CHAPTER 3 Cellular Growth and Neoplasia
MECHANISMS OF NORMAL CELL HOMEOSTASIS
CELLULAR PROLIFERATION
Neoplasia is the ultimate result of the disruption of exquisite mechanisms regulating normal cell growth. Growth is determined by the balance of cellular proliferation, differentiation, senescence, and programmed cell death. Proliferation occurs as cells traverse the cell cycle (Fig. 3-1). In preparation for cell division, there is a period of deoxyribonucleic acid (DNA) synthesis, designated the S phase. After an intervening gap period, designated the G2 phase, actual mitosis occurs in the M phase. After another intervening gap period, the G1 phase, DNA replication can begin again.
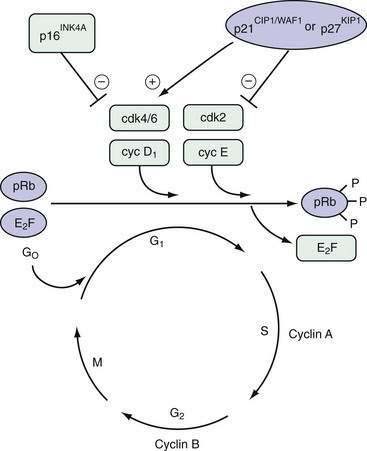
The commitment to proceed through DNA replication and cell division occurs during the G1 phase at the so-called start or restriction (R) point. Cells may exit this cycle of active proliferation before reaching the R point and enter a quiescent phase, G0. Cells can subsequently re-enter the cell cycle from the G0 state (see Fig. 3-1). The duration of each cell cycle phase as well as the overall length of the cycle vary among cell types.
Regulation of cell cycle progression appears to be achieved principally by cyclins and cyclin-dependent kinase activity at the G1/S and G2/M phase transitions. Cyclin proteins are classified on the basis of their structural features and temporal expression patterns during the cell cycle (see Fig. 3-1). Cyclins A and B are expressed predominantly during the S and G2 phases. In contrast, cyclins D and E proteins are most active during the G1 phase.1 Overexpression of cyclin D1 in fibroblasts results in more rapid entry of cells into the S phase. Cyclin D1 is frequently overexpressed in a number of GI and non-GI malignancies, including those originating from the oral cavity, esophagus, breast, and bladder.2
Each cyclin forms a complex with a cyclin-dependent kinase (cdk) in a cell cycle–dependent fashion. Cyclins function as catalysts for cdk activity (see Fig. 3-1). Cdks physically associate with cyclins through their catalytic domains. The cyclin-cdk complexes regulate cell cycle progression through phosphorylation of key target proteins, including the retinoblastoma gene product (pRb) as well as the Rb family members p130 and p107.3 The final result is progression out of G1 into the S phase of the cell cycle.
The cell cycle is also regulated by multiple cdk inhibitors; p21CIP1/WAF1 and p27KIP1 are inhibitors of cyclin E/cdk2. Originally discovered to be part of the complex containing cyclin D1 and cdk4/6, p21CIP1/WAF1 is transcriptionally activated by the TP53 tumor suppressor gene product (see Fig. 3-1).4 p16INK4A is another cdk inhibitor that specifically inhibits cdk4 and cdk65 and is part of a larger family of related inhibitors that includes p14, p15, and p18. p16INK4A is frequently inactivated in esophageal squamous cell cancers and pancreatic ductal adenocarcinomas, a finding that is consistent with its function as a tumor suppressor gene.6,7 p16INK4A disrupts the complex of cyclin D1 and cdk 4/6, thereby freeing p21CIP1/WAF1 and p27KIP1 to inhibit the activity of cyclin E/cdk2.8
PROGRAMMED CELL DEATH AND SENESCENCE
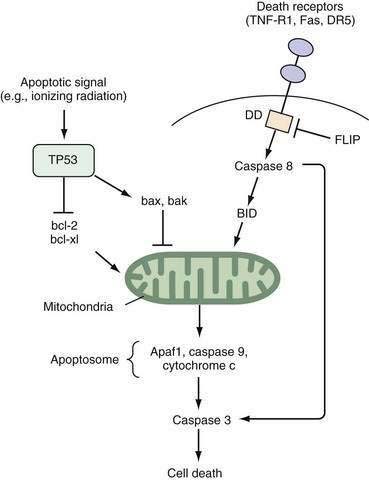
Studies of the roundworm Caenorhabditis elegans have led to the initial identification of the gene ced-3, a protease that is the major effector of apoptosis. Two key regulators of ced-3, designated ced-9 and ced-4, were found to prevent or induce apoptosis, respectively.9 The mammalian oncogene bcl-2 shares homology with ced-9 and protects lymphocytes and neurons from apoptosis10; bcl-2 complexes with bax, a protein that by itself contributes to apoptosis.11 Of note, both bcl-2 and bax are part of larger gene families, and the stoichiometric relationships among different combinations of the encoded proteins can determine the balance between cell survival and cell death.12
Two well-defined pathways that trigger apoptosis have been described in detail. One pathway is mediated through membrane-bound death receptors, which include tumor necrosis factor (TNF) receptors, Fas, and DR5, whereas the other pathway involves activation of TP53 expression by environmental insults such as ionizing radiation, hypoxia, or growth factor withdrawal, with a subsequent increase in the bax-to-bcl-2 ratio. Both pathways converge to disrupt mitochondrial integrity and release of cytochrome c (Fig. 3-2). The so-called apoptosome complex (cytochrome c, caspase 9, and Apaf1) then activates downstream caspases, such as caspase 3, eventuating in cell death. Activation of caspases, intracellular cysteine proteases that cleave their substrates at aspartate residues, is a key step in programmed cell death in mammalian cells.
Replicative senescence also plays a role in determining overall growth in cell populations. Most primary cells when grown in vitro have a limited replicative potential and eventually undergo senescence.13 In contrast, malignant cells can replicate indefinitely. Up-regulation of the telomerase enzyme is essential to escape from replicative senescence. Telomeres are repetitive DNA sequences at the ends of all chromosomes that regulate chromosomal stability. Telomeres shorten with each cell division and, when they have been reduced to a certain critical length, senescence occurs. Cancer cells are able to maintain their telomere length despite multiple cell divisions through the reactivation of telomerase enzyme activity.14
SIGNALING PATHWAYS THAT REGULATE CELLULAR GROWTH
Cellular proliferation is achieved through transition of cells from G0 arrest into the active cell cycle (see Fig. 3-1). Although progression through the cell cycle is controlled by the regulatory proteins just described, overall proliferation is modulated by external stimuli. Growth factors that bind to specific transmembrane receptors on the cell surface may be especially important. The cytoplasmic tails of these transmembrane receptor proteins produce an intracellular signal after ligand binding.
Other receptors on the cell surface possess kinase activity directed toward serine or threonine residues rather than tyrosine. These receptors also phosphorylate a variety of cellular proteins, leading to a cascade of biological responses. Multiple sites of serine and threonine phosphorylation are present on many growth factor receptors, including the tyrosine kinase receptors, suggesting the existence of significant interactions among various receptors present on a single cell.15 The transforming growth factor-β (TGF-β) receptor complex is one important example of a serine-threonine kinase–containing transmembrane receptor.
Many receptors are members of the so-called seven-membrane–spanning receptor family. These receptors are coupled to guanine nucleotide binding proteins, and are designated G proteins. G proteins undergo a conformational change that is dependent on the presence of guanosine phosphates.16 Activation of G proteins can trigger a variety of intracellular signals, including stimulation of phospholipase C and the generation of phosphoinositides (most importantly, inositol 1,4,5-triphosphate) and diacylglycerol through hydrolysis of membrane phospholipids, as well as modulation of the second messengers cyclic AMP and GMP.17 Somatostatin receptors exemplify a G protein–coupled receptor prevalent in the GI tract.
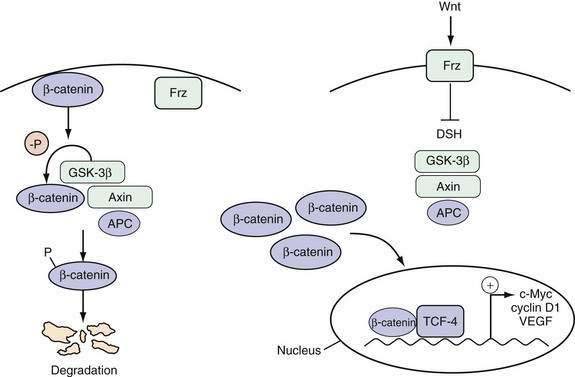
The Wnt pathway is one important example of a signaling pathway that regulates the cell cycle machinery to control the proliferation of intestinal epithelial cells (Fig. 3-3). Although the details of the specific interactions between the Wnt ligand and its receptor Frz, a member of the seven-membrane receptor family, in the GI tract are not fully clarified, an active Wnt signal ultimately results in the accumulation of β-catenin in the nucleus, where it binds with the transcription factor TCF-4 to activate a set of target genes.18 Inhibition of the Wnt signal in mice can be achieved by deletion of TCF-4 or overexpression of a Wnt inhibitor designated Dickkopf1, which results in dramatic hypoproliferation of the intestinal epithelium.19,20 This hypoproliferation appears to be mediated by decreased expression of the TCF-4 target gene c-MYC, which directly represses p21CIP1/WAF1.21 Thus, a Wnt signal stimulates proliferation of intestinal epithelial cells by repressing the cell cycle inhibitor p21CIP1/WAF1.
Cyclin D1 has an extremely short half-life (<20 minutes) and is a rate-limiting factor for progression through the G1 phase of the cell cycle (see Fig. 3-1). Consequently, it is one of the most tightly regulated of all cell cycle proteins. Extracellular signals from growth factors, including EGF, colony-stimulating factor 1 (CSF-1), platelet-derived growth factor (PDGF), and insulin-like growth factor (IGF), can regulate cellular proliferation by rapidly inducing the expression of the cyclin D1 gene.22
Tissue homeostasis is also maintained by growth-inhibiting signals that counterbalance proliferative signals. TGF-β is a potent growth-inhibiting factor that mediates arrest of the cell cycle at the G1 phase. TGF-β not only induces the transcription of the cell cycle inhibitors p15INK4B and p21CIP1/WAF1, but also enhances the inhibitory activity of p27KIP1 on the cyclin E/cdk2 complex (see Fig. 3-1).23 These effects are mediated intracellularly through the Smad family of proteins.
INTESTINAL TUMOR DEVELOPMENT: MULTISTEP FORMATION AND CLONAL EXPANSION
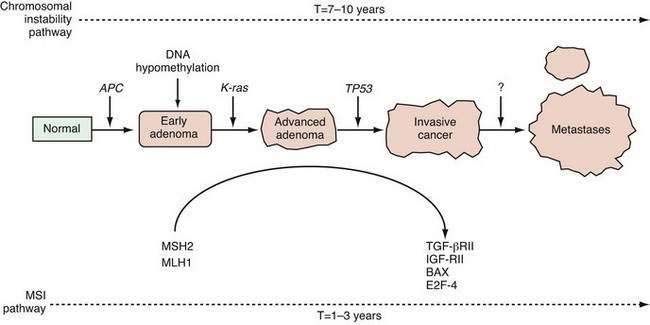
Multiple sequential genetic alterations are required for the transformation of normal intestinal epithelium to frank malignancy. This multistep nature of tumorigenesis is most directly illustrated by the changes that accrue in the development of colonic neoplasia (see Chapters 122 and 123). The accumulation of genetic alterations roughly parallels the progression from normal epithelium through adenomatous polyps (or, in the case of ulcerative colitis, flat dysplastic mucosa) to malignant neoplasia. Studies on the molecular pathogenesis of colon cancer have served as a paradigm for the elucidation of genetic alterations in other GI cancers. For example, a similar progression is also seen in the transition from normal squamous epithelium to metaplastic mucosa (Barrett’s esophagus) through dysplasia to adenocarcinoma of the esophagus. Gastric and pancreatic oncogenesis are each thought to proceed through similar multistep pathways.
Models of the multistep or multiple hit process of tumor formation have largely superseded earlier concepts of oncogenesis that discriminated between tumor initiation and subsequent promotion. Initiation was attributed to a single change in a cell that converted it from a normal to a malignant cell. Promotion reflected all the factors that acted after the initiating event to enhance tumor growth. However, oncogenesis occurs through a series of events that result in incremental changes in cell behavior until the cell eventually passes some threshold associated with the malignant phenotype. Nevertheless, there is still some merit in a more limited concept of tumor promotion. A number of factors promote the likelihood of malignant transformation through the stimulation of increased cellular turnover, which increases opportunities for somatic mutations to occur.24 In the GI tract, these promoting factors include dietary constituents (see later) as well as chronic inflammation, which are associated with increased cell proliferation. Thus, a number of chronic inflammatory conditions increase the site-specific risk of cancer, such as ulcerative colitis (see Chapter 112), chronic gastritis (see Chapters 51 and 54), chronic pancreatitis (see Chapters 59 and 60), Barrett’s esophagus (see Chapters 44 and 46), and chronic hepatitis (see Chapter 94). Although the mechanisms whereby inflammatory processes elicit eventual tumor development are incompletely understood, cytokines produced by inflammatory cells can stimulate tumor cells, leading to activation of nuclear factor-κB (NF-κB) in the tumor cells that can serve to inhibit apoptosis and stimulate proliferation.25
Clonal expansion is also essential to tumor development.26 Whereas germline mutations may lead to altered expression of a gene in all cells in a tissue, subsequent additional somatic mutations generally occur only in a small, largely random subpopulation of cells. Clonal expansion occurs if a specific gene mutation results in a survival advantage for the cell. A second round of clonal expansion occurs when a cell within this population sustains still another genetic alteration, which further enhances its growth properties. After several iterations, a final genetic alteration eventually confers a property that, together with the preceding genetic alterations, makes a cell malignant.
Recent evidence has led to the suggestion that cancer stem cells in tumors may play a central role in tumorigenesis. These cells are defined by the capacity for self-renewal and the ability to generate progeny that lack this capacity but manifest the characteristics of the heterogeneous lineages that comprise the tumor.27 Failure to eradicate the cancer stem cell population is posited to underlie tumor recurrences after chemotherapy. Precise identification of the cancer stem cell compartment has been possible for hematologic malignancies. However, comparable definitive proof of their presence in solid tumors, such as intestinal cancers, remains a challenge.28
A genetically unstable environment was thought to be necessary for the development of the multiple alterations that ultimately result in cancer. Genomic instability is observed in almost all cancers, regardless of organ site. Instability of the genome may result from several mechanisms. In colon cancer, there are at least two well-recognized forms of genetic instability, and they have been termed chromosomal instability and microsatellite instability.29 Chromosomal instability results in tumor cells that display frequent aneuploidy, large chromosomal deletions, and chromosomal duplications. In contrast, tumors that display microsatellite instability are often diploid or near-diploid on a chromosomal level but harbor frequent alterations in smaller tracts of microsatellite DNA (see later discussion on DNA repair). Thus, there are at least two distinct routes to the formation of a colorectal cancer, depending on the nature of the underlying genetic instability (Fig. 3-4).
NEOPLASIA-ASSOCIATED GENES
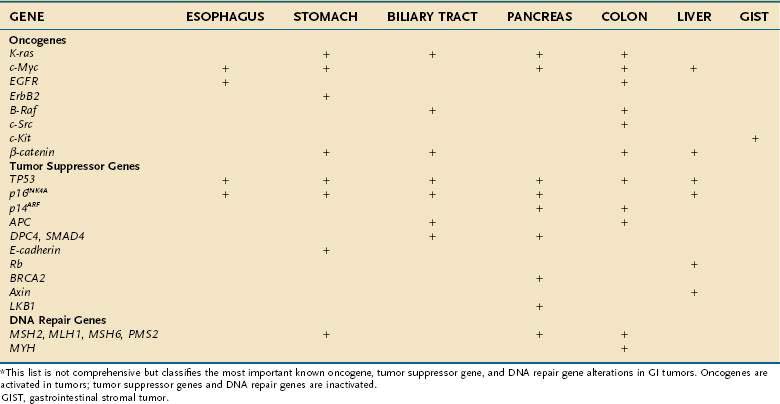
The genes that collectively play an important role in oncogenesis generally lead to disruption of the orderly mechanisms of normal cell proliferation. Insofar as normal cell proliferation appears to depend on a wide variety of genes, it is not surprising that alterations in diverse genes confer part or all the phenotypic features of transformation. Despite this diversity, all these genes that become altered appear to belong to one of three distinct groups: (1) oncogenes, which actively confer a growth-promoting property; (2) tumor suppressor genes, the products of which normally restrain growth or proliferation; and (3) DNA repair genes, which contribute to transformation by fostering genomic instabilbity and facilitating mutations in other genes. Activation of oncogenes or inactivation of tumor suppressor genes and DNA repair genes contributes to malignant transformation (Table 3-1).
Table 3-1 Oncogenes, Tumor Suppressor Genes, and DNA Repair Genes Altered in Gastrointestinal Tumors*
ONCOGENES
Oncogenes and Peptide Growth Factors
The transforming effects of enhanced expression of a variety of growth factors have been demonstrated both in vitro and in vivo. Several growth factor–related proteins encoded by oncogenes have now been recognized, including the family of Wnt proteins and Sis, which encodes the β chain of platelet-derived growth factor. It is axiomatic that cells that produce high levels of a growth factor must also express specific receptors activated by that growth factor to yield an autocrine growth-stimulating loop that is often present in neoplasms. For example, several colon-derived cancer cell lines express TGF-β and IGF I and II, as well as their receptors. This autocrine mechanism may be contrasted with overproduction of a growth factor that exerts its influence at a remote cellular target rather than within the tumor itself, exemplified by gastrin produced by gastrinoma cells, which exerts trophic effects on the gastric mucosa but not on the tumor itself (see Chapter 32).
Protein Kinase–Related Oncogenes
A brief consideration of receptor protein tyrosine kinase HER2/Neu/ERBB2, which is related to the EGF receptor, is particularly illustrative. There are four EGF receptor family members (ERBB1-4). The viral v-erb-b2 encodes a truncated form of the EGF receptor that lacks most of the external EGF-binding domain.30 As a result, the receptor no longer requires the presence of the ligand for activation and remains continuously activated, stimulating proliferation. The neu oncogene is derived from a rat cellular proto-oncogene closely related to the EGF receptor. The oncogene differs from its normal counterpart by a point mutation that changes a single residue (valine to glutamic acid) within the transmembrane domain, thereby causing activation of the 185-kd tyrosine kinase protein (p185neu).31 The human counterpart (ERBB2) of the neu oncogene is not mutated but is overexpressed or amplified in a variety of adenocarcinomas, including those arising in the stomach, breast, and prostate.32 In addition, ERBB2 expression increases progressively in the transition from normal esophageal mucosa through the dysplastic state characteristic of Barrett’s esophagus to esophageal adenocarcinoma.33
In contrast with the receptor type of tyrosine kinase that possesses intrinsic catalytic activity, many other receptors and membrane proteins lack self-contained signaling activity. Instead, they are coupled to nonreceptor tyrosine kinases on the cytoplasmic side of the plasma membrane that act as signal transducers. A number of oncogenes associated with neoplasms of the GI tract, most notably the colon, are members of the src family of nonreceptor tyrosine kinases. Members of the src family are approximately 60-kd phosphoproteins (v-src) that possess inherent tyrosine kinase activity and associate with the inner surface of the plasma membrane. Autophosphorylation of the normal c-src leads to attenuation of its kinase activity, thereby providing inherent regulation to limit unrestrained activity.34 Increased levels of c-src activity have been found in colonic cancer tissue and colon cancer–derived cell lines.35 Activating mutations of c-src have been identified in a subset of advanced, metastatic colon cancers.36
Signal Transduction–Related Oncogenes (Membrane-Associated G Proteins)
Almost all ras mutations in GI malignancies that have been identified occur in the K-ras oncogene, and the frequency of mutations varies greatly among different GI tumor types. The highest frequency is found in tumors of the exocrine pancreas; more than 90% of these tumors possess mutations in the K-ras gene.37 Ras genes activated through point mutation have been identified in approximately 50% of colonic cancers as well as large benign colonic polyps.38 In contrast, fewer than 10% of colonic adenomas smaller than 1 cm have K-ras mutations (see Fig. 3-4).
Most oncogenic mutations in ras cause biochemical changes that maintain it in the active, GTP-bound state by reducing guanosine triphosphatase (GTPase) activity or by destabilizing the inactive GDP-bound form. However, several ras mutants retain significant GTPase activity; therefore, other mechanisms that convert ras to a transforming protein may be involved.39 The GTPase-activating protein (GAP) induces a 500-fold increase in the GTPase activity of the normal ras protein, and some mutant ras proteins are resistant to this modifying protein.40 In the presence of GAP, ras oncogenic activity correlates strongly with its reduced GTPase activity.
A functional consequence of ras activation is the phosphorylation of key serine and threonine kinases. One important downstream signaling target of ras is B-raf. In colon cancers without an identifiable K-ras mutation, 20% possess an activating B-raf mutation,41 consistent with the concept that activation of an oncogenic pathway can be achieved through an alteration in any of several sequential components of a particular pathway.
Nuclear Oncogenes
The role of nuclear oncogenes that encode transcriptional regulatory proteins and that are involved in protein-protein interactions is illustrated by the myc family. The c-Myc protein product is involved in critical cellular functions, such as proliferation, differentiation, apoptosis, transformation, and transcriptional activation of key genes.42 Frequently, c-Myc is overexpressed in many GI cancers. The protein contains several important domains. The carboxy terminal contains a helix-loop-helix motif that mediates binding to other proteins, such as Max.43 These heterodimers bind DNA through the basic domain of c-Myc. The amino terminal of c-Myc contains regions critical for transcriptional activation of genes, transformation, and apoptosis.44 c-Myc has been found to be a transcriptional target of the β-catenin/TCF-4 complex in colorectal cancers (see Fig. 3-3), which may explain the overexpression of c-Myc observed in this cancer type.45
TUMOR SUPPRESSOR GENES
The products of tumor suppressor genes prevent the acquisition of the transformed phenotype in vitro and have similar functional properties in vivo. Mutations that disrupt the biological function of these genes are associated with all GI cancers. Germline mutations of this class of gene underlie most of the known inherited cancer syndromes in which a specific gene has been implicated. A number of these genes and their products have been identified and characterized (Table 3-2).
Table 3-2 Chromosomal Localization and Function of Several Key Tumor Suppressor Genes in Gastrointestinal (GI) Cancers
| CHROMOSOME | GENE* | FUNCTION |
|---|---|---|
| 5q | APC | Inhibition of Wnt signaling |
| 9p | p16INK4A | Cell cycle inhibition |
| 11q | MEN1 | Regulation of histone methyltransferase |
| 16q | E-cadherin | Maintenance of cell-cell interactions |
| 17p | TP53 | Regulation of DNA repair and apoptosis |
| 18q | DPC-4, SMAD4 | Transduction of transforming growth factor-β signal |
* Clinical GI disorders associated with defects in some of these genes are listed in Table 3-4.
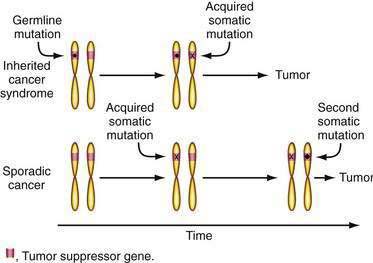
These observations led Knudson to hypothesize that tumors in familial cancer syndromes might derive from independent mutations in the two alleles of a specific tumor suppressor gene (Fig. 3-5)—that the first mutation was present in one copy of the gene inherited in the germline and therefore present in all cells in affected family members.46 A somatic mutation of the remaining normal allele of the tumor suppressor gene that might occur in any cell would then lead to tumor development, explaining the high incidence of cancer and multiple tumors. The same gene might play a role in the development of the same tumor type in the general population (sporadic cancer), but two independent somatic mutations of each of the two alleles would be required. However, this combination of events should be uncommon and would explain the lower frequency and later age of diagnosis of similar tumors in the general population. Comings was the first to suggest that the relevant gene in a familial cancer syndrome might encode a tumor-suppressing gene product.47 Although this two-hit hypothesis has been generally accepted, there are exceptions. For example, there are data suggesting that a single alteration in just one allele of the Lkb1 tumor suppressor gene that underlies the Peutz-Jeghers syndrome may be sufficient for intestinal polyp formation.48
Loss of Heterozygosity, Allelic Deletion, and Tumor Suppressor Gene Inactivation
Three types of polymorphisms are recognized—single nucleotide polymorphisms (SNPs), restriction fragment length polymorphisms (RFLPs), and microsatellite polymorphisms. SNPs represent single base pair alterations that are typically silent; these are the most abundant type of polymorphism, occurring in approximately every 1000 base pairs throughout the genome.49 RFLPs are a unique type of SNP in which the single nucleotide change alters a recognition site for a restriction endonuclease. Thus, digestion of DNA with a restriction endonuclease allows the two different alleles of the same genetic segment inherited from the subject’s two parents to be distinguished (Fig. 3-6). A more widely applicable approach uses polymorphisms present within DNA microsatellite markers. Microsatellite DNA sequences are short repetitive mononucleotide or dinucleotide repeats, such as a poly-A or poly-CA sequence. Microsatellite polymorphisms are much more common than RFLPs in the genome. There is a wide variation in the number of repeats in different alleles, and these differences can be detected through a polymerase chain reaction (PCR; see Fig. 3-6). Although SNPs or microsatellites can be used to detect deletions, SNPs will likely supersede other techniques. Either approach provides a means of assessing whether a specific region of a chromosome is deleted in tumor tissue when compared with normal tissue from the same individual.
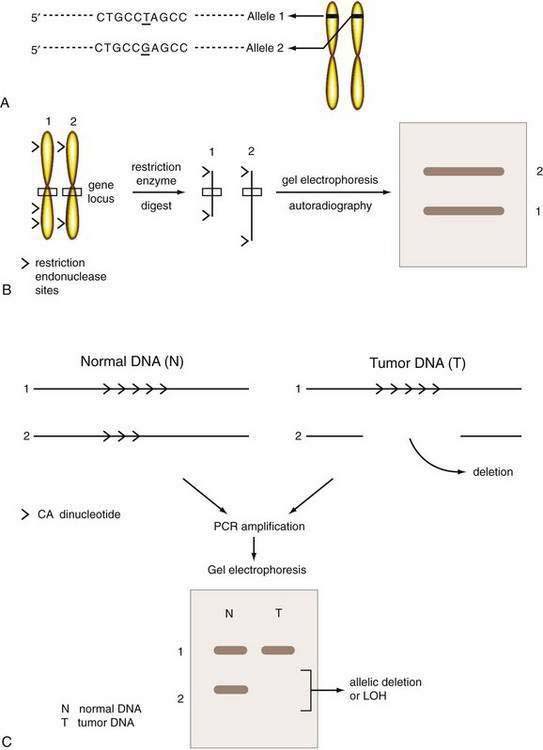
These losses, termed loss of heterozygosity or an allelic deletion (loss of an allele from one parent), represent an important mechanism of inactivation of one copy of tumor suppressor genes (see Fig. 3-6). When coupled with a preexisting germline mutation, such allelic deletions provide the second hit, which results in a loss of function of both tumor suppressor gene copies. Other mechanisms of tumor suppressor gene inactivation include point mutation or small intragenic deletions that result in premature truncation of the protein product, or promoter hypermethylation. Transcriptional silencing can result from methylation of CpG islands in gene promoters; this has been demonstrated to occur in the gene encoding p16INK4A in esophageal and pancreatic cancers and the gene encoding E-cadherin in gastric cancer.50
Adenomatous Polyposis Coli Gene
Genetic linkage analysis has revealed markers on chromosome 5q21 that are tightly linked to polyp development in affected members of kindreds with the familial adenomatous polyposis (FAP) and Gardner’s syndrome.51 Further work led to the identification of the gene responsible for FAP, the adenomatous polyposis coli (APC) gene.52 As predicted, germline mutations of APC were found in affected patients, and the germline mutations segregate with the disease within a given family.53,54 The full spectrum of adenomatous polyposis syndromes attributable to APC is discussed in detail in Chapter 122. Although these syndromes are relatively rare, studies identifying genetic factors that contribute to these syndromes have provided insight into mechanisms essential to the development of common sporadic colon cancers as well as to tumorigenesis in general.55 Somatic mutations in APC have been found in most sporadic colon polyps and cancers.56,57 Mutations in APC are characteristically identified in the earliest adenomas, indicating that APC plays a critical role as the gatekeeper in the multistep progression from normal epithelial cell to colon cancer (see Fig. 3-4).
The APC gene comprises 15 exons and encodes a predicted protein of 2843 amino acids, or approximately 310 kd. Most germline and somatic APC gene mutations result in a premature stop codon and therefore a truncated protein product. Although mutations are most common in exon 15 of the APC gene, they may occur throughout the gene. Those occurring in the APC amino terminal are associated with a rare variant of FAP, attenuated familial adenomatous polyposis (AFAP).58 Studies have revealed a segregation of certain APC mutations with the phenotype of congenital hypertrophy of the retinal pigment epithelium (CHRPE).59
APC mutations result in functional changes in key protein-protein interactions. As discussed noted, APC is a negative regulator of the Wnt signaling pathway (see Fig. 3-3). Mutant APC proteins are unable to interact with β-catenin, resulting in uncontrolled activation of the Wnt signaling pathway and the subsequent oncogenic phenotype.
TP53 Gene
p53, named for a 53-kD sized gene product, is a nuclear phosphoprotein that plays a key role in cell cycle regulation and apoptosis.60 The p53 protein was first detected in tumors as the product of a mutated gene that was mapped to chromosome 17p, a region found to exhibit loss of heterozygosity in many tumors. Point mutations in TP53 have been identified in as many as 50% to 70% of sporadic colon cancers but only a small subset of colonic adenomas (see Fig. 3-4).61 Point mutations in TP53 have also been found in esophageal squamous carcinoma and adenocarcinoma, gastric carcinoma, pancreatic adenocarcinoma, and hepatocellular carcinoma.60 Interestingly, aflatoxin appears to induce a mutation in a single hot spot codon (codon 249) of TP53 in many hepatocellular carcinomas.62 In addition to the TP53 point mutations in sporadic cancers, germline TP53 mutations have been observed in the Li-Fraumeni syndrome, an autosomal dominant familial disorder in which breast carcinoma, soft tissue sarcoma, osteosarcoma, leukemia, brain tumor, and adrenocortical carcinoma can develop in affected persons.63
p53 is a sequence-specific transcription factor that is induced in conditions of cellular stress, such as ionizing radiation, growth factor withdrawal, or cytotoxic therapy (see Fig. 3-2). As a consequence of genotoxic damage, p53 arrests cells at the G1 phase to facilitate DNA repair or trigger apoptosis. p53 mediates some of these responses through the induction of the p21CIP1/WAF1 inhibitor of the cell cycle or pro-apoptotic genes, including PUMA, and c-Myc appears to play a role in this cell fate decision.64 The functional importance of p53 in colon cancer has been underscored by experiments in which wild-type TP53 was reintroduced into colon cancer cells that had only mutant TP53.65 Repleting cells with p53, the product of TP53, can arrest growth in a cell cycle phase-specific manner.
SMAD4 Gene
SMAD4, also designated deleted in pancreas cancer-4 (DPC-4), is a tumor suppressor gene located on chromosome 18q and is deleted or mutated in most pancreatic adenocarcinomas and a subset of colon cancers. This gene encodes Smad4, an essential intracellular mediator of the growth inhibitory effects of TGF-β. The Smad4 protein has two important domains, the mad homology domains 1 and 2 (MH1 and MH2), which are essential for DNA binding and for oligomerization with other Smad proteins, respectively.66 Mutant Smad4 blocks TGF-β–induced inhibition of proliferation. Germline mutations in SMAD4 result in the juvenile polyposis syndrome. Other genes on chromosome 18q may also be important in colon carcinogenesis.67
DNA REPAIR GENES
Cellular mechanisms have evolved to preserve the fidelity of DNA. Errors can be introduced into the genome through the spontaneous mismatching of nucleotides during normal DNA replication. This occurs most commonly from slippage in microsatellite DNA, which involves regions of mononucleotide (e.g., poly-A) or dinucleotide (e.g., poly-CA) repeats.68 The DNA mismatch repair system corrects these errors. The components of this system have been studied most extensively in prokaryotes and lower eukaryotes, most notably yeast. The enzymes bind mismatched DNA, cut the DNA strand with the mismatched nucleotide, unwind the DNA fragment, fill in the gap with the correct nucleotide, and finally reseal the remaining nick. The human homologs of these DNA mismatch repair genes include hMSH2, hMSH3, hMSH4, hMSH5, hMSH6, hMLH1, hMLH3, hPMS1, and hPMS2, and likely others.
The genes hMSH2 and hMLH1 are the two DNA mismatch repair genes that are most frequently mutated at the germline level in the hereditary nonpolyposis colorectal cancer (HNPCC) syndrome, also known as Lynch syndrome.69,70 Mutations can lead to functional alterations that allow strand slippage during replication. Affected cells are called replication error (RER)–positive, in contrast to the RER-negative phenotype.71,72 Because microsatellite DNA sequences are primarily affected by this type of genetic instability, the tumor cells are said to display microsatellite instability (MSI). DNA mismatch repair genes are mutated not only in Lynch syndrome, but also in a subset of sporadic GI cancers, including many arising in the esophagus, stomach, pancreas, and colon. Mechanistically, the absence of DNA repair does not cause cancer directly. Rather, the DNA repair defect creates a milieu that permits the accumulation of mutations in a variety of other genes that contain microsatellite DNA sequences, such as the TGF-β type II receptor, BAX, IGF type II receptor, and E2F-4. This MSI pathway represents a novel mechanism for the accumulation of mutations within a tumor (see Fig. 3-4). It is characteristic of all Lynch-related tumors and is observed in approximately 15% of all sporadic colon cancers.
Errors can also be introduced when individual nucleotides are damaged by chemical factors; the base excision repair system corrects these types of errors. 8-Oxoguanine residues can result from oxidative DNA damage, and these altered bases will inappropriately pair with adenines, ultimately leading to G:C→T:A mutations if uncorrected. MYH is a DNA glycosylase that participates in the repair of these oxidized guanine nucleotides. An autosomal recessive adenomatous polyposis syndrome caused by germline mutations in the MYH repair gene has been identified.73,74 Interestingly, G:C→T:A mutations in the APC gene were almost universally found in the polyps of patients with germline MYH mutations, indicating that there are important similarities in the molecular pathogenesis of polyps in the MYH and FAP syndromes.
ONCOGENIC SIGNALING PATHWAYS
Individual oncogenes or tumor suppressor genes do not necessarily induce cellular transformation directly but typically function as components of larger oncogenic signaling pathways. Some of the pathways that are particularly relevant for gastrointestinal tumorigenesis include the Wnt and ras signaling pathways. These are pathways that regulate normal tissue homeostasis but become oncogenic when the signals are transduced in an aberrant or amplified manner. The key features of Wnt signaling are illustrated in Figure 3-4. β-Catenin is translocated from the plasma membrane to the cytoplasm. There, it forms a macromolecular complex with the APC protein, glycogen synthase kinase-3β (GSK-3β), and Axin. Phosphorylation of β-catenin by GSK-3β triggers its degradation. In the presence of an active Wnt signal, β-catenin is stabilized, and it enters the nucleus where it interacts with the transcription factor TCF-4 to upregulate a number of key target genes, including c-Myc, cyclin D1, and VEGF. As discussed earlier, Wnt signaling is essential for regulating proliferation of normal intestinal epithelium, and dysregulated Wnt signaling is an almost universal feature of all colon cancers. The latter can result from a mutation in the APC, Axin, or β-catenin genes, but alterations in the APC tumor suppressor gene are the most common. An alteration in just one of these components is sufficient to activate the entire pathway. Thus, it is essential to consider individual genetic alterations in the context of the overall signaling pathway in which they function.
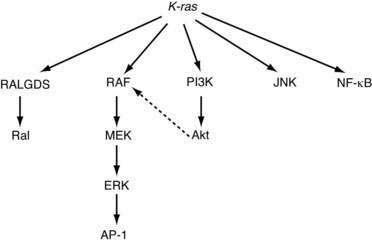
Because pathways are typically not linear, additional levels of complexity arise. There is frequent overlap among pathways, and the distinction between pathways can be somewhat arbitrary. For example, mutations in the K-ras oncogene result in the activation of multiple distinct signaling pathways, including Raf/ERK/MAPK, PI3K/Akt, and NF-κB, all of which play an important role in tumorigenesis (Fig. 3-7). Crosstalk between these effector pathways serves to modulate the cellular responses further. Akt, a target of PI3K, can phosphorylate Raf and thereby regulate signaling through the MAPK pathway.75 Finally, each of these signaling pathways regulates multiple biological processes related to tumorigenesis,76 including cell cycle progression, apoptosis, senescence, angiogenesis, and invasion.
Another pathway that plays a particularly important role in gastrointestinal tumors is the cyclooxygenase-2 (COX2) pathway. The enzyme COX-2 is a key regulator of prostaglandin synthesis that is induced in inflammation and neoplasia. Although no mutations of COX-2 have been described, overexpression of COX-2 in colonic adenomas and cancers is associated with tumor progression and angiogenesis, primarily through the induction of synthesis of prostaglandin E2. Inhibition of COX-2 with a variety of agents (aspirin, nonsteroidal anti-inflammatory drugs, or COX-2 selective inhibitors) is associated with a reduced risk of colorectal adenomas and cancer.77
ENVIRONMENTAL MUTAGENESIS
CHEMICAL CARCINOGENESIS
Metabolic activation by the host is a key determinant of the carcinogenic potential of many compounds. The initial compound, the procarcinogen, is converted by host enzymes to an electrophilic derivative, which then chemically modifies DNA. Mutations result from errors that occur during DNA replication as a result of distorted base pairs. These mutations, in conjunction with other tumor-promoting factors, facilitate or cause the development of malignancy. Factors that influence the potency of any chemical carcinogen include the equilibrium between the activation of the procarcinogen and deactivation or degradation of the carcinogen.78 Deactivation typically occurs through a conjugation reaction, usually in the liver.
DIETARY FACTORS
Chemical mutagenesis may be especially important in the development of cancers within the GI tract and related organs. The mucosal surfaces from which most primary cancers in the GI tract develop are exposed to a complex mixture of dietary constituents that are potential carcinogens or procarcinogens. The ability of dietary factors to act as mutagens in humans was demonstrated directly in 1995. The frequency of contamination of foodstuffs with aflatoxins, a fungal metabolite, parallels the incidence of hepatocellular carcinoma in various areas of the world.79 Studies demonstrating that aflatoxins cause mutations in the TP53 gene in hepatocellular carcinoma have provided a compelling link between genes and the environment.79
Nitrates present in many foods appear to be additional dietary constituents that may act as procarcinogens in the GI tract. Diet-derived nitrates can be converted by bacterial action in a hypochlorhydric stomach to nitrites and subsequently to mutagenic nitrosamines.80 These events may underlie the documented correlation between dietary intake of foods high in nitrates and the incidence of gastric cancer in different populations.
These mechanisms could explain well-documented correlations between the intake of various dietary constituents and the incidence of colon cancer in certain populations. Populations that have a high fiber intake and resulting fast colonic transit times generally exhibit a lower incidence of colon cancer than populations with low fiber intake and delayed transit. The incidence of colon cancer in Japanese immigrants to the United States who consume a Western diet is much higher than that of native Japanese who consume a traditional Japanese diet.81
BIOLOGICAL FEATURES OF TUMOR METASTASIS
The establishment of distant metastasis requires multiple processes, many of which involve alterations in interactions between tumor cells and normal host cells. To metastasize, a cell or group of cells must detach from the primary tumor, gain access to the lymphatic or vascular space, adhere to the endothelial surface at a distant site and penetrate the vessel wall to invade the second tissue site and, finally, proliferate as a second tumor focus. Angiogenesis is necessary for proliferation of the primary tumor and tumor metastases. Tumor cells must also overcome host immune cell killing. As a result, few circulating tumor cells (less than 0.01%) successfully initiate metastatic foci. A “survival of the fittest” view of metastasis has been proposed, in which selective competition favors metastasis of a subpopulation of cells from the primary site.82 Clonal expansion occurs again after formation of a metastatic focus.
EPITHELIAL-MESENCHYMAL TRANSITION
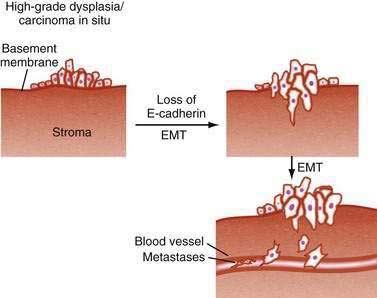
Modulation of tumor cell interactions with adjacent cells and with the extracellular matrix is an essential step as tumor cells invade through the basement membrane and ultimately metastasize to distant sites. A similar process occurs during normal embryogenesis, when polarized epithelial cells no longer recognize the boundaries imposed by adjacent epithelial cells or their basement membrane and adopt features of migratory, mesenchymal cells. This phenomenon, designated epithelial-mesenchymal transition (EMT), has provided a new model for understanding tumor progression (Fig. 3-8). E-cadherin is a critical component of adherens junctions that maintain cell-cell interactions, and loss of E-cadherin is one of the key features of the EMT phenotype.83 Mutations in E-cadherin are common in many GI cancers, particularly gastric cancer. E-cadherin gene expression can be down-regulated by the transcriptional repressors Snail, SIP1, and Twist,84–86 but it is not yet clear whether these are relevant in GI cancers.
ANGIOGENESIS AND LYMPHANGIOGENESIS
Angiogenesis is essential to sustain continued growth of the primary tumor. If new vessels are not developed as the primary tumor expands, cells most distant from available vessels are deprived of an adequate source of nutrition and central necrosis occurs. Neovascularization is also an important permissive factor in facilitating metastatic dissemination of tumors.87 A number of protein growth factors, produced by malignant tumor cells and stromal cells, have been found to be potent stimuli of angiogenesis, including vascular endothelial growth factor A (VEGF-A), basic fibroblast growth factor (bFGF) and TGF-β. VEGF-A is perhaps the most critical factor that is up-regulated in most tumor types, including colon cancer. Multiple genetic pathways modulate VEGF-A expression, including Wnt and mutant ras.88 Therapeutic strategies that inhibit VEGF-A have demonstrated some promise for patients with advanced colon cancer.89
In addition to angiogenesis, lymphangiogenesis plays an important role in tumor metastasis. Some important clues into the molecular basis of tumor lymphangiogenesis have been obtained. VEGF-C or VEGF-D bind to the VEGF receptor-3 on lymphatic endothelial cells to stimulate the formation of new lymphatic vessels.90 This results in the development of new lymphatic channels within the tumor mass and, consequently, the enhanced dissemination of tumor cells to regional lymph nodes.91 Strategies to inhibit tumor lymphangiogenesis are being actively pursued.
METASTASIS GENES
It is likely that properties important to the development of metastasis reflect the effects of genes distinct from oncogenes involved in the initial formation of the tumor. Although no gene specifically associated with metastasis has yet been identified, one gene, designated nm-23, may be a potential metastasis suppressor gene. Levels of nm-23 are reduced in a variety of metastatic tumors and in cell lines with high metastatic potential, compared with primary tumors and cell lines with low metastatic potential, respectively.92 The function of nm-23 is uncertain but may be inferred from its predicted sequence homology to nucleoside diphosphate (NDP) kinases. NDP kinases are involved in microtubule assembly and signal transduction through G proteins, functions that may be important in the formation of metastases.
MOLECULAR MEDICINE: CURRENT AND FUTURE APPROACHES IN GASTROINTESTINAL ONCOLOGY
DNA-BASED APPROACHES
Progress in the identification of cancer-associated genes coupled with the inherent power of molecular biological techniques to analyze exquisitely small amounts of DNA and protein are leading to more effective diagnostic markers (Table 3-3). The most immediate application is assessment of cancer risk in members of cancer-prone kindreds. Strategies have been developed to identify germline mutations in patients with a variety of inherited GI cancer syndromes, including FAP, HNPCC, and hereditary gastric cancer (Table 3-4). In most of these conditions, there is no consensus hot spot mutational site, so these tests analyze the full gene through a variety of analytic techniques (see Table 3-3). Genetic testing is a powerful tool to identify high-risk families and to define the cancer risk for individual family members. Application of genetic testing must take into consideration the sensitivity and specificity of the assay as well as issues of patient confidentiality and potential impact on medical insurability. For these reasons, genetic counseling is an essential component of the genetic testing process.
Table 3-3 Molecular Diagnostic Techniques for Detection of Cancer-Associated DNA Mutations or Altered Proteins
| TECHNIQUE | PURPOSE OR STRATEGY |
|---|---|
| PCR-Based Strategies to Detect DNA Mutations | |
| Single-strand conformational polymorphism (SSCP) | Detection of alteration of secondary structure of single-stranded DNA caused by single base mutation |
| Denaturing gradient gel electrophoresis (DGGE) | Detection of strand dissociation of double-stranded DNA altered by mutations |
| Heteroduplex analysis | Detection of altered electrophoretic migration caused by mutations |
| Heteroduplex mismatch cleavage | Detection of chemical cleavage of mismatches in heteroduplexes |
| Direct DNA sequencing | Direct detection of altered DNA nucleotide sequence |
| PCR-Based Strategies to Detect Known Mutations in Genes* | |
| Restriction enzyme digestion | Detection of mismatched primers followed by enzymatic cleavage |
| Allele-specific oligonucleotide hybridization | Hybridization of specific oligonucleotides with wild-type or mutant sequence |
| Protein-Based Strategies | |
| In vitro translation (IVT) | Detection of truncated protein resulting from nonsense mutation and a premature stop codon |
| Yeast and bacterial colorimetric assays | Detection of altered colorimetric assay caused by mutation |
| Immunohistochemistry | Determination of presence or absence of gene product in tumor sample |
Table 3-4 Applications of Molecular Diagnostics for Gastrointestinal Cancers
| DISORDER | GENE(S) DETECTED |
|---|---|
| Germline DNA Analysis for Hereditary GI Cancer Syndromes | |
| FAP, AFAP | APC |
| Lynch, HNPCC | hMSH2, hMLH1, hMSH6, hPMS2 |
| MYH polyposis | MYH |
| Peutz-Jeghers syndrome | LKB1 |
| Cowden’s disease | PTEN |
| Juvenile polyposis | SMAD4, BMPR1A |
| Hereditary gastric cancer | E-cadherin |
| Hereditary pancreatic cancer | p16INK4A, BRCA2 |
| MEN1 | Menin |
| Molecular Analysis for the Diagnosis of Sporadic GI Cancers | |
| Colon cancer | |
| Stool DNA testing | K-ras, APC, TP53 |
| Tumor DNA MSI testing | |
| Tumor immunohistochemistry for hMSH2, hMLH1, hMSH6, hPMS2 protein | |
APC, adenomatous polyposis coli; FAP, familial adenomatous polyposis; AFAP, attenuated FAP; HNPCC, hereditary nonpolyposis colorectal cancer; MEN1, multiple endocrine neoplasia, type 1; MSI, microsatellite instability.
Improved detection of sporadic GI cancers and their precursor lesions has also been the focus of research studies. Small numbers of shed cells obtained from stool can be assessed for the presence of mutations in specific tumor-associated genes (K-ras, APC, and TP53) using the PCR assay.93 Detection of ras mutations in DNA extracted from the pancreatic ductal fluid obtained at the time of endoscopic retrograde cholangiopancreatographic evaluation for pancreatic cancer has also been reported.94
The MSI test can be performed on archived colon tumor samples and serves as a useful screening test to identify individuals whose colon cancers may have developed as a manifestation of the Lynch syndrome.95 Loss of hMSH2, hMLH1, or hMSH6 protein by immunohistochemical staining may provide similar information. Studies have suggested that the MSI status of a colon tumor may be predictive of the response to 5-fluorouracil–based chemotherapy.96 Therapies that target specific signaling pathways are likely to increase as our molecular understanding of GI cancers increases. Antibodies that target EGF receptors and block the EGF receptor signaling pathway have proven therapeutic benefit in colon cancer, and their role in treatment strategies has been evolving.97 In addition, small molecule tyrosine kinase inhibitors of the c-KIT oncogene now constitute routine treatment of gastrointestinal stromal tumors (see Chapter 30).98 Molecular techniques may also find a role in the staging of disease. For example, the PCR assay has been used to detect lymph node micrometastases from colon cancer.99 Finally, as more tests for genetic markers become available, monitoring for disease recurrence after surgery may become another important application.
ONCOFETAL PROTEINS
Characterization of malignant and transformed cells has led to the identification of markers that may be useful for the early detection and diagnosis of GI cancers. The most productive approaches have exploited the antigenicity of distinctive cell surface glycoconjugates to prepare antisera or monoclonal antibodies directed against tumor-associated determinants. The first useful marker developed through this approach was the carcinoembryonic antigen (CEA), which was identified by Gold and coworkers after immunization of rabbits with colorectal cancer tissue.100 The resulting antisera were found to recognize a determinant present in tumor tissue and circulating in blood from patients with colorectal cancer but largely absent from normal colonic mucosa and normal serum. This oncofetal determinant is also expressed in nonmalignant mucosa in association with increased proliferation. On a practical level, however, the CEA concentration is falsely elevated in a variety of inflammatory conditions associated with increased cell turnover, such as ulcerative colitis. In addition, it was noted that CEA could be produced by tumors arising from many sites, particularly those elsewhere in the GI tract (e.g., gastric and pancreatic cancers). This finding underscores the relatively limited tissue specificity of transformation-associated alterations in cell surface determinants. Future strategies to identify new protein markers that may be useful for diagnosis, therapy, or prognosis will rely on emerging proteomic techniques.
Brown JR, DuBois RN. COX-2: A molecular target for colorectal cancer prevention. J Clin Oncol. 2005;23:2840-55. (Ref 77.)
Cech TR. Beginning to understand the end of the chromosome. Cell. 2004;116:273-9. (Ref 14.)
Clarke MF, Dick JE, Dirks PB, et al. Cancer stem cells—perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer Res. 2006;66:9339-44. (Ref 27.)
Erichsen HC, Chanock SJ. SNPs in cancer research and treatment. Br J Cancer. 2004;90:747-51. (Ref 49.)
Giardiello FM, Brensinger JD, Petersen GM. AGA technical review on hereditary colorectal cancer and genetic testing. Gastroenterology. 2001;121:198-213. (Ref 95.)
Herman JG, Baylin SB. Gene silencing in cancer in association with promoter hypermethylation. N Engl J Med. 2003;349:2042-54. (Ref 50.)
Hurwitz H, Fehrenbacher L, Novotny W, et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med. 2004;350:2335-42. (Ref 89.)
Lin WW, Karin M. A cytokine-mediated link between innate immunity, inflammation, and cancer. J Clin Invest. 2007;117:1175-83. (Ref 25.)
Reed JC. Mechanisms of apoptosis. Am J Pathol. 2000;157:1415-30. (Ref 12.)
Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2000;103:211-25. (Ref 15.)
Sherr CJ. The Pezcoller lecture: Cancer cell cycles revisited. Cancer Res. 2000;60:3689-95. (Ref 1.)
Siegel PM, Massague J. Cytostatic and apoptotic actions of TGF-beta in homeostasis and cancer. Nat Rev Cancer. 2003;3:807-21. (Ref 23.)
Thiery JP. Epithelial-mesenchymal transitions in tumour progression. Nat Rev Cancer. 2002;2:442-54. (Ref 83.)
Ulku AS, Der CJ. Ras signaling, deregulation of gene expression and oncogenesis. Cancer Treat Res. 2003;115:189-208. (Ref 76.)
van de Wetering M, Sancho E, Verweij C, et al. The beta-catenin/TCF-4 complex imposes a crypt progenitor phenotype on colorectal cancer cells. Cell. 2002;111:241-50. (Ref 21.)
1. Sherr CJ. The Pezcoller lecture: Cancer cell cycles revisited. Cancer Res. 2000;60:3689-95.
2. Motokura T, Bloom T, Kim HG, et al. A novel cyclin encoded by a bcl1-linked candidate oncogene. Nature. 1991;350:512-15.
3. Weinberg RA. The retinoblastoma protein and cell cycle control. Cell. 1995;81:323-30.
4. el-Deiry WS, Tokino T, Velculescu VE, et al. WAF1, a potential mediator of p53 tumor suppression. Cell. 1993;75:817-25.
5. Serrano M, Hannon GJ, Beach D. A new regulatory motif in cell-cycle control causing specific inhibition of cyclin D/CDK4. Nature. 1993;366:704-7.
6. Liu Q, Yan YX, McClure M, et al. MTS-1 (CDKN2) tumor suppressor gene deletions are a frequent event in esophagus squamous cancer and pancreatic adenocarcinoma cell lines. Oncogene. 1995;10:619-22.
7. Caldas C, Hahn SA, da Costa LT, et al. Frequent somatic mutations and homozygous deletions of the p16 (MTS1) gene in pancreatic adenocarcinoma. Nat Genet. 1994;8:27-32.
8. Muraoka RS, Lenferink AE, Law B, et al. ErbB2/Neu-induced, cyclin D1-dependent transformation is accelerated in p27-haploinsufficient mammary epithelial cells but impaired in p27-null cells. Mol Cell Biol. 2002;22:2204-19.
9. Yuan J, Shaham S, Ledoux S, et al. The C. elegans cell death gene ced-3 encodes a protein similar to mammalian interleukin-1 beta-converting enzyme. Cell. 1993;75:641-52.
10. Hengartner MO, Horvitz HR. C. elegans cell survival gene ced-9 encodes a functional homolog of the mammalian proto-oncogene bcl-2. Cell. 1994;76:665-76.
11. Oltvai ZN, Milliman CL, Korsmeyer SJ. Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that accelerates programmed cell death. Cell. 1993;74:609-19.
12. Reed JC. Mechanisms of apoptosis. Am J Pathol. 2000;157:1415-30.
13. Hayflick L. Mortality and immortality at the cellular level. A review. Biochemistry. 1997;62:1180-90.
14. Cech TR. Beginning to understand the end of the chromosome. Cell. 2004;116:273-9.
15. Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2000;103:211-25.
16. McCormick F. Signalling networks that cause cancer. Trends Cell Biol. 1999;9:M53-6.
17. Parekh DB, Ziegler W, Parker PJ. Multiple pathways control protein kinase C phosphorylation. Embo J. 2000;19:496-503.
18. Chung DC. The genetic basis of colorectal cancer: Insights into critical pathways of tumorigenesis. Gastroenterology. 2000;119:854-65.
19. Korinek V, Barker N, Moerer P, et al. Depletion of epithelial stem-cell compartments in the small intestine of mice lacking Tcf-4. Nat Genet. 1998;19:379-83.
20. Pinto D, Gregorieff A, Begthel H, Clevers H. Canonical Wnt signals are essential for homeostasis of the intestinal epithelium. Genes Dev. 2003;17:1709-13.
21. van de Wetering M, Sancho E, Verweij C, et al. The beta-catenin/TCF-4 complex imposes a crypt progenitor phenotype on colorectal cancer cells. Cell. 2002;111:241-50.
22. Pestell RG, Albanese C, Reutens AT, et al. The cyclins and cyclin-dependent kinase inhibitors in hormonal regulation of proliferation and differentiation. Endocr Rev. 1999;20:501-34.
23. Siegel PM, Massague J. Cytostatic and apoptotic actions of TGF-beta in homeostasis and cancer. Nat Rev Cancer. 2003;3:807-21.
24. Thompson TC, Southgate J, Kitchener G, Land H. Multistage carcinogenesis induced by ras and myc oncogenes in a reconstituted organ. Cell. 1989;56:917-30.
25. Lin WW, Karin M. A cytokine-mediated link between innate immunity, inflammation, and cancer. J Clin Invest. 2007;117:1175-83.
26. Nowell PC. The clonal evolution of tumor cell populations. Science. 1976;194:23-8.
27. Clarke MF, Dick JE, Dirks PB, et al. Cancer stem cells—perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer Res. 2006;66:9339-44.
28. Shmelkov SV, Butler JM, Hooper AT, et al. CD133 expression is not restricted to stem cells, and both CD133+ and CD133− metastatic colon cancer cells initiate tumors. J Clin Invest. 2008;118:2111-20.
29. Lengauer C, Kinzler KW, Vogelstein B. Genetic instabilities in human cancers. Nature. 1998;396:643-9.
30. Downward J, Yarden Y, Mayes E, et al. Close similarity of epidermal growth factor receptor and v-erb-B oncogene protein sequences. Nature. 1984;307:521-7.
31. Brandt-Rauf PW, Pincus MR, Carney WP. The c-erbB-2 protein in oncogenesis: Molecular structure to molecular epidemiology. Crit Rev Oncog. 1994;5:313-29.
32. Yokota J, Yamamoto T, Miyajima N, et al. Genetic alterations of the c-erbB-2 oncogene occur frequently in tubular adenocarcinoma of the stomach and are often accompanied by amplification of the v-erbA homologue. Oncogene. 1988;2:283-7.
33. Miller CT, Moy JR, Lin L, et al. Gene amplification in esophageal adenocarcinomas and Barrett’s with high-grade dysplasia. Clin Cancer Res. 2003;9:4819-25.
34. Cooper JA, Gould KL, Cartwright CA, Hunter T. Tyr527 is phosphorylated in pp60c-src: Implications for regulation. Science. 1986;231:1431-4.
35. Cartwright CA, Kamps MP, Meisler AI, et al. pp60c-src activation in human colon carcinoma. J Clin Invest. 1989;83:2025-33.
36. Irby RB, Mao W, Coppola D, et al. Activating SRC mutation in a subset of advanced human colon cancers. Nat Genet. 1999;21:187-90.
37. Sigal I. The ras oncogene: Structure and some function. Nature. 1989;332:485.
38. Bos JL, Fearon ER, Hamilton SR, et al. Prevalence of ras gene mutations in human colorectal cancers. Nature. 1987;327:293-7.
39. Bourne HR, Sanders DA, McCormick F. The GTPase superfamily: A conserved switch for diverse cell functions. Nature. 1990;348:125-32.
40. McCormick F. Activators and effectors of ras p21 proteins. Curr Opin Genet Dev. 1994;4:71-6.
41. Rajagopalan H, Bardelli A, Lengauer C, et al. Tumorigenesis: RAF/RAS oncogenes and mismatch-repair status. Nature. 2002;418:934.
42. Luscher B, Eisenman RN. New light on Myc and Myb. Part I. Myc. Genes Dev. 1990;4:2025-35.
43. Blackwood EM, Eisenman RN. Max: A helix-loop-helix zipper protein that forms a sequence-specific DNA-binding complex with Myc. Science. 1991;251:1211-17.
44. Evan GI, Wyllie AH, Gilbert CS, et al. Induction of apoptosis in fibroblasts by c-myc protein. Cell. 1992;69:119-28.
45. He TC, Sparks AB, Rago C, et al. Identification of c-MYC as a target of the APC pathway. Science. 1998;281:1509-12.
46. Knudson AGJr. Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci U S A. 1971;68:820-3.
47. Comings DE. A general theory of carcinogenesis. Proc Natl Acad Sci U S A. 1973;70:3324-8.
48. Miyoshi H, Nakau M, Ishikawa TO, et al. Gastrointestinal hamartomatous polyposis in Lkb1 heterozygous knockout mice. Cancer Res. 2002;62:2261-6.
49. Erichsen HC, Chanock SJ. SNPs in cancer research and treatment. Br J Cancer. 2004;90:747-51.
50. Herman JG, Baylin SB. Gene silencing in cancer in association with promoter hypermethylation. N Engl J Med. 2003;349:2042-54.
51. Leppert M, Dobbs M, Scambler P, et al. The gene for familial polyposis coli maps to the long arm of chromosome 5. Science. 1987;238:1411-3.
52. Kinzler KW, Nilbert MC, Vogelstein B, et al. Identification of a gene located at chromosome 5q21 that is mutated in colorectal cancers. Science. 1991;251:1366-70.
53. Groden J, Thliveris A, Samowitz W, et al. Identification and characterization of the familial adenomatous polyposis coli gene. Cell. 1991;66:589-600.
54. Nishisho I, Nakamura Y, Miyoshi Y, et al. Mutations of chromosome 5q21 genes in FAP and colorectal cancer patients. Science. 1991;253:665-9.
55. Rustgi AK. Hereditary gastrointestinal polyposis and nonpolyposis syndromes. N Engl J Med. 1994;331:1694-702.
56. Powell SM, Zilz N, Beazer-Barclay Y, et al. APC mutations occur early during colorectal tumorigenesis. Nature. 1992;359:235-7.
57. Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61:759-67.
58. Spirio L, Olschwang S, Groden J, et al. Alleles of the APC gene: An attenuated form of familial polyposis. Cell. 1993;75:951-7.
59. Olschwang S, Tiret A, Laurent-Puig P, et al. Restriction of ocular fundus lesions to a specific subgroup of APC mutations in adenomatous polyposis coli patients. Cell. 1993;75:959-68.
60. Greenblatt MS, Bennett WP, Hollstein M, Harris CC. Mutations in the p53 tumor suppressor gene: Clues to cancer etiology and molecular pathogenesis. Cancer Res. 1994;54:4855-78.
61. Nigro JM, Baker SJ, Preisinger AC, et al. Mutations in the p53 gene occur in diverse human tumour types. Nature. 1989;342:705-8.
62. Bressac B, Kew M, Wands J, Ozturk M. Selective G to T mutations of p53 gene in hepatocellular carcinoma from southern Africa. Nature. 1991;350:429-31.
63. Malkin D, Li FP, Strong LC, et al. Germline p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science. 1990;250:1233-8.
64. Seoane J, Le HV, Massague J. Myc suppression of the p21(Cip1) Cdk inhibitor influences the outcome of the p53 response to DNA damage. Nature. 2002;419:729-34.
65. Baker SJ, Markowitz S, Fearon ER, et al. Suppression of human colorectal carcinoma cell growth by wild-type p53. Science. 1990;249:912-15.
66. Liu F, Pouponnot C, Massague J. Dual role of the Smad4/DPC4 tumor suppressor in TGFbeta-inducible transcriptional complexes. Genes Dev. 1997;11:3157-67.
67. Fearon ER, Cho KR, Nigro JM, et al. Identification of a chromosome 18q gene that is altered in colorectal cancers. Science. 1990;247:49-56.
68. Chung DC, Rustgi AK. DNA mismatch repair and cancer. Gastroenterology. 1995;109:1685-99.
69. Fishel R, Lescoe MK, Rao MR, et al. The human mutator gene homolog MSH2 and its association with hereditary nonpolyposis colon cancer. Cell. 1993;75:1027-38.
70. Leach FS, Nicolaides NC, Papadopoulos N, et al. Mutations of a mutS homolog in hereditary nonpolyposis colorectal cancer. Cell. 1993;75:1215-25.
71. Parsons R, Li GM, Longley MJ, et al. Hypermutability and mismatch repair deficiency in RER+ tumor cells. Cell. 1993;75:1227-36.
72. Aaltonen LA, Peltomaki P, Mecklin JP, et al. Replication errors in benign and malignant tumors from hereditary nonpolyposis colorectal cancer patients. Cancer Res. 1994;54:1645-8.
73. Al-Tassan N, Chmiel NH, Maynard J, et al. Inherited variants of MYH associated with somatic G:C→T:A mutations in colorectal tumors. Nat Genet. 2002;30:227-32.
74. Sieber OM, Lipton L, Crabtree M, et al. Multiple colorectal adenomas, classic adenomatous polyposis, and germ-line mutations in MYH. N Engl J Med. 2003;348:791-9.
75. Zimmermann S, Moelling K. Phosphorylation and regulation of Raf by Akt (protein kinase B). Science. 1999;286:1741-4.
76. Ulku AS, Der CJ. Ras signaling, deregulation of gene expression and oncogenesis. Cancer Treat Res. 2003;115:189-208.
77. Brown JR, DuBois RN. COX-2: A molecular target for colorectal cancer prevention. J Clin Oncol. 2005;23:2840-55.
78. Miller EC, Miller JA. Searches for ultimate chemical carcinogens and their reactions with cellular macromolecules. Cancer. 1981;47:2327-45.
79. Ozturk M. p53 mutations in nonmalignant human liver: Fingerprints of aflatoxins? Hepatology. 1995;21:600-1.
80. Bortsch H. N-nitroso-compounds and human cancer: Where do we stand? IARC Sci Publ. 1991;103:105.
81. Haenszel W, Kurihara M. Studies of Japanese migrants. I. Mortality from cancer and other diseases among Japanese in the United States. J Natl Cancer Inst. 1968;40:43-68.
82. Fidler IJ, Radinsky R. Genetic control of cancer metastasis. J Natl Cancer Inst. 1990;82:166-8.
83. Thiery JP. Epithelial-mesenchymal transitions in tumour progression. Nat Rev Cancer. 2002;2:442-54.
84. Yang J, Mani SA, Donaher JL, et al. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell. 2004;117:927-39.
85. Comijn J, Berx G, Vermassen P, et al. The two-handed E box binding zinc finger protein SIP1 downregulates E-cadherin and induces invasion. Mol Cell. 2001;7:1267-78.
86. Batlle E, Sancho E, Franci C, et al. The transcription factor snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nat Cell Biol. 2000;2:84-9.
87. Liotta LA, Steeg PS, Stetler-Stevenson WG. Cancer metastasis and angiogenesis: an imbalance of positive and negative regulation. Cell. 1991;64:327-36.
88. Zhang X, Gaspard JP, Chung DC. Regulation of vascular endothelial growth factor by the Wnt and K-ras pathways in colonic neoplasia. Cancer Res. 2001;61:6050-4.
89. Hurwitz H, Fehrenbacher L, Novotny W, et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med. 2004;350:2335-42.
90. Jeltsch M, Kaipainen A, Joukov V, et al. Hyperplasia of lymphatic vessels in VEGF-C transgenic mice. Science. 1997;276:1423-5.
91. Skobe M, Hawighorst T, Jackson DG, et al. Induction of tumor lymphangiogenesis by VEGF-C promotes breast cancer metastasis. Nat Med. 2001;7:192-8.
92. Steeg PS, Bevilacqua G, Kopper L, et al. Evidence for a novel gene associated with low tumor metastatic potential. J Natl Cancer Inst. 1988;80:200-4.
93. Ahlquist DA, Skoletsky JE, Boynton KA, et al. Colorectal cancer screening by detection of altered human DNA in stool: feasibility of a multitarget assay panel. Gastroenterology. 2000;119:1219-27.
94. Tada M, Omata M, Kawai S, et al. Detection of ras gene mutations in pancreatic juice and peripheral blood of patients with pancreatic adenocarcinoma. Cancer Res. 1993;53:2472-4.
95. Giardiello FM, Brensinger JD, Petersen GM. AGA technical review on hereditary colorectal cancer and genetic testing. Gastroenterology. 2001;121:198-213.
96. Ribic CM, Sargent DJ, Moore MJ, et al. Tumor microsatellite-instability status as a predictor of benefit from fluorouracil-based adjuvant chemotherapy for colon cancer. N Engl J Med. 2003;349:247-57.
97. Cunningham D, Humblet Y, Siena S, et al. Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan-refractory metastatic colorectal cancer. N Engl J Med. 2004;351:337-45.
98. Demetri GD, Benjamin RS, Blanke CD, et al. NCCN Task Force report: Management of patients with gastrointestinal stromal tumor (GIST)—update of the NCCN clinical practice guidelines. J Natl Compr Canc Netw. 2007;5(Suppl 2):S1-29.
99. Liefers GJ, Cleton-Jansen AM, van de Velde CJ, et al. Micrometastases and survival in stage II colorectal cancer. N Engl J Med. 1998;339:223-8.
100. Gold P, Shuster J, Freedman SO. Carcinoembryonic antigen (CEA) in clinical medicine: historical perspectives, pitfalls and projections. Cancer. 1978;42:1399-405.