to the tumor and its metastases and are not found in the surrounding normal tissue. The molecular alterations include dominant gain-of-function mutations in oncogenes, recessive loss-of-function mutations in tumor-suppressor genes and DNA repair genes, gene amplification, and chromosome rearrangements. Rarely, a single mutation in certain genes may be sufficient to transform a normal cell into a malignant cell. In most cancers, however, the development of a malignant phenotype requires several genetic alterations for the gradual progression from a normal cell to a cancerous cell, a phenomenon termed multistep carcinogenesis (Chaps. 101e and 102e). Genome-wide analyses of cancers using deep sequencing often reveal somatic rearrangements resulting in fusion genes and mutations in multiple genes. Comprehensive sequence analyses provide further insight into genetic heterogeneity within malignancies; these include intratumoral heterogeneity among the cells of the primary tumor, intermetastatic and intrametastatic heterogeneity, and interpatient differences. These analyses further support the notion of cancer as an ongoing process of clonal evolution, in which successive rounds of clonal selection within the primary tumor and metastatic lesions result in diverse genetic and epigenetic alterations that require targeted (personalized) therapies. The heterogeneity of mutations within a tumor can also lead to resistance to target therapies because cells with mutations that are resistant to the therapy, even if they are a minor part of the tumor population, will be selected as the more sensitive cells are killed. Most human tumors express telomerase, an enzyme formed of a protein and an RNA component, which adds telomere repeats at the ends of chromosomes during replication. This mechanism impedes shortening of the telomeres, which is associated with senescence in normal cells and is associated with enhanced replicative capacity in cancer cells. Telomerase inhibitors provide a novel strategy for treating advanced human cancers.
In many cancer syndromes, there is an inherited predisposition to tumor formation. In these instances, a germline mutation is inherited in an autosomal dominant fashion inactivating one allele of an autosomal tumor-suppressor gene. If the second allele is inactivated by a somatic mutation or by epigenetic silencing in a given cell, this will lead to neoplastic growth (Knudson two-hit model). Thus, the defective allele in the germline is transmitted in a dominant mode, although tumorigenesis results from a biallelic loss of the tumor-suppressor gene in an affected tissue. The classic example to illustrate this phenomenon is retinoblastoma, which can occur as a sporadic or hereditary tumor. In sporadic retinoblastoma, both copies of the retinoblastoma (RB) gene are inactivated through two somatic events. In hereditary retinoblastoma, one mutated or deleted RB allele is inherited in an autosomal dominant manner and the second allele is inactivated by a subsequent somatic mutation. This two-hit model applies to other inherited cancer syndromes such as MEN 1 (Chap. 408) and neurofibromatosis type 2 (Chap. 118).
NUCLEOTIDE REPEAT EXPANSION DISORDERS Several diseases are associated with an increase in the number of nucleotide repeats above a certain threshold (Table 82-4). The repeats are sometimes located within the coding region of the genes, as in Huntington’s disease or the X-linked form of spinal and bulbar muscular atrophy (SBMA; Kennedy’s syndrome). In other instances, the repeats probably alter gene regulatory sequences. If an expansion is present, the DNA fragment is unstable and tends to expand further during cell division. The length of the nucleotide repeat often correlates with the severity of the disease. When repeat length increases from one generation to the next, disease manifestations may worsen or be observed at an earlier age; this phenomenon is referred to as anticipation. In Huntington’s disease, for example, there is a correlation between age of onset and length of the triplet codon expansion (Chap. 444e). Anticipation has also been documented in other diseases caused by dynamic mutations in trinucleotide repeats (Table 82-4). The repeat number may also vary in a tissue-specific manner. In myotonic dystrophy, the CTG repeat may be tenfold greater in muscle tissue than in lymphocytes (Chap. 462e).
|
SELECTED TRINUCLEOTIDE REPEAT DISORDERS |
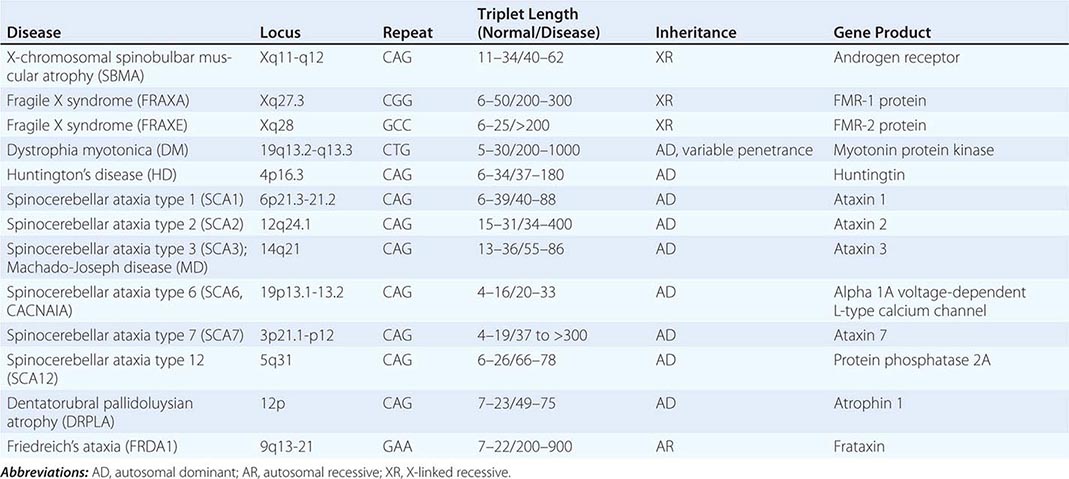
Complex Genetic Disorders The expression of many common diseases such as cardiovascular disease, hypertension, diabetes, asthma, psychiatric disorders, and certain cancers is determined by a combination of genetic background, environmental factors, and lifestyle. A trait is called polygenic if multiple genes contribute to the phenotype or multifactorial if multiple genes are assumed to interact with environmental factors. Genetic models for these complex traits need to account for genetic heterogeneity and interactions with other genes and the environment. Complex genetic traits may be influenced by modifier genes that are not linked to the main gene involved in the pathogenesis of the trait. This type of gene-gene interaction, or epistasis, plays an important role in polygenic traits that require the simultaneous presence of variations in multiple genes to result in a pathologic phenotype.
Type 2 diabetes mellitus provides a paradigm for considering a multifactorial disorder, because genetic, nutritional, and lifestyle factors are intimately interrelated in disease pathogenesis (Table 82-5) (Chap. 417). The identification of genetic variations and environmental factors that either predispose to or protect against disease is essential for predicting disease risk, designing preventive strategies, and developing novel therapeutic approaches. The study of rare monogenic diseases may provide insight into some of the genetic and molecular mechanisms important in the pathogenesis of complex diseases. For example, the identification of the genes causing monogenic forms of permanent neonatal diabetes mellitus or maturity-onset diabetes defined them as candidate genes in the pathogenesis of diabetes mellitus type 2 (Tables 82-2 and 82-5). Genome scans have identified numerous genes and loci that may be associated with susceptibility to development of diabetes mellitus in certain populations. Efforts to identify susceptibility genes require very large sample sizes, and positive results may depend on ethnicity, ascertainment criteria, and statistical analysis. Association studies analyzing the potential influence of (biologically functional) SNPs and SNP haplotypes on a particular phenotype are providing new insights into the genes involved in the pathogenesis of these common disorders. Large variants ([micro]deletions, duplications, and inversions) present in the human population also contribute to the pathogenesis of complex disorders, but their contributions remain poorly understood.
|
GENES AND LOCI INVOLVED IN MONO- AND POLYGENIC FORMS OF DIABETES |
Linkage and Association Studies There are two primary strategies for mapping genes that cause or increase susceptibility to human disease: (1) classic linkage can be performed based on a known genetic model or, when the model is unknown, by studying pairs of affected relatives; or (2) disease genes can be mapped using allelic association studies (Table 82-6).
|
GENETIC APPROACHES FOR IDENTIFYING DISEASE GENES |
GENETIC LINKAGE Genetic linkage refers to the fact that genes are physically connected, or linked, to one another along the chromosomes. Two fundamental principles are essential for understanding the concept of linkage: (1) when two genes are close together on a chromosome, they are usually transmitted together, unless a recombination event separates them (Figs. 82-6); and (2) the odds of a crossover, or recombination event, between two linked genes is proportional to the distance that separates them. Thus, genes that are farther apart are more likely to undergo a recombination event than genes that are very close together. The detection of chromosomal loci that segregate with a disease by linkage can be used to identify the gene responsible for the disease (positional cloning) and to predict the odds of disease gene transmission in genetic counseling.
Polymorphisms are essential for linkage studies because they provide a means to distinguish the maternal and paternal chromosomes in an individual. On average, 1 out of every 1000 bp varies from one person to the next. Although this degree of variation seems low (99.9% identical), it means that >3 million sequence differences exist between any two unrelated individuals and the probability that the sequence at such loci will differ on the two homologous chromosomes is high (often >70–90%). These sequence variations include variable number of tandem repeats (VNTRs), short tandem repeats (STRs), and SNPs. Most STRs, also called polymorphic microsatellite markers, consist of di-, tri-, or tetranucleotide repeats that can be characterized readily using the polymerase chain reaction (PCR). Characterization of SNPs, using DNA chips or beads, permits comprehensive analyses of genetic variation, linkage, and association studies. Although these sequence variations often have no apparent functional consequences, they provide much of the basis for variation in genetic traits.
In order to identify a chromosomal locus that segregates with a disease, it is necessary to characterize polymorphic DNA markers from affected and unaffected individuals of one or several pedigrees. One can then assess whether certain marker alleles cosegregate with the disease. Markers that are closest to the disease gene are less likely to undergo recombination events and therefore receive a higher linkage score. Linkage is expressed as a lod (logarithm of odds) score—the ratio of the probability that the disease and marker loci are linked rather than unlinked. Lod scores of +3 (1000:1) are generally accepted as supporting linkage, whereas a score of –2 is consistent with the absence of linkage.
ALLELIC ASSOCIATION, LINKAGE DISEQUILIBRIUM, AND HAPLOTYPES Allelic association refers to a situation in which the frequency of an allele is significantly increased or decreased in individuals affected by a particular disease in comparison to controls. Linkage and association differ in several aspects. Genetic linkage is demonstrable in families or sibships. Association studies, on the other hand, compare a population of affected individuals with a control population. Association studies can be performed as case-control studies that include unrelated affected individuals and matched controls or as family-based studies that compare the frequencies of alleles transmitted or not transmitted to affected children.
Allelic association studies are particularly useful for identifying susceptibility genes in complex diseases. When alleles at two loci occur more frequently in combination than would be predicted (based on known allele frequencies and recombination fractions), they are said to be in linkage disequilibrium. Evidence for linkage disequilibrium can be helpful in mapping disease genes because it suggests that the two loci are tightly linked.
Detecting the genetic factors contributing to the pathogenesis of common complex disorders remains a great challenge. In many instances, these are low-penetrance alleles (e.g., variations that individually have a subtle effect on disease development, and they can only be identified by unbiased GWAS) (Catalog of Published Genome-Wide Association Studies; Table 82-1) (Fig. 82-14). Most variants occur in noncoding or regulatory sequences but do not alter protein structure. The analysis of complex disorders is further complicated by ethnic differences in disease prevalence, differences in allele frequencies in known susceptibility genes among different populations, locus and allelic heterogeneity, gene-gene and gene-environment interactions, and the possibility of phenocopies. The data generated by the HapMap Project are greatly facilitating GWAS for the characterization of complex disorders. Adjacent SNPs are inherited together as blocks, and these blocks can be identified by genotyping selected marker SNPs, so-called Tag SNPs, thereby reducing cost and workload (Fig. 82-4). The availability of this information permits the characterization of a limited number of SNPs to identify the set of haplotypes present in an individual (e.g., in cases and controls). This, in turn, permits performing GWAS by searching for associations of certain haplotypes with a disease phenotype of interest, an essential step for unraveling the genetic factors contributing to complex disorders.
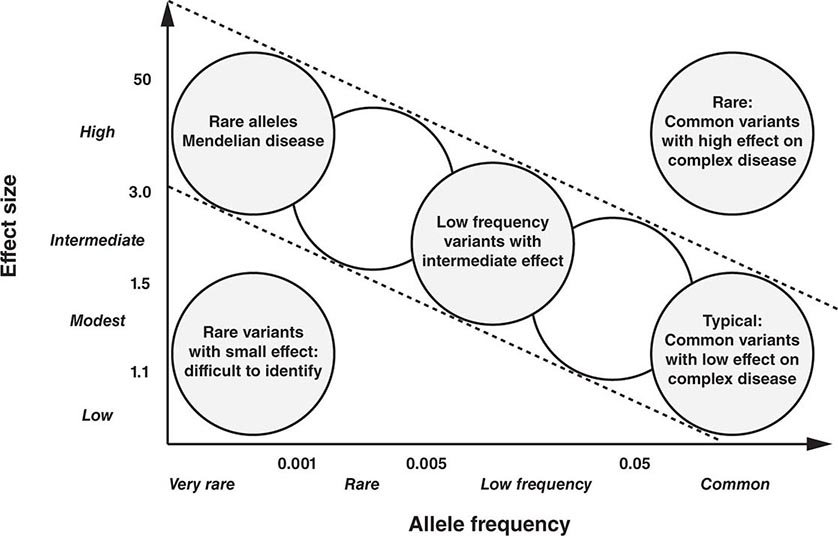
FIGURE 82-14 Relationship between allele frequency and effect size in monogenic and polygenic disorders. In classic Mendelian disorders, the allele frequency is typically low but has a high impact (single gene disorder). This contrasts with polygenic disorders that require the combination of multiple low impact alleles that are frequently quite common in the general population.
POPULATION GENETICS In population genetics, the focus changes from alterations in an individual’s genome to the distribution pattern of different genotypes in the population. In a case where there are only two alleles, A and a, the frequency of the genotypes will be p2 + 2pq + q2 = 1, with p2 corresponding to the frequency of AA, 2pq to the frequency of Aa, and q2 to aa. When the frequency of an allele is known, the frequency of the genotype can be calculated. Alternatively, one can determine an allele frequency if the genotype frequency has been determined.
Allele frequencies vary among ethnic groups and geographic regions. For example, heterozygous mutations in the CFTR gene are relatively common in populations of European origin but are rare in the African population. Allele frequencies may vary because certain allelic variants confer a selective advantage. For example, heterozygotes for the sickle cell mutation, which is particularly common in West Africa, are more resistant to malarial infection because the erythrocytes of heterozygotes provide a less favorable environment for Plasmodium parasites. Although homozygosity for the sickle cell mutation is associated with severe anemia and sickle crises (Chap. 127), heterozygotes have a higher probability of survival because of the reduced morbidity and mortality from malaria; this phenomenon has led to an increased frequency of the mutant allele. Recessive conditions are more prevalent in geographically isolated populations because of the more restricted gene pool.
APPROACH TO THE PATIENT:
Inherited Disorders
For the practicing clinician, the family history remains an essential step in recognizing the possibility of a hereditary predisposition to disease. When taking the history, it is useful to draw a detailed pedigree of the first-degree relatives (e.g., parents, siblings, and children), because they share 50% of genes with the patient. Standard symbols for pedigrees are depicted in Fig. 82-11. The family history should include information about ethnic background, age, health status, and deaths, including infants. Next, the physician should explore whether there is a family history of the same or related illnesses to the current problem. An inquiry focused on commonly occurring disorders such as cancers, heart disease, and diabetes mellitus should follow. Because of the possibility of age-dependent expressivity and penetrance, the family history will need intermittent updating. If the findings suggest a genetic disorder, the clinician should assess whether some of the patient’s relatives may be at risk of carrying or transmitting the disease. In this circumstance, it is useful to confirm and extend the pedigree based on input from several family members. This information may form the basis for genetic counseling, carrier detection, early intervention, and disease prevention in relatives of the index patient (Chap. 84).
In instances where a diagnosis at the molecular level may be relevant, it is important to identify an appropriate laboratory that can perform the appropriate test. Genetic testing is available for a rapidly growing number of monogenic disorders through commercial laboratories. For uncommon disorders, the test may only be performed in a specialized research laboratory. Approved laboratories offering testing for inherited disorders can be identified in continuously updated online resources (e.g., GeneTests; Table 82-1). If genetic testing is considered, the patient and the family should be counseled about the potential implications of positive results, including psychological distress and the possibility of discrimination. The patient or caretakers should be informed about the meaning of a negative result, technical limitations, and the possibility of false-negative and inconclusive results. For these reasons, genetic testing should only be performed after obtaining informed consent. Published ethical guidelines address the specific aspects that should be considered when testing children and adolescents. Genetic testing should usually be limited to situations in which the results may have an impact on medical management.
IDENTIFYING THE DISEASE-CAUSING GENE
Genomic medicine aims to enhance the quality of medical care through the use of genotypic analysis (DNA testing) to identify genetic predisposition to disease, to select more specific pharmacotherapy, and to design individualized medical care based on genotype. Genotype can be deduced by analysis of protein (e.g., hemoglobin, apoprotein E), mRNA, or DNA. However, technologic advances have made DNA analysis particularly useful because it can be readily applied.
DNA testing is performed by mutational analysis or linkage studies in individuals at risk for a genetic disorder known to be present in a family. Mass screening programs require tests of high sensitivity and specificity to be cost-effective. Prerequisites for the success of genetic screening programs include the following: that the disorder is potentially serious; that it can be influenced at a presymptomatic stage by changes in behavior, diet, and/or pharmaceutical manipulations; and that the screening does not result in any harm or discrimination. Screening in Jewish populations for the autosomal recessive neurodegenerative storage disease Tay-Sachs has reduced the number of affected individuals. In contrast, screening for sickle cell trait/disease in African Americans has led to unanticipated problems of discrimination by health insurers and employers. Mass screening programs harbor additional potential problems. For example, screening for the most common genetic alteration in cystic fibrosis, the ΔF508 mutation with a frequency of ~70% in northern Europe, is feasible and seems to be effective. One has to keep in mind, however, that there is pronounced allelic heterogeneity and that the disease can be caused by about 2000 other mutations. The search for these less common mutations would substantially increase costs but not the effectiveness of the screening program as a whole. Next-generation genome sequencing permits comprehensive and cost-effective mutational analyses after selective enrichment of candidate genes. For example, tests that sequence all the common genes causing hereditary deafness are already commercially available. Occupational screening programs aim to detect individuals with increased risk for certain professional activities (e.g., α1 antitrypsin deficiency and smoke or dust exposure). Integrating genomic data into electronic medical records is evolving and may provide significant decision support at the point of care, for example, by providing the clinician with genomic data and decision algorithms for the prescription of drugs that are subject to pharmacogenetic influences.
Mutational Analyses DNA sequence analysis is now widely used as a diagnostic tool and has significantly enhanced diagnostic accuracy. It is used for determining carrier status and for prenatal testing in monogenic disorders (Chap. 84). Numerous techniques, discussed in previous versions of this chapter, are available for the detection of mutations. In a very broad sense, one can distinguish between techniques that allow for screening of known mutations (screening mode) or techniques that definitively characterize mutations. Analyses of large alterations in the genome are possible using classic methods such as cytogenetics, fluorescent in situ hybridization (FISH), and Southern blotting (Chap. 83e), as well as more sensitive novel techniques that search for multiple single exon deletions or duplications.
More discrete sequence alterations rely heavily on the use of PCR, which allows rapid gene amplification and analysis. Moreover, PCR makes it possible to perform genetic testing and mutational analysis with small amounts of DNA extracted from leukocytes or even from single cells, buccal cells, or hair roots. DNA sequencing can be performed directly on PCR products or on fragments cloned into plasmid vectors amplified in bacterial host cells. Sequencing of all exons of the genome or selected chromosomes, or sequencing of numerous candidate genes in a single run, is now possible with next-generation sequencing platforms.
The majority of traditional diagnostic methods were gel-based. Novel technologies for the analysis of mutations, genotyping, large-scale sequencing, and mRNA expression profiles are undergoing rapid evolution. DNA chip technologies allow hybridization of DNA or RNA to hundreds of thousands of probes simultaneously. Microarrays are being used clinically for mutational analysis of several human disease genes, as well as for the identification of viral or bacterial sequence variations. With advances in high-throughput DNA sequencing technology, complete sequencing of the genome or an exome has entered the clinical realm. Although comprehensive sequencing of large genomic regions or multiple genes is already a reality, the subsequent bioinformatics analysis, assembly of sequence fragments, and comparative alignments remains a significant and commonly underestimated challenge. The discovery of incidental (or secondary) findings that are unrelated to the indication for the sequencing analysis but indicators of other disorders of potential relevance for patient care can pose a difficult ethical dilemma. It can lead to the detection of undiagnosed medically actionable genetic conditions, but can also reveal deleterious mutations that cannot be influenced, as numerous sequence variants are of unknown significance.
A general algorithm for the approach to mutational analysis is outlined in Fig. 82-15. The importance of a detailed clinical phenotype cannot be overemphasized. This is the step where one should also consider the possibility of genetic heterogeneity and phenocopies. If obvious candidate genes are suggested by the phenotype, they can be analyzed directly. After identification of a mutation, it is essential to demonstrate that it segregates with the phenotype. The functional characterization of novel mutations is labor intensive and may require analyses in vitro or in transgenic models in order to document the relevance of the genetic alteration.
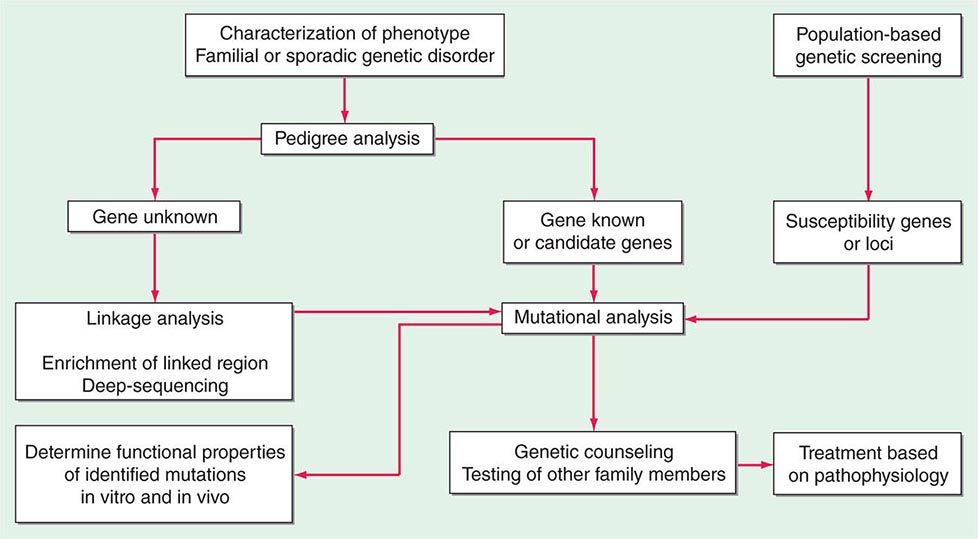
FIGURE 82-15 Approach to genetic disease.
Prenatal diagnosis of numerous genetic diseases in instances with a high risk for certain disorders is now possible by direct DNA analysis. Amniocentesis involves the removal of a small amount of amniotic fluid, usually at 16 weeks of gestation. Cells can be collected and submitted for karyotype analyses, FISH, and mutational analysis of selected genes. The main indications for amniocentesis include advanced maternal age (>35 years), an abnormal serum triple marker test (α-fetoprotein, β human chorionic gonadotropin, pregnancy-associated plasma protein A, or unconjugated estriol), a family history of chromosomal abnormalities, or a Mendelian disorder amenable to genetic testing. Prenatal diagnosis can also be performed by chorionic villus sampling (CVS), in which a small amount of the chorion is removed by a transcervical or transabdominal biopsy. Chromosomes and DNA obtained from these cells can be submitted for cytogenetic and mutational analyses. CVS can be performed earlier in gestation (weeks 9–12) than amniocentesis, an aspect that may be of relevance when termination of pregnancy is a consideration. Later in pregnancy, beginning at about 18 weeks of gestation, percutaneous umbilical blood sampling (PUBS) permits collection of fetal blood for lymphocyte culture and analysis. Recently, the entire fetal genome has been determined prenatally from cells taken from the mother’s plasma through deep sequencing and the counting of parental haplotypes, or by inferring it from DNA sequences obtained from blood samples from the mother, father, and umbilical cord. These approaches enable screening for clinically relevant and deleterious alleles inherited from the parents, as well as for de novo germline mutations, and they may have the potential to change the diagnosis of genetic disorders in the prenatal setting.
In combination with in vitro fertilization (IVF) techniques, it is even possible to perform genetic diagnoses in a single cell removed from the four- to eight-cell embryo or to analyze the first polar body from an oocyte. Preconceptual diagnosis thereby avoids therapeutic abortions but is costly and labor intensive. It should be emphasized that excluding a specific disorder by any of these approaches is never equivalent to the assurance of having a normal child.
Mutations in certain cancer susceptibility genes such as BRCA1 and BRCA2 may identify individuals with an increased risk for the development of malignancies and result in risk-reducing interventions. The detection of mutations is an important diagnostic and prognostic tool in leukemias and lymphomas. The demonstration of the presence or absence of mutations and polymorphisms is also relevant for the rapidly evolving field of pharmacogenomics, including the identification of differences in drug treatment response or metabolism as a function of genetic background. For example, the thiopurine drugs 6-mercaptopurine and azathioprine are commonly used cytotoxic and immunosuppressive agents. They are metabolized by thiopurine methyltransferase (TPMT), an enzyme with variable activity associated with genetic polymorphisms in 10% of whites and complete deficiency in about 1 in 300 individuals. Patients with intermediate or deficient TPMT activity are at risk for excessive toxicity, including fatal myelosuppression. Characterization of these polymorphisms allows mercaptopurine doses to be modified based on TPMT genotype. Pharmacogenomics may increasingly permit individualized drug therapy, improve drug effectiveness, reduce adverse side effects, and provide cost-effective pharmaceutical care (Chap. 5).
ETHICAL ISSUES
Determination of the association of genetic defects with disease, comprehensive data of an individual’s genome, and studies of genetic variation raise many ethical and legal issues. Genetic information is generally regarded as sensitive information that should not be readily accessible without explicit consent (genetic privacy). The disclosure of genetic information may risk possible discrimination by insurers or employers. The scientific components of the Human Genome Project have been paralleled by efforts to examine ethical, social, and legal implications. An important milestone emerging from these endeavors consists in the Genetic Information Nondiscrimination Act (GINA), signed into law in 2008, which aims to protect asymptomatic individuals against the misuse of genetic information for health insurance and employment. It does not, however, protect the symptomatic individual. Provisions of the U.S. Patient Protection and Affordable Care Act, effective in 2014, will fill this gap and prohibit exclusion from, or termination of, health insurance based on personal health status. Potential threats to the maintenance of genetic privacy consist in the emerging integration of genomic data into electronic medical records, compelled disclosures of health records, and direct-to-consumer genetic testing.
It is widely accepted that identifying disease-causing genes can lead to improvements in diagnosis, treatment, and prevention. However, the information gleaned from genotypic results can have quite different impacts, depending on the availability of strategies to modify the course of disease (Chap. 84). For example, the identification of mutations that cause MEN 2 or hemochromatosis allows specific interventions for affected family members. On the other hand, at present, the identification of an Alzheimer’s or Huntington’s disease gene does not currently alter therapy and outcomes. Most genetic disorders are likely to fall into an intermediate category where the opportunity for prevention or treatment is significant but limited (Chap. 84). However, the progress in this area is unpredictable, as underscored by the finding that angiotensin II receptor blockers may slow disease progression in Marfan’s syndrome. Genetic test results can generate anxiety in affected individuals and family members. Comprehensive sequence analyses are particularly challenging because most individuals can be expected to harbor several serious recessive gene mutations.
The impact of genetic testing on health care costs is currently unclear. It is likely to vary among disorders and depend on the availability of effective therapeutic modalities. A significant problem arises from the marketing of genetic testing directly to consumers by commercial companies. The validity of these tests has not been defined, and there are numerous concerns about the lack of appropriate regulatory oversight, the accuracy and confidentiality of genetic information, the availability of counseling, and the handling of these results.
Many issues raised by the genome project are familiar, in principle, to medical practitioners. For example, an asymptomatic patient with increased low-density lipoprotein (LDL) cholesterol, high blood pressure, or a strong family history of early myocardial infarction is known to be at increased risk of coronary heart disease. In such cases, it is clear that the identification of risk factors and an appropriate intervention are beneficial. Likewise, patients with phenylketonuria, cystic fibrosis, or sickle cell anemia are often identified as having a genetic disease early in life. These precedents can be helpful for adapting policies that relate to genetic information. We can anticipate similar efforts, whether based on genotypes or other markers of genetic predisposition, to be applied to many disorders. One confounding aspect of the rapid expansion of information is that our ability to make clinical decisions often lags behind initial insights into genetic mechanisms of disease. For example, when genes that predispose to breast cancer such as BRCA1 are described, they generate tremendous public interest in the potential to predict disease, but many years of clinical research are still required to rigorously establish genotype and phenotype correlations.
Genomics may contribute to improvements in global health by providing a better understanding of pathogens and diagnostics, and through contributions to drug development. There is, however, concern about the development of a “genomics divide” because of the costs associated with these developments and uncertainty as to whether these advances will be accessible to the populations of developing countries. The World Health Organization has summarized the current issues and inequities surrounding genomic medicine in a detailed report titled “Genomics and World Health.”
Whether related to informed consent, participation in research, or the management of a genetic disorder that affects an individual or his or her family, there is a great need for more information about fundamental principles of genetics. The pervasive nature of the role of genetics in medicine makes it important for physicians and other health care professionals to become more informed about genetics and to provide advice and counseling in conjunction with trained genetic counselors (Chap. 84). The application of screening and prevention strategies will therefore require intensive patient and physician education, changes in health care financing, and legislation to protect patient’s rights.
83e |
Chromosome Disorders |
CHROMOSOME DISORDERS
Alterations of the chromosomes (numerical and structural) occur in about 1% of the general population, in 8% of stillbirths, and in close to 50% of spontaneously aborted fetuses. The 3 × 109 base pairs that encode the human genome are packaged into 23 pairs of chromosomes, which consist of discrete portions of DNA, bound to several classes of regulatory proteins. Technical advances that led to the ability to analyze human chromosomes immediately translated into the revelation that human disorders can be caused by an abnormality of chromosome number. In 1959, the clinically recognizable disorder, Down syndrome, was demonstrated to result from having three copies of chromosome 21 (trisomy 21). Very soon thereafter, in 1960, a small, structurally abnormal chromosome was recognized in the cells of some patients with chronic myelogenous leukemia (CML), and this abnormal chromosome is now known as the Philadelphia chromosome.
Since these early discoveries, the techniques for analysis of human chromosomes, and DNA in general, have gone through several revolutions, and with each technical advancement, our understanding of the role of chromosomal abnormalities in human disease has expanded. While early studies in the 1950s and 1960s easily identified abnormalities of chromosome number (aneuploidy) and large structural alterations such as deletions (chromosomes with missing regions), duplications (extra copies of chromosome regions), or translocations (where portions of the chromosomes are rearranged), many other types of structural alterations could only be identified as techniques improved. The first important technical advance was the introduction of chromosome banding in the late 1960s, a technique that allowed for the staining of the chromosomes, so that each chromosome could be recognized by its pattern of alternating dark and light (or fluorescent and nonfluorescent) bands. Other technical innovations ranged from the introduction of fluorescence in situ hybridization in the 1980s to use of array-based and sequencing technologies in the early 2000s. Currently, we can appreciate that many types of chromosome abnormalities contribute to human disease including aneuploidy; structural alterations such as deletions and duplications, translocations, or inversions; uniparental disomy, where two copies of one chromosome (or a portion of a chromosome) are inherited from one parent; complex alterations such as isochromosomes, markers, and rings; and mosaicism for all of the aforementioned abnormalities. The first chromosome disorders identified had very striking and generally severe phenotypes, because the abnormalities involved large regions of the genome, but as methods have become more sensitive, it is now possible to recognize many more subtle phenotypes, often involving smaller genomic regions.
METHODS FOR CHROMOSOME ANALYSIS
STANDARD CYTOGENETIC ANALYSIS
Standard cytogenetic analysis refers to the examination of banded human chromosomes. Banded chromosome analysis allows for both the determination of the number and identity of chromosomes in the cell and recognition of abnormal banding patterns associated with a structural rearrangement. A stained band is defined as the part of a chromosome that is clearly distinguishable from its adjacent segments by appearing darker or lighter with one or more banding techniques. Cytogenetic analysis is most commonly carried out on cells in mitosis, requiring dividing cells. Actively growing cells are most often obtained from peripheral blood; however, it is only a small subset of the blood cells that are actually used for cytogenetic analysis. Often, chemicals, like phytohemagglutinin (PHA), are used to specially stimulate growth of T cells in a blood sample. Other sources of dividing cells include skin-derived fibroblasts, amniotic fluid or placental tissue (for prenatal diagnosis), or tumor tissue (for cancer diagnosis). After culturing, cells are treated with a mitotic spindle inhibitor, which prevents the separation of the chromatids during metaphase. Halting mitosis in metaphase is essential, because chromosomes are at their most condensed state during this stage of mitosis. The banding pattern of a metaphase chromosome is easily recognizable and is ideal for karyotyping. There are several different types of chromosome staining techniques, including R-banding, C-banding, and quinacrine staining, but the most commonly used is G-banding. G-banding is accomplished by treatment of the chromosomes with a proteolytic enzyme, such as trypsin, which digests some of the proteins holding DNA in a three-dimensional structure, followed by staining with a dye (Giemsa) that binds DNA. The resulting patterns have both dark and light bands; in general, the light bands occur in regions on the chromosome in which genes are actively being transcribed, and dark bands are in regions of less active transcription.
The banded human karyotype has now been standardized based on an internationally agreed upon system for designating not only individual chromosomes but also chromosome regions, providing a way in which structural rearrangements and variants can be described in terms of their composition. The normal human female karyotype is referred to as 46,XX (46 chromosomes, with 22 pairs of autosomes and two of the same type of sex chromosomes [two Xs], indicating this is a female); and the normal human male karyotype is referred to as 46,XY (46 chromosomes, with 22 pairs of autosomes and one of each type of sex chromosome [one × and one Y], indicating this is a male). The anatomy of a chromosome includes the central constriction, known as the centromere, which is critical for movement of the chromosomes during mitosis and meiosis; the two chromosome arms (p for the smaller or petite arm, and q for the longer arm); and the chromosome ends, which contain the telomeres. The telomeres are made up of a hexanucleotide repeat (TTAGGG)n, and unlike the centromere, they are not visible at the light microscope level. Telomeres are functionally important because they confer stability to the end of the chromosome. Broken chromosomes tend to fuse end to end, whereas a normal chromosome with an intact telomere structure is stable. To create the standard chromosome-banding map, each chromosome is divided into segments that are numbered, and then further subdivided. The precise band names are recorded in an international document so that each band has a distinct number. Figure 83e-1 shows an ideogram (chromosome map with bands) of the × chromosome and a G-banded × chromosome. This system provides a way for a chromosome abnormality to be written, with an indication of which band is deleted, duplicated, or rearranged.
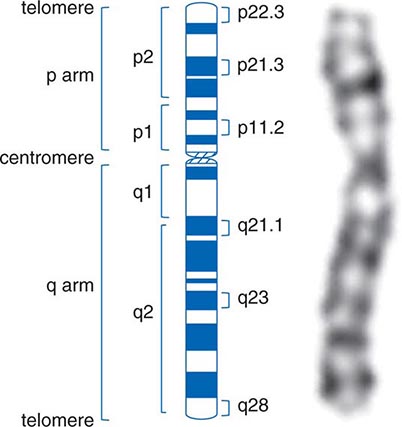
FIGURE 83e-1 Ideogram of the × chromosome and a G-banded × chromosome. The labeling of the × ideogram shows the positioning of the p and q arms, the centromere, and the telomeres. The numbering of the bands is also demonstrated, indicating the broadest subbands (p1, p2, q1, q2) and the further subdivisions to the right. Numbering begins at the centromere and moves out along each arm toward the telomeres.
MOLECULAR CYTOGENETICS
Molecular cytogenetics provides a link between chromosome and molecular analysis and overcomes some of the limitations of standard cytogenetics. Deletions smaller than several million base pairs are not routinely detectable by standard G-banding techniques, and chromosomal abnormalities with indistinct or novel banding patterns can be difficult or impossible to interpret. To carry out cytogenetic analysis, cells must be dividing, which is not always possible to obtain (e.g., in autopsy or tumor material that has already been fixed). Finally, growth selection or bias may occasionally cause the results of cytogenetic studies to be misleading because cells that proliferate in vitro may not be representative of the original population, as is often the case with tumor specimens.
Fluorescence in situ hybridization (FISH) is a combined cytogenetic-molecular technique that solves many of the aforementioned problems. FISH permits determination of the number and location of specific DNA sequences in human cells. FISH can be performed on metaphase chromosomes, as with G-banding, but can also be performed on cells not actively progressing through mitosis. FISH performed on nondividing cells is referred to as interphase or nuclear FISH (Fig. 83e-2). The FISH procedure relies on the complementarity between the two strands of the DNA double helix and uses a molecular probe, which can be a pool of sequences across an entire chromosome, a DNA sequence for a repetitive part of the genome (e.g., centromeres or telomeres), or a specific DNA sequence found only once in the genome (e.g., a disease-associated gene). The choice of probes for FISH studies is important and will vary with the information needed for the diagnosis of a particular disorder. The most common type of probes are locus-specific probes, which are used to determine if a critical gene or region is absent (indicating a deletion), or present in the normal number of copies, or if an additional copy of the region is present. FISH on metaphase chromosomes will give the additional information of the location of the additional copy, which is necessary information to determine whether a structural rearrangement, such as a translocation, is present. FISH can also be performed with probes that bind to repeated sequences, such as DNA found in centromeres or telomeres, or with probes that bind to an entire chromosome (“painting” probes), to determine the chromosome composition of an abnormal chromosome. Interphase FISH studies can also help to identify structural alterations when probes are used that map to both sides of a translocation breakpoint. Each side of the breakpoint is labeled in a different color, and when no translocation is present the two probes appear to be overlapping. When a translocation is present, the two probes appear separate from one another. These set of probes, called “break-apart” probes, are commonly used to detect recurrent translocations in cancer cells.
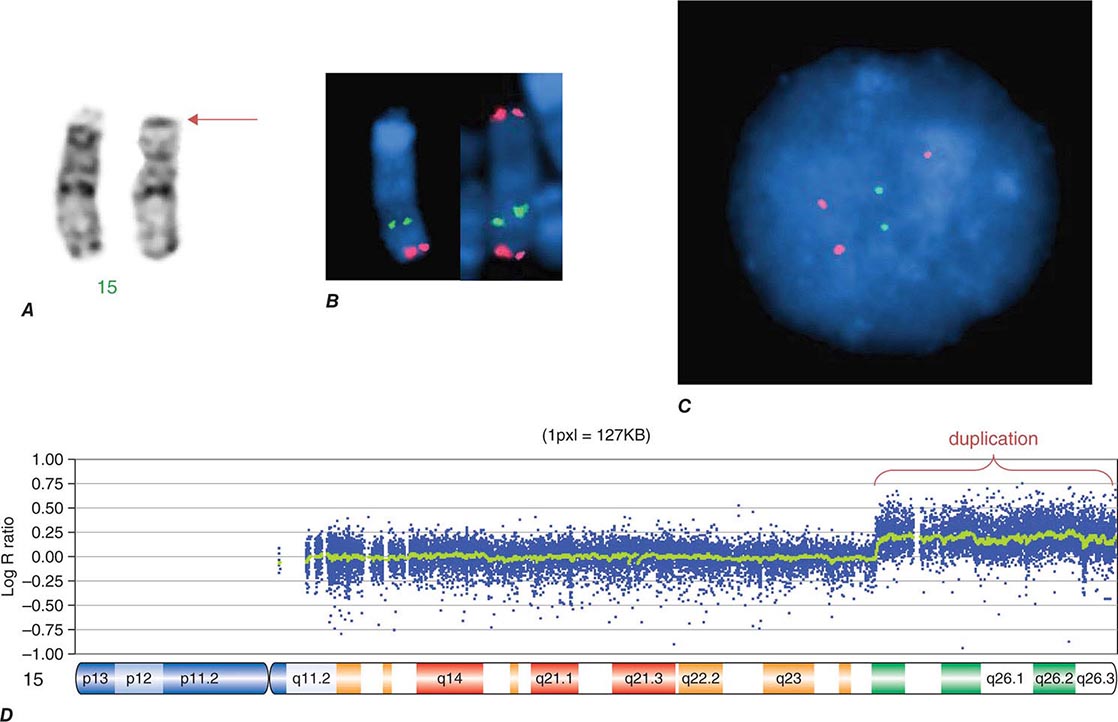
FIGURE 83e-2 G-banding, fluorescence in situ hybridization (FISH), and single nucleotide polymorphism (SNP) array demonstrate an abnormal chromosome 15. A. G-banding shows an abnormal chromosome 15, with unrecognizable material in place of the p arm in the chromosome on the right (top arrow). B. Metaphase FISH (only chromosome 15s are shown) using a probe from the 15q telomere region (red) and a control probe that maps outside of the duplicated region (green). C. Interphase FISH demonstrates three copies of the 15q tel probe in red, and two copies of the 15q control probe (green). D. Genome-wide SNP array demonstrates the increased copy number for a portion of 15q. Note that the G-banding alone indicates the abnormal chromosome 15, but the origin of the extra material can only be demonstrated by FISH or array. The FISH analysis requires additional information about possible genetic causes to select the correct probe. The array can exactly identify the origin of the extra material, but by itself would not provide positional information.
ARRAY-BASED METHODOLOGIES (CYTOGENOMICS)
Array-based methods were introduced into the clinical lab beginning in 2003 and quickly revolutionized the field of cytogenetics. These techniques used arrays (collections of DNA segments from the entire genome) which could be interrogated with respect to copy number. With standard cytogenetics, the missing or extra pieces of DNA have to be big enough to see in the microscope on banded chromosomes (usually larger than 5 Mb). FISH requires a preselection of an informative molecular probe prior to analysis. In contrast, array-based techniques permit analysis of many regions of the genome in a single analysis, with greatly increased resolution over standard cytogenetics. Array-based techniques allow for scanning of the genome for small deletions or duplications quickly and accurately. The resolution of the test is a function of the number of probes or DNA sequences present on the array. Arrays may use probes of different sizes (ranging from 50 to 200,000 base pairs of DNA) and different probe densities depending on the requirements of the application. Low-resolution platforms can have hundreds of probes, targeted to known disease regions, whereas high-resolution platforms can have millions of probes spread across the entire genome. Depending on the size of the probes and the probe placement across the genome, array-based testing may be able to detect single exon deletions or duplications.
Comparative Genomic Hybridization (CGH) and Single Nucleotide Polymorphism (SNP) Analysis CGH and SNP-based genotyping arrays can both be used for the analysis of genomic deletions and duplications. For both techniques, oligonucleotide probes are placed onto a slide or chip in a grid format. Each of these probes is specific for a particular genomic region. In array CGH, the amount of DNA from a patient is compared to that in a clinically normal control, or pool of controls, for each of the probes present on the array. DNA from a patient is fluorescently labeled with a dye of one color, and DNA from a control individual is labeled with another color. These DNA samples are then hybridized at the same time to the array. The resulting fluorescent signal will vary depending on whether both the control and patient DNA are present in equal amounts or if one has a different copy number than the other. SNP platforms use arrays targeting SNPs that are distributed across the genome. SNP arrays vary in density of markers and in the technology used for genotyping, depending on the manufacturer of the array. SNP arrays were initially designed to determine genotypes at a biallelic, polymorphic base (e.g., CC, CT, or TT) and have been increasingly used in genome-wide association studies to identify disease susceptibility genes. SNP arrays were subsequently adapted to identify genomic deletions and duplications (Fig. 83e-2). SNP arrays, in addition to identifying copy number changes, can also detect regions of the genome that have an excess of homozygous genotypes and absence of heterozygous genotypes (e.g., CC and TT genotypes only, with no CT genotypes). Absence of heterozygosity is sometimes associated with uniparental disomy (discussed later in this chapter) but is also observed when an individual’s parents are related to one another (identity by descent). Regions of homozygosity have been used to help identify genes in which homozygous mutations result in disease phenotypes in families with known consanguinity.
Array-based techniques (which we will now refer to as cytogenomic analysis) have proven superior to chromosome analysis in the identification of clinically significant deletions or duplications. It is estimated that for a deletion or duplication to be visualized by standard cytogenetics it must be minimally between 5 and 10 million base pairs in size. In almost all cases, deletions and duplications of this size contain multiple genes, and these deletions and duplications are disease causing. However, utilization of array-based cytogenomic testing, which can routinely identify deletions and duplications smaller than 50,000 base pairs, reveals that clinically normal individuals all have some deletions and duplications. This presents a dilemma for the analyst to discern which smaller copy number variations (CNVs) are disease causing (pathogenic) and which are likely benign polymorphisms. Although initially burdensome, the cytogenomics community has been curating these CNVs for almost a decade, and databases have been created reporting CNVs routinely seen in clinically normal individuals and those routinely seen in individuals with clinical abnormalities. Nevertheless, each copy number variant that is identified in an individual undergoing genomic testing must be evaluated for gene content and overlap with CNVs in other patients and in controls.
Array technologies are DNA based, unlike cytogenetic technologies, which are cell based. Although resolution of gains and losses are greatly increased with array technology, this technique cannot identify structural changes. When DNA is extracted for array studies, chromosomal structure is lost because the DNA is fragmented for better hybridization to the slides. As an example, the array may be able to detect a duplication of a small region of a chromosome, but no information on the location of this extra material can be determined from this test. The location of this extra copy in the genome may be critical, as the chromosomal material may be involved in a translocation, insertion, marker, or other complex rearrangement. Depending on the chromosomal position of this extra material, the patient may have different clinical outcomes, and recurrence risks for the family can be significantly different. Often, combinations of array-based and cytogenetic-based techniques are required to fully characterize chromosomal abnormalities (see Table 83e-1 for comparison of these technologies).
|
COMPARISON OF CYTOGENETIC AND CYTOGENOMIC TECHNIQUES |
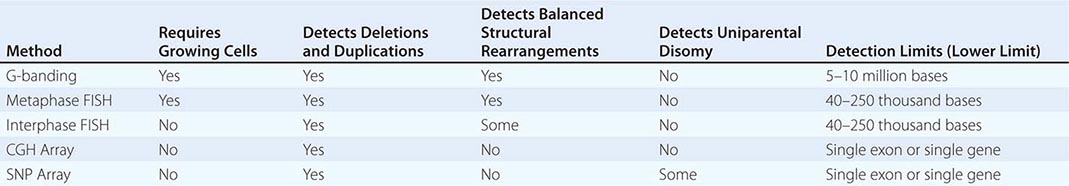
NEXT-GENERATION SEQUENCING—BASED METHODOLOGIES
Recent advances in genomic sequencing, known as next-generation sequencing (NGS), have vastly increased the speed and throughput of DNA sequence analysis. NGS is rapidly finding its way into the diagnostic lab for detection of clinically relevant intragenic mutations, and new bioinformatic tools for analysis of genomic deletions and duplications are being developed. It is anticipated that NGS will soon allow the complete analysis of a patient’s genome, with identification of intragenic mutations as well as chromosome abnormalities resulting in gain or loss of genetic material. Identification of completely balanced translocations is the most challenging for NGS, but recent reports of successes in this area suggest that in a matter of time, sequencing will be used for all types of genomic analysis.
INDICATIONS FOR CHROMOSOME/CYTOGENOMIC ANALYSIS
Cytogenetic analysis is most commonly used for (1) examination of the fetal chromosomes or genome during pregnancy (prenatal diagnosis) or in the event of a spontaneous miscarriage; (2) examination of chromosomes in the neonatal or pediatric population to look for an underlying diagnosis in the case of congenital or developmental anomalies, including short stature and abnormalities of sexual differentiation or progression; (3) chromosome analysis in adults who are facing fertility problems; or (4) examination of cancer cells to look for alterations that aid in establishing a diagnosis or contributing to the prognosis of a tumor (Table 83e-2).
|
INDICATIONS FOR CYTOGENETIC AND CYTOGENOMIC ANALYSIS ACROSS THE LIFESPAN |
PRENATAL DIAGNOSIS
Prenatal diagnosis is carried out by analysis of samples obtained by four techniques: amniocentesis, chorionic villous sampling, fetal blood sampling, and analysis of cell free DNA from maternal serum. Amniocentesis, which has been the most commonly used test to date, is usually performed between 15 and 17 weeks of gestational age and carries a small but significant risk for miscarriage. Amniocentesis can be performed as early as 12 weeks, but because there is a lower volume of fluid, the risks for fetal injury or miscarriage are greater. Chorionic villous sampling (CVS) or placental biopsy is routinely carried out earlier than amniocentesis, between 10 and 12 weeks, but a reported increase in limb defects when the procedure is carried out earlier than 10 weeks has resulted in reduced use of this test in some centers. Fetal blood sampling (percutaneous umbilical blood sampling [PUBS]) is a riskier procedure that is carried out in the second or third trimester of pregnancy, usually to follow up on an unclear finding from an amniocentesis (such as mosaicism) or an ultrasound abnormality that was detected later in pregnancy. One of the far-reaching recent advances in prenatal diagnosis of chromosome and other genetic disorders is the utilization of cell free fetal DNA that can be identified in maternal serum. The obvious advantages of using fetal DNA obtained from maternal serum is that the DNA can be obtained at minimal risk to the pregnancy, because it requires a maternal blood sample, rather than amniotic fluid which is obtained by puncturing the uterine membranes and carries a risk of miscarriage or infection. Although cell free fetal DNA screening, also called noninvasive prenatal screening, has started to be offered clinically, it requires further confirmation of fetal tissues when an abnormal result is identified. Furthermore, ethical concerns have been raised, because it is feared that the ease of doing this test may encourage testing for individuals who are not truly prepared to deal with the choices that accompany diagnosis of a genetic disease and this testing may change the ethical implications of prenatal testing. Nevertheless, this is an active of area of research, both in terms of the technology and the utilization and implications.
Common Indications Common indications for prenatal diagnosis by cytogenetic or cytogenomic analysis are (1) advanced maternal age, (2) presence of an abnormality of the fetus on ultrasound examination, and (3) abnormalities in maternal serum screening that reveal an increased risk for chromosome abnormality.
Maternal age is well known to be an important risk factor for having a fetus with trisomy. At a maternal age less than 25 years, 2% of all clinically recognized pregnancies are trisomic, but by a maternal age of 36 years, this figure increases to 10%, and by the maternal age of 42 years, the figure increases to >33%. Based on the risk of having a chromosomally abnormal fetus in comparison to the risk for an adverse event from amniocentesis or CVS, the recommendation is that women over the age of 35 consider prenatal testing if they want to know the chromosomal status of their fetus. The precise mechanism for the maternal age effect is not known, but it is believed that it involves a breakdown in the process of chromosome segregation. A similar effect is not seen for trisomy and paternal age. This difference may reflect the fact that oocytes are generated early in ovary development in the female, whereas spermatogonia are generated continuously after puberty in the male.
Abnormalities on prenatal ultrasound are the second most frequent indication for prenatal genetic screening. Ultrasound screening can reveal structural or functional anomalies in the fetus, which might be associated with chromosome or genomic disorders. Follow-up chromosome studies may therefore be recommended.
Maternal serum screening results are the third most frequent indication for prenatal chromosome analysis. There have been several versions of maternal serum screening offered over the past few decades. Currently, the “quad” screen analyzes levels of α fetoprotein (AFP), human chorionic gonadotropin (hCG), estriol, and inhibin-A. The values of these analytes are used to adjust the maternal age–predicted risk of a trisomy 21 or trisomy 18 fetus.
POSTNATAL INDICATIONS
Postnatal indications for cytogenetic or cytogenomic analysis in neonates or children are varied, and the list has been growing with the increasing ability to diagnose smaller genomic alterations via array-based techniques. Common indications include multiple congenital anomalies, suspicion of a known cytogenetic or cytogenomic syndrome, intellectual disability or developmental delay both with and without accompanying dysmorphic features, autism, failure to thrive in infancy or short stature during childhood, and disorders of sexual development. The ability to detect smaller genomic alterations with involvement of fewer genes, sometimes as few as a single gene, suggests that a wider range of phenotypes could be investigated by cytogenomic analysis. Reasons for chromosome testing in adults include recurrent miscarriages or infertility, where balanced chromosome rearrangements such as reciprocal translocations may occur. Additionally, some adults with anomalies who were not diagnosed when they were children are referred for cytogenetic analysis, often when other members of their family want to understand any potential genetic implications, as they plan their own families.
TYPES OF CHROMOSOME ABNORMALITIES
NUMERICAL CHROMOSOME ABNORMALITIES
Aneuploidy (extra or missing chromosomes) is the most common type of abnormality, occurring in 3/1000 newborns and at much higher frequency (about 35%) in spontaneously aborted fetuses. The only autosomal trisomies that are compatible with being live born in humans are trisomies 13, 18, and 21, although there are several chromosomes that can be trisomic in mosaic form. Trisomy 21 is associated with the relatively common disorder Down syndrome. Down syndrome has characteristic features including recognizable facial features, along with intellectual disability and abnormalities of multiple other organ systems including the heart. Both trisomy 13 and trisomy 18 are much more severe disorders than Down syndrome, with low frequency of patients surviving past 1 year of age. Trisomy 13 is characterized by low birth weight, postaxial polydactyly, microcephaly, ocular malformations such as anophthalmia or microphthalmia, cleft lip and palate, cardiac defects, and renal malformations. Trisomy 18 neonates have distinct facial characteristics at birth accompanied by an abnormal neurologic exam, underdeveloped genitalia, general lack of responsiveness, and structural birth defects such as congenital heart disease, esophageal atresia, and omphalocele.
Mosaicism refers to the presence of two or more populations of cells with distinct chromosome constitutions: for example, an individual with a normal female karyotype in some cells (46,XX) and trisomy 21 in other cells (47,XX,+21). In general, individuals who are mosaic for a chromosomal abnormality have less severe phenotypes than individuals with that same finding in every cell. The severity and presentation of phenotypes are related to the mosaic levels and the tissue distribution of the abnormal cells. There are a number of trisomies that have been reported in mosaic form including mosaic trisomies for chromosomes 8, 9, 14, 17, and 22. A number of trisomies have also been reported in spontaneous abortions (SABs) that have not been seen in live-born individuals, including trisomy 16, which is the most common trisomy in SABs. Monosomy for human chromosomes is very rare, with the single exception being monosomy for the × chromosome, associated with Turner syndrome (45,X). Monosomy for the × chromosome occurs in 1% of all conceptions, yet 98% of these conceptions do not go to term and result in SABs. Trisomies for the sex chromosomes also occur, with 47,XXX (trisomy × or triple × syndrome), 47,XXY (Klinefelter syndrome), and 47,XYY all reported in individuals with relatively mild phenotypes (Chap. 410). Klinefelter syndrome is the most common clinically recognized sex chromosome abnormality, and clinical features include gynecomastia, azoospermia, small testes, and hypogonadism. The 47,XYY karyotype is most often found in boys with developmental delay and or behavioral difficulties, but population-based studies have shown that intelligence for individuals with this karyotype is generally within the normal range, although slightly lower than that found in siblings.
STRUCTURAL CHROMOSOME ABNORMALITIES
Structural chromosome abnormalities include deletions, duplications, translocations, inversions, as well as other types of abnormalities, each relatively rare, but nonetheless contributing to clinical disease resulting from chromosome anomalies. These rare alterations include isochromosomes, ring chromosomes, dicentric chromosomes, and marker chromosomes (structurally abnormal chromosomes that cannot be identified based on cytogenetics alone). Both translocations and inversions can be completely balanced in some cases, such that there is no disruption of coding regions of the genome, with a completely normal clinical phenotype; however, carriers are at risk for unbalanced forms of these rearrangements in their offspring.
Reciprocal translocations are found in approximately 1/500–1/600 individuals in the general population and result from the exchange of chromosomal segments between at least two chromosomes. These usually occur between nonhomologous chromosomes and can be identified based on an altered banding pattern on G-banding. Balanced translocation carriers are at risk for abnormal chromosome segregation during meiosis and therefore have a higher risk for infertility, SAB, and live-born offspring with multiple congenital malformations. These phenotypes are observed when only one of the pairs of chromosomes involved in a translocation is inherited from a parent, resulting in an unbalanced genotype (Fig. 83e-3). Sometimes the exchanged segments are so small that they cannot be appreciated by banding (cryptic translocation), and these are sometimes recognized when a phenotypically affected child with an unbalanced form is born. Parental chromosomes can then be studied by FISH to determine if the rearrangement is inherited from a parent with a balanced form of the translocation. The majority of reciprocal, apparently balanced translocations occur in phenotypically normal individuals. The risk for a clinical abnormality when a new reciprocal translocation is identified (usually during prenatal diagnostic studies) is about 7%. Analysis of cytogenetically reciprocal translocations using arrays has demonstrated that translocations in clinically normal individuals are more likely to show no deletions or duplications at the breakpoint, whereas translocations in clinically affected individuals are more likely to have breakpoint-associated deletions or duplications. Most reciprocal translocations occur uniquely, at apparently random positions throughout the genome; however, there are a few exceptions with multiple cases of recurrent translocations occurring. These recurrent translocations include t(11;22), which results in Emanuel syndrome in the unbalanced form, and several translocations involving a region on 4p, 8p, and 12p. These recurrent translocations occur in regions of the genome that contain specific types of AT-rich repeats, or other repeat sequences, that are prone to rearrangement. A special category of translocations is the Robertsonian translocations, which involve the acrocentric chromosomes. An acrocentric chromosome has unique genetic material only on the long arm of the chromosomes, whereas the short arm contains repetitive DNA. The acrocentric chromosomes are 13, 14, 15, 21, and 22. Robertsonian translocations occur when an entire long arm of an acrocentric chromosome is translocated onto the short arm of another acrocentric chromosome. Balanced carriers of a Robertsonian translocation contain only 45 chromosomes, with one chromosome consisting of two long arms of an acrocentric chromosome. Technically, this is an unbalanced translocation, as two short arms of the acrocentric chromosomes are missing; however, because the short arms are repetitive, there is no phenotypic consequence. Unbalanced Robertsonian carriers have 46 chromosomes, but have three copies of the long arm of an acrocentric chromosome. The most common Robertsonian translocation involves chromosomes 13 and 14. Unbalanced Robertsonian translocations involving chromosomes 13 and 21 result in trisomy 13 and Down syndrome, respectively. Approximately 4% of patients with Down syndrome have a translocation, and because recurrence risks are different for families of these individuals, all patients with clinically identified Down syndrome should have a karyotype to look for translocations.
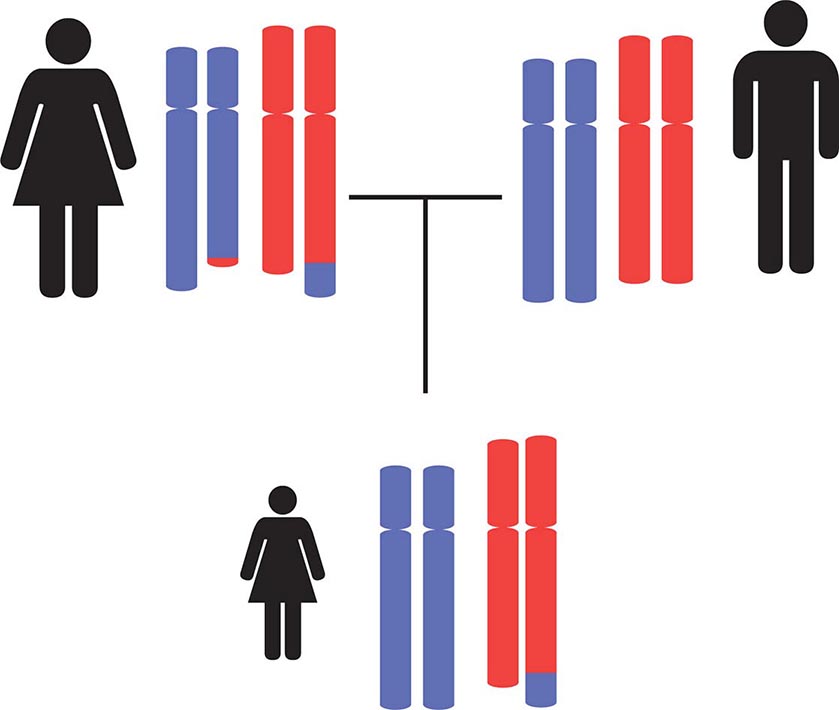
FIGURE 83e-3 Segregation of a balanced translocation in a mother, with inheritance of an unbalanced form in her child. Note that the mother has two rearranged chromosomes, but her child only received one of these, resulting in extra copies of a region of the blue chromosome, with loss of some material from the red chromosome.
Inversions are another type of chromosome abnormality involving rearranged segments, where there are two breaks within a chromosome, with the intervening chromosomal material inserted in an inverted orientation. As with reciprocal translocations, if a break occurs within a gene or control region for a gene, a clinical phenotype may result, but often there are no consequences for the inversion carrier; however, there is risk for abnormalities in the offspring of carriers, as recombinant chromosomes may result after crossing over between a normal chromosome and an inverted chromosome during meiosis.
Deletion refers to the loss of a chromosomal segment, which results in the presence of only a single copy of that region in an individual’s genome. A deletion can be at the end of a chromosome (terminal), or it can be within the chromosome (interstitial). Deletions that are visible at the microscopic level in standard cytogenetic analysis are generally greater than 5 Mb in size. Smaller deletions have been identified by FISH and by chromosomal microarray. The clinical consequences of a deletion depend on the number and function of genes in the deleted region. Genes that cause a phenotype when a single copy is deleted are known as haploinsufficient genes (one copy is not sufficient), and it is estimated that less than 10% of genes are haploinsufficient. Genes associated with disease that are not haploinsufficient include genes for known recessive disorders, such as cystic fibrosis or Tay-Sachs disease.
The first chromosome deletion syndromes were diagnosed clinically and were subsequently demonstrated to be caused by a chromosome deletion on cytogenetic analysis. Examples of these disorders include the Wolf-Hirschhorn syndrome, which is associated with deletions of a small region of the short arm of chromosome 4 (4p); the cri-du-chat syndrome, associated with deletion of a small region of the short arm of chromosome 5 (5p); Williams syndrome, which is associated with interstitial deletions of the long arm of chromosome 7 (7q11.23); and the DiGeorge/velocardiofacial syndromes, associated with interstitial deletions of the long arm of chromosome 22 (22q11.2). Initial cytogenetic studies were able to provide a rough localization of the deletions in different patients, but with the increased usage of arrays, precise mapping of the extent and gene content of these deletions has become much easier. In many cases, one or two genes that are critical for the phenotype associated with these deletions have been identified. In other cases, the phenotype stems from the deletion of multiple genes. The increased utilization of genomic testing by array, which can identify deletions that are much smaller than those detectable by standard cytogenetic analysis, has resulted in the discovery of several new cytogenomic disorders. These include the 1q21.1, 15q13.3, 16p11.2, and 17q21.31 microdeletion syndromes.
Duplication of genomic regions is better tolerated than deletion, as evidenced by the viability of several autosomal trisomies (whole chromosome duplications) but no autosomal monosomies (whole chromosome deletions). There are several duplication syndromes where the duplicated region of the genome is present as a supernumerary chromosome. Utilization of chromosome microarray analysis has made analysis of the origins of duplicated chromosome material straightforward (Fig. 83e-2). Recurrent syndromes associated with supernumerary chromosomes include the inverted duplication 15 (inv dup 15) syndrome, caused by the presence of a marker chromosome derived from chromosome 15, with two copies of proximal 15q resulting in tetrasomy (four copies) of this region. The inv dup 15 syndrome has a distinct phenotype and is associated with hypotonia, developmental delay, intellectual disability, epilepsy, and autistic behavior. Another syndrome is the cat eye syndrome, named for the “cat-eye-like” appearance of the pupil, resulting from a coloboma of the iris. This syndrome results from a supernumerary chromosome derived from a portion of chromosome 22, and the marker chromosomes can vary in size and are often mosaic. Consistent with expectations of a mosaic disorder, the phenotype of this syndrome is highly variable and includes renal malformations, urinary tract anomalies, congenital heart defects, anal atresia with fistula, imperforate anus, and mild to moderate intellectual disability. Another rare duplication syndrome is the Pallister-Killian syndrome (PKS), which illustrates the principle of tissue-specific mosaicism. Individuals with PKS have coarse facial features with pigmentary skin anomalies, localized alopecia, profound intellectual disability, and seizures. The disorder is caused by a supernumerary isochromosome for the short arm of chromosome 12 (isochromosome 12p). Isochromosomes consist of two copies of one chromosome arm (p or q), rather than one copy of each arm. This isochromosome is not generally seen in peripheral blood lymphocytes when they are analyzed by G-banding, but it is detected in fibroblasts. Array technology has been reported to detect the isochromosome in uncultured peripheral blood in some patients, and it has been hypothesized that a growth bias against cells with the isochromosome prevents their identification in cytogenetic studies.
Numerical abnormalities, translocations, and deletions are the most common chromosome alterations observed in the diagnostic laboratory, but in addition to inversions and duplications, several other types of abnormal chromosomes have been reported, including ring chromosomes, where the two ends of the chromosome fuse to form a circle, and insertions, where a piece of one chromosome is inserted into another chromosome or elsewhere into the same chromosome.
Uniparental disomy (UPD) is the inheritance of a pair of chromosomes (or part of a chromosome) from only one parent. This usually occurs as a result of nondisjunction during meiosis, with a gamete missing or having an extra copy of a chromosome. A resulting fertilized egg would then have only one parental contribution for a given chromosome pair, or a trisomy for a given chromosome. If the monosomy or trisomy is not compatible with life, the embryo may undergo a “rescue” to normal copy number. If a monosomy is rescued, the single chromosome may be duplicated, resulting in a cell with two identical chromosomes (monosomy rescue) (Fig. 83e-4). In the case of trisomies, a subsequent nondisjunction can result in cells where one of the extra chromosomes is lost (trisomy rescue) (Fig. 83e-4). For trisomy rescue, there is a one in three chance that the lost chromosome will be the sole chromosome from one parent, resulting in a cell with two chromosomes from the same parent. UPD is sometimes associated with clinical abnormalities, and this can occur by two mechanisms. UPD can cause disease when there is an imprinted gene on the involved chromosome, resulting in altered gene expression. Imprinting is the chemical marking of the parental origin of a chromosome, and genes that are imprinted are only expressed from either the maternal or paternal chromosome (Chap. 82). Imprinting therefore results in the differential expression of affected genes, based on parent of origin. Imprinting usually occurs through differential modification of the chromosome from one of the parents, and methylation is one of several epigenetic mechanisms (others include histone acetylation, ubiquitylation, and phosphorylation). Imprinted chromosomes that are associated with phenotypes include paternal UPD6 (associated with neonatal diabetes), maternal UPD7 and UPD11 (associated with Russell-Silver syndrome), paternal UPD11 (associated with Beckwith-Wiedemann syndrome), paternal UPD14, maternal UPD15 (Angelman syndrome), and paternal UPD15 (Prader-Willi syndrome). UPD can also result in disease if the two copies from the same parent are the same chromosome (uniparental isodisomy), and the chromosome contains an allele involving a pathogenic mutation associated with a recessive disorder. Two copies of the deleterious allele would result in the associated disease, even though only one parent is a disease carrier.
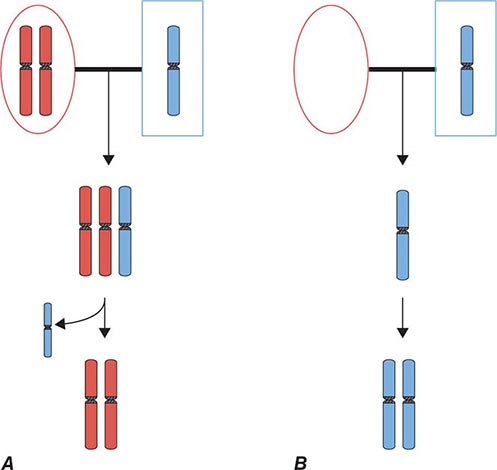
FIGURE 83e-4 Mechanisms of formation of uniparental disomy. Panel A demonstrates nondisjunction in one parent (mother, represented in red), with trisomy in the zygote. A subsequent nondisjunction, with loss of the paternal chromosome (represented in blue), restores the diploid karyotype but leaves two copies of the maternal chromosome (maternal uniparental disomy [UPD]). Panel B demonstrates nondisjunction in one parent (mother, indicated by red oval), resulting in only one copy of this chromosome in the zygote. Subsequent nondisjunction duplicates the single chromosome, rescuing the monosomy, but resulting in two copies of the paternal chromosome (represented in blue; paternal UPD).
ACQUIRED CHROMOSOME ABNORMALITIES IN CANCER
Chromosome changes can occur during meiosis or mitosis and can occur at any point across the lifespan. Mosaicism for a developmental disorder is one consequence of mitotic chromosome abnormalities, and another consequence is cancer, when the chromosome change confers a growth or proliferation advantage on the cell. The types of chromosome abnormalities seen in cancer are similar to those seen in the developmental disorders (e.g., aneuploidy, deletion, duplication, translocation, isochromosomes, rings, inversion). Tumor cells often have multiple chromosome changes, some of which happen early in the development of a tumor, and may contribute to its selective advantage, whereas others are secondary effects of the deregulation that characterizes many tumors. Chromosome changes in cancer have been studied extensively and have been shown to provide important diagnostic, classification, and prognostic information. The identification of cancer type–specific translocation breakpoints has led to the identification of a number of cancer-associated genes. For example, the small abnormal chromosome found to be associated with chronic myelogenous leukemia (CML) in 1960 was shown to be the result of translocation between chromosomes 9 and 22 once techniques for analysis of banded chromosomes were introduced, and subsequently, the translocation breakpoint was cloned to reveal the c-abl oncogene on chromosome 9. This translocation produces a fusion protein, which has been targeted for treatment of CML. For detailed discussion of cancer genetics, see Chap. 101e.




