[level-membership-for-neurosurgery-category]
CHAPTER 56 Chemotherapy and Experimental Medical Therapies for Meningiomas
INTRODUCTION
Current therapies for meningiomas include surgery, radiation therapy and stereotactic radiosurgery.1–9 For the majority of patients with benign meningiomas (World Health Organization [WHO] grade I) and a subset of patients with atypical meningiomas (WHO grade II), these therapies are effective in achieving tumor control. However, there is an important group of patients with inoperable or higher grade tumors who develop recurrent disease after surgery and radiation therapy. The treatment options for these patients are currently inadequate.
The WHO classification scheme for meningiomas is based on the degree of anaplasia, number of mitoses, and presence of necrosis.10 Histological grading is important because it helps to predict the likelihood of recurrence. Benign meningiomas account for more than 90% of tumors and have a low risk of recurrence (7%–20%).10–12 Atypical meningiomas are much less common. They account for 4.7% to 7.2% of meningiomas and are associated with a 40% recurrence rate despite surgical resection.12 Malignant meningiomas represent only 1% to 2.8% of meningiomas but recur in 50% to 80% of cases and usually result in death within 2 years of diagnosis.10,11 Patients with meningiomas of any histological grade that recur after surgery and radiation therapy are frequently considered for chemotherapy or experimental medical therapies.
CYTOTOXIC CHEMOTHERAPY
To date, chemotherapy has had only a very limited role in the treatment of meningiomas. Data from small clinical trials and case series suggest that most chemotherapeutic agents have minimal activity against meningiomas.5,6,13,14 The evaluation of chemotherapy has also been complicated by the lack of data regarding the natural history of untreated meningiomas. Many chemotherapy studies report variable periods of disease stabilization, but it is difficult to know whether this represents an improvement because benign meningiomas grow slowly and may appear radiographically stable for prolonged periods.15,16
In general, chemotherapeutic regimens (such as dacarbazine and Adriamycin) that have activity in other soft tissue tumors have produced disappointing results in patients with meningiomas.6 Hydroxyurea, an oral ribonucleotide reductase inhibitor, arrests meningioma cell growth in the S phase of the cell cycle and induces apoptosis.17 In a preliminary report, hydroxyurea (1000–1500 mg/day; 20 mg/kg/day) decreased tumor size in three patients with recurrent benign meningiomas and prevented recurrent disease for 24 months in a patient with a completely resected malignant meningioma.18 Several more recent studies suggest that hydroxyurea has modest activity; responses are uncommon but some patients appear to have disease stabilization.19–23 The Southwest Oncology Group conducted a phase II study to further evaluate the role of hydroxyurea in meningiomas (SWOG-S9811). This study is closed to accrual but the final results are not yet available.
There have been reports of small numbers of patients with malignant meningiomas who responded to recombinant interferon alpha-2b.24,25 Temozolomide (Temodar, TMZ), an alkylating agent with activity in malignant gliomas, was evaluated in 16 patients with refractory meningiomas and showed negligible activity.26 The topoisomerase inhibitor irinotecan (Camptosar, CPT-11) caused moderate toxicity in 16 patients with benign meningiomas and had no demonstrable activity.27 A large number of cytotoxic agents are under evaluation for sarcomas and other systemic malignancies.28 Most of these have not been evaluated in meningiomas, and it is possible that some may have modest activity. However, it is likely that the more novel therapeutic approaches discussed in the text that follows will provide a greater chance of improving the outcome for patients with recurrent meningiomas.
CHALLENGES IN THE DEVELOPMENT OF EFFECTIVE MEDICAL THERAPIES FOR MENINGIOMAS
In contrast to the extensive understanding of the molecular pathogenesis and biology of systemic malignancies, and even brain tumors such as malignant gliomas, relatively little is known about the molecular pathogenesis of meningiomas and the critical molecular changes driving tumor growth.11,12,29,30 Overexpression of various growth factors including platelet-derived growth factor (PDGF), epidermal growth factor (EGF), and vascular endothelial growth factor (VEGF) and their receptors, and signal transduction pathways such as the Ras/mitogen-activated protein kinase (MAPK), phosphatidylinositol-3-kinase (PI3K)-Akt, and phospholipase C (PLC)-γ1-protein kinase C (PKC) pathways have been implicated, but their relative significance is largely unknown.29,30 As a result, the most important molecular targets may remain to be elucidated.
Another factor limiting progress in the development of more effective therapies for meningiomas is the lack of robust cell lines and animal models. There is a need for animal models that replicate the genetic changes in meningiomas with a high frequency of spontaneous meningioma development, benign meningioma lines for in vitro and in vivo studies, and meningeal specific promoters. Many of the existing meningioma cell lines are derived from malignant meningiomas and likely contain culture-induced artifacts and lack progesterone receptors.12 There are some orthotopic31,32 and genetic models33 in development that appear promising. Recently, two cell lines were developed from benign meningioma specimens via immortalization with human telomerase reverse transcriptase and SV40 large T antigen. Orthotopic tumors with similar immunostaining patterns to human meningiomas were established from both cell lines in athymic nude mice.31 Another model uses a Cre recombinase technology to inactivate NF2 in arachnoid cells, resulting in intracranial meningothelial hyperplasia and meningiomas in 30% of mice.33 These models may aid in the preclinical evaluation of novel therapies.
A final factor limiting progress is the relatively small number of patients with meningiomas who require additional therapies after treatment with surgery and radiation therapy. In general, there is little incentive for pharmaceutical companies to evaluate their therapies in meningiomas because of the small potential market. Hopefully as the molecular pathogenesis of these tumors becomes better understood, a compelling case can be made for evaluating specific agents directed at critical molecular targets. In the next section, targeted molecular drugs that have a potential role against meningiomas are reviewed in detail. These therapies have also been discussed in recent review articles.2,5,30,34
EXPERIMENTAL THERAPIES: TARGETED MOLECULAR AGENTS
Recently, it has become apparent that many human diseases result from aberrations in cell signaling pathways. Protein-tyrosine kinases play a fundamental role in signal transduction, and deregulated activity of these enzymes has been observed in many cancers. Therefore, specific inhibitors of tyrosine kinases could have potential therapeutic applications in the treatment of cancer, with potentially lower toxicity and/or higher, prolonged response rates.35,36 The prototypical targeted molecular agent is imatinib mesylate (Gleevec), which has shown significant benefit in chronic myeloid leukemia (CML)37 and gastrointestinal stromal tumors (GIST).38 There is also a growing experience with targeted molecular agents in malignant gliomas.39–41 However, to date, there have been minimal data on the use of these agents in meningiomas.
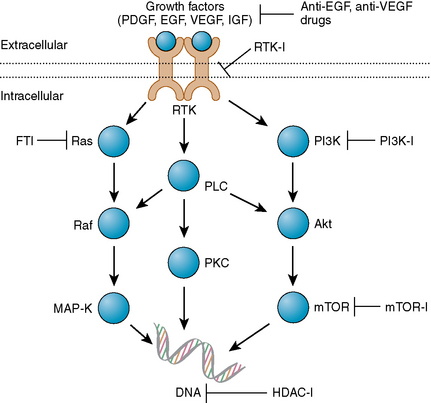
In contrast to the extensive work aimed at understanding the genetics of meningiomas, relatively little work has been conducted to understand the growth factors and their receptors, and the signal transduction pathways that are critical to meningioma growth.2,5,30 PDGF, EGF, VEGF, insulin-like growth factor (IGF), transforming growth factor-beta (TGF-β), and their receptor tyrosine kinases, together with their downstream signaling pathways including the Ras/MAPK pathway, the PI3K/Akt pathway, the PLC-γ1-PKC pathway, and the TGF-β-SMAD pathways, are all thought to be important in meningioma growth (Fig. 56-1).30
Platelet-Derived Growth Factor Receptor
PDGF is a fundamental driver of cell proliferation in normal development and in a variety of pathological conditions, including cancer.42 Accumulating evidence suggests that PDGF plays an important role in meningioma growth.43–47 Most meningiomas of all histologic grades express PDGF ligands AA and BB and the PDGF-beta receptor (PDGF-βR).43–47 Expression levels may be higher in atypical and malignant meningiomas than in benign meningiomas.45 Laboratory data suggest that an autocrine PDGF loop supports meningioma cell growth and maintenance.47 When PDGF-BB is applied to cultured meningioma cells, MAPK48 and c-fos49 are activated and tumor cell proliferation is enhanced. Conversely, anti-PDGF-BB antibodies inhibit cell growth.50 These data provide sound rationale for testing PDGF inhibitors in meningioma patients.
Imatinib is a potent inhibitor of the Bcr-Abl, PDGF-α and β receptors, and c-Kit tyrosine kinases.51 Its ability to inhibit PDGFR with an IC50 of 0.1 μM suggested that it may have therapeutic potential in meningiomas. The North American Brain Tumor Consortium (NABTC) conducted a phase II study of imatinib in patients with recurrent meningiomas (NABTC 01-08).52 Patients were stratified into two cohorts: (1) benign meningiomas or (2) atypical and malignant meningiomas. As imatinib is metabolized by the cytochrome P450 system (3A4), patients could not be receiving enzyme inducing antiepileptic drugs (EIAEDs). Patients initially received 600 mg/day of imatinib; the dose was increased in the second cycle to 800 mg/day if no significant toxicity was observed in the first cycle. Twenty-three patients were enrolled into the study (13 with meningiomas, 5 with atypical meningiomas, and 5 with malignant meningiomas). Though the treatment was generally well tolerated, imatinib had minimal activity. Of the 19 patients evaluable for response, 10 progressed at the first scan and 9 were stable. There were no radiographic responses. Overall median progression-free survival (PFS) was 2 months (range 0.7 to 18 months); 6-month PFS was 29.4%. For benign meningiomas, median PFS was 3 months; 6-month PFS was 45%. For the atypical and malignant meningiomas, median PFS was 2 months; 6-month PFS was 0%. Several other inhibitors of PDGF are undergoing evaluation such as sunitinib, MLN518, dasatinib, AMN 107, pazopanib, CP673451 and CHIR 265. Some of these, such as MLN518, are more potent PDGFR inhibitors than imatinib, whereas others target additional kinases that are potentially important in meningiomas. For example, pazopanib also inhibits VEGF receptors (VEGFR) 1, 2, and 3 as well as c-Kit, while CHIR 265 inhibits VEGFR, c-Kit and Raf. These drugs may be more effective than imatinib as monotherapy against meningiomas.
There is also interest in combining imatinib with hydroxyurea, the cytotoxic chemotherapy agent with the most activity in meningiomas. Though a recent phase I/II trial of imatinib as monotherapy for recurrent malignant glioma showed minimal activity,53 a study of 33 recurrent glioblastoma (GBM) patients treated with the combination of imatinib mesylate (400 mg/day or 500 mg twice/day, depending on concurrent EIAED use) and hydroxyurea (500 mg twice/day), had encouraging results.54 After a median follow-up period of 58 weeks, 1 patient achieved a complete response, 2 achieved a partial response, and 14 achieved stable disease. Six-month PFS was 27%, and the median PFS was 14.4 weeks. In light of these results in patients with recurrent GBM, a multicenter phase II trial of the combination for patients with recurrent or progressive meningiomas after surgery is currently in progress (ClinicalTrials.gov identifier NCT00354913). Twenty-one patients are expected to enroll, and monitoring includes magnetic resonance imaging (MRI) scans and clinical examinations every 8 weeks.
Epidermal Growth Factor Receptor
The EGFR is overexpressed in more than 60% of meningiomas.55–61 EGF and TGF-β activate these receptors and stimulate meningioma growth in vitro,30,56 supporting the concept that activation of EGFRs in human meningiomas by autocrine/paracrine stimulation may contribute to their proliferation. Increased TGF-β immunoreactivity in meningiomas has been associated with aggressive growth.30,61,62
In addition to gefitinib and erlotinib, there are a large number of other agents currently undergoing evaluation. These inhibit EGFR alone, or together with other receptor tyrosine kinases, which may have therapeutic potential in meningiomas (Table 56-1). For example, lapatinib inhibits EGFR and HER2, HKI-272 inhibits all subtypes of the EGFR, and ZD6474 (Zactima) inhibits EGFR and VEGFR. Although EGFR monoclonal antibodies have been effective for some systemic malignancies (e.g., cetuximab in colorectal cancer), they have generally not been used for brain tumors because of the concern regarding the ability of these agents to pass through the blood–brain barrier (BBB) in sufficient concentrations to produce a therapeutic effect. Because the BBB is not a factor in most meningiomas, it is possible that these antibodies may be effective in these tumors. To date very few studies have evaluated the therapeutic potential of these agents in meningiomas. In a phase I study of a murine monoclonal antibody against EGFR in nine patients with either gliomas or meningiomas, treatment was reasonably well tolerated. No radiographic responses were detected, but efficacy data is difficult to interpret in a study with so few subjects.63 Currently, several anti-EGFR antibodies are undergoing evaluation for other malignancies such as cetuximab, panitumumab, EMD 72000, nimotuzumab, and mAb 806. Trials of these agents in meningiomas may be worthwhile, especially if combined with correlative studies examining whether the antibodies can achieve therapeutic concentrations in meningiomas and inhibit EGFR in vivo.
TABLE 56-1 Selected potential targeted molecular drugs for meningiomas
| Class/agent | Alternative name(s) | Mechanism(s) |
|---|---|---|
| Apoptosis enhancers | ||
| ABT737 | Bcl-2 inhibitor | |
| AT101 | Bcl-2 inhibitor | |
| Fenretinide | Multiple targets | |
| GX15-070 | Bcl-2 inhibitor | |
| Integrin inhibitor | ||
| Cilengitide | αvβ3 and αvβ5 integrin inhibitor | |
| Cell cycle inhibitors | ||
| AG024322 | pan-CDK inhibitor | |
| AZ703 | CDK 2, 1 inhibitor | |
| BMS-387032 | CDK 2, 1 inhibitor | |
| CINK4 | CDK 4, 6 inhibitor | |
| E7070 | Unknown | |
| Flavopiridol | pan-CDK inhibitor | |
| PD-0332991 | CDK 4, 6 inhibitor | |
| Seliciclib | CDK 2, 1 inhibitor | |
| c-MET (HGF/SF) inhibitors | ||
| AMG-102 | HGF/SF antibody | |
| XL880 | c-MET, VEGFR2, PDGFR, c-Kit, Tie-2 inhibitor | |
| EGFR inhibitors | ||
| AEE788 | EGFR, VEGFR inhibitor | |
| BIBW 2992 | EGFR, HER2 inhibitor | |
| Cetuximab | Erbitux | EGFR antibody |
| EMD 72000 | EGFR antibody | |
| Erlotinib | OSI-774, Tarceva | EGFR inhibitor |
| Gefitinib | ZD1839, Iressa | EGFR inhibitor |
| HKI-272 | pan-EGFR inhibitor | |
| Lapatinib | GW-572016 | EGFR, HER2 inhibitor |
| mAb 806 | EGFR antibody | |
| Nimotuzumab | TheraCIM | EGFR antibody |
| Panitumumab | Vectibix | EGFR antibody |
| ZD6474 | Zactima | EGFR, VEGFR inhibitor |
| Endothelin-A inhibitors | ||
| Astrasentan | Xinlay | ETA inhibitor |
| ZD4054 | ETA inhibitor | |
| Farnesyltransferase inhibitors | ||
| Lonafarnib | SCH 66336, Sarasar | FT inhibitor |
| Tipifarnib | R115777, Zarnestra | FT inhibitor |
| Histone deacetylase inhibitors | ||
| Depsipeptide | HDAC inhibitor | |
| Voronistat | Suberoylanilide hydroxamic acid (SAHA) | HDAC inhibitor |
| Valproic acid | HDAC inhibitor | |
| HSP-90 inhibitors | ||
| 17AAG | HSP-90 inhibitor | |
| 17DMAG | HSP-90 inhibitor | |
| IPI504 | HSP-90 inhibitor | |
| MEK inhibitors | ||
| AZD6244 | MEK inhibitor | |
| PD0325901 | MEK inhibitor | |
| mTOR inhibitors | ||
| AP23573 | mTOR inhibitor | |
| Everolimus | RAD001 | mTOR inhibitor |
| Rapamycin | mTOR inhibitor | |
| Temsirolimis | CCI-779, Torisel | mTOR inhibitor |
| PDGFR inhibitors | ||
| AMN 107 | PDGFR, c-Kit, Bcr-Abl inhibitor | |
| CHIR 265 | PDGFR, Raf, VEGFR, c-Kit inhibitor | |
| CP-673-451 | ||
| Dasatinib | PDGFR, Src, c-Kit, ephrin A inhibitor | |
| Imatinib mesylate | Gleevec | |
| MLN518 | PDGFR, c-Kit inhibitor | |
| Pazopanib | GW786034 | PDGFR, VEGFR, c-Kit inhibitor |
| Sunitinib | Sutent | PDGFR, VEGFR, c-Kit inhibitor |
| Vatalanib | PTK787 | PDGFR, VEGFR inhibitor |
| XL-999 | PDGFR, VEGFR, FGFR inhibitor | |
| PKC β2 inhibitor | ||
| Enzastaurin | LY31761 | PKC β2 inhibitor |
| PI3K inhibitor | ||
| BEZ235 | PI3K inhibitor | |
| Proteasome inhibitor | ||
| Bortezomib | Velcade | Proteasome inhibitor |
| Raf kinase inhibitor | ||
| Sorafenib | Nexavar, BAY 43-9006 | Raf kinase, VEGFR, PDGFR inhibitor |
| Src inhibitor | ||
| AZD0530 | Src inhibitor | |
| TGF-β inhibitors | ||
| AP 12009 | TGF-β2 antisense oligonucleotide | |
| GC1008 | TGF-β antibody | |
| SB-431542 | TGF-β receptor inhibitor | |
| VEGF inhibitors | ||
| Bevacizumab | Avastin | VEGF antibody |
| VEGF trap | VEGF soluble decoy receptor | |
| VEGFR inhibitors | ||
| AEE788 | VEGFR, EGFR inhibitor | |
| AG013736 | VEGFR inhibitor | |
| AMG 706 | VEGFR inhibitor | |
| AZD2171 | VEGFR inhibitor | |
| CEP-7055 | VEGFR inhibitor | |
| CHIR 265 | VEGFR, Raf, PDGFR, c-Kit inhibitor | |
| CHIR-258 | VEGFR, PDGFR, FGFR, c-Kit, FLT-3 inhibitor | |
| GW786034 | Pazopanib | VEGFR, PDGFR, c-Kit inhibitor |
| Sorafenib | Nexavar, BAY 43-9006 | Raf kinase, VEGFR, PDGFR inhibitor |
| Sunitinib | Sutent | PDGFR, VEGFR, c-Kit inhibitor |
| Vatalanib | PTK787 | PDGFR, VEGFR inhibitor |
| XL-999 | PDGFR, VEGFR, FGFR inhibitor | |
| ZD6474 | Zactima | EGFR, VEGFR inhibitor |
bcl-2, B-cell CLL/lymphoma 2; CDK, cyclin-dependent kinase; c-Kit, tyrosine kinase proto-oncogene; EGFR, epidermal growth factor receptor; ETA, endothelin-A; FLT-3, FMS-like tyrosine kinase 3; FT, farnesyltransferase; FGFR, fibroblast growth factor receptor; HDAC, histone deacetylase; HGF/SF, hepatocyte growth factor/scatter factor; HSP-90, heat shock protein 90; mTOR, mammalian target of rapamycin; MEK, mitogen-activated protein kinase; PI3K, phosphoinositol 3-kinase; PDGFR, platelet derived growth factor receptor; PKC β2, protein kinase C β2; VEGFR, vascular endothelial growth factor receptor.
Mitogen-Activated Protein (MAP) Kinase Pathway
Signal transduction from activated tyrosine kinases such as EGFR and PDGFR is mediated in part by the Ras/Raf/MAPK pathway and the PI3K/Akt pathways. PDGF-BB stimulates meningioma growth in vitro via the MAPK pathway, which is constitutively activated in benign meningiomas and meningioma cell cultures.48 Treatment with PD098059, a MEK inhibitor, reduces MAPK phosphorylation, inhibits meningioma growth in vitro, and prevents PDGF-BB stimulation of meningioma growth.48 The MAPK pathway is also activated in some, but not all atypical and malignant meningiomas,64 suggesting that other signal transduction pathways may also be involved. A number of Raf inhibitors (e.g., sorafenib) and MEK inhibitors (e.g., PD098059 and AZD6244) are undergoing clinical evaluation and may have role in meningiomas. Activation of Ras requires localization to the cytoplasmic surface of the cell membrane.65 This subcellular localization is dependent on the addition of a hydrophobic farnesyl group to the ras protein, catalyzed by the enzyme farnesyltransferase. Farnesyltransferase inhibitors such as tipifarnib (Zarnestra) and lonarfarnib (Sarasar) inhibit the Ras pathway, and may have therapeutic potential in meningiomas. However, preliminary studies suggest that the activity of these agents may be limited in benign meningiomas.30
PI3K/Akt Pathway
The PI3K/Akt pathway plays a central role in many malignancies.66,67 Akt and p70S6K are expressed and activated (phosphorylated) in benign meningiomas, and play a role in signal transduction from PDGFR stimulated by PDGF-BB.68 Treatment with a PI3K inhibitor produced a dose-dependent inhibition of PDGF-BB stimulation, with a concomitant attenuation of Akt and p70S6K phosphorylation.30,68 Phospho-Akt is present in higher levels in atypical and malignant meningiomas compared to benign meningiomas.64 Inhibition of the PI3K resulted in reduction in phospho-Akt activity in atypical and malignant meningiomas.64 These results suggest that the PI3K/Akt pathway may play a central role in meningiomas, especially in atypical and malignant meningiomas, Inhibitors of PI3K (e.g., BEZ235), inhibitors of Akt (e.g., perifosine), and mTOR inhibitors, located downstream of Akt (e.g., sirolimus, temsirolimus [CCI-779], everolimus [RAD001], and AP23573), may have therapeutic potential in these tumors.
PLC-γ1-PKC Pathway
In addition to activation of the MAPK and the PI3K/Akt pathway, receptor tyrosine kinases such as EGFR and PDGFR also activate PLC-γ1-PKC.30 This results in hydrolysis of phosphatidylinositol-4,5-diacylglycerol to inositol 1,4,5-triphosphate and 1,2 diacylglycerol (1,2-DAG). 1,2-DAG activates PKC, the MAPK and the PI3K/Akt pathways.30,59 PKC enters the nucleus and activates c-fos and c-jun, which leads to cell proliferation and inhibition of apoptosis.30 Activation of EGFR on meningioma cells results in phosphorylation of PLC-γ1.59 The interaction of PLC-γ1 with the MAPK and the PI3K/Akt pathways underscores the complexities of the signaling pathways and the likelihood that inhibition of multiple targets will be necessary.
TGF-β-SMAD Signaling Pathways
The precise role of TGF-β in meningiomas remains to be defined. Meningiomas secrete TGF-β1, 2, and 3, and possess functional TGF-β type I and II receptors.30,69 TGF-β1 inhibits proliferation of leptomeningeal and benign meningioma cells via signal transduction through the Smad 2/3 pathway.30,69 In other tumors, including gliomas, higher grade tumors change from being inhibited to being stimulated by TGF-β.30 Whether a similar process occurs in meningiomas is unclear. If TGF-β plays an activating role in high-grade meningiomas, evaluating inhibitors of TGF-β such as SB-431542, AP12009, and GC1008 may be worthwhile.
Cell Cycle Inhibitors
Recently, there has been progress in identifying agents that target the cell cycle to treat cancer.70,71 Cyclin-dependent kinase (cdk) activity can be inhibited by agents that competitively inhibit cdk ATP-binding pockets or that allosterically modulate cdk or endogenous cdk inhibitor complexes. Single-agent activity has been modest to date, but newer oral agents that allow chronic dosing, and combinations of cdk inhibitors with other targeted agents or cytotoxic agents may hold greater promise.70,71
Apoptosis
Defects in programmed cell death (apoptosis) mechanisms play an important role in tumor pathogenesis and resistance to therapy.41,72–74 Apoptosis occurs via two main mechanisms. The extrinsic pathway is characterized by activation of death receptors with subsequent activation and cleavage of caspase 8. The intrinsic pathway is characterized by depolarization of the mitochondrial membrane, activation of caspase 9 and then caspase 3 and other executioner caspases.72–74 There has been increasing interest in identifying targeted agents that modulate apoptosis to destroy tumor cells.72–75 This can be achieved either by inhibiting pro-survival pathways such as the AKT and MAPK pathways, and NFκB, or by inducing apoptosis. The activation of cell surface receptors by the tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) leads to stimulation of the extrinsic pathway.74 Agents that activate these receptors, such as monoclonal antibodies to TRAIL receptors and recombinant TRAIL, are being evaluated as monotherapies and in combination with chemotherapeutic agents.74 The Bcl-2 family of proteins plays a central regulatory role in the intrinsic pathway. Overexpression of Bcl-2 or Bcl-XL renders tumor cells resistant to apoptotic stimuli, including many cytotoxic agents. Strategies to inhibit these proteins with small molecules such as ABT-737,76 Bcl-2 antisense oligonucleotides,77 and BH3 mimetic peptides78 are being evaluated. Inhibitors of apoptosis proteins (IAPs) are endogenous apoptosis suppressors, many of which function as caspase inhibitors.73 A number of inhibitors of IAPs are in development; they represent a promising class of agents with antitumor activity that may be synergistic with conventional cytotoxic therapies and other targeted molecular agents.73,79,80 To date these agents have not been evaluated in meningiomas. Another class of agents that may have therapeutic potential in meningiomas is synthetic retinoids, such as fenretinide, that induce apoptosis in tumor cells.81 In an in vitro study, fenretinide induced apoptosis in all three histologic subtypes of meningioma and exerted diverse cellular effects, including DR5 up-regulation, modulation of retinoid receptor levels, and inhibition of IGF-1–induced proliferation.81
Inhibition of Angiogenesis
Meningiomas are highly vascular tumors that derive their blood supply predominantly from meningeal vessels supplied by the external carotid artery, with additional supply from cerebral pial vessels.11 Inhibition of angiogenesis has become an increasingly important approach to treating cancer.82 Studies evaluating inhibitors of angiogenesis in meningiomas are limited. One early study found that TNP-470, a fumigillin analog, inhibits the growth of benign and malignant meningioma xenografts in nude mice.83
VEGF plays a central role in tumor angiogenesis, and there is increasing evidence that inhibition of VEGF or VEGF receptors (VEGFR) can lead to significant antitumor effects.82 Inhibition of VEGF with the anti-VEGF antibody bevacizumab (Avastin) has significantly improved survival in several malignancies including colorectal, lung, and breast cancer.84 Inhibitors of VEGFR such as sorafenib (Nexavar) and sunitinib (Sutent) have also prolonged survival in renal cell carcinoma and GIST.84 VEGF and VEGFR are expressed in meningiomas, and the level of expression increases with tumor grade.85–87 VEGF expression is increased 2-fold in atypical meningiomas, and 10-fold in malignant meningiomas compared to benign meningiomas.85 VEGF also plays an important role in the formation of peritumoral edema, which adds to the morbidity of these tumors.86,87 Inhibitors of VEGF and VEGFR are promising agents in meningiomas, with the potential not only to inhibit angiogenesis, but also to decrease peritumoral edema. A multicenter phase II study of sunitinib in patients with recurrent or inoperable meningiomas is expected to open soon at Dana-Farber/Brigham and Women’s Cancer Center and Memorial Sloan-Kettering Cancer Center. Sunitinib is a theoretically appealing agent for meningiomas because it is a multiple tyrosine kinase inhibitor that targets both VEGFR and PDGFR (in addition to c-Kit and the FLT3 and RET kinases). During each 6-week cycle, patients will receive sunitinib (50 mg/day) daily for 4 weeks followed by 2 weeks without treatment. Potential imaging (perfusion MRI) and serum biomarkers as well as tumor expression profiling will be obtained with the goal of developing predictors of response. A trial of sorafenib, which also targets VEGFR and PDGFR, is in progress.
Other angiogenic factors implicated in meningiomas include fibroblast growth factor (FGF),85 placental growth factor,88 and possibly hepatocyte growth factor/scatter factor (HGF/SF), although the role of the latter is less clear.85,89 The presence of HGF/SF and its receptor cMET appears to be associated with an increased proliferation index and rate of recurrence.89
Endothelins (ETs) are peptides that promote tumor progression by several mechanisms, including angiogenesis, cell proliferation, inhibition of apoptosis, and matrix remodeling. Several isoforms are known including ET-1, ET-2, and ET-3. ETs function via 2 G-protein–coupled receptors ET-A and ET-B.90,91 ET-1 and ET-A are overexpressed in meningiomas.91,92 The growth of primary meningioma cultures can be blocked by the ET-A inhibitor BQ-123 but not by the ET-B inhibitor RES-701-3, which suggests that the effects of ET-1 on meningioma growth are mediated by ET-A receptors.92 Several ET-A receptor antagonists are under development such as astrasentan (Xinlay) and ZD4054. These have not yet been tested in meningiomas.
Inhibition of Invasion
Brain invasion is a fundamental characteristic of high-grade meningiomas.11,12 Meningiomas frequently overexpress molecules that facilitate brain invasion including matrix metalloproteinases, such as MMP-2 and 9,93,94 SPARC, tenascin, and stremelysin-3.12 In many cases, expression of pro-invasion proteins correlates with invasive behavior. Malignant meningiomas typically fail to express tissue factor pathway inhibitor 2 (TFPI-2).95 TFPI-2 is an extracellular matrix-associated kunitz-type serine proteinase inhibitor secreted by all vascular cells and by benign meningiomas. It plays a role in tumor invasion and metastasis, presumably by plasmin-mediated matrix remodeling. Previous studies showed that expression of TFPI-2 is lost in high-grade tumors, including gliomas. Transfection of TFPI-2 mRNA into the human meningioma cell line IOMM-Lee inhibited tumor growth in vitro and in vivo, which suggests that TFPI-2 may have therapeutic potential in malignant meningiomas.95 Other invasion inhibitors have been studied for systemic cancers and gliomas, and some of these agents may have a therapeutic effect in meningiomas.96–99 One potential agent is cilengitide, an inhibitor of αvβ3 and αvβ5 integrins, both of which are expressed in meningiomas100 and are potentially important for angiogenesis and invasion. The drug is in clinical trials for gliomas, where it has been well tolerated and appears to show evidence of activity.101
Other Molecular Targets
Other potentially attractive therapeutic targets in meningiomas include IGFR-2,102,103 histone deacetylase,104 NFκB,105 heat shock protein 90 (HSP90),106 JAK/STAT,107 checkpoint kinase,108 and possibly Src kinase,109 focal adhesion kinase,110 and hypoxia-inducible factor 1α.111 As with gliomas and other solid tumors, the complexity of the molecular abnormalities in meningiomas and the redundancy of the signaling pathways make it improbable that single agents will achieve the same success as imatinib in CML.41 Nonetheless, the use of targeted molecular agents remains a potentially promising and largely unexplored area in meningiomas. It will be important to improve our understanding of the molecular pathogenesis of meningiomas and to identify the critical molecular abnormalities driving tumor growth that can be targeted. Multitargeted “dirty” drugs, combinations of targeted agents inhibiting complementary molecular targets, or the combination of targeted agents with conventional cytotoxic agents and especially radiation therapy, will lead to greater antitumor effects than single agents alone.41,112,113
Preclinical studies suggest that radiation sensitivity can be regulated by growth factors (EGFR, IGFR), signal transduction pathways (Ras/MAPK, PI3K/Akt), checkpoint activation and DNA repair (ATM, Chk1, Rad 51), and apoptosis related proteins (Fas, BcL2).112 Many of these can be inhibited by targeted molecular agents.
HSP90 acts as molecular chaperone protein that is required for maturation and stability of various client proteins including EGFR, Akt, Raf, p53, and cdk4. By blocking HSP90, 17AAG enhances radiation sensitivity in several tumor cell lines. This agent and other HSP90 inhibitors may have therapeutic potential in meningiomas.106,113
There is also increasing evidence that angiogenesis inhibitors may enhance radiation sensitivity.41,114 Possible mechanisms for the beneficial effects of angiogenesis inhibition include direct antitumor effects, endothelial cell radiosensitization resulting in damaged tumor vasculature, and improved oxygenation as a result of elimination of ineffective tumor vessels and decreased interstitial pressure.115 Specific angiogenesis inhibitors with potential radiosensitizing effects include vandetinib (ZD6474), an inhibitor of VEGFR and EGFR; vatalanib (PTK787), an inhibitor of VEGFR and PDGFR; enzastaurin (LY317615), an inhibitor of PKC-β2 and PI3K/Akt; and bevacizumab, a monoclonal antibody against VEGF.
HORMONAL THERAPIES
There has been longstanding interest in the possible role of sex hormones in meningioma growth. This is the result of several observations: (1) Meningiomas are twice as common in women, (2) increased growth of meningiomas may occur in pregnancy and in the luteal phase of the menstrual cycle, and (3) the incidence of meningiomas is slightly increased in breast cancer patients.11,116 Estrogen receptors (ERs) are present at low levels in approximately 10% of meningiomas, while progesterone and androgen receptors are present in approximately two thirds of meningiomas, and are more frequently expressed in women than in men.11,29,117,118 Progesterone receptors (PRs) are expressed predominantly in benign meningiomas with low proliferation indices119; they are infrequently expressed in atypical and malignant gliomas. Because meningiomas express only low levels of ERs, PR expression is likely not regulated in an estrogen-dependent manner, as is the case in breast cancer.12
Over the past two decades there have been several studies evaluating antihormonal agents in meningiomas. Given the low level of estrogen receptor expression, it is not surprising that studies with anti-estrogens did not show any efficacy.120,121 In one study of tamoxifen for refractory meningioma, partial or minor responses were observed in 3 of 19 patients.120 Because of the high likelihood of PR expression in most meningiomas, there has been substantial interest in PR inhibitors.122 Initial studies of the anti-progesterone mifepristone (RU486) were encouraging. In one study, 4 of 14 patients had a minor decrease in the size of the tumor, and 1 patient had objective clinical improvement.123 In another study mifepristone produced stable disease in 3 of 10 patients and minimally decreased tumor size in another 3.124 However, a prospective randomized multicenter study conducted by SWOG failed to demonstrate any treatment effect; the median survival was 31 months, and 42 of 45 patients progressed with a median time-to-progression of 6 months.125 In a more recent phase II study of 28 patients with unresectable meningiomas, 8 patients achieved minor responses with maximal reduction in tumor area of 10%.126 Of interest is the fact that most responders were males or premenopausal females, which suggests a potential subgroup for further evaluation.
There has been interest in the role of growth hormone (GH) on meningiomas since the initial observation that the incidence of meningiomas may be increased in patients with acromegaly.2 GH secreted by the pituitary gland stimulates the synthesis of IGF-1 in the liver, and together they facilitate normal growth.2 GHRs are ubiquitous in meningiomas, and inhibition of the GHR decreases tumor growth.2,127–129 Pegvisomant, a pegylated GH analog that acts as a competitive antagonist of the GHR, significantly inhibited the growth of meningioma xenografts in nude mice.129 Tumor IGF-1 concentrations did not vary with pegvisomant treatment, and there was no autocrine IGF-1 production by the tumors. The antitumor effect was thought to be a consequence of decreased IGF-1 in the circulation and/or surrounding host tissues. Direct blockade of the GHR on tumor cells may also contribute to the antitumor effect. Whether pegvisomant can inhibit meningioma growth in patients remains to be established. In a single patient with acromegaly and a meningioma, the tumor continued to grow despite several years of treatment with pegvisomant.130
Somatostatin receptors, especially the sst2A subtype, are present on most meningiomas, although their functional role remains unclear. The addition of somatostatin inhibits meningioma growth in vitro in some studies, but increases meningioma proliferation in others.131 Radiolabeled octreotide, a long-acting somatostatin agonist, has been used to image meningiomas.132,133 There have been anecdotal reports of octreotide inhibiting growth in human meningiomas,134 but the results are difficult to interpret in light of very small numbers. In a recent study, 16 patients with recurrent meningiomas were treated with a sustained-release somatostatin preparation (Sandostatin LAR).135 Indium 111-octreotide gamma scanning was used to confirm the presence of somatostatin receptors in the tumors. Patients received monthly injections of sustained-release somatostatin (20–40 mg/month). No patients withdrew from the treatment because of toxicity. After 3 months, 31% of patients achieved partial response, 31% had stable disease, and 38% had progressive disease. The 6-month PFS rate was 44%. A large scale trial evaluating this approach is currently in progress.
SOM230 (pasireotide) is a novel, orally administered, somatostatin analog with a wider somatostatin receptor spectrum (including subtypes 1, 2, 3, and 5) and higher affinity (particularly for subtypes 1, 3, and 5) than the sustained-release somatostatin described in the preceding text.136 A phase II trial for patients with recurrent or progressive meningiomas is planned. Biomarkers that may serve as predictors of efficacy or toxicity will be evaluated in all patients.
[1] Whittle I.R., Smith C., Navoo P., Collie D. Meningiomas. Lancet. 2004;363:1535.
[2] Ragel B., Jensen R.L. New approaches for the treatment of refractory meningiomas. Cancer Control. 2003;10:148.
[3] D’Ambrosio A.L., Bruce J.N. Treatment of meningioma: an update. Curr Neurol Neurosci Rep. 2003;3:206.
[4] Chamberlain M.C. Intracerebral meningiomas. Curr Treat Options Neurol. 2004;6:297.
[5] McMullen K.P., Stieber V.W. Meningioma: current treatment options and future directions. Curr Treat Options Oncol. 2004;5:499.
[6] Chamberlain M.C., Blumenthal D.T. Intracranial meningiomas: diagnosis and treatment. Expert Rev Neurother. 2004;4:641.
[7] Drummond K.J., Zhu J.J., Black P.M. Meningiomas: updating basic science, management, and outcome. Neurologist. 2004;10:113.
[8] Modha A., Gutin P.H. Diagnosis and treatment of atypical and anaplastic meningiomas: a review. Neurosurgery. 2005;57:538.
[9] Goldsmith B., McDermott M.W. Meningioma. Neurosurg Clin N Am. 2006;17:111.
[10] Louis D.N., Scheithauer B.W., Budka H. Meningeal tumors. Lyon: IARC, 2000.
[11] Lamszus K. Meningioma pathology, genetics, and biology. J Neuropathol Exp Neurol. 2004;63:275.
[12] Perry A., Gutmann D.H., Reifenberger G. Molecular pathogenesis of meningiomas. J Neurooncol. 2004;70:183.
[13] Chamberlain M.C. Adjuvant combined modality therapy for malignant meningiomas. J Neurosurg. 1996;84:733.
[14] Kyritsis A.P. Chemotherapy for meningiomas. J Neurooncol. 1996;29:269.
[15] Herscovici Z., Rappaport Z., Sulkes J., et al. Natural history of conservatively treated meningiomas. Neurology. 2004;63:1133.
[16] Zeidman L.A., Ankenbrandt W.J., et al. Analysis of growth rate in non-operated meningiomas. Neurology. 2006;66:A400.
[17] Schrell U.M., Rittig M.G., Anders M., et al. Hydroxyurea for treatment of unresectable and recurrent meningiomas. I. Inhibition of primary human meningioma cells in culture and in meningioma transplants by induction of the apoptotic pathway. J Neurosurg. 1997;86:845.
[18] Schrell U.M., Rittig M.G., Anders M., et al. Hydroxyurea for treatment of unresectable and recurrent meningiomas. II. Decrease in the size of meningiomas in patients treated with hydroxyurea. J Neurosurg. 1997;86:840.
[19] Mason W.P., Gentili F., Macdonald D.R., et al. Stabilization of disease progression by hydroxyurea in patients with recurrent or unresectable meningioma. J Neurosurg. 2002;97:341.
[20] Newton H.B., Scott S.R., Volpi C. Hydroxyurea chemotherapy for meningiomas: enlarged cohort with extended follow-up. Br J Neurosurg. 2004;18:495.
[21] Rosenthal M.A., Ashley D.L., Cher L. Treatment of high risk or recurrent meningiomas with hydroxyurea. J Clin Neurosci. 2002;9:156.
[22] Loven D., Hardoff R., Sever Z.B., et al. Non-resectable slow-growing meningiomas treated by hydroxyurea. J Neurooncol. 2004;67:221.
[23] Cusimano M.D. Hydroxyurea for treatment of meningioma. J Neurosurg. 1998;88:938.
[24] Kaba S.E., DeMonte F., Bruner J.M., et al. The treatment of recurrent unresectable and malignant meningiomas with interferon alpha-2B. Neurosurgery. 1997;40:271.
[25] Muhr C., Gudjonsson O., Lilja A., et al. Meningioma treated with interferon-alpha, evaluated with [(11)C]-L-methionine positron emission tomography. Clin Cancer Res. 2001;7:2269.
[26] Chamberlain M.C., Tsao-Wei D.D., Groshen S. Temozolomide for treatment-resistant recurrent meningioma. Neurology. 2004;62:1210.
[27] Chamberlain M.C., Tsao-Wei D.D., Groshen S. Salvage chemotherapy with CPT-11 for recurrent meningioma. J Neurooncol. 2006;78:271.
[28] Mocellin S., Rossi C.R., Brandes A., Nitti D. Adult soft tissue sarcomas: conventional therapies and molecularly targeted approaches. Cancer Treat Rev. 2006;32:9.
[29] Sanson M., Cornu P. Biology of meningiomas. Acta Neurochir (Wien). 2000;142:493.
[30] Johnson M., Toms S. Mitogenic signal transduction pathways in meningiomas: novel targets for meningioma chemotherapy? J Neuropathol Exp Neurol. 2005;64:1029.
[31] Cargioli T.G., Ugur H.C., Ramakrishna N., et al. Establishment of an in vivo meningioma model with human telomerase reverse transcriptase. Neurosurgery. 2007;60:750.
[32] McCutcheon I.E., Friend K.E., Gerdes T.M., et al. Intracranial injection of human meningioma cells in athymic mice: an orthotopic model for meningioma growth. J Neurosurg. 2000;92:306.
[33] Kalamarides M., Niwa-Kawakita M., Leblois H., et al. Nf2 gene inactivation in arachnoidal cells is rate-limiting for meningioma development in the mouse. Genes Dev. 2002;16:1060.
[34] Wen P.Y., Drappatz J. Novel therapies for meningiomas. Expert Rev Neurother. 2006;6:1447.
[35] Adjei A.A., Hidalgo M. Intracellular signal transduction pathway proteins as targets for cancer therapy. J Clin Oncol. 2005;23:5386.
[36] Dy G.K., Adjei A.A. Obstacles and opportunities in the clinical development of targeted therapeutics. Prog Drug Res. 2005;63:19.
[37] Druker B.J., Talpaz M., Resta D.J., et al. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med. 2001;344:1031.
[38] Demetri G.D., von Mehren M., Blanke C.D., et al. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med. 2002;347:472.
[39] Chi A.S., Wen P.Y. Inhibiting kinases in malignant gliomas. Expert Opin Ther Targets. 2007;11:473.
[40] Kesari S., Ramakrishna N., Sauvageot C., et al. Targeted molecular therapy of malignant gliomas. Curr Oncol Rep. 2006;8:58.
[41] Wen P.Y., Kesari S., Drappatz J. Malignant gliomas: strategies to increase the effectiveness of targeted molecular treatment. Expert Rev Anticancer Ther. 2006;6:733.
[42] Pietras K., Sjoblom T., Rubin K., et al. PDGF receptors as cancer drug targets. Cancer Cell. 2003;3:439.
[43] Black P.M., Carroll R., Glowacka D., et al. Platelet-derived growth factor expression and stimulation in human meningiomas. J Neurosurg. 1994;81:388.
[44] Wang J.L., Nister M., Hermansson M., et al. Expression of PDGF beta-receptors in human meningioma cells. Int J Cancer. 1990;46:772.
[45] Yang S.Y., Xu G.M. Expression of PDGF and its receptor as well as their relationship to proliferating activity and apoptosis of meningiomas in human meningiomas. J Clin Neurosci. 2001;8(Suppl. 1):49.
[46] Nagashima G., Asai J., Suzuki R., Fujimoto T. Different distribution of c-myc and MIB-1 positive cells in malignant meningiomas with reference to TGFs, PDGF, and PgR expression. Brain Tumor Pathol. 2001;18:1.
[47] Maxwell M., Galanopoulos T., Hedley-Whyte E.T., et al. Human meningiomas co-express platelet-derived growth factor (PDGF) and PDGF-receptor genes and their protein products. Int J Cancer. 1990;46:16.
[48] Johnson M.D., Woodard A., Kim P., Frexes-Steed M. Evidence for mitogen-associated protein kinase activation and transduction of mitogenic signals by platelet-derived growth factor in human meningioma cells. J Neurosurg. 2001;94:293.
[49] Kirsch M., Wilson J.C., Black P. Platelet-derived growth factor in human brain tumors. J Neurooncol. 1997;35:289.
[50] Todo T., Adams E.F., Fahlbusch R., et al. Autocrine growth stimulation of human meningioma cells by platelet-derived growth factor. J Neurosurg. 1996;84:852.
[51] Capdeville R., Buchdunger E., Zimmermann J., Matter A. Glivec (STI571, imatinib), a rationally developed, targeted anticancer drug. Nat Rev Drug Discov. 2002;1:493.
[52] Wen P.Y., Yung W.K.A., Lamborn K.R., et al. Phase II study of imatinib mesylate for patients with recurrent meningiomas (NABTC 01-08), 2006. Society for Neuro-Oncology Annual Meeting
[53] Wen P.Y., Yung W.K., Lamborn K.R., et al. Phase I/II study of imatinib mesylate for recurrent malignant gliomas: North American Brain Tumor Consortium Study 99-08. Clin Cancer Res. 2006;12:4899.
[54] Reardon D.A., Egorin M.J., Quinn J.A., et al. Phase II study of imatinib mesylate plus hydroxyurea in adults with recurrent glioblastoma multiforme. J Clin Oncol. 2005;23:9359.
[55] Andersson U., Guo D., Malmer B., et al. Epidermal growth factor receptor family (EGFR, ErbB2-4) in gliomas and meningiomas. Acta Neuropathol (Berl). 2004;108:135.
[56] Weisman A.S., Raguet S.S., Kelly P.A. Characterization of the epidermal growth factor receptor in human meningioma. Cancer Res. 1987;47:2172.
[57] Carroll R.S., Black P.M., Zhang J., et al. Expression and activation of epidermal growth factor receptors in meningiomas. J Neurosurg. 1997;87:315.
[58] Jones N.R., Rossi M.L., Gregoriou M., Hughes J.T. Epidermal growth factor receptor expression in 72 meningiomas. Cancer. 1990;66:152.
[59] Johnson M.D., Horiba M., Winnier A.R., Arteaga C.L. The epidermal growth factor receptor is associated with phospholipase C-gamma 1 in meningiomas. Hum Pathol. 1994;25:146.
[60] Sanfilippo J.S., Rao C.V., Guarnaschelli J.J., et al. Detection of epidermal growth factor and transforming growth factor alpha protein in meningiomas and other tumors of the central nervous system in human beings. Surg Gynecol Obstet. 1993;177:488.
[61] Linggood R.M., Hsu D.W., Efird J.T., Pardo F.S. TGF alpha expression in meningioma–tumor progression and therapeutic response. J Neurooncol. 1995;26:45.
[62] Hsu D.W., Efird J.T., Hedley-Whyte E.T. MIB-1 (Ki-67) index and transforming growth factor-alpha (TGF alpha) immunoreactivity are significant prognostic predictors for meningiomas. Neuropathol Appl Neurobiol. 1998;24:441.
[63] Crombet T., Torres O., Rodriguez V., et al. Phase I clinical evaluation of a neutralizing monoclonal antibody against epidermal growth factor receptor in advanced brain tumor patients: preliminary study. Hybridoma. 2001;20:131.
[64] Mawrin C., Sasse T., Kirches E., et al. Different activation of mitogen-activated protein kinase and Akt signaling is associated with aggressive phenotype of human meningiomas. Clin Cancer Res. 2005;11:4074.
[65] Adjei A.A. Farnesyltransferase inhibitors. Cancer Chemother Biol Response Modif. 2005;22:123.
[66] Engelman J.A., Luo J., Cantley L.C. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet. 2006;7:606.
[67] Cully M., You H., Levine A.J., Mak T.W. Beyond PTEN mutations: the PI3K pathway as an integrator of multiple inputs during tumorigenesis. Nat Rev Cancer. 2006;6:184.
[68] Johnson M.D., Okedli E., Woodard A., et al. Evidence for phosphatidylinositol 3-kinase-Akt-p7S6K pathway activation and transduction of mitogenic signals by platelet-derived growth factor in meningioma cells. J Neurosurg. 2002;97:668.
[69] Johnson M.D., Okediji E., Woodard A. Transforming growth factor-beta effects on meningioma cell proliferation and signal transduction pathways. J Neurooncol. 2004;66:9.
[70] Shapiro G.I. Cyclin-dependent kinase pathways as targets for cancer treatment. J Clin Oncol. 2006;24:1770.
[71] Schwartz G.K., Shah M.A. Targeting the cell cycle: a new approach to cancer therapy. J Clin Oncol. 2005;23:9408.
[72] Newton H.B. Molecular neuro-oncology and the development of targeted therapeutic strategies for brain tumors. Part 5: apoptosis and cell cycle. Expert Rev Anticancer Ther. 2005;5:355.
[73] Reed J.C. Apoptosis-targeted therapies for cancer. Cancer Cell. 2003;3:17.
[74] Rowinsky E.K. Targeted induction of apoptosis in cancer management: the emerging role of tumor necrosis factor-related apoptosis-inducing ligand receptor activating agents. J Clin Oncol. 2005;23:9394.
[75] Ghobrial I.M., Witzig T.E., Adjei A.A. Targeting apoptosis pathways in cancer therapy. CA Cancer J Clin. 2005;55:178.
[76] Oltersdorf T., Elmore S.W., Shoemaker A.R., et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435:677.
[77] Piro L.D. Apoptosis, Bcl-2 antisense, and cancer therapy. Oncology (Williston Park). 2004;18:5.
[78] Letai A., Bassik M.C., Walensky L.D., et al. Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell. 2002;2:183.
[79] Schimmer A.D., Dalili S. Targeting the IAP family of caspase inhibitors as an emerging therapeutic strategy. Hematol Am Soc Hematol Educ Prog. 2005:215-219.
[80] Schimmer A.D., Dalili S., Batey R.A., Riedl S.J. Targeting XIAP for the treatment of malignancy. Cell Death Differ. 2006;13:179.
[81] Puduvalli V.K., Li J.T., Chen L., McCutcheon I.E. Induction of apoptosis in primary meningioma cultures by fenretinide. Cancer Res. 2005;65:1547.
[82] Folkman J. Angiogenesis. Annu Rev Med. 2006;57:1.
[83] Yazaki T., Takamiya Y., Costello P.C., et al. Inhibition of angiogenesis and growth of human non-malignant and malignant meningiomas by TNP-470. J Neurooncol. 1995;23:23.
[84] Jain R.K., Duda D.G., Clark J.W., Loeffler J.S. Lessons from phase III clinical trials on anti-VEGF therapy for cancer. Nat Clin Pract Oncol. 2006;3:24.
[85] Lamszus K., Lengler U., Schmidt N.O., et al. Vascular endothelial growth factor, hepatocyte growth factor/scatter factor, basic fibroblast growth factor, and placenta growth factor in human meningiomas and their relation to angiogenesis and malignancy. Neurosurgery. 2000;46:938.
[86] Provias J., Claffey K., delAguila L., et al. Meningiomas: role of vascular endothelial growth factor/vascular permeability factor in angiogenesis and peritumoral edema. Neurosurgery. 1997;40:1016.
[87] Goldman C.K., Bharara S., Palmer C.A., et al. Brain edema in meningiomas is associated with increased vascular endothelial growth factor expression. Neurosurgery. 1997;40:1269.
[88] Donnini S., Machein M.R., Plate K.H., Weich H.A. Expression and localization of placenta growth factor and PlGF receptors in human meningiomas. J Pathol. 1999;189:66.
[89] Martinez-Rumayor A., Arrieta O., Guevara P., et al. Coexpression of hepatocyte growth factor/scatter factor (HGF/SF) and its receptor cMET predict recurrence of meningiomas. Cancer Lett. 2004;213:117.
[90] Bek E.L., McMillen M.A. Endothelins are angiogenic. J Cardiovasc Pharmacol. 2000;36:S135.
[91] Harland S.P., Kuc R.E., Pickard J.D., Davenport A.P. Expression of endothelin(A) receptors in human gliomas and meningiomas, with high affinity for the selective antagonist PD156707. Neurosurgery. 1998;43:890.
[92] Pagotto U., Arzberger T., Hopfner U., et al. Expression and localization of endothelin-1 and endothelin receptors in human meningiomas. Evidence for a role in tumoral growth. J Clin Invest. 1995;96:2017.
[93] Nordqvist A.C., Smurawa H., Mathiesen T. Expression of matrix metalloproteinases 2 and 9 in meningiomas associated with different degrees of brain invasiveness and edema. J Neurosurg. 2001;95:839.
[94] Siddique K., Yanamandra N., Gujrati M., et al. Expression of matrix metalloproteinases, their inhibitors, and urokinase plasminogen activator in human meningiomas. Int J Oncol. 2003;22:289.
[95] Kondraganti S., Gondi C.S., Gujrati M., et al. Restoration of tissue factor pathway inhibitor inhibits invasion and tumor growth in vitro and in vivo in a malignant meningioma cell line. Int J Oncol. 2006;29:25.
[96] Nakada M., Nakada S., Demuth T., et al. Molecular targets of glioma invasion. Cell Mol Life Sci. 2007;64:458.
[97] Rao J.S. Molecular mechanisms of glioma invasiveness: the role of proteases. Nat Rev Cancer. 2003;3:489.
[98] Salhia B., Tran N.L., Symons M., et al. Molecular pathways triggering glioma cell invasion. Expert Rev Mol Diagn. 2006;6:613.
[99] Lefranc F., Brotchi J., Kiss R. Possible future issues in the treatment of glioblastomas: special emphasis on cell migration and the resistance of migrating glioblastoma cells to apoptosis. J Clin Oncol. 2005;23:2411.
[100] Bello L., Zhang J., Nikas D.C., et al. Alpha(v)beta3 and alpha(v)beta5 integrin expression in meningiomas. Neurosurgery. 2000;47:1185.
[101] Nabors L.B., Mikkelsen T., Rosenfeld S.S., et al. Phase I and correlative biology study of cilengitide in patients with recurrent malignant glioma. J Clin Oncol. 2007;25:1651.
[102] Nordqvist A.C., Peyrard M., Pettersson H., et al. A high ratio of insulin-like growth factor II/insulin-like growth factor binding protein 2 messenger RNA as a marker for anaplasia in meningiomas. Cancer Res. 1997;57:2611.
[103] Nordqvist A.C., Mathiesen T. Expression of IGF-II, IGFBP-2, -5, and -6 in meningiomas with different brain invasiveness. J Neurooncol. 2002;57:19.
[104] Liu T., Kuljaca S., Tee A., Marshall G.M. Histone deacetylase inhibitors: multifunctional anticancer agents. Cancer Treat Rev. 2006;32:157.
[105] Nakanishi C., Toi M. Nuclear factor-kappaB inhibitors as sensitizers to anticancer drugs. Nat Rev Cancer. 2005;5:297.
[106] Graner M.W., Bigner D.D. Therapeutic aspects of chaperones/heat-shock proteins in neuro-oncology. Expert Rev Anticancer Ther. 2006;6:679.
[107] Magrassi L., De-Fraja C., Conti L., et al. Expression of the JAK and STAT superfamilies in human meningiomas. J Neurosurg. 1999;91:440.
[108] Luo Y., Leverson J.D. New opportunities in chemosensitization and radiosensitization: modulating the DNA-damage response. Expert Rev Anticancer Ther. 2005;5:333.
[109] Homsi J., Cubitt C., Daud A. The Src signaling pathway: a potential target in melanoma and other malignancies. Expert Opin Ther Targets. 2007;11:91.
[110] McLean G.W., Carragher N.O., Avizienyte E., et al. The role of focal-adhesion kinase in cancer – a new therapeutic opportunity. Nat Rev Cancer. 2005;5:505.
[111] O’Donnell J.L., Joyce M.R., Shannon A.M., et al. Oncological implications of hypoxia inducible factor-1alpha (HIF-1alpha) expression. Cancer Treat Rev. 2006;32:407.
[112] Camphausen K., Tofilon P.J. Combining radiation and molecular targeting in cancer therapy. Cancer Biol Ther. 2004;3:247.
[113] Russell J.S., Burgan W., Oswald K.A., et al. Enhanced cell killing induced by the combination of radiation and the heat shock protein 90 inhibitor 17-allylamino-17-demethoxygeldanamycin: a multitarget approach to radiosensitization. Clin Cancer Res. 2003;9:3749.
[114] Citrin D., Menard C., Camphausen K. Combining radiotherapy and angiogenesis inhibitors: clinical trial design. Int J Radiat Oncol Biol Phys. 2006;64:15.
[115] Winkler F., Kozin S.V., Tong R.T., et al. Kinetics of vascular normalization by VEGFR2 blockade governs brain tumor response to radiation: role of oxygenation, angiopoietin-1, and matrix metalloproteinases. Cancer Cell. 2004;6:553.
[116] Bondy M., Ligon B.L. Epidemiology and etiology of intracranial meningiomas: a review. J Neurooncol. 1996;29:197.
[117] Hsu D.W., Efird J.T., Hedley-Whyte E.T. Progesterone and estrogen receptors in meningiomas: prognostic considerations. J Neurosurg. 1997;86:113.
[118] McCutcheon I.E. The biology of meningiomas. J Neurooncol. 1996;29:207.
[119] Wolfsberger S., Doostkam S., Boecher-Schwarz H.G., et al. Progesterone-receptor index in meningiomas: correlation with clinico-pathological parameters and review of the literature. Neurosurg Rev. 2004;27:238.
[120] Goodwin J.W., Crowley J., Eyre H.J., et al. A phase II evaluation of tamoxifen in unresectable or refractory meningiomas: a Southwest Oncology Group study. J Neurooncol. 1993;15:75.
[121] Markwalder T.M., Seiler R.W., Zava D.T. Antiestrogenic therapy of meningiomas–a pilot study. Surg Neurol. 1985;24:245.
[122] Grunberg S.M. Role of antiprogestational therapy for meningiomas. Hum Reprod. 1994;9(Suppl. 1):202.
[123] Grunberg S.M., Weiss M.H., Spitz I.M., et al. Treatment of unresectable meningiomas with the antiprogesterone agent mifepristone. J Neurosurg. 1991;74:861.
[124] Lamberts S.W., Tanghe H.L., Avezaat C.J., et al. Mifepristone (RU 486) treatment of meningiomas. J Neurol Neurosurg Psychiatry. 1992;55:486.
[125] Grunberg S.M., Rankin C., Townsend J., et al. Phase III double-blind randomized placebo-controlled study of mifepristone (RU) for the treatment of unresectable meningioma. Proc Am Soc Clin Oncol. 2001;20:222.
[126] Grunberg S.M., Weiss M.H., Russell C.A., et al. Long-term administration of mifepristone (RU486): clinical tolerance during extended treatment of meningioma. Cancer Invest. 2006;24:727.
[127] Friend K.E., Radinsky R., McCutcheon I.E. Growth hormone receptor expression and function in meningiomas: effect of a specific receptor antagonist. J Neurosurg. 1999;91:93.
[128] Friend K.E. Cancer and the potential place for growth hormone receptor antagonist therapy. Growth Horm IGF Res. 2001;11(Suppl. A):S121.
[129] McCutcheon I.E., Flyvbjerg A., Hill H., et al. Antitumor activity of the growth hormone receptor antagonist pegvisomant against human meningiomas in nude mice. J Neurosurg. 2001;94:487.
[130] Drake W.M., Grossman A.B., Hutson R.K. Effect of treatment with pegvisomant on meningioma growth in vivo. Eur J Endocrinol. 2005;152:161.
[131] Cavalla P., Schiffer D. Neuroendocrine tumors in the brain. Ann Oncol. 2001;12(Suppl. 2):S131.
[132] Henze M., Dimitrakopoulou-Strauss A., Milker-Zabel S., et al. Characterization of 68Ga-DOTA-D-Phe1-Tyr3-octreotide kinetics in patients with meningiomas. J Nucl Med. 2005;46:763.
[133] Klutmann S., Bohuslavizki K.H., Brenner W., et al. Somatostatin receptor scintigraphy in postsurgical follow-up examinations of meningioma. J Nucl Med. 1998;39:1913.
[134] Garcia-Luna P.P., Relimpio F., Pumar A., et al. Clinical use of octreotide in unresectable meningiomas. A report of three cases. J Neurosurg Sci. 1993;37:237.
[135] Chamberlain M.C., Glantz M.J., Fadul C.E. Recurrent meningioma: salvage therapy with long-acting somatostatin analogue. Neurology. 2007;69:969.
[136] Bruns C., Lewis I., Briner U., et al. SOM230: a novel somatostatin peptidomimetic with broad somatotropin release inhibiting factor (SRIF) receptor binding and a unique antisecretory profile. Eur J Endocrinol. 2002;146:707.
[/level-membership-for-neurosurgery-category][not-level-membership-for-neurosurgery-category]
CHAPTER 56 Chemotherapy and Experimental Medical Therapies for Meningiomas
INTRODUCTION
Current therapies for meningiomas include surgery, radiation therapy and stereotactic radiosurgery.1–9 For the majority of patients with benign meningiomas (World Health Organization [WHO] grade I) and a subset of patients with atypical meningiomas (WHO grade II), these therapies are effective in achieving tumor control. However, there is an important group of patients with inoperable or higher grade tumors who develop recurrent disease after surgery and radiation therapy. The treatment options for these patients are currently inadequate.
The WHO classification scheme for meningiomas is based on the degree of anaplasia, number of mitoses, and presence of necrosis.10 Histological grading is important because it helps to predict the likelihood of recurrence. Benign meningiomas account for more than 90% of tumors and have a low risk of recurrence (7%–20%).10–12 Atypical meningiomas are much less common. They account for 4.7% to 7.2% of meningiomas and are associated with a 40% recurrence rate despite surgical resection.12 Malignant meningiomas represent only 1% to 2.8% of meningiomas but recur in 50% to 80% of cases and usually result in death within 2 years of diagnosis.10,11 Patients with meningiomas of any histological grade that recur after surgery and radiation therapy are frequently considered for chemotherapy or experimental medical therapies.
CYTOTOXIC CHEMOTHERAPY
To date, chemotherapy has had only a very limited role in the treatment of meningiomas. Data from small clinical trials and case series suggest that most chemotherapeutic agents have minimal activity against meningiomas.5,6,13,14 The evaluation of chemotherapy has also been complicated by the lack of data regarding the natural history of untreated meningiomas. Many chemotherapy studies report variable periods of disease stabilization, but it is difficult to know whether this represents an improvement because benign meningiomas grow slowly and may appear radiographically stable for prolonged periods.15,16
In general, chemotherapeutic regimens (such as dacarbazine and Adriamycin) that have activity in other soft tissue tumors have produced disappointing results in patients with meningiomas.6 Hydroxyurea, an oral ribonucleotide reductase inhibitor, arrests meningioma cell growth in the S phase of the cell cycle and induces apoptosis.17 In a preliminary report, hydroxyurea (1000–1500 mg/day; 20 mg/kg/day) decreased tumor size in three patients with recurrent benign meningiomas and prevented recurrent disease for 24 months in a patient with a completely resected malignant meningioma.18 Several more recent studies suggest that hydroxyurea has modest activity; responses are uncommon but some patients appear to have disease stabilization.19–23 The Southwest Oncology Group conducted a phase II study to further evaluate the role of hydroxyurea in meningiomas (SWOG-S9811). This study is closed to accrual but the final results are not yet available.
There have been reports of small numbers of patients with malignant meningiomas who responded to recombinant interferon alpha-2b.24,25 Temozolomide (Temodar, TMZ), an alkylating agent with activity in malignant gliomas, was evaluated in 16 patients with refractory meningiomas and showed negligible activity.26 The topoisomerase inhibitor irinotecan (Camptosar, CPT-11) caused moderate toxicity in 16 patients with benign meningiomas and had no demonstrable activity.27 A large number of cytotoxic agents are under evaluation for sarcomas and other systemic malignancies.28 Most of these have not been evaluated in meningiomas, and it is possible that some may have modest activity. However, it is likely that the more novel therapeutic approaches discussed in the text that follows will provide a greater chance of improving the outcome for patients with recurrent meningiomas.
CHALLENGES IN THE DEVELOPMENT OF EFFECTIVE MEDICAL THERAPIES FOR MENINGIOMAS
In contrast to the extensive understanding of the molecular pathogenesis and biology of systemic malignancies, and even brain tumors such as malignant gliomas, relatively little is known about the molecular pathogenesis of meningiomas and the critical molecular changes driving tumor growth.11,12,29,30 Overexpression of various growth factors including platelet-derived growth factor (PDGF), epidermal growth factor (EGF), and vascular endothelial growth factor (VEGF) and their receptors, and signal transduction pathways such as the Ras/mitogen-activated protein kinase (MAPK), phosphatidylinositol-3-kinase (PI3K)-Akt, and phospholipase C (PLC)-γ1-protein kinase C (PKC) pathways have been implicated, but their relative significance is largely unknown.29,30 As a result, the most important molecular targets may remain to be elucidated.
Another factor limiting progress in the development of more effective therapies for meningiomas is the lack of robust cell lines and animal models. There is a need for animal models that replicate the genetic changes in meningiomas with a high frequency of spontaneous meningioma development, benign meningioma lines for in vitro and in vivo studies, and meningeal specific promoters. Many of the existing meningioma cell lines are derived from malignant meningiomas and likely contain culture-induced artifacts and lack progesterone receptors.12 There are some orthotopic31,32 and genetic models33 in development that appear promising. Recently, two cell lines were developed from benign meningioma specimens via immortalization with human telomerase reverse transcriptase and SV40 large T antigen. Orthotopic tumors with similar immunostaining patterns to human meningiomas were established from both cell lines in athymic nude mice.31 Another model uses a Cre recombinase technology to inactivate NF2 in arachnoid cells, resulting in intracranial meningothelial hyperplasia and meningiomas in 30% of mice.33 These models may aid in the preclinical evaluation of novel therapies.
A final factor limiting progress is the relatively small number of patients with meningiomas who require additional therapies after treatment with surgery and radiation therapy. In general, there is little incentive for pharmaceutical companies to evaluate their therapies in meningiomas because of the small potential market. Hopefully as the molecular pathogenesis of these tumors becomes better understood, a compelling case can be made for evaluating specific agents directed at critical molecular targets. In the next section, targeted molecular drugs that have a potential role against meningiomas are reviewed in detail. These therapies have also been discussed in recent review articles.2,5,30,34
EXPERIMENTAL THERAPIES: TARGETED MOLECULAR AGENTS
Recently, it has become apparent that many human diseases result from aberrations in cell signaling pathways. Protein-tyrosine kinases play a fundamental role in signal transduction, and deregulated activity of these enzymes has been observed in many cancers. Therefore, specific inhibitors of tyrosine kinases could have potential therapeutic applications in the treatment of cancer, with potentially lower toxicity and/or higher, prolonged response rates.35,36 The prototypical targeted molecular agent is imatinib mesylate (Gleevec), which has shown significant benefit in chronic myeloid leukemia (CML)37 and gastrointestinal stromal tumors (GIST).38 There is also a growing experience with targeted molecular agents in malignant gliomas.39–41 However, to date, there have been minimal data on the use of these agents in meningiomas.
In contrast to the extensive work aimed at understanding the genetics of meningiomas, relatively little work has been conducted to understand the growth factors and their receptors, and the signal transduction pathways that are critical to meningioma growth.2,5,30 PDGF, EGF, VEGF, insulin-like growth factor (IGF), transforming growth factor-beta (TGF-β), and their receptor tyrosine kinases, together with their downstream signaling pathways including the Ras/MAPK pathway, the PI3K/Akt pathway, the PLC-γ1-PKC pathway, and the TGF-β-SMAD pathways, are all thought to be important in meningioma growth (Fig. 56-1).30
Platelet-Derived Growth Factor Receptor
PDGF is a fundamental driver of cell proliferation in normal development and in a variety of pathological conditions, including cancer.42 Accumulating evidence suggests that PDGF plays an important role in meningioma growth.43–47 Most meningiomas of all histologic grades express PDGF ligands AA and BB and the PDGF-beta receptor (PDGF-βR).43–47 Expression levels may be higher in atypical and malignant meningiomas than in benign meningiomas.45 Laboratory data suggest that an autocrine PDGF loop supports meningioma cell growth and maintenance.47 When PDGF-BB is applied to cultured meningioma cells, MAPK48 and c-fos49 are activated and tumor cell proliferation is enhanced. Conversely, anti-PDGF-BB antibodies inhibit cell growth.50 These data provide sound rationale for testing PDGF inhibitors in meningioma patients.
Imatinib is a potent inhibitor of the Bcr-Abl, PDGF-α and β receptors, and c-Kit tyrosine kinases.51 Its ability to inhibit PDGFR with an IC50 of 0.1 μM suggested that it may have therapeutic potential in meningiomas. The North American Brain Tumor Consortium (NABTC) conducted a phase II study of imatinib in patients with recurrent meningiomas (NABTC 01-08).52 Patients were stratified into two cohorts: (1) benign meningiomas or (2) atypical and malignant meningiomas. As imatinib is metabolized by the cytochrome P450 system (3A4), patients could not be receiving enzyme inducing antiepileptic drugs (EIAEDs). Patients initially received 600 mg/day of imatinib; the dose was increased in the second cycle to 800 mg/day if no significant toxicity was observed in the first cycle. Twenty-three patients were enrolled into the study (13 with meningiomas, 5 with atypical meningiomas, and 5 with malignant meningiomas). Though the treatment was generally well tolerated, imatinib had minimal activity. Of the 19 patients evaluable for response, 10 progressed at the first scan and 9 were stable. There were no radiographic responses. Overall median progression-free survival (PFS) was 2 months (range 0.7 to 18 months); 6-month PFS was 29.4%. For benign meningiomas, median PFS was 3 months; 6-month PFS was 45%. For the atypical and malignant meningiomas, median PFS was 2 months; 6-month PFS was 0%. Several other inhibitors of PDGF are undergoing evaluation such as sunitinib, MLN518, dasatinib, AMN 107, pazopanib, CP673451 and CHIR 265. Some of these, such as MLN518, are more potent PDGFR inhibitors than imatinib, whereas others target additional kinases that are potentially important in meningiomas. For example, pazopanib also inhibits VEGF receptors (VEGFR) 1, 2, and 3 as well as c-Kit, while CHIR 265 inhibits VEGFR, c-Kit and Raf. These drugs may be more effective than imatinib as monotherapy against meningiomas.
There is also interest in combining imatinib with hydroxyurea, the cytotoxic chemotherapy agent with the most activity in meningiomas. Though a recent phase I/II trial of imatinib as monotherapy for recurrent malignant glioma showed minimal activity,53 a study of 33 recurrent glioblastoma (GBM) patients treated with the combination of imatinib mesylate (400 mg/day or 500 mg twice/day, depending on concurrent EIAED use) and hydroxyurea (500 mg twice/day), had encouraging results.54 After a median follow-up period of 58 weeks, 1 patient achieved a complete response, 2 achieved a partial response, and 14 achieved stable disease. Six-month PFS was 27%, and the median PFS was 14.4 weeks. In light of these results in patients with recurrent GBM, a multicenter phase II trial of the combination for patients with recurrent or progressive meningiomas after surgery is currently in progress (ClinicalTrials.gov identifier NCT00354913). Twenty-one patients are expected to enroll, and monitoring includes magnetic resonance imaging (MRI) scans and clinical examinations every 8 weeks.
Epidermal Growth Factor Receptor
The EGFR is overexpressed in more than 60% of meningiomas.55–61 EGF and TGF-β activate these receptors and stimulate meningioma growth in vitro,30,56 supporting the concept that activation of EGFRs in human meningiomas by autocrine/paracrine stimulation may contribute to their proliferation. Increased TGF-β immunoreactivity in meningiomas has been associated with aggressive growth.30,61,62
In addition to gefitinib and erlotinib, there are a large number of other agents currently undergoing evaluation. These inhibit EGFR alone, or together with other receptor tyrosine kinases, which may have therapeutic potential in meningiomas (Table 56-1). For example, lapatinib inhibits EGFR and HER2, HKI-272 inhibits all subtypes of the EGFR, and ZD6474 (Zactima) inhibits EGFR and VEGFR. Although EGFR monoclonal antibodies have been effective for some systemic malignancies (e.g., cetuximab in colorectal cancer), they have generally not been used for brain tumors because of the concern regarding the ability of these agents to pass through the blood–brain barrier (BBB) in sufficient concentrations to produce a therapeutic effect. Because the BBB is not a factor in most meningiomas, it is possible that these antibodies may be effective in these tumors. To date very few studies have evaluated the therapeutic potential of these agents in meningiomas. In a phase I study of a murine monoclonal antibody against EGFR in nine patients with either gliomas or meningiomas, treatment was reasonably well tolerated. No radiographic responses were detected, but efficacy data is difficult to interpret in a study with so few subjects.63 Currently, several anti-EGFR antibodies are undergoing evaluation for other malignancies such as cetuximab, panitumumab, EMD 72000, nimotuzumab, and mAb 806. Trials of these agents in meningiomas may be worthwhile, especially if combined with correlative studies examining whether the antibodies can achieve therapeutic concentrations in meningiomas and inhibit EGFR in vivo.
TABLE 56-1 Selected potential targeted molecular drugs for meningiomas
| Class/agent | Alternative name(s) | Mechanism(s) |
|---|---|---|
| Apoptosis enhancers | ||
| ABT737 | Bcl-2 inhibitor | |
| AT101 | Bcl-2 inhibitor | |
| Fenretinide | Multiple targets | |
| GX15-070 | Bcl-2 inhibitor | |
| Integrin inhibitor | ||
| Cilengitide | αvβ3 and αvβ5 integrin inhibitor | |
| Cell cycle inhibitors | ||
| AG024322 | pan-CDK inhibitor | |
| AZ703 | CDK 2, 1 inhibitor | |
| BMS-387032 | CDK 2, 1 inhibitor | |
| CINK4 | CDK 4, 6 inhibitor | |
| E7070 | Unknown | |
| Flavopiridol | pan-CDK inhibitor | |
| PD-0332991 | CDK 4, 6 inhibitor | |
| Seliciclib | CDK 2, 1 inhibitor | |
| c-MET (HGF/SF) inhibitors | ||
| AMG-102 | HGF/SF antibody | |
| XL880 | c-MET, VEGFR2, PDGFR, c-Kit, Tie-2 inhibitor | |
| EGFR inhibitors | ||
| AEE788 | EGFR, VEGFR inhibitor | |
| BIBW 2992 | EGFR, HER2 inhibitor | |
| Cetuximab | Erbitux | EGFR antibody |
| EMD 72000 | EGFR antibody | |
| Erlotinib | OSI-774, Tarceva | EGFR inhibitor |
| Gefitinib | ZD1839, Iressa | EGFR inhibitor |
| HKI-272 | pan-EGFR inhibitor | |
| Lapatinib | GW-572016 | EGFR, HER2 inhibitor |
| mAb 806 | EGFR antibody | |
| Nimotuzumab | TheraCIM | EGFR antibody |
| Panitumumab | Vectibix | EGFR antibody |
| ZD6474 | Zactima | EGFR, VEGFR inhibitor |
| Endothelin-A inhibitors | ||
| Astrasentan | Xinlay | ETA inhibitor |
| ZD4054 | ETA inhibitor | |
| Farnesyltransferase inhibitors | ||
| Lonafarnib | SCH 66336, Sarasar | FT inhibitor |
| Tipifarnib | R115777, Zarnestra | FT inhibitor |
| Histone deacetylase inhibitors | ||
| Depsipeptide | HDAC inhibitor | |
| Voronistat | Suberoylanilide hydroxamic acid (SAHA) | HDAC inhibitor |
| Valproic acid | HDAC inhibitor | |
| HSP-90 inhibitors | ||
| 17AAG | HSP-90 inhibitor | |
| 17DMAG | HSP-90 inhibitor | |
| IPI504 | HSP-90 inhibitor | |
| MEK inhibitors | ||
| AZD6244 | MEK inhibitor | |
| PD0325901 | MEK inhibitor | |
| mTOR inhibitors | ||
| AP23573 | mTOR inhibitor | |
| Everolimus | RAD001 | mTOR inhibitor |
| Rapamycin | mTOR inhibitor | |
| Temsirolimis | CCI-779, Torisel | mTOR inhibitor |
| PDGFR inhibitors | ||
| AMN 107 | PDGFR, c-Kit, Bcr-Abl inhibitor | |
| CHIR 265 | PDGFR, Raf, VEGFR, c-Kit inhibitor | |
| CP-673-451 | ||
| Dasatinib | PDGFR, Src, c-Kit, ephrin A inhibitor | |
| Imatinib mesylate | Gleevec | |
| MLN518 | PDGFR, c-Kit inhibitor | |
| Pazopanib | GW786034 | PDGFR, VEGFR, c-Kit inhibitor |
| Sunitinib | Sutent | PDGFR, VEGFR, c-Kit inhibitor |
| Vatalanib | PTK787 | PDGFR, VEGFR inhibitor |
| XL-999 | PDGFR, VEGFR, FGFR inhibitor | |
| PKC β2 inhibitor | ||
| Enzastaurin | LY31761 | PKC β2 inhibitor |
| PI3K inhibitor | ||
| BEZ235 | PI3K inhibitor | |
| Proteasome inhibitor | ||
| Bortezomib | Velcade | Proteasome inhibitor |
| Raf kinase inhibitor | ||
| Sorafenib | Nexavar, BAY 43-9006 | Raf kinase, VEGFR, PDGFR inhibitor |
| Src inhibitor | ||
| AZD0530 | Src inhibitor | |
| TGF-β inhibitors | ||
| AP 12009 | TGF-β2 antisense oligonucleotide | |
| GC1008 | TGF-β antibody | |
| SB-431542 | TGF-β receptor inhibitor | |
| VEGF inhibitors | ||
| Bevacizumab | Avastin | VEGF antibody |
| VEGF trap | VEGF soluble decoy receptor | |
| VEGFR inhibitors | ||
| AEE788 | VEGFR, EGFR inhibitor | |
| AG013736 | VEGFR inhibitor | |
| AMG 706 | VEGFR inhibitor | |
| AZD2171 | VEGFR inhibitor | |
| CEP-7055 | VEGFR inhibitor | |
| CHIR 265 |
