[level-membership-for-cardiovascular-category]
Chapter 7 Central nervous system control of cardiovascular function
The brain exerts powerful effects on cardiovascular behaviour, predominantly by altering sympathetic motor drive to the heart and to vascular smooth muscle. While you may read about sympathetic activation specifically in relation to cardiac function or arteriolar resistance or venous capacitance, depending on the topic being discussed, the truth is that centrally mediated sympathetic activation almost always involves increased drive to both heart and blood vessels and in non-selective increases in vasomotor drive to arteries and veins. Therefore, heart rate and blood pressure usually change in parallel. This parallelism is convenient because it means that changes in any of these parameters can be used as an (approximate) index of sympathetic activation. In practice, the parameter used routinely is the change in heart rate, because ECGs can be recorded continually in moving subjects so much more easily than blood pressure.
ORGANIZATION OF CENTRAL CARDIOVASCULAR CONTROL
Although sympathetic outflow to the cardiovascular system can be affected by pathways arising in many areas of the brain, there are for our purposes three primary sources of this activation (Fig. 7.1). The first is the cardiovascular control centre lying in the medulla of the hindbrain, which is the final common pathway for all descending sympathetic information. This hindbrain centre receives reflex inputs directly from peripheral sensory nerves, but also receives descending inputs from two areas of the forebrain. The most important of these are the limbic system, which includes the hypothalamus and several neighbouring nuclei, mediates emotional arousal and determines levels of consciousness, and the motor cortex from which all instructions for muscle movement must originate.
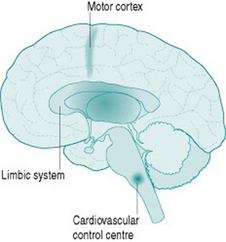
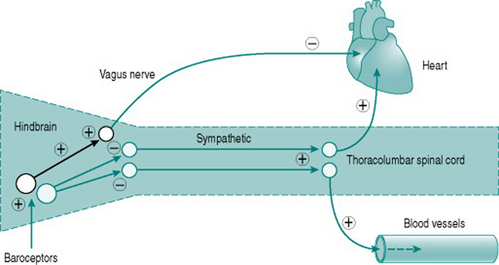
Neuroscientists are still unravelling the specific neural connections and neurotransmitters involved in mediation of hindbrain regulation of the cardiovascular system. However, we can regard the cardiovascular control centre as consisting of two sets of tonically active descending neurons, one producing continual activation of the sympathetic outflows to heart and blood vessels and one producing continual inhibition of heart rate via the vagus (Fig. 7.2). As a result of this ongoing activity, there is usually some degree of vasoconstrictor tone in both arterial and venous vessels, while the heart rate at rest reflects the algebraic sum of inputs to the sinoatrial node of both sympathetic and vagal activity.
LOW-PRESSURE AND HIGH-PRESSURE BAROREFLEXES
Baroreflex effects on sympathetic cardiovascular control
The moment-to-moment regulation of blood pressure is via reflexes that operate through inputs to the hindbrain centre from mechanosensitive sensory axons monitoring pressure in the aorta and the carotid arteries (high-pressure baroreceptors) and in the atria (low-pressure baroreceptors). In all cases, increased distension results in increased sensory firing and the sensory neurons inhibit activity of the hindbrain centre sympathetic neurons. In consequence, either increased blood pressure or increased central blood volume will reduce peripheral resistance, while falls in blood pressure or central blood volume have the opposite effects.
Low-pressure baroreflexes have a distinct pattern of cardiac and vascular responses, with reflex falls in peripheral resistance caused by increased atrial filling being usually accompanied by tachycardia. However, increases in atrial volume that raise stroke volume will also induce an arterial baroreflex and bradycardia. This means that heart rate changes may be an uncertain index of baroreflex activity since their magnitude will vary depending on the algebraic sum of low-pressure and high-pressure receptor inputs.
Baroreflex effects on hormonal cardiovascular control
Activation of low-pressure baroreceptors by atrial distension sends inhibitory inputs to the hypothalamus that depress release of vasopressin (antidiuretic hormone, ADH) from the posterior pituitary gland. As a result, less water is reabsorbed from the renal collecting ducts and plasma volume is reduced. Conversely, reduced atrial baroreceptor activity due to lower atrial pressure will increase vasopressin release, retaining water and elevating plasma volume.
AROUSAL AND SYMPATHETIC CARDIOVASCULAR CONTROL: THE LIMBIC SYSTEM
The association of sympathetic activity with arousal means that performing a mental task will produce significant elevation of heart rate and blood pressure in a resting subject. This provides a simple laboratory technique for assessing the gain of sympathetic cardiovascular drive.
In man, emotionally oriented arousal can cause profound forearm muscle vasodilation and this response has been shown in some studies to be dependent on sympathetic nerve integrity and abolished by acetylcholine antagonists, such as atropine. Other studies, by contrast, have found the response to be independent of local sympathetic innervation and sensitive to β-adrenoreceptor antagonists or inhibitors of nitric oxide, but not to atropine. Other studies again report that emotional stimuli evoke only generalized vasoconstriction. The basis of arousal-linked muscle dilatation in man is, therefore, still uncertain, except in so far that it is clearly not as straightforward as in some other species (Joyner & Dietz 2003). More importantly, it remains unknown whether such a response would confer any physiological advantage to muscle perfusion on initiation of exercise over and above that provided by local dilator factors released from the contracting muscle cells (see Chapter 6, p. 64). If there were a functional role, then it might be postulated to be most likely of significance for short, repetitive, sub-tetanic activity that does not release sufficient local factors from the muscle cells to induce optimal dilatation. So, for example, a role in playing musical instruments is possible. However, a role in whole-body exercise seems unlikely, although the rise in blood pressure induced by limbic arousal may itself provide some instantaneous benefit for muscle perfusion over the first few seconds of an anticipated exercise.
SYMPATHETIC CARDIOVASCULAR CONTROL DURING EXERCISE
Pressor effect of exercise
Sites of peripheral resistance increase
During exercise, absolute blood flow to skeletal and cardiac muscles rises with work intensity, reflecting the increased local metabolic needs and due in the main to a variety of local dilator processes (see Chapter 6, pp. 72–74). The maximum levels of flow achieved reflect the increments in tissue activity that occur, being 10–20-fold for skeletal muscle (from 1 l/min to 10–20 l/min) and around four-fold for the coronary circulation (from 250 ml/min to 1 l/min). However, the sympathetic activation that accompanies exercise is not region-specific, so there is increased vasoconstrictor drive to skeletal muscle and the heart as well as to splanchnic and renal beds. Thus, the maximum nutritional perfusion that can be achieved in these tissues are less than would be predicted on the basis of the dilator stimuli alone.
Several aspects of this interaction between dilator and constrictor processes bear mention. First, the presence of sympathetic vasoconstriction in skeletal muscle contributes to total peripheral resistance during intense dynamic exercise, and appears to be essential in order to allow maintenance of adequate blood pressure. Second, there is evidence that at near-maximum dynamic exercise intensities, muscle oxygenation may become limited by the sympathetic constrictor effect (Mortensen et al 2005). Third, although vasoconstriction limits the blood flow to exercising skeletal muscle to levels substantially lower than would exist if there were unopposed dilatation, nutritional capillary perfusion during exercise is significantly greater than indicated by the fall in muscle vascular resistance. This is because only around 30% of the capillary bed is perfused in resting skeletal muscle (see Chapter 6, p. 72). Recruitment of the remainder of the capillary bed by metabolite-induced reduction of metarteriolar critical closing pressure provides a threefold elevation in tissue perfusion before there is any requirement for reduced arteriolar resistance.
There is evidence that in the coronary circulation also, sympathetic activation has less effect on nutritional perfusion than is suggested by the absolute effect on regional resistance. This is due to the fact that, in the coronary vasculature, the α-adrenoreceptors are localized primarily to the largest arterioles and small arteries. The constrictor effect of sympathetic activation on these larger vessels stiffens them and actually increases flow to the deeper regions of myocardium, presumably because it reduces external compression during cardiac contraction (Tune et al 2004).
Origin of signals for sympathetic activation
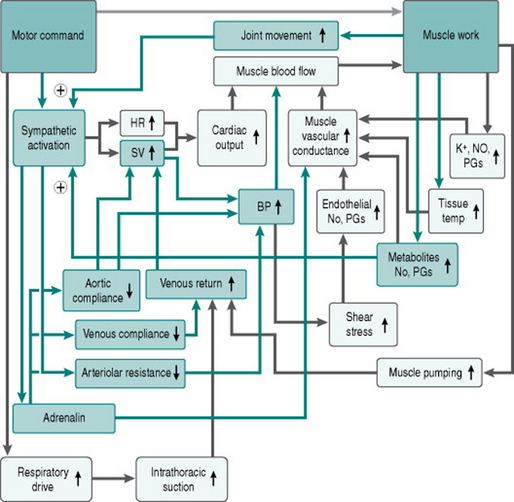
All forms of exercise depend on descending information from the upper motor neurons of motor cortex to the lower motor neurons controlling muscle contraction. The upper motor neurons also send excitatory inputs to the cardiovascular control centre, so that peripheral vasoconstriction, tachycardia and increased cardiac contraction occur proportionately to both the amount of musculature activated and the intensity of activation. This input to cardiovascular regulation from the motor cortex is known as central command. Although it has traditionally been viewed as originating solely from the cortex, recent studies using imaging and electrophysiological recording indicate that areas of the limbic system are also involved (Green et al 2007).
Two additional signals provide additional input to the cardiovascular control centre. Mechanoreceptors in the limb joints and in the limb musculature give information on the amount and speed of muscle contraction. As well, chemoreceptors within the muscle interstitium detect accumulation of metabolites. Both these signals have the capacity to enhance the sympatho-excitatory effect of central command during any modality of exercise, but limb mechanoreceptors will be activated predominantly during dynamic exercise, while chemoreceptors are activated far more during static exercise when muscle perfusion is minimized by external compression. Evidence from studies of static exercise indicates also that the effect of chemoreceptor activation (the so-called metaboreflex) is preferentially on vasoconstrictor function, with relatively little effect on cardiac function.
Figure 7.3 adds the factors discussed above to our flow chart of the exercise response.
Comparison of dynamic and static exercise
The different contributions from different signals of motor activity, together with the circulatory effects of the exercise itself, result in distinct patterns of pressor and cardioaccelerator responses to dynamic and to static exercise. During dynamic exercise, sympathetic activation is determined primarily by central command, so that maximal tachycardia and blood pressure are achieved only when a large proportion of the whole-body musculature is involved. Under these conditions, heart rate will attain its age-dependent maximum (see Chapter 4, p. 32) and SBP will rise substantially due to the combined effects of tachycardia, increased stroke volume and reduced aortic compliance. DBP is not usually elevated during dynamic exercise because of the large fall in total peripheral resistance. At low-to-moderate workloads, the reduced resistance is balanced by tachycardia and DBP remains unchanged from the resting value (Fig. 7.4A). At high workloads in the upright posture, by contrast, DBP often begins to fall because of the profound reduction in total peripheral resistance caused by maximal muscle vasodilation.
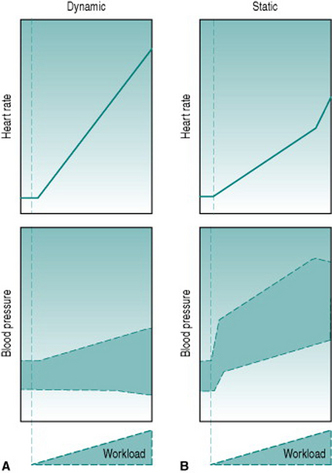
The picture seen during static exercise is strikingly different (Fig. 7.4B). For one thing, substantial elevation of SBP and heart rate occurs not only with whole-body exercise, but is seen with contraction of quite small muscle groups, such as those controlling handgrip. This is due in a large part to metaboreflex input, secondary to mechanical compression of the muscle vascular supply. In addition, since peripheral resistance does not fall during static exercise, but actually rises due to compression of the muscle vasculature, DBP rises with workload. Finally, increased intra-abdominal and intra-thoracic pressures during whole-body static activity progressively reduce venous return and stroke volume, leading to blood pressure actually falling slightly at peak workload.
Comparison of upper-body and lower-body exercise
Selective upper-body exercise constitutes the basis of fitness training for patients with limited mobility or spinal cord injury. The heart rate achieved at maximum workload is similar to that seen with whole-body exercise, but the absolute achievable workload and the maximum cardiac output are lower because of the smaller muscle mass involved. In addition, heart rate increments are greater for given increments of work than when whole-body exercise takes place, because the absence of muscle pumping in the legs reduces the venous pressure gradient and limits ventricular filling (see Chapter 3, p. 22).
BAROREFLEX RESETTING IN EXERCISE
It is clear from the preceding discussion that exercise is associated with maintenance of blood pressure at levels significantly higher than that during rest. This pressure elevation serves two purposes. First, it optimizes nutritional perfusion of the various organ systems in the face of increased regional vasoconstrictor tone. Second, suddenly stopping exercise would immediately stop muscle pumping in the legs, causing a fall in venous return and cardiac output that might prejudice cerebral perfusion in the upright posture. The pre-existence of elevated sympathetic vasoconstriction makes this less likely.
POST-EXERCISE HYPOTENSION
Green AL, Wang S, Purvis, et al. Identifying cardiorespiratory neurocircuitry involved in central command during exercise in humans. Journal of Physiology. 2007;578:605-612.
Joyner MJ, Dietz NM. Sympathetic vasodilation in human muscle. Acta Physiologica Scandinavica. 2003;177:329-336.
Mortensen SP, Dawson EA, Yoshiga CC, et al. Limitations to systemic and locomotor limb muscle oxygen delivery and uptake during maximal exercise in humans. Journal of Physiology. 2005;566:273-285.
Tune JD, Gorman MW, Feigl EO. Matching coronary blood flow to myocardial oxygen consumption. Journal of Applied Physiology. 2004;97:404-415.
Biaggioni I. Autonomic/metabolic interactions modulating the exercise pressor reflex: the purinergic hypothesis. Journal of Physiology. 2007;578:5-6.
Rezk CC, Marrache RCB, Tinucci T, Mion DJr, Forjaz CLM. Post-resistance exercise hypotension, hemodynamics, and heart rate variability: influence of exercise intensity. European Journal of Applied Physiology. 2006;98:105-112.
Rowell LB. Ideas about control of skeletal and cardiac muscle blood flow (1876–2003): cycles of revision and new vision. Journal of Applied Physiology. 2004;97:384-392.
[/level-membership-for-cardiovascular-category][not-level-membership-for-cardiovascular-category]
Chapter 7 Central nervous system control of cardiovascular function
The brain exerts powerful effects on cardiovascular behaviour, predominantly by altering sympathetic motor drive to the heart and to vascular smooth muscle. While you may read about sympathetic activation specifically in relation to cardiac function or arteriolar resistance or venous capacitance, depending on the topic being discussed, the truth is that centrally mediated sympathetic activation almost always involves increased drive to both heart and blood vessels and in non-selective increases in vasomotor drive to arteries and veins. Therefore, heart rate and blood pressure usually change in parallel. This parallelism is convenient because it means that changes in any of these parameters can be used as an (approximate) index of sympathetic activation. In practice, the parameter used routinely is the change in heart rate, because ECGs can be recorded continually in moving subjects so much more easily than blood pressure.
ORGANIZATION OF CENTRAL CARDIOVASCULAR CONTROL
Although sympathetic outflow to the cardiovascular system can be affected by pathways arising in many areas of the brain, there are for our purposes three primary sources of this activation (Fig. 7.1). The first is the cardiovascular control centre lying in the medulla of the hindbrain, which is the final common pathway for all descending sympathetic information. This hindbrain centre receives reflex inputs directly from peripheral sensory nerves, but also receives descending inputs from two areas of the forebrain. The most important of these are the limbic system, which includes the hypothalamus and several neighbouring nuclei, mediates emotional arousal and determines levels of consciousness, and the motor cortex from which all instructions for muscle movement must originate.
Neuroscientists are still unravelling the specific neural connections and neurotransmitters involved in mediation of hindbrain regulation of the cardiovascular system. However, we can regard the cardiovascular control centre as consisting of two sets of tonically active descending neurons, one producing continual activation of the sympathetic outflows to heart and blood vessels and one producing continual inhibition of heart rate via the vagus (Fig. 7.2). As a result of this ongoing activity, there is usually some degree of vasoconstrictor tone in both arterial and venous vessels, while the heart rate at rest reflects the algebraic sum of inputs to the sinoatrial node of both sympathetic and vagal activity.
LOW-PRESSURE AND HIGH-PRESSURE BAROREFLEXES
Baroreflex effects on sympathetic cardiovascular control
The moment-to-moment regulation of blood pressure is via reflexes that operate through inputs to the hindbrain centre from mechanosensitive sensory axons monitoring pressure in the aorta and the carotid arteries (high-pressure baroreceptors) and in the atria (low-pressure baroreceptors). In all cases, increased distension results in increased sensory firing and the sensory neurons inhibit activity of the hindbrain centre sympathetic neurons. In consequence, either increased blood pressure or increased central blood volume will reduce peripheral resistance, while falls in blood pressure or central blood volume have the opposite effects.
Low-pressure baroreflexes have a distinct pattern of cardiac and vascular responses, with reflex falls in peripheral resistance caused by increased atrial filling being usually accompanied by tachycardia. However, increases in atrial volume that raise stroke volume will also induce an arterial baroreflex and bradycardia. This means that heart rate changes may be an uncertain index of baroreflex activity since their magnitude will vary depending on the algebraic sum of low-pressure and high-pressure receptor inputs.
Baroreflex effects on hormonal cardiovascular control
Activation of low-pressure baroreceptors by atrial distension sends inhibitory inputs to the hypothalamus that depress release of vasopressin (antidiuretic hormone, ADH) from the posterior pituitary gland. As a result, less water is reabsorbed from the renal collecting ducts and plasma volume is reduced. Conversely, reduced atrial baroreceptor activity due to lower atrial pressure will increase vasopressin release, retaining water and elevating plasma volume.
