CHAPTER 87 CHANNELOPATHIES OF MUSCLE (INCLUDING THE MYOTONIC DYSTROPHIES)
Channelopathies are disorders resulting from alterations in function of the ion channels found in cell membranes throughout the body. Disorders such as episodic ataxia types 1 and 2, spinocerebellar ataxia type 6, familial hemiplegic migraine, and benign familial neonatal convulsions are examples of channelopathies affecting the central nervous system. These disorders are outside the scope of this chapter.
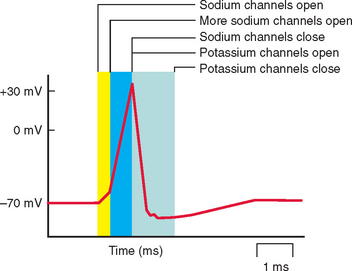
Each ion channel type has a different role in sarcolemmal function (Fig. 87-1). Chloride channels are important in stabilizing the membrane potential at the resting level; inward flow of ions through sodium channels induces membrane depolarization and hence action potential; outflow through a voltage-gated subset of potassium channels is involved in the depolarization of the action potential. Calcium channels may be involved in action potential generation or in the regulation of other channel types. Action potential generation in the membrane is coupled to activation of the contractile machinery in the skeletal muscle and to muscle contraction.
Electromyography in patients with myotonia congenita or myotonic dystrophy should demonstrate repetitive generation of muscle cell membrane action potentials (Fig. 87-2) in resting muscle, although the test may be unnecessary in clinically evident cases, particularly of myotonic dystrophy. The myotonic discharges are described as high-frequency repetitive biphasic spikes or positive waves with varying frequency or amplitude. Electromyography in periodic paralysis will be normal between attacks in patients without a myopathy. During an attack, features such as reduced compound muscle action potential amplitude are seen.
NONDYSTROPHIC MYOTONIAS AND PERIODIC PARALYSES
Chloride Channel Disorders (CLCN1)
Epidemiology
Thomsen’s and Becker’s myotonia congenita are allelic disorders due to mutations in the CLCN1 gene, which encodes the voltage-gated chloride channel. Myotonia congenita, Thomsen type, first described in 1876, is an autosomal dominant condition with a prevalence of about 1 : 400,000. Becker’s myotonia congenita, described in 1957, is an autosomal recessive condition, more common than Thomsen’s myotonia, with a prevalence of between 1 : 23,000 and 1 : 50,000.1
Molecular Pathophysiology
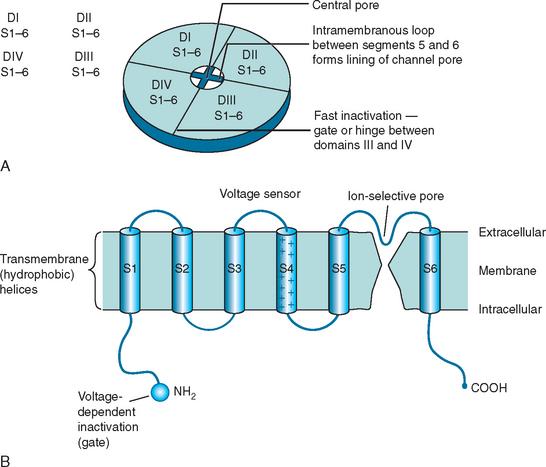
CLCN1 mutations are spread throughout the sequence of the gene (Pusch) and are predicted to produce functional changes in the chloride channel protein (CIC-1) with faulty assembly of subunits, impaired ion channel formation, and channel dysfunction (Fig. 87-3). The resulting reduction in chloride ion conductance lowers the threshold for depolarization through sodium channel activation and hence leads to sustained excitability, that is, myotonia.
Treatment
To alleviate the myotonia, mexiletine is currently the first-line agent; it acts as a “membrane-stabilizing” agent, reducing sodium channel activation, but it has no direct effect on chloride channels. Mexilitene should be used with care as toxicity may develop at higher doses. If mexilitene is not effective, it may be worth switching to carbamazepine or phenytoin. Acetazolamide is not beneficial (although it may be in sodium channel myotonias, see later) and—along with other diuretics—may worsen symptoms of myotonia. Many patients believe that treatment for their myotonia is not necessary as they have adapted to their condition and manage their activities without undue disruption. However, even mild untreated myotonia may lead to subsequent muscle weakness.
Care should be taken that depolarizing agents and anticholinesterases are avoided during anesthesia.
Sodium Channel Disorders (SCN4A)
Clinical Features
Myotonia fluctuans is a mild version of potassium-aggravated myotonia in which the muscle stiffness is variable but not generally severe and there is no weakness. The myotonia may show a warm-up phenomenon. Myotonia permanens is at the severe end of the spectrum for potassium-aggravated myotonia and produces a condition of continuous depolarization at the cellular level with consequent sustained muscle contraction, myotonia, and hypertrophy. In severe cases, respiratory muscle compromise and hypoventilation may ensue. The situation may be worsened by exercise or potassium loads. There is an intermediate-severity potassium-aggravated myotonia called potassium- and cold-aggravated myotonia congenita, which clinically resembles Thomsen’s myotonia congenita and probably explains earlier overestimates of its frequency. The two may be distinguishable only on molecular testing.
Calcium Channel Disorders (CACNL1A3)
MYOTONIC DYSTROPHIES
Recognition of myotonic dystrophy as an autosomal dominantly inherited condition was straightforward. However, it was not so easy to understand why successive generations seemed to become progressively more severely affected. Current genetic concepts and theories of pathogenesis are outlined in the following sections. These are suggested strategies for targeted therapies, which may lead to a cure.
Clinical Features
Myotonic Dystrophy Type 2
Patients with DM2 have in common with DM1 patients the presence of myotonia, early cataracts, and autosomal dominant family history but tend to have proximal more than distal weakness. Table 87-1 compares the salient features of the two conditions.
| DM1 | DM2 | |
|---|---|---|
| Prevalence | ||
| Congenital cases | Yes | No |
| Onset | Earlier | Later |
| Inheritance | Autosomal dominant | Autosomal dominant |
| Genetic mutation | CTG triplet repeat exp | CCTG quad repeat exp |
| Affected gene | DMPK (3′ UTR) | ZNF9 (intron 1) |
| Chromosomal locus | 19q13 | 3q21 |
| Limb myotonia | Frequent | Less frequent |
| Myotonia on electromyography | Yes | Yes |
| Distribution of weakness | Distal | Proximal |
| Presence of myalgia | Common | Common |
| Facial weakness | Yes | No |
| Bulbar weakness | Yes | No |
| Cardiac involvement | Yes | Less common |
| Respiratory involvement | Yes | No |
| Anesthetic risks | Yes | No |
| Cognitive abilities | Affected | Normal |
| Somnolence | Frequent | Infrequent |
Investigations
In general, serum creatine kinase is only slightly raised in patients with DM1 and DM2. Electromyography shows myotonic discharges in both conditions. Muscle biopsy is not usually indicated but may show the presence of fiber size variation and central nuclei.2
Theories of Molecular Pathogenesis
In DM1, the expanded CTG repeat within the 3′ untranslated region of the myotonic dystrophy protein kinase (DMPK) gene is unstable through successive generations and thus explains the anticipation phenomenon whereby offspring have increased repeat numbers (and earlier onset) than their parents. Extreme numbers of repeats (in excess of 1000) may occur in infants with congenital DM1. It seems that the phenotype is maintained in populations by the “premutation” repeat size, which is prone to expansion and maintains the population prevalence.
Principles of Care
Personality changes are well recognized in patients with DM1 and may be best described as a lack of motivation,3 although it is not clear how these cognitive changes relate to changes seen on brain imaging.
Harper P, et al, editors. Myotonic Dystrophy. Present Management. Future Therapy. 2004.
Lehmann-Horn F, Rudel R, Jurkat-Rott K: Nondystrophic myotonias and periodic paralyses. In Engel A, Franzini-Armstrong C, eds: Myology, 3rd ed. 2004.
Ranum L, Day J. Pathogenic RNA repeats: an expanding role in genetic disease. Trends Genet. 2004;20:506-512.
Surtees R. Inherited ion channel disorders. Eur J Pediatr. 2000;159(Suppl 3):S199-S203.
van Engelen B, Eymard B, Wilcox D. 123rd ENMC International Workshop: Management and Therapy in Myotonic Dystrophy. Neuromuscular Disord. 2005;15:389-394.
1 Lehmann-Horn F, Rudel R, Jurkat-Rott K: Nondystrophic myotonias and periodic paralyses. In Engel A, Franzini-Armstrong C, eds: Myology, 3rd ed. 2004.
2 Schoser BG, Schneider-Gold C, Kress W, et al. Muscle pathology in 57 patients with myotinic dystrophy type 2. Muscle Nerve. 2004;29:275-281.
3 Winblad S, Lindberg C, Hansen S. Temperament and character in patients with classical myotonic dystrophy type 1 (DM-1). Neuromuscul Disord. 2005;15:287-292.






