[level-membership-for-cardiovascular-category]29
Cell Biology of the Specialized Cardiac Conduction System
The framework of the cardiac conduction system is laid down early during heart development. Paff et al.1 noted that the chick electrocardiogram (ECG) transforms from a sinusoidal waveform to the mature configuration before identifiable components of the cardiac conduction system are formed (Figure 29-1).1 They also noted that atrioventricular (AV) block was achievable with digitalis at the 18-somite stage before a discernible PR interval was evident, suggesting that the AV nodal primordium develops within the early heart tube (see Figure 29-1, B).1 After the 18-somite stage, cardiac chamber formation ensues and the ECG begins to manifest evidence of fast conduction by the presence of high frequency P waves and QRS complexes (see Figure 29-1, C and D). The evolution of the chick ECG clearly demonstrates that the slow conducting nodal elements are present in the early looped heart and that the fast conducting elements are incorporated during chamber formation. The attainment of a mature ECG configuration before structural maturation of the CCS is achieved by interposing slowly conducting AV canal myocardium between the fast conducting atrial and ventricular chamber myocardium (Figure 29-1, E). The fast-conducting components are enriched in high-conductance gap junction proteins, connexin40 (Cx40), and the α-subunit of the cardiac sodium channel, Nav1.5 (encoded by Scn5a), whereas the AV canal expresses low-conductance gap junction proteins, Cx30.2 and Cx45.2 How these electrophysiologically distinct regions are specified and what defines the boundaries between slow and fast conduction have been the focus of intense research over the past 50 years. This chapter discusses the developmental origins of the cardiac conduction system and the transcriptional networks that govern its formation.
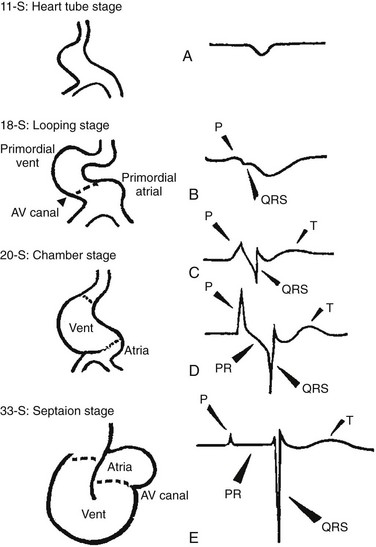
Figure 29-1 Schematic of chick heart development with corresponding electrocardiograms at the somite stage. A, 11. B, 18. C and D, 20. E, 33.1
Histologic Analysis of the Developing Mammalian Cardiac Conduction System
Viragh and Challice3–5 performed meticulous histologic analysis of the developing cardiac conduction system in mouse embryos between 8 and 12 days after coitus (E8-E12). Conduction cells were distinguished from working cardiomyocytes by the following characteristics: (1) periodic acid–Schiff (PAS) positive staining, (2) poorly organized contractile apparatus; (3) enriched glycogen content, and (4) reduced number of T-tubules. Using these features, the temporal-spatial distribution of conduction cells was then tracked during cardiac development.3–5
In the developing mouse embryo, the contractile sequence of the heart is established by E9, well before the appearance of the sinoatrial node primordium at E11.5 The origin of contraction was noted to be in the right sinus horn. Within the dorsolateral wall of the sinus horns, loose mesenchymal cells were noted to transform into the early sinus musculature, which extends along the sinus side of the sinoatrial venous valves. This aggregation of early sinus muscle tissue was the presumed site of SAN development and corresponded to the site that Wenink termed the “sinoatrial (SA) ring.”6 The SAN primordium was recognizable at E11 in the medioanterior wall of the right superior vena cava within the early sinus muscle. A left-sided SAN develops simultaneously in the medioanterior region of the left common cardinal vein, but ultimately resorbs and incorporates into the wall of the left atrium.5 In humans, the SAN primordium is a long structure located within the subepicardial cleft at the site of invagination of the sinus venosus and the right atrium, which forms the right venous valve.5
The sinoatrial (SA) and AV conduction system develop simultaneously. At E9 to E10, the AV canal (AVC; i.e., AV ring) is a well-defined constriction, and the inner cell layer of the AVC makes numerous interconnections with the trabecular compartment, which is the source of the His-Purkinje system (HPS).3,4 At E11, the primordium of the AVN was identified as a PAS+ cell cluster in the inner, dorsal AVC. These PAS+ AVC cells were contiguous with the crest of the developing interventricular septum (primary ring), positioning the AV nodal anlage in direct communication with the primordial His bundle and bundle branches. During this time, the outer cell layer of the AVC was undergoing apoptosis. In the trabecular region, glycogen-rich, PAS+ cells were seen immediately subjacent to the endocardium; these nascent Purkinje cells formed extensive connections with the developing bundle branches.3,4 Therefore, all components of the AV conduction system are in contact with each other throughout cardiogenesis, indicating that an initial framework for the mature conduction system is in place in the early heart.
The work by Viragh and Challice3–5 clearly demonstrated that conduction system development is inextricably linked to cardiogenesis. Yet, significant questions remained regarding the cellular origins of the conduction system, the mechanism by which the pool of conduction cells expands, and the factors that dictate CCS specification and patterning.
Cellular Origins of the Cardiac Conduction System
The neuronal qualities of the cardiac conduction system led many to believe that its cellular origins were from neural crest derivatives. However, lineage-tracing studies in the chick and mouse demonstrated that all conductive components of the CCS are myocardial in origin.7–9 Using a replication-defective retrovirus expressing LacZ, individual myocytes were labeled in developing chick hearts before neural crest immigration, and the expression of β-galactosidase (β-gal) was traced in daughter cells.7 Single myocyte clones gave rise to both conduction cells and working cardiomyocytes. Labeling of neural crest cells, however, failed to show any incorporation into the CCS.7,8 Similar findings were reported in the murine heart using retrospective clonal analysis, where individual myocyte progenitor clones gave rise to conduction and nonconduction cells.9 Similar to the chick, lineage-tracing studies in mice have failed to identify a direct contribution of neural crest cells to the specialized conduction system.10,11
Models of Cardiac Conduction System Development
The prevailing models of conduction system development are the ring model,6 the inductive recruitment or in-growth model,7 the early specification or outgrowth model,12 and the biphasic model (Figure 29-2). The ring hypothesis was based on early observations that the specialized conduction system formed within four constriction points in the D-looped heart (see Figure 29-2, A).6 Four rings were noted to form from the venous pole to the arterial pole: the SA ring, AV ring, primary (interventricular) ring, and ventriculoarterial (VA) ring. These constriction areas are created by differential proliferation rates that exist between ring myocardial cells and flanking chamber cardiomyocytes. Although these areas of constriction exist within the developing heart during chamber formation, the ring hypothesis has largely been discredited, as there is no evidence to support a preexisting template for ring formation within the tubular heart. It is now known that the linear heart tube is composed of clonally related myocardial cells from the first heart field.13
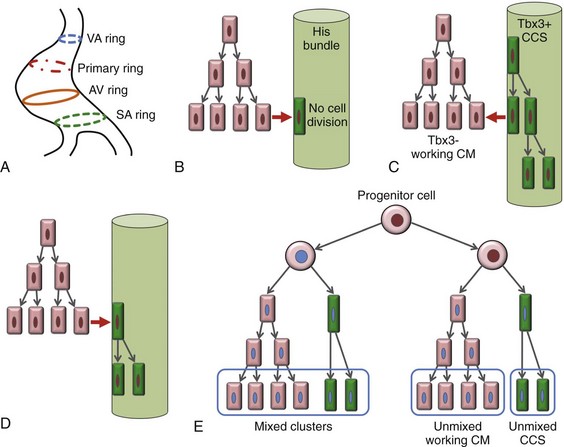
Figure 29-2 Models of cardiac conduction system development. A, Ring model. Prespecification of conduction system components within the linear heart tube. B, Inductive recruitment or ingrowth model. Undefined inductive signals recruit proliferating cardiomyocytes to a conduction lineage, then cease to proliferate (red arrow). C, Early specification or outgrowth model. Tbx3+ CCS cells expand from primitive myocardium that retains a conduction phenotype. Loss of Tbx3 (or Tbx2) expression results in a phenotypic change from a CCS cell to a working cardiomyocyte, the default pathway (red arrow). D, The biphasic model incorporates both the ingrowth and outgrowth models. E, Retrospective clonal analysis of LacZ-labeled, cardiomyocyte clones (blue nuclei). Mixed clusters of conduction and working cardiomyocytes are consistent with a common myocardial progenitor. Unmixed clusters of CCS-only cells demonstrate the potential for limited rounds of cell proliferation. (From Miquerol L, Beyer S, Kelly RG: Establishment of the mouse ventricular conduction system. Cardiovasc Res 91:232–242, 2011.)
Cheng et al.7 put forth the recruitment model or “in-growth model” based on several observations noted during chick VCS development (see Figure 29-2, B).7 First, the proliferative capacity of developing VCS components was found to be significantly lower than working myocytes based on pulse labeling experiments using [3H]-thymidine. Once specified, conduction cells appeared to exit the cell cycle and become quiescent. Second, lineage tracing studies showed that individually labeled myocyte clones gave rise to conduction cells and working myocytes. Third, cell birth dating experiments demonstrated that new conductive cells were added to the developing His bundle in lamellar fashion, analogous to tree rings. These observations led the authors to conclude that the specialized conduction system expands through a process of inductive recruitment of neighboring myocytes.7 However, it was not speculated what constituted the early framework upon which new conduction cells were added or the nature of the molecular signal used for inductive recruitment.
The early specification model, or outgrowth model, states that conduction cells expand from a progenitor pool that retains its specialized conduction phenotype (see Figure 29-2, C).12,14 The conduction gene programming is retained by the expression of transcriptional repressors that suppress a working myocardial phenotype, which is the default pathway. In support of this hypothesis, persistent expression of repressive transcription factors (Tbx2, Tbx3, Msx2, and Id2) has been identified within primordial conduction regions.12,14,15 Tbx3 is expressed as a continuous band linking the SAN and AVN, internodal tracts, and the proximal ventricular conduction system.12 Heterologous expression of Tbx2 or Tbx3 is able to suppress chamber-type myocardial genes (Nppa [ANF], Gja1 [Cx43], Gja5 [Cx40]), inhibit cardiac chamber formation, and in the case of Tbx3 elicit ectopic pacemaker formation.12,14–16 Consistent with these findings, Tbx3 and Cx43 exhibit complementary expression patterns in the developing heart.12
Most recently a biphasic model of conduction system development has been proposed (Figure 29-2D).9 In this model, once conduction cells are recruited from myocardial precursors, they retain the capacity to undergo limited rounds of cell division. Analysis of labeled myocyte clones revealed two classes of conductive clusters, mixed and unmixed (see Figure 29-2, E). The mixed clusters represented single myocyte clones that gave rise to both conductive and working myocytes (recruitment). The unmixed clones were composed of either working myocytes or conduction myocytes, but not both. Conduction-only, unmixed clones were identified throughout the central and peripheral VCS, indicating that once specified, all components of the conduction system are capable of approximately four to five rounds of cell division. Based on these findings, the authors concluded that mammalian VCS development appears to use both in-growth and outgrowth modes of expansion.9 However, these findings are not incompatible with the early specification/outgrowth model, because mixed clusters might represent conduction cells that have lost Tbx2/Tbx3 expression and have defaulted to the chamber pathway.
Molecular Markers of the Cardiac Conduction System
Visualization of the developing CCS has been greatly enhanced by the development of conduction system reporter mice (Figure 29-3). Each reporter mouse delineates different components of the CCS at various developmental time points using LacZ or green fluorescent protein (GFP) expression. The CCS-LacZ and minK-LacZ mouse lines are representative examples of well-established markers of the specialized conduction system.17–19 The CCS-LacZ mouse was created serendipitously through a complex genomic rearrangement involving the MC4/engrailed-2-LacZ cassette (see Figure 29-3, A).18 CCS-LacZ reporter expression can first be detected at E8.5 in the SAN primordium within the venous pole. At subsequent stages, β-gal expression is detected in the developing and mature AVN and His-Purkinje system. All CCS reporter lines have some degree of cardiac expression outside of the conduction system. In the adult CCS-LacZ heart, significant β-gal expression is seen within the right atrium.18
Figure 29-3 Cardiac conduction system reporter mice. A, CCS-LacZ. B, Contactin2-eGFP. C, Connexin-40–eGFP.20
The minK-LacZ reporter mouse was created by replacing the minK gene with a nuclear-targeted LacZ cassette.17 Early in development, β-gal expression was noted in the SA ring, AV ring, interventricular ring, and the VA ring. Subsequently, β-gal expression was confined to the AVN and the proximal conduction system, as well as in the venous valves, AV ring, and VA valves.17,19
The Cx40-eGFP reporter mouse has become a widely used tool to characterize normal and abnormal patterning of the mature His-Purkinje system (see Figure 29-3, C).20 Developmentally, Cx40 expression is not restricted to the VCS, with significant expression in the trabecular myocardium. In addition, Cx40 is not expressed in the distal AVN or His bundle before E14.5. In the mature heart, Cx40 is enriched in atrial myocardium and in coronary endothelial cells.20
Contactin-2 (Cntn-2) was recently identified as a CCS-enriched factor using differential gene profiling of adult mouse Purkinje fibers versus working myocytes (see Figure 29-3, B).21 Cntn-2 is a cell adhesion molecule that has a role in neuronal patterning and ion channel clustering. Both Cntn2-LacZ knock-in mice and Cntn2-EGFP BAC transgenic reporter mice delineated the entire cardiac conduction system in postnatal hearts. Currently, a functional role for Cntn-2 in the CCS has not been identified.21
Transcription Factor Regulatory Networks
A rich hierarchy of gene networks dictates the specification and patterning of conduction components (Figure 29-4). Unifying all these networks is the balance struck between prochamber myocardial programming versus antichamber programming. As mentioned previously, the T-box transcription factors dictate much of this equilibrium, tilting the scales toward or away from a conduction lineage. The T-box factors can function as transcriptional activators or repressors and are known to be critical regulators of cardiac specification and differentiation.22 Seven TBX family members are expressed in the developing heart, four of which (TBX1, TBX5, TBX20, TBX3) have been linked to human congenital heart disease.23–25 The major cardiac transcriptional activators, Tbx5 and Tbx20, act through Nkx2-5 and Gata4 to drive prochamber myocardial gene expression, such as Nppa (ANF) and Gja5 (Cx40).26–28 The transcriptional repressors Tbx2, Tbx3, and Tbx18 compete with Tbx5 for Nkx2-5 binding to suppress chamber-specific gene expression, thus maintaining a conduction gene profile.12,14,15,29,30
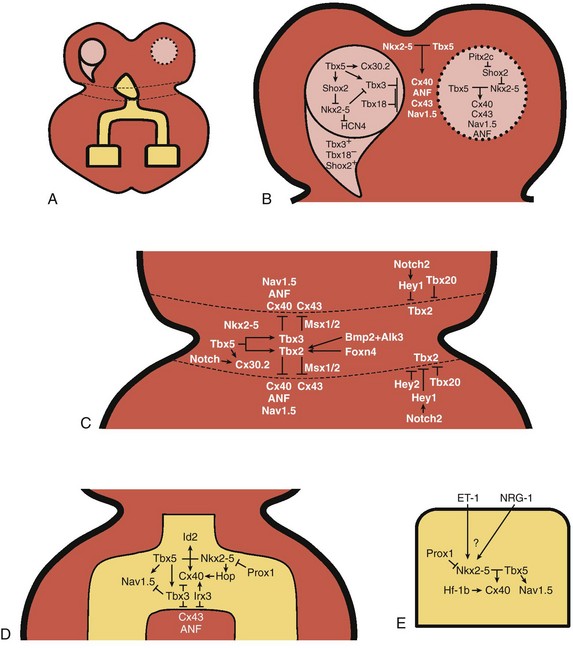
Figure 29-4 Transcription factor regulatory networks. A, Schematic of the cardiac conduction system. B, Right sinoatrial node (head and tail domains), atrial myocardium, and left sinoatrial node (stippled circle). C, Atrioventricular canal/node region flanked by atrial and ventricular myocardium. D, His bundle and bundle branches and ventricular myocardium. E, Purkinje fiber.
The delicate balance between Tbx activators and repressors and the expression of cofactors dictates the rich conduction system phenotypes seen in the mature CCS. Sitting on top of the CCS specification hierarchy is Tbx5, a critical determinant of many elements of the slow and fast conduction system.31–33 Mutations in Tbx5 result in Holt-Oram syndrome, an autosomal dominant condition characterized by preaxial radial ray limb deformities and cardiac septation defects.22 The septal defects are typically ostium secundum atrial septal defects, muscular ventricular septal defects, and atrioventricular canal defects. Patients with Holt-Oram syndrome manifest variable degrees of CCS dysfunction, including sinus bradycardia and AV block, even in the absence of overt structural heart disease.22 Analysis of Tbx5 transgenic mice demonstrated an exquisitely sensitive, dose-dependent correlation of Tbx5 expression level with severity of congenital heart defects and conduction disease.34
Interestingly, the temporal-spatial expression of Tbx5 correlates well with developmental timing of conduction system components.35 At E8.5, Tbx5 expression is enriched in a posterior-anterior axis, with highest levels of expression in the atria and sinus venosus, the site of SA node specification. During chamber formation, Tbx5 expression is graded in a left-right axis with highest levels in the left ventricle, particularly in the AV canal, ventricular conduction system and the trabecular myocardium (the site of Purkinje fiber development). Tbx5 expression is scant in the right ventricle except in the developing AV bundle, right bundle branch, and trabecular region.35
Both the dose–phenotype correlation and the temporospatial expression pattern of Tbx5 implicate it as a master regulator of conduction system development. However, the broad expression pattern of Tbx5 beyond the borders of the CCS, and the ability of Tbx5 with its binding partners, Nkx2-5 and Gata4, to elicit chamber myocardial gene programming suggests that it functions with CCS-restricted cofactors to elicit conduction specification.26,28 Numerous regulatory feedback loops have been identified in CCS components that enhance the conduction phenotype while simultaneously repressing chamber-specific gene programming (see Figure 29-4). These conduction-restricted cofactors will be discussed in their regional context.
The Sinoatrial Node
The mammalian sinoatrial node is a large comma-shaped structure with its head region located at the junction between the right superior vena cava and the right atrium, and the tail region situated along the crista terminalis. These regions represent distinct cellular lineages, as evidenced by their unique expression profiles. The SAN head, which constitutes up to 75% of SAN volume, develops from sinus venosus (SV) myocardium and retains the SV signature, Shox2+;Tbx3+;Tbx18+;Nkx2-5−.36,37 In contrast, working atrial myocytes derive from second heart field mesodermal progenitors and express Nkx2-5+;Shox2−;Tbx3−;Tbx18−. The SAN tail domain has an expression profile in between that of the head region and the atrial myocardium, expressing Shox2+;Tbx3+;Tbx18−;Nkx2-5weakly +. Lineage tracing studies have shown that the SAN tail originates from SV myocardium, but loses Tbx18 expression during development.37
Proper SAN development is dependent on the appropriate expression of Tbx5, Shox2, Tbx18, and Tbx3 (see Figure 29-4, B). Tbx5 expression in the sinus venosus is a critical regulator of the SAN signature through its actions on the transcriptional repressor, Tbx3, and the homeobox transcription factor, Shox2.34,38 Homozygous Tbx5del/del mice die embryonically at E10.5 because of severe hypoplasia of the sinoatrial region and of the primitive LV.27 Microarray analysis of Tbx5 heterozygous hearts, identified Tbx3 and Shox2 as significantly downregulated targets, and both factors showed reduced expression in the sinoatrial region.34,38
As stated earlier, Tbx3 is expressed throughout the developing and mature CCS (except the Purkinje network) and represses chamber-specific programming. Like Tbx5, Tbx3 displays critical dose dependency for proper differentiation and homeostatic maintenance of the conduction system.39 Analysis of Tbx3 mutant mice revealed a dose-dependent SAN phenotype with variable degrees of sinus node dysfunction and inappropriate expression of chamber-specific genes (Cx43, Cx40, Nppa, Scn5a) within the SAN region. Although the overall structure of the SAN is normal, SAN volume was reduced by approximately 45% to 60%, although this phenotype has not been consistently seen in all Tbx3 mutants when adjusted for weight.16,29,37,39 Ectopic overexpression of Tbx3 in atrial myocardium results in inappropriate suppression of chamber genes and upregulation of the SAN gene profile.29 These data suggest that Tbx3 is not essential for SAN formation, but is important for establishing and maintaining proper pacemaker gene programming.
Shox2 is essential for formation of the sinoatrial valves and development of the sinoatrial node.40 Mice deficient in Shox2 are embryonic lethal because of severe bradycardia in the setting of SAN hypoplasia.40,41 Evaluation of the Shox2−/− SAN region revealed reduced levels of Tbx3 and the pacemaker channel, HCN4, indicating a failure of differentiation.41 Downregulation of Tbx3 resulted in ectopic expression of chamber myocardial genes (Nppa, Cx43, Cx40) within the SAN. Furthermore, Nkx2-5, which is normally absent in SAN myocardium, was expressed ectopically in the SAN region.40 Luciferase reporter assays revealed that Shox2 negatively regulates expression of Nkx2-5, a known inhibitor of Tbx3 and Hcn4 expression.16 Thus, Shox2 promotes SAN development by repressing Nkx2-5 expression, thereby preventing atrial myocardialization of the SAN region.41
The left-sided sinoatrial node (L-SAN) forms in parallel with right-sided SAN (R-SAN), but regresses during development and incorporates into the left atrium.5 Abnormal persistence of L-SAN remnants has been implicated as a source for left atrial arrhythmic triggers.42 The homeodomain transcription factor Pitx2c, which regulates left-right asymmetry, directly regulates L-SAN resorption.43 Mice deficient in Pitx2c invariably develop a persistent L-SAN, which shares an identical gene expression profile with the R-SAN.16 Therefore, L-SAN specification is dependent on Shox2 signaling. Pitx2c directly represses Shox2 expression in the developing sinus venous myocardium and in the adult left atrium, leading to L-SAN regression.42 Genome-wide association studies (GWAS) have identified a region near PITX2 as a key susceptibility locus for atrial fibrillation.44 Interestingly, Pitx2c haploinsufficiency increased the propensity for atrial arrhythmias, corroborating the GWAS data.42
The importance of Tbx18 in SAN formation was identified in knockout mice, which revealed a marked reduction in the volume of the SAN head, because of either failure to expand the mesenchymal precursor pool or failure to differentiate into SAN cells.37 The tail region, however, was unaffected.37 Despite this reduction in SAN volume, sinus node function was reportedly normal owing to residual SAN tail tissue.37 Tbx3 and Tbx18 double heterozygous embryos demonstrated normal SAN development, indicating a lack of interaction on a genetic level. In addition, Tbx3 deficiency had no effect on Tbx18 and Shox2 levels, suggesting that Tbx3 functions downstream to these factors.45
Similar to Tbx3, heterologous expression of Tbx18 leads to transcriptional repression of Cx43.45 In summary, Shox2 and Tbx18 are critical regulators of SAN formation, whereas Tbx3 together with Tbx18 maintain the appropriate pacemaker signature, assuring optimal function.
Atrioventricular Canal and Atrioventricular Node
The mammalian AVN is a complex, heterogeneous structure that serves as the only conduction pathway between the atria and ventricles. Histologic and electrophysiologic (EP) evaluation of the AVN revealed three distinct layers, from subendocardial to deep layers, termed atrionodal (AN), compact nodal (N), and nodo-His (NH).46 The compact or true nodal region has characteristic nodal action potentials, such as slow upstroke of phase 0, lower peak amplitudes, and diastolic depolarization of phase 4. The AN and NH transition regions exhibit hybrid action potential morphologies intermediate to nodal cells and atrial myocytes or His bundle, respectively.47 These transition zones serve a similar purpose as in SAN cells, to insulate core nodal cells from the hyperpolarizing influence of working myocytes. The AN and NH regions are enriched in Scn5a (Nav1.5), Cx40, and Cx43, whereas the N region has low levels of Nav1.5 and high levels of Cx45/30.2 resulting in the different action potential morphologies and conduction velocities (CV).46
Lineage tracing studies and differential gene expression analysis indicate that the compact AVN (N) derives from AV canal myocardium, whereas the lower nodal cells (NH) are ventricular in origin.48,49 In the adult heart, remnants of the AVC persist as rings of myocardial tissue that possess distinct electrophysiological layers, mirroring that seen in the AVN.50 These results lend support to the histologic observations that the AVN derives from dorsal AV canal cells.3,4
During development, the AVC can be distinguished from flanking atrial and ventricular myocardium by the enrichment of Tbx2, Tbx3, and Bmp2 (see Figure 29-4, C). This transcriptional signature maintains the slow conduction properties (Cx45+;Cx30.2+) and low proliferative capacity of the AVC. As the embryonic heart matures, the outer cell layer of the AVC undergoes apoptosis. Paracrine signals from AVC myocytes induce epicardial cells to undergo epithelial-to-mesenchymal transformation and invade the AV junction to create the annulus fibrosus. This process electrically isolates the atrium and ventricle, except at the dorsal wall of the AVC where the lower AV nodal transitional cells penetrate the fibrous insulation. Defective gene dosing of Tbx2, Tbx3, and Bmp2 leads to developmental abnormalities of the AVC, which can manifest as AV block or as ventricular pre-excitation (i.e., Wolff-Parkinson-White) because of defective annulus fibrosus formation.
The AVC is established in part by the expression of Tbx5 and Nkx2-5, whereas Notch signaling delimits the AVC to its confined region within the developing heart. Tbx5 is enriched in the AVC during development and drives the local expression of Tbx3 and Cx30.2.31,34 Mice haploinsufficient for Tbx5 exhibit sinus node dysfunction, P wave widening, and first- and second-degree AV block.27,33 Significant maturation defects were noted in the AVC and AVN of Tbx5+/− mice.33 The immature configuration of the AV canal resulted in significant slowing of AV nodal conduction time, as measured by prolonged Atrio-His (A-H) intervals during invasive EP testing.33
Nkx2-5, a member of the homeodomain family, plays a central role in cardiac development. Loss of the Nkx2-5 homolog, tinman, in the fruit fly results in failure of cardiogenesis.51 Mice deficient in Nkx2-5 die in utero at E9 to E10 because of arrested cardiac development in the linear heart tube stage. The hearts of these mice undergo partial looping morphogenesis, lack endocardial cushions and trabeculae, and have underdeveloped AV canals.52 Nkx2-5 null embryos lacked minK-LacZ reporter staining in the region of AVN primordium.53 Nkx2-5+/neo mice exhibited an overall reduction in the size of the VCS from the AVN to the distal Purkinje network. Histologic evaluation of the Nkx2-5+/neo AVN revealed that compact nodal cells (N region, Cx40−/Cx45+) are markedly reduced whereas the nodo-His (NH) region (Cx40+/Cx45+) remains intact.53 Consistent with these findings, Nkx2-5+/neo mice had prolonged PR intervals on surface ECG and displayed abnormal AVN physiology on EP testing.53
Tbx2 and Tbx3 are essential for maintaining the slow conduction phenotype of the AV canal/AVN.12,14,15 Tbx2 and Tbx3 are differentially expressed in the AVC, with Tbx2 being more dominant in the left AVC and Tbx3 being more dominant in the right AVC.54 Evaluation of a Tbx3 allelic series demonstrated an exquisite dose–phenotype correlation, with diminishing Tbx3 levels directly correlating with the severity of AV conduction disease and fetal demise.39 AV block was exclusively seen in embryos with the lowest Tbx3 gene dosage. Consistent with its known repressive function, Tbx3-deficient embryos showed ectopic expression of chamber genes within the SAN, AVC, AV bundle, and bundle branches.39,55 Regional deletion of Tbx3 within the AV bundle primordium or the AV canal caused AV block. Therefore, Tbx3 expression is required throughout the multitiered AV conduction axis to maintain normal AV conduction. Furthermore, conditional deletion of Tbx3 at an adult time point resulted in AV block, indicating that Tbx3 is critical for proper maintenance of the AV conduction axis.39
Tbx2 deficiency results in embryonic lethality because of defects in AVC differentiation and outflow tract septation.56 Inappropriate expression of chamber-type genes was also noted in the AVC of Tbx2-deficient hearts, with the left AVC being more affected than the right.54,56 Accordingly, Tbx2 deficiency had a minimal impact on AVN formation, which is predominantly a right-sided structure. However, loss of myocardial Tbx2 resulted in structural defects in the left annulus fibrosus resulting in accessory pathway formation and ventricular preexcitation.54 Therefore, Tbx2 and Tbx3 share nonredundant functions in AVC development and maturation, which may be in part due to unequal distribution of the T-box proteins. Tbx2 expression appears to be critical for maturation of the left annulus fibrosus, which is the most common location of accessory pathways in humans.
A transcriptional regulatory network involving Bmp2, Foxn4, Tbx20, and Notch2/Hey signaling has been identified that regulates the expression of Tbx2 within the AVC. Bmp2 and Foxn4 function as activators of Tbx2 within the AVC, whereas Tbx20 and Notch2/Hey signaling are negative regulators of Tbx2 that define the boundaries of the AVC. The bone morphogenetic factor Bmp2 is enriched in the AVC during development and drives the local expression of Tbx2.57 Bmp2 upregulates Tbx2 promoter activity through a Bmp receptor (Alk3)/Smad dependent pathway.58 Deletion of Bmp2 in the AV canal resulted in loss of Tbx2 expression and ectopic expression of chamber genes.57 Furthermore, AVC-restricted loss of Alk3 led to abnormal annulus fibrosus formation, ventricular preexcitation, and structural and functional defects in the AVN.59,60 A human correlate has been identified in a rare familial form of Wolff-Parkinson-White syndrome associated with a microdeletion involving the BMP2 gene.61
Foxn4, an upstream regulator of tbx2b, was recently identified from a zebrafish mutagenesis screen.62 Foxn4 mutants displayed structural and functional defects in the AV canal. Several AV canal restricted genes were mislocated, including bmp4 and endocardial notch1b, and tbx2b was completely absent from the AVC. Highly conserved Foxn4 and Tbx5 binding sites were identified in the tbx2b promoter, and tbx2b expression in the AVC proved highly sensitive to mutagenesis of the Foxn4 or Tbx5 binding sites.62
In the chick heart, Notch2, Hey1, and Hey2 signaling delimits the AV canal myocardium. Hey1 (expressed in atrium and ventricle) and Hey2 (expressed in ventricle only) are expressed in complementary fashion to Bmp2 in the looped heart.63 Notch2 acts directly through Hey1 to suppress Bmp2, whereas Hey2 suppresses Bmp2 in a Notch-independent manner.63 Mouse and zebrafish hearts deficient in Hey2 displayed abnormally expanded AVC regions that were enriched in Bmp2/bmp4, respectively.63 Tbx2 expression in the AVC is also delimited by atrial and ventricular chamber expression of Tbx20. Observations of Tbx20 knockout embryos showed precocious upregulation and ectopic expression of Tbx2 throughout the cardiac crescent and early heart tube. In vitro reporter assays demonstrated that Tbx20 inhibits Tbx2 promoter activity through its actions on the Alk3/Smad signaling pathway.57
Notch signaling in endocardial cells is critical for the formation of slow conducting AVC myocytes in zebrafish and trabecular myocardium in mice.64,65 Notch2 signaling in chamber myocytes restricts Tbx2 expression within the AVC.63 Whether Notch signaling has a role within developing conduction cells was explored using loss-of-function and gain-of-function Notch mutants overexpressed under the control Mlc2vCre/+.66 The dominant negative Notch mutant, DNMAML, driven by Mlc2vCre/+ displayed reduced AV nodal volumes owing to loss of Cx30.2-expressing cells. Mlc2vCre/+/DNMAML had shortened PR and AH intervals on EP testing, consistent with loss of slow AV nodal conduction.66 Constitutive Notch activation, however, resulted in enlarged AVNs and accessory pathway formation because of abnormal boundary formation between the AVC and ventricular myocardium.66 Subsequent work using gain-of-function Notch mutants demonstrated a robust upregulation of conduction system markers, such as Cntn-2, Tbx5, and Nkx2-5. Notch activation also upregulated nodal genes Gjc3 (Cx30.2) and HCN1 and Purkinje gene Scn5a. Notch activation was able to reprogram ventricular myocytes in vivo and newborn ventricular myocytes in vitro into a Purkinje-type lineage. These results suggest that Notch signaling fate-restricts cardiomyocytes toward a conduction lineage.67
Proximal Ventricular Conduction System
The proximal ventricular conduction system consists of the His bundle and bundle branches. These fast conducting components of the CCS derive from ventricular myocardial precursors. Formation of the proximal VCS is critically dependent on the coexpression of Nkx2-5 and Tbx5, as combined haploinsufficiency of these two factors results in developmental failure of the His bundle and bundle branches.32 During development, the expression levels of Nkx2-5 and Tbx5 increase significantly in the proximal VCS, which drives the expression of a unique set of genes that impart fast conduction properties (Cx40+, Nav1.5+) while also maintaining phenotypic distinctiveness from working myocytes (Tbx3+, Id2+, Cx43−; see Figure 29-4, D). Both Nkx2-5 and Tbx5 have cell-autonomous roles in VCS development and perturbation of either factor leads to significant CCS structural and functional abnormalities.
Nkx2-5 haploinsufficient mice exhibit marked hypoplasia of the ventricular conduction system resulting in PR and QRS interval prolongation.53,68,69 Ventricular-restricted, Nkx2-5 knockout mice display progressive AV conduction disease, advancing to complete heart block by 6 months to 1 year. Histologic analysis revealed a diminutive AVN and an atrophic His-Purkinje system that worsened with age.53,68,70,71 These mice accurately phenocopy the progressive postnatal AV conduction disease seen in patients with NKX2-5 mutations.68 The phenotypic manifestations of Nkx2-5 haploinsufficiency are highly pleotropic in man and mouse models, which suggests that Nkx2-5 is subject to upstream regulatory control by genetic modifiers. One such modifier is prospero-related homeobox protein 1 (Prox-1), which functions in concert with HDAC3 to regulate Nkx2-5 expression.72 Combined haploinsufficiency of Nkx2-5cre/+;Prox1loxP/+ rescued the Nkx2-5cre/+ conduction phenotype on a structural and functional level. Compound heterozygotes rescued the hypoplastic phenotype of the AVN and significantly restored cellularity of the His-Purkinje system, resulting in normalization of ECG parameters.72 In light of these findings, Prox1 appears to function as an upstream regulator of Nkx2-5 gene dosage, which ensures accurate gene expression within the ventricular conduction system.
Mice haploinsufficient for Tbx5 exhibited patterning and maturation defects of the ventricular conduction system. The His bundle and left bundle branches remained immature in all Tbx5del/+ mice, and many of the mice had absent right bundle branches. The expression of Tbx5-responsive genes, Nppa and Cx40, were significantly downregulated.33 Nkx2-5 and Tbx5 coordinately drive the expression of Nppa, Cx40, and inhibitor of differentiation 2 (Id2) within the proximal VCS.32 Id2 is believed to function as an inhibitor of muscle differentiation allowing specification towards a conduction lineage.32 Id2 null mice displayed structural and functional VCS abnormalities similar to those seen in Tbx+/− mice, suggesting that they function within the same transcriptional regulatory network. Indeed, combined haploinsufficiency of Tbx5+/−;Id2+/− resulted in developmental failure of the His bundle and bundle branches.68 Therefore, Nkx2-5, Tbx5, and Id2 function as a ventricular conduction system transcriptional unit that imparts fast conduction properties while also inhibiting myocardial gene programming.
Tbx5 is essential for maintaining the fast conduction properties of the ventricular conduction system. Using a VCS-specific, tamoxifen-inducible Cre driver (minKCreERT2), Tbx5 was knocked out of the adult mouse AVN and HPS, resulting in sudden death as early as 5 weeks after Cre induction.73 Tbx5minKCreERT2 VCS conduction was severely slowed, leading to spontaneous arrhythmias, including Mobitz type II AV block and ventricular tachycardia. Selective ablation of Tbx5 in the VCS resulted in a corresponding loss of Gja5 (Cx40) and Scn5a expression.73,74 SCN5a mutations have been identified in patients with progressive cardiac conduction disease that can exist alone or as overlap syndromes with Brugada or LQT3.75 Inherited progressive cardiac conduction disease is associated with a high risk of complete AV block and Stoke-Adams syncope.75–77
GWAS focused on CCS parameters have identified several loci (TBX5, TBX3, NKX2-5, SCN10A, and SCN5A) that modulate PR and QRS duration in the general population.78–82 Given the importance of Tbx5 and Tbx3 in CCS specification and function, the Scn5a locus was screened for Tbx-responsive elements. A Tbx-responsive enhancer was identified approximately 15 kb downstream of Scn5a and mirrored Scn5a expression in the AV bundle and bundle branches.74 Using another approach, the genome-wide occupancy profile of Tbx3 was performed using chromatin immunoprecipitation-massive parallel sequencing (ChIP-Seq) analysis.83 Two Tbx3/Tbx5 responsive enhancers were located within the Scn5a/Scn10a locus. The orthologous human fragments had expression patterns similar to their mouse counterparts within the developing ventricular conduction system, providing evidence that these enhancers were functionally conserved in humans.83 Furthermore, the GWAS single nucleotide polymorphism, rs6801957, was positioned in a conserved enhancer site in the SCN10A locus. Interestingly, the single nucleotide polymorphism itself alters a highly conserved residue in the consensus T-box binding site, altering its ability to respond to Tbx5 activation and Tbx3 repression.83 These data reaffirm the fidelity of GWAS in identifying gene regulatory networks that underlie phenotypic variations in humans on a population scale.
The maintenance of Cx40 in the proximal VCS is vital for proper function of the His bundle and bundle branches. Mice deficient in Cx40 exhibit slowed conduction through the His-Purkinje system and display bundle branch block.84,85 During development, Cx40 is expressed in the ventricular trabeculae, the site of His-Purkinje formation.3,4,86 Cx40 expression in the ventricular trabeculae dictates the switch in ventricular activation from a pattern of activation to the mature apical-to-basal sequence.86 The regional localization of Cx40 within the proximal VCS is dependent on several transcription factors that function downstream of Nkx2-5 and Tbx5: Hf-1b, Hop, and Irx3.
Hf-1b is a zinc-finger transcription factor with enriched expression within the compact myocardial layers and the ventricular conduction system.87 Mice deficient in Hf-1b manifest sinus node dysfunction, intermittent AV block, and sudden cardiac death due to spontaneous ventricular arrhythmias. Hf-1b−/− hearts demonstrated a reduced number of Cx40+ distal Purkinje fibers and as well as an abnormal intracellular distribution of Cx40, suggesting a defect in trafficking. This phenotype was seen predominantly in the ventricular apex; apical working myocytes were also smaller and had significantly reduced levels of Cx43.88 In addition, the coronary arterial structure and function were also perturbed within the apical region.88 Therefore, the defects in Cx40 and Cx43 expression within the ventricular apex could be multifactorial and will need further investigation.
Homeodomain-only protein (Hop) is a unique member of the homeobox transcription factors that does not directly bind DNA.89 It is known to function downstream of Nkx2-5 and inhibits serum response factor (SRF)-dependent transcription.68 Hop is expressed in the cardiac conduction system, with significant enrichment beyond the neonatal time period. Hop−/− mice have structurally normal hearts and CCS anatomy but manifest slowed conduction in the HPS on electrophysiological testing. Expression of Cx40 was significantly reduced in the atria and the HPS, whereas levels of Cx43 remained normal.89 Therefore, Hop is not required for CCS specification and patterning, but it has an important role in optimizing fast conduction in the HPS through its action on Cx40 expression.
The Iroquois homeobox 3 (Irx3) transcription factor is expressed in the VCS and regulates the fast conduction properties of the His-Purkinje system.90 Irx3 knockout mice exhibit prolonged QRS intervals on surface ECG. Bundle branch cells deficient in Irx3 had reduced levels of Cx40 and ectopic expression of Cx43. The direct coupling of His-bundle cells to working myocytes presumably resulted in source-sink mismatch, leading to charge dissipation and conduction block. Irx3 represses Cx43 expression by directly binding a putative Irx3 responsive element that overlaps with an Nkx2-5 binding site near a conserved T-box binding element. The activation of Cx40, on the other hand, is through an indirect mechanism.90 Therefore, Hf-1b, Hop, and Irx3 function in a nonredundant manner to enhance Cx40 expression in the VCS, whereas simultaneously Irx3 and Tbx3 repress Cx43 expression in specialized conduction cells.
Purkinje Fiber Network
The Purkinje fiber network (PFN) represents the most distal aspect of the ventricular conduction system. In mammals, the PFN derives from the trabecular myocardium likely through a process of recruitment; therefore, the complex patterning of the PFN directly mirrors the complexity of the trabecular network.8 Paracrine signals from the overlying endocardium, such as endothelin-1 (ET-1) and neuregulin-1 (NRG-1), direct underlying trabecular myocytes towards a conduction path (see Figure 29-4, E).91 The involvement of ET-1 was discovered in the chick, where unlike in mammals, avian Purkinje fibers are found endocardially and periarterially.8 Periarterial Purkinje fibers (PFs) develop in a pattern that strictly follows coronary vascular anatomy. It was shown subsequently that exposure of cultured embryonic myocytes to ET-1 was able to convert these cells to a PF lineage.8,91 Exposure of mouse embryonic stem cells to ET-1, but not NRG-1, was able to upregulate CCS markers.92 Retroviral-mediated coexpression of both the endothelin-1 precursor (preproendothelin-1) and ECE-1 in the chick embryonic myocardium induced myocytes to express PF genes in vivo.93
ET-1 is converted to its active form by endothelin-converting enzyme (ECE), which is highly expressed in endothelial cells and in developing PFs.94 ECE-1 expression is regulated by hemodynamic load. Conotruncal banding, a model of ventricular pressure overload, resulted in significant upregulation of ECE-1 expression and concomitant precocious expansion of Cx40+ PFs.95 As discussed before, the shift in ventricular activation to an apex-to base pattern directly correlates with Cx40 expression. Banding resulted in an earlier shift in ventricular activation to an apex-to-base pattern, whereas pressure unloading delayed this process. Therefore, ventricular hemodynamics is a key epigenetic factor in the regulation of HPS development.94–96
Neuregulin-1 (Nrg-1) is another endothelial-derived factor with a significant role in PF specification. Using an in vitro embryonic culture system, the response of developing CCS-LacZ hearts to Nrg-1 and ET-1 treatment was studied.97 Nrg-1 induced a profound upregulation of LacZ+ cells in the developing heart in a time window between E8.5 and E10.5. The increase in LacZ+ cells could not be explained by changes in proliferation or apoptosis, and suggested that Nrg-1 induced embryonic cardiomyocytes toward a conduction phenotype.97 The marked upregulation of CCS-LacZ–positive cells in Nrg-1–treated hearts was associated with changes in the ventricular activation pattern on optical mapping. The effect of ET-1 on CCS-LacZ upregulation, however, was more modest.97
The ability of NRG-1 and ET-1 to induce a Purkinje gene profile was evaluated in dissociated mouse embryonic ventricular myocytes.98 Both Nrg-1 and ET-1 increased the expression of Nkx2-5, Gata4, Irx4, Hop, HF-1b, minK, Cx40, and Cx45. There was no cumulative effect of coadministration of NRG-1 and ET-1 on Purkinje gene programming.98 The ability of ET-1 and NRG-1 to activate Nkx2-5, Gata4, HF-1b, and Hop suggests that these secreted factors activate a potent gene regulatory network for PF specification.
Nkx2-5 gene dosage is critical for proper maturation of the PFN.99 Inappropriately high or low levels of Nkx2-5 can negatively affect proper Purkinje development.99 Consistent with other models of Nkx2-5 deficiency, adult Nkx2-5+/−/Cx40eGFP/+ mice showed marked reduction in HPS cellularity, despite the trabecular region appearing qualitatively normal during development.69 Evaluation of the remaining PFs revealed that they were of normal size and had normal electrophysiologic properties.69 Therefore, the conduction defects seen in Nkx2-5+/−/Cx40eGFP/+ hearts appears to be due to HPS patterning abnormalities rather than electrophysiological defects at the cellular level.69 The reduction of Purkinje fibers in Nkx2-5+/− hearts occurs mainly after birth and is due to either reduced PF recruitment or loss of PF in the postnatal period.69 Based on chimeric analysis, postnatal development of the PFN was critically dependent on the dose of Nkx2-5, which behaved in a cell-autonomous manner.69
References
1. Paff, GH, Boucek, RJ, Harrell, TC. Observations on the development of the electrocardiogram. Anat Rec. 1968; 160:575–582.
2. Kreuzberg, MM, Willecke, K, Bukauskas, FF. Connexin-mediated cardiac impulse propagation: connexin 30. 2 slows atrioventricular conduction in mouse heart. Trends Cardiovasc Med. 2006; 16:266–272.
3. Viragh, S, Challice, CE. The development of the conduction system in the mouse embryo heart. II. Histogenesis of the atrioventricular node and bundle. Dev Biol. 1977; 56:397–411.
4. Viragh, S, Challice, CE. The development of the conduction system in the mouse embryo heart. I. The first embryonic A-V conduction pathway. Dev Biol. 1977; 56:382–396.
5. Viragh, S, Challice, CE. The development of the conduction system in the mouse embryo heart. Dev Biol. 1980; 80:28–45.
6. Wenink, AC. Development of the human cardiac conducting system. J Anat. 1976; 121:617–631.
7. Cheng, G, Litchenberg, WH, Cole, GJ, et al. Development of the cardiac conduction system involves recruitment within a multipotent cardiomyogenic lineage. Development. 1999; 126:5041–5049.
8. Gourdie, RG, Mima, T, Thompson, RP, et al. Terminal diversification of the myocyte lineage generates Purkinje fibers of the cardiac conduction system. Development. 1995; 121:1423–1431.
9. Miquerol, L, Moreno-Rascon, N, Beyer, S, et al. Biphasic development of the mammalian ventricular conduction system. Circ Res. 2010; 107:153–161.
10. Kitajima, S, Miyagawa-Tomita, S, Inoue, T, et al. Mesp1-nonexpressing cells contribute to the ventricular cardiac conduction system. Dev Dyn. 2006; 235:395–402.
11. Poelmann, RE, Jongbloed, MR, Molin, DG, et al. The neural crest is contiguous with the cardiac conduction system in the mouse embryo: a role in induction? Anat Embryol (Berl). 2004; 208:389–393.
12. Hoogaars, WM, Tessari, A, Moorman, AF, et al. The transcriptional repressor Tbx3 delineates the developing central conduction system of the heart. Cardiovasc Res. 2004; 62:489–499.
13. Meilhac, SM, Kelly, RG, Rocancourt, D, et al. A retrospective clonal analysis of the myocardium reveals two phases of clonal growth in the developing mouse heart. Development. 2003; 130:3877–3889.
14. Christoffels, VM, Hoogaars, WM, Tessari, A, et al. T-box transcription factor Tbx2 represses differentiation and formation of the cardiac chambers. Dev Dyn. 2004; 229:763–770.
15. Boogerd, KJ, Wong, LY, Christoffels, VM, et al. Msx1 and Msx2 are functional interacting partners of T-box factors in the regulation of Connexin43. Cardiovasc Res. 2008; 78:485–493.
16. Mommersteeg, MT, Hoogaars, WM, Prall, OW, et al. Molecular pathway for the localized formation of the sinoatrial node. Circ Res. 2007; 100:354–362.
17. Kupershmidt, S, Yang, T, Anderson, ME, et al. Replacement by homologous recombination of the minK gene with lacZ reveals restriction of minK expression to the mouse cardiac conduction system. Circ Res. 1999; 84:146–152.
18. Rentschler, S, Vaidya, DM, Tamaddon, H, et al. Visualization and functional characterization of the developing murine cardiac conduction system. Development. 2001; 128:1785–1792.
19. Kondo, RP, Anderson, RH, Kupershmidt, S, et al. Development of the cardiac conduction system as delineated by minK-lacZ. J Cardiovasc Electrophysiol. 2003; 14:383–391.
20. Miquerol, L, Meysen, S, Mangoni, M, et al. Architectural and functional asymmetry of the His-Purkinje system of the murine heart. Cardiovasc Res. 2004; 63:77–86.
21. Pallante, BA, Giovannone, S, Fang-Yu, L, et al. Contactin-2 expression in the cardiac Purkinje fiber network. Circ Arrhythm Electrophysiol. 2010; 3:186–194.
22. Basson, CT, Bachinsky, DR, Lin, RC, et al. Mutations in human TBX5 [corrected] cause limb and cardiac malformation in Holt-Oram syndrome. Nat Genet. 1997; 15:30–35.
23. Stennard, FA, Harvey, RP. T-box transcription factors and their roles in regulatory hierarchies in the developing heart. Development. 2005; 132:4897–4910.
24. Linden, H, Williams, R, King, J, et al. Ulnar Mammary syndrome and TBX3: expanding the phenotype. Am J Med Genet A. 2009; 149A:2809–2812.
25. Meneghini, V, Odent, S, Platonova, N, et al. Novel TBX3 mutation data in families with ulnar-mammary syndrome indicate a genotype-phenotype relationship: mutations that do not disrupt the T-domain are associated with less severe limb defects. Eur J Med Genet. 2006; 49:151–158.
26. Hiroi, Y, Kudoh, S, Monzen, K, et al. Tbx5 associates with Nkx2-5 and synergistically promotes cardiomyocyte differentiation. Nat Genet. 2001; 28:276–280.
27. Bruneau, BG, Nemer, G, Schmitt, JP, et al. A murine model of Holt-Oram syndrome defines roles of the T-box transcription factor Tbx5 in cardiogenesis and disease. Cell. 2001; 106:709–721.
28. Garg, V, Kathiriya, IS, Barnes, R, et al. GATA4 mutations cause human congenital heart defects and reveal an interaction with TBX5. Nature. 2003; 424:443–447.
29. Hoogaars, WM, Engel, A, Brons, JF, et al. Tbx3 controls the sinoatrial node gene program and imposes pacemaker function on the atria. Genes Dev. 2007; 21:1098–1112.
30. Habets, PE, Moorman, AF, Clout, DE, et al. Cooperative action of Tbx2 and Nkx2. 5 inhibits ANF expression in the atrioventricular canal: implications for cardiac chamber formation. Genes Dev. 2002; 16:1234–1246.
31. Munshi, NV, McAnally, J, Bezprozvannaya, S, et al. Cx30. 2 enhancer analysis identifies Gata4 as a novel regulator of atrioventricular delay. Development. 2009; 136:2665–2674.
32. Moskowitz, IP, Kim, JB, Moore, ML, et al. A molecular pathway including Id2, Tbx5, and Nkx2-5 required for cardiac conduction system development. Cell. 2007; 129:1365–1376.
33. Moskowitz, IP, Pizard, A, Patel, VV, et al. The T-Box transcription factor Tbx5 is required for the patterning and maturation of the murine cardiac conduction system. Development. 2004; 131:4107–4116.
34. Mori, AD, Zhu, Y, Vahora, I, et al. Tbx5-dependent rheostatic control of cardiac gene expression and morphogenesis. Dev Biol. 2006; 297:566–586.
35. Bruneau, BG, Logan, M, Davis, N, et al. Chamber-specific cardiac expression of Tbx5 and heart defects in Holt-Oram syndrome. Dev Biol. 1999; 211:100–108.
36. Mommersteeg, MT, Dominguez, JN, Wiese, C, et al. The sinus venosus progenitors separate and diversify from the first and second heart fields early in development. Cardiovasc Res. 2010; 87:92–101.
37. Wiese, C, Grieskamp, T, Airik, R, et al. Formation of the sinus node head and differentiation of sinus node myocardium are independently regulated by Tbx18 and Tbx3. Circ Res. 2009; 104:388–397.
38. Puskaric, S, Schmitteckert, S, Mori, AD, et al. Shox2 mediates Tbx5 activity by regulating Bmp4 in the pacemaker region of the developing heart. Hum Mol Genet. 2010; 19:4625–4633.
39. Frank, DU, Carter, KL, Thomas, KR, et al. Lethal arrhythmias in Tbx3-deficient mice reveal extreme dosage sensitivity of cardiac conduction system function and homeostasis. Proc Natl Acad Sci U S A. 2012; 109:E154–163.
40. Blaschke, RJ, Hahurij, ND, Kuijper, S, et al. Targeted mutation reveals essential functions of the homeodomain transcription factor Shox2 in sinoatrial and pacemaking development. Circulation. 2007; 115:1830–1838.
41. Espinoza-Lewis, RA, Yu, L, He, F, et al. Shox2 is essential for the differentiation of cardiac pacemaker cells by repressing Nkx2-5. Dev Biol. 2009; 327:376–385.
42. Wang, J, Klysik, E, Sood, S, et al. Pitx2 prevents susceptibility to atrial arrhythmias by inhibiting left-sided pacemaker specification. Proc Natl Acad Sci U S A. 2010; 107:9753–9758.
43. Ammirabile, G, Tessari, A, Pignataro, V, et al. Pitx2 confers left morphological, molecular, and functional identity to the sinus venosus myocardium. Cardiovasc Res. 2012; 93:291–301.
44. Gudbjartsson, DF, Arnar, DO, Helgadottir, A, et al. Variants conferring risk of atrial fibrillation on chromosome 4q25. Nature. 2007; 448:353–357.
45. Kapoor, N, Galang, G, Marban, E, et al. Transcriptional suppression of connexin43 by TBX18 undermines cell-cell electrical coupling in postnatal cardiomyocytes. J Biol Chem. 2011; 286:14073–14079.
46. Ko, YS, Yeh, HI, Ko, YL, et al. Three-dimensional reconstruction of the rabbit atrioventricular conduction axis by combining histological, desmin, and connexin mapping data. Circulation. 2004; 109:1172–1179.
47. Efimov, IR, Mazgalev, TN. High-resolution, three-dimensional fluorescent imaging reveals multilayer conduction pattern in the atrioventricular node. Circulation. 1998; 98:54–57.
48. Aanhaanen, WT, Brons, JF, Dominguez, JN, et al. The Tbx2+ primary myocardium of the atrioventricular canal forms the atrioventricular node and the base of the left ventricle. Circ Res. 2009; 104:1267–1274.
49. Horsthuis, T, Buermans, HP, Brons, JF, et al. Gene expression profiling of the forming atrioventricular node using a novel tbx3-based node-specific transgenic reporter. Circ Res. 2009; 105:61–69.
50. Aanhaanen, WT, Mommersteeg, MT, Norden, J, et al. Developmental origin, growth, and three-dimensional architecture of the atrioventricular conduction axis of the mouse heart. Circ Res. 2010; 107:728–736.
51. Ranganayakulu, G, Elliott, DA, Harvey, RP, et al. Divergent roles for NK-2 class homeobox genes in cardiogenesis in flies and mice. Development. 1998; 125:3037–3048.
52. Tanaka, M, Chen, Z, Bartunkova, S, et al. The cardiac homeobox gene Csx/Nkx2. 5 lies genetically upstream of multiple genes essential for heart development. Development. 1999; 126:1269–1280.
53. Jay, PY, Harris, BS, Maguire, CT, et al. Nkx2-5 mutation causes anatomic hypoplasia of the cardiac conduction system. J Clin Invest. 2004; 113:1130–1137.
54. Aanhaanen, WT, Boukens, BJ, Sizarov, A, et al. Defective Tbx2-dependent patterning of the atrioventricular canal myocardium causes accessory pathway formation in mice. J Clin Invest. 2011; 121:534–544.
55. Bakker, ML, Boukens, BJ, Mommersteeg, MT, et al. Transcription factor Tbx3 is required for the specification of the atrioventricular conduction system. Circ Res. 2008; 102:1340–1349.
56. Harrelson, Z, Kelly, RG, Goldin, SN, et al. Tbx2 is essential for patterning the atrioventricular canal and for morphogenesis of the outflow tract during heart development. Development. 2004; 131:5041–5052.
57. Ma, L, Lu, MF, Schwartz, RJ, et al. Bmp2 is essential for cardiac cushion epithelial-mesenchymal transition and myocardial patterning. Development. 2005; 132:5601–5611.
58. Singh, R, Horsthuis, T, Farin, HF, et al. Tbx20 interacts with smads to confine tbx2 expression to the atrioventricular canal. Circ Res. 2009; 105:442–452.
59. Gaussin, V, Morley, GE, Cox, L, et al. Alk3/Bmpr1a receptor is required for development of the atrioventricular canal into valves and annulus fibrosus. Circ Res. 2005; 97:219–226.
60. Stroud, DM, Gaussin, V, Burch, JB, et al. Abnormal conduction and morphology in the atrioventricular node of mice with atrioventricular canal targeted deletion of Alk3/Bmpr1a receptor. Circulation. 2007; 116:2535–2543.
61. Lalani, SR, Thakuria, JV, Cox, GF, et al. 20p12. 3 microdeletion predisposes to Wolff-Parkinson-White syndrome with variable neurocognitive deficits. J Med Genet. 2009; 46:168–175.
62. Chi, NC, Shaw, RM, De Val, S, et al. Foxn4 directly regulates tbx2b expression and atrioventricular canal formation. Genes Dev. 2008; 22:734–739.
63. Rutenberg, JB, Fischer, A, Jia, H, et al. Developmental patterning of the cardiac atrioventricular canal by Notch and Hairy-related transcription factors. Development. 2006; 133:4381–4390.
64. Milan, DJ, Giokas, AC, Serluca, FC, et al. Notch1b and neuregulin are required for specification of central cardiac conduction tissue. Development. 2006; 133:1125–1132.
65. Grego-Bessa, J, Luna-Zurita, L, del Monte, G, et al. Notch signaling is essential for ventricular chamber development. Dev Cell. 2007; 12:415–429.
66. Rentschler, S, Harris, BS, Kuznekoff, L, et al. Notch signaling regulates murine atrioventricular conduction and the formation of accessory pathways. J Clin Invest. 2011; 121:525–533.
67. Rentschler, S, Yen, AH, Lu, J, et al. Myocardial Notch signaling reprograms cardiomyocytes to a Conduction-Like Phenotype. Circulation. 2012.
68. Pashmforoush, M, Lu, JT, Chen, H, et al. Nkx2-5 pathways and congenital heart disease; loss of ventricular myocyte lineage specification leads to progressive cardiomyopathy and complete heart block. Cell. 2004; 117:373–386.
69. Meysen, S, Marger, L, Hewett, KW, et al. Nkx2. 5 cell-autonomous gene function is required for the postnatal formation of the peripheral ventricular conduction system. Dev Biol. 2007; 303:740–753.
70. Wakimoto, H, Kasahara, H, Maguire, CT, et al. Developmentally modulated cardiac conduction failure in transgenic mice with fetal or postnatal overexpression of DNA nonbinding mutant Nkx2. 5. J Cardiovasc Electrophysiol. 2002; 13:682–688.
71. Kasahara, H, Wakimoto, H, Liu, M, et al. Progressive atrioventricular conduction defects and heart failure in mice expressing a mutant Csx/Nkx2. 5 homeoprotein. J Clin Invest. 2001; 108:189–201.
72. Risebro, CA, Petchey, LK, Smart, N, et al. Epistatic rescue of Nkx2. 5 adult cardiac conduction disease phenotypes by prospero-related homeobox protein 1 and HDAC3. Circ Res. 2012; 111:e19–31.
73. Arnolds, DE, Moskowitz, IP. Inducible recombination in the cardiac conduction system of minK: CreERT(2) BAC transgenic mice. Genesis. 2011; 49:878–884.
74. Arnolds, DE, Liu, F, Fahrenbach, JP, et al. TBX5 drives Scn5a expression to regulate cardiac conduction system function. J Clin Invest. 2012; 122:2509–2518.
75. Probst, V, Kyndt, F, Potet, F, et al. Haploinsufficiency in combination with aging causes SCN5A-linked hereditary Lenegre disease. J Am Coll Cardiol. 2003; 41:643–652.
76. Kyndt, F, Probst, V, Potet, F, et al. Novel SCN5A mutation leading either to isolated cardiac conduction defect or Brugada syndrome in a large French family. Circulation. 2001; 104:3081–3086.
77. Schott, JJ, Alshinawi, C, Kyndt, F, et al. Cardiac conduction defects associate with mutations in SCN5A. Nat Genet. 1999; 23:20–21.
78. Sotoodehnia, N, Isaacs, A, de Bakker, PI, et al. Common variants in 22 loci are associated with QRS duration and cardiac ventricular conduction. Nat Genet. 2010; 42:1068–1076.
79. Chambers, JC, Zhao, J, Terracciano, CM, et al. Genetic variation in SCN10A influences cardiac conduction. Nat Genet. 2010; 42:149–152.
80. Holm, H, Gudbjartsson, DF, Arnar, DO, et al. Several common variants modulate heart rate, PR interval and QRS duration. Nat Genet. 2010; 42:117–122.
81. Pfeufer, A, van Noord, C, Marciante, KD, et al. Genome-wide association study of PR interval. Nat Genet. 2010; 42:153–159.
82. Smith, JG, Magnani, JW, Palmer, C, et al. Genome-wide association studies of the PR interval in African Americans. PLoS Genet. 2011; 7:e1001304.
83. van den Boogaard, M, Wong, LY, Tessadori, F, et al. Genetic variation in T-box binding element functionally affects SCN5A/SCN10A enhancer. J Clin Invest. 2012; 122:2519–2530.
84. van Rijen, HV, van Veen, TA, van Kempen, MJ, et al. Impaired conduction in the bundle branches of mouse hearts lacking the gap junction protein connexin40. Circulation. 2001; 103:1591–1598.
85. Tamaddon, HS, Vaidya, D, Simon, AM, et al. High-resolution optical mapping of the right bundle branch in connexin40 knockout mice reveals slow conduction in the specialized conduction system. Circ Res. 2000; 87:929–936.
86. Sankova, B, Benes, J, Jr., Krejci, E, et al. The effect of connexin40 deficiency on ventricular conduction system function during development. Cardiovasc Res. 2012; 95:469–479.
87. Nguyen-Tran, VT, Kubalak, SW, Minamisawa, S, et al. A novel genetic pathway for sudden cardiac death via defects in the transition between ventricular and conduction system cell lineages. Cell. 2000; 102:671–682.
88. Hewett, KW, Norman, LW, Sedmera, D, et al. Knockout of the neural and heart expressed gene HF-1b results in apical deficits of ventricular structure and activation. Cardiovasc Res. 2005; 67:548–560.
89. Ismat, FA, Zhang, M, Kook, H, et al. Homeobox protein Hop functions in the adult cardiac conduction system. Circ Res. 2005; 96:898–903.
90. Zhang, SS, Kim, KH, Rosen, A, et al. Iroquois homeobox gene 3 establishes fast conduction in the cardiac His-Purkinje network. Proc Natl Acad Sci U S A. 2011; 108:13576–13581.
91. Gourdie, RG, Wei, Y, Kim, D, et al. Endothelin-induced conversion of embryonic heart muscle cells into impulse-conducting Purkinje fibers. Proc Natl Acad Sci U S A. 1998; 95:6815–6818.
92. Gassanov, N, Er, F, Zagidullin, N, et al. Endothelin induces differentiation of ANP-EGFP expressing embryonic stem cells towards a pacemaker phenotype. FASEB J. 2004; 18:1710–1712.
93. Takebayashi-Suzuki, K, Yanagisawa, M, Gourdie, RG, et al. In vivo induction of cardiac Purkinje fiber differentiation by coexpression of preproendothelin-1 and endothelin converting enzyme-1. Development. 2000; 127:3523–3532.
94. Sedmera, D, Harris, BS, Grant, E, et al. Cardiac expression patterns of endothelin-converting enzyme (ECE): Implications for conduction system development. Dev Dyn. 2008; 237:1746–1753.
95. Hall, CE, Hurtado, R, Hewett, KW, et al. Hemodynamic-dependent patterning of endothelin converting enzyme 1 expression and differentiation of impulse-conducting Purkinje fibers in the embryonic heart. Development. 2004; 131:581–592.
96. Reckova, M, Rosengarten, C, deAlmeida, A, et al. Hemodynamics is a key epigenetic factor in development of the cardiac conduction system. Circ Res. 2003; 93:77–85.
97. Rentschler, S, Zander, J, Meyers, K, et al. Neuregulin-1 promotes formation of the murine cardiac conduction system. Proc Natl Acad Sci U S A. 2002; 99:10464–10469.
98. Patel, R, Kos, L. Endothelin-1 and Neuregulin-1 convert embryonic cardiomyocytes into cells of the conduction system in the mouse. Dev Dyn. 2005; 233:20–28.
99. Harris, BS, Spruill, L, Edmonson, AM, et al. Differentiation of cardiac Purkinje fibers requires precise spatiotemporal regulation of Nkx2-5 expression. Dev Dyn. 2006; 235:38–49.
[/level-membership-for-cardiovascular-category][not-level-membership-for-cardiovascular-category]29
Cell Biology of the Specialized Cardiac Conduction System
The framework of the cardiac conduction system is laid down early during heart development. Paff et al.1 noted that the chick electrocardiogram (ECG) transforms from a sinusoidal waveform to the mature configuration before identifiable components of the cardiac conduction system are formed (Figure 29-1).1 They also noted that atrioventricular (AV) block was achievable with digitalis at the 18-somite stage before a discernible PR interval was evident, suggesting that the AV nodal primordium develops within the early heart tube (see Figure 29-1, B).1 After the 18-somite stage, cardiac chamber formation ensues and the ECG begins to manifest evidence of fast conduction by the presence of high frequency P waves and QRS complexes (see Figure 29-1, C and D). The evolution of the chick ECG clearly demonstrates that the slow conducting nodal elements are present in the early looped heart and that the fast conducting elements are incorporated during chamber formation. The attainment of a mature ECG configuration before structural maturation of the CCS is achieved by interposing slowly conducting AV canal myocardium between the fast conducting atrial and ventricular chamber myocardium (Figure 29-1, E). The fast-conducting components are enriched in high-conductance gap junction proteins, connexin40 (Cx40), and the α-subunit of the cardiac sodium channel, Nav1.5 (encoded by Scn5a), whereas the AV canal expresses low-conductance gap junction proteins, Cx30.2 and Cx45.2 How these electrophysiologically distinct regions are specified and what defines the boundaries between slow and fast conduction have been the focus of intense research over the past 50 years. This chapter discusses the developmental origins of the cardiac conduction system and the transcriptional networks that govern its formation.
Figure 29-1 Schematic of chick heart development with corresponding electrocardiograms at the somite stage. A, 11. B, 18. C and D, 20. E, 33.1
Histologic Analysis of the Developing Mammalian Cardiac Conduction System
Viragh and Challice3–5 performed meticulous histologic analysis of the developing cardiac conduction system in mouse embryos between 8 and 12 days after coitus (E8-E12). Conduction cells were distinguished from working cardiomyocytes by the following characteristics: (1) periodic acid–Schiff (PAS) positive staining, (2) poorly organized contractile apparatus; (3) enriched glycogen content, and (4) reduced number of T-tubules. Using these features, the temporal-spatial distribution of conduction cells was then tracked during cardiac development.3–5
In the developing mouse embryo, the contractile sequence of the heart is established by E9, well before the appearance of the sinoatrial node primordium at E11.5 The origin of contraction was noted to be in the right sinus horn. Within the dorsolateral wall of the sinus horns, loose mesenchymal cells were noted to transform into the early sinus musculature, which extends along the sinus side of the sinoatrial venous valves. This aggregation of early sinus muscle tissue was the presumed site of SAN development and corresponded to the site that Wenink termed the “sinoatrial (SA) ring.”6 The SAN primordium was recognizable at E11 in the medioanterior wall of the right superior vena cava within the early sinus muscle. A left-sided SAN develops simultaneously in the medioanterior region of the left common cardinal vein, but ultimately resorbs and incorporates into the wall of the left atrium.5 In humans, the SAN primordium is a long structure located within the subepicardial cleft at the site of invagination of the sinus venosus and the right atrium, which forms the right venous valve.5
The sinoatrial (SA) and AV conduction system develop simultaneously. At E9 to E10, the AV canal (AVC; i.e., AV ring) is a well-defined constriction, and the inner cell layer of the AVC makes numerous interconnections with the trabecular compartment, which is the source of the His-Purkinje system (HPS).3,4 At E11, the primordium of the AVN was identified as a PAS+ cell cluster in the inner, dorsal AVC. These PAS+ AVC cells were contiguous with the crest of the developing interventricular septum (primary ring), positioning the AV nodal anlage in direct communication with the primordial His bundle and bundle branches. During this time, the outer cell layer of the AVC was undergoing apoptosis. In the trabecular region, glycogen-rich, PAS+ cells were seen immediately subjacent to the endocardium; these nascent Purkinje cells formed extensive connections with the developing bundle branches.3,4 Therefore, all components of the AV conduction system are in contact with each other throughout cardiogenesis, indicating that an initial framework for the mature conduction system is in place in the early heart.
The work by Viragh and Challice3–5 clearly demonstrated that conduction system development is inextricably linked to cardiogenesis. Yet, significant questions remained regarding the cellular origins of the conduction system, the mechanism by which the pool of conduction cells expands, and the factors that dictate CCS specification and patterning.
Cellular Origins of the Cardiac Conduction System
The neuronal qualities of the cardiac conduction system led many to believe that its cellular origins were from neural crest derivatives. However, lineage-tracing studies in the chick and mouse demonstrated that all conductive components of the CCS are myocardial in origin.7–9 Using a replication-defective retrovirus expressing LacZ, individual myocytes were labeled in developing chick hearts before neural crest immigration, and the expression of β-galactosidase (β-gal) was traced in daughter cells.7 Single myocyte clones gave rise to both conduction cells and working cardiomyocytes. Labeling of neural crest cells, however, failed to show any incorporation into the CCS.7,8 Similar findings were reported in the murine heart using retrospective clonal analysis, where individual myocyte progenitor clones gave rise to conduction and nonconduction cells.9 Similar to the chick, lineage-tracing studies in mice have failed to identify a direct contribution of neural crest cells to the specialized conduction system.10,11
Models of Cardiac Conduction System Development
The prevailing models of conduction system development are the ring model,6 the inductive recruitment or in-growth model,7 the early specification or outgrowth model,12 and the biphasic model (Figure 29-2). The ring hypothesis was based on early observations that the specialized conduction system formed within four constriction points in the D-looped heart (see Figure 29-2, A).6 Four rings were noted to form from the venous pole to the arterial pole: the SA ring, AV ring, primary (interventricular) ring, and ventriculoarterial (VA) ring. These constriction areas are created by differential proliferation rates that exist between ring myocardial cells and flanking chamber cardiomyocytes. Although these areas of constriction exist within the developing heart during chamber formation, the ring hypothesis has largely been discredited, as there is no evidence to support a preexisting template for ring formation within the tubular heart. It is now known that the linear heart tube is composed of clonally related myocardial cells from the first heart field.13
Figure 29-2 Models of cardiac conduction system development. A, Ring model. Prespecification of conduction system components within the linear heart tube. B, Inductive recruitment or ingrowth model. Undefined inductive signals recruit proliferating cardiomyocytes to a conduction lineage, then cease to proliferate (red arrow). C, Early specification or outgrowth model. Tbx3+ CCS cells expand from primitive myocardium that retains a conduction phenotype. Loss of Tbx3 (or Tbx2) expression results in a phenotypic change from a CCS cell to a working cardiomyocyte, the default pathway (red arrow). D, The biphasic model incorporates both the ingrowth and outgrowth models. E, Retrospective clonal analysis of LacZ-labeled, cardiomyocyte clones (blue nuclei). Mixed clusters of conduction and working cardiomyocytes are consistent with a common myocardial progenitor. Unmixed clusters of CCS-only cells demonstrate the potential for limited rounds of cell proliferation. (From Miquerol L, Beyer S, Kelly RG: Establishment of the mouse ventricular conduction system. Cardiovasc Res 91:232–242, 2011.)
Cheng et al.7 put forth the recruitment model or “in-growth model” based on several observations noted during chick VCS development (see Figure 29-2, B).7 First, the proliferative capacity of developing VCS components was found to be significantly lower than working myocytes based on pulse labeling experiments using [3H]-thymidine. Once specified, conduction cells appeared to exit the cell cycle and become quiescent. Second, lineage tracing studies showed that individually labeled myocyte clones gave rise to conduction cells and working myocytes. Third, cell birth dating experiments demonstrated that new conductive cells were added to the developing His bundle in lamellar fashion, analogous to tree rings. These observations led the authors to conclude that the specialized conduction system expands through a process of inductive recruitment of neighboring myocytes.7 However, it was not speculated what constituted the early framework upon which new conduction cells were added or the nature of the molecular signal used for inductive recruitment.
The early specification model, or outgrowth model, states that conduction cells expand from a progenitor pool that retains its specialized conduction phenotype (see Figure 29-2, C).12,14 The conduction gene programming is retained by the expression of transcriptional repressors that suppress a working myocardial phenotype, which is the default pathway. In support of this hypothesis, persistent expression of repressive transcription factors (Tbx2, Tbx3, Msx2, and Id2) has been identified within primordial conduction regions.12,14,15 Tbx3 is expressed as a continuous band linking the SAN and AVN, internodal tracts, and the proximal ventricular conduction system.12 Heterologous expression of Tbx2 or Tbx3 is able to suppress chamber-type myocardial genes (Nppa [ANF], Gja1 [Cx43], Gja5 [Cx40]), inhibit cardiac chamber formation, and in the case of Tbx3 elicit ectopic pacemaker formation.12,14–16 Consistent with these findings, Tbx3 and Cx43 exhibit complementary expression patterns in the developing heart.12
Most recently a biphasic model of conduction system development has been proposed (Figure 29-2D).9 In this model, once conduction cells are recruited from myocardial precursors, they retain the capacity to undergo limited rounds of cell division. Analysis of labeled myocyte clones revealed two classes of conductive clusters, mixed and unmixed (see Figure 29-2, E). The mixed clusters represented single myocyte clones that gave rise to both conductive and working myocytes (recruitment). The unmixed clones were composed of either working myocytes or conduction myocytes, but not both. Conduction-only, unmixed clones were identified throughout the central and peripheral VCS, indicating that once specified, all components of the conduction system are capable of approximately four to five rounds of cell division. Based on these findings, the authors concluded that mammalian VCS development appears to use both in-growth and outgrowth modes of expansion.9 However, these findings are not incompatible with the early specification/outgrowth model, because mixed clusters might represent conduction cells that have lost Tbx2/Tbx3 expression and have defaulted to the chamber pathway.
Molecular Markers of the Cardiac Conduction System
Visualization of the developing CCS has been greatly enhanced by the development of conduction system reporter mice (Figure 29-3). Each reporter mouse delineates different components of the CCS at various developmental time points using LacZ or green fluorescent protein (GFP) expression. The CCS-LacZ and minK-LacZ mouse lines are representative examples of well-established markers of the specialized conduction system.17–19 The CCS-LacZ mouse was created serendipitously through a complex genomic rearrangement involving the MC4/engrailed-2-LacZ cassette (see Figure 29-3, A).18 CCS-LacZ reporter expression can first be detected at E8.5 in the SAN primordium within the venous pole. At subsequent stages, β-gal expression is detected in the developing and mature AVN and His-Purkinje system. All CCS reporter lines have some degree of cardiac expression outside of the conduction system. In the adult CCS-LacZ heart, significant β-gal expression is seen within the right atrium.18
Figure 29-3 Cardiac conduction system reporter mice. A, CCS-LacZ. B, Contactin2-eGFP. C, Connexin-40–eGFP.20
The minK-LacZ reporter mouse was created by replacing the minK gene with a nuclear-targeted LacZ cassette.17 Early in development, β-gal expression was noted in the SA ring, AV ring, interventricular ring, and the VA ring. Subsequently, β-gal expression was confined to the AVN and the proximal conduction system, as well as in the venous valves, AV ring, and VA valves.17,19
The Cx40-eGFP reporter mouse has become a widely used tool to characterize normal and abnormal patterning of the mature His-Purkinje system (see Figure 29-3, C).20 Developmentally, Cx40 expression is not restricted to the VCS, with significant expression in the trabecular myocardium. In addition, Cx40 is not expressed in the distal AVN or His bundle before E14.5. In the mature heart, Cx40 is enriched in atrial myocardium and in coronary endothelial cells.20
Contactin-2 (Cntn-2) was recently identified as a CCS-enriched factor using differential gene profiling of adult mouse Purkinje fibers versus working myocytes (see Figure 29-3, B).21 Cntn-2 is a cell adhesion molecule that has a role in neuronal patterning and ion channel clustering. Both Cntn2-LacZ knock-in mice and Cntn2-EGFP BAC transgenic reporter mice delineated the entire cardiac conduction system in postnatal hearts. Currently, a functional role for Cntn-2 in the CCS has not been identified.21
Transcription Factor Regulatory Networks
A rich hierarchy of gene networks dictates the specification and patterning of conduction components (Figure 29-4). Unifying all these networks is the balance struck between prochamber myocardial programming versus antichamber programming. As mentioned previously, the T-box transcription factors dictate much of this equilibrium, tilting the scales toward or away from a conduction lineage. The T-box factors can function as transcriptional activators or repressors and are known to be critical regulators of cardiac specification and differentiation.22 Seven TBX family members are expressed in the developing heart, four of which (TBX1, TBX5, TBX20, TBX3) have been linked to human congenital heart disease.23–25 The major cardiac transcriptional activators, Tbx5 and Tbx20, act through Nkx2-5 and Gata4 to drive prochamber myocardial gene expression, such as Nppa (ANF) and Gja5 (Cx40).26–28 The transcriptional repressors Tbx2, Tbx3, and Tbx18 compete with Tbx5 for Nkx2-5 binding to suppress chamber-specific gene expression, thus maintaining a conduction gene profile.12,14,15,29,30
Figure 29-4 Transcription factor regulatory networks. A, Schematic of the cardiac conduction system. B, Right sinoatrial node (head and tail domains), atrial myocardium, and left sinoatrial node (stippled circle). C, Atrioventricular canal/node region flanked by atrial and ventricular myocardium. D, His bundle and bundle branches and ventricular myocardium. E, Purkinje fiber.
The delicate balance between Tbx activators and repressors and the expression of cofactors dictates the rich conduction system phenotypes seen in the mature CCS. Sitting on top of the CCS specification hierarchy is Tbx5, a critical determinant of many elements of the slow and fast conduction system.31–33 Mutations in Tbx5 result in Holt-Oram syndrome, an autosomal dominant condition characterized by preaxial radial ray limb deformities and cardiac septation defects.22 The septal defects are typically ostium secundum atrial septal defects, muscular ventricular septal defects, and atrioventricular canal defects. Patients with Holt-Oram syndrome manifest variable degrees of CCS dysfunction, including sinus bradycardia and AV block, even in the absence of overt structural heart disease.22 Analysis of Tbx5 transgenic mice demonstrated an exquisitely sensitive, dose-dependent correlation of Tbx5 expression level with severity of congenital heart defects and conduction disease.34
Interestingly, the temporal-spatial expression of Tbx5 correlates well with developmental timing of conduction system components.35 At E8.5, Tbx5 expression is enriched in a posterior-anterior axis, with highest levels of expression in the atria and sinus venosus, the site of SA node specification. During chamber formation, Tbx5 expression is graded in a left-right axis with highest levels in the left ventricle, particularly in the AV canal, ventricular conduction system and the trabecular myocardium (the site of Purkinje fiber development). Tbx5 expression is scant in the right ventricle except in the developing AV bundle, right bundle branch, and trabecular region.35
Both the dose–phenotype correlation and the temporospatial expression pattern of Tbx5 implicate it as a master regulator of conduction system development. However, the broad expression pattern of Tbx5 beyond the borders of the CCS, and the ability of Tbx5 with its binding partners, Nkx2-5 and Gata4, to elicit chamber myocardial gene programming suggests that it functions with CCS-restricted cofactors to elicit conduction specification.26,28 Numerous regulatory feedback loops have been identified in CCS components that enhance the conduction phenotype while simultaneously repressing chamber-specific gene programming (see Figure 29-4). These conduction-restricted cofactors will be discussed in their regional context.
The Sinoatrial Node
The mammalian sinoatrial node is a large comma-shaped structure with its head region located at the junction between the right superior vena cava and the right atrium, and the tail region situated along the crista terminalis. These regions represent distinct cellular lineages, as evidenced by their unique expression profiles. The SAN head, which constitutes up to 75% of SAN volume, develops from sinus venosus (SV) myocardium and retains the SV signature, Shox2+;Tbx3+;Tbx18+;Nkx2-5−.36,37 In contrast, working atrial myocytes derive from second heart field mesodermal progenitors and express Nkx2-5+;Shox2−;Tbx3−;Tbx18−. The SAN tail domain has an expression profile in between that of the head region and the atrial myocardium, expressing Shox2+;Tbx3+;Tbx18−;Nkx2-5weakly +. Lineage tracing studies have shown that the SAN tail originates from SV myocardium, but loses Tbx18 expression during development.37
Proper SAN development is dependent on the appropriate expression of Tbx5, Shox2, Tbx18, and Tbx3 (see Figure 29-4, B). Tbx5 expression in the sinus venosus is a critical regulator of the SAN signature through its actions on the transcriptional repressor, Tbx3, and the homeobox transcription factor, Shox2.34,38 Homozygous Tbx5del/del mice die embryonically at E10.5 because of severe hypoplasia of the sinoatrial region and of the primitive LV.27 Microarray analysis of Tbx5 heterozygous hearts, identified Tbx3 and Shox2 as significantly downregulated targets, and both factors showed reduced expression in the sinoatrial region.34,38
As stated earlier, Tbx3 is expressed throughout the developing and mature CCS (except the Purkinje network) and represses chamber-specific programming. Like Tbx5, Tbx3 displays critical dose dependency for proper differentiation and homeostatic maintenance of the conduction system.39 Analysis of Tbx3 mutant mice revealed a dose-dependent SAN phenotype with variable degrees of sinus node dysfunction and inappropriate expression of chamber-specific genes (Cx43, Cx40, Nppa, Scn5a) within the SAN region. Although the overall structure of the SAN is normal, SAN volume was reduced by approximately 45% to 60%, although this phenotype has not been consistently seen in all Tbx3 mutants when adjusted for weight.16,29,37,39 Ectopic overexpression of Tbx3 in atrial myocardium results in inappropriate suppression of chamber genes and upregulation of the SAN gene profile.29 These data suggest that Tbx3 is not essential for SAN formation, but is important for establishing and maintaining proper pacemaker gene programming.
Shox2 is essential for formation of the sinoatrial valves and development of the sinoatrial node.40
[/not-level-membership-for-cardiovascular-category]
