Cholinergic and Constitutive Regulation of Atrial Potassium Channel
Stimulation of the vagal nerve, the principal cardiac arm of the parasympathetic nervous system, reduces heart rate and slows conduction in the atrioventricular node, thereby tuning the heart rate to “rest-and-digest” activities. In 1921, Otto Loewi found that the vagal effects on the heart are mediated by release of acetylcholine (ACh) from the parasympathetic synapses, and ACh became the first neurotransmitter ever discovered.1 However, it took more than 50 years until it was suggested that ACh activates a specific population of K+ channels (ACh-gated IK,ACh; Figure 38-1, A) leading to hyperpolarization of the cell membrane, thereby decreasing pacemaker activity in sinoatrial (SA) node cells.2 It was also found that IK,ACh conductance is voltage dependent, with high K+ conductance at hyperpolarized membrane potentials (at which the current is typically inward) and small conductance at depolarized membrane potentials associated with outward current. This typical current-voltage (IV) relationship designates IK,ACh as an inward-rectifier K+ current (see Figure 38-1, B and C) similar to IK1, which is active in the absence of any receptor agonists, and IK,ATP, which is activated by reduced intracellular ATP levels. Cardiac IK,ACh channels are heterotetramers, usually consisting of two Kir3.1 and two Kir3.4 channel subunits.3

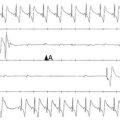
Figure 38-1 Basal and muscarinic (M2)-receptor activated inward-rectifier potassium (K+) currents in sinus rhythm (SR) and chronic atrial fibrillation (cAF). A, Left panel, Physiologically G-protein–activated inward-rectifier K+ (GIRK) channels are supposed to be closed in the absence of M-receptor agonists and only the basal inward-rectifier potassium current IK1 is active. B and C, Representative single-channel (upper tracings) and whole-cell recordings (lower tracings) in patients with SR (B) and cAF (C). The single-channel recordings were performed at –100 mV in a cell-attached configuration. Filled and empty arrow heads indicate closed and open levels of IK,ACh, respectively. In the absence of M-receptor agonists (basal conditions), myocytes from the cAF patients (C) exhibit both IK1 (#) and constitutive IK,ACh openings (*), whereas the latter is a rare event in myocytes from patients with SR (B). Besides increased expression of IK1 channels, constitutive IK,ACh,c activity can contribute to the increased whole-cell basal inward-rectifier current in cAF (compare left lower tracings in B and C) (Replotted with permission from Dobrev D, Friedrich A, Voigt N, et al: The G protein-gated potassium current IK,ACh is constitutively active in patients with chronic atrial fibrillation. Circulation 112:3697–3706, 2005. Figure was produced using Servier Medical Art.)
IK,ACh channels are activated by ACh binding to type-2 muscarinic receptors (M2-receptors), which causes dissociation of inhibitory Gi-proteins, thereby increasing IK,ACh open probability via direct interaction of G-protein βγ-subunits with the channel (Figure 38-2, A, B).4,5 After current activation by M2-receptor stimulation, atrial IK,ACh shows a characteristic biphasic desensitization that starts within a few seconds (Figure 38-3, middle). Although both Kir3.1 and Kir3.4 channel homomers produce similar peak-current densities upon M2-receptor stimulation when expressed in HEK-cells, IK,ACh desensitization was observed in Kir3.1 homomers only. Moreover, the desensitization process depends on the presence of the Kir3.1 subunit within the channel complex.6 Besides channel subunit composition, kinetics of G-protein cycle, membrane content of the anionic phospholipid phosphatidylinositol-4,5-bisphosphate (PIP2), and phosphorylation processes also contribute IK,ACh regulation and desensitization (see Figure 38-3).
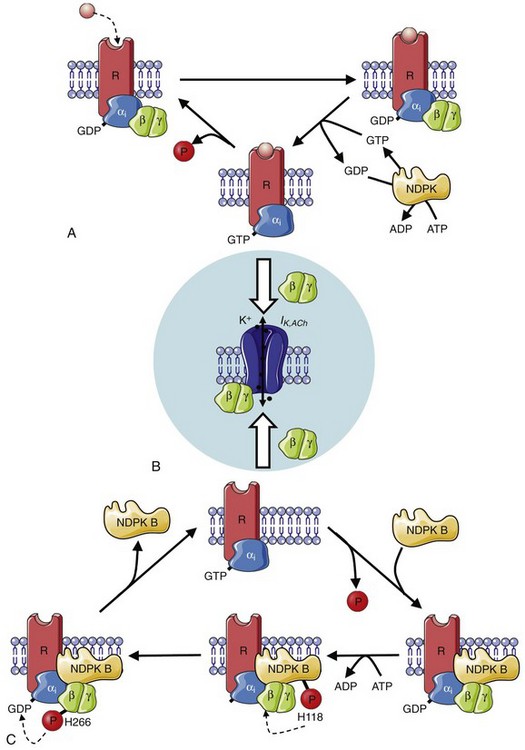
Figure 38-2 Muscarinic (M2)-receptor and nucleoside diphosphate kinase (NDPK)-dependent activation of Gi-proteins. A, Agonist binding to M2-receptors triggers GDP/GTP (guanosine-5′-triphosphate and guanosine-5′-diphosphate) exchange at the Gαi-subunit of the heterotrimeric Gi-proteins. Consecutive dissociation of the Gαi– and Gβγ-subunits leads to activation of IK,ACh by binding of the Gβγ-subunit to the IK,ACh channel (B). Intrinsic GTPase activity of the Gαi-subunit hydrolyses GTP into GDP resulting in reassociation of the heterotrimeric G-protein and restorage of the initial state. NDPKs provide the GTP, which is necessary for G-protein dissociation, by a phosphotransfer from ATP to GDP. C, In the subpopulation of heterotrimeric Gi-proteins complexed with NDPK B, a phosphotransfer from ATP to His118 in NDPK-B, and subsequently onto His266 of the Gβ, results in a high energetic phosphate, which promotes the formation of GTP and leads to receptor independent G-protein activation. This can contribute to the development of receptor-independent constitutive IK,ACh activity. (Figure was produced using Servier Medical Art.)
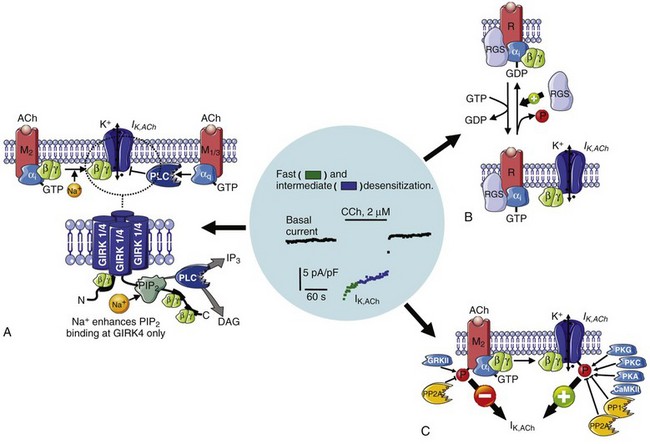
Figure 38-3 Mechanisms contributing to IK,ACh desensitization. Upon muscarinic (M2)-receptor activation, IK,ACh is activated by direct binding of liberated Gβγ-subunits to the IK,ACh channel, followed by a biphasic desensitization (fast and intermediate desensitization). A, The binding of Gβγ-subunits to the IK,ACh channel subunits Kir3.1 and Kir3.4 strengthens their interaction with cell membrane–located phosphatidyl inositol 4,5-bisphosphate (PIP2), thereby increasing IK,ACh open probability. Similarly, increased intracellular Na+ also strengthens the interaction of PIP2 with the channel leading to receptor-independent current activation. In contrast, stimulation of Gq-coupled M1/3-receptors activates phospholipase-C (PLC), thereby lowering the PIP2 membrane content resulting in IK,ACh inhibition and contributing to fast IK,ACh desensitization. B, Regulator of G-protein signaling proteins (RGS) accelerate the GTP hydrolysis rate of the Gαi-subunit, thereby contributing to faster desensitization and reducing agonist-independent constitutive IK,ACh,c activity (see also Figure 38-2, A). C, The phosphorylation of muscarinic (M) receptors and IK,ACh channels is controlled by various kinases and phosphatases, whereby channel phosphorylation increases IK,ACh, while concomitant phosphorylation of M-receptors reduces IK,ACh. Whereas channel dephosphorylation is supposed to contribute to the fast phase (green), the intermediate phase (blue) involves progressive receptor phosphorylation by a G-protein–coupled receptor kinase (GRK), which uncouples the receptor from the G-protein. (Figure was produced using Servier Medical Art.)
Apart from canonical M2-receptor–mediated activation of IK,ACh, purinergic A1,7,8 and sphingolipid Edg-39 receptors (also coupled to Gi-proteins) can activate cardiac IK,ACh channels. Based on their regulation and biophysics, IK,ACh channels are also designated G-protein–activated inwardly rectifying K+ (GIRK) channels. In contrast to ventricular tissue, IK,ACh channels are highly expressed in the atria, including SA and atrioventricular nodes, where they contribute to vagal regulation of heart rate (negative chronotropic effect) and conduction from the atria to the ventricles (negative dromotropic effects).10–12
M2-Receptor–Dependent IK,ACh Facilitates Initiation and Maintenance of Atrial Fibrillation
In atrial myocytes, IK,ACh channel activation leads to hyperpolarization of the resting membrane potential and shortening of the action-potential duration (APD).13 ACh-induced APD shortening and resting membrane potential hyperpolarization create an arrhythmogenic substrate facilitating the induction of atrial fibrillation (AF; see also chapter 45 of this book).14 It is well known that vagal nerve stimulation promotes AF in animal models and patients by facilitating the initiation and maintenance of reentry circuits.15 Reentry, the most established basic mechanism of AF, is described as continuous impulse propagation around a functional barrier or an anatomical obstacle. An important requirement for reentry is that the initially activated tissue zone regains excitability while the electrical impulse propagates around the reentry circuit, explaining why reduced effective refractory period or decreased conduction velocity can provide a substrate for AF maintenance. APD shortening induced by IK,ACh activation reduces effective refractory period, thereby favoring AF initiation by vagal stimulation. In knockout mice lacking the Kir3.4 channel subunit, M-receptor stimulation does not induce AF, clearly suggesting that the AF facilitating effects of vagal nerve activation are exclusively mediated by IK,ACh.16 In addition, mathematical modeling studies suggest that increases in an inward-rectifier K+ current such as IK,ACh have a significant role in AF promotion.17 Because of their ability to hyperpolarize atrial cardiomyocytes and remove voltage-dependent Na+-current (INa) inactivation, enhanced inward-rectifier K+ currents are more effective in stabilizing and accelerating AF-sustaining reentry circuits (rotors) than are changes in other ionic currents (e.g., reduced L-type Ca2+ currents) that produce a similar degree of APD shortening. In vivo evidence has been obtained to support the validity of these modeling data.18
Interestingly, AF-related atrial remodeling is associated with reduced maximum activation of IK,ACh upon M-receptor stimulation,13,19–22 which might partly result from reduced expression levels of Kir3.1 and Kir3.4 subunits in AF patients.13,21 The reduced agonist-dependent IK,ACh in AF patients could be a protective mechanism against the profibrillatory effects of vagal nerve stimulation.
Left-to-right atrial gradients of inward-rectifier K+ currents contribute to AF pathophysiology. There is clinical and experimental evidence that certain cases of paroxysmal AF (pAF) and chronic AF (cAF) are maintained by high-frequency reentrant sources (rotors) with a consistent left-to-right dominant frequency gradient, particularly in pAF.23–25 Because increased inward-rectifier currents enhance rotor frequency, the left-to-right atrial gradient in the basal inward-rectifier current present in pAF, but not in cAF or sinus rhythm (SR), can contribute to these dominant frequency gradients.22 In a sheep model of ACh-mediated, pacing-induced AF, the left-to-right (LA-RA) dominant frequency gradient parallels a left-to-right IK,ACh gradient, supporting the hypothesis that an unequal LA-RA distribution of inward-rectifier K+ currents can contribute to AF maintenance.26 However, in atria from patients with SR, there is a right-to-left atrial gradient of IK,ACh current that is absent in pAF and cAF.22 The lack of RA-dominant agonist-activated IK,ACh could have a permissive role for LA-dominant drivers in pAF and cAF, particularly in vagal contexts.
Agonist-Independent, Constitutively Active IK,ACh Can Contribute to Atrial Fibrillation Maintenance
It is well recognized that IK,ACh can possess agonist-independent “resting” activity, with a much lower opening frequency than agonist-induced IK,ACh.27 Constitutive IK,ACh current (IK,ACh,c) may underlie a major part of basal K+ conductance in SA node cells, which lack IK1, creating an important role in regulating heart rate.28 In the normal heart, constitutive IK,ACh activity is low in atrial myocytes, but can increase substantially with cardiac pathology. Agonist-independent IK,ACh,c activity increases in atrial myocytes from patients and animal models of AF, whereas maximum M-receptor activation of IK,ACh is reduced (see Figure 38-1, C).19,20,22,29 IK,ACh,c might contribute to APD shortening, which is a hallmark of the AF-related electrical remodeling14 (see also Chapter 45). Therefore, agonist-independent IK,ACh,c is expected to increase atrial vulnerability to tachyarrhythmias and to promote persistence of AF. Accordingly, inhibition of IK,ACh with the highly-selective IK,ACh blocker tertiapin reverses the APD abbreviation and prevents the AF promotion in dogs with atrial tachycardia remodeling (ATR) that also develop increased IK,ACh,c.30 IK,ACh is almost absent in ventricles; therefore, it is a promising atrial-selective anti-AF target that lacks proarrhythmic side effects in the ventricles.31
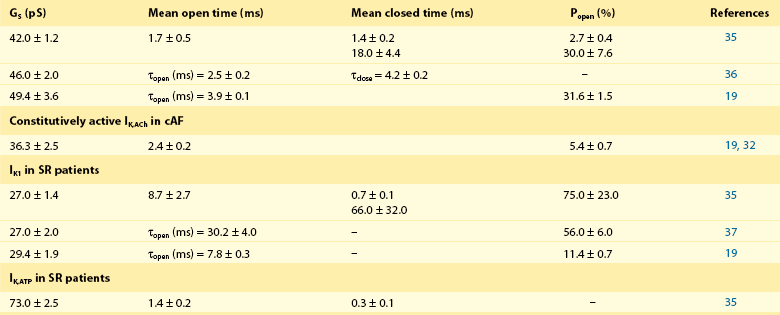
In whole-cell patch-clamp experiments, electrophysiological properties of IK,ACh (i.e., current-voltage relationship) are comparable with other inward-rectifier currents such as IK1 or IK,ATP; therefore, single-channel recordings are often used as a direct index of increased constitutive IK,ACh,c activity in atrial myocytes from patients with cAF and dogs with ATR.19,32–34 Due to their short opening-times and a characteristic single-channel conductance of approximately 40 pS IK,ACh single-channel openings are clearly different from openings of IK1 or IK,ATP (Table 38-1). Figure 38-1 shows representative recordings of IK,ACh in the presence and absence of a muscarinic-receptor agonist in atrial myocytes from patients with SR and cAF. Inclusion of the nonselective M-receptor agonist carbachol (10 µM) in the pipette solution strongly activated IK,ACh in both groups, causing frequent channel openings. In the absence of M-receptor agonists, constitutive IK,ACh,c openings are apparent in cAF, whereas they occur only sporadically in myocytes from SR patients.19
Table 38-1
Single-Channel Characteristics of Inward-Rectifier K+ Currents in Human Atrial Myocytes: Acetylcholine/Carbachol (10 µM) Activated IK,ACh in Patients with Sinus Rhythm
In dogs subjected to AF mimicking, atrial tachycardia remodeling IK,ACh also develops agonist-independent constitutive IK,ACh,c activity, suggesting that the development of IK,ACh,c in patients with cAF can result from the high atrial rate rather than from the underlying heart disease.29,30,34 In addition, the alterations in single-channel properties of IK,ACh,c are comparable between dogs with ATR and cAF patients, suggesting a common molecular basis.34 The complex regulation of IK,ACh points to several possible mechanisms that could contribute to the development of IK,ACh,c. The following discussion summarizes the current knowledge about the molecular regulation of agonist-dependent IK,ACh in atrial myocytes, particularly focusing on the putative mechanisms underlying constitutive IK,ACh activity in AF.
G-Protein Cycle Can Contribute to the Generation of Constitutive IK,ACh
M2-Receptor–Dependent IK,ACh
Inward-rectifier IK,ACh channels are activated through stimulation of appropriate Gi-protein–coupled receptors (M2-receptors), resulting in the dissociation of heterotrimeric Gi-proteins and consecutive binding of Gβγ-subunits to the channel (see Figure 38-2, A, B).38 Heterotrimeric G-proteins are composed of two functional units, the guanosine-5′-diphosphate and guanosine-5′-triphosphate (GDP/GTP) binding Gα-subunit and the Gβγ-dimer. Upon stimulation of G-protein–coupled receptors, GDP (bound to the Gα-subunit under resting conditions) is released and replaced by GTP. The resulting conformational changes lead to a dissociation of Gα and Gβγ subunits, which then initiate and regulate multiple intracellular pathways. Intrinsic GTPase activity of the Gα-subunit results in GTP hydrolysis into GDP followed by reassembly of the G-protein subunits and reestablishment of the initial inactive state.39 Gα-subunits are subdivided into Gαs-subunits, which stimulate adenylate cyclases, Gαi-subunits, which inhibit adenylate cyclases, and Gαq-subunits, which activate phospholipase-C (see later). IK,ACh channels are activated by direct binding of βγ-subunits originating from Gi-protein.4,38 It is unclear whether Gβγ-subunits originating from Gs– or Gq-proteins also activate IK,ACh channels under physiological conditions.
Although Gαi-subunits do not directly activate IK,ACh channels, they certainly have an important role in IK,ACh regulation. Under resting conditions, the GDP-bound Gαi-subunit is associated with the Kir3.1 channel subunit, thereby creating a “preformed complex” between the heterotrimeric G-protein and the IK,ACh channel.40 In this condition, Gβγ-subunits are always in close proximity to the channel and can thus effectively and rapidly activate IK,ACh upon Gαi-dissociation. The interaction between GαI and Kir3.1 is therefore an important contributor to the specificity between receptor stimulation and IK,ACh activation. In addition, GDP-bound Gαi can act as a Gβγ-scavenger, continuously chelating Gβγ-subunits from IK,ACh channels, thereby preventing agonist-independent IK,ACh activity.41 It is unknown whether reduced Gαi-expression or a higher Gβγ-subunit availability contribute to the enhanced constitutive IK,ACh activity in patients with AF.
The cycling of Gi-proteins is fine-tuned by multiple mechanisms. Regulator of G-protein signaling (RGS) proteins, which accelerate the GTPase activity of the Gα-protein subunit, accelerate the inactivation kinetics of receptor-activated IK,ACh (see Figure 38-3, B).42 [Na+]i also accelerates the G-protein cycle,43,44 although direct binding of Na+ to Kir3.4 could activate IK,ACh channels (see Figure 38-3, A).45–47,47a In addition, nucleoside diphosphate kinases (NDPKs), which classically transfer phosphates from ATP to GDP by a ping-pong mechanism involving the formation of a high-energy phosphate intermediate on histidine-118, might also contribute to the replenishment of the GTP necessary for the G-protein cycle (see Figure 38-2, B, C).39
Constitutively Active IK,ACh
Previous studies showed that the M-receptor antagonist atropine does not abolish constitutive IK,ACh,c activity in atrial myocytes from cAF patients or ATR-dogs, suggesting that a receptor-independent mechanism might be implicated in the formation of constitutive IK,ACh,c.19 Whether increased receptor-independent dissociation of Gαi– and Gβγ-subunits contributes to generation of constitutive IK,ACh,c in cAF patients is not known. Neither pertussis toxin, which uncouples Gi-proteins from the receptor, nor the absence of GTP, which is necessary for the action of G-proteins, affected the IK,ACh-like current component of the basal inward-rectifier K+ current in atrial myocytes from ATR dogs and atrial myocytes from cAF patients,19,29 suggesting that receptor-independent dissociation of Gαi– and Gβγ-subunits might not be a major contributor to constitutive IK,ACh,c in AF. However, a fraction of G-protein βγ-subunits forms complexes with the B-isoform of NDPKs and these complexes may cause receptor-independent G-protein activation (see Figure 38-2, B).39 This process involves NDPK-B–mediated phosphorylation of the Gβ-subunit at histidine-266. Subsequently the phosphate is transferred onto GDP bound to the Gα-subunit, and the formed GTP leads to receptor-independent G-protein activation. Because NDPK-B accumulates in the atrial membrane fraction of cAF patients48 and is involved in IK,ACh regulation,49 this receptor-independent mechanism of G-protein activation could be a major contributor to the development of constitutive IK,ACh,c in patients with cAF, although the NDPK hypothesis has also been challenged as a possible explanation for agonist-independent channel activation.50
Regulation of IK,ACh Gating by PIP2
M2-Receptor Dependent IK,ACh
IK,ACh channels require PIP2 to maintain physiological properties and removal of PIP2 completely runs down the IK,ACh current.51 Under resting conditions, the interaction between IK,ACh-channels and PIP2 is weak, and IK,ACh activity is low. Binding of Gβγ-subunits to IK,ACh channels strengthen the PIP2-channel subunit interaction, strongly increasing IK,ACh activity (see Figure 38-3, A). In atrial myocytes, the membrane PIP2 levels close to the IK,ACh channel are dynamically regulated by Gαq-protein–coupled receptors, the stimulation of which results in PIP2 breakdown by activating the PIP2 hydrolyzing enzyme phospholipase-C (see Figure 38-3, A). IK,ACh is also regulated by Gq-protein–coupled α1-adrenergic-, angiotensin-II type-1 (AT1)-, and endothelin-A (ETA) receptors, suggesting that GIRK-channels integrate hormone and neurotransmitter signals from different pathways.52,53 In addition, there is also evidence for the existence of Gαq-coupled M1– and M3-receptors in atria.54,55 Activation of Gαq-coupled M1– and M3-receptors and the associated PIP2 depletion are suggested to contribute to the fast desensitization of M2-receptor–activated IK,ACh current (see Figure 38-3, B).56,57 However, the existence of functional non–M2-receptors in atria is still controversial.58,59
Constitutively Active IK,ACh
Activation of IK,ACh channels via Gβγ-subunits is mediated via stronger binding of PIP2 (see Figure 38-3, A), and enhanced PIP2 levels in the IK,ACh channel microdomain activate the IK,ACh channel, even in the absence of Gβγ-subunits.51 As a result, agonist-independent IK,ACh,c activity can result from a Gβγ-independent increase in the PIP2-IK,ACh channel interaction. In addition, IK,ACh channels containing Kir3.4 subunits can be activated by an increase in [Na+]i in a Gβγ-subunit–independent manner (see Figure 38-3, A).45–47 Na+ ions are thought to neutralize a negatively charged aspartate in the C-terminus of Kir3.4, but not in Kir3.1, allowing stronger binding of PIP2 (see Figure 38-3, A). Mintert et al46 found that increases in [Na+]i resulted in higher constitutive IK,ACh,c activity in rat atrial cardiomyocytes overexpressing the Kir3.4 subunit.46 However, it is unknown whether changes in PIP2 membrane content or the subunit composition of the IK,ACh-channel contribute to clinically relevant constitutive IK,ACh,c activity and AF-associated IK,ACh remodeling. Nevertheless, rapid atrial rate-related increases in subsarcolemmal [Na+]i are likely to occur during AF in vivo, potentially contributing to constitutive IK,ACh,c activity.60
Phosphorylation-Dependent IK,ACh Regulation
M2-Receptor–Dependent IK,ACh
The GIRK channel forms a macromolecular complex that is composed of the catalytic subunits of protein kinase A (PKA) and C (PKC), the Ca2+/calmodulin-regulated protein kinase II (CaMKII) and the type-1 and type-2A protein phosphatases (PP1 and PP2A).61 In addition, the Kir3.1 and Kir3.4 channel subunits possess phosphorylation sites for PKA, PKC, CaMKII, and possibly for PKG,61,62 suggesting that channel phosphorylation status might play a crucial role in the regulation of IK,ACh activity (see Figure 38-3, C).
Inhibition of CaMKII, PKC, and PKG reduced agonist-activated IK,ACh current in human atrial myocytes, suggesting that CaMKII-, PKC- and PKG-mediated channel phosphorylation may stabilize the binding of Gβγ-subunits to the channel, thereby increasing IK,ACh.20 The regulation of IK,ACh by PKC is isoform specific. The PKC family contains many isoforms, including conventional isoforms (cPKCs), which are activated by Ca2+ and DAG, and novel isoforms (nPKCs), which require DAG but not Ca2+ for activation.63 There are at least five major PKC isoforms in the heart: cPKCα, cPKCβI, cPKCβII, nPKCδ, and nPKCε. Purified cPKC isoforms strongly reduce GTPγS-activated IK,ACh channel, whereas nPKC isoforms have the opposite effect.33 The relative expression-levels of stimulatory and inhibitory PKC isoforms can vary depending on species, diseases, or other conditions, potentially accounting for discrepant results between neonatal rat and canine atrial myocytes, in which PKC activation inhibits IK,ACh and human atrial myocytes, for which PKC appears to stimulate IK,ACh.61,64 PKA activity is expected to be reduced via Gαi-coupled M2-receptor activation consequent to ACh-application.
Upon application of ACh, direct binding of liberated Gβγ-subunits to the IK,ACh channel causes rapid activation of IK,ACh, followed by desensitization with a typical biphasic pattern (see Figure 38-3, middle).62 Although other mechanisms such as RGS proteins and PIP2 depletion owing to activation of Gq-protein–coupled M1/M3-receptors could also contribute, it has been suggested that the fast phase of desensitization is due to channel dephosphorylation,65,66 whereas the slower (intermediate) phase involves progressive M-receptor phosphorylation by a G-protein–coupled receptor type-2 kinase, which uncouples the receptor from the G-protein (see Figure 38-3, C).67
Because there is evidence for increased PKA, CaMKII, and PKC activity in cAF patients,20,68 IK,ACh phosphorylation might also be involved in disease-associated remodeling of IK,ACh. However, inhibition of CaMKII, PKC, PKG, and PKA does not affect M-receptor–activated IK,ACh in patients with cAF.20 In addition, activities of PP1 and PP2A are higher in cAF than in control SR patients and do not translate into altered protein phosphorylation.69–71 Thus, the lack of contribution of the these kinases to M-receptor–activated IK,ACh in cAF might result from opposing increases in channel dephosphorylation. Alternatively, the channel could be hyperphosphorylated because of abnormal control of kinase–phosphatase signaling in the macromolecular complex,61 and inhibition of a single kinase might not reduce the channel’s phosphorylation state below the threshold required to impair agonist-activated IK,ACh. Further work is needed to verify these hypotheses.
Constitutively Active IK,ACh,c
Because activation of IK,ACh requires ATP, modified phosphorylation-dependent channel regulation could contribute to constitutive IK,ACh,c activity.62,66 In single-channel patch-clamp experiments, excision of the patch and removal from the intact cell produces a cell-free piece of sarcolemma attached to the pipette and containing the IK,ACh channel (inside-out configuration), with the cytosolic side facing the bath solution. Under these conditions, IK,ACh,c open probability is strongly reduced, suggesting that an intracellular component necessary for constitutive IK,ACh,c activity is washed out.33 In the presence of phosphatase inhibitors, excision of the patch only slightly reduces the open probability of constitutive IK,ACh,c. These experiments point to a crucial role of phosphorylation in IK,ACh,c. Inhibition of PKC reduces basal current, which contains constitutive IK,ACh,c activity, in patients with cAF but not SR, further suggesting that PKC-mediated phosphorylation of IK,ACh channels is involved in maintaining constitutive IK,ACh,c.20 The expression level of PKCε, which stimulates IK,ACh,33 is increased in patients with cAF, whereas PKCα, PKCβI, and PKCδ remains unchanged.20 These data indicate the involvement of PKC isoform imbalance in the development of agonist-independent IK,ACh,c. Consistent with this hypothesis, Makary et al.33 showed that changes in the balance between stimulatory nPKC and inhibitory cPKC isoforms contribute to increased constitutive IK,ACh,c activity in AF.33 The cPKC (inhibitory) phenotype predominates in control dogs, whereas the nPKC (stimulatory) phenotype prevails in ATR dogs (Figure 38-4).33 This idea is supported by a reduced total expression of PKCα (cPKC isoform) and increased membrane translocation of PKCε (nPKC isoform), in atrial myocytes from ATR dogs. In control myocytes, blocking cPKC isoforms increases constitutive IK,ACh,c, suggesting tonic cPKC-mediated inhibition of IK,ACh because of predominance of inhibitory PKCα isoforms. cPKC inhibition has no effect in ATR dogs. In contrast, blocking PKCε does not affect constitutive IK,ACh,c in control myocytes, but it reduces IK,ACh,c in ATR myocytes to levels seen in control dogs. These data indicate that a reduced tonic inhibitory effect of PKCα and an increased stimulatory effect of PKCε are the major determinants of enhanced constitutive IK,ACh,c activity in AF.
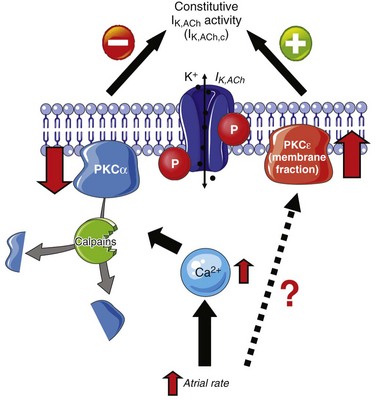
Figure 38-4 Protein kinase C (PKC) isoform shift contributes to development of constitutively active IK,ACh (IK,ACh,c). Atrial tachycardia induces Ca2+-independent translocation of PKCε from the cytosol to the membrane and causes Ca2+/calpain–dependent breakdown of PKCα. Both reduced PKCα expression and PKCε membrane translocation contribute to increased IK,ACh,c. (Figure was produced using Servier Medical Art.)
Rapid in vitro pacing of isolated atrial myocytes from control dogs reproduces the phenotype seen in ATR dogs, with a PKC isoform switch and increased constitutive IK,ACh,c activity, demonstrating that the PKC isoform switch in ATR could be attributable to the rapid atrial activity per se.33 In addition, the cellular in vitro pacing experiments revealed that PKCα downregulation likely results from atrial tachycardia-induced Ca2+ loading and subsequent activation of the Ca2+-dependent protease calpain, which might increase the degradation of PKCα isoforms. In contrast, the increased membrane translocation of PKCε in ATR myocytes seems to be Ca2+ independent (see Figure 38-4), suggesting that two independent mechanisms combine to cause constitutive IK,ACh,c activity in AF. Although there is only indirect evidence that phosphorylation-dependent mechanisms contribute to the increase of constitutive IK,ACh,c in human AF,20 these posttranslational mechanisms could provide interesting new therapeutic targets for atrial-selective and pathology-specific treatment of AF.
IK,ACh Channels as Potential Therapeutic Targets in Atrial Fibrillation
Constitutive IK,ACh activity has emerged as a potentially interesting therapeutic target for AF treatment. Because of much higher IK,ACh expression in the atria in comparison to the ventricles, selective IK,ACh inhibition could have antiarrhythmic effects in the atria, without ventricular side effects.72 Because some drugs with anti-AF efficacy (e.g., amiodarone, flecainide, quinidine, verapamil) are also inhibitors of IK,ACh,4,21,73,74 their clinical effectiveness may result in part from blockade of IK,ACh,c.
Recent research on structural motifs in K+ channel drug interactions can allow the development of more specific IK,ACh-channel blocking agents for AF therapy. In addition to a central conduction pathway formed by four channel subunits, both the amino and carboxyl termini of the subunits form a second cytoplasmic pore that extends the ion permeation pathway toward the intracellular side of the membrane.75,76 Tertiapin, a peptide toxin from the honey bee venom, inhibits IK,ACh-channel selectively77–79 by binding to the external vestibule of the central conduction pathway.80,81 In contrast, chloroquine blocks inward-rectifier K+ channels by interacting with the center of the cytoplasmic conduction pore via interference with negatively charged aromatic residues within a central cavity.82 These results suggest that compounds targeting the specific cytoplasmic domain residues of ACh-dependent inward-rectifier K+ channels might constitute chemical leads for the design of novel selective IK,ACh channel blockers.
During the past few years, several IK,ACh-selective drugs have been developed (e.g., NIP-142, NTC-801). However off-target effects on IK,ACh channels in the SA node and peripheral tissues (i.e., gastrointestinal tract, genitourinary system) could limit the value of directly targeting the channel pore.72 An improved understanding of the molecular basis for increased constitutive IK,ACh,c activity in cAF might allow for atrial-selective and disease-specific targeting of IK,ACh in AF. However, such strategies are still in their infancy, and much more work is needed before this approach can be tested for efficacy and value in clinical AF.
References
1. Loewi, O. Über humorale Übertragbarkeit der Herznervenwirkung. Pflügers Arch. 1921; 189(1):239–242.
2. Noma, A, Trautwein, W. Relaxation of the ACh-induced potassium current in the rabbit sinoatrial node cell. Pflügers Arch. 1978; 377(3):193–200.
3. Krapivinsky, G, Gordon, EA, Wickman, K, et al. The G-protein-gated atrial K+ channel IK,ACh is a heteromultimer of two inwardly rectifying K+-channel proteins. Nature. 1995; 374(6518):135–141.
4. Hibino, H, Inanobe, A, Furutani, K, et al. Inwardly rectifying potassium channels: their structure, function, and physiological roles. Physiol Rev. 2010; 90(1):291–366.
5. Yamada, M, Inanobe, A, Kurachi, Y. G protein regulation of potassium ion channels. Pharmacol Rev. 1998; 50(4):723–760.
6. Bender, K, Wellner-Kienitz, MC, Inanobe, A, et al. Overexpression of monomeric and multimeric GIRK4 subunits in rat atrial myocytes removes fast desensitization and reduces inward rectification of muscarinic K+ current (IK,ACh). Evidence for functional homomeric GIRK4 channels. J Biol Chem. 2001; 276(31):28873–28880.
7. Dobrev, D, Wettwer, E, Himmel, HM, et al. G-Protein beta(3)-subunit 825T allele is associated with enhanced human atrial inward rectifier potassium currents. Circulation. 2000; 102(6):692–697.
8. Kurachi, Y, Nakajima, T, Sugimoto, T. On the mechanism of activation of muscarinic K+ channels by adenosine in isolated atrial cells: involvement of GTP-binding proteins. Pflugers Arch. 1986; 407(3):264–274.
9. Himmel, HM, Meyer Zu Heringdorf, D, Graf, E, et al. Evidence for Edg-3 receptor-mediated activation of IK,ACh by sphingosine-1-phosphate in human atrial cardiomyocytes. Mol Pharmacol. 2000; 58(2):449–454.
10. Verkerk, AO, Geuzebroek, GS, Veldkamp, MW, et al. Effects of acetylcholine and noradrenalin on action potentials of isolated rabbit sinoatrial and atrial myocytes. Front Physiol. 2012; 3:174.
11. Gaborit, N, Le Bouter, S, Szuts, V, et al. Regional and tissue specific transcript signatures of ion channel genes in the non-diseased human heart. J Physiol. 2007; 582(Pt 2):675–693.
12. Koumi, S, Wasserstrom, JA. Acetylcholine-sensitive muscarinic K+ channels in mammalian ventricular myocytes. Am J Physiol. 1994; 266(5 Pt 2):H1812–H1821.
13. Dobrev, D, Graf, E, Wettwer, E, et al. Molecular basis of downregulation of G-protein-coupled inward rectifying K+ current (IK,ACh) in chronic human atrial fibrillation: decrease in GIRK4 mRNA correlates with reduced IK,ACh and muscarinic receptor-mediated shortening of action potentials. Circulation. 2001; 104(21):2551–2557.
14. Wakili, R, Voigt, N, Kaab, S, et al. Recent advances in the molecular pathophysiology of atrial fibrillation. J Clin Invest. 2011; 121(8):2955–2968.
15. Chen, PS, Tan, AY. Autonomic nerve activity and atrial fibrillation. Heart Rhythm. 2007; 4(3 Suppl):S61–S64.
16. Kovoor, P, Wickman, K, Maguire, CT, et al. Evaluation of the role of IK,ACh in atrial fibrillation using a mouse knockout model. J Am Coll Cardiol. 2001; 37(8):2136–2143.
17. Pandit, SV, Berenfeld, O, Anumonwo, JM, et al. Ionic determinants of functional reentry in a 2-D model of human atrial cells during simulated chronic atrial fibrillation. Biophys J. 2005; 88(6):3806–3821.
18. Katsouras, G, Sakabe, M, Comtois, P, et al. Differences in atrial fibrillation properties under vagal nerve stimulation versus atrial tachycardia remodeling. Heart Rhythm. 2009; 6(10):1465–1472.
19. Dobrev, D, Friedrich, A, Voigt, N, et al. The G protein-gated potassium current IK,ACh is constitutively active in patients with chronic atrial fibrillation. Circulation. 2005; 112(24):3697–3706.
20. Voigt, N, Friedrich, A, Bock, M, et al. Differential phosphorylation-dependent regulation of constitutively active and muscarinic receptor-activated IK,ACh channels in patients with chronic atrial fibrillation. Cardiovasc Res. 2007; 74(3):426–437.
21. Voigt, N, Rozmaritsa, N, Trausch, A, et al. Inhibition of IK,ACh current may contribute to clinical efficacy of class I and class III antiarrhythmic drugs in patients with atrial fibrillation. Naunyn Schmiedebergs Arch Pharmacol. 2010; 381(3):251–259.
22. Voigt, N, Trausch, A, Knaut, M, et al. Left-to-right atrial inward rectifier potassium current gradients in patients with paroxysmal versus chronic atrial fibrillation. Circ Arrhythm Electrophysiol. 2010; 3(5):472–480.
23. Lazar, S, Dixit, S, Marchlinski, FE, et al. Presence of left-to-right atrial frequency gradient in paroxysmal but not persistent atrial fibrillation in humans. Circulation. 2004; 110(20):3181–3186.
24. Sanders, P, Berenfeld, O, Hocini, M, et al. Spectral analysis identifies sites of high-frequency activity maintaining atrial fibrillation in humans. Circulation. 2005; 112(6):789–797.
25. Swartz, MF, Fink, GW, Lutz, CJ, et al. Left versus right atrial difference in dominant frequency, K+ channel transcripts, and fibrosis in patients developing atrial fibrillation after cardiac surgery. Heart Rhythm. 2009; 6(10):1415–1422.
26. Sarmast, F, Kolli, A, Zaitsev, A, et al. Cholinergic atrial fibrillation: IK,ACh gradients determine unequal left/right atrial frequencies and rotor dynamics. Cardiovasc Res. 2003; 59(4):863–873.
27. Sakmann, B, Noma, A, Trautwein, W. Acetylcholine activation of single muscarinic K+ channels in isolated pacemaker cells of the mammalian heart. Nature. 1983; 303(5914):250–253.
28. Ito, H, Ono, K, Noma, A. Background conductance attributable to spontaneous opening of muscarinic K+ channels in rabbit sino-atrial node cells. J Physiol. 1994; 476(1):55–68.
29. Ehrlich, JR, Cha, TJ, Zhang, L, et al. Characterization of a hyperpolarization-activated time-dependent potassium current in canine cardiomyocytes from pulmonary vein myocardial sleeves and left atrium. J Physiol. 2004; 557(Pt 2):583–597.
30. Cha, TJ, Ehrlich, JR, Chartier, D, et al. Kir3-based inward rectifier potassium current: potential role in atrial tachycardia remodeling effects on atrial repolarization and arrhythmias. Circulation. 2006; 113(14):1730–1737.
31. Dobrev, D, Nattel, S. New antiarrhythmic drugs for treatment of atrial fibrillation. Lancet. 2010; 375(9721):1212–1223.
32. Voigt, N, Makary, S, Nattel, S, et al. Voltage-clamp-based methods for the detection of constitutively active acetylcholine-gated IK,ACh channels in the diseased heart. Methods Enzymol. 2010; 484:653–675.
33. Makary, S, Voigt, N, Maguy, A, et al. Differential protein kinase C isoform regulation and increased constitutive activity of acetylcholine-regulated potassium channels in atrial remodeling. Circ Res. 2011; 109(9):1031–1043.
34. Voigt, N, Maguy, A, Yeh, YH, et al. Changes in IK,ACh single-channel activity with atrial tachycardia remodelling in canine atrial cardiomyocytes. Cardiovasc Res. 2008; 77(1):35–43.
35. Heidbüchel, H, Vereecke, J, Carmeliet, E. Three different potassium channels in human atrium. Contribution to the basal potassium conductance. Circ Res. 1990; 66(5):1277–1286.
36. Sato, R, Hisatome, I, Wasserstrom, JA, et al. Acetylcholine-sensitive potassium channels in human atrial myocytes. Am J Physiol. 1990; 259(6 Pt 2):H1730–H1735.
37. Koumi, S-i, Backer, CL, Arentzen, CE. Characterization of inwardly rectifying K+ channel in human cardiac myocytes : alterations in channel behavior in myocytes isolated from patients with idiopathic dilated cardiomyopathy. Circulation. 1995; 92(2):164–174.
38. Logothetis, DE, Kurachi, Y, Galper, J, et al. The beta gamma subunits of GTP-binding proteins activate the muscarinic K+ channel in heart. Nature. 1987; 325(6102):321–326.
39. Wieland, T. Interaction of nucleoside diphosphate kinase B with heterotrimeric G protein betagamma dimers: consequences on G protein activation and stability. Naunyn Schmiedebergs Arch Pharmacol. 2007; 374(5-6):373–383.
40. Riven, I, Iwanir, S, Reuveny, E. GIRK channel activation involves a local rearrangement of a preformed G protein channel complex. Neuron. 2006; 51(5):561–573.
41. Yamada, M, Ho, YK, Lee, RH, et al. Muscarinic K+ channels are activated by beta gamma subunits and inhibited by the GDP-bound form of alpha subunit of transducin. Biochem Biophys Res Commun. 1994; 200(3):1484–1490.
42. Doupnik, CA, Davidson, N, Lester, HA, et al. RGS proteins reconstitute the rapid gating kinetics of gbetagamma-activated inwardly rectifying K+ channels. Proc Natl Acad Sci U S A. 1997; 94(19):10461–10466.
43. Rishal, I, Keren-Raifman, T, Yakubovich, D, et al. Na+ promotes the dissociation between Galpha GDP and Gbeta gamma, activating G protein-gated K+ channels. J Biol Chem. 2003; 278(6):3840–3845.
44. Yakubovich, D, Rishal, I, Dascal, N. Kinetic modeling of Na+-induced, Gbetagamma-dependent activation of G protein-gated K+ channels. J Mol Neurosci. 2005; 25(1):7–19.
45. Ho, IH, Murrell-Lagnado, RD. Molecular mechanism for sodium-dependent activation of G protein-gated K+ channels. J Physiol. 1999; 520(Pt 3):645–651.
46. Mintert, E, Bosche, LI, Rinne, A, et al. Generation of a constitutive Na+-dependent inward-rectifier current in rat adult atrial myocytes by overexpression of Kir3. 4. J Physiol. 2007; 585(Pt 1):3–13.
47. Rosenhouse-Dantsker, A, Sui, JL, Zhao, Q, et al. A sodium-mediated structural switch that controls the sensitivity of Kir channels to PtdIns(4,5)P(2). Nat Chem Biol. 2008; 4(10):624–631.
47a. Voigt, N, Heijman, J, Trausch, A, et al. Impaired Na+-dependent regulation of acetylcholine-activated inward-rectifier K+ current modulates action potential rate dependence in patients with chronic atrial fibrillation. J Mol Cell Cardiol. 2013.
48. Abu-Taha, I, Voigt, N, Nattel, S, et al. Nucleoside diphosphate kinase B is a novel receptor-independent activator of G-protein signaling in clinical and experimental atrial fibrillation. Heart Rhythm. 2012; 9(5 (Supplement):S397.
49. Kaibara, M, Nakajima, T, Irisawa, H, et al. Regulation of spontaneous opening of muscarinic K+ channels in rabbit atrium. J Physiol. 1991; 433:589–613.
50. Sorota, S, Chlenov, M, Du, XY, et al. ATP-dependent activation of the atrial acetylcholine-induced K+ channel does not require nucleoside diphosphate kinase activity. Circ Res. 1998; 82(9):971–979.
51. Huang, CL, Feng, S, Hilgemann, DW. Direct activation of inward rectifier potassium channels by PIP2 and its stabilization by Gbetagamma. Nature. 1998; 391(6669):803–806.
52. Cui, S, Ho, WK, Kim, ST, et al. Agonist-induced localization of Gq-coupled receptors and G protein-gated inwardly rectifying K+ (GIRK) channels to caveolae determines receptor specificity of phosphatidylinositol 4,5-bisphosphate signaling. J Biol Chem. 2010; 285(53):41732–41739.
53. Choisy, SC, James, AF, Hancox, JC. Acute desensitization of acetylcholine and endothelin-1 activated inward rectifier K+ current in myocytes from the cardiac atrioventricular node. Biochem Biophys Res Commun. 2012.
54. Wang, H, Han, H, Zhang, L, et al. Expression of multiple subtypes of muscarinic receptors and cellular distribution in the human heart. Mol Pharmacol. 2001; 59(5):1029–1036.
55. Perez, CC, Tobar, ID, Jimenez, E, et al. Kinetic and molecular evidences that human cardiac muscle express non-M2 muscarinic receptor subtypes that are able to interact themselves. Pharmacol Res. 2006; 54(5):345–355.
56. Jan, LY, Jan, YN. Heartfelt crosstalk: desensitization of the GIRK current. Nat Cell Biol. 2000; 2(9):E165–E167.
57. Kobrinsky, E, Mirshahi, T, Zhang, H, et al. Receptor-mediated hydrolysis of plasma membrane messenger PIP2 leads to K+-current desensitization. Nat Cell Biol. 2000; 2(8):507–514.
58. Hellgren, I, Mustafa, A, Riazi, M, et al. Muscarinic M3 receptor subtype gene expression in the human heart. Cell Mol Life Sci. 2000; 57(1):175–180.
59. Oberhauser, V, Schwertfeger, E, Rutz, T, et al. Acetylcholine release in human heart atrium: influence of muscarinic autoreceptors, diabetes, and age. Circulation. 2001; 103(12):1638–1643.
60. Grandi, E, Pandit, SV, Voigt, N, et al. Human atrial action potential and Ca2+ model: sinus rhythm and chronic atrial fibrillation. Circ Res. 2011; 109(9):1055–1066.
61. Nikolov, EN, Ivanova-Nikolova, TT. Coordination of membrane excitability through a GIRK1 signaling complex in the atria. J Biol Chem. 2004; 279(22):23630–23636.
62. Medina, I, Krapivinsky, G, Arnold, S, et al. A switch mechanism for G beta gamma activation of IK,ACh. J Biol Chem. 2000; 275(38):29709–29716.
63. Steinberg, SF. Structural basis of protein kinase C isoform function. Physiol Rev. 2008; 88(4):1341–1378.
64. Yeh, YH, Ehrlich, JR, Qi, X, et al. Adrenergic control of a constitutively active acetylcholine-regulated potassium current in canine atrial cardiomyocytes. Cardiovasc Res. 2007; 74(3):406–415.
65. Kim, D. Mechanism of rapid desensitization of muscarinic K+ current in adult rat and guinea pig atrial cells. Circ Res. 1993; 73(1):89–97.
66. Shui, Z, Boyett, MR, Zang, WJ. ATP-dependent desensitization of the muscarinic K+ channel in rat atrial cells. J Physiol. 1997; 505(Pt 1):77–93.
67. Shui, Z, Boyett, MR, Zang, WJ, et al. Receptor kinase-dependent desensitization of the muscarinic K+ current in rat atrial cells. J Physiol. 1995; 487(Pt 2):359–366.
68. Voigt, N, Li, N, Wang, Q, et al. Enhanced sarcoplasmic reticulum Ca2+ leak and increased Na+-Ca2+ exchanger function underlie delayed afterdepolarizations in patients with chronic atrial fibrillation. Circulation. 2012; 125(17):2059–2070.
69. Christ, T, Boknik, P, Wohrl, S, et al. L-type Ca2+ current downregulation in chronic human atrial fibrillation is associated with increased activity of protein phosphatases. Circulation. 2004; 110(17):2651–2657.
70. El-Armouche, A, Boknik, P, Eschenhagen, T, et al. Molecular determinants of altered Ca2+ handling in human chronic atrial fibrillation. Circulation. 2006; 114(7):670–680.
71. Greiser, M, Halaszovich, CR, Frechen, D, et al. Pharmacological evidence for altered src kinase regulation of ICa,L in patients with chronic atrial fibrillation. Naunyn Schmiedebergs Arch Pharmacol. 2007; 375(6):383–392.
72. Dobrev, D, Carlsson, L, Nattel, S. Novel molecular targets for atrial fibrillation therapy. Nat Rev Drug Discov. 2012; 11(4):275–291.
73. Kurachi, Y, Nakajima, T, Ito, H, et al. AN-132, a new class I anti-arrhythmic agent, depresses the acetylcholine-induced K+ current in atrial myocytes. Eur J Pharmacol. 1989; 165(2-3):319–322.
74. Kurachi, Y, Nakajima, T, Sugimoto, T. Quinidine inhibition of the muscarine receptor-activated K+ channel current in atrial cells of guinea pig. Naunyn Schmiedebergs Arch Pharmacol. 1987; 335(2):216–218.
75. Furutani, K, Ohno, Y, Inanobe, A, et al. Mutational and in silico analyses for antidepressant block of astroglial inward-rectifier Kir4. 1 channel. Mol Pharmacol. 2009; 75(6):1287–1295.
76. Whorton, MR, MacKinnon, R. Crystal structure of the mammalian GIRK2 K+ channel and gating regulation by G proteins, PIP2, and sodium. Cell. 2011; 147(1):199–208.
77. Jin, W, Klem, AM, Lewis, JH, et al. Mechanisms of inward-rectifier K+ channel inhibition by tertiapin-Q. Biochemistry. 1999; 38(43):14294–14301.
78. Jin, W, Lu, Z. A novel high-affinity inhibitor for inward-rectifier K+ channels. Biochemistry. 1998; 37(38):13291–13299.
79. Jin, W, Lu, Z. Synthesis of a stable form of tertiapin: a high-affinity inhibitor for inward-rectifier K+ channels. Biochemistry. 1999; 38(43):14286–14293.
80. Ramu, Y, Klem, AM, Lu, Z. Short variable sequence acquired in evolution enables selective inhibition of various inward-rectifier K+ channels. Biochemistry. 2004; 43(33):10701–10709.
81. Ramu, Y, Xu, Y, Lu, Z. Engineered specific and high-affinity inhibitor for a subtype of inward-rectifier K+ channels. Proc Natl Acad Sci U S A. 2008; 105(31):10774–10778.
82. Noujaim, SF, Stuckey, JA, Ponce-Balbuena, D, et al. Specific residues of the cytoplasmic domains of cardiac inward rectifier potassium channels are effective antifibrillatory targets. FASEB J. 2010; 24(11):4302–4312.