[level-membership-for-basic-science-category]
Chapter 22 Cholinergic and antimuscarinic (anticholinergic) mechanisms and drugs
Acetylcholine antagonists that block the nicotine-like effects (neuromuscular blockers and autonomic ganglion blockers) are described elsewhere (Ch. 19).








Cholinergic drugs (cholinomimetics)
• For myasthenia gravis, both to diagnose (edrophonium) and to treat symptoms (neostigmine, pyridostigmine, distigmine).
• To lower intraocular pressure in chronic simple glaucoma (pilocarpine).
• To bronchodilate patients with airflow obstruction (ipratropium, tiotropium).
• To improve cognitive function in Alzheimer’s disease (rivastigmine, donepezil).
Classification
Direct-acting (receptor agonists)
• Choline esters (bethanechol, carbachol), which act at all sites, like acetylcholine, but are resistant to degradation by acetylcholinesterases (AChE; see Fig. 22.1). Muscarinic effects are much more prominent than nicotinic (see p. 373).
• Alkaloids (pilocarpine, muscarine) act selectively on end-organs of postganglionic, cholinergic neurones. Effects are exclusively muscarinic.
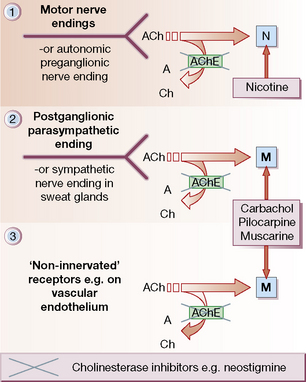
Sites of action (Fig. 22.1)
• Autonomic nervous system (Fig. 22.1, sites 1 and 2).
• Neuromuscular junction (Fig. 22.1, site 1).
• Central nervous system (CNS).
• Non-innervated sites: blood vessels, chiefly arterioles (Fig. 22.1, site 3).
Acetylcholine is released from nerve terminals to activate a post-synaptic receptor, except on blood vessels, where the action of cholinergic drugs is unrelated to cholinergic ‘vasodilator’ nerves. It is also produced in tissues unrelated to nerve endings, e.g. placenta and ciliated epithelial cells, where it acts as a local hormone (autacoid) on local receptors.
Pharmacology
Autonomic nervous system
• Eye: miosis and spasm of the ciliary muscle occur so that the eye is accommodated for near vision. Intraocular pressure falls.
• Exocrine glands: there is increased secretion most noticeably from salivary, lachrymal, bronchial and sweat glands. The last are cholinergic, but anatomically part of the sympathetic system; some sweat glands, e.g. axillary, may be adrenergic.
• Heart: bradycardia occurs with atrioventricular block, and eventually cardiac arrest.
• Bronchi: there is bronchoconstriction and mucosal hypersecretion that may be clinically serious in asthmatic subjects, in whom cholinergic drugs should be avoided if possible.
• Gut: motor activity is increased and may cause colicky pain. Exocrine secretion is also increased. Tone in sphincters falls, which may cause defaecation (anal sphincter) or acid reflux/regurgitation (oesophageal sphincter).
• Urinary bladder and ureters contract and the drugs promote micturition.
Choline esters
Acetylcholine
Acetylcholine was first injected intravenously as a therapeutic convulsant in 1939, in the reasonable expectation that the fits would be less liable to cause fractures than those following therapeutic leptazol (pentylenetetrazole) convulsions. Recovery rates of up to 80% were claimed in various psychotic conditions. Enthusiasm began to wane, however, when it was shown that the fits were due to anoxia resulting from cardiac arrest and not to pharmacological effects on the brain.1
The following description is typical:
Lachrymation, sweating and borborygmi were prominent. The deep reflexes became diminished. The patient then relaxed and ‘lay quietly in bed – cold moist and gray. In about 90 seconds, flushing of the face marked the return of the pulse’. The respiratory rate rose and consciousness returned in about 125 seconds. The patients sometimes micturated but did not defaecate. They ‘tended to lie quietly in bed after the treatment’. ‘Most of the patients were reluctant to be retreated.’2
Alkaloids with cholinergic effects
(see also p. 151) is a social drug that lends its medicinal use as an adjunct to stopping its own abuse as tobacco. It is available as gum to chew, dermal patches, a nasal spray or an inhalator. These deliver a lower dose of nicotine than cigarettes and appear to be safe in patients with ischaemic heart disease. The patches are slightly better tolerated than the gum, which releases nicotine in a more variable fashion depending on the rate at which it is chewed and the salivary pH, which is influenced by drinking coffee and carbonated drinks. Nicotine treatment is reported to be nearly twice as effective as placebo in achieving sustained withdrawal from smoking (18% versus 11% in one review).3 Treatment is much more likely to be successful if it is used as an aid to, not a substitute for, continued counselling. Bupropion is possibly more effective than the nicotine patch4 (see also p. 152) and the partial nicotinic agonist, varenicline, slightly more effective still. The efficacy of varenicline is tempered by its ability to cause suicidal ideation and behaviour.
from a South American plant (Pilocarpus spp.), acts directly on muscarinic receptors (see Fig. 22.1); it also stimulates and then depresses the CNS. The chief clinical use of pilocarpine is to lower intraocular pressure in primary open-angle glaucoma (also called chronic simple or wide-angle glaucoma), as an adjunct to a topical β-blocker; it produces miosis, opens drainage channels in the trabecular network and improves the outflow of aqueous humour. Oral pilocarpine is available for the treatment of xerostomia (dry mouth) in Sjögren’s syndrome, or following irradiation of head and neck tumours. The commonest adverse effect is sweating, an effect actually exploited in a diagnostic test for cystic fibrosis.
Poisoning with these fungi may present with antimuscarinic, cholinergic or GABAergic effects. All have CNS actions. Happily, poisoning by Amanita muscaria is seldom serious, but species of Inocybe contain substantially larger amounts of muscarine (see Ch. 10). The lengths to which humans are prepared to go in taking ‘chemical vacations’ when life is hard are shown by the inhabitants of eastern Siberia, who used Amanita muscaria recreationally for its cerebral stimulant effects. They were apparently prepared to put up with the autonomic actions to escape briefly from reality – so much so that when the fungus was scarce in winter they were even prepared to drink their own urine to prolong the experience. Sometimes, in generous mood, they would even offer their urine to others as a treat.
Anticholinesterases
At cholinergic nerve endings and in erythrocytes there is a specific enzyme that destroys acetylcholine, true cholinesterase or acetylcholinesterase. In various tissues, especially plasma, there are other esterases that are not specific for acetylcholine but that also destroy other esters, e.g. suxamethonium, procaine (and cocaine) and bambuterol (a prodrug that is hydrolysed to terbutaline). Hence, they are called pseudocholinesterases. Chemicals that inactivate these esterases (anticholinesterases) are used in medicine and in agriculture as pesticides. They act by allowing naturally synthesised acetylcholine to accumulate instead of being destroyed. Their effects are explained by this accumulation in the CNS, neuromuscular junction, autonomic ganglia, postganglionic cholinergic nerve endings (which are principally in the parasympathetic nervous system) and in the walls of blood vessels, where acetylcholine has a paracrine5 role not necessarily associated with nerve endings. Some of these effects oppose one another, e.g. the effect of anticholinesterase on the heart will be the result of stimulation at sympathetic ganglia and the opposing effect of stimulation at parasympathetic (vagal) ganglia and at postganglionic nerve endings.
is an alkaloid, obtained from the seeds of the West African Calabar bean (spp. Physostigma), which has had long use both as a weapon and as an ordeal poison.6 It acts for a few hours. It has been shown to have some efficacy in improving cognitive function in Alzheimer-type dementia.
A more recent use of anticholinesterase drugs has been to improve cognitive function in patients with Alzheimer’s disease (see p. 344), where both the degree of dementia and amyloid plaque density correlate with the impairment of brain cholinergic function. Donepezil, galantamine (which has additional nicotinic agonist properties) and rivastigmine are licensed in the UK for this indication. They are all reversible inhibitors that are orally active and cross the blood–brain barrier readily (see p. 81).
Anticholinesterase poisoning
Substances of this type have also been developed and used in war, especially the three G agents: GA (tabun), GB (sarin) and GD (soman). Although called nerve ‘gas’, they are actually volatile liquids, which facilitates their use.7 Where there is known risk of exposure, prior use of pyridostigmine, which occupies cholinesterases reversibly for a few hours (the lesser evil), competitively protects them from access by the irreversible warfare agent (the greater evil); soldiers during the Gulf Wars expecting attack were provided with preloaded syringes (of the same design as the Epipen) for delivering adrenaline/epinephrine as antidote therapy (see below). Organophosphate agents are absorbed through the skin, the gastrointestinal tract and by inhalation. Diagnosis depends on observing a substantial part of the list of actions below.
• Atropine is the mainstay of treatment; 2 mg is given i.m. or i.v. as soon as possible and repeated every 15–60 min until dryness of the mouth and a heart rate exceeding 70 beats per minute indicate that its effect is adequate. A poisoned patient may require 100 mg or more for a single episode. Atropine antagonises the muscarinic parasympathomimetic effects of the poison, i.e. due to the accumulated acetylcholine stimulating postganglionic nerve endings (excessive secretion and vasodilatation), but has no effect on the neuromuscular block, which is nicotinic.
• Mechanical ventilation may therefore be needed to assist the respiratory muscles; special attention to the airway is vital because of bronchial constriction and excessive secretion.
• Diazepam may be needed for convulsions.
• Atropine eye drops may relieve the headache caused by miosis.
• Enzyme reactivation. The organophosphate (OP) pesticides inactivate cholinesterase by irreversibly phosphorylating the active centre of the enzyme. Substances that reactivate the enzyme hasten the destruction of the accumulated acetylcholine and, unlike atropine, they have both antinicotinic and antimuscarinic effects. The principal agent is pralidoxime, which should be given by slow intravenous injection (diluted) over 5–10 min, initially 30 mg/kg repeated every 4–6 h or by intravenous infusion, 8 mg/kg/h; usual maximum 12 g in 24 h. Its efficacy is greatest if administered within 12 h of poisoning, then falls off steadily as the phosphorylated enzyme is further stabilised by ‘ageing’. If significant reactivation occurs, muscle power improves within 30 min.
Disorders of neuromuscular transmission
Myasthenia gravis
Neostigmine was introduced in 1931 for its stimulant effects on intestinal activity. In 1934 it occurred to Dr Mary Walker that, as the paralysis of myasthenia had been (erroneously) attributed to a curare-like substance in the blood, physostigmine (eserine), an anticholinesterase drug known to antagonise curare, might be beneficial. It was, and she reported this important observation in a short letter.8 Soon after this she used neostigmine by mouth, with greater benefit. The sudden appearance of an effective treatment for a hitherto untreatable chronic disease must always be a dramatic event for its victims. One patient described the impact of the discovery of the action of neostigmine, as follows:
My myasthenia started in 1925, when I was 18. For several months it consisted of double vision and fatigue … An ophthalmic surgeon … prescribed glasses with a prism. However, soon more alarming symptoms began. [Her limbs became weak and she] was sent to an eminent neurologist. This was a horrible experience. He … could find no physical signs … declared me to be suffering from hysteria and asked me what was on my mind. When I answered truthfully, that nothing except anxiety over my symptoms, he replied ‘my dear child, I am not a perfect fool …’, and showed me out. [She became worse and at times she was unable to turn over in bed. Eating and even speaking were difficult. Eventually, her fiancé, a medical student, read about myasthenia gravis and she was correctly diagnosed in 1927.] There was at that time no known treatment and therefore many things to try. [She had gold injections, thyroid, suprarenal extract, lecithin, glycine and ephedrine. The last had a slight effect.] Then in February 1935, came the day that I shall always remember. I was living alone with a nurse … It was one of my better days, and I was lying on the sofa after tea … My fiancé came in rather late saying that he had something new for me to try. My first thought was ‘Oh bother! Another injection, and another false hope’. I submitted to the injection with complete indifference and within a few minutes began to feel very strange … when I lifted my arms, exerting the effort to which I had become accustomed, they shot into the air, every movement I attempted was grotesquely magnified until I learnt to make less effort … it was strange, wonderful and at first, very frightening … we danced twice round the carpet. That was my first meeting with neostigmine, and we have never since been separated.9
involves immunosuppression, thymectomy (unless contraindicated) and symptom relief with drugs:
• Immunosuppressive treatment is directed at eliminating the acetylcholine receptor autoantibody. Prednisolone induces improvement or remission in 80% of cases. The dose should be increased slowly using an alternate-day regimen until the minimum effective amount is attained; an immunosuppressive improvement may take several weeks. Azathioprine may be used as a steroid-sparing agent. Prednisolone is effective for ocular myasthenia, which is fortunate, for this variant of the disease responds poorly to thymectomy or anticholinesterase drugs. Some acute and severe cases respond poorly to prednisolone with azathioprine and, for these, intermittent plasmapheresis or immunoglobulin i.v. (to remove circulating anti-receptor antibody) can provide dramatic short-term relief.
• Thymectomy should be offered to those with generalised myasthenia gravis under 40 years of age, once the clinical state allows and unless there are powerful contraindications to surgery. Most cases benefit and about 25% can discontinue drug treatment. Thymectomy should also be undertaken in all myasthenic patients who have a thymoma, but the main reason is to prevent local infiltration, as the procedure is less likely to relieve the myasthenia.
• Symptomatic drug treatment is decreasingly used. Its aim is to increase the concentration of acetylcholine at the neuromuscular junction with anticholinesterase drugs. The mainstay is usually pyridostigmine, starting with 60 mg by mouth 4-hourly. It is preferred because its action is smoother than that of neostigmine, but the latter is more rapid in onset and can with advantage be given in the mornings to get the patient mobile. Either drug can be given parenterally if bulbar paralysis makes swallowing difficult. An antimuscarinic drug, e.g. propantheline (15–30 mg t.i.d.), should be added if muscarinic effects are troublesome.
Drugs that oppose acetylcholine
• Antimuscarinic drugs, which act principally at postganglionic cholinergic (parasympathetic) nerve endings, i.e. atropine-related drugs (see Fig. 22.1). Muscarinic receptors can be subdivided according to their principal sites, namely in the brain (M1), heart (M2) and glandular, gastric parietal cells and smooth muscle cells (M3). As with many receptors, the molecular basis of the subtypes has been defined together with two further cloned subtypes M4 and M5, the precise functional roles of which remain to be clarified.


Antimuscarinic drugs
Atropine
Atropine is an alkaloid from the deadly nightshade, Atropa belladonna.10 It is a racemate (DL-hyoscyamine), and almost all of its antimuscarinic effects are attributable to the l-isomer alone. Atropine is more stable chemically as the racemate, which is the preferred formulation. In general, the effects of atropine are inhibitory but in large doses it stimulates the CNS (see poisoning, below). Atropine also blocks the muscarinic effects of injected cholinergic drugs, both peripherally and on the CNS. The clinically important actions of atropine at parasympathetic postganglionic nerve endings are listed below; they are mostly the opposite of the activating effects on the parasympathetic system produced by cholinergic drugs.
Mydriasis occurs with a rise in intraocular pressure due to the dilated iris blocking drainage of the intraocular fluid from the angle of the anterior chamber. An attack of glaucoma may be induced in eyes predisposed to primary angle (also called acute closed-angle or narrow-angle) closure and is a medical emergency. There is no significant effect on pressure in normal eyes. The ciliary muscle is paralysed and so the eye is accommodated for distant vision. After atropinisation, normal pupillary reflexes may not be regained for 2 weeks. Atropine use is a cause of unequally sized and unresponsive pupils.11
with atropine (and other antimuscarinic drugs) presents with the more obvious peripheral effects: dry mouth (with dysphagia), mydriasis, blurred vision, hot, flushed, dry skin, and, in addition, hyperthermia (CNS action plus absence of sweating), restlessness, anxiety, excitement, hallucinations, delirium, mania. The cerebral excitation is followed by depression and coma or, as it has been described with characteristic American verbal felicity, ‘hot as a hare, blind as a bat, dry as a bone, red as a beet and mad as a hen’.12 Poisoning is typically seen (especially in children) following ingestion of the rather attractive berries of solanaceous plants, e.g. deadly nightshade and henbane. Treatment involves activated charcoal to adsorb the drug, sponging to cool the patient and diazepam for the central excitement.
Other antimuscarinic drugs
• For their central actions – some (trihexyphenidyl (benzhexol) and orphenadrine) are used against the rigidity and tremor of parkinsonism, especially drug-induced parkinsonism, where doses higher than the usual therapeutic amounts are often needed and tolerated. They are used as antiemetics (principally hyoscine, promethazine). Their sedative action is used in anaesthetic premedication (hyoscine).
• For their peripheral actions – atropine, homatropine and cyclopentolate are used in ophthalmology to dilate the pupil and to paralyse ocular accommodation. Patients should be warned of a transient, but unpleasant, stinging sensation, and that they cannot read or drive (at least without dark glasses) for at least 3–4 h. Tropicamide is the shortest acting of the mydriatics. If it is desired to dilate the pupil and to spare accommodation, a sympathomimetic, e.g. phenylephrine, is useful.
In anaesthesic premedication, atropine, and hyoscine* block the vagus and reduce mucosal secretions; hyoscine also has useful sedative effects. Glycopyrronium* is frequently used during anaesthetic recovery to block the muscarinic effects of neostigmine given to reverse a non-depolarising neuromuscular blockade.
In the respiratory tract, ipratropium* is a useful bronchodilator in chronic obstructive pulmonary disease and acute asthma.
• For their actions on the gut, against muscle spasm and hypermotility, e.g. against colic (pain due to spasm of smooth muscle), and to reduce morphine-induced smooth muscle spasm when the analgesic is used against acute colic.
• In the urinary tract, flavoxate, oxybutynin, propiverine, tolterodine, trospium and propantheline* are used to relieve muscle spasm accompanying infection in cystitis, and for detrusor instability.
• In disorders of the cardiovascular system, atropine is useful in bradycardia following myocardial infarction.
• In cholinergic poisoning, atropine is an important antagonist of both central nervous, parasympathomimetic and vasodilator effects, though it has no effect at the neuromuscular junction and will not prevent voluntary muscle paralysis. It is also used to block muscarinic effects when cholinergic drugs, such as neostigmine, are used for their effect on the neuromuscular junction in myasthenia gravis.
(Urispas) is used for urinary frequency, tenesmus and urgency incontinence because it increases bladder capacity and reduces unstable detrusor contractions (see p. 400).
Oral antimuscarinics have occasional use in the treatment of hyperhidrosis.
• Acetylcholine is the most important receptor agonist neurotransmitter in both the brain and the peripheral nervous system.
• It acts on neurones in the CNS and at autonomic ganglia, on skeletal muscle at the neuromuscular junction, and at a variety of other effector cell types, mainly glandular or smooth muscle.
• The effector response is terminated rapidly through enzymatic destruction by acetylcholinesterase.
• Outside the CNS, acetylcholine has two main classes of receptor: those on autonomic ganglia and skeletal muscle responding to stimulation by nicotine, and the rest which respond to stimulation by muscarine.
• Drugs that mimic or oppose acetylcholine have a wide variety of uses. For instance, the muscarinic agonist pilocarpine lowers intraocular pressure and antagonist atropine reverses vagal slowing of the heart.
• The main use of drugs at the neuromuscular junction is to relax muscle in anaesthesia, or to inhibit acetylcholinesterase in diseases where nicotinic receptor activation is reduced, e.g. myasthenia gravis.
Cohen L.H., Thale T., Tissenbaum M.J. Acetylcholine treatment of schizophrenia. Arch. Neurol. Psychiatry. 1944;51:171–175.
Costa L.G. Current issues in organophosphate toxicology. Clin. Chim. Acta. 2006;366:1–13.
Hawkins J.R., Tibbetts R.W. Intravenous acetylcholine therapy in neurosis. A controlled clinical trial. J.Ment. Sci.. 1956;102:43–51. (In the same issue see also: Carbon dioxide inhalation therapy in neurosis. A controlled clinical trial (p. 52); The placebo response (p. 60))
Medical Manual of Defence Against Chemical Agents. (no. 0117725692) (JSP 312). London:HMSO; 1987.
Meriggioli M.N., Sanders D.B. Autoimmune myasthenia gravis: emerging clinical and biological heterogeneity. Lancet Neurol.. 2009;8:475–490.
Morita H., Yanigasawa T., Shimizu M., et al. Sarin poisoning in Matsumoto, Japan. Lancet. 1995;346:290–293.
Morton H.G. Atropine intoxication: its manifestation in infants and children. J. Pediatr.. 1939;14(6):755–760.
Weitz G. Love and death in Wagner’s Tristan und Isolde – an epic anticholinergic crisis. Br. Med. J. 2003;327:1469–1471. (and the Commentary by Jeff Aronson, pp. 1471–1472)
1 Harris M et al 1943 Archives of Neurology and Psychiatry 50:304.
2 Cohen L H et al 1944 Archives of Neurology and Psychiatry 51:171.
3 Drug and Therapeutics Bulletin 1999; 37 (July issue).
4 Jorenby D E, Leischow S J, Nides M A et al 1999 A controlled trial of sustained-release bupropion, a nicotine patch, or both for smoking cessation. New England Journal of Medicine 340:685–692.
5 A hormone function that is restricted to the local environment.
6 To demonstrate guilt or innocence according to whether the accused died or lived after the judicial dose. The practice had the advantage that the demonstration of guilt provided simultaneous punishment.
7 In recent times, there have been major instances of use against populations by both military and terrorist bodies (in the field and in an underground transport system).
8 Walker M B 1934 Lancet i:1200.
9 Disabilities and How to Live with Them. (1952) Lancet Publications, London.
10 The first name commemorates its success as a homicidal poison, for it is derived from the senior of three legendary Fates, Atropos, who cuts with shears the thread of life spun out by her sister Clothos, of a length determined by her other sister, Lachesis (there is a minor synthetic atropine-like drug called lachesine). The term belladonna (Italian: beautiful woman) refers to the once fashionable female practice of using an extract of the plant to dilate the pupils (incidentally blocking ocular accommodation) as part of the process of improving attractiveness.
11 A doctor, after working in his garden greenhouse, was alarmed to find that the vision in his left eye was blurred and the pupil was grossly dilated. Physical examination failed to reveal a cause and the pupil gradually and spontaneously returned to normal, suggesting that the explanation was exposure to some exogenous agent. The doctor then recalled that his greenhouse contained flowering plants called angel’s trumpet (sp. Brugmansia, syn. Datura, of the nightshade family), and he may have brushed against them. Angel’s trumpet is noted for its content of scopolamine (hyoscine), and is very toxic if ingested. The plant is evidently less angelic than the name suggests (Merrick J, Barnett S 2000 British Medical Journal 321:219).
12 Cohen H L et al 1944 Archives of Neurology and Psychiatry 51:171.
[/level-membership-for-basic-science-category][not-level-membership-for-basic-science-category]
Chapter 22 Cholinergic and antimuscarinic (anticholinergic) mechanisms and drugs
Acetylcholine antagonists that block the nicotine-like effects (neuromuscular blockers and autonomic ganglion blockers) are described elsewhere (Ch. 19).
Cholinergic drugs (cholinomimetics)
• For myasthenia gravis, both to diagnose (edrophonium) and to treat symptoms (neostigmine, pyridostigmine, distigmine).
• To lower intraocular pressure in chronic simple glaucoma (pilocarpine).
• To bronchodilate patients with airflow obstruction (ipratropium, tiotropium).
• To improve cognitive function in Alzheimer’s disease (rivastigmine, donepezil).
Classification
Direct-acting (receptor agonists)
• Choline esters (bethanechol, carbachol), which act at all sites, like acetylcholine, but are resistant to degradation by acetylcholinesterases (AChE; see Fig. 22.1). Muscarinic effects are much more prominent than nicotinic (see p. 373).
• Alkaloids (pilocarpine, muscarine) act selectively on end-organs of postganglionic, cholinergic neurones. Effects are exclusively muscarinic.
Sites of action (Fig. 22.1)
• Autonomic nervous system (Fig. 22.1, sites 1 and 2).
• Neuromuscular junction (Fig. 22.1, site 1).
• Central nervous system (CNS).
• Non-innervated sites: blood vessels, chiefly arterioles (Fig. 22.1, site 3).
Acetylcholine is released from nerve terminals to activate a post-synaptic receptor, except on blood vessels, where the action of cholinergic drugs is unrelated to cholinergic ‘vasodilator’ nerves. It is also produced in tissues unrelated to nerve endings, e.g. placenta and ciliated epithelial cells, where it acts as a local hormone (autacoid) on local receptors.
Pharmacology
Autonomic nervous system
• Eye: miosis and spasm of the ciliary muscle occur so that the eye is accommodated for near vision. Intraocular pressure falls.
• Exocrine glands: there is increased secretion most noticeably from salivary, lachrymal, bronchial and sweat glands. The last are cholinergic, but anatomically part of the sympathetic system; some sweat glands, e.g. axillary, may be adrenergic.
• Heart: bradycardia occurs with atrioventricular block, and eventually cardiac arrest.
• Bronchi: there is bronchoconstriction and mucosal hypersecretion that may be clinically serious in asthmatic subjects, in whom cholinergic drugs should be avoided if possible.
• Gut: motor activity is increased and may cause colicky pain. Exocrine secretion is also increased. Tone in sphincters falls, which may cause defaecation (anal sphincter) or acid reflux/regurgitation (oesophageal sphincter).
• Urinary bladder and ureters contract and the drugs promote micturition.
Choline esters
Acetylcholine
Acetylcholine was first injected intravenously as a therapeutic convulsant in 1939, in the reasonable expectation that the fits would be less liable to cause fractures than those following therapeutic leptazol (pentylenetetrazole) convulsions. Recovery rates of up to 80% were claimed in various psychotic conditions. Enthusiasm began to wane, however, when it was shown that the fits were due to anoxia resulting from cardiac arrest and not to pharmacological effects on the brain.1
The following description is typical:
Lachrymation, sweating and borborygmi were prominent. The deep reflexes became diminished. The patient then relaxed and ‘lay quietly in bed – cold moist and gray. In about 90 seconds, flushing of the face marked the return of the pulse’. The respiratory rate rose and consciousness returned in about 125 seconds. The patients sometimes micturated but did not defaecate. They ‘tended to lie quietly in bed after the treatment’. ‘Most of the patients were reluctant to be retreated.’2
Alkaloids with cholinergic effects
(see also p. 151) is a social drug that lends its medicinal use as an adjunct to stopping its own abuse as tobacco. It is available as gum to chew, dermal patches, a nasal spray or an inhalator. These deliver a lower dose of nicotine than cigarettes and appear to be safe in patients with ischaemic heart disease. The patches are slightly better tolerated than the gum, which releases nicotine in a more variable fashion depending on the rate at which it is chewed and the salivary pH, which is influenced by drinking coffee and carbonated drinks. Nicotine treatment is reported to be nearly twice as effective as placebo in achieving sustained withdrawal from smoking (18% versus 11% in one review).3 Treatment is much more likely to be successful if it is used as an aid to, not a substitute for, continued counselling. Bupropion is possibly more effective than the nicotine patch4 (see also p. 152) and the partial nicotinic agonist, varenicline, slightly more effective still. The efficacy of varenicline is tempered by its ability to cause suicidal ideation and behaviour.
from a South American plant (Pilocarpus spp.), acts directly on muscarinic receptors (see Fig. 22.1); it also stimulates and then depresses the CNS. The chief clinical use of pilocarpine is to lower intraocular pressure in primary open-angle glaucoma (also called chronic simple or wide-angle glaucoma), as an adjunct to a topical β-blocker; it produces miosis, opens drainage channels in the trabecular network and improves the outflow of aqueous humour. Oral pilocarpine is available for the treatment of xerostomia (dry mouth) in Sjögren’s syndrome, or following irradiation of head and neck tumours. The commonest adverse effect is sweating, an effect actually exploited in a diagnostic test for cystic fibrosis.
Poisoning with these fungi may present with antimuscarinic, cholinergic or GABAergic effects. All have CNS actions. Happily, poisoning by Amanita muscaria is seldom serious, but species of Inocybe contain substantially larger amounts of muscarine (see Ch. 10). The lengths to which humans are prepared to go in taking ‘chemical vacations’ when life is hard are shown by the inhabitants of eastern Siberia, who used Amanita muscaria recreationally for its cerebral stimulant effects. They were apparently prepared to put up with the autonomic actions to escape briefly from reality – so much so that when the fungus was scarce in winter they were even prepared to drink their own urine to prolong the experience. Sometimes, in generous mood, they would even offer their urine to others as a treat.
Anticholinesterases
At cholinergic nerve endings and in erythrocytes there is a specific enzyme that destroys acetylcholine, true cholinesterase or acetylcholinesterase. In various tissues, especially plasma, there are other esterases that are not specific for acetylcholine but that also destroy other esters, e.g. suxamethonium, procaine (and cocaine) and bambuterol (a prodrug that is hydrolysed to terbutaline). Hence, they are called pseudocholinesterases. Chemicals that inactivate these esterases (anticholinesterases) are used in medicine and in agriculture as pesticides. They act by allowing naturally synthesised acetylcholine to accumulate instead of being destroyed. Their effects are explained by this accumulation in the CNS, neuromuscular junction, autonomic ganglia, postganglionic cholinergic nerve endings (which are principally in the parasympathetic nervous system) and in the walls of blood vessels, where acetylcholine has a paracrine5 role not necessarily associated with nerve endings. Some of these effects oppose one another, e.g. the effect of anticholinesterase on the heart will be the result of stimulation at sympathetic ganglia and the opposing effect of stimulation at parasympathetic (vagal) ganglia and at postganglionic nerve endings.
is an alkaloid, obtained from the seeds of the West African Calabar bean (spp. Physostigma), which has had long use both as a weapon and as an ordeal poison.6 It acts for a few hours. It has been shown to have some efficacy in improving cognitive function in Alzheimer-type dementia.
[/not-level-membership-for-basic-science-category]
