Chapter 3 Challenge
Albayram et al. 2002 Albayram S, Melham E, Mori S, et al. Holoprosencephaly in children: diffusion tensor MR imaging of white matter tracts of the brainstem. Initial experience. Radiology. 2002;223:645-651.
Barkovich et al. 2002 Barkovich A, Simon E, Clegg N, Kinsman S, Hahn J. Analysis of the cerebral cortex in holoprosencephaly with attention to the sylvian fissures. AJNR Am J Neuroradiol. 2002;23:143-150.
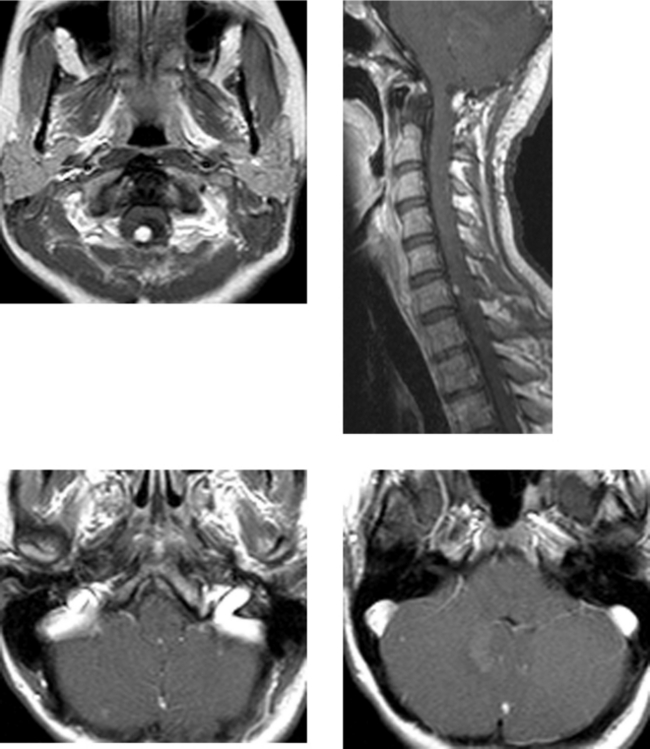
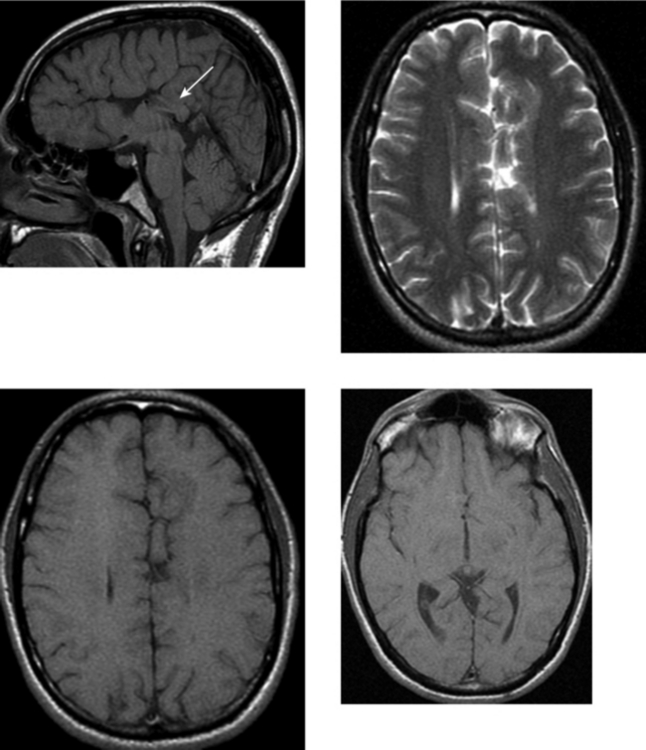
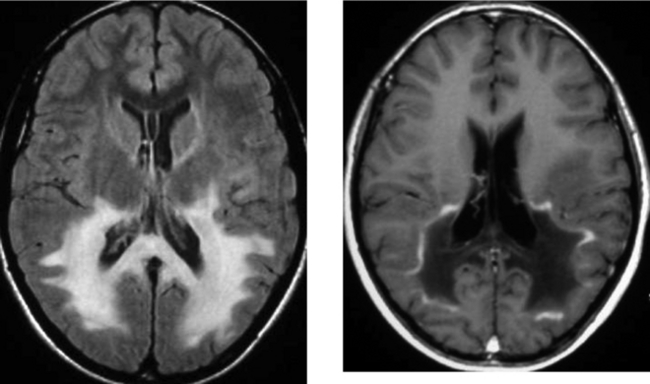
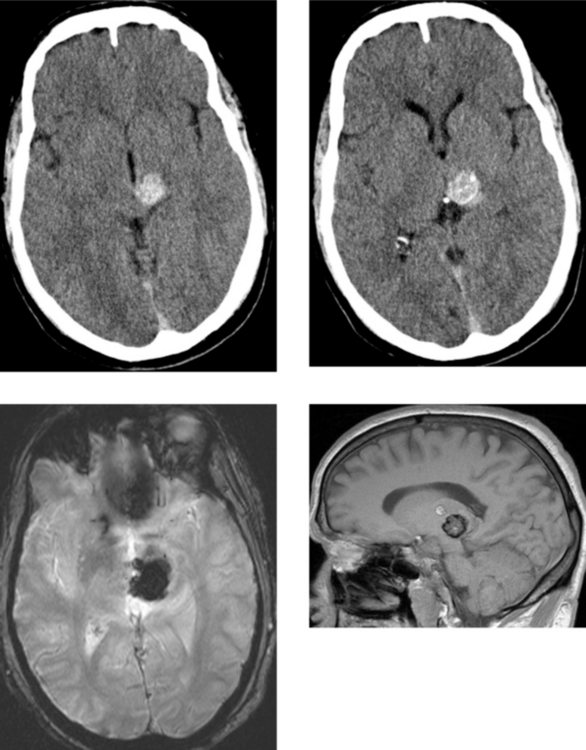
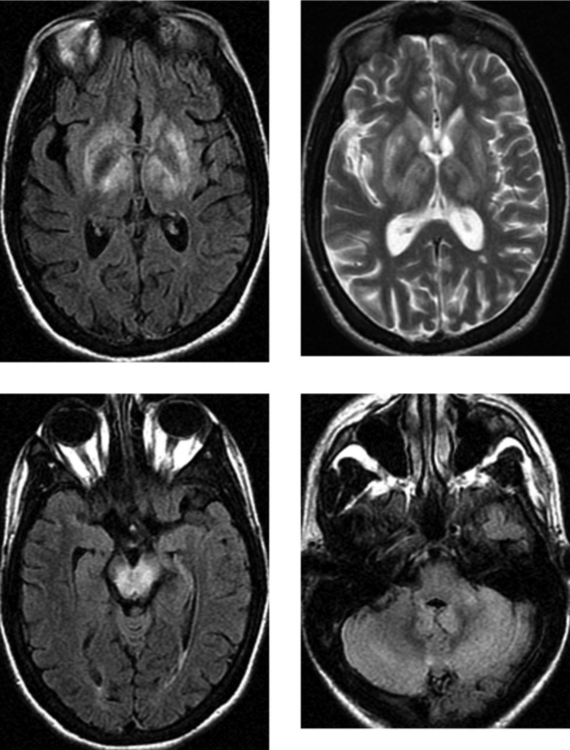
CASE 152 Olivopontocerebellar Degeneration
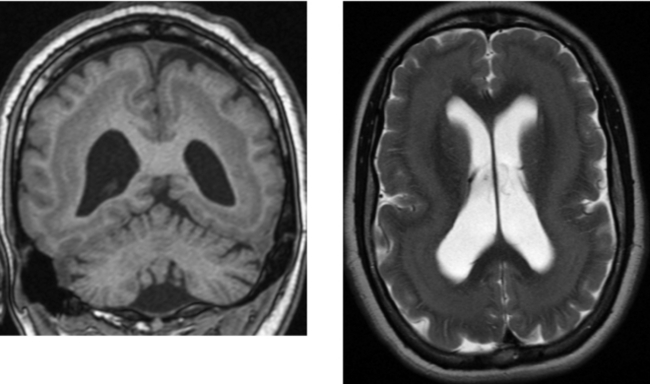
CASE 154 Huntington’s Disease
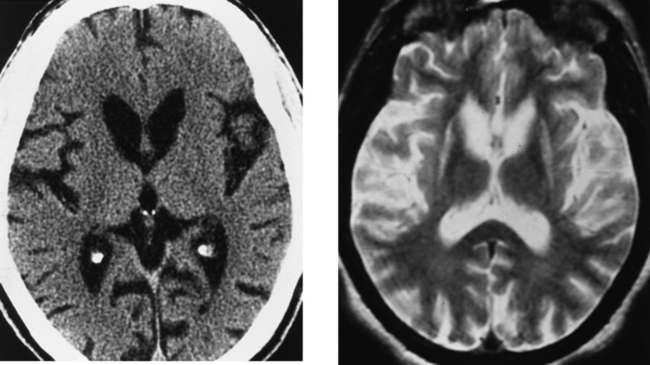
Krausz et al. 1996 Krausz Y, Bonne O, Marciano R, Yaffe S, Lerer B, Chisin R. Brain SPECT imaging of neuropsychiatric disorders. Eur J Radiol. 1996;21:183-187.
Montoya et al. 2006 Montoya A, Price BH, Menear M, Lepage M. Brain imaging and cognitive dysfunctions in Huntington’s disease. J Psychiatry Neurosci. 2006;31:21-29.
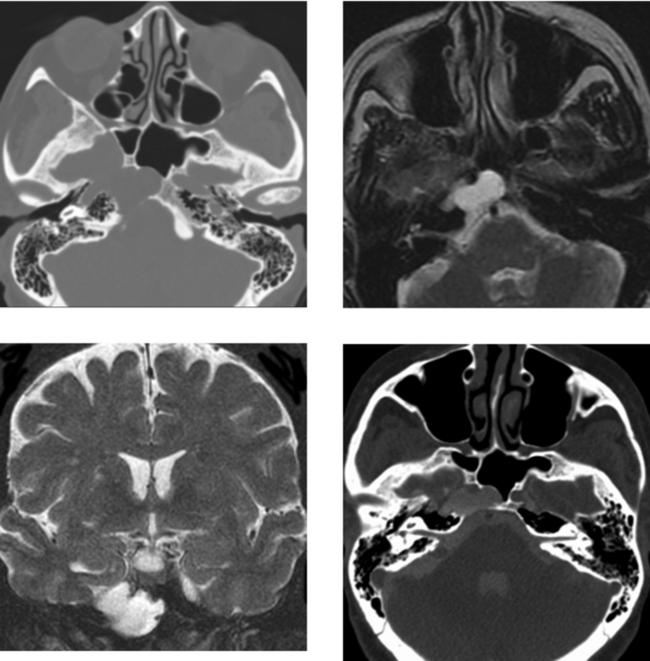
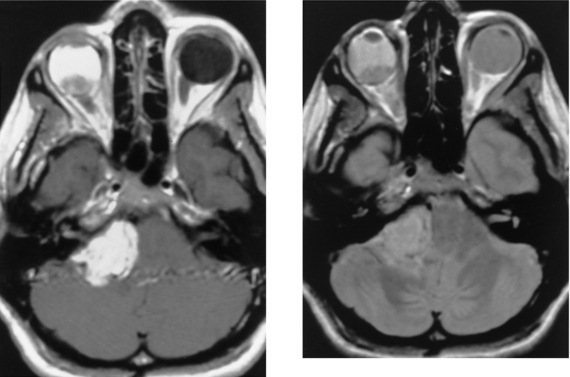
CASE 155 Meningocele and Pseudomeningocele of the Skull Base (Petrous Apex)
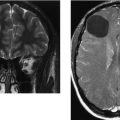
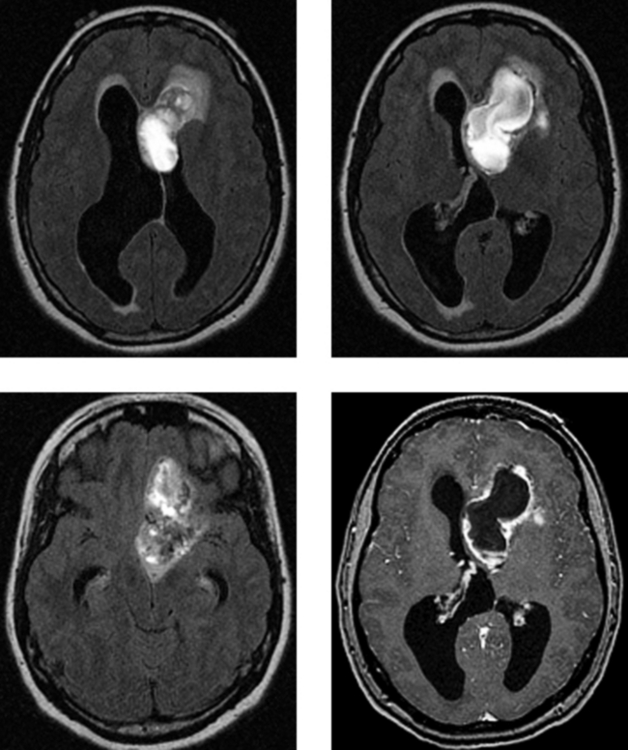
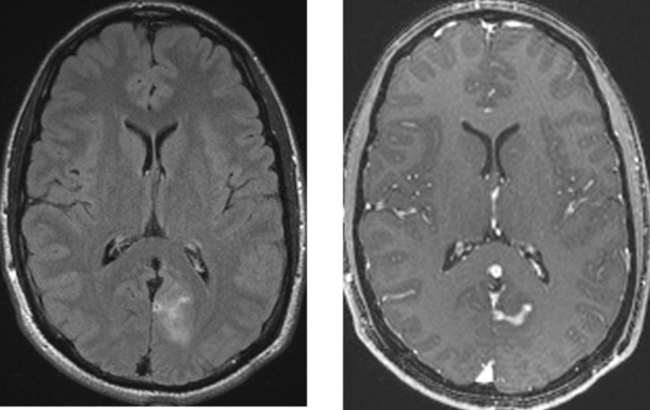
CASE 156 Right Thalamic Glioblastoma
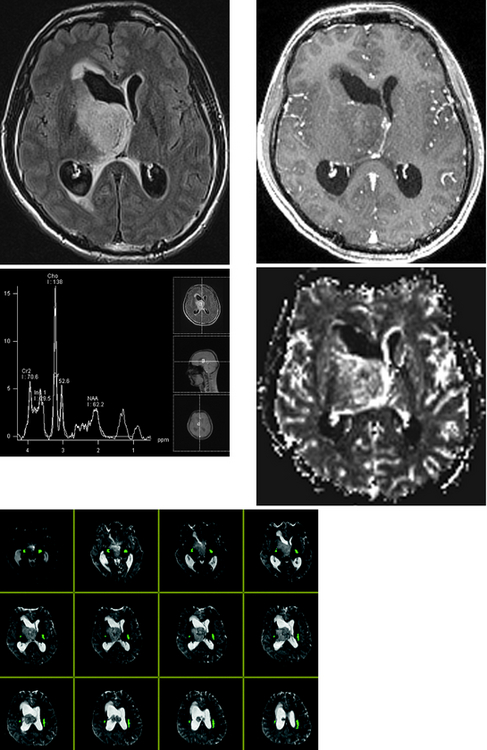
Earnest FIV, Kelly PJ, Scheithauer BW, et al. Cerebral astrocytomas: histopathologic correlation of MR and CT contrast enhancement with stereotactic biopsy. Radiology. 1988;166:823-827.
Lupo JM, Cha S, Chang SM, Nelson SJ. Analysis of metabolic indices in regions of abnormal perfusion in patients with high grade glioma. AJNR Am J Neuroradiol. 2007;28:1455-1461.
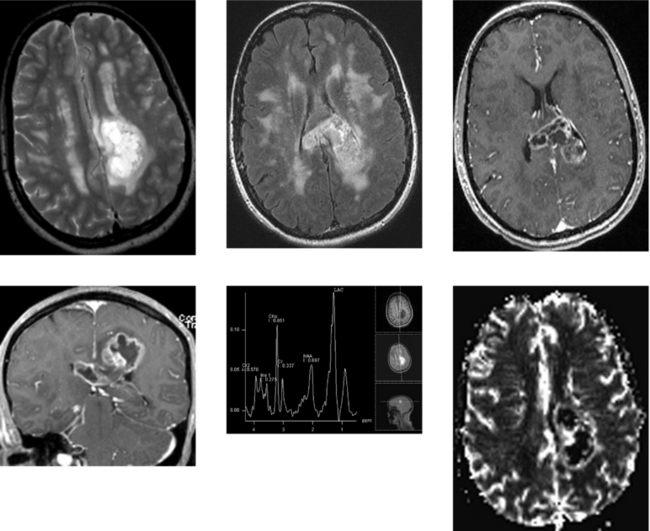
CASE 157 Multiple Sclerosis and Glioblastoma
CASE 158 Adrenoleukodystrophy
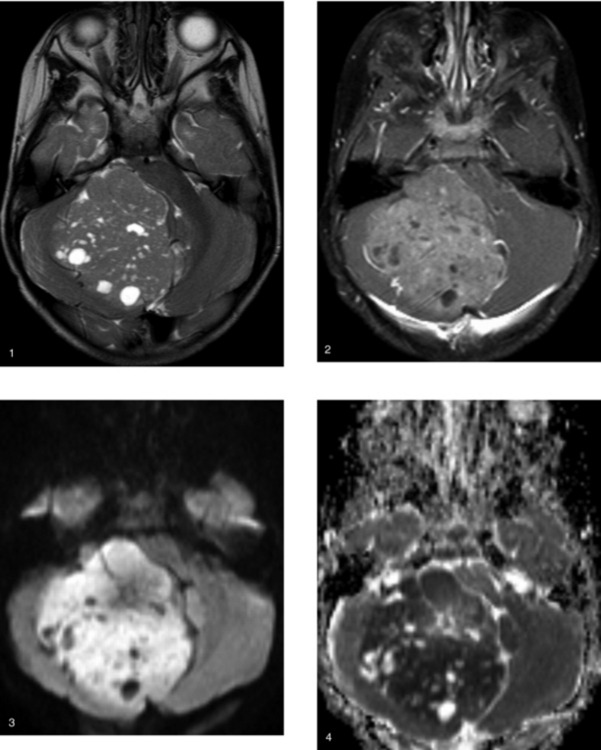
CASE 159 Medulloblastoma
CASE 160 Middle Cerebral Artery Territory Stroke Mimicking a Bone Metastasis
CASE 161 Cytomegalovirus Meningitis and Ependymitis in a Patient with AIDS
Boska MD, Mosley RL, Nawab M, et al. Advances in neuroimaging for HIV-1 associated neurological dysfunction: clues to diagnosis. pathogenesis and therapeutic monitoring, Curr HIV Res. 2004;2:61-68.
Vinters HV, Kwok MK, Ho HW, et al. Cytomegalovirus in the nervous system of patients with the acquired immune deficiency syndrome. Brain. 1989;112:245-268.
CASE 162 Upper Brainstem and Thalamic Cavernous Malformation
Porter RW, Detwiler PW, Spetzler RF, et al. Cavernous malformations of the brainstem: experience with 100 patients. J Neurosurg. 1999;90:50-58.
Zausinger S, Yousry I, Brueckmann H, Schmid-Elsaesser R, Tonn JC. Cavernous malformations of the brainstem: three-dimensional-constructive interference in steady-state magnetic resonance imaging for improvement of surgical approach and clinical results. Neurosurgery. 2006;58:322-330.
CASE 163 Capillary Telangiectasia and Developmental Venous Anomaly of the Brainstem
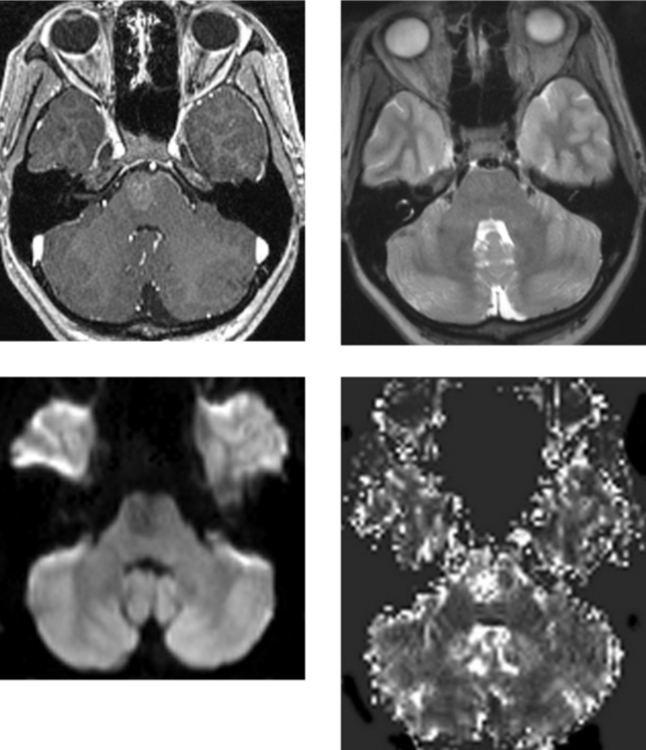
CASE 164 Central Neurocytoma
CASE 165 Marchiafava-Bignami Disease
Arbelaez A, Pajon A, Castillo M. Acute Marchiafava-Bignami disease: MR findings in two patients. AJNR Am J Neuroradiol. 2003;24:1955-1957.
Kawarabuki K, Sakakibara T, Hirai M, Yoshioko Y, Yamamoto Y, Yamaki T. Marchiafava-Bignami disease: magnetic resonance imaging findings in corpus callosum and subcortical white matter. Eur J Radiol. 2003;48:175-177.
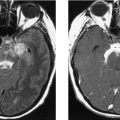
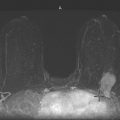
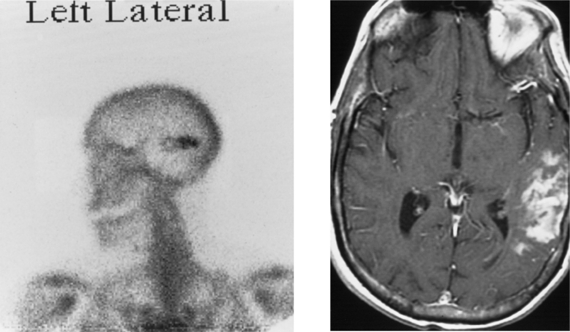
CASE 166 Metastatic Carcinoid to the Orbital Extraocular Muscles
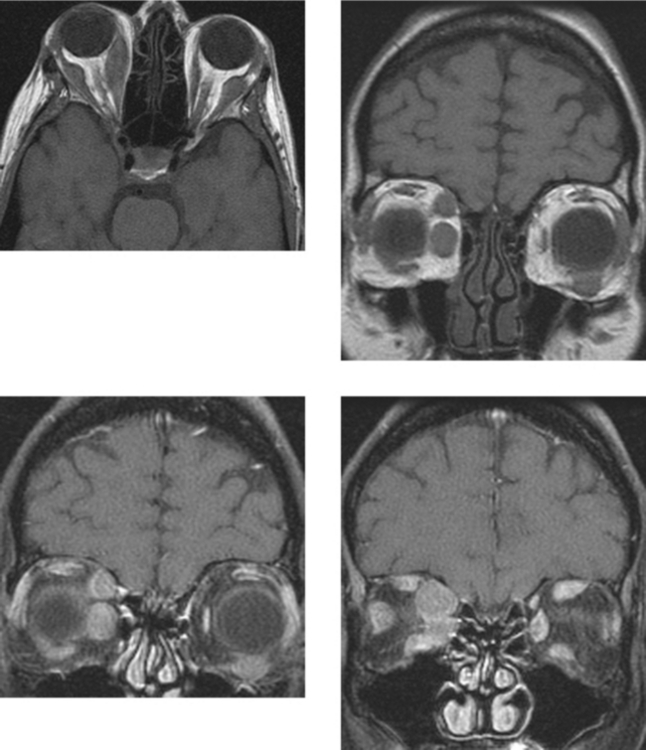
Borota OC, Kloster R, Lindal S. Carcinoid tumour metastatic to the orbit with infiltration to the extraocular orbital muscle. APMIS. 2005;113:135-139.
Hanson MW, Schneider AM, Enterline DS, Feldman JM, Gockerman JP. Iodine-131-mataiodobenzylguanidine uptake in metastatic carcinoid tumor to the orbit. J Nucl Med. 1998;39:647-650.
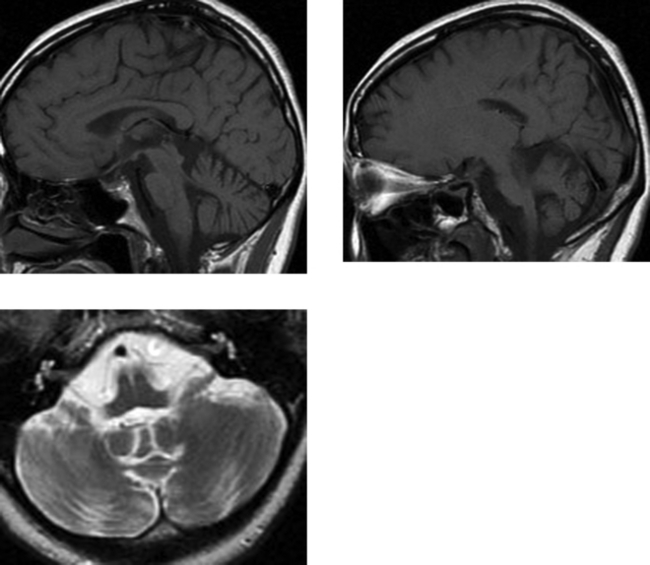
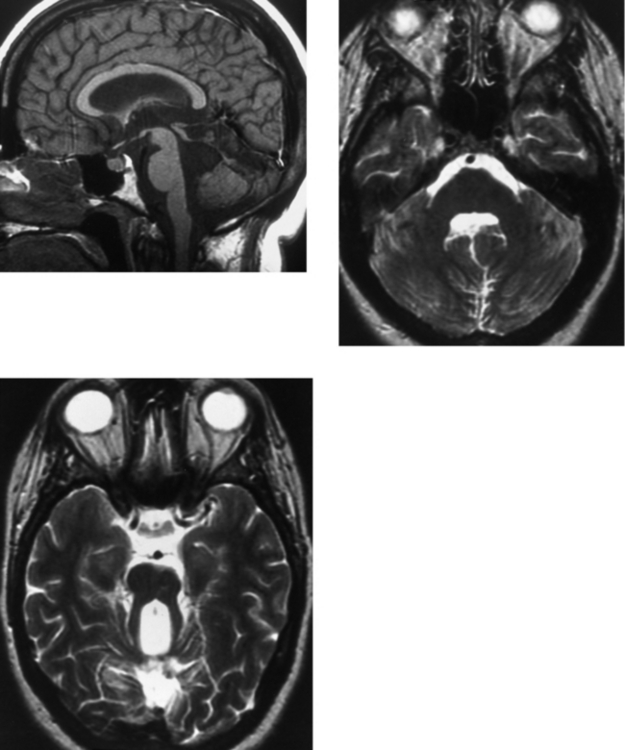
CASE 167 Joubert’s Syndrome
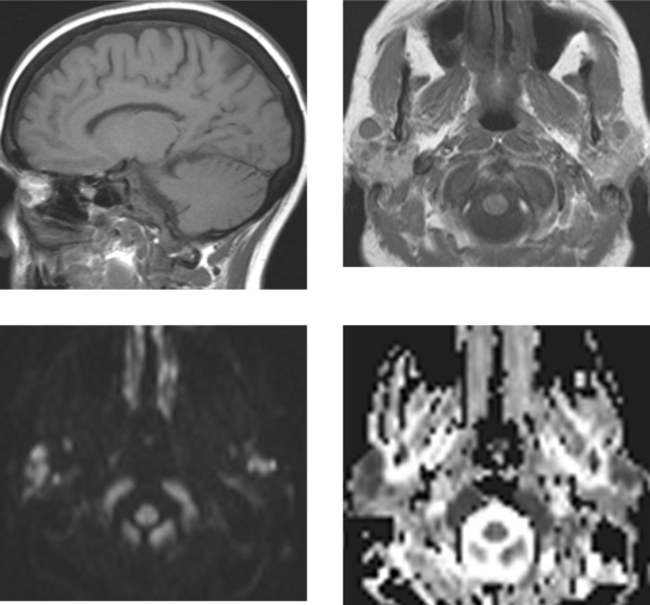
CASE 168 Systemic Non-Hodgkin’s Lymphoma
CASE 169 Abnormal Iron Deposition in Deep Gray Matter Nuclei
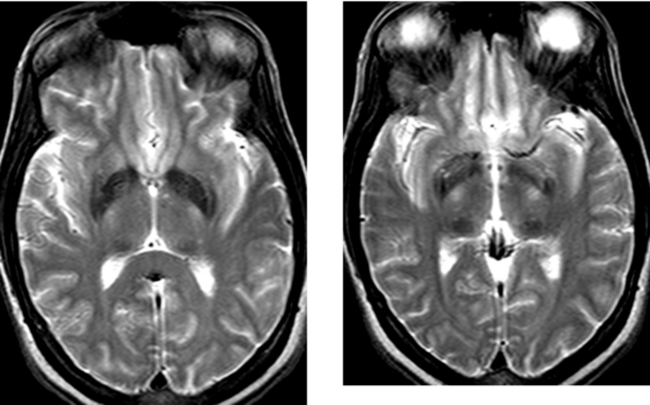
Loevner LA, Shapiro RM, Grossman RI, Overhauser J, Kamholz J. White matter changes associated with partial deletion of the long-arm of chromosome 18 (18q-syndrome): a dysmyelinating disorder? AJNR Am J Neuroradiol. 1996;17:1843-1848.
Milton WJ, Atlas SW, Lexa FJ, Mozley PD, Gur RE. Deep gray matter hypointensity patterns with aging in healthy adults: MR imaging at 1.5 T. Radiology. 1991;181:715-719.
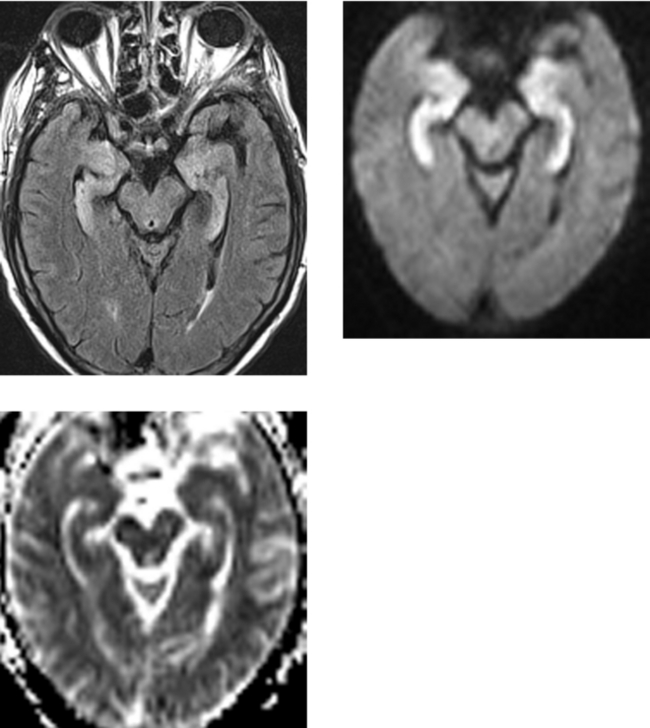
CASE 171 Jakob-Creutzfeldt Disease
Hori M, Ishigame K, Aoki S, Araki T. Creutzfeldt-Jacob disease shown by line scan diffusion-weighted imaging. Am J Roentgenol. 2003;180:1481-1482.
Mao-Drayer Y, Braff SP, Nagle KJ, Pendlebury W, Penar PL, Shapiro RE. Emerging patterns of diffusion-weighted MR imaging in Creutzfeldt-Jakob disease: case report and review of the literature. AJNR Am J Neuroradiol. 2002;23:550-556.
CASE 173 Carotid–Cavernous Fistula—Part I
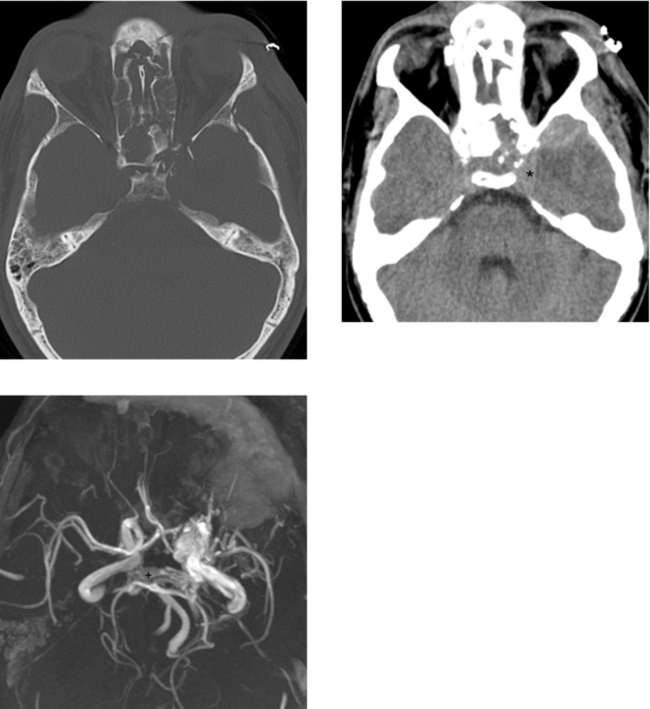
CASE 174 Carotid–Cavernous Fistula—Part II
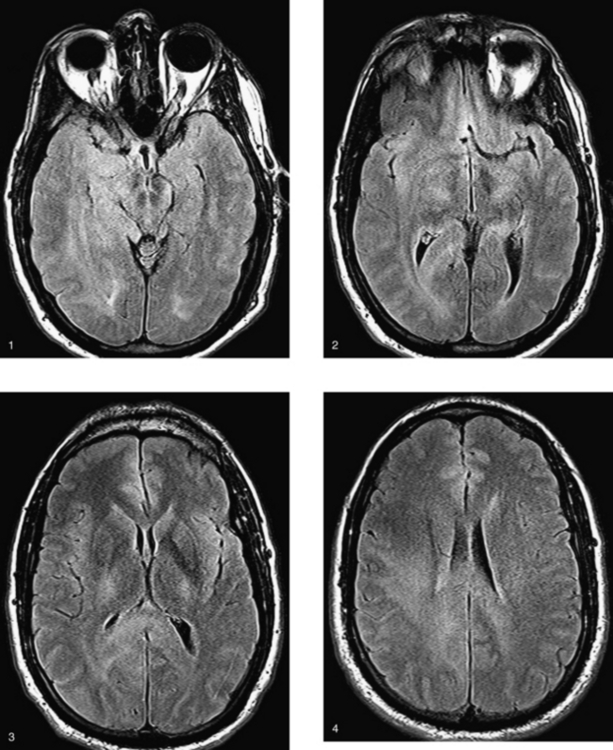
CASE 175 Gliomatosis Cerebri
CASE 176 Canavan Disease
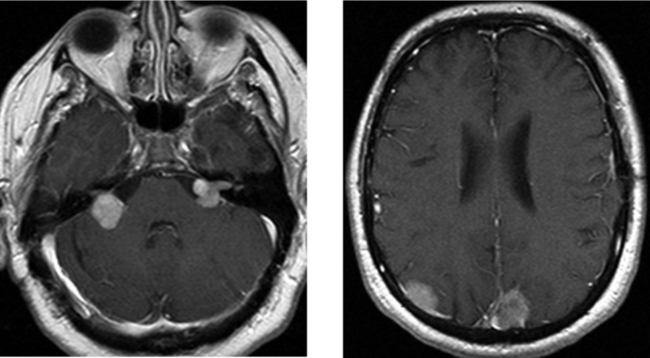
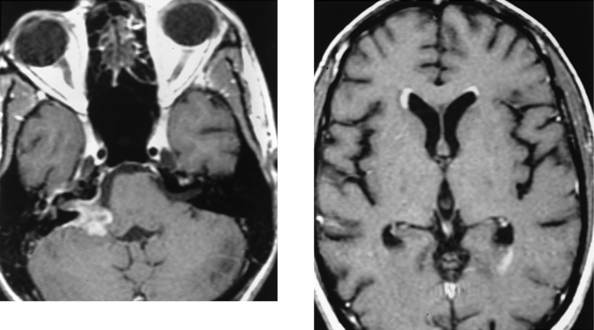
CASE 178 Metastasis to the Cerebellopontine Angle—Ocular Melanoma
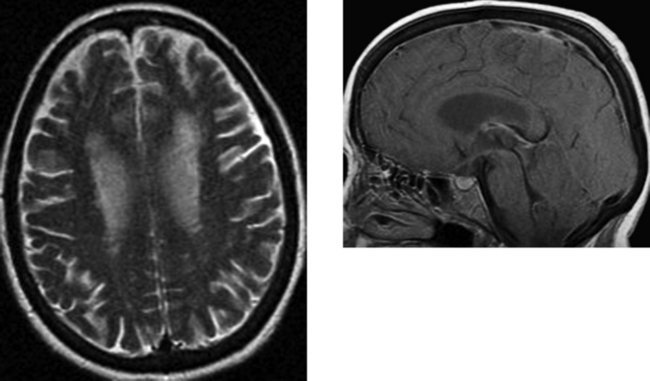
CASE 179 Retained Gadolinium—Renal Failure
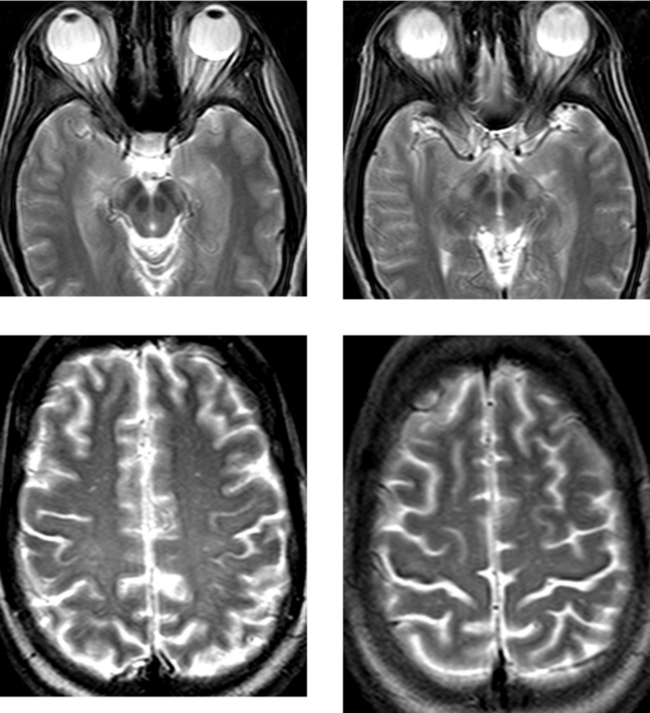
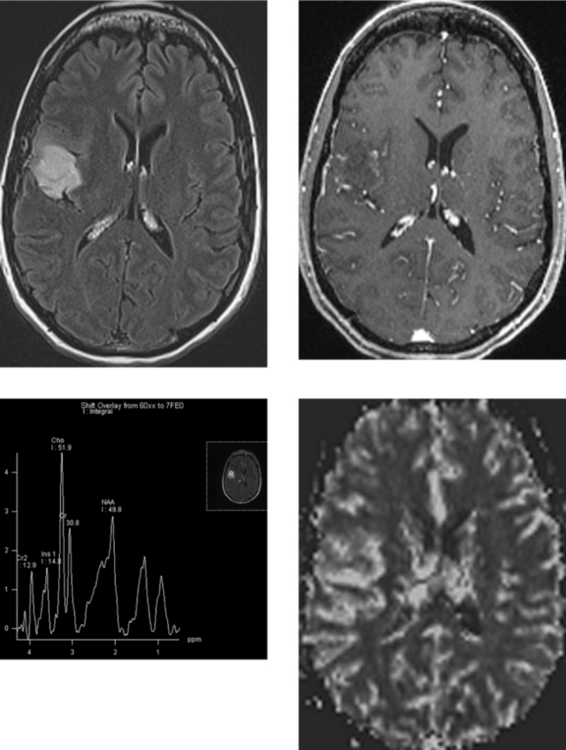
CASE 181 High-Grade Anaplastic Astrocytoma
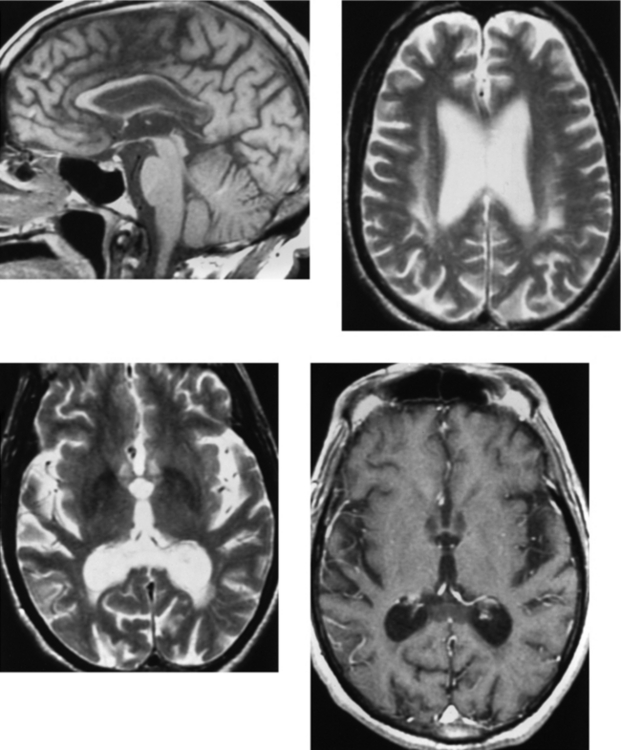
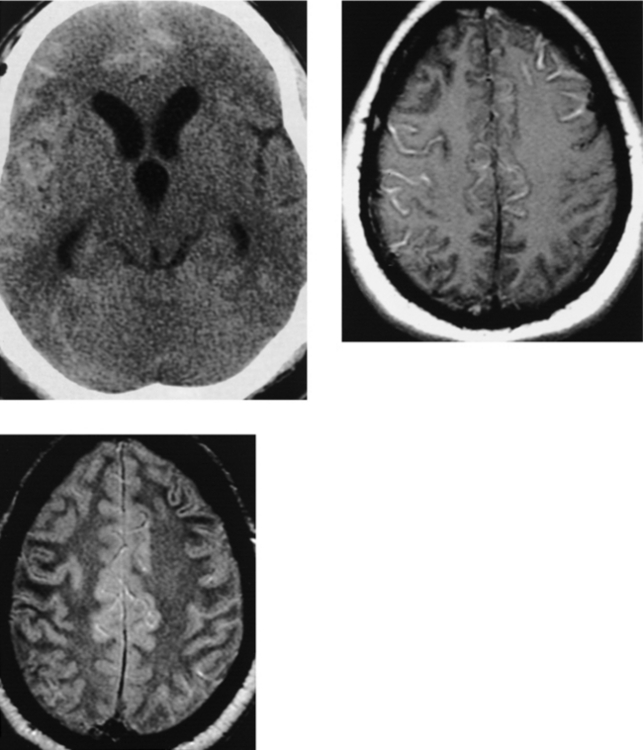
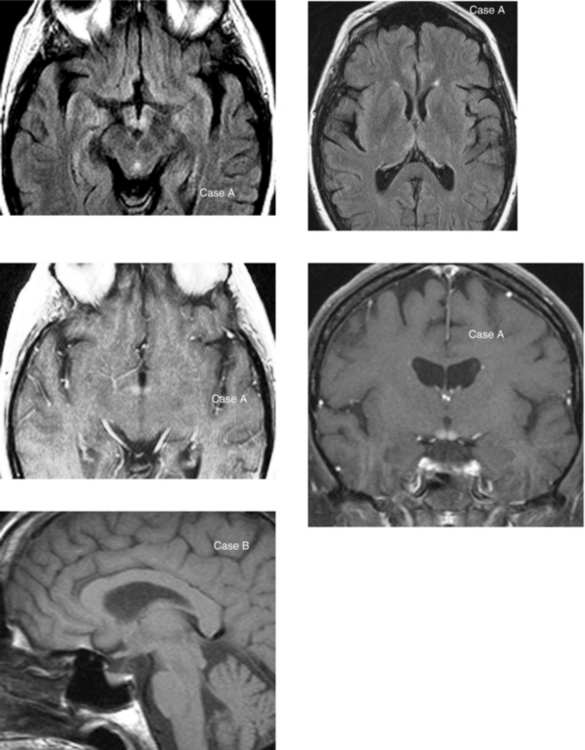
CASE 182 Wilson’s Disease
Akhan O, Akpinar, Oto A, et al. Unusual findings in Wilson’s disease. Eur Radiol. 2002;12(Suppl 3):S66-S69.
van Wassenaer-van Hall HN, van den Heuvel AG, Algra A, Hoogenrad TU, Mali WP. Wilson’s disease: findings at MR imaging and CT of the brain with clinical correlation. Radiology. 1996;198:531-536.
CASE 183 Pleomorphic Xanthoastrocytoma
CASE 184 Primary Leptomeningeal Melanosis
Byrd SE, Reyes-Mugica M, Darling CF, Chou P, Tomita T. MR of leptomeningeal melanosis in children. Eur J Radiol. 1995;20:93-99.
Hayashi M, Maeda M, Majji T, et al. Diffuse leptomeningeal hyperintensity on fluid attenuated inversion recovery MR images in neurocutaneous melanosis. AJNR Am J Neuroradiol. 2004;25:138-141.
CASE 185 Dural Arteriovenous Fistula—Part I
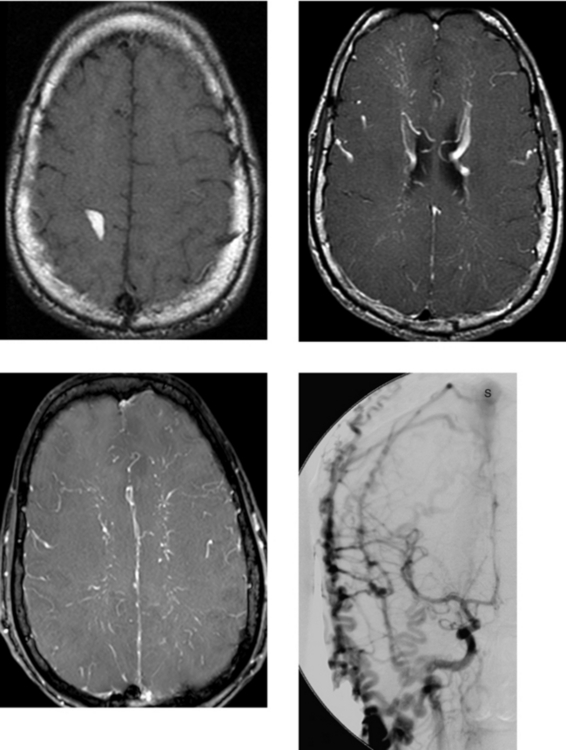
CASE 186 Dural Arteriovenous Fistula—Part II
CASE 187 Fetal Schizencephaly
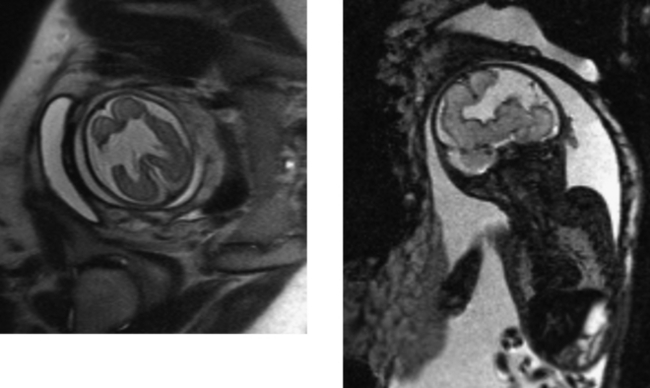
Ceccherini AF, Twining P, Varied S. Schizencephaly: antenatal detection using ultrasound. Clin Radiol. 1999;54:620-622.
Glenn OA, Barkovich J. Magnetic resonance imaging of the fetal brain and spine: an increasingly important tool in prenatal diagnosis: part 2. AJNR Am J Neuroradiol. 2006;27:1807-1814.
CASE 188 Wernicke Encephalopathy
Mascalchi M, Belli G, Guerrini L, Nistri M, Del Seppia I, Villari N. Proton MR spectroscopy of Wernicke encephalopathy. AJNR Am J Neuroradiol. 2002;23:1803-1806.
Weidauer S, Nichtweiss M, Lanfermann H, Zanella FE. Wernicke encephalopathy: MR findings and clinical presentation. Eur Radiol. 2003;13:1001-1009.
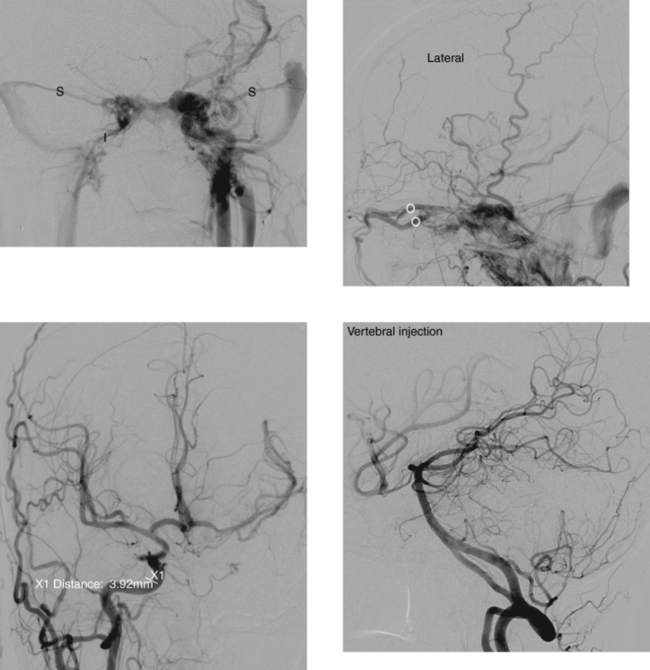
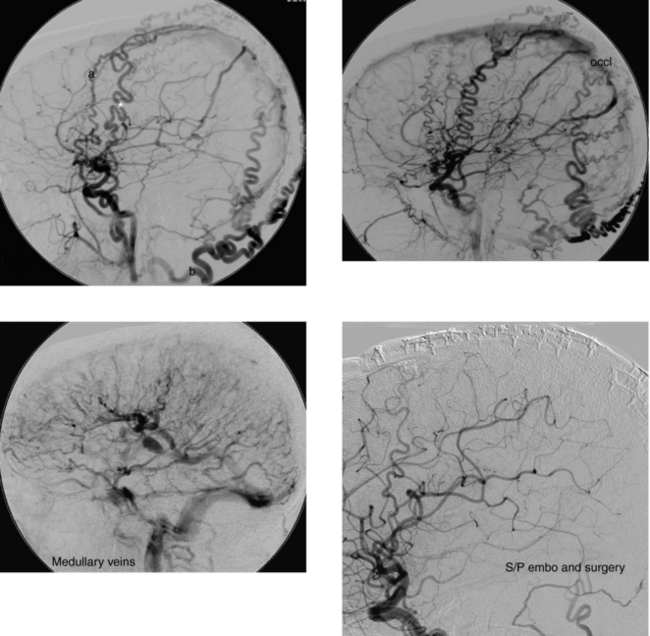
CASE 189 Ruptured Mycotic Aneurysm
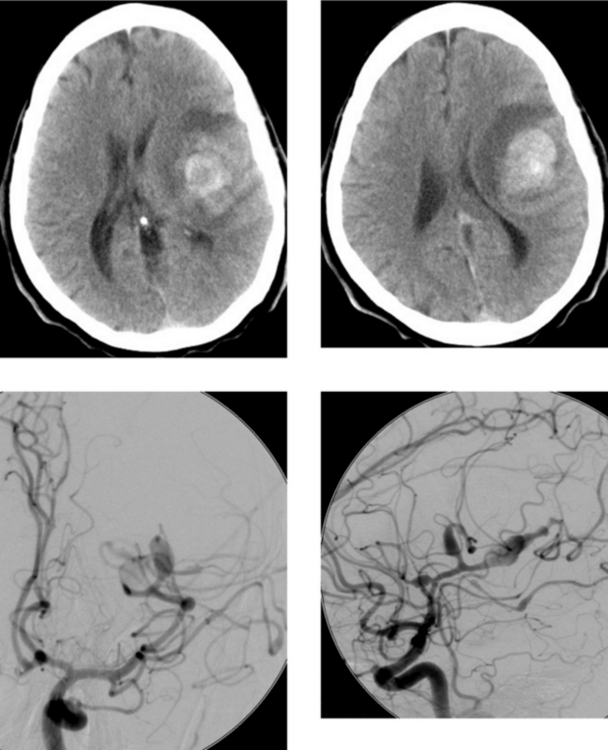
Cloft HJ, Kallmes DF, Jensen ME, Lanzino G, Dion JE. Endovascular treatment of ruptured, peripheral cerebral aneurysms: parent artery occlusion with short Guglielmi detachable coils. AJNR Am J Neuroradiol. 1999;20:308-310.
Loevner LA, Ting TY, Hurst RW, Goldberg H, Schut L. Spontaneous thrombosis of a basilar artery traumatic aneurysm in a child. AJNR Am J Neuroradiol. 1998;19:386-388.
CASE 190 Osmotic Demyelination
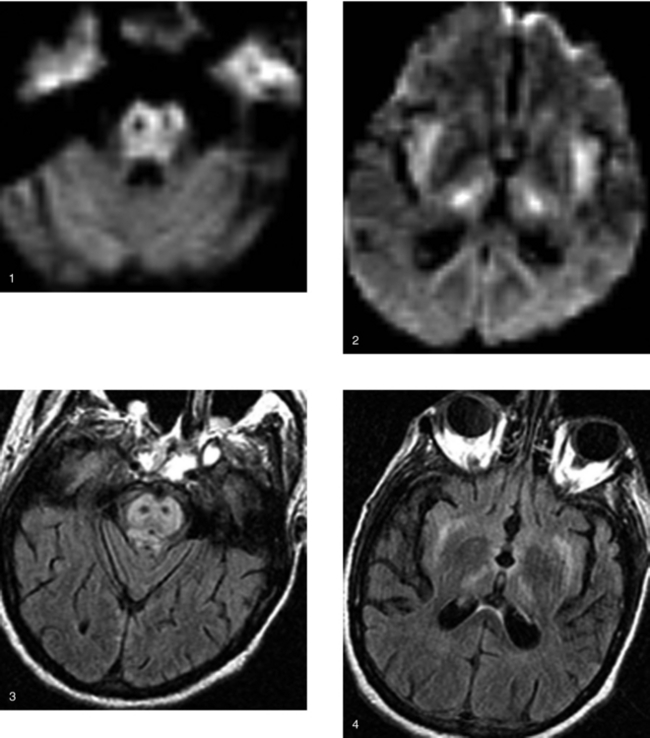
CASE 191 Rosai-Dorfman Disease—Benign Sinus Histiocytosis
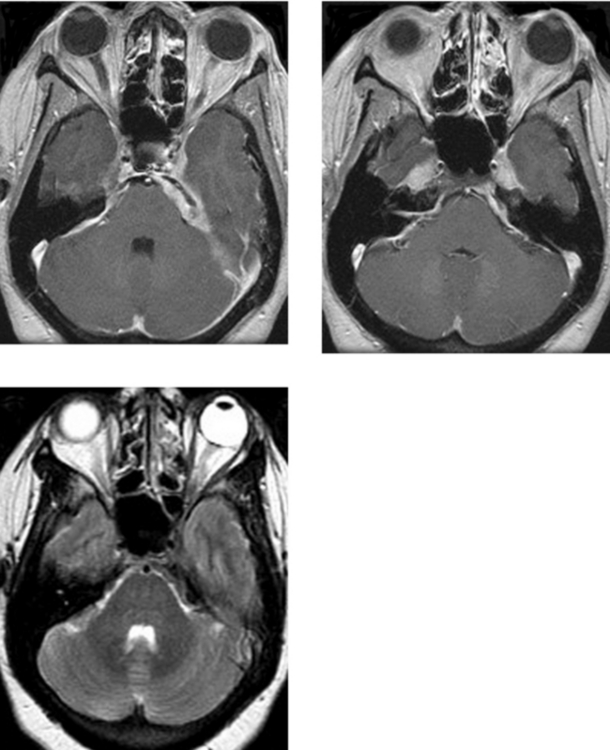
CASE 192 Cystic Necrotic Brain Mass—Part I
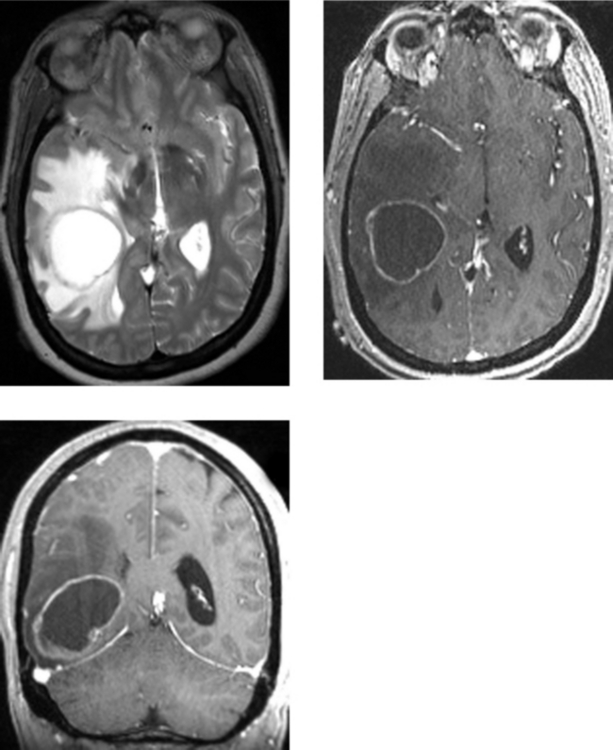
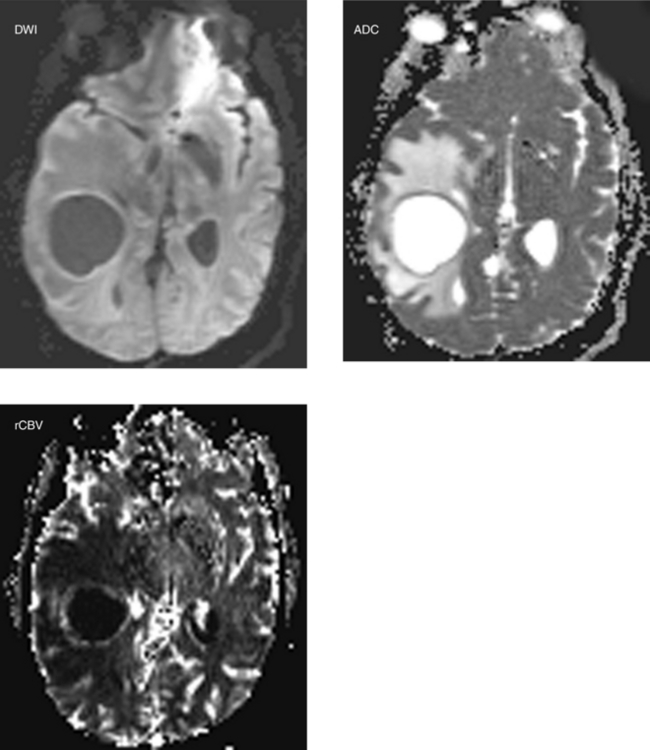
CASE 193 Cystic Necrotic Brain Mass—Part II—Metastatic Adenocarcinoma of the Lung
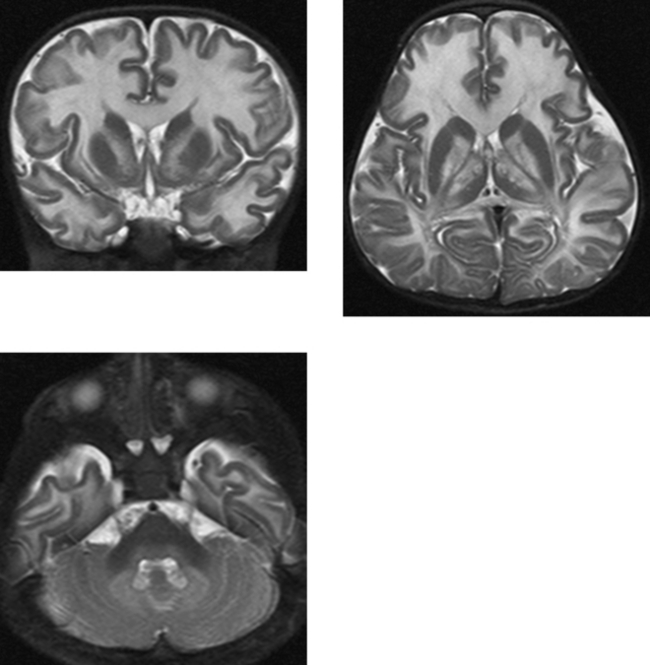
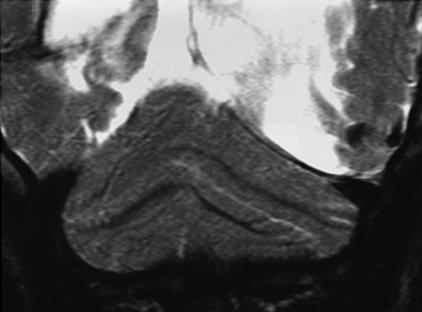
CASE 194 Rhombencephalosynapsis
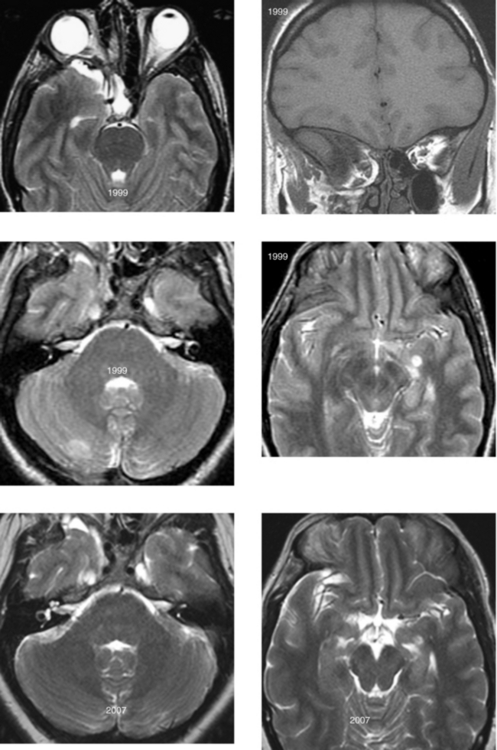
CASE 195 Progressive Multifocal Leukoencephalopathy
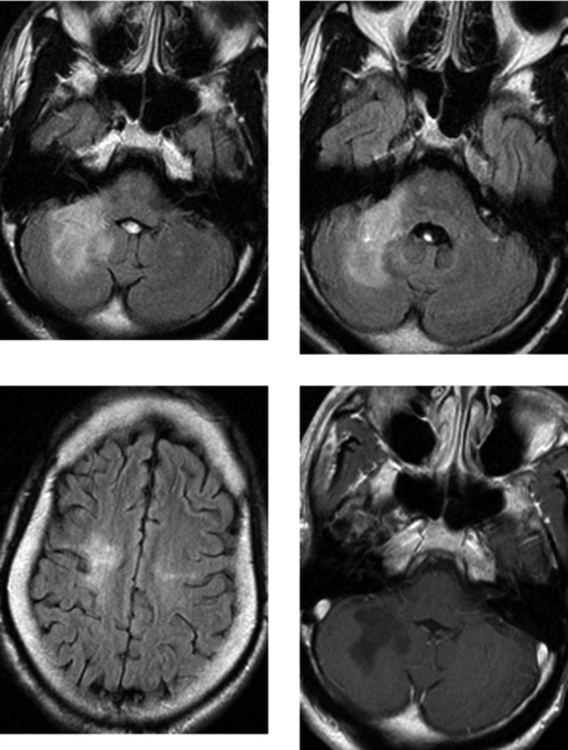
Mader I, Herrlinger U, Klose U, Schmidt F, Kuker W. Progressive multifocal leukoencephalopathy: analysis of lesion development with diffusion-weighted MRI. Neuroradiology. 2003;45:717-721.
Thurnher MM, Post MJ, Rieger A, Kleibl-Popov C, Loewe C, Schindler E. Initial and follow-up MR imaging findings in AIDS-related progressive multifocal leukoencephalopathy treated with highly active antiretroviral therapy. AJNR Am J Neuroradiol. 2001;22:977-984.
CASE 196 Sphenoid Dysplasia and Brain Dysplasia—Neurofibromatosis Type 1 (von Recklinghausen’s Disease)
CASE 197 Panhypopituitarism with Absence of the Infundibulum
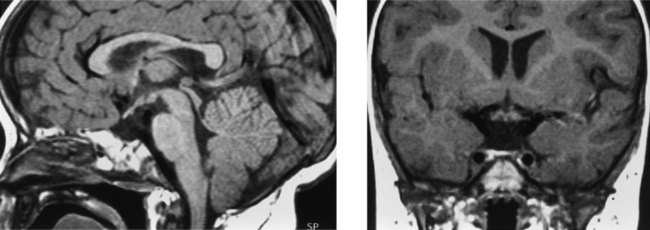
Caruso RD, Postel GC, McDonald CS, et al. High signal on T1-weighted MR images of the head: a pictorial essay. Clin Imaging. 2001;25:312-319.
Saeki N, Hayasaka M, Murai H, et al. Posterior pituitary bright spot in large adenomas: MR assessment of its disappearance or relocation along the stalk. Radiology. 2003;226:359-365.
CASE 198 Progressive External Ophthalmoplegia Syndrome—due to Mitochondrial Defect
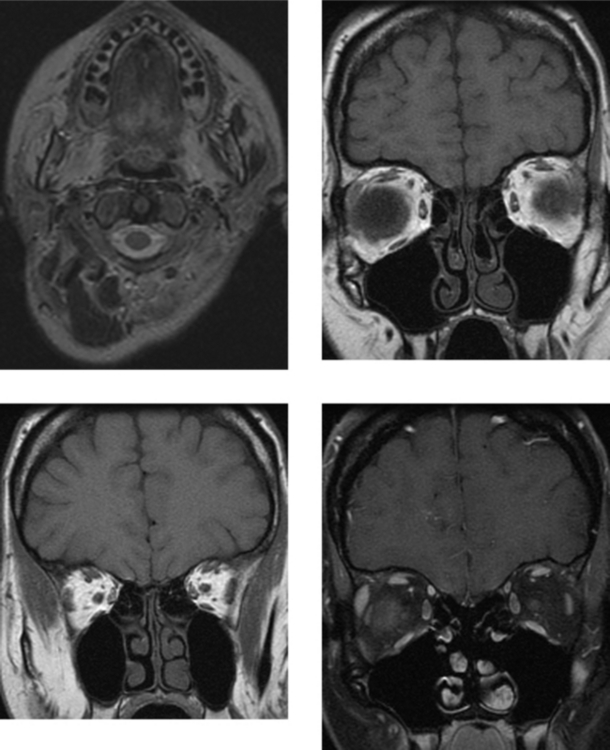
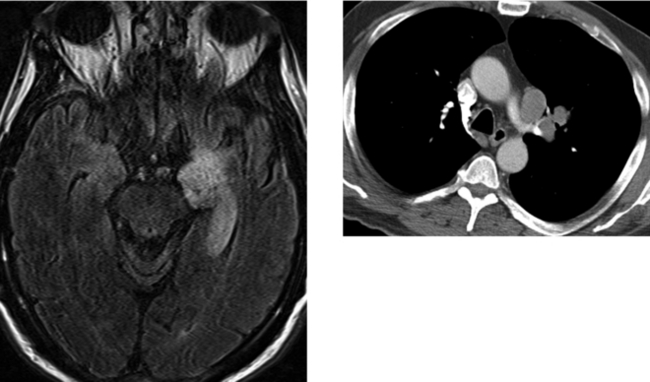
CASE 199 Limbic Encephalitis—Paraneoplastic Syndrome (Small Cell Lung Carcinoma)
Aguirre-Cruz et al. 2005 Aguirre-Cruz L, Charuel JL, Carpentier AF, et al. Clinical relevance of non-neuronal auto-antibodies in patients with anti-Hu or anti-Yo paraneoplastic diseases. J Neurooncol. 2005;71:39-41.
Ances et al. 2005 Ances BM, Vitaliani R, Taylor RA, et al. Treatment-responsive limbic encephalitis identified by neuropil antibodies: MRI and PET correlates. Brain. 2005;128:1764-1777.