4 Chemical Modifiers of Radiation Response
This chapter deals with chemical agents that have been used solely as modifiers of radiation response and with methods to detect hypoxic cells in tumors. For a discussion of the interaction of cytotoxic cancer chemotherapeutic agents with radiation (actinomycin D, 5-fluorouracil, doxorubicin [Adriamycin], hydroxyurea, and paclitaxel [Taxol]), the reader is referred to Chapter 6.
Rationale for the Use of Radiosensitizers and Radioprotectors
Radiosensitizers
Radiosensitizers are compounds that, when combined with radiation, achieve greater tumor inactivation than would have been expected from the additive effect of each modality. The application of chemical agents that simply have an additive effect in normal tissues is equivalent to the administration of an increment of radiation dose with no differential benefit. The toxicities of the chemical agent and the radiation overlap with no major gain.1 The addition of a chemical modifier to a course of radiation to improve treatment results should be considered only in those tumor sites where there is already evidence that an increase in dose intensity by 20% to 30% will translate into an increase in tumor control, because most of the sensitizer enhancement ratios (SERs) are in the range of 1.2 to 1.3 (a 20% to 30% increase in the effective dose to the tumor). Examples of such tumors are head and neck tumors and carcinoma of the cervix, in which it has been demonstrated that there is a high fraction of hypoxic tumor cells and also a rapid cell turnover.2,3
Chemical Radiosensitizers of Hypoxic Cells and the Tumor Microenvironment
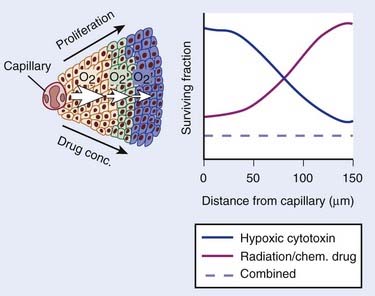
The role of the oxygen effect in promoting tumor cell inactivation by ionizing radiation has been well demonstrated in vitro and in animal tumor models for several decades. Mounting evidence of this influence in tumor control and patient survival has become available.2–7 Clearly, the microenvironment, the nutritional state of the tumor, and the presence of hypoxia are only some of the many factors that contribute to tumor radioresistance, and it is likely that this resistance is multifactorial, that is, caused by tumor hypoxia, proliferation rates, and inherent cell radioresistance, as well as other biologic microenvironmental factors such as the presence of cytokines. Nonetheless, the clinical data that hypoxia, by conferring radiation resistance, is a prognostic indicator of poor response to standard radiotherapy, at least for some tumors, is reasonably clear. Hypoxia, however, has other consequences beyond conferring radiation resistance. First, hypoxia causes cells to slow their rate of proliferation and to come out of cycle. Because most anticancer drugs are more effective against rapidly proliferating than slowly or nonproliferating cells, this slowing of cell proliferation leads to decreased cell killing in the hypoxic cells. In addition, because the concentration of anticancer drugs is higher closer to blood vessels than further away, both as a consequence of geometry and the reactivity of the drugs, there is less killing of the hypoxic cells, which are invariably the farthest from the blood vessels.
In summary, there are a number of well-established phenomena that cause a gradient of reduced cell killing by most anticancer agents as a function of distance from the vasculature (Fig. 4-1). Such a gradient has been shown in experimental tumors and in spheroid systems.8,9
Recent studies have also shown that hypoxia in solid tumors has an important consequence in addition to conferring a direct resistance to radiation and chemotherapy. Graeber and colleagues10 have demonstrated that low oxygen levels cause apoptosis in minimally transformed mouse embryo fibroblasts, and by selecting for mutant p53, might predispose tumors to a more malignant phenotype. Clinical data support this conclusion: Studies both with soft tissue sarcomas5 and with carcinoma of the cervix11,12 have shown that hypoxia is an independent and highly significant prognostic factor predisposing tumors to metastatic spread. Thus a more hypoxic tumor, in addition to being more difficult to control locally, is also more likely to have spread to distant sites and hence be more difficult to cure. In addition, hypoxia stabilizes the transcription factor HIF-1α, which increases levels of various survival factors such as vascular endothelial growth factor, which can protect against radiation damage to the tumor vasculature and hence, at least theoretically, protect the tumor from radiation damage.13,14
The rationale for developing hypoxic cell sensitizers has been based on the assumption that sensitizing hypoxic cells to radiation killing would improve the outcome of radiotherapy. The possibility of doing this was based on pioneering studies by Adams and Chapman and colleagues on the use of electron-affinic drugs to sensitize hypoxic bacteria and mammalian cells in vitro.15–17 The first drug of this class to show significant activity in sensitizing mouse tumors was the 5-nitroimidazole metronidazole, a drug that was already in clinical use.18,19 Data on this and other such compounds are discussed in the following text.
Nitroimidazole Compounds
Under hypoxic tissue conditions, electron-affinic nitroimidazoles oxidize the radiation-induced free radicals on deoxyribonucleic acid (DNA), thereby mimicking oxygen for the fixation of DNA damage. However, unlike oxygen, nitroimidazoles are not rapidly metabolized by the cells through which they penetrate and are thus able to reach areas beyond the oxygen-diffusion distance. They were shown by various groups to be effective in preclinical studies with transplanted mouse tumors in radiosensitizing the tumors to large single radiation doses without sensitizing normal tissues.20,21
The first compound to be investigated clinically in terms of oral and intravenous pharmacokinetics, toxicity, and efficacy in patients with solid tumors was metronidazole (a 5-nitroimidazole). The plasma β-half-life of the drug was 9.8 hours and the absolute oral availability was estimated to approximate 100%.19 The dose-limiting toxicity was manifested in gastrointestinal and peripheral neuropathic effects; therefore, this compound reached an estimated SER of only 1.2. Given this limitation, the first randomized control clinical trial of a hypoxic radiosensitizer showing efficacy was performed in patients with glioblastoma multiforme. This study demonstrated the relevance of tumor hypoxia in terms of patient survival, showing that the results with a less-than-optimal radiation fractionation regimen approached the level of the results obtained with conventional fractionation.22
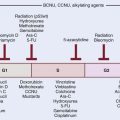
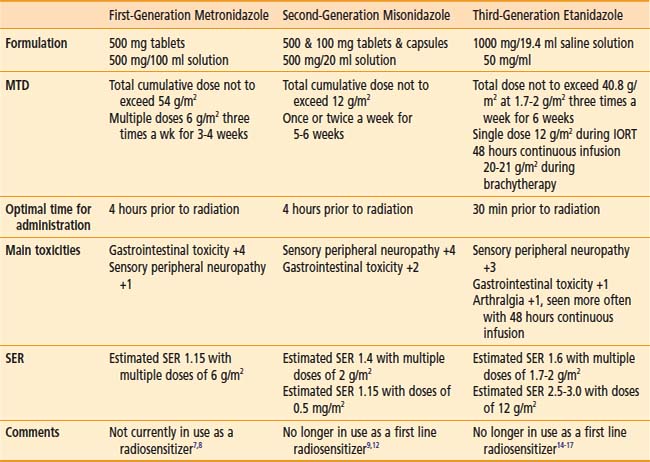
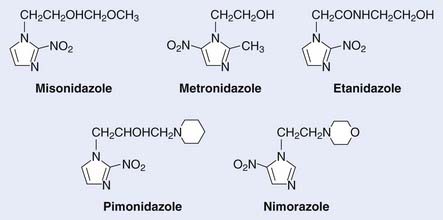
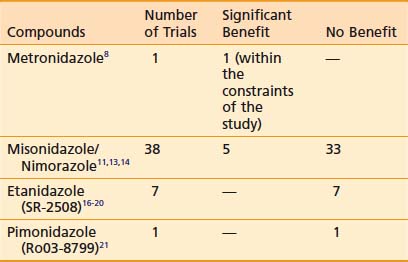
Of interest, a major spin-off of these early investigations with the pharmacokinetics of metronidazole, particularly the use of a high-dose intravenous route, was not in the field of oncology but in the practice of abdominal surgery and infectious diseases, in which the compound was used as a parenteral agent for anaerobic bacteria. These initial investigations prompted a level of high activity in the investigation of nitroimidazole compounds in human solid tumors and the search for new and better nitroimidazole compounds with less toxicity and higher SER. The first clinical studies on the second generation of these drugs, misonidazole, a 2-nitroimidazole, were initiated in both Europe and North America during the early 1980s (Table 4-1). The structures of the hypoxic sensitizers discussed are shown in Fig. 4-2.
Table 4-1 The Evaluation from First- to Third-Generation Nitroimidazole Compounds During the Past 25 Years
Misonidazole and Nimorazole
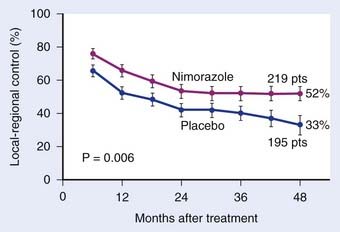
Misonidazole, a 2-nitroimidazole, was developed as a more efficient radiosensitizer because of its known increased electron affinity. In the oral form, the dose-limiting toxicity was again manifested in gastrointestinal effects (nausea, vomiting) and peripheral neuropathy, which limits the effective SER.23,24 Not surprisingly, almost all of the clinical trials of radiotherapy combined with misonidazole turned out to be negative,25 an outcome consistent with the small degree of radiosensitization expected with the clinically used low doses.26 Once more the inability to deliver a sufficient dose may have been one of the reasons that a significant proportion of large international clinical trials showed no benefit to misonidazole (Table 4-2). However, it has been shown that selected populations of patients with specific tumors did benefit by using this compound.27 This observation was made in patients with head and neck cancers. Overgaard and colleagues28 reported that not only with misonidazole but also with nimorazole (a 5-nitroimidazole), there was a significant benefit in a cohort of patients with stage T1 and T4 pharynx carcinoma in terms of local regional control (Fig. 4-3). Patients with hemoglobin levels lower than 9 mmol/L showed particular improvement. These two studies also showed that the compounds had a similar benefit, despite the fact that fewer fractions of radiation were “sensitized” in the misonidazole groups than in the nimorazole groups, in which the drug was administrated with every fraction of radiation (first 30 fractions). The significant improvement of the effect of the radiotherapeutic management of supraglottic and pharynx tumors was again demonstrated in a randomized double-blind phase III study of 414 patients receiving nimorazole or placebo in association with conventional primary radiotherapy (62-68 Gy, 2 Gy per fraction, 5 fractions per week).28 Of interest is that in this study the nimorazole could be given without major side effects. Nimorazole is currently standard treatment for patients in Denmark receiving radiation therapy for head and neck cancer.29 The benefit of misonidazole was not observed in another large clinical trial conducted in North America under the Radiation Therapy Oncology Group (RTOG) in patients with stage III and IV head and neck tumors, in which misonidazole was used with two of the five radiation fractions per week. In this study, the administration of the radiosensitizer was limited by neurologic complications, and the use of efficient doses was prevented.30 A prospective randomized trial was initiated afterward in the same cooperative group with a newer third-generation compound, etanidazole. A significantly greater dose of etanidazole than misonidazole could be administered for a sensitizer effect with acceptable gastrointestinal and neurologic toxicity.31
Etanidazole
Etanidazole (originally known as SR2508) was developed by a team led by Brown and Lee with the aim of reducing the neurotoxicity seen with misonidazole.31 It was postulated that because etanidazole is less lipophilic, it would be associated with a lower incidence of neuropathies. This was confirmed in a phase I pharmacokinetic and toxicity study in which doses three times higher than with misonidazole were delivered and fewer peripheral neuropathies were observed.31
Based on the encouraging results of the Danish Group in pharynx carcinoma with misonidazole and nimorazole, the RTOG initiated a two-part study in advanced head and neck cancer. The first part, a toxicity and logistic study in which etanidazole was used three times a week for 17 doses in combination with standard radiation, was completed with acceptable toxicity (although the 22% incidence of peripheral neuropathies seen in that study still presented a problem). Subsequently, a phase III study in a similar patient population and with the same frequency of sensitizer administration was completed in 1992. The results showed that adding etanidazole to conventional radiotherapy produced no benefit for patients with advanced head and neck carcinomas, except for a suggestion of benefit in a subset of patients with early stages (N0-N1 disease).32 A randomized study of 374 patients from 27 European centers, conducted between 1987 and 1990, adding etanidazole to conventional radiotherapy did not afford any benefit for patients with head and neck carcinoma. Furthermore the study failed to confirm the hypothesis of benefit for patients with early disease.33
A similar study in patients with locally advanced cancer of the prostate—T2b, T3, and T4—was also initiated in North America (RTOG). This study was designed to deliver the sensitizer with as many fractions of conventional radiation as possible. Nineteen doses of 1.8 g/m2 were delivered three times a week during the course of radiation, with tolerable toxicity. The results of this trial with regard to prostatic-specific antigen response and clinical disappearance of tumor are similar to those of historical control subjects, and are not considered to represent an improvement.34
Evidence exists that prolonged exposure to severe hypoxia can lead to increased sensitization beyond the oxygen effect, owing to the formation of reactive reduced metabolic species. Based on this evidence, an evaluation was made of etanidazole administered in 48-hour and 96-hour continuous intravenous infusions to patients undergoing brachytherapy.35 The use of etanidazole under these particular conditions is considered worth pursing.
Hypoxia marker studies (see the following text) that used 3H-misonidazole or 123I-iodoazomycin arabinoside showed evidence of tumor hypoxia in more than 50% of patients with small cell carcinoma (SCC) of the lung and indicated that tumor hypoxia could be one of the causes of chemoresistance and radioresistance in these patients. Therefore, a phase I and II clinical prospective study was initiated in patients with limited stage of small cell lung cancer, in which etanidazole was given in doses of 1.7 g/m2 three times a week with concomitant chemotherapy and thoracic irradiation. In patients with limited-stage disease, the median and the crude rates of survival at 5 years with no evidence of disease were superior to the best results reported in the literature from similar radiotherapy and chemotherapy regimens in which etanidazole was not used.36
Pimonidazole (a 2-nitroimidazole) was developed in Europe at the same time that etanidazole was developed as a third-generation sensitizer, and was considered to be more potent than misonidazole because of its potential to be concentrated in tumors as a result of its pH-dependent sidechain charge. The maximum tolerated dose, when administered with a daily 20-fraction course of radiotherapy, was established at 750 mg/m2. The dose-limiting toxicity has been in the central nervous system, manifesting as disorientation and malaise. A randomized clinical trial in advanced carcinoma of the cervix was conducted by 16 centers in Western Europe under the guidance of the Medical Research Council of the UK. Patient accrual was completed in May 1989. Overall and disease-free survival rates were found to be poor among the patients who received pimonidazole in combination with external radiation.37
Conclusions
The current status after  decades of clinical investigations with hypoxic cell sensitizers can be summarized by stating that these compounds have not become part of the standard practice of radiotherapy, at least in North America. In Denmark, however all head and neck cancer patients routinely are given nimorazole based on the positive phase III randomized trial of this drug with conventional radiotherapy.28 The only patients who appear to have benefited significantly from this approach are those with advanced pharyngeal and supraglottic carcinomas treated with radiation and either misonidazole or nimorazole. In the immediate future and with the advent of noninvasive markers of tumor hypoxia, it should be possible to select patients for the use of these agents. In addition, the increased use of stereotactic body radiotherapy is likely to make these compounds more attractive in the future because hypoxia is a bigger problem for large single or a limited number of large radiation doses and the individual doses of the particular sensitizers that can be delivered will be higher.
decades of clinical investigations with hypoxic cell sensitizers can be summarized by stating that these compounds have not become part of the standard practice of radiotherapy, at least in North America. In Denmark, however all head and neck cancer patients routinely are given nimorazole based on the positive phase III randomized trial of this drug with conventional radiotherapy.28 The only patients who appear to have benefited significantly from this approach are those with advanced pharyngeal and supraglottic carcinomas treated with radiation and either misonidazole or nimorazole. In the immediate future and with the advent of noninvasive markers of tumor hypoxia, it should be possible to select patients for the use of these agents. In addition, the increased use of stereotactic body radiotherapy is likely to make these compounds more attractive in the future because hypoxia is a bigger problem for large single or a limited number of large radiation doses and the individual doses of the particular sensitizers that can be delivered will be higher.
Possible Explanation for the Inconclusive Therapeutic Benefits Seen With Nitroimidazoles in Most Solid Tumors
Of interest is an updated review of all randomized clinical trials on approximately 10,108 patients combining tumor hypoxia modifiers (hyperbaric and normobaric oxygen plus carbogen and hypoxic radiosensitizers) with curative radiation, showing an improved effect of radiotherapy on local-regional control, without evidence of increase of radiation side effects in normal tissues.40
Current Status and Future Directions in Tumor Hypoxia
Microinvasive Methods of Measuring Tumor Hypoxia
One possible interpretation of the clinical sensitizer trials is that hypoxia is less important for the therapy of human tumors than for murine tumors and other model systems. Arguing against this is a series of studies using a newly developed semiautomatic needle electrode (Eppendorf Histograph).41–44 These studies not only confirm the radiation resistance of hypoxic human tumors, but demonstrate their more “aggressive” phenotype for other types of treatment failure (chemotherapy–resistance, metastasis, surgical). The overall “aggressiveness” of hypoxic tumors determined by this pioneering work is now much better understood since the discovery and subsequent elucidation of the central role played by hypoxia-inducible factor-1 (HIF-1) in many aspects of tumor behavior.45 The resolution of the Eppendorf system is roughly 0.7 mm and several assays capable of much finer resolution have been developed for immunohistochemical analysis.
Some of these microassays involve endogenous molecular changes (often HIF-mediated46) or inherent radiation resistance.47 The most extensively developed microscopic assay of hypoxia, also central to the development of noninvasive assays (see the following text), was based on the finding that 2-nitroimidazole metabolism led to the hypoxia-dependent formation of adducts between the metabolized drug (activated by nitro-reduction) and cellular macromolecules, later shown to be primarily thiol-containing proteins.48,49 Chapman first proposed that this metabolism could be used for the practical measurement of tissue hypoxia, and one resulting technique (autoradiography after excision of the labeled tumor) progressed to a clinical trial.50 The measurement of tritiated misonidazole metabolism by autoradiography was problematic for a general-use assay, but this problem was solved through the development of antibody-based assays for the detection of 2-nitroimidazole adducts, particularly using pimonidazole or EF5 as the hypoxia-detecting agent.51,52
One endogenous hypoxia marker currently capable of predicting outcome is carbonic anhydrase IX (CA-IX). CA-IX is one of several carbonic anhydrase enzymes, and is strongly upregulated under hypoxic conditions through the HIF-1α mechanism. CA-IX is expressed on the surface of cells and therefore can be detected by antibodies. However, its correlation with gold-standard measures of hypoxia is suboptimal.53
Noninvasive Methods of Measuring Tumor Hypoxia
Determination of therapy-relevant tumor hypoxia using nuclear medicine techniques holds many potential benefits, including timely stratification of treatment, prediction of outcome, and even the spatial optimization of radiation therapies using image-guided radiation therapy (IGRT), intensity-modulated radiation therapy (IMRT), and protons.54 It has become clear that the first of these is much more important than previously considered—that is, heterogeneity of the extent and degree of hypoxia between tumors or patients with otherwise similar characteristics can ultimately stifle the development and interpretation of hypoxia-specific therapies, resulting in the waste of clinical trial resources on patient cohorts for which the hypoxia-specific therapy is inappropriate. With the exception of Copper(II)-diacetyl-bis(N4-methylthiosemicarbazone) (Cu-ATSM) and the antibody developed against CA-IX, most agents being considered for the noninvasive detection of hypoxia are also 2-nitroimidazoles. Thus, it is fitting that the first agent developed for noninvasive imaging of hypoxia [18F]fluoromisonidazole (18F-FMISO) was first used to demonstrate the predictive value of the first clinically developed hypoxic cell cytotoxin (tirapazamine [TPZ]).55,56 Nevertheless, this delay in the successful application of 18F-FMISO imaging has resulted in the extensive development of other possible noninvasive hypoxia markers.
Half-Life of Isotope versus Drug
Chapman and colleagues17 suggested that the relatively short isotope half-life of 18F (≈110 minutes) was suboptimal in detecting tumor hypoxia because, first, this does not allow adequate clearance (pharmacologic half-life of drug) of nonmetabolized drug (which forms the image background), and second, binding to hypoxic cells should increase with time, so that inherent limitations exist in generating optimal tumor versus normal tissue contrast during the relatively short imaging times suitable for 18F.57 Use of iodine as the detecting isotope was considered superior because of its variety of half-lives and decay types. However, directly iodinating the imidazole ring of misonidazole did not lead to suitably stable compounds, so a variety of nucleoside derivatives were devised, with the iodine conjugated to the sugar moiety rather than the 2-nitroimidazole. These compounds (exemplified by iodoazomycin arabinoside [IAZA] and iodoazomycin galactopyranose [IAZGP]) were also more polar than FMISO (see next paragraph). Both single photon emission computed tomography (SPECT) (138I) and positron emission tomography (PET) (139I) isotopes have been used to evaluate the utility of these drugs. Clinical trials of IAZA successfully imaged hypoxia but because of deiodination this agent was not considered optimal.57 IAZGP is under current investigation clinically at the Memorial Sloan Kettering Institute. Although the positive aspects of longer-lived isotopes seem clear, there are also negative aspects that must be mentioned. First, to provide a suitable image quality, the number of counts and imaging time must be specified. For these to be equivalent for long- versus short-lived isotopes, the former must necessarily cause a substantially higher total radiation dose burden to the patient. This is exaggerated for many PET isotopes (e.g., Cu and I), because the fraction of positron emissions per decay can be much less than one. This problem cannot be resolved by use of SPECT isotopes, because, other factors being equal, SPECT imaging has an inherently lower resolution than does PET imaging. Ultimate resolution of PET depends on the energy spectrum of the positron, and 18F is optimal in this respect.
Drug Polarity (Octanol-to-Water Partition Coefficient)
Because of the potential high resolution and low patient radiation dose from 18F, several other fluorinated drugs have been developed. Some of these compounds have been significantly more polar than FMISO. The goal of minimizing neurotoxicity seems “off-target” for the use of noninvasive imaging agents, because their concentration is orders of magnitude lower than was used in the sensitizer trials. Additionally, there are data suggesting that highly polar nitroimidazoles do not achieve suitable access to brain and possibly other tissues.58 Thus, it is possible that polar imaging agents will have to be evaluated much more thoroughly with respect to their biodistribution and ability to diffuse into actively metabolizing tissue.
Drug Biodistribution and Stability
EF5’s design was based on specific properties of oxygen dependence of bioreduction, drug stability, and biodistribution determined in former pharmacologic and biochemical studies. It was hypothesized that emphasis on the uniform biodistribution allowed by a lipophilic molecule, in effect minimizing excretion, might allow relatively low hypoxia-dependent bioreduction to be detected above the uniform background of the parent drug.59 These properties were validated at the relatively high concentration used for immunohistochemistry (≈100 µm—using monoclonal antibodies against EF5 adducts) and appear to be maintained at approximately 1000-fold lower drug concentrations used for PET.60 Recently, two other 18F-containing hypoxia markers have been tested in humans: [18F]fluoroazomycin arabinoside,61 the fluorinated equivalent of IAZA; and EF3,62 a tri-fluoro analog of EF5. These drugs are somewhat more lipophilic than FMISO, with octanol-to-water partition coefficients close to 1.
Several imaging agents have been found to concentrate in tumors, but their precise mechanism binding is not fully understood. The best characterized of these is Cu-ATSM. ATSM is a copper chelator related to the perfusion marker copper pyruvaldehyde bis (N4-methylthiosemicarbazone).63 ATSM is very lipophilic but unexpectedly shows rapid biodistribution and binding. Like iodine, copper has a number of radioactive isotopes with selectable properties. Thus this hypoxia marker has many appealing properties.
Manipulation of the Tumor Microenvironment
Increasing the Oxygen-Carrying Capacity of Blood: Hyperbaric Oxygen and Fluosol
Clinical studies have been completed in which hyperbaric oxygen,64 carbogen,65 packed red cell transfusions,66,67 and oxygen-carrier substances such as perfluorocarbons (Fluosol-DA) have being used.68,69 Although the use of the hyperbaric oxygen chamber at three atmospheres presented technical difficulties, such as barotrauma and the limitations of its use to only a few high-dose fractions of radiation, 3 out of 10 clinical trials showed significant positive results. The beneficial effect was seen particularly in patients with advanced cancer of the head and neck and of the cervix.70
A phase II study of Fluosol-DA and 100% oxygen in combination with radiotherapy in advanced head and neck tumors has shown promising results.68,69 Investigators have also been assessing the compound 2-(4-[{3,5-dimethylanilino}carbonyl]methyl]phenoxy)-2-methylproprionic acid derivative, an allosteric hemoglobin modifier, and preliminary reports are encouraging.71
An alternative approach is to increase the level of hypoxia in tumor cells and to treat them with hypoxic cytotoxic agents. Attempts have been made in the past to reduce tumor perfusion by the use of agents like hydralazine. This has not proved to be of value because of the potential systemic side effects. Another approach to increase the level of tumor hypoxia by modifying the oxyhemoglobin disassociation curve with a specific chemical agent such as 5-(2-formyl-3-hydroxyphenoxy) pentanoic acid, combining this approach with the hypoxic cytotoxic agent, mitomycin C. This approach was investigated in patients with advanced gastrointestinal cancer.72
Increasing Tumor Blood Flow: Nicotinamide and Carbogen/Arcon
Improvement in tumor PO2 following carbogen breathing (95% O2, 5% CO2) has been shown in both animal and human tumors. Tumor tissue oxygenation was measured in humans with the polarographic electrode system (Eppendorf) PO2 histograph. In one study, in 12 out of 17 patients with solid tumors there was a significant increase in median tumor PO2 during the first 10 minutes of carbogen breathing. Measurements were taken in accessible superficial tumors; 15 were epithelial tumors (most of them breast and lung carcinomas) and 2 were soft tissue sarcomas.73
It is hypothesized that nicotinamide decreases the presence of acute intermittent hypoxia and that carbogen breathing reoxygenates the chronic hypoxic cells.74,75 The benefit of this combination could be further enhanced in tumors with rapidly proliferating stem cells if accelerated radiotherapy schedules are used. This approach, accelerated radiotherapy with carbogen, nicotinamide (ARCON), was initiated originally in a pilot study in the United Kingdom. A multiple-institution ARCON phase I trial was conducted under the European Organization for Research and Treatment of Cancer. In this three-step study, 115 patients with glioblastoma multiforme were registered. The overall survival was not different when compared with results of other series using radiotherapy alone.76 Nicotinamide produced gastrointestinal toxicity, necessitating dose reduction. Two other phase I and II clinical trials using ARCON in non-small cell lung cancer (NSCLC)77 and in 215 patients with advanced head and neck squamous cell carcinoma78,79 were conducted recently with encouraging results in regard to tumor responses, but continued to require a reduction in the dose of nicotinamide because of the incidence of gastrointestinal acute toxicity. The use of ARCON in advanced bladder cancer has been studied to assess its potential gain, with encouraging results and no overt increase in normal tissue radiosensitivity.80
The use of carbogen (without nicotinamide) has recently been explored in a randomized study comparing definitive hyperfractionated radiation therapy to the same radiation plus carbogen in T2-T4 head and neck tumors. This study did not appear to improve the local tumor control.81 However, another trial using carbogen breathing combined with radical radiotherapy also in advanced head and neck cancer patients suggested that carbogen breathing may be an alternative form for patients who are unfit to receive concomitant chemotherapy with the radiation treatments.82
Hypoxic Cytotoxins
Tirapazamine
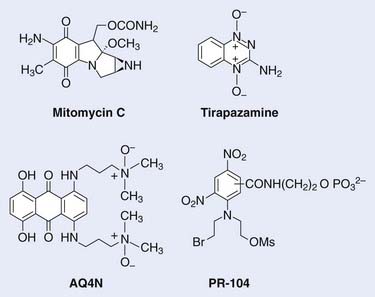
The development of hypoxic radiosensitizers led to that of hypoxic cytotoxins, also known as bioreductive drugs. These are agents that can sensitize a solid tumor to radiotherapy or to chemotherapy by killing, rather than sensitizing, the resistant hypoxic cells. Hypoxic cells in tumors are not only resistant to radiation, they are also resistant to most anticancer drugs. This is because hypoxic cells, by definition, must be those farthest from functioning blood vessels, and also because cells at low oxygen levels divide much less rapidly than when fully oxygenated. These two factors lead to resistance to anticancer drugs, first because the majority of anticancer drugs are only effective against rapidly proliferating cells, and second because drugs have to reach all tumor cells regardless of their distance from blood vessels. Thus hypoxic cytotoxins are fundamentally different from conventional agents in that they target a different subpopulation of cells within the tumor. Typically, hypoxic cytotoxins have maximum cytotoxicity to the cells at maximum distance from tumor blood vessels, thereby complementing the pattern of cytotoxicity for both radiation and anticancer drugs, which is maximum for the cells immediately adjacent to the blood vessels (see Fig. 4-1). Thus these agents have the potential of overcoming a major cause of resistance of solid tumors to conventional therapies, namely that resulting from the inadequate oxygenation and drug delivery to tumor cells distant to blood vessels. The structures of the hypoxic cytotoxins discussed in this Chapter are shown in Fig. 4-4.
Mitomycin C
Mitomycin C, a quinone antibiotic that requires reductive metabolism for activity, is the prototype bioreductive agent. Introduced into clinical use in 1958, mitomycin C has demonstrated activity toward a number of different tumors in combination with other chemotherapeutic drugs and radiation. Sartorelli and colleagues suggested that the lower oxidation reduction (redox) potential of tumor relative to normal tissue might be exploited to obtain greater activation of this compound to its cytotoxic species.83 Although tumor redox potential did not turn out to be key for the activity of mitomycin C, Sartorelli and Rockwell were able to show that this drug preferentially kills hypoxic rather than aerobic cells in vitro.84 However, the differential toxicity is modest: The ratio of drug concentrations under aerobic to hypoxic conditions for the same level of cell kill (hypoxic cytotoxicity ratio [HCR]) is in the range of 1 (no differential) to 5.85 Nonetheless, this can be sufficient to overcome the resistance of hypoxic cells in animal tumors, and clinical trials have reported higher cure rates for head and neck cancers by adding mitomycin C to radiotherapy compared with radiotherapy alone,86 although because mitomycin C is a chemotherapy drug with toxicity toward all cells, it is not clear whether the improved cure rates over radiotherapy alone were the result of selective killing of hypoxic cells.
Tirapazamine
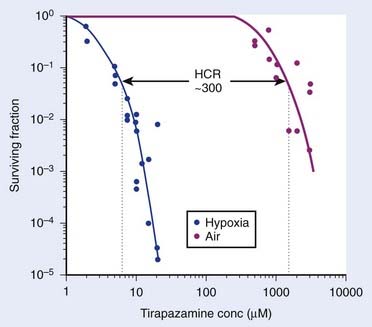
A group led by Brown and Lee introduced a third class of bioreductive drugs in 1975. The compound introduced, SR 4233, now known as tirapazamine (TPZ), a benzotriazene di-N-oxide, had an HCR of 50 to 300 for different cell lines87 (Fig. 4-5), and (unlike the classic hypoxic radiosensitizers) is active when combined with fractionated radiation at doses comparable with those used clinically.88 The mechanism for the selective toxicity of TPZ (and other members of this class) toward hypoxic cells is that the drug is reduced (an electron is added) by intracellular reductases to form a highly reactive radical that produces both single- and double-strand breaks in DNA that result in cell death, although the exact mechanism of this is quite complex.89,90 However, under aerobic conditions, oxygen removes the electron from the TPZ radical, thereby back-oxidizing it to the nontoxic parent with a concomitant production of superoxide radical. Thus the differential hypoxic cytotoxicity results from the fact that the TPZ radical is much more cytotoxic than the superoxide radical. In addition to its toxicity to hypoxic cells, TPZ was shown to be remarkably efficient at enhancing the cytotoxicity of some chemotherapeutic agents, notably cisplatin (CIS), in experimental animal tumors.91 Following favorable results in phase I and II studies with the combination of CIS and TPZ, a phase III, multicenter, randomized clinical trial with TPZ combined with CIS in patients with advanced NSCLC showed a doubling of the overall response when TPZ was combined with CIS compared with CIS only and a significant increase in the median survival time of the patients.92 This increase of antitumor activity occurred without any evidence of increased systemic toxicity of the anticancer drug CIS, as was also seen in experimental animal systems. Promising results of phase I and II trials of TPZ combined with both CIS and fractionated irradiation have been reported for cervix and ovarian cancer93–95 and for head and neck cancer.96 Currently, there are phase III trials underway with TPZ combined either with chemotherapy in NSCLC or with radiotherapy and CIS in head and neck cancer, and the results are awaited.96 However, the results of the large (861-patient) multicenter phase III trial of SCC were recently reported at the 2008 American Society of Clinical Oncology (ASCO) meeting.97 Patients with previously untreated stage III or IV SCC of the oral cavity, oropharynx, hypopharynx, or larynx were randomized to receive definitive radiotherapy (70 Gy in 7 weeks) concurrently with either CIS (100 mg/m2) on day 1 of weeks 1, 4, and 7; or CIS (75 mg/m2) plus TPZ (290 mg/m2/day) on day 1 of weeks 1, 4, and 7, and TPZ alone (160 mg/m2/day) on days 1, 3, and 5 of weeks 2 and 3 (CIS/TPZ). There were no significant differences in failure-free survival or time to locoregional failure (LRF) between the two groups. Interestingly, however, 20% of patients were found to have major deviations in the radiotherapy plan, which was associated with an increased risk of death (hazard ratio [HR] = 1.56; p = <0.0001), and LRF (HR = 1.82; p = 0.0002), and there was a trend of a benefit with the addition of TPZ in patients without major radiotherapy deviations, HR for risk of LRF (CIS/TPZ:CIS) 0.74, 95% CI: 0.53 to 1.04. As well, there is an ongoing phase III study under the Gynecological Oncology Group (GOG) studying the role of TPZ in combination with CIS and radiation in the management of patients with advanced cervical cancer. One of the drawbacks to the use of TPZ is muscle and gastrointestinal toxicities,95,96 although with the chemoradiotherapy studies, myelotoxicity is the dose-limiting toxicity. TPZ remains the most widely studied hypoxic cytotoxin at this time. One of the major conclusions from the TPZ trials is the importance of selection of those patients with hypoxic tumors for trials with hypoxic cytotoxins. This was not done and there is evidence that in patients with hypoxic tumors there is a substantial benefit of the addition of the drug.98
AQ4N (Banoxantrone)
The anthraquinone AQ4N was designed specifically as a hypoxia selective cytotoxin. It resembles TPZ in being a di-N-oxide, but has a distinct mechanism of activation and cytotoxicity. AQ4N is a prodrug of a potent DNA intercalator/topoisomerase poison, AQ4, which is formed by reduction of the two tertiary amine N-oxide groups that mask DNA binding in the prodrug form.99 AQ4N has substantial activity against hypoxic cells in a variety of transplanted tumors,100 and has recently completed phase I and II clinical trials with lymphomas and leukemias and a phase I and II trial in combination with radiotherapy and temozolomide, with glioblastoma multiforme in progress.
PR-104
PR-104 is a dinitrobenzamide mustard, developed by Wilson and colleagues at the University of Auckland, with considerable advantages over TPZ or AQ4N.101 The prototype for this class is the prodrug CB 1954, which first came to attention because of its dramatic curative activity against the Walker rat tumor.102 It was subsequently shown to be a bioreductive prodrug, activated within the tumors by rat DT-diaphorase, which reduces its 4-nitro group to the corresponding hydroxylamine, a potent DNA crosslinking agent.103,104
PR-104 is a phosphate ester that is in effect a “pre-prodrug”; systemic or tumor phosphatases generate the corresponding alcohol (prodrugs), which is subsequently activated by reduction of one or more of the nitro groups by nitroreductases, including one-electron reductases, to produce hypoxia-selective cytotoxicity by virtue of its ability to form DNA interstrand crosslinks.105
Modifiers of Hemoglobin Levels
Erythropoietin
Erythropoietin is a growth factor that has been synthesized in the laboratory. It has shown efficacy in the treatment of anemia related to systemic chemotherapy,106 as well as in combined chemoradiation. A significant increase in hemoglobin levels compared with controls has been shown in patients receiving radiotherapy.107,108 These studies did not address the question of whether the increase in hemoglobin levels seen when administering erythropoietin results also in improvement in local tumor control. A randomized phase III trial to assess the effect of erythropoietin on local-regional control in anemic patients treated with radiotherapy for advanced carcinoma of the head and neck was initiated by the RTOG. This study was terminated before completion of patient accrual because of negative results. Further, a review of all phase III studies have shown mixed results, with some studies reporting a decrease in patient survival despite an improvement in hemoglobin levels.109–111 One phase III study evaluating erythropoietin in cervical cancer closed prematurely because of potential concerns with thromboembolic events with the use of this compound. The tumor recurrence status between treatment regimens were not statistically significant (GOG 191).112 Also, similar results, with no improvement in tumor control, have been reported on head and neck cancer.113
Glutathione Depletion
Sulfhydryls are scavengers of free radicals, protecting chemical damage induced by either ionizing radiation or alkylating agents. It has been postulated and demonstrated in the laboratory that one approach to increasing the efficacy of the nitroimidazoles as sensitizers is to decrease the levels of the competing endogenous sulfhydryls. Glutathione is one of the major endogenous sulfhydryls. Buthionine sulfoximine was developed as a specific inhibitor of glutathione synthesis. It has been shown to deplete glutathione levels in both in vitro and in vivo systems, therefore making misonidazole a more effective sensitizer. Earlier studies in laboratory experiments showed little or no increase in the toxicity of misonidazole in normal tissue, but showed an increased sensitizing efficacy in the tumor tissue.114
The use of buthionine sulfoximine as a modulator for hypoxic cell sensitizers in combination with radiation and as a modulator of chemotherapeutic drugs such as alkylating agents and bioreductive hypoxic cytotoxics has been of interest in the laboratory.115–118
An Alternative to Nitroimidazole Hypoxic Cell Radiosensitizers
Nitric Oxide
The hypoxic cell radiosensitization properties and vasodilator effects on tumor vasculature of nitric oxide gas have been described, and there is continued laboratory interest on the possible practical therapeutic use of nitric oxide–releasing compounds (NONOates) under specific physiologic conditions. Studies done in the laboratory under in vitro conditions has shown a marked radiosensitizer effect under hypoxic conditions.119 Further studies are being conducted in in vivo models. For the time being, this approach is still limited to the laboratory level.
Nonhypoxic Cell Sensitizers
The cancer chemotherapy agents hydroxyurea and 5-fluorouracil are not discussed in this chapter. The reader is referred to Chapter 6 for description of these agents as possible nonhypoxia-specific cell radiosensitizers.
Importance of Cell Labeling and DNA Incorporation
Clinical Investigations
The means of achieving an optimal incorporation of these compounds in the cell in the clinical situation has been extensively explored over the years, particularly the route of administration and length of drug exposure. Early on, BUDR was used intra-arterially both to avoid dehalogenation by the liver and to increase the drug tumor concentration.120 However, the necessary prolonged use of this route in patients over several weeks was laborious and had a high incidence of complications. Although there have been reports of rapid debromination of halopyrimidines occurring after intravenous therapy, Goffinet and Brown121 showed that following intravenous infusion, enough halopyrimidine apparently passes through the hepatic vessels to permit tumor radiosensitization, despite dilution of the drug by the systemic circulation. This has also been shown in human studies. There was a renewed interest in the 1980s and 1990s in the use of continuous intravenous infusion of halopyrimidines. It was observed that adequate steady-state arterial plasma levels could be maintained with this route of administration with acceptable systemic toxicities.122 A large study was reported by the Northern California Oncology Group in 160 patients with glioblastoma treated with 96-hour infusion of BrUdR at 800 mg/m2 a day for a total of 6 weeks, in combination with 60 Gy irradiation directed to tumor plus a margin. The patients in this series received chemotherapy with procarbazine, lomustine (CCNU), and vincristine (PCV) for 1 year following radiotherapy. The median survival time was 12.8 months. Patients with anaplastic astrocytoma had a median survival time of almost 5 years, and the observation was made that the use of pyrimidine analogues in combination with radiation may be of greater benefit in this group of patients.123 However a randomized study in anaplastic astrocytoma conducted by the RTOG using radiation and PCV chemotherapy compared with radiation and PCV plus BrUdR was terminated earlier because of the inferior time to tumor recurrence and survival observed in the arm using BrUdR.124
In addition, because of the toxicities and limited tumor efficacy, the use of this drug with radiation was not warranted in patients with glioblastoma.125
Chemical Radioprotectors
The first chemical radioprotector compounds intended for use on humans were developed for the purpose of protecting individuals from whole-body irradiation, such as in the event of nuclear warfare. The sulfhydryl compounds, including β-mercaptoethylamine and thiophosphates, were considered. An extensive drug developmental program was initiated by the U.S. Department of Defense (Walter Reed Army Research Institute [WR]). Out of 4000 screened compounds, the thiophosphate WR-2721 was the most promising. Clearly, these compounds were designed to protect all tissues, a very different requirement from their possible use in the field of oncology, in which protection of normal tissues to the exclusion of tumor tissues is essential to improve the therapeutic ratio. Initially, there were serious questions about whether these agents could also protect the tumor from the effects of radiation.126,127
The assumption that the tumor tissues are not protected to the same degree as normal tissues is based on the probability that there is poor drug penetration in tumors because of their poor blood perfusion, the partition coefficient of the drug, and the probability that there is a higher concentration of the drug in normal tissues because of their higher pH. In addition, for WR-2721 to be active, the phosphate group must be cleaved by the enzyme alkaline phosphatase to form the dephosphorylated free thiol WR-1065. This enzyme is not as abundant in tumor tissues as in normal tissues; therefore, the levels of alkaline phosphatase and pH in tissues determine the uptake of WR-2721. There is also a final assumption that thiol compounds could have less protective effect on hypoxic cells. The mechanisms of radioprotection fit the competition-model, dual-action theory. Once inside the cell, the active free thiol WR-1065 can chemically reduce free radicals. However because protectors will have maximum effect at intermediate oxygen levels, the possibility of tumor protection can occur presumably at levels of intermediate hypoxia. It also takes part in the repair reaction of DNA damage. Of interest in the field of cancer chemotherapy is the fact the dephosphorylated WR-2721 can bind to the active species of alkylating agents, as well as prevent the formation of CIS-DNA adducts.128
Biologic agents such as interleukin-1 have been shown to protect normal tissues in animal systems.129
Amifostine WR-2721 (Ethyol)
The dose-modifying factor (DMF) of amifostine WR-2127 for both normal and tumor tissues was studied in animals carrying solid tumors.130,131 For normal tissues, the greatest protection is found in bone marrow, with a DMF of 2.7 to 3, and in the gastrointestinal tract, with a DMF of 1.6. The lowest DMF, at 1.2, is in lung tissue. However, this drug also protects tumor tissues, with DMFs ranging from 1.3 for cure of EMT-6 carcinoma to 2.2 for mean survival time of P-388 leukemia. Once more, the degree of protection to tumors appears to be related to the tumor blood perfusion, degree of hypoxia, the tissue pH, and the levels of alkaline phosphatase. A cautionary note is that most of these experiments were performed with single-radiation doses. It is possible that the differential protective effect between normal and tumor tissues would be less if multiple, daily, small doses in combination with radiation were used.
Experimental work has also been done in which amifostine is used as a chemoprotector of normal tissues. Three studies were performed with different animal tumor models and all three showed protection of bone marrow and intestine with no protection of the tumor when amifostine was used with melphalan132 or in combined chemotherapy regimes with nitrogen mustard, cyclophosphamide, carmustine, CIS, and 5-fluorouracil.133,134
It should be noted that amifostine does not protect the central nervous system tissues from radiation effects because of the blood-brain barrier.135
Clinical Experience
Amifostine has been approved for clinical use and is available for intravenous route in a sterile lyophilized powder mixture with mannitol, requiring reconstitution for intravenous administration. It is administered over a 15-minute period prior to radiation or chemotherapy. Initial single-dose toxicity and pharmacokinetic studies were performed in 1983. The plasma initial-half-life is 9 minutes, and it is assumed that the protective concentrations are maintained in normal tissues for approximately 2 hours.136,137
The maximum tolerated dose for multiple doses is 340 mg/m2 given 4 days a week for 5 weeks, 15 minutes prior to external radiation. Toxicities at this dose level are manifested as nausea, vomiting, anorexia, malaise, transient moderate hypotension, and occasional hypocalcemia. It is recommended that the amifostine infusion be interrupted if there is a 25% decrease in systolic blood pressure. The maximum tolerated dose with single doses is 740 mg/m2, although recently the dose was increased to 910 mg/m2 given twice a week for at least five treatments, and this dose has been considered acceptable.138
Amifostine in combination with chemoradiation therapy for small cell lung cancer and NSCLC has been studied in several randomized phase III clinical trials, which have shown a statistically significant decrease in esophagitis and pneumonitis with no observed tumor protection.139–142 A meta-analysis of all published clinical trials (seven randomized involving 601 patients, with locally advanced NSCLC treated with radiotherapy with or without chemotherapy) revealed that amifostine has not protected the tumor from the therapeutic effect of either radiation or chemoradiation.143 However, amifostine toxicity consisting of hypotension, nausea, and vomiting with the use of the intravenous route prevented a rather large proportion of patients from receiving the full dose according to protocol.142 To avoid the amifostine toxicity seen with intravenous use, studies have been initiated to explore the subcutaneous administration based on previous phase I and II trials demonstrating a reduction of amifostine toxicities when using this route.144–147
Head and neck tumors treated with chemoradiotherapy is another area in which a large number of randomized phase III clinical trials were initiated over the past 7 years using amifostine as a radioprotector. Most of the reported clinical trials on this disease site have demonstrated a decreased incidence of xerostomia and mucositis, with no clear evidence of tumor protection as measured by local regional tumor control.148 A significant reduction in the incidence of radiation-induced xerostomia at 12 months posttreatment was reported with the use of amifostine in a multinational phase III trial of 315 patients with head and neck cancer.149 An update of this study at 18 and 24 months after initial treatment indicated that amifostine reduces the severity and duration of xerostomia 2 years after treatment and does not seem to compromise local-regional tumor control rates, progression-free survival, or overall survival.150 Amifostine-related nausea, vomiting, and hypotension at a dose of 200 mg IV daily with each radiation fraction led to discontinuation of the drug in approximately 20% of patients with, in some cases, a delay of radiotherapy.151
Early phase II efficacy trials have been initiated using amifostine during chemoradiotherapy of pelvic tumors in an attempt to protect rectal mucosa, indicating significant protective effect in patients with cancers of the prostate, cervix, and rectum using either the intravenous or subcutaneous routes as well as with the intrarectal aqueous solution in a 48-ml enema.152,153
Concerns about amifostine toxicity as well as the question of tumor tissue protection have been raised by some authors.154 However, contradicting this view, a review of all clinical studies done up to 2003 have clearly indicated no evidence of amifostine tumor protection.155
The practical application of amifostine in the standard practice of radiation oncology continues to be a controversial one. Up to 2008, the use of amifostine in the chemoradiotherapy of head and neck tumors to protect from xerostomia has been recommended by the ASCO and is approved by the U.S. Food and Drug Administration for (1) reduction of the incidence of moderate to severe xerostomia in patients undergoing postoperative radiation treatment for head and neck cancer, and (2) reduction in the cumulative renal toxicity associated with CIS in patients with advanced ovarian cancer.156,157 Recent observations of the efficacy of IMRT in reducing xerostomia have raised questions about the expense of using amifostine for this purpose.
Chemoprotection
The clinical effectiveness of amifostine as a chemoprotector was first assessed with the use of bone marrow as the normal tissue endpoint and cyclophosphamide as the chemotherapeutic agent. Definitive evidence of bone marrow protection was observed. Tumor protection was not assessed.158 In another study, renal damage and peripheral neuropathy were assessed as endpoints and CIS was used; normal tissue protection with no tumor protection was observed.159 Another clinical study demonstrated the protective effect of amifostine in bone marrow, kidney, and peripheral nerves when cyclophosphamide is used in combination with CIS.160 Protection of carboplatin myelotoxicity was observed in one study,161 but findings were inconclusive in another preliminary study.162 The use of amifostine in combination with chemotherapy and radiation is currently being used in clinical research protocols for both adult and pediatric patient populations.
Tempol
Nitroxide superoxide dismutase mimic (Tempol), a nonthio nitroxide, has been shown both in vitro and in vivo to be an effective radioprotector with the absences of tumor radioprotection. In addition, nitroxide compounds are known for their modest hypoxic cell radiosensitization. The potential of Tempol as an aerobic cell radioprotector has been described by Mitchell and colleagues.163
Conclusions on Radioprotectors and Chemoprotectors
A spin-off of the work on radioprotectors is manifested in the increasing interest in the use of these compounds as chemoprotectors in the field of medical oncology.164 The protective effects of amifostine have been seen in bone marrow during the use of alkylating and platinum compound agents. Phase III large clinical trials are ongoing to demonstrate conclusively normal tissue protection without tumor protection in esophageal mucosa, salivary glands, and bone marrow when these protectors are combined with radiation, and in kidney, peripheral nerves, and bone marrow when they are used with alkylating agents and platinum compounds.
An exciting evolving potential is in the combination of chemical protectors (amifostine or Tempol) with biologic stimulators (cytokines). Furthermore, the effect of thiol compounds might be even greater in the laboratory, based on their promising use as a probe in examining the mechanisms of radiation cell damage. It is likely that vital new information will be available in the near future on the molecular mechanisms of radiation protection.164
1 Steel GG. Combined radiotherapy-chemotherapy: principles. In: Steel GG, Adams GE, Horwich E, editors. The biological basis of radiotherapy. ed 2. Amsterdam: Elsevier; 1989:267.
2 Gatenby RA, Kessler HB, Rosenblum JS, et al. Oxygen distribution of squamous cell carcinoma metastases and its relationship to outcome of radiation therapy. Int J Radiat Oncol Biol Phys. 1988;14(5):831.
3 Hockel M, Knoop C, Schlenger K, et al. Intratumoral Po2 predicts the survival in advanced cancer of the uterine cervix: updated analysis of an open prospective clinical study. Radiother Oncol. 1992;26:45.
4 Bush RS, Jenkin RDT, Eltt WEC, et al. Definitive evidence for hypoxic cells influencing cure in cancer therapy. Br J Cancer. 1978;37(3):302.
5 Brizel DM, Scully SP, Harrelson JM, et al. Tumor oxygenation predicts for the likelihood of distant metastases in human soft tissue sarcoma. Cancer Res. 1996;56:941.
6 Nordsmark M, Overgaard M, Overgaard J. Pretreatment oxygenation predicts radiation response in advanced squamous cell carcinoma of the head and neck. Radiother Oncol. 1996;41:31.
7 Nordsmark M, Bentzen SM, Rudat V, et al. prognostic value of tumor oxygenation in 396 head and neck tumors after primary radiation therapy. An international multi-center study. Radiother Oncol. 2005;77(1):18-24.
8 Durand RE. Distribution and activity of antineoplastic drugs in a tumor model. J Natl Cancer Inst. 1989;81:146-152.
9 Sutherland RM. Cell and environment interaction in tumor microregions. The multicell spheroid model. Science. 1988;24:177.
10 Graeber TG, Osmanian C, Jacks T, et al. Hypoxia-mediated selection of cells with diminished apoptotic potential in solid tumours. Nature. 1996;379:88.
11 Hockel M, Schlenger K, Aral B, et al. Association between tumor hypoxia and malignant progression in advanced cancer of the uterine cervix. Cancer Res. 1996;56:4509.
12 Sundfor K, Lyng H, Rofstad EK. Tumour hypoxia and vascular density as predictors of metastasis in squamous cell carcinoma of the uterine cervix. Br J Cancer. 1998;78:822-827.
13 Wachsberger P, Burd R, Dicker AP. Tumor response to ionizing radiation combined with antiangiogenesis or vascular targeting agents: exploring mechanisms of interaction. Clin Cancer Res. 2003;9(6):1957.
14 Moeller BJ, Cao Y, Li CY, Dewhirst MW. Radiation activates HIF-1 to regulate vascular radiosensitivity in tumors: role of reoxygenation, free radicals, and stress granules. Cancer Cell. 2004;5(5):429.
15 Adams GE, Cooke MS. Electron-affinic sensitization. I. A structural basis for chemical radiosensitizers in bacteria. Int J Radiat Biol. 1969;15:457.
16 Adams GE, Asquith JC, Watts ME, et al. Radiosensitization of hypoxic cells in vitro: a water-soluble derivative of paranitroacetophenone. Nature [New Biol]. 1972;239:23.
17 Chapman JD, Reuvers AP, Borsa J, et al. Chemical radioprotection and radiosensitization of mammalian cells growing in vitro. Radiat Res. 1973;56(2):291.
18 Foster JL, Willson RL. Radiosensitization of anoxic cells by metronidazole. Br J Radiol. 1973;46:234.
19 Rabin HR, Urtasun RC, Partington J, et al. Pharmacokinetics and bioavailability of metronidazole using an IV preparation and application of its use as a radiosensitizer. Cancer Treatment Reports. 1980;64:1087.
20 Brown JM. Selective radiosensitization of the hypoxic cells of mouse tumors with the nitroimidazoles metronidazole and Ro 7-0582. Radiat Res. 1975;64(3):633.
21 Denekamp J, Harris SR. Tests of two electron-affinic radiosensitizers in vivo using regrowth of an experimental carcinoma. Radiat Res. 1975;61:191.
22 Urtasun RC, Band P, Chapman JD, et al. Radiation and high dose metronidazole in supratentorial glioblastomas. N Engl J Med. 1976;294:1364.
23 Urtasun RC, Chapman JD, Feldstein L, et al. Peripheral neuropathy related to misonidazole: Incidence and pathology. Br J Cancer. 1978;3:271.
24 Dische S, Saunders MI, Anderson P, et al. Neurotoxicity of misonidazole, pooling of data from five centers. Brit J Radiol.. 1978;51:1023.
25 Dische S. Chemical sensitizers for hypoxic cells: A decade of experience in clinical radiotherapy. Radiother Oncol. 1985;3:97.
26 Brown JM. Clinical trials of radiosensitizers: What should we expect? Int J Radiat Oncol Biol Phys. 1984;10:425.
27 Overgaard J. Clinical evaluation of nitroimidazoles as modifiers of hypoxia in solid tumors. Oncol Res. 1994;6:509.
28 Overgaard J, Sand Hansen H, Overgaard M, et al. A randomized double-blind phase III study on nimorazole as a hypoxic radiosensitizer of primary radiotherapy in supraglottic larynx and pharynx. Results of the Danish Head and Neck Cancer Study (Dahanca). Radiother Oncol. 1998;46(2):135.
29 Horsman MR, Bohm L, Margison GP, et al. Tumor radiosensitizers—current status of development of various approaches: Report on an International Atomic Energy Agency Meeting. Int J Radiat Oncol Biol Phys. 2006;64(2):551.
30 Fazekas J, Pagak TF, Wasserman TH, et al. Failure of misonidazole-sensitized radiotherapy to impact upon outcome among stage III/IV squamous cancers of the head and neck. Int J Radiat Oncol Biol Phys. 1987;13(8):1155.
31 Wasserman TH, Lee DJ, Cosmatos D, et al. Clinical trials with etanidazole (SR-2508) by the Radiation Oncology Group (RTOG). Radiother Oncol. 1991;20:129.
32 Lee DJ, Cosmatos D, Marcial VA, et al. Results of an RTOG phase III trial (RTOG 85-27) comparing radiotherapy plus etanidazole with radiotherapy alone for locally advanced head and neck carcinomas. Int J Radiat Oncol Biol Phys. 1995;32(3):567.
33 Eschwege R, Sancho-Garnier H, Chassagne D, et al. Results of a European randomized trial of etanidazole combined with radiotherapy in head and neck carcinomas. Int J Radiat Oncol Biol Phys. 1997;39(2):275.
34 Lawton CA, Coleman CN, Buzydlowsky JW, et al. Results of a phase II trial of external beam radiation with etanidazole for the treatment of locally advanced prostatic cancer (RTOG protocol 90-20). Int J Radiat Oncol Biol Phys. 1996;36(3):673.
35 Coleman CN, Noll L, Howes AE, et al. Initial results of the phase I trial of the continuous infusion of SR-2508 (etanidazole). Int J Radiat Oncol Biol Phys. 1989;16:1085.
36 Urtasun RC, Palmer M, Kinney B, et al. Intervention with the hypoxic tumor cell sensitizer etanidazole in the combined modality treatment of limited stage small-cell lung cancer. A one-institution study. Int J Rad Oncol Biol Phys. 1998;40:337.
37 Dische S. Radiotherapy, carcinoma of cervix and the radiosensitizer Ro 03-8799 (pimonidazole). In: Dewey WC, Edington M, Fry MRG, et al, editors. Radiation Research: A 20th Century Perspective, vol 2. Toronto: Academic Press; 1992.
38 Wouters BG, Brown JM. Cells at intermediate oxygen levels can be more important than the “hypoxic fraction” in determining tumor response to fractionated radiotherapy. Radiat Res. 1997;147:541.
39 Koch CJ, Howell RL. Combined radiation-protective and radiation-sensitizing agents: II Radiosensitivity of hypoxic or aerobic Chinese hamster fibroblasts in the presence of cysteamine and misonidazole: implications for the “oxygen effect” (with appendix on calculation of dose-modifying factors). Radiat Res. 1981;87(2):265.
40 Overgaard J. Hypoxic radiosensitization: Adored and Ignored. J Clin Oncol. 2007;25(26):4066.
41 Höckel M, Knoop C, Schlenger K, et al. Intratumoral PO2 predicts survival in advanced cancer of the uterine cervix. Radiother Oncol. 1993;26(1):45.
42 Brizel DM, Rosner GL, Harrelson J, et al. Pretreatment oxygenation profiles of human soft tissue sarcomas. Int J Radiat Oncol Biol Phys. 1994;30(3):635.
43 Fyles AW, Milosevic M, Wong R, et al. Oxygenation predicts radiation response and survival in patients with cervix cancer. Radiother Oncol. 1998;48(2):149.
44 Nordsmark M, Overgaard J. Tumor hypoxia is independent of hemoglobin and prognostic for loco-regional tumor control after primary radiotherapy in advanced head and neck cancer. Acta Oncol. 2004;43(4):396.
45 Wang GL, Semenza GL. Characterization of hypoxia-inducible factor 1 and regulation of DNA binding activity by hypoxia. J Biol Chem. 1993;268(29):21513.
46 Olive PL, Aquino-Parsons C, MacPhail SH, et al. Carbonic anhydrase 9 as an endogenous marker for hypoxic cells in cervical cancer. Cancer Res. 2001;61(24):8924.
47 Olive PL, Johnston PJ, Banáth JP, et al. The comet assay: a new method to examine heterogeneity associated with solid tumors. Nat Med. 1998;4(1):103.
48 Varghese AJ, Whitmore GF. Binding of nitroreduction products of misonidazole to nucleic acids and protein. Cancer Clin Trials. 1980;3(1):43.
49 Raleigh JA, Koch CJ. Importance of thiols in the reductive binding of 2-nitroimidazoles to macromolecules. Biochem Pharmacol. 1990;40(11):2457.
50 Urtasun RC, Chapman JD, Raleigh JA, et al. Binding of 3H-misonidazole to solid human tumors as a measure of tumor hypoxia. Int J Radiat Oncol Biol Phys. 1986;12(7):1263.
51 Kennedy AS, Raleigh JA, Perez GM, et al. Proliferation and hypoxia in human squamous cell carcinoma of the cervix: first report of combined immunohistochemical assays. Int J Radiat Oncol Biol Phys. 1997;37(4):897.
52 Evans SM, Hahn S, Pook DR, et al. Detection of hypoxia in human squamous cell carcinoma by EF5 binding. Cancer Res. 2000;60(7):2018.
53 Mayer A, Höckel M, Vaupel P. Endogenous hypoxia markers: case not proven!. Adv Exp Med Biol. 2008;614:127.
54 Tatum JL, Kelloff GJ, Gillies RJ, et al. Hypoxia: importance in tumor biology, noninvasive measurement by imaging, and value of its measurement in the management of cancer therapy. Int J Radiat Biol. 2006;82(10):699.
55 Koh WJ, Rasey JS, Evans ML, et al. Imaging of hypoxia in human tumors with [F-18]fluoromisonidazole. Int J Radiat Oncol Biol Phys. 1992;22(1):199.
56 Rischin D, Hicks RJ, Fisher R, et al. Prognostic significance of [18F]-misonidazole positron emission tomography-detected tumor hypoxia in patients with advanced head and neck cancer randomly assigned to chemoradiation with or without tirapazamine: a substudy of Trans-Tasman Radiation Oncology Group Study 98.02. J Clin Oncol. 2006;24(13):2098.
57 Parliament MB, Chapman JD, Urtasun RC, et al. Non-invasive assessment of human tumour hypoxia with 123I-iodoazomycin arabinoside: Preliminary report of a clinical study. Br J Cancer. 1992;65(1):90.
58 Brown JM, Workman P. Partition coefficient as a guide to the development of radiosensitizers which are less toxic than misonidazole. Radiat Res. 1980;82(1):171.
59 Laughlin KM, Evans SM, Jenkins WT, et al. Biodistribution of the nitroimidazole EF5 (2-[2-nitro-1H-imidazol-1-yl]-N-[2,2,3,3,3-pentafluoropropyl] acetamide) in mice bearing subcutaneous EMT6 tumors. J Pharmacol Exp Ther. 1996;277(2):1049.
60 Ziemer LS, Evans SM, Kachur AV, et al. Noninvasive imaging of tumor hypoxia in rats using the 2-nitroimidazole 18F-EF5. Eur J Nucl Med Mol Imaging. 2003;30(2):259.
61 Piert M, Machulla HJ, Picchio M, et al. Hypoxia-specific tumor imaging with 18F-fluoroazomycin arabinoside. J Nucl Med. 2005;46(1):106.
62 Christian N, Bol A, De Bast M, et al. Determination of tumour hypoxia with the PET tracer [18F]EF3: improvement of the tumour-to-background ratio in a mouse tumour model. Eur J Nucl Med Mol Imaging. 2007;34(9):1348.
63 Matsumoto K, Szajek L, Krishna MC, et al. The influence of tumor oxygenation on hypoxia imaging in murine squamous cell carcinoma using [64Cu]Cu-ATSM or [18F] Fluoromisonidazole positron emission tomography. Int J Oncol. 2007;30(4):873.
64 Hank JM. Does hyperbaric oxygen have a future in radiation therapy? Int J Rad Oncol Biol Phys. 1981;7:1125.
65 Rubin P, Hanley J, Keys HM, et al. Carbogen breathing during radiation therapy. Radiation Oncology Group study. Int J Radiat Oncol Biol Phys. 1979;5:1963.
66 Bush RS. The significance of anemia in clinical radiation therapy. Int J Radiat Oncol Biol Phys. 1986;12:2047.
67 Dische S, Saunders MI, Warvurton MF. Hemoglobin, radiation, morbidity and survival. Int J Radiat Oncol Biol Phys. 1986;12:1335.
68 Rose C, Lustig R, McIntosh LN, et al. A clinical trial of fluosol-DA 20% in advanced squamous cell carcinoma of the head and neck. Int J Radiat Oncol Biol Phys. 1988;12:1325.
69 Lustig R, McIntosh LN, Rose C, et al. Phase I/II, study of fluosol-DA and 100% oxygen as adjuvant to radiation in the treatment of advanced squamous cell tumors of the head and neck. Int J Radiat Oncol Biol Phys. 1989;16(6):1587.
70 Dische S. Hypoxia in local tumor control: Part 2. Radiother Oncol. 1991;20:9.
71 Amorino GP, Lee H, Holburn GE, et al. Enhancement of tumor oxygenation and radiation response by the allosteric effector of hemoglobin, RSR13. Radiat Res. 2001;156:294.
72 Ramsay JRS, Bleehan NM, Dennis I, et al. Phase I: A study of BW12C in combination with mitomycin C in patients with advanced gastrointestinal cancer. Int J Rad Oncol Biol Phys. 1992;22:21.
73 Falk SJ, Ward R, Bleehan NM. The influence of carbogen breathing on tumor tissue oxygenation in man evaluated by computerized PO2 histography. Br J Cancer. 1992;66:919.
74 Rojas A. Radiosensitization with normobaric oxygen and carbogen. Radiother Oncol. 1991;20:65.
75 Horsman MR, Kristjansen PEG, Mizuno M, et al. Biochemical and physiological changes induced by nicotinamide in a C3H mouse mammary carcinoma and CDF1 mice. Int J Radiat Oncol Biol Phys. 1992;22:451.
76 Miralbell R, Mornex F, Greiner R, et al. Accelerated radiotherapy, carbogen and nicotinamide in glioblastoma. J Clin Oncol. 1999;17:3143.
77 Bernier J, Denekamp J, Rojas A, et al. ARCON: Accelerated radiotherapy with carbogen and nicotinamide in non-small cell lung cancer: A phase I/II study by the EORTC. Radiotherapy and Oncology. 1999;52(2):149-156.
78 Hoogsteen IJ, Pot LAM, Marres HAH. Oxygen modifying treatment with ARCON reduces the prognostic significance of hemoglobin in squamous cell carcinoma of the head and neck. Int J Radiat Oncol Biol Phys. 2006;61(1):83.
79 Kaanders JH, Pop LA, Marres HA, et al. ARCON experience into 115 patients with advanced head and neck cancer. Int J Radiat Oncol Biol Phys. 2002;52(3):769.
80 Hoskin PJ, Rojas AM, Phillips H, et al. Acute and late morbidity in the treatment of advanced bladder carcinoma with accelerated radiotherapy, carbogen, and nicotinamide. Cancer. 2005;103(11):2287.
81 Mendenhall WM, Morris CG, Amdur RS, et al. Radiotherapy alone or combined with carbogen breathing for squamous cell carcinoma of the head and neck: A prospective randomized trial. Cancer. 2005;104(2):332.
82 Vees H, Allal AS. Carbogen breathing combined with radical radiotherapy in advanced head and neck cancer patients, Clinical Oncology. Royal College of Radiologists. 2006;18(6):493-496.
83 Lin AJ, Cosby LA, Shansky CW, et al. Potential bioreductive alkylating agents. 1. Benzoquinone derivatives. J Med Chem. 1972;15:1247.
84 Rockwell S, Kennedy KA, Sartorelli AC. Mitomycin-C as a prototype bioreductive alkylating agent: in vitro studies of metabolism and cytotoxicity. Int J Radiat Oncol Biol Phys. 1982;8:753.
85 Brown JM, Siim BG. Hypoxia-specific cytotoxins in cancer therapy. Seminars in Rad Onc. 1996;6:22.
86 Haffty BG, Son YH, Sasaki CT, et al, Mitomycin C as an adjunct to postoperative radiation therapy in squamous cell carcinoma of the head and neck: results from two randomized clinical trials, Int J Radiat Oncol Biol Phys; 27; 1993:241.
87 Zeman EM, Brown JM, Lemmon MJ, et al. SR-4233: a new bioreductive agent with high selective toxicity for hypoxic mammalian cells. Int J Radiat Oncol Biol Phys. 1986;12:1239.
88 Brown JM, Lemmon MJ. SR-4233: a tumor specific radiosensitizer active in fractionated radiation regimes. Radiother Oncol. 1991;1:151.
89 Evans JW, Chernikova SB, Kachnic LA, et al. Homologous recombination is the principal pathway for the repair of DNA damage induced by tirapazamine in mammalian cells. Cancer Res. 2008;68(1):257.
90 Peters KB, Brown JM. Tirapazamine: a hypoxia-activated topoisomerase II poison. Cancer Res. 2002;62(18):5248.
91 Dorie MJ, Brown JM. Tumor-specific, schedule-dependent interaction between tirapazamine (SR 4233) and cisplatin. Cancer Research. 1993;53:4644.
92 von Pawel J, von Roemeling R, Gatzemeier U, et al, Tirapazamine plus cisplatin versus cisplatin in advanced non-small-cell lung cancer: A report of the international CATAPULT I study group, [In Process Citation]; J Clin Oncol; 48; 2000:791.
93 Craighead PS, Pearcey R, Stuart G. A phase I/II evaluation of tirapazamine administered intravenously concurrent with cisplatin and radiotherapy in woman with locally advanced cervical cancer. Int J Radiat Oncol Biol Phys. 2000;48:791.
94 Maluf FC, Leiser AL, Aghajanian, et al. Phase II study of Tirapazamine plus cisplatin in patients with advanced or recurrent cervical cancer. Int J Gynecological Cancer. 2006;16(3):1165.
95 Covens A, Blessing J, Bender D, et al. A phase II evaluation of Tirapazamine plus cisplatin-sensitive ovarian or primary peritoneal cancer: A Gynecological Oncology Group Study. Gynecologic Oncology. 2006;100(3):585.
96 Rischin D, Peters L, Fisher R, et al. Tirapazamine, Cisplatin, and Radiation versus Fluorouracil, Cisplatin and Radiation in patients with locally advanced head and neck cancer: A randomized phase II trial of the Trans-Tasman Radiation Oncology Group (TROG 98-02). J Clin Oncol. 2005;23:79.
97 Rischin D, Peters L, O’Sullivan B, et al, Phase III study of tirapazamine, cisplatin and radiation versus cisplatin and radiation for advanced squamous cell carcinoma of the head and neck, J Clin Oncol; 26; 2008, (May 20 suppl; abstr LBA6008)
98 Peters LJ, Rischin D, Fisher R, et al: Identification and therapeutic targeting of hypoxia in H&N cancer. Abstract PS1042 International Congress of Radiation Research, July 2007.
99 Patterson LH. Rationale for the use of aliphatic N-oxides of cytotoxic anthraquinones as prodrug DNA binding agents: a new class of bioreductive agent. Cancer Metastasis Rev. 1993;12(2):119.
100 Patterson LH, McKeown SR, Ruparelia K, et al. Enhancement of chemotherapy and radiotherapy of murine tumours by AQ4N, a bioreductively activated anti-tumour agent. Br J Cancer. 2000;82(12):1984.
101 Wilson WR, Edmunds SJ, Valentine S, et al: Mechanism of action and antitumour activity of PR-104, A dinitrobenzamide mustard pre-prodrug that is activated selectively under hypoxia. Poster at National Cancer Research Institute Conference, Birmingham, Oct 3, 2005.
102 Cobb LM, Connors TA, Elson LA, et al. 2,4-Dinitro-5-ethyleneiminobenzamide (CB 1954): A potent and selective inhibitor of the growth of the Walker carcinoma 256. Biochem Pharmacol. 1969;18:1519.
103 Knox RJ, Friedlos F, Jarman M, Roberts JJ. A new cytotoxic, DNA interstrand crosslinking agent, 5-(Aziridin-1-YL)-4-hydroxylamino-2-nitrobenzamide, is formed from 5-(Aziridin-1-YL)-2,4-dinitrobenzamide (CB 1954) by a nitroreductase enzyme in Walker carcinoma cells. Biochem Pharmacol. 1988;37(24):4661.
104 Knox RJ, Boland MP, Friedlos F, et al. The nitroreductase enzyme in Walker cells that activates 5-(Aziridin-1-YL)-2,4-dinitrobenzamide (CB 1954) to 5-(Aziridin-1-YL)-4-hydroxylamino-2-nitrobenzamide is a form of NAD(P)H dehydrogenase (quinone) (EC 1.6.99.2). Biochem Pharmacol. 1988;37(24):4671.
105 Patterson AV, Ferry DM, Edmunds SJ, et al. Mechanism of action and preclinical antitumor activity of the novel hypoxia-activated DNA cross-linking agent PR-104. Clin Cancer Res. 2007;13(13):3922.
106 Bunn H. Recombinant erythropoietin therapy in cancer patients. J Clin Oncol. 1990;8:949.
107 Vijayakumar S, Roach N, Wara W, et al. Effects of subcutaneous recombinant human erythropoietin in cancer patients receiving radiotherapy: Preliminary results of a randomized, open-label phase II trial. Int J Radiat Oncol Biol Phys. 1993;26:721.
108 Lavey RS, Dempsey WH. Erythropoietin increases hemoglobin in cancer patients during radiation therapy. Int J Radiat Oncol Biol Phys. 1993;27:1147.
109 Lavoy RS, Liu PY, Greer BE, et al. Recombinant human erythropoietin as a adjunct to radiation therapy and cisplatin for stage IIB-IVA carcinoma of the cervix. A Southwest Oncology Group Study. Gynecol Oncol. 2004;95(1):145.
110 Varlotto J, Stevenson MA. Anemia, tumor hypoxia and the cancer patient (Review). Int J Radiat Oncol Biol Phys. 2005;63(1):25.
111 Thomas G, Ali S, Hoevers FJ, et al. Phase III trial to evaluate the efficacy of maintaining hemoglobin levels above 12.0 g/dl with erythropoietin above 10.0 g/dl without erythropoietin in anemic patients receiving concurrent radiation and cisplatin for cervical cancer. Gynecol Oncol. 2008;108(2):317.
112 Jacobson GM, Kamth RS, Smith BJ, et al. Thromboembolic events in patients treated with definitive chemotherapy and radiation therapy for invasive cervical cancer. Gynecol Oncol. 2005;6(2):470.
113 Nordsmark M, Overgaard J. Tumor hypoxia is independent of hemoglobin and prognostic for loco-regional tumor control after primary radiotherapy in advanced head and neck cancer. Acta Oncol. 2004;43(4):396.
114 Yu NY, Brown MJ. Depletion of glutathione in vivo as a method of improving the therapeutic ratio of misonidazole and SR 2508. Int J Radiat Oncol Biol Phys. 1984;10(8):1265.
115 Mitchell JB, Cook JA, DeGraff W, et al. Glutathione modulation in cancer treatment: Will it work? Int J Radiat Oncol Biol Phys. 1989;16(5):1289.
116 Kramer RA, Soble M, Howes AE. The effect of glutathione (GSH) depletion in vivo by buthionine sulfoximine (BSO) in the radiosensitization SR2508. Int J Radiat Oncol Biol Phys. 1989;16(5):1325.
117 Allalunis-Turner MJ, Barrone GM, Day RS, et al. Heterogeneity in response to treatment with buthionine or sulfoximine or interferon in human malignant glioma cells. Int J Radiat Oncol Biol Phys. 1992;22:765.
118 Britten RA, Warenius HM, White R. BSO-induced reduction of glutathione levels increases the cellular radiosensitivity of drug-resistant human tumor cells. Int J Radiat Oncol Biol Phys. 1992;22:769.
119 Mitchell JB, Wink DA, DeGraff W, et al. Hypoxic mammalian cell radiosensitization by nitric oxide. Cancer Res. 1993;53:5845.
120 Bagsaw MA, Doggett RSL, Smith KC, et al. Intra-arterial 5-bromodeoxyuridine and x-ray therapy. Radiology. 1967;99:886.
121 Goffinet DR, Brown JM. Comparison of intravenous and intra-arterial pyrimidine infusion as a means of radiosensitizing tumors. Radiology. 1977;124:819.
122 Kinsella TJ, Collins J, Rowald J, et al. Pharmacology in phase I/II study of continuous infusions of iododeoxyuridine and hyperfractionated radiotherapy in patients with glioblastoma multiforme. J Clin Oncol. 1988;6:871.
123 Phillips TL, Levin VA, Ahn DK. Evaluation of bromodeoxyuridine in glioblastoma multiforme, a NCOG phase II study. Int J Radiat Oncol Biol Phys. 1991;21:709.
124 Prados M, Scott C, Sander H, et al. A phase III randomized study of radiotherapy with or without BUDR plus procarbazine, CCNU and Vincristine (PCV) for the treatment of anaplastic astrocytoma: A preliminary report of RTOG 94-04 radiation and BUDR in malignant gliomas. Int J Radiat Oncol Biol Phys. 1999;45(5):1109-1115.
125 Urtasun RC, Cosmatos D, DelRowe J, et al. Iododeoxyuridine (IUdR) combined with radiation in the treatment of malignant glioma: A comparison of short vs long intravenous dose schedules (RTOG86-12). Int J Radiat Oncol Biol Phys. 1993;27:207.
126 Yuhas MJ, Storer JB. Differential chemo protection of normal and malignant tissues. J Nat Cancer Inst. 1969;32:331.
127 Denekamp J, Rojas A, Stewart FA. Is radioprotection by WR-2731 restricted to normal tissues? In: Nygaard OF, Simic MG, editors. Radioprotectors and Anticarcinogens. New York: Academic Press; 1983:655.
128 Tresher M, Nitjmans LG, Shepman A, et al. Effects of modulating agent WR-2721 and its main metabolites on the formation and stability of cisplatin/DNA adducts in vitro. Bio Chem Pharmacol. 1992;33:1013.
129 Neta R, Oppenheim JJ, Douche S. Interdependence of the radioprotective effects of human recombinant interleukin 1 alpha, tumor necrosis factor alpha, granulocyte colony-stimulating factor, and murine recombinant granulocyte-macrophage colony-stimulating factor. J Immunol. 1988;140:108.
130 Phillips TL, Kane L, Utley JF. Radioprotection of tumor and normal tissues by thiol phosphate compounds. Cancer. 1973;32:528.
131 Milas L. Improving radiotherapy by reducing normal tissue damage with radioprotectors. Cancer Bull. 1986;38:223.
132 Millar JL, McElwain TJ, Clutterbuck RD, et al. Demodification of melphalan toxicity as single agent in tumor bearing mice by WR 2721. Am J Clin Oncol (CCT). 1992;5:321.
133 Wasserman TH, Phillips TL, Ross G, et al. Differential protection against cytotoxic chemotherapeutic effects on bone marrow CFU’s by WR-2721. Cancer Clin Trials. 1981;4:3.
134 Peters GJ, Vanderwilt CL, Gyergyay F, et al. Protection by WR-2721 of the toxicity induced by the combination of cisplatin and 5-fluorouracil. Int J Radiat Oncol Biol Phys. 1992;22:785.
135 Washburn LC, Rafter JJ, Hayes RL:. Prediction of the effective radioprotective dose of WR-2721 in humans through an interspecies tissue distribution study. Radiat Res. 1976;66:100-101.
136 Kligerman MN, Glover DJ, Turrisi AT, et al. Toxicity of WR-2721 administered in single and multiple doses. Int J Radiat Oncol Biol Phys. 1984;10:1773.
137 Turrisi AT, Glover DJ, Hurwitz, et al. Final report of the phase I trial of single dose WR-2721. Can Treat RTTS. 1986;70:1389.
138 Coia L, Crigel R, Hanks G, et al. Phase I study WR-2721 in combination with total body radiation in patients with refractory lymphoid malignancies. Int J Rad Oncol Biol Phys. 1992;22:791.
139 Garces YI, Okuno SH, Schild SE, et al. Phase I North Central Cancer Treatment Group Trial—N9923 of escalating dose of twice daily thoracic radiation therapy with amifostine with alternating chemotherapy in limited stage small cell lung cancer. Int J Radiat Oncol Biol Phys. 2007;67(4):995.
140 Antonadu D, Coliarakis N, Synodinou M, et al. Randomized Phase III trial of radiation treatment +/− Amifostine in patients with advanced-stage lung cancer. Int J Radiat Oncol Biol Phys. 2001;51:915.
141 Komaki R, Lee JS, Kaplan B, et al. Randomized Phase III study of chemoradiation with or without Amifostine for patients with favorable performance status inoperable Stage II-III non-small cell lung cancer: Preliminary results. Semin Rad Oncol. 2002;12:S46.
142 Movsas B, Scott C, Langer C, et al. Phase III study of Amifostine in patients with locally advanced NSCLC receiving chemotherapy and hyperfractionated radiation. RTOG 98-01. Proc Am Soc Clin Oncol. 2003;22:636. (Abstract 2559)
143 Mell LK, Malik R, Komaki R, et al. Effect of Amifostine on response rates in locally advanced non-small cell lung cancer patients treated with unrandomized control trials: A meta-analysis. Int J Radiat Oncol Biol Phys. 2007;68(1):111.
144 Werner-Wasik M, Langer C, Movsas B. Amifostine in chemoradiation therapy for non-small cell lung cancer: Review of experience and design of a phase II trial assessing subcutaneous and intravenous bolus administration. Semin Oncol. 2005;32(Suppl 3):S105.
145 Law A, Kennedy T, Pellitteri P, et al. Efficacy and safety of subcutaneous Amifostine in minimizing radiation-induced toxicity in patients receiving combined-modality treatment for a squamous cell carcinoma of the head and neck. Int J Radiat Oncol Biol Phys. 2007;69(5):1361.
146 Koukourakis MI, Kyrias G, Kakolyris S, et al. Subcutaneous administration of Amifostine during fractionated radiotherapy: A randomized Phase II study. J Clin Oncol. 2000;18:2226.
147 Anne PR, Machtay M, Rosenthal DI, et al. A Phase II trial of subcutaneous amifostine and radiation therapy in patients with head and neck cancer. Int J Radiat Oncol Biol Phys. 2007;67(2):445.
148 Jellema AP, Slotmann BJ, Muller MJ, et al. Radiotherapy alone, versus radiotherapy with amifostine 3 times weekly, versus radiotherapy with amifostine 5 times weekly: A prospective randomized study in squamous cell head and neck cancer. Cancer. 2006;107(3):544.
149 Brizel DM, Wasserman TH, Henke M, et al. Phase III randomized trial of Amifostine as a radioprotector in head and neck cancer. J Clin Oncol. 2000;18:3339.
150 Wasserman TH, Brizel DM, Henke M, et al. Influence of IV Amifostine on xerostomia, tumor control and survival after radiotherapy for head and neck cancer: Two year follow-up of a prospective randomized phase III trial. Int J Radiat Oncol Biol Phys. 2005;63(4):985.
151 Rades D, Fehlauer F, Bajrovic A, et al. Serious adverse effects of amifostine during radiotherapy in head and neck cancer patients. Radiother Oncol. 2004;70(3):261.
152 Kouvaris JR, Kouloulias V, Malaise E, et al. Amifostine as radioprotective agent for the rectal mucosa during radiation of pelvic tumors. A Phase II randomized study using various toxicities scales and rectal sigmoidoscopy. Sonderb Z Strahlenther Onkol. 2003;179(3):167.
153 Small WJr. Cytoprotection/radioprotection with amifostine: Potential role in cervical cancer and early findings in the Radiation Therapy Oncology Group C-0116 Trial. Semin Oncol. 2003;30((6), Suppl 18):68.
154 Overgaard J. Does Amifostine have a role in chemoradiation treatment? Lancet. 2003;4:380.
155 Koukourakis MI. Amifostine: Is there evidence of tumor protection? Semin Oncol. 2003;30((6)Suppl 18):18.
156 Schuchter LM, Hensley ML, Meropol NJ, et al. 2002 Update of recommendations for the use of chemotherapy and radiotherapy protectants: Clinical practice guidelines of the American Society of Clinical Oncology. J Clin Oncol. 2002;20:2895.
157 Kouvaris JR, Kouloulias VE, Vlahos LJ. Amifostine: The first selective-target and broad-spectrum radioprotector. Oncologist. 2007;12:738.
158 Glick JH, Glover D, Weiler C, et al. Phase I controlled trials of WR-2721 and cyclophosphamide. Int J Radiat Oncol Biol Phys. 1984;10:1777.
159 Glover D, Glick JH, Weiler C, et al. Phase I/II trials of WR-2721 and cis-platinum. Int J Radiat Oncol Biol Phys. 1986;12:1509.
160 Glick J, Kemp J, Rose P, et al. A randomized trial of cyclophosphamide and cisplatin plus or minus WR-2721 in the treatment of advanced epithelial ovarian cancer. ASCO. 1992;11:122.
161 Muggia F, Parker R, Reed E, et al. WR-2721 pretreatment protects against the bone marrow toxicity of carboplatin and cisplatin. ASCO. 1992;11:132.
162 Luginbuhl W, Tester W, Shaw L, et al. One or two doses of WR-2721: Does it protect patients receiving carboplatin? ASCO. 1992;11:312.
163 Mitchell JB, DeGraff W, Kaufman D, et al. Inhibition of oxygen-dependent radiation-induced damage by nitroxide superoxide dismutase mimic, Tempol. Arch-Biochem Biophys. 1991;289(1):62.
164 Coleman CN, Mitchell JB. Radiation modifiers. In: Chabner BA, Longo DL, editors. Cancer chemotherapy and biotherapy: Principles and practice. 3. Lippincott Williams and Wilkins; 2001:707-751.