3 CELL SIGNALING
Cells respond to extracellular signals produced by other cells or by themselves. This mechanism, called cell signaling, allows cell-cell communication and is necessary for the functional regulation and integration of multicellular organisms. Our discussion in this chapter not only provides the basis for understanding normal cell function but serves also as an introduction to the role of abnormal cell signaling in human disease.
Cell signaling mechanisms
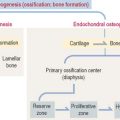
Five major types of cell-cell signaling are considered (Figure 3-1):
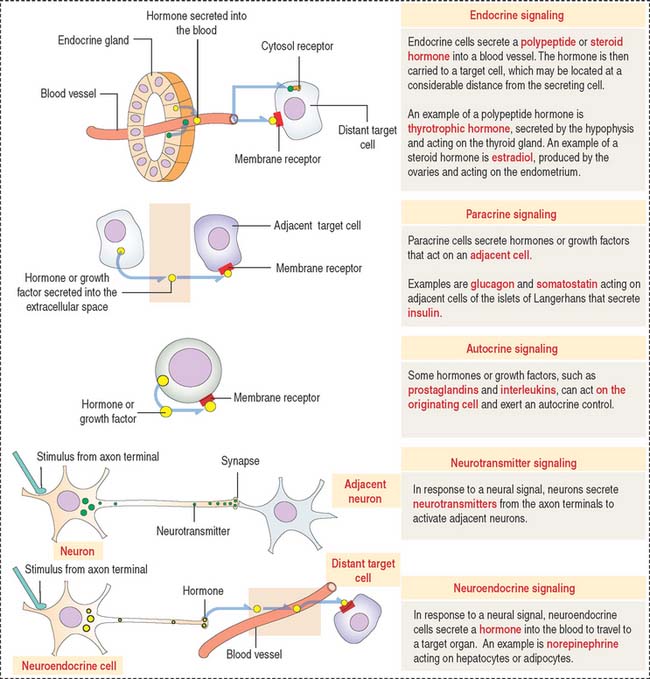
Box 3-A Paracrine cell signaling
Mechanisms of action of cell signaling molecules
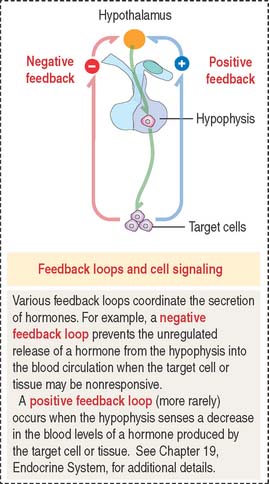
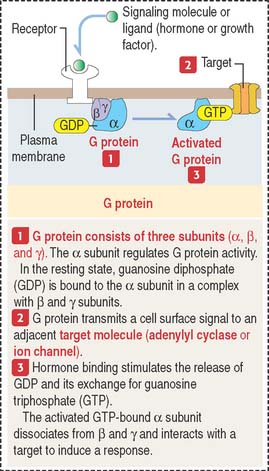
Cell signaling molecules exert their action after binding to receptors expressed by their target cells. Target cells, in turn, can determine either a negative or positive feedback action to regulate the release of the targeting hormone (Figure 3-2).
Cell receptors can be expressed on the cell surface of the target cells. Some receptors are intracellular proteins in the cytosol or the nucleus of target cells. Intracellular receptors require that the signaling molecules diffuse across the plasma membrane (Figure 3-3).
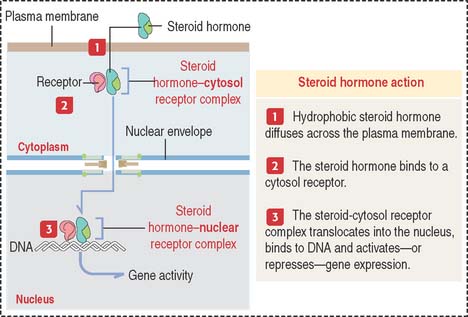
Steroid hormones (Box 3-B) belong to this class of signaling molecules. Steroid hormones are synthesized from cholesterol and include testosterone, estrogen, progesterone, and corticosteroids.
Testosterone, estrogen, and progesterone are sex steroids and are produced by the gonads. Corticosteroids are produced by the cortex of the adrenal gland and include two major classes: glucocorticoids, which stimulate the production of glucose, and mineralocorticoids, which act on the kidneys to regulate water and salt balance.
Steroid receptors are members of the steroid receptor superfamily. They act as transcription factors through their DNA binding domains, which have transcription activation or repression functions. Steroid hormones and related molecules can therefore regulate gene expression.
In the androgen insensitivity syndrome (also known as the testicular feminization syndrome [Tfm]), there is a mutation in the gene expressing the testosterone receptor such that the receptor cannot bind the hormone, and hence the cells do not respond to the hormone. Although genetically male, the individual develops the secondary sexual characteristics of a female. We discuss the androgen insensitivity syndrome in Chapter 21, Sperm Transport and Maturation.
Nitric oxide
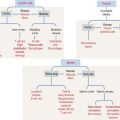
Cell signaling molecules bind to cell surface receptors
A large variety of signaling molecules bind to cell surface receptors. Several groups are recognized
Pathways of intracellular signaling by cell surface receptors
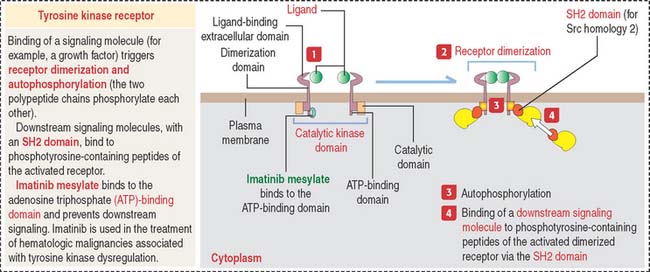
Binding of a ligand (a growth factor) to the extracellular domain of these receptors induces receptor dimerization that results in receptor autophosphorylation (the two polypeptide chains phosphorylate one another). The autophosphorylation of the receptors determines the binding of the tyrosine kinase domain to downstream signaling molecules. Downstream signaling molecules bind to phosphotyrosine residues through domains called SH2 domains (for Src homology 2). Src (for sarcoma) is a gene present in the tumor-producing Rous sarcoma virus and encodes a protein that functions as a protein tyrosine kinase.
Clinical significance: Tyrosine kinases, targets for therapeutic agents
There are two main classes of tyrosine kinases: (1) receptor tyrosine kinases are transmembrane proteins with a ligand-binding extracellular domain and a catalyic intracellular kinase domain (see Figure 3-5), and (2) nonreceptor tyrosine kinases found in the cytosol, nucleus, and inner side of the plasma membrane.
In the absence of a ligand, receptor tyrosine kinases are unphosphorylated and monomeric. The nonreceptor tyrosine kinase is maintained in an inactive state by cellular inhibitor proteins. Activation occurs when the inhibitors are dissociated or by recruitment to transmembrane receptors that trigger autophosphorylation. Tyrosine kinase activity terminates when tyrosine phosphatases hydrolyze tyrosyl phosphates and by induction of inhibitory molecules.
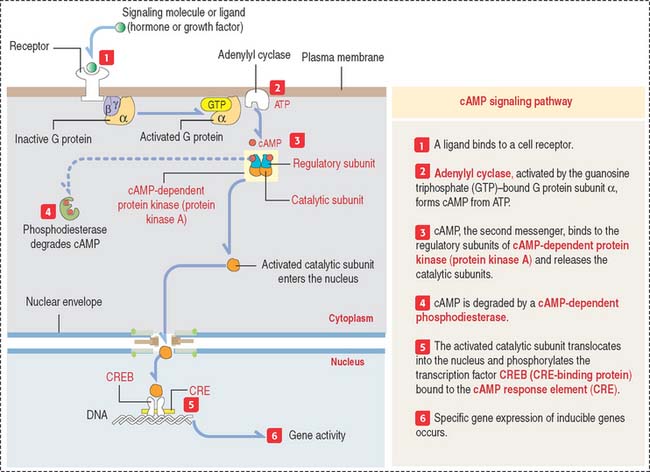
The cAMP pathway
The intracellular signaling effects of cAMP (Figure 3-6) are mediated by the enzyme cAMP-dependent protein kinase (or protein kinase A). In its inactive form, protein kinase A is a tetramer composed of two regulatory subunits (to which cAMP binds) and two catalytic subunits. Binding of cAMP results in the dissociation of the catalytic subunits. Free catalytic subunits can phosphorylate serine residues on target proteins.
The cGMP pathway
The best characterized role of cGMP is in photoreceptor rod cells of the retina, where it converts light signals to nerve impulses. Chapter 9, Sensory Organs: Vision and Hearing, in the eye section, provides a detailed description of this cell signaling process.
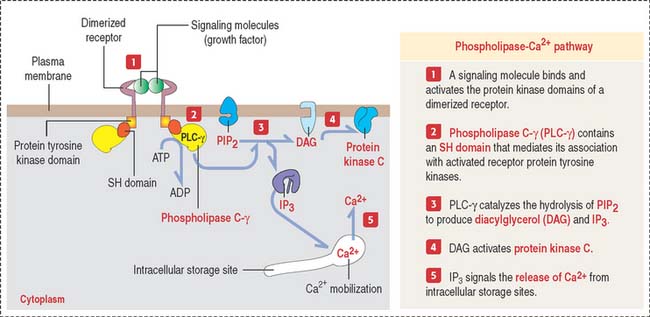
Phospholipase C–Ca2+ pathway
Another second messenger involved in intracellular signaling derives from the phospholipid phosphatidylinositol 4,5-bisphosphate (PIP2) present in the inner leaflet of the plasma membrane (Figure 3-7).
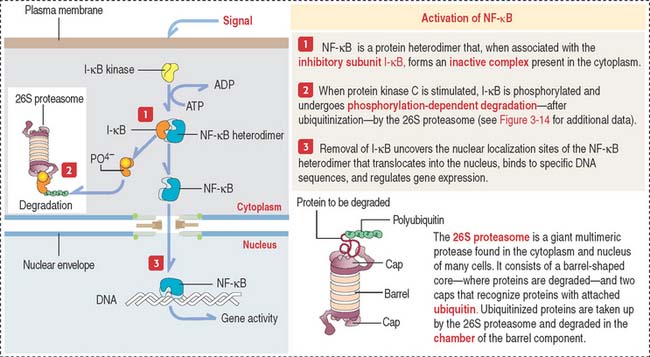
NF-κB transcription factor pathway
NF-κB is a transcription factor involved in immune responses in several cells and is stimulated by protein kinase C (Figure 3-8).
Ca2+-calmodulin pathway
Although the second messenger diacylglycerol remains associated with the plasma membrane, the other second messenger IP3, derived from PIP2, is released into the cytosol to activate ion pumps and free Ca2+ from intracellular storage sites. High cytosolic Ca2+ concentrations (from a basal level of 0.1 μM to an increased 1.0 μM concentration after cytosolic release) activate several Ca2+-dependent protein kinases and phosphatases.
MAP kinase pathway
This pathway involves evolutionarily conserved protein kinases (yeast to humans) with roles in cell growth and differentiation. MAP kinases are protein serine and threonine kinases activated by growth factors and other signaling molecules (Figure 3-9).
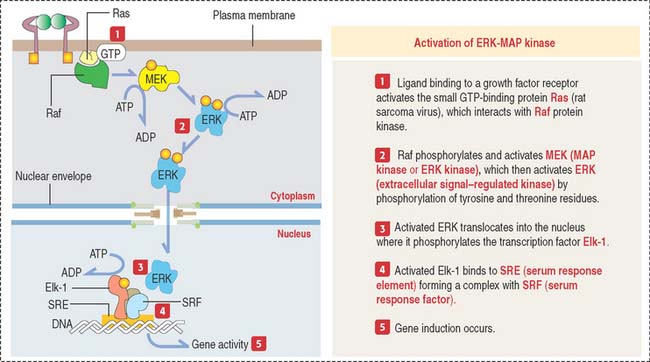
In addition to ERK, mammalian cells contain two other MAP kinases called JNK and p38 MAP kinases. Cytokines and ultraviolet irradiation stimulate JNK and p38 MAP kinase activation mediated by small GTP-binding proteins different from Ras. These kinases are not activated by MEK but by a distinct dual kinase called MKK (for MAP kinase kinase).
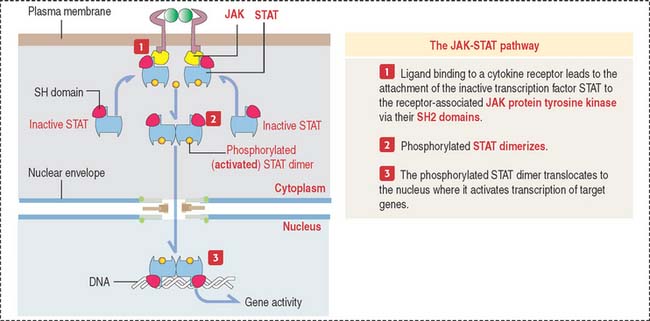
JAK-STAT pathway
The JAK-STAT pathway provides a close connection between protein tyrosine kinases and transcription factors by directly affecting transcription factors (Figure 3-10).
Transcription factor genes: SOX9
The HMG domain of Sox proteins can bend DNA, and facilitate the interaction of enhancers with a distantly located promoter region of a target gene. Several SOX genes act in different developmental pathways. For example, Sox9 protein is expressed in the gonadal ridges of both genders but is up-regulated in males and down-regulated in females before gonadal differentiation. Sox9 also regulates chondrogenesis and the expression of type II collagen (see Chapter 4, Connective Tissue). Mutations of the SOX9 gene cause skeletal defects (campomelic dysplasia), and sex reversal (XY females).
Stem cells: A multipotent cell population
Stem cells have the potential to generate a large number of mature cells continuously throughout life. When stem cells divide by mitosis, some of the progeny differentiates into a specific cell type. Other progeny remains as stem cells (Figure 3-11). The intestinal epithelium, the epidermis of the skin, the hematopoietic system, and spermatogenic cells of the seminiferous epithelium share this property. We discuss in detail the significance of stem cells in each of these tissues in the appropriate chapters.
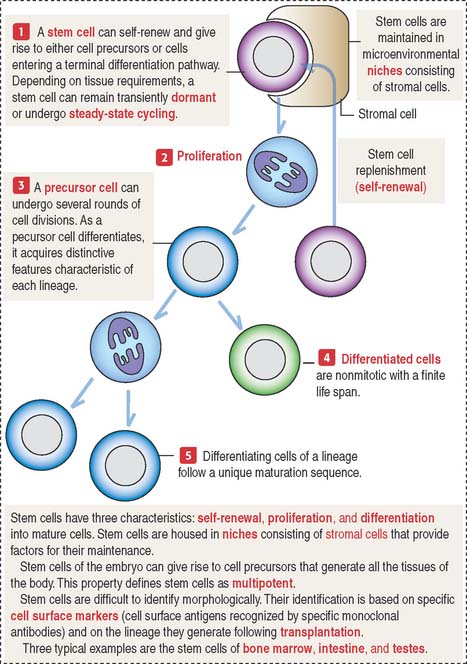
Following stress and injury, other tissues, such as the liver, muscle, and the nervous system, can regenerate mature cells. For example, it has been shown that bone marrow stem cells can produce muscle tissue as well as hematopoietic tissue in an appropriate host system (see Chapter 7, Muscle Tissue). Cultured stem cells of the central nervous system are capable of hematopoiesis in transplanted irradiated mouse recipients.
In vitro cell proliferation, senescence, and telomerase
In our discussion of mitosis (see Figure 1-53 in Chapter 1, Epithelium), we call attention to the role of telomerase, an enzyme that maintains the ends of chromosomes, or telomeres.
Apoptosis, or programmed cell death
Apoptosis (Figure 3-12) is different from necrosis. Necrosis is a nonphysiologic process that occurs after acute injury (for example, in an ischemic stroke). Necrotic cells lyse and release cytoplasmic and nuclear contents into the environment, thus triggering an inflammatory reaction.
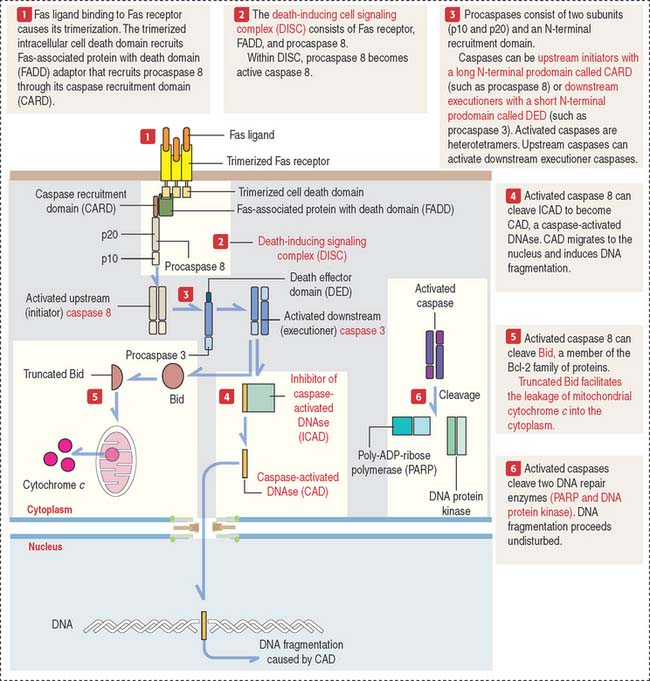
Apoptotic cell death is observed during fetal development. For example, the formation of fingers and toes of the fetus requires the elimination by apoptosis of the tissue between them. During fetal development of the central nervous system, an excess of neurons, eliminated later by apoptosis, is required to establish appropriate connections or synapses between them (see Chapter 8, Nervous Tissue). Mature granulocytes in peripheral blood have a life span of 1 to 2 days before undergoing apoptosis. The clonal selection of T cells in the thymus (to eliminate self-reactive lymphocytes to prevent autoimmune diseases; see Chapter 10, Immune-Lymphatic System) and cellular immune responses involve apoptosis.
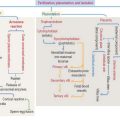
External signals trigger apoptosis: Fas receptor/Fas ligand
Procaspase 8 autoactivated at DISC becomes active caspase 8. Active caspase 8 can do two things:
As we will discuss in Chapter 10, Immune-Lymphatic System, a cytotoxic T cell destroys a target cell (for example, a virus-infected cell) by first binding to the target cell and then releasing Fas ligand. Fas ligand binds to Fas receptor on the surface of the target cell and triggers the cell death cascade.
Caspases: Initiators and executioners of cell death
Procaspases consist of two sub units (p10 and p20) and an N-terminal recruitment domain (see Figure 3-12). Activated caspases are heterotetramers consisting of two p10 sub units and two p20 sub units derived from two procaspases.
Completion of the cell death process occurs when executioner caspases activate the DNA degradation machinery. Caspases cleave two DNA repair enzymes (poly-ADP-ribose polymerase [PARP], and DNA protein kinase), and unrestricted fragmentation of chromatin occurs.
Bcl-2 regulates the release of mitochondrial cytochrome c through Bax
In the cytoplasm, leaking cytochrome c, in the presence of ATP, soluble internal membrane proteins (SIMPs), and procaspase 9, binds to Apaf-1 to form a complex called an apoptosome.
The apoptosome determines the activation of caspase 9, an upstream initiator of apoptosis (Figure 3-13). Caspase 9 activates caspase 3 and caspase 7, leading to cell death.
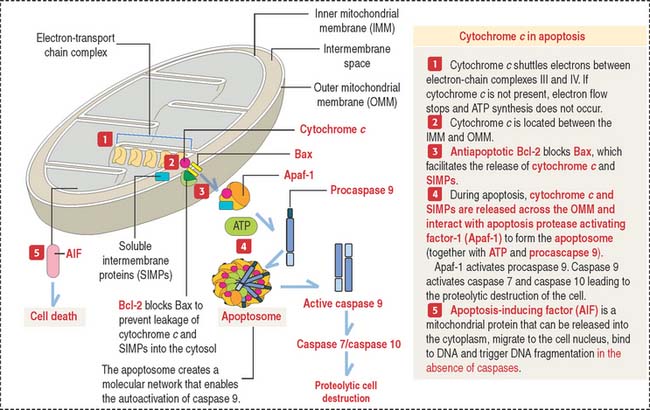
Clinical significance of apoptosis: Neurodegenerative diseases
ALS consists in the progressive loss of motor neurons in the brain, brainstem, and spinal cord. A mutation in the gene encoding superoxide dismutase 1 (SID1) has been identified in patients with familial ALS. Activated caspase 1 and caspase 3 have been found in spinal cord samples of patients with ALS. Motor neurons and axons die and reactive microglia and astrocytes are present. We come back to ALS in Chapter 9, Nervous Tissue.
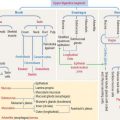
Three major cellular mechanisms are involved in proteolysis
In addition to the procaspase-caspase pathway activated by Fas ligand (see Figure 3-12), the intracellular degradation of residual or misfolded proteins (proteolysis) can occur by the classic endosomal-lysosomal pathway (see Figure 2-19), the apoptosis pathway (see Figure 3-12), and the ubiquitin-proteasome pathway (Figure 3-14). We have already seen that the endosomal-lysosomal mechanism operates within a membrane-bound acidic compartment. In contrast, the procaspase-caspase pathway and the ubiquitin-proteasome pathway carry out proteolysis in the cytosol.
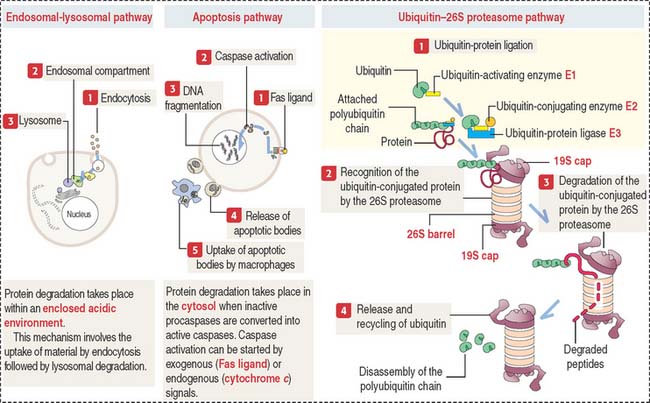
The ubiquitin–26S proteasome pathway involves four successive regulated steps:
Proto-oncogenes and oncogenes
Genes that cause cancer are called oncogenes (Greek onkos, bulk, mass; genos, birth). Most oncogenes originate from proto-oncogenes (Greek prõtos, first). Proto-oncogenes (Box 3-E) are involved in the four basic regulatory mechanisms of cell growth by expressing growth factors, growth factor receptors, signal transduction molecules, and nuclear transcription factors.
The classification of genes as proto-oncogenes is based on the understanding that mutant forms of these genes participate in the development of cancer (see Box 3-F). However, proto-oncogenes serve different biochemical functions in the control of normal growth and development. They can also undergo a variety of mutations that convert them to dominant genes capable of inducing cancers in the absence of viruses.
Box 3-F Proto-oncogenes and tumor suppressor proteins in human cancers
p53: Inactivation of this tumor suppressor protein, a transcription factor expressed in response to DNA damage (see Figure 1-54), is associated with 50% to 60% of human cancers. Inactive p53 enables the progression of cells containing damaged DNA through the cell cycle.
Cell Signaling