CHAPTER 104 Celiac Disease and Refractory Celiac Disease
DEFINITIONS
Celiac disease is characterized by small intestinal malabsorption of nutrients after the ingestion of wheat gluten or related proteins from rye and barley, villus atrophy of the small intestinal mucosa, prompt clinical and histologic improvement following strict adherence to a gluten-free diet, and clinical and histologic relapse when gluten is reintroduced.1 The many other names used to identify patients with this condition, including nontropical sprue, celiac syndrome, adult celiac disease, idiopathic steatorrhea, and primary malabsorption, among others, are testimony to the confusion of the past. The term celiac disease is recognized widely and is used in this chapter; celiac sprue and gluten-sensitive enteropathy are acceptable alternative terms.
Celiac disease exhibits a spectrum of clinical presentations (Fig. 104-1). Atypical celiac disease is fully expressed gluten-sensitive enteropathy manifest only by extraintestinal symptoms and signs including short stature, anemia, osteoporosis, and infertility. Silent celiac disease is fully expressed gluten-sensitive enteropathy usually found after serologic screening in asymptomatic patients. The atypical and silent variants are more common than classic or typical celiac disease, which is fully expressed gluten-sensitive enteropathy found in association with the classic gastrointestinal symptoms of malabsorption.
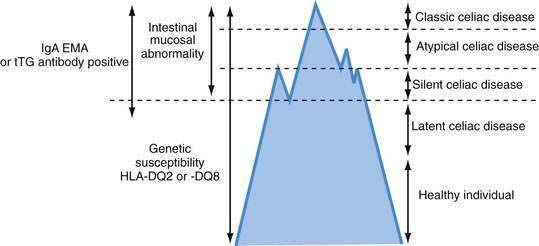
A combination of serologic, genetic, and histologic data also has led to the identification of two other types of celiac disease. Patients with latent celiac disease have normal villus architecture on a gluten-containing diet but, at another time, have had or will have gluten-sensitive villus atrophy. For example, a patient who had celiac disease in childhood and recovered completely on a gluten-free diet might have latent celiac disease later in life on resumption of a normal diet. Patients with potential celiac disease have never had a biopsy consistent with celiac disease but show immunologic abnormalities characteristic for the disease, such as a positive immunoglobulin (Ig)A antibody to endomysium (or tissue transglutaminase [tTG]) or increased intraepithelial lymphocytes (IELs) in the small intestine. These patients often have a genetic predisposition to celiac disease, especially human leukocyte antigen class II DQ (HLA-DQ2), an affected first-degree relative, or both. The probability of their eventually developing celiac disease is unpredictable.2
Refractory celiac disease, also known as unclassified or intractable celiac sprue, is defined as symptomatic, severe small intestinal villus atrophy that mimics celiac disease but does not respond to at least six months of a strict gluten-free diet. This is a diagnosis of exclusion that is not accounted for by inadvertent gluten ingestion, other causes of villus atrophy, or overt intestinal lymphoma.1,3
HISTORY OF CELIAC DISEASE
Celiac disease was recognized as a clinical entity by Aretaeus the Cappadocian in the first century ad.4 The name sprue was coined in the 18th century and is derived from the Dutch word spruw, which means “aphthous disease,” so named because of the high prevalence of aphthous mouth ulcers in these patients. In 1888, Samuel Gee published his paper “On the Coeliac Affection,” which described many of the clinical features of celiac disease in patients of all age groups and concluded, “If the patient can be cured at all it must be by means of the diet.”5 It was not until the middle of the 20th century, however, that the link between certain cereals and celiac disease was made by Willem Karel Dicke, a Dutch pediatrician. He became convinced that the consumption of bread and wheat flour was directly responsible for the deterioration in patients suffering from this condition.6 During World War II, cereals used to make bread were particularly scarce in the Netherlands, and during this time, children with celiac disease improved, only to relapse after the supply of cereal was re-established at the end of the war. It was this serendipitous observation that led to the finding that wheat ingestion exacerbated celiac disease. Subsequent work by van de Kamer and coworkers showed that it was the water-insoluble portion, or gluten moiety, of wheat that produced intestinal injury in patients with celiac disease.7
In 1954, Paulley provided the first accurate description of the characteristic intestinal lesion in patients with celiac disease.8 With the development of effective peroral suction biopsy instruments in the late 1950s, Rubin and coworkers demonstrated that celiac disease in children and idiopathic or nontropical sprue in adults were identical diseases with the same clinical and pathologic features.9
Since the 1980s, we have seen substantial advances in our understanding of the genetic, immune, and molecular mechanisms fundamental to the pathogenesis of celiac disease. In 1986, Howell and associates observed that celiac disease was associated with specific HLA-DQ2 haplotypes.10 In 1993, Lundin and colleagues demonstrated that the DQ2 gene products preferentially present gluten-derived gliadin peptides to intestinal mucosal T cells in celiac patients.11 Subsequently, the enzyme tTG (more specifically tTG type 2 [tTG-2]) was identified as a celiac autoantigen, leading to more accurate serologic diagnostic tests.12
In 1998, Molberg and colleagues reported that modification of gliadin by host tTG enhances gliadin-specific celiac disease T-cell responses.13 The identification of specific tTG-modified deamidated gliadin peptides (DGPs) as dominant α-gliadin T-cell epitopes has highlighted the pivotal role played by tTG in the pathogenesis of celiac disease.14 This discovery already has led to more accurate antigliadin antibody serologic testing using DGPs as capture antigens, and it might pave the way for antigen-specific immunotherapy.
The key role played by IELs in the development of refractory celiac disease and enteropathy-associated T cell lymphoma (EATL) continues to evolve.15 Studies also point to the importance of interleukin (IL)-15, a potent proinflammatory cytokine at the interface between innate and adaptive immunity in the pathogenesis of both celiac disease and refractory celiac disease.16
Epidemiologic studies using endomysial antibody (EMA) and tTG serology have substantially increased estimates of celiac disease prevalence in the United States and elsewhere.17 This in turn has led to renewed interest in potential nondietary treatments including glutenases, modifiers of tight junction function, tTG inhibitors and immune-based interventions, bringing celiac disease therapy into a new era.18,19
EPIDEMIOLOGY
The term celiac iceberg was coined to describe the wide variations in the nature and intensity of clinical presentation of which overt celiac disease is only the emerging peak (see Fig. 104-1). The discovery of the large immersed part of the celiac iceberg has transformed the status of celiac disease, long considered a rare disease, particularly in adults, to that of a common health problem. Because we are uncertain of the depth and breadth of the celiac iceberg, the true prevalence of celiac disease remains unknown.
Serologic testing has demonstrated that silent celiac disease, characterized by positive serology and villus atrophy with few or no symptoms, is approximately seven times more common than symptomatic celiac disease.20 A Finnish study of 3654 schoolchildren of ages 7 to 16 years, using two serologic screens with antiendomysial and tTG antibodies, demonstrated the heterogeneity of the celiac iceberg, with one of every 99 children having biopsy-proved celiac disease.21 Only 10 of 56 subjects with a positive serology had overt symptoms of celiac disease. Two subjects with positive antibodies and at risk for celiac disease because of HLA-DQ2 haplotype had normal mucosa, but both had increased epithelial expression of HLA-DR suggesting mild intestinal inflammation, and one had high counts of IELs; these patients might represent cases of potential disease susceptible to evolving into overt celiac disease. Five patients who had HLA-DQ2 and positive antibodies when studied in 1994 had negative antibodies on repeat testing in 2001; their intestinal biopsies were normal, but all had increased HLA-DR expression, and four of the five had markedly increased numbers of IELs. This latter finding might indicate a variation in the natural history of celiac disease, occasionally seen in teenagers, in whom gluten sensitivity fluctuates with time.
Celiac disease shows a marked geographic variation, with the highest incidence in Western Europe. The condition is more common in Scandinavian and Celtic populations, where the prevalence has been reported to be as high as 1 in 9921 and 1 in 122,22 respectively. The prevalence is similarly high in Italy20 and the southeastern region of Austria.23 The prevalence in Denmark is 40-fold lower than that in Sweden,24 suggesting considerable variation in prevalence among geographically proximate populations. Factors such as predominant HLA haplotype, timing of introduction of gluten into the diet, differences in the gliadin concentration of infant formulas, and interobserver variation in interpreting small intestinal biopsy findings might explain the differences in prevalence.25 Celiac disease also is found in areas to which Europeans have emigrated, notably North America, South America, and Australia.
Epidemiologic studies in the United States, where the disease only recently has attracted much attention, underscore the varying clinical presentation of celiac disease and indicate that the prevalence in the United States is comparable with that in Western Europe. A large multicenter study by Fasano and coworkers17 determined the prevalence of antiendomysial antibodies in more than 13,000 at-risk and not-at-risk American subjects and found the prevalence of antiendomysial antibodies to be 1 in 22 and 1 in 39 among first-degree and second-degree relatives of subjects with celiac disease, respectively.17 A prevalence of 1 in 56 was documented among patients with celiac-like gastrointestinal symptoms or with associated disorders. Of most significance, these investigators found a prevalence of antiendomysial antibodies of 1 : 133 among 4126 “not-at-risk” subjects.
Although celiac disease is rare in the predominantly rice-eating area of southern India, it is prevalent in the Bengal and Punjab provinces of northwest India, where wheat rather than rice has, for many generations, has been a staple of the diet. The condition has been reported in blacks, Arabs, Hispanics, Israeli Jews, Sudanese of mixed Arab-black descent, and Cantonese and is particularly high among the Saharawi population in northwest Africa.26 The condition rarely affects people of purely sub-Saharan African, African-Caribbean, Chinese, or Japanese descent. Some authors have noted a female-to-male ratio of 2 : 1, whereas others have reported ratios as low as 1.3 : 1 but still suggesting a female predominance.
PATHOLOGY
Celiac disease affects the mucosa of the small intestine; the submucosa, muscularis propria, and serosa usually are not involved. The mucosal lesion of the small intestine in celiac disease can vary considerably in severity and extent.9 This spectrum of pathologic involvement might contribute to the striking variations in the clinical manifestations of the disease.
Examination under magnification of the small intestinal mucosal surface in severe untreated celiac disease reveals a flat mucosal surface with complete absence of normal intestinal villi. Histologic examination of tissue sections confirms this loss of normal villus structure (Fig. 104-2A). The intestinal crypts are markedly elongated and open onto a flat absorptive surface. The total thickness of the mucosa is reduced only slightly in most cases, because crypt hyperplasia compensates for the absence or shortening of the villi. These architectural changes decrease the amount of epithelial surface available for digestion and absorption.9
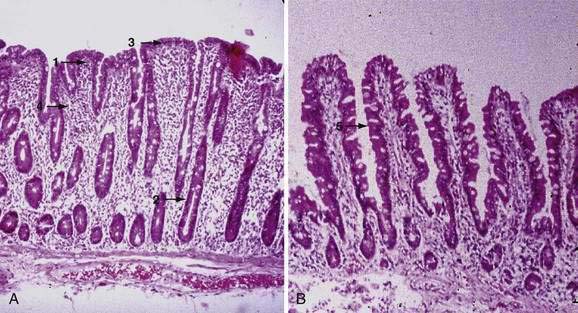
Structural abnormalities of tight junctions between damaged absorptive cells provide a morphologic explanation for the increased permeability of the mucosal barrier in celiac disease.27 The endoplasmic reticulum is sparse, reflecting the low level of synthesis of digestive enzymes, including disaccharidases and peptidases. Thus, mature absorptive cells are reduced in number and functionally compromised.
Unlike the absorptive cells, the undifferentiated crypt cells are markedly increased in number in patients with severe untreated celiac disease, and the crypts are therefore lengthened. Moreover, the number of mitoses in crypts is strikingly increased. Cytologic features and histochemistry of the crypt cells are normal by both light and electron microscopy. Studies of epithelial cell kinetics in untreated celiac disease suggest that “villus atrophy” is a misnomer because there is evidence for an actual increase in enteropoiesis in the crypts. Wright and colleagues28 estimated that intestinal mucosa from patients with celiac disease produces six times as many cells per hour per crypt as does normal small intestine and that the cell cycle time is halved, reflecting premature shedding. The experimental evidence suggests, therefore, that the central mechanism of villus shortening in celiac disease is a gliadin-associated toxic effect on maturing enterocytes that results in their premature loss into the intestinal lumen and a compensatory increase in enterocyte replication in the crypts. Such a mechanism would explain many of the histologic abnormalities described earlier.
The cellularity of the lamina propria is increased in the involved small intestine. The cellular infiltrate consists largely of plasma cells and lymphocytes. The number of IgA-, IgM-, and IgG-producing cells is increased two-fold to six-fold, but, as in normal mucosa, IgA-producing cells predominate.29 Polymorphonuclear leukocytes, eosinophils, and mast cells also can contribute substantially to the increased cellularity of the lamina propria. The number of IELs per unit area of absorptive epithelium (often reported as number of IELs per 100 enterocytes) is increased in untreated celiac disease.9 In the normal small intestinal mucosa, lamina propria T cells are predominantly CD4+ (helper/inducer) cells, whereas the IELs are mainly CD8+ (cytotoxic/suppressor) cells. In untreated celiac disease, this distribution of lamina propria T cells is maintained, but the density of cells in both compartments is increased.
Marsh30 pioneered the theory of a sequence of progression of the celiac lesion in the small intestinal mucosa. Starting with a normal, preinfiltrative (stage 0) mucosa, the initial observed event is an increase in IELs, followed by infiltration of the lamina propria with lymphocytes (stage 1). Crypt hyperplasia (stage 2) precedes villus atrophy (stage 3) and is observed only in the presence of lamina propria lymphocytosis, suggesting that IELs are not sufficient to induce intestinal architectural changes in celiac disease. Finally, total mucosal atrophy (stage 4) develops and is characterized by complete loss of villi, enhanced apoptosis, and crypt hyperplasia.
In untreated patients, the length of small intestinal involvement by the celiac disease lesion varies among individual patients and correlates with the severity of clinical symptoms. Thus, the patient with a severe lesion that involves the full length of the small intestine has more severe malabsorption than the patient with a severe duodenal lesion, a milder jejunal lesion, and a normal ileum. When the intestinal lesion does not involve the entire length of small bowel, the proximal intestine is usually the most severely involved; sparing of proximal intestine with involvement of the distal small intestine can occur, but it is rare. In some untreated patients with clinically mild celiac disease, even the proximal intestine shows only mild partial villus atrophy.9 It is important to note that an increase in IEL count alone is not sufficient to support the histologic diagnosis of celiac disease. This finding is nonspecific and is seen in many other conditions including bacterial overgrowth, mild peptic duodenitis, H. pylori infection, and in other autoimmune disorders. Thus, some shortening of the villi, crypt hyperplasia, cytologically abnormal surface cells, and increased lamina propria cellularity must be present to establish the diagnosis firmly.
Treatment with a gluten-free diet results in significant improvement in intestinal structure (see Fig. 104-2B). The cytologic appearance of the surface absorptive cells improves first, often within a few days. Tall, columnar absorptive cells with basal nuclei and well-developed brush borders replace the abnormal, immature cuboidal surface cells; the ratio of IELs to absorptive cells decreases. Subsequently, villus architecture reverts toward normal, with lengthening of the villi and shortening of the crypts; the lamina propria decreases in cellularity. The mucosa of the distal small intestine improves more rapidly than that of the more severely involved proximal bowel.30,31 In some patients, months or even years of gluten withdrawal may be required before the mucosa reverts to normal; indeed, some residual abnormality, which may be striking or subtle, often persists, possibly because of inadvertent gluten ingestion.32 In the debilitated patient with severe untreated celiac disease and associated nutritional deficiency states, pathologic changes may be present in many other organ systems besides the digestive tract. Finally, the mucosal lesion of celiac disease can be identical histologically to the mucosal response to injury typical of a wide range of other enteropathies (see “Differential Diagnosis”).
PATHOGENESIS
ENVIRONMENTAL FACTORS
Celiac disease is a model for autoimmune diseases with a defined environmental trigger. Early work involving physiologic digestion with pepsin and trypsin, followed by separation according to solubility properties, identified several wheat proteins as being responsible for the grain’s toxicity in celiac disease. Wheat protein exists in a number of storage forms that can be categorized into four general groups based on their solubility characteristics: prolamins (soluble in ethanol), glutenins (partially soluble in dilute acid or alkali solutions), globulins (soluble in 10% NaCl), and minor albumins (soluble in water). The term gluten encompasses both the prolamins and the glutenins. Although most toxicity studies have been performed with prolamins, there are data to suggest that glutenins also can damage the celiac intestinal mucosa.33
The prolamins of wheat are referred to as gliadins. Prolamins from other cereals also are considered to be gluten and are named according to their source (secalins from rye, hordeins from barley, avenins from oats, and zeins from corn). The taxonomic relationships of the major cereal grain families provide a framework on which their toxicities in celiac disease can be predicted (Fig. 104-3).34 Wheat, rye, and barley belong to the tribe known as Triticeae, and oats belong to a neighboring tribe known as Aveneae. Avenin is genetically less similar to gliadin than gliadin is to secalin and hordein. Despite their genetic differences, however, prolamins from oats, barley, wheat, and rye still have immunologic cross-reactivity because of their common ancestry.35 Grains that do not activate disease (rice, corn, sorghum, and millet) are separated still further from wheat, rye, and barley in terms of their derivation from the primitive grasses.
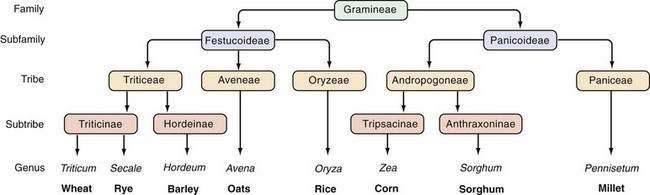
Figure 104-3. Taxonomic relationships of the major cereal grains.
(From Kasarda DD, Okita TW, Bernardin JE, et al. Nucleic acid [cDNA] and amino acid sequences of α-type gliadin from wheat [Triticum aestivum]. Proc Natl Acad Sci U S A 1984; 81:4712.)
Gliadin can be separated electrophoretically into four major fractions that range in molecular weight from 20 to 75 kd and exist as single polypeptide chains. These have been designated α-, β-, γ-, and ω-gliadins, and all four fractions appear to be toxic to patients with celiac disease.36 The complete amino acid sequences of several of the gliadins and related prolamins in grains other than wheat are known.33 In 2000, Anderson and colleagues14 identified a partially deamidated peptide, consisting of amino acids 56 to 75 of α-gliadin as a dominant epitope, responsible for activation of T cells in celiac disease. The complexity and diversity of the gliadin-specific T-cell response, however, is far greater than was previously appreciated, and persons with celiac disease can respond to a diverse repertoire of gluten peptides.37 Furthermore, the release of intracellular tTG leads to the deamidation of gluten proteins and an enhancement of T-cell responses to the resulting DGPs.14
In organ cultures, a synthetic peptide corresponding to amino acids 31 to 49 of α-gliadin has been shown to be toxic to intestinal mucosa and to induce epithelial lesions via recruitment of IELs. Peptide 31-49 does not activate intestinal CD4+ T cells from patients with celiac disease in vitro, but a related peptide corresponding to amino acids 31 to 43 is capable of activating peripheral CD4+ T cells isolated from patients with celiac disease and of inducing epithelial cell apoptosis and activating macrophages, thereby indicating a likely role for innate immune responses in disease pathogenesis.38 Gianfrani and colleagues39 reported that the α-gliadin-derived peptide corresponding to amino acids 123 to 132 is recognized by CD8+ T lymphocytes from patients with celiac disease and is associated with cytotoxic activity. By contrast, another peptide corresponding to amino acids 57 to 68 appears to function in adaptive immunity via stimulation of intestinal T cells in vivo but does not appear to be directly toxic to the intestinal mucosa of patients in vitro.37
It also is possible that immunologic similarities between gliadin protein motifs and enteric pathogens may be involved in the pathogenesis of an immunologic response to gluten antigens. This hypothesis was supported by a study in which analysis of α-gliadin demonstrated an amino acid region that was homologous to the 54-kd E1b protein coat of adenovirus 12, suggesting that exposure to the virus in a susceptible person could be involved in pathogenesis of celiac disease.40 Although patients with celiac disease have been reported to have a significantly higher prevalence of past adenovirus 12 infection than do control subjects,41 the role of adenovirus molecular mimicry in the pathogenesis of celiac disease has not been confirmed.
The reason oats are tolerated by almost all patients with celiac disease is not obvious, because the prolamin fraction of oats contains the same amino acid sequences (QQQPF, where Q = glutamine, P = proline, and F = phenylalanine) that in wheat gliadin have been shown to be toxic.42 A possible explanation for this paradox is that oats contain a relatively smaller proportion of this toxic prolamin moiety than do toxic gluten-containing cereals. Although a feature common to prolamins of wheat, rye, and barley is a high content of glutamine (∼30%) and proline (∼15%), the prolamins of oats have an intermediate content of these amino acids, and the nontoxic prolamins of rice, corn, and millet have an even lower content of them.43 This hypothesis is supported by collectively considering the studies on oat challenge in patients with celiac disease; these studies suggest that tolerance to oats might depend at least in part on the total amount consumed.44 Daily oats consumption of less than 40 to 60 g/day by patients whose celiac disease is in remission appears to be well tolerated.
The data on oats also highlight the important relationship between the amount of gluten consumed and the severity of disease manifestation. A 5- to 10-fold higher incidence of overt celiac disease in children from Sweden compared with Denmark (two populations with similar genetic backgrounds) has long been cited as evidence of the importance of environmental over genetic factors in pathogenesis of celiac disease. Subsequent studies found as much as a 40-fold difference in the gliadin concentration of Swedish compared with Danish infant formula.25 This finding suggests that early exposure of the immature immune system to significant amounts of gliadin is a prominent cofactor for the development of overt celiac disease, possibly by skewing the intestinal immune response to gliadin toward a T-helper 1 (Th1) T-cell response.
The age at which gluten is first introduced into an infant’s diet might also play a pivotal role in facilitating gluten tolerance or intolerance. In one study, early exposure to dietary gluten (within three months of birth) was associated with a five-fold increased risk for celiac disease compared with later gluten introduction (four to six months).45 In the same study, delaying gluten introduction (after 7 months of age) also was associated with a slightly increased risk for subsequent celiac disease (1.9-fold compared with the nadir at introduction at four to six months).
GENETIC FACTORS
Family studies that demonstrate frequent intrafamilial occurrence of celiac disease reflect the importance of genetic factors in its pathogenesis.44 Concordance for celiac disease in first-degree relatives ranges between 8% and 18% and reaches 70% in monozygotic twins.46 Our understanding of the nature of this genetic predisposition began with the significant observation by Howell and coworkers10 that celiac disease was associated with specific HLA-DQ2 haplotypes. HLA class II molecules are glycosylated transmembrane heterodimers (α and β chains) that are organized into three related subregions—DQ, DR, and DP—and encoded within the HLA class II region of the major histocompatibility complex on chromosome 6p. An important link to a genetic predisposition was provided by the isolation of gliadin-specific HLA-DQ2-restricted T-cell clones from celiac disease mucosa.11,47
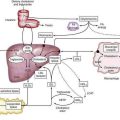
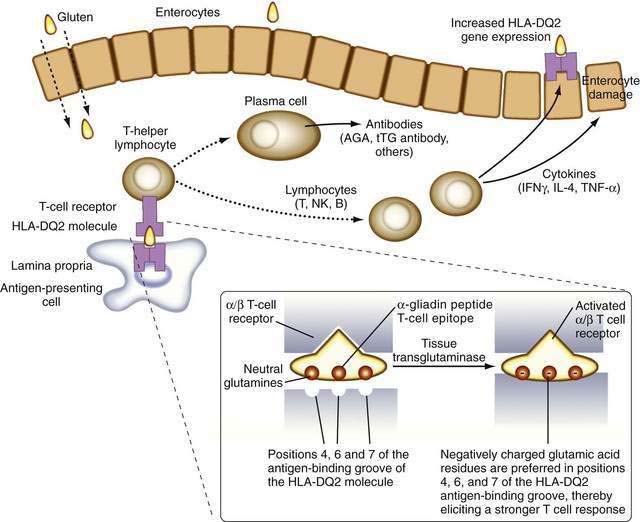
It is now known that after gluten is absorbed, lamina propria antigen-presenting cells (probably dendritic cells) that express HLA-DQ2 or HLA-DQ8, present gliadin peptides on their α/β heterodimer antigen-presenting grooves to sensitized T lymphocytes expressing the α/β T cell receptor (TCR). These lymphocytes then activate B lymphocytes to generate immunoglobulins and other T lymphocytes to secrete cytokines, including interferon (IFN)-γ, as well as IL-4, IL-5, IL-6, IL-10, tumor necrosis factor (TNF)-α, and transforming growth factor (TGF)-β.48 These cytokines induce not only enterocyte injury but also expression of aberrant HLA class II cell-surface antigens on the luminal surface of enterocytes, possibly facilitating additional direct antigen presentation by these cells to the sensitized lymphocytes (Fig. 104-4).
Only a minority of persons who express DQ2 actually develop celiac disease. HLA-DQ2 is expressed by approximately 35% of Europeans and their descendants, but it is rare in other populations (e.g., in sub-Saharan Africa or far eastern Asia). Thus, much of the genetic predisposition to celiac disease is conferred by genes other than those encoding HLA DQ molecules. The search for other genes that confer susceptibility to celiac disease has revealed numerous loci of interest on several different chromosomes, some of which also are associated with susceptibility to type 1 diabetes.49–52
IMMUNE FACTORS
There is substantial evidence implicating both humoral- and cell-mediated immune responses to gliadin and related prolamins in the pathogenesis of celiac disease. There is a two- to six-fold increase in the numbers of immunoglobulin-producing B cells in the lamina propria of the small intestine in untreated celiac disease patients.28 In addition, IgA and IgG serum antibodies to purified gliadin, all major fractions of gliadin, and DGPs can be detected in the sera of most patients with untreated celiac disease.14,53–56 Antigliadin antibodies (AGAs), however, do not appear to be essential for the pathogenesis of celiac disease and might simply reflect a nonspecific response to the passage of incompletely digested antigenic gluten proteins across an abnormally permeable intestinal epithelium. Many normal persons have increased IgA or IgG antigliadin.57 The frequency of elevated IgA or IgG DGP antibodies in healthy controls, however, is very low, possibly reflecting the antigenic potency of DGPs and their more central role in disease pathogenesis.14,54–56 Many persons with celiac disease have increased levels of serum antibodies against other food proteins, such as β-lactoglobulin, casein, and ovalbumin.58 It is unclear whether this reflects a general aberrant immune responsiveness to food antigens in patients with celiac disease or enhanced systemic exposure to these proteins because of increased small intestinal permeability. Gluten can be absorbed across normal epithelium, but it is unclear if this results in immune tolerance in persons who are not genetically predisposed to develop celiac disease.
The identification of more-specific autoantibody responses has altered our understanding of the pathogenesis of celiac disease. IgA antibodies to endomysium, a connective tissue structure surrounding smooth muscle, are virtually pathognomonic for celiac disease and are found only rarely in the absence of disease.59 It is now known that the target autoantigen contained within the endomysium is the enzyme tTG-2.12 Gliadin is a preferred substrate for this ubiquitous calcium-dependent intracellular enzyme, and it has been shown that tTG deamidates key neutral glutamine residues in gliadin and converts them into negatively charged glutamic acid residues, which are preferred in positions 4, 6, and 7 of the nonapeptide antigen-binding groove of the HLA-DQ2 heterodimer (see Fig. 104-4),13,14,60 thereby facilitating antigen presentation. Thus, tTG-mediated modification of gliadin to generate DGPs plays a pivotal role in eliciting a stronger proliferative response by gliadin-specific T-cell clones, or, stated differently, tTG makes gliadin tastier for the T cells.
With gliadin serving as the glutamine donor, tTG also can generate additional novel antigenic epitopes by cross-linking molecules of the extracellular matrix with gliadin or with tTG-gliadin complexes.61 As evidence of the fundamental role of tTG in celiac disease pathogenesis, one of the dominant epitopes responsible for the T-cell response contains a deamidated glutamine residue (Q65E) of α-gliadin.14 It also has been observed that tTG is necessary for the bioactivation of TGF-β that is required for epithelial differentiation. In a T84-crypt epithelial cell culture system, autoantibodies to tTG-blocked TGF-β–mediated enterocyte differentiation,62 a finding that suggests that release of tTG from cells during inflammation potentiates gliadin presentation by HLA-DQ2 and HLA-DQ8 and that local production of autoantibodies to tTG might contribute to the lack of epithelial differentiation observed in the active celiac lesion.
Given the marked infiltration of lymphocytes into the small intestinal mucosal epithelium and lamina propria in active disease, it is not surprising that cell-mediated immune responses also are important in the pathogenesis of celiac disease. Many findings support interplay between adaptive immunity, characterized by a specific and memory T-cell response to gluten peptides, and innate immunity, involving less-specific mechanisms. Many of the T cells in the small intestinal mucosa are activated in untreated celiac disease and release potent proinflammatory mediators such as IFN-γ, TNF-α, IL-2, IL-6, and TGF-β.48 Activated T lymphocytes, most of which are CD4+ cells, are abundant in the lamina propria of the small intestine.63 In contrast, IELs, which are present in large numbers in untreated celiac disease, are predominantly CD8+ T cells.64
There is an influx of primed memory T cells, marked by high CD45RO expression, in the mucosa of untreated celiac disease patients.65 In healthy persons, more than 90% of IELs express the α/β TCR, whereas expression of the γ/δ TCR by IELs in patients with untreated celiac disease is increased as much as six-fold (to 35%) and is considered a hallmark of the disease.66 These primitive lymphocytes recognize bacterial nonpeptide antigens and unprocessed stress-related proteins. They appear to act as mucosal guardians and might protect the intestinal mucosa from chronic exposure to dietary gluten in gluten-tolerant persons by secreting IL-4, which dampens Th1 in favor of Th2 reactivity.67 Their continuous presence in patients on a gluten-free diet might indicate inadvertent gluten ingestion. Patients with refractory celiac disease also have aberrant IELs with restricted γ/δ TCR gene rearrangements indicating oligoclonality. The pathogenetic role of these lymphocytes, compared with lamina propria lymphocytes, continues to evolve (see “Refractory Celiac Disease”).68
Studies suggest that IL-15 plays a key role in bridging the innate and adaptive immune responses in pathogenesis of celiac disease.15,16,69 This enterocyte- and macrophage-derived proinflammatory cytokine is increased massively in the mucosa of patients with active celiac disease and refractory celiac disease. Although the mechanisms that lead to its overproduction remain unknown, IL-15 regulates IEL homeostasis by promoting migration, preventing apoptosis, and enhancing the capacity of dendritic cells to function as antigen-presenting cells.69 In response to gliadin peptides, IL-15 triggers an adaptive CD4+ T-cell response in the lamina propria and also is capable of inducing direct epithelial cell injury by inducing IEL secretion of IFN-γ.16
CLINICAL FEATURES
Samuel Gee’s classic description, with its evocative account, was concerned largely with the gross manifestations of the disorder.5 This florid presentation, however, is now unusual in the Western world, constituting only the extreme tip of the celiac iceberg. Although some patients still present with severe illness, most have few, subtle, or no symptoms at diagnosis. Such cases may be identified by screening relatives of patients during research studies or from screening patients with associated disorders, such as type 1 diabetes mellitus, autoimmune thyroid disease, or Down syndrome. Incidental hematologic abnormalities (e.g., iron deficiency anemia) or biochemical abnormalities (e.g., elevated serum aminotransferase levels) also can lead to a diagnosis of celiac disease.
CHILDHOOD PRESENTATION
The classic presentation of celiac disease in infancy is not easily missed. The typical history is of steatorrhea with or without vomiting and occasional cramping abdominal pain that can occur anytime after weaning when cereals are introduced into the diet, but especially in the first and second years of life. Classically, the child fails to thrive, is apathetic and irritable, and has muscle wasting, hypotonia, and abdominal distention. Watery diarrhea or occasionally constipation may be reported. Diagnosis is more difficult when gastrointestinal features are less prominent, and the possibility of gluten sensitivity should be considered in all children who present with short stature or failure to thrive, even when there are no other symptoms to suggest an enteropathy. Once a gluten-free diet is commenced, catch-up growth is well documented.70 Nutritional deficiencies, particularly anemia, are another common mode of presentation, especially in older children. With earlier diagnosis, clinical rickets now is an uncommon complication but is seen occasionally, especially among Asian children with untreated celiac disease. Many pediatric patients enjoy a temporary, spontaneous remission of symptoms during adolescence, and it is unusual for celiac disease to manifest during the teens.
Considerable debate continues as to why celiac disease tends to be diagnosed later and with milder signs and symptoms than in the past. A number of studies suggest that breast-feeding can significantly delay the onset of symptoms,71,72 but not all studies support this conclusion.73 In one study of at-risk children, the introduction of gluten into the diet during the first three months of life or after seven months of age was associated with a significantly increased risk for celiac disease (hazard ratio [HR] 23.0; 95% confidence interval [CI]: 4.6-115.9; P = 0.001) compared with introduction at four to six months (HR 3.98; 95% CI: 1.18-13.46; P = 0.04).74
ADULTHOOD PRESENTATION
In the past, celiac disease was perceived to be a pediatric disorder, but the diagnosis now is being made increasingly in adults; currently, the overall mean age at presentation is approximately 45 years. Symptoms also have changed during the past 50 years. Diarrhea now is reported less often, and many patients now present with higher body mass indices and even with obesity. The unmasking of asymptomatic disease by surgery that induces rapid gastric emptying (e.g., gastric resection, pyloroplasty) or the finding of the typical lesion in asymptomatic relatives of celiac disease patients suggests that adults can have silent celiac disease for some time. A proportion of these adult patients have short stature or give a history consistent with unrecognized celiac disease in childhood. In many, however, there is nothing to suggest previous disease, and it is possible that celiac disease can develop for the first time in adult life. Celiac disease also is being diagnosed increasingly in later life, with approximately 25% of cases diagnosed in patients older than 60 years.75
GASTROINTESTINAL FEATURES
Several factors contribute to the diarrhea associated with celiac disease. The stool volume and osmotic load delivered to the colon are increased by the malabsorption of fat,76 carbohydrate, protein, electrolytes, and other nutrients. In addition, the delivery of excessive dietary fat into the large bowel results in the production by bacteria of hydroxy fatty acids, which are potent cathartics. Electrolytes actually are secreted into, rather than absorbed from, the lumen of the severely damaged upper small intestine in symptomatic patients. This secretion further increases luminal fluid in an intestine with an already compromised absorptive capacity. There also is evidence that secretin and cholecystokinin release in response to a meal are impaired in celiac disease, diminishing delivery of bile and pancreatic secretions into the gut lumen and possibly compromising intraluminal digestion.77 Alterations in the secretion of other intestinal peptides have been noted and can contribute to the observed diarrhea. Finally, if the disease extends to and involves the ileum, patients can experience the direct cathartic action of malabsorbed bile salts on the colon.76
Vague abdominal discomfort and especially abdominal bloating are extremely common and can lead to a mistaken diagnosis of irritable bowel syndrome (IBS). Because of the difficulty in distinguishing celiac disease with mild gastrointestinal manifestations from symptomatic IBS, serologic testing of IgA EMAs or IgA tTG should be considered in patients with symptoms suggesting diarrhea-predominant IBS. In a UK study, Sanders and colleagues78 evaluated 300 consecutive new patients who fulfilled Rome II criteria for IBS and 300 healthy age- and sex-matched controls for celiac disease using IgA AGA (antigliadin antibody), IgG AGA, and EMA; two controls (0.7%) (both EMA positive) and 14 IBS patients (4.6%) had celiac disease (P = 0.004; odds ratio [OR], 7.0; 95% confidence interval [CI]: 1.7-28.0). Severe abdominal pain can occur but is uncharacteristic in uncomplicated celiac disease; its occurrence can suggest the presence of complications such as intussusception, ulcerative jejunitis, or intestinal lymphoma. Abdominal distention with excessive amounts of malodorous flatus is a common complaint. Conversely, nausea and vomiting are uncommon in uncomplicated celiac disease. Recurrent severe aphthous stomatitis affects many celiac patients and may be their sole presenting complaint. It is important to exclude celiac disease in cases of recurrent aphthous stomatitis because a significant proportion of these patients respond well to dietary treatment.79
EXTRAINTESTINAL FEATURES
As patients with celiac disease get older, they tend to present with complaints not directly referable to the gastrointestinal tract. These extraintestinal symptoms and clinical findings often result from nutrient malabsorption and can involve virtually all organ systems (Table 104-1).80 Extraintestinal features, including anemia, osteopenia, neurologic symptoms, and menstrual abnormalities, often prove more distressing to the patient than do the gastrointestinal symptoms.
Table 104-1 Extraintestinal Manifestations of Celiac Disease
| MANIFESTATION | PROBABLE CAUSE(S) |
|---|---|
| Cutaneous | |
| Ecchymoses and petechiae | Vitamin K deficiency; rarely, thrombocytopenia |
| Edema | Hypoproteinemia |
| Dermatitis herpetiformis | Unknown |
| Follicular hyperkeratosis and dermatitis | Vitamin A malabsorption, vitamin B complex malabsorption |
| Endocrinologic | |
| Amenorrhea, infertility, impotence | Malnutrition, hypothalamic-pituitary dysfunction |
| Secondary hyperparathyroidism | Calcium and/or vitamin D malabsorption causing hypocalcemia |
| Hematologic | |
| Anemia | Iron, folate, vitamin B12, or pyridoxine deficiency |
| Hemorrhage | Vitamin K deficiency; rarely, thrombocytopenia due to folate deficiency |
| Thrombocytosis, Howell-Jolly bodies | Hyposplenism |
| Hepatic | |
| Elevated liver biochemical test levels | Unknown |
| Muscular | |
| Atrophy | Malnutrition due to malabsorption |
| Tetany | Calcium, vitamin D, and/or magnesium malabsorption |
| Weakness | Generalized muscle atrophy, hypokalemia |
| Neurologic | |
| Peripheral neuropathy | Deficiencies of vitamins such as vitamin B12 and thiamine |
| Ataxia | Cerebellar and posterior column damage |
| Demyelinating central nervous system lesions | Unknown |
| Seizures | Unknown |
| Skeletal | |
| Osteopenia | Malabsorption of calcium and vitamin D |
| Osteoarthropathy | Unknown |
| Pathologic fractures | Osteopenia |
Anemia
Anemia is a common manifestation of celiac disease in children and adults and usually is caused by impaired iron or folate absorption from the proximal intestine; in severe disease with ileal involvement, vitamin B12 absorption also is impaired. Patients with extensive disease can bleed into the skin or mucous membranes or can develop hematuria, epistaxis, or vaginal or gastrointestinal bleeding. Bleeding can aggravate pre-existing anemia and most often is caused by a coagulopathy resulting from impaired intestinal absorption of fat-soluble vitamin K. Evidence of hyposplenism of unknown cause, with thrombocytosis, deformed erythrocytes, and splenic atrophy, occurs in up to 50% of adults with celiac disease but only rarely is seen in children.81 In many patients, evidence of hyposplenism disappears with elimination of gluten from the diet.81
Osteopenia
Osteopenia is the most common complication of celiac disease, and its prevalence increases with age at diagnosis. More than 70% of patients with untreated celiac disease have osteopenia,82 and osteoporosis occurs in more than one quarter of all celiac disease patients.83 Osteopenia develops as a result of impaired calcium absorption (secondary to defective calcium transport by the diseased small intestine), vitamin D deficiency (caused by impaired absorption of this fat-soluble vitamin), and binding of intraluminal calcium and magnesium to unabsorbed dietary fatty acids (forming insoluble soaps, which are then excreted in the feces). Chronic intestinal inflammation also can contribute to bone loss through release of inflammatory mediators.
Osteopenia is less common in patients with silent celiac disease, in whom prevalence rates between 30% and 40% have been reported.84 Whereas bone disease generally is more severe among patients with symptomatic disease, severe osteopenia has been reported in up to one third of symptom-free adults whose celiac disease was diagnosed during childhood and who resumed a normal diet during adolescence.85
A key unanswered question is the functional consequence of osteopenia. An increased risk of fractures was observed in patients with overt celiac disease in one study84 but not in another.86 The fracture risk among patients with silent celiac also remains unclear.
Neurologic Symptoms
Neurologic symptoms caused by lesions of the central or peripheral nervous system occasionally occur in patients with celiac disease and are poorly understood. Celiac disease often is found in patients presenting with nonhereditary ataxia, and progressive gait and limb ataxia may be the sole manifestations of disease in some patients. These abnormalities, referred to as gluten ataxia, are believed to result from immunologic damage to the cerebellum, posterior columns of the spinal cord, and peripheral nerves.87 Muscle weakness and paresthesias with sensory loss also are encountered occasionally, and pathologic evidence of peripheral neuropathy and patchy demyelinization of the spinal cord, cerebellar atrophy, and capillary proliferation suggestive of Wernicke’s encephalopathy have been described rarely.
Although potential causative roles for specific vitamin deficiencies (including vitamin B12, thiamine, riboflavin, and pyridoxine) have not been established, neurologic symptoms have been reported to improve in some patients who are given multivitamins, including vitamins A, B, and E, and calcium. Night blindness is a clear indication for vitamin A therapy. Peripheral neuropathy and ataxia, however, often appear unrelated to specific vitamin deficiency states and usually do not respond to gluten withdrawal.88
The associations of celiac disease and epilepsy, frequently complex partial seizures, and bilateral parieto-occipital cerebral calcification are well recognized.89 In one series, epilepsy was reported in approximately 5% of children and young adults with celiac disease.90 The cause of the epilepsy remains unclear, and the prognosis might depend on how early in the course of the disease a gluten-free diet is started.
Although most patients with celiac disease do not appear psychologically abnormal, many affected subjects report a striking improvement in mood after commencing a gluten-free diet.91
Gynecologic and Fertility Problems
Gynecologic and obstetric problems are common in women with untreated celiac disease.92 Amenorrhea occurs in one third of women of childbearing age and menarche is often delayed, typically by one year, in untreated subjects. Women with untreated celiac disease can present with infertility, and it is common for infertile women with celiac disease to become pregnant shortly after commencing a gluten-free diet.93
A high prevalence of silent celiac disease has been reported in women with recurrent spontaneous abortions, intrauterine fetal growth retardation, and unfavorable outcomes of pregnancy, underlining the need to test for celiac disease in these situations.94
Infertility secondary to impotence or an abnormally low sperm count can occur in men with untreated celiac disease.95 Although malnutrition, including folate deficiency related to malabsorption, can contribute to male infertility, abnormalities in hypothalamic-pituitary regulation of gonadal function and gonadal androgen resistance that disappears on gluten withdrawal also have been incriminated.95
Physical Examination
Examination of the mouth may show aphthous stomatitis, angular cheilosis, and glossitis with decreased papillation of the tongue. Dental enamel defects are common.96 The abdomen may be protuberant and tympanitic, with a characteristic doughy consistency, owing to distention of intestinal loops with fluid and gas. Hepatomegaly and abdominal tenderness are uncommon, but ascites may be detected in patients with significant hypoproteinemia. Peripheral lymphadenopathy is unusual in the absence of complicating lymphoma.
DIAGNOSIS
Laboratory findings in celiac disease, like the symptoms and signs, vary with the extent and severity of the intestinal lesion. Serum IgA EMA or tTG antibody and small intestinal biopsy are the most reliable diagnostic tests for celiac disease. Stool studies, hematologic and biochemical tests, and radiologic studies may be abnormal, but they seldom provide a specific diagnosis because similar abnormalities often are seen in patients with other malabsorptive diseases (see Chapter 101).
SEROLOGY
In current clinical practice, there are many serologic studies to aid in the diagnosis of celiac disease; however, the most powerful and clinically useful are the IgA EMA and IgA tTG assays. IgA EMA and IgA tTG are based on the target antigen tTG, and IgA and IgG AGAs are based on the target antigen gliadin.97 A second-generation AGA test has been developed based on DGPs with improved diagnostic accuracy.14,54–56
The approximate sensitivity and specificity of commonly used serum antibody tests are outlined in Table 104-2.98–102 A working group of 13 European laboratories has attempted to improve standardization by establishing standard curves on reference sera and protocols for calibration of quality controls. This collaboration has reported that IgA EMA has a sensitivity of 90%, specificity of 99%, and reproducibility of 93% and currently remains the gold standard. IgA antihuman tTG is slightly less reliable (sensitivity 93%, specificity 95%, reproducibility 83%), and the anti-IgG AGA and IgA AGA are the least reliable.98
Table 104-2 Sensitivity, Specificity, and Positive and Negative Predictive Values of Serologic Tests for Untreated Celiac Disease
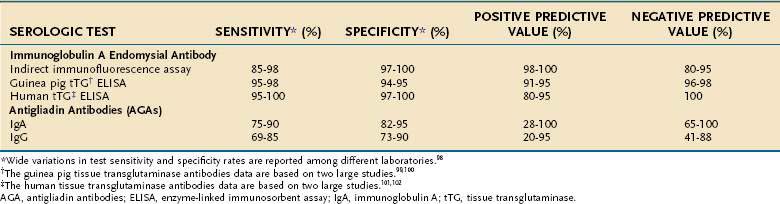
In addition to laboratory variation, the reported sensitivities and specificities of these tests depend on the prevalence of the disease in the tested population and the severity of the disease. In one study of 101 patients with biopsy-proven celiac disease, the sensitivity of IgA EMA among patients with total villus atrophy was 100% compared with only 31% in those with partial villus atrophy.103
Immunoglobulin A Endomysial Antibody
Serum IgA EMA binds to connective tissue (endomysium), surrounding smooth muscle cells, producing a characteristic staining pattern that is identified by indirect immunofluorescence.59 The target antigen has been identified as tTG. Frozen sections of monkey esophagus initially were used for the assay, but currently most laboratories use sections of human umbilical cord, which are more readily available.104 The test result is reported simply as positive or negative because even low titers of serum IgA EMA are highly specific for celiac disease. IgA EMA has a sensitivity of 90% or greater and a specificity approaching 100% in untreated celiac disease.59,105 Antibody levels fall on a gluten-free diet, the test often becoming negative in treated patients.106 The clinical applications of EMA and other serologic tests are discussed in the following sections.
Tissue Transglutaminase Antibodies
The epitope against which EMA is directed has been identified as type 2 tTG (tTG-2).12 Type 1 tTG, which has different structure, enzymatic activities, and tissue distributions, is not implicated in celiac disease. Type 3 (epidermal) tTG plays a distinct role in DH (discussed later).
An IgA enzyme-linked immunosorbent assay (ELISA) that used guinea pig tTG was used initially, and it proved less costly and easier to perform than the immunofluorescence assay used to detect IgA EMA. IgA guinea pig tTG assays are sensitive and specific for the diagnosis of celiac disease.99,100 In one study, anti-tTG was present in 98% of patients with biopsy-proven celiac disease compared with 5% of controls.99 In another study that included 136 patients with celiac disease and 207 controls, the sensitivity and specificity of IgA guinea pig tTG were 95% and 94%, respectively.100 IgA guinea pig tTGs, however, are responsible for some false-positive results, particularly in at-risk persons with autoimmune diseases or liver disease and in patients with other inflammatory bowel diseases.98
ELISAs using human tTG are now preferred because they have proved to be more specific than those using guinea pig tTG.101,102 In contrast to the false-positive results of IgA guinea pig tTG, a false-positive IgA human tTG result is unlikely (especially at high titer). A false-positive EMA also is highly unlikely and in the setting of a normal biopsy might indicate a future predisposition to development of clinical celiac disease—that is, potential celiac disease.
Antigliadin Antibodies
Serum IgA and IgG AGA levels often are elevated in untreated celiac disease. Unfortunately, these tests have only moderate sensitivity and their specificity is substantially lower than those of IgA EMA or tTG tests.1,53,97,107 Thus, testing for AGA no longer is recommended as a primary test for untreated celiac disease; IgA EMA or IgA tTG testing is preferable. The sensitivity and specificity of IgA AGA are marginally superior to those of IgG AGA; however, many clinicians test simultaneously for both IgA and IgG AGA, an approach that gives a small incremental increase in sensitivity but reduces specificity even further. IgG AGA testing may be useful in the 2% of patients with celiac disease who have IgA deficiency. The positive predictive value of AGA in a general population is relatively poor. In one series, for example, the positive predictive value for IgG AGA, corrected for the expected prevalence in the general population, was less than 2%.105 AGA test results are reported as a titer—a high titer of AGA is somewhat more specific for celiac disease than a low titer—but some normal persons have high AGA levels.57
It is now known that tTG-2 catalyses the deamidation of gliadin peptides. This increases their binding to the antigen groove of DQ2, thereby increasing their toxicity.14 Based on this knowledge, a second-generation AGA test has been developed using synthetic deamidated gliadin peptides (DGP) that replicate the structure of tTG-modified gliadin antigens. IgA and IgG DGP assays are available with sensitivities, in early studies, similar to those obtained using tTG assays.54–56 The specificity of a positive DGP test result is substantially greater than that of standard AGA assays, presumably indicating that celiac-associated AGAs recognize DGP, whereas AGAs in persons who do not have celiac disease recognize other epitopes less specific for the disease.
DGP testing is not yet in widespread use, but potential applications include the confirmation or exclusion of celiac disease in persons who have small bowel biopsy findings consistent with celiac disease but a negative IgA-tTG test result (including those with IgA deficiency where DGP-IgG can be used) and in persons with positive AGA test results but a negative IgA-tTG. DGP concentrations fall on a gluten-free diet in a similar fashion to AGA or anti-tTG concentrations.54 A more controversial application of DGP testing is to use anti-tTG and anti-DGP assays in combination for diagnosis or exclusion of disease without small bowel biopsy.
Clinical Application of Serologic Tests
An approach to diagnosing celiac disease is outlined in Figure 104-5.108 When the index of suspicion is low—the pretest probability is less than 10%—a negative result for IgA EMA or IgA tTG has a high negative predictive value and can obviate the need for small bowel biopsy. Falsely negative IgA EMA and IgA tTG test results are more likely to occur in very young children (<2 years of age), those with mild celiac enteropathy, and, of course, in IgA deficiency. The AGA tests have lower diagnostic accuracy and are no longer recommended for initial diagnosis of celiac disease.105 Because the specificities of IgA EMA and IgA tTG tests are high, their positive predictive values are high even in low-risk populations.105,109,110
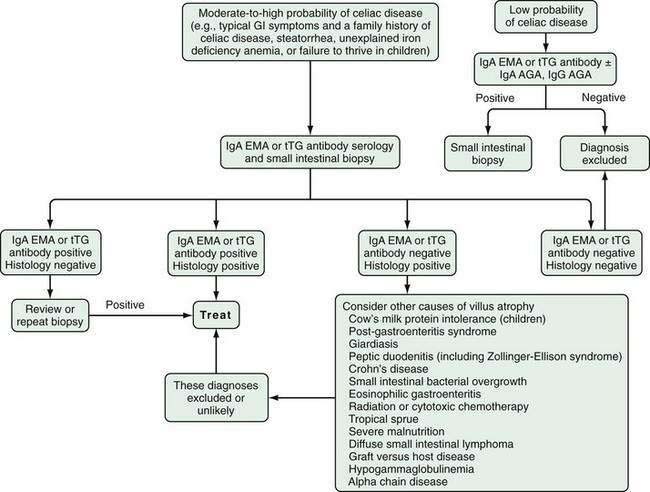
Similar to IgA AGA levels, IgA tTG levels decrease in the months following a gluten-free diet and are useful in assessing dietary compliance and excluding inadvertent gluten ingestion.53,54,111 IgA AGA and IgA tTG currently are the most widely used tests for monitoring adherence and response to a gluten-free diet among patients whose antibody levels are elevated before therapy.54,107,111 Hence, a pretreatment antibody level should be determined at the time of diagnosis. A normal baseline value typically is reached within 3 to 12 months depending on the pretreatment antibody concentration (IgA tTG concentrations fall with a half-life of approximately six to eight weeks) and on the degree of success in avoiding gluten ingestion. If the levels do not fall as anticipated, the patient may be continuing to ingest gluten either intentionally or inadvertently.
The advent of highly sensitive and specific serologic tests has changed the epidemiology of celiac disease radically by revealing the high incidence of silent celiac disease; this awareness in turn has led to debate on the merits of mass screening. To date, the benefit of screening for asymptomatic celiac disease, usually using IgA tTG or EMA, has not been demonstrated.112 The potential advantages of screening for asymptomatic celiac disease are a reduction in risk for malignancy including enteropathy-associated T-cell lymphoma (EATL); a reversal of unrecognized nutritional deficiency states; resolution of mild or ignored intestinal symptoms; possible reduction in T-cell activation and “antigenic drift” to other autoantigens, thereby reducing the onset of other autoimmune disorders; and an improvement in general well-being.113,113A
All of these hypothetical benefits, however, depend on compliance with a challenging dietary regimen, and asymptomatic patients might not be motivated sufficiently to adhere to a strict gluten-free diet.114 There also may be adverse psychological effects when asymptomatic individuals are given the diagnosis of celiac disease. Furthermore, the natural history of undetected celiac disease and the consequences of screening and treating silent celiac disease are unknown. For these reasons, mass screening of asymptomatic persons generally is not advocated at this time, even in populations in which the prevalence of celiac disease is high.
The current standard of care is a case-finding approach, which targets at-risk subjects such as patients with mild gastrointestinal symptoms, iron deficiency anemia, or IBS-like symptoms, all instances in which the value of serologic testing for celiac disease is accepted widely. Simply by case finding among at-risk subjects in a primary care setting, Hin and colleagues115 observed a four-fold increase in the number of celiac disease diagnoses during a one-year period. Thus, for now, increased awareness of the typical and atypical presentations of celiac disease, coupled with a low threshold for serologic testing in at-risk subjects, can uncover a substantial portion of the submerged iceberg.
GENETIC TESTING
As discussed earlier, almost all patients with celiac disease are positive for HLA DQ2 or DQ8. As a result, HLA testing may be helpful in excluding celiac disease in specific clinical situations, mainly before embarking on a gluten challenge (see later) or evaluating a patient who has a celiac-like enteropathy but negative IgA tTG, EMA, and DGP serologies, in which case a negative result indicates the need to investigate for an alternative diagnosis. Approximately 40% of persons of European ancestry are DQ2 or DQ8 positive, however, so a positive result is of little diagnostic value.97,116–118
SMALL INTESTINE BIOPSY
Although the diagnosis of celiac disease may be suspected on clinical grounds or as a result of abnormal serologic tests, biopsy of the small intestine has remained the standard test to establish the diagnosis. Biopsies usually are performed during endoscopic examination of the upper gastrointestinal tract, an examination that may be indicated for reasons related or unrelated to celiac disease, such as investigation of iron deficiency anemia or upper abdominal discomfort.119 Multiple biopsies should be obtained (e.g., a total of six to eight biopsies from the second and third parts of the duodenum). Biopsies taken from the duodenal bulb also may be diagnostic, but might show mucosal architectural distortion produced by Brunner’s glands and changes caused by peptic duodenitis, both of which can cause difficulty in histopathologic diagnosis.9
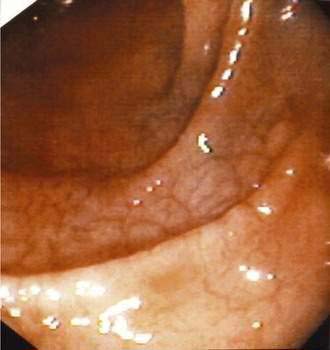
Scalloping or absence of duodenal folds has been noted in some patients with celiac disease (Fig. 104-6).120 Scalloping is not specific for celiac disease, however, and may be seen in eosinophilic enteritis, giardiasis, amyloidosis, tropical disease, and human immunodeficiency virus enteropathy.121 Other endoscopic features include multiple fissures or a mosaic-like appearance where the fissures circumscribe areas of mucosal nodularity in a manner similar to the grouting around a mosaic tile. The mucosa of celiac disease, however, often appears normal at endoscopy, and absence of the previously described macroscopic features does not obviate the need for biopsy and histologic examination if celiac disease is suspected, based on clinical grounds or serologic testing.
GLUTEN CHALLENGE
A gluten challenge should be considered in patients who began a gluten-free diet empirically without documentation of a characteristic intestinal lesion or the presence of IgA EMA antibody. In such patients, symptomatic response to a gluten-free diet might indicate the presence of gluten-sensitive enteropathy or simply reflect a change in gastrointestinal function in response to a major dietary change. Mild and nonspecific gastrointestinal symptoms after ingesting gluten are not a reliable way to diagnose celiac disease. For example, patients with IBS might experience improvement in symptoms, including abdominal bloating, cramping, or diarrhea, after beginning a gluten-free diet.122 Gluten challenge also should be considered if a diagnosis of celiac disease was made during childhood based on small intestinal biopsy abnormalities in the absence of a positive IgA EMA or IgA tTG, because a number of transient childhood enteropathies can mimic the celiac lesion (see later).
Gluten challenge must be initiated with caution because occasionally patients are exquisitely sensitive to small amounts of gluten123; other patients might require prolonged challenge before symptoms or significant histologic abnormalities recur. If a small amount of gluten, such as a cracker or one quarter of a slice of bread, is tolerated, the amount can be doubled every three days until the patient is ingesting the equivalent of at least four slices of bread daily. The challenge should be continued for at least six weeks or until more severe symptoms redevelop, at which time both serum IgA EMA or IgA tTG and small bowel biopsy should be performed. A total of 10 g of gluten daily for a period of six to eight weeks is usually sufficient to result in definite histologic deterioration.
HEMATOLOGY AND BIOCHEMISTRY TESTS
A variety of hematologic and biochemical abnormalities may be found in persons with untreated celiac disease, including deficiencies of iron, folic acid, and vitamin D. These abnormalities reflect nutritional deficiency states secondary to enteropathy-induced malabsorption. Iron deficiency anemia is common in both children and adults with celiac disease, and combined iron and folate deficiency is characteristic, especially in children. With the exception of pregnancy, severe anemia is uncommon, usually develops with extensive disease, and should raise the suspicion of a complication, such as lymphoma. The peripheral blood film might reveal target cells, siderocytes, Heinz bodies, crenated red blood cells, and Howell-Jolly bodies, which suggest splenic atrophy.81
DIFFERENTIAL DIAGNOSIS
Malabsorption and steatorrhea can result from pancreatic insufficiency, cholestatic liver disease, terminal ileal disease or resection, or small intestinal bacterial overgrowth. In some patients, microscopic colitis or bacterial overgrowth may be present concurrently with celiac disease. It is important to exclude these disorders in patients who do not respond to treatment with a gluten-free diet (see later).118,124
In adults, celiac disease is histologically distinguished easily from Whipple’s disease and from malabsorption because of infiltration of the mucosa with Mycobacterium avium complex. Changes in mucosal morphology can be seen in parasitic infections other than Giardia, including strongyloidiasis, coccidiosis, and hookworm disease, but these changes rarely include villus atrophy. Although villus atrophy is characteristic of untreated celiac disease, it is by no means pathognomonic and may be seen in varying degrees in a wide variety of other enteric disorders (see Fig. 104-2); thus villus atrophy on small intestinal biopsy is not in itself sufficient to diagnose celiac disease.
In infants and young children, cow’s milk or soy protein intolerance also can result in biopsy findings identical to those of celiac disease.125,126 Soy protein often is used as a substitute for milk protein in cow’s milk protein intolerance, but some children also develop mucosal abnormalities resembling those of celiac disease following ingestion of soy protein.126
A rare condition that can cause diagnostic confusion is collagenous sprue. Patients with collagenous sprue might present initially with symptoms and biopsy findings consistent with celiac disease, but their symptoms fail to respond to gluten withdrawal, and with time, extensive deposition of collagen in the lamina propria develops just beneath the absorptive epithelium.127 The relationship between celiac disease and both collagenous sprue and the microscopic colitides (lymphocytic and collagenous colitis) is discussed later.
DISEASES ASSOCIATED WITH CELIAC DISEASE
A large number of diseases occur more commonly among patients with celiac disease and are delineated in Table 104-3.128 In addition to an association with autoimmune disorders, some of the associated diseases also have similar HLA haplotype associations.
Modified from Mulder CJ, Tytgat GN. Coeliac disease and related disorders. Neth J Med 1987;31:286.
Dermatitis Herpetiformis
DH is a skin disease characterized by papulovesicular lesions that occur symmetrically over the extensor surfaces of the extremities and the buttocks, trunk, neck, and scalp. Unlike celiac disease, DH rarely is diagnosed in childhood and usually manifests in early or middle adult life. DH is slightly more common in men (3 : 2), but in patients younger than 20 years, women predominate (3 : 2).129 The rash is intensely pruritic, and scratching off the vesicle relieves the itching; hence, intact vesicles might not be present except for the earliest lesions.
The diagnosis of DH requires demonstration by immunofluorescence of granular or speckled IgA deposits in an area of perilesional skin—that is, skin close to a lesion but not affected by blistering.129 DH is associated with a mild patchy enteropathy indistinguishable from celiac disease; because it has a patch distribution, multiple intestinal biopsies may be required for diagnosis.129 DH-associated enteropathy tends to be less severe than celiac disease, and only a minority of patients has intestinal symptoms. An increased risk of intestinal malignancy has been reported, however, as in celiac disease,130,131 and most lymphomas occur in patients whose DH was not controlled by a strict gluten-free diet or in those who had been treated with a gluten-free diet for a period less than 5 years.131,132
Approximately 5% to 15% of patients with DH-like skin lesions have linear IgA deposits along the dermoepidermal junction. This condition has been termed linear IgA disease and is distinguished from DH on the basis of its unique immunofluorescent finding; the presence of circulating IgA anti–basement membrane antibody, which binds to a 97-kd protein found in normal human skin133; the absence of circulating IgA EMA or tTG; different HLA susceptibility genes; and, most important, the lack of any associated gluten-sensitive enteropathy.59,134
Sardy and colleagues shed light on the pathogenesis of DH by demonstrating that epidermal (type 3) transglutaminase (eTG) is the dominant autoantigen in DH (rather than the type 2 tTG autoantigen of celiac disease).135 This helps to explain why DH skin lesions appear in only a minority of patients having celiac disease. They also showed that the IgA precipitates in the papillary dermis of patients with DH, the defining manifestation of the disease, contain eTG but not tTG or keratinocyte transglutaminase. In DH, the prevalences of HLA-DQ2, circulating AGA, antireticulin, and EMA parallel those observed in patients with celiac disease without DH.129 Although many patients with DH have elevated IgA tTG antibodies, confirming its pathogenic relationship with celiac disease, its prevalence in DH (75%) is lower than that found in celiac disease (95% to 98%).136
Thus, DH and celiac disease are two very closely related gluten-sensitive disorders but nonetheless distinct clinical disease entities. Most, if not all, patients with DH also have at least latent celiac disease, whereas less than 10% of patients with celiac disease have DH. Dapsone treatment at a dose of 1 to 2 mg/kg daily is effective and often diagnostic in its ability to heal the rash of DH and to relieve the pruritus rapidly, but the enteropathy associated with DH does not improve with dapsone. Six to 12 months of gluten withdrawal, however, usually reverses not only the intestinal but also the skin lesions in most patients with DH, and a strict gluten-free diet allows most patients to reduce or discontinue dapsone.137 Iodine can also exacerbate DH and should be avoided, especially in refractory cases. Patients with DH, just like those with celiac disease, can include moderate amounts of oats in their gluten-free diet without deleterious effects to their skin or intestine.138
Other Disease Associations
The strong association between celiac disease and type 1 diabetes mellitus (T1DM) reflects, in part, the increased frequency of the celiac-associated DQ alleles in patients with T1DM. The frequency of celiac disease in T1DM patients is approximately 5% (ranging from 3% to 8%)139–141 and the frequency of T1DM in celiac disease is also approximately 5%.142 Most patients with T1DM who have celiac disease are asymptomatic from the point of view of their celiac disease, but unexpected episodes of hypoglycemia or diarrhea in patients with T1DM should alert clinicians to the possibility of coexisting celiac disease. Control of diabetes in patients with celiac disease can be difficult because of varying nutrient absorption.
There also is a high prevalence of autoimmune thyroid disease among patients with celiac disease, hypothyroidism being more common than hyperthyroidism.143 Celiac disease also can be associated with a variety of other autoimmune connective tissue diseases, including inflammatory bowel disease, chronic hepatitis, sclerosing cholangitis, primary biliary cirrhosis, IgA nephropathy, interstitial lung disease (including chronic fibrosing alveolitis), idiopathic pulmonary hemosiderosis, systemic lupus erythematosus, Sjögren’s syndrome, and polymyositis.1,52,97,117,128,142,144,145
Although the relationship between celiac disease and many autoimmune disorders has been explained by the sharing of a common genetic factor, Ventura and colleagues146 suggested an increased incidence of autoimmune disease with increased age at diagnosis and lack of diet therapy. The role of a gluten-free diet in preventing the subsequent development of autoimmune disease, however, has been challenged by more recent studies.147
Hyposplenism and splenic atrophy have been noted frequently in patients with celiac disease; the incidence increases with advancing age, duration of exposure to dietary gluten, and disease activity.81 The underlying mechanism is unknown, but affected patients may be at increased risk of developing bacterial infections148 and might benefit from vaccination for pneumonia.
There is a well-established relationship of celiac disease with inflammatory bowel disease and the microscopic colitides (see Chapter 124).149–151 Mild to moderate small intestinal lymphocytosis and, occasionally, partial or subtotal villus atrophy are common in both lymphocytic and collagenous colitis,150 and mild colonic lymphocytosis occurs in many patients with untreated celiac disease and has improved on a gluten-free diet.151,152 Rectal gluten challenge in patients with celiac disease has been shown to induce a mild proctitis characterized by lymphocytosis of the rectal lamina propria and epithelium.153 Furthermore, a gluten-free diet has been reported to be an effective therapy in some patients with refractory collagenous colitis.154 The demonstration that patients with celiac disease and microscopic colitis share a set of predisposing HLA-DQ genes155 underscores the overlap between both diseases.
Confusion also can arise in patients with refractory celiac disease, who have a higher prevalence of colonic lymphocytosis than patients with responsive celiac disease. Colonic lymphocytosis can be difficult to distinguish from lymphocytic colitis, although most colonic IELs in lymphocytic colitis are CD8+, whereas those in the colonic lymphocytosis of refractory celiac disease rarely are CD8+.124
TREATMENT
GLUTEN-FREE DIET
Removal of gluten from the diet is essential for treating patients with celiac disease (Table 104-4).80 The importance of gluten withdrawal was established by Dicke’s, van de Kamer’s, and Weijers’ astute studies in the early 1950s when the toxicity of wheat protein in children with celiac disease was demonstrated.6,7 In 1962, Rubin and colleagues31 showed that instillation of wheat, barley, and rye flour into the histologically normal small intestine of persons with treated celiac disease rapidly induced celiac-like symptoms and that these symptoms were accompanied by the development of the typical celiac lesions in the exposed mucosa.
Table 104-4 Principles of Initial Dietary Therapy for Patients with Celiac Disease
ppm, parts per million.
Modified from Trier JS. Celiac sprue and refractory sprue. In: Feldman M, Scharschmidt BF, Sleisenger MH, editors. Gastrointestinal and Liver Disease. 6th ed. Philadelphia: WB Saunders; 1997. p 1557.
Because a gluten-free diet represents a lifetime commitment for patients with celiac disease, is more expensive than a normal diet, and carries a social liability, it should not be undertaken casually or as a therapeutic trial. In reality, complete dietary elimination of all gluten-containing cereal grains is a challenge for most patients to achieve and maintain. Hidden gluten is present in a wide variety of processed foods, because wheat flour is used widely in the food industry as a thickener and inexpensive filler for many commercial products, precooked meals, and convenience foods, including ice cream, pasta, sausages, fish sticks, cheese spreads, salad dressings, soups, sauces, mixed seasonings, mincemeat for mince pies, and even some medications156 and vitamin preparations (Table 104-5). Furthermore, grains that are naturally gluten-free can become contaminated with wheat, particularly when mills use the same production lines and equipment to process both gluten-containing and gluten-free products. Beers, lagers, ales, and stout should be avoided (apart from the few available specifically gluten-free products), but wines, many liqueurs, and ciders as well as spirits, including brandy, malt, and scotch whiskey, can be consumed (unless gluten-containing flavorings have been added after distillation).
Table 104-5 Some Potential Sources of Hidden Gluten
Helpful recipes as well as detailed instructions regarding gluten-free diets have been published in excellent, inexpensive books that are of great value to patients with celiac disease.157 National celiac societies in many countries publish regularly updated handbooks that list the available gluten-free products. Food lists are applicable for use only in the country in which they were compiled. Similar foods with well-known brand names may be made under franchise using slightly different recipes in different countries and may be gluten-free in one country and not in others. Consequently, patient education is crucial, and the institution of an effective gluten-free diet requires extensive and repeated instruction of the patient by the physician and dietitian, as well as a motivated and basically suspicious, label-reading patient. The importance of patient education and support by a multidisciplinary team of health care providers was emphasized in the recently published National Institutes of Health consensus development conference statement (Table 104-6).158
Table 104-6 Key Elements in the Management of Celiac Disease
From the National Institutes of Health Consensus Development Conference Statement on Celiac Disease, June 28-30, 2004. Gastroenterology 2005;128:S1-S9.
There is considerable variation among patients with celiac disease in their ability to tolerate gluten. Some patients can ingest small amounts of gluten without developing symptoms. Others are exquisitely sensitive to ingestion of even minute amounts of gluten and can develop massive watery diarrhea reminiscent of acute cholera within hours of eating very small amounts of gluten. Occasionally, the diarrhea is so severe that it can induce acute dehydration, termed gliadin shock or celiac crisis.123
It is now clear that moderate amounts of oats are not toxic to the vast majority of patients with celiac disease. In a carefully conducted randomized clinical trial, adults with celiac disease who consumed 50 to 70 g of oats per day for 6 to 12 months did not differ in regard to symptoms, nutritional status, or duodenal mucosal histology compared with patients maintained on an oat-restricted, gluten-free diet159; in a smaller follow-up study, the same investigators showed no harm in celiac disease patients from five years of oat ingestion.160 Patients with DH also can include moderate amounts of oats in their gluten-free diet without deleterious effects to the skin or intestine,130 and an open-labeled study showed that a six-month trial of commercial oat breakfast cereal also was safe for children with newly diagnosed celiac disease beginning a gluten-free diet.161 It should be stressed, however, that oat products obtained from the grocery store shelf can contain significant amounts of other grains, especially wheat, because of contamination in the field, during harvesting or transport, or at the mill. Consequently, oats often are avoided initially in patients with newly diagnosed celiac disease until remission is achieved on a gluten-free diet. Subsequently, up to 2 ounces of oats per day from a reliable source may be introduced and continued if tolerated.
A gluten-free diet can be rich in nutrition and conducive to excellent general health. Careful attention is needed, however, to avoid certain pitfalls of the gluten-free diet. The gluten-free diet often contains inadequate iron, calcium, vitamin D, and B vitamins.162 Hence nutritional counseling and monitoring together with a daily gluten-free multivitamin supplement is recommended to avoid deficiency states. Avoiding wheat, barley, and rye often leads to inadequate fiber intake and constipation unless steps are taken to replace these with other sources of dietary fiber. Some weight gain is common after starting a gluten-free diet as malabsorption resolves; however, excess weight gain or obesity can easily ensue, especially if fat- and calorie-rich gluten-free processed foods and snacks are consumed in excess. Preparing meals using fresh ingredients including gluten-free grains and reducing the use of prepared or processed foods are often the keys to a healthy gluten-free diet.
After starting a gluten-free diet, most patients improve within a few weeks. In many, substantial symptomatic improvement is noticed within 48 hours, although it can take weeks or months to achieve full clinical remission. Pink and Creamer163 reported that 70% of patients with celiac disease who started a gluten-free diet returned quickly to normal health and reported improvement in their symptoms within two weeks. The speed and eventual degree of histologic improvement are unpredictable, but they invariably lag behind the clinical response.
Although an increase in enterocyte height may be evident within a week of gluten withdrawal, the return of villus architecture toward normal takes considerably longer and might not be evident on rebiopsy for two or three months. In some patients, histologic resolution can take up to two years; the main reason for this slow or partial recovery is inadvertent exposure to gluten.31 Although a return to normal is common in children, in approximately 50% of adults on a gluten-free diet, biopsies show only partial improvement; the less severely damaged distal intestine recovers more rapidly than the maximally damaged proximal intestine.164 The investigation and management of patients who have celiac disease and whose symptoms or signs do not respond to dietary gluten avoidance are discussed later (see the discussions of nonresponsive and refractory celiac disease).
DIETARY SUPPLEMENTS
The risks of osteopenia and osteoporosis should be explained to all patients with celiac disease and general advice should be given about weight-bearing exercises, dietary calcium and vitamin D intake, and the adverse effects of smoking and excessive alcohol consumption. There is compelling evidence that strict adherence to a gluten-free diet protects against further bone loss and initially is associated with an increase in bone mineral density (BMD).165 A total daily calcium intake of 1500 mg should be ensured—a cup of skim milk provides 300 mg. If dietary calcium is inadequate, 500 to 1500 mg of supplemental calcium should be given. Vitamin D deficiency should be sought and treated (particularly in patients with significant steatorrhea), until the malabsorption has responded to gluten withdrawal, to prevent mobilization of skeletal calcium.
One year of gluten withdrawal has been shown to reverse osteopenia in most patients, including postmenopausal women and patients with incomplete mucosal recovery,166 but patients who have secondary hyperparathyroidism at the time celiac disease is diagnosed tend to have more refractory osteopenia, and their BMD might not normalize even after several years of gluten withdrawal.167
Clinicians often rely on serum calcium, phosphate, and alkaline phosphatase measurements, but osteomalacia still can exist even if these tests are normal. If these tests are normal and osteomalacia still is suspected, a serum 25-hydroxy vitamin D level can be determined; this test is expensive, however, and the less expensive parathormone assay may be considered. A low-normal calcium and an elevated parathormone level indicates secondary hyperparathyroidism, and calcium (500 to 1000 mg daily) together with vitamin D (400 to 800 units daily) should be given.168 The serum calcium level must be monitored and supplementation promptly discontinued if hypercalcemia develops.
All patients should have their BMD measured one year after diagnosis. Recent guidelines suggest that patients with treated celiac disease who have progressive osteoporosis despite correction of calcium and vitamin D deficiency should be offered oral biphosphonate therapy and have their BMD checked every one to two years.168
Vitamin A, thiamine, riboflavin, niacin, pyridoxine, vitamin C, and vitamin E, all in the form of a multivitamin preparation, probably should be taken daily by all patients with celiac disease in light of their predisposition to malabsorption and the risks for nutritional deficiencies associated with a gluten-free diet.162 Some patients have reported symptomatic improvement with correction of magnesium, copper, and zinc deficiencies.169
GLUCOCORTICOIDS
In vitro studies have shown that the addition of glucocorticoids prevented the harmful effects of gluten on biopsy specimens from patients with celiac disease.170 Although celiac disease can be treated with glucocorticoids, with rapid improvement in symptoms, the effect rarely persists once treatment is stopped, and significant side effects are common.171 Therefore, glucocorticoids are not indicated in the routine management of celiac disease but are reserved for severely ill patients who present with acute celiac crisis manifested by severe diarrhea, dehydration, weight loss, acidosis, hypocalcemia, and hypoproteinemia.172 These few patients might benefit from a short course of glucocorticoids until the gluten-free diet takes effect. A brief course of glucocorticoids also may be used in the rare instances of gliadin shock that occurs occasionally in treated patients who are subject to gluten challenge.123
NONRESPONSIVE CELIAC DISEASE
Nonresponsive celiac disease (NRCD) is a clinical diagnosis defined by the persistence of symptoms, signs, or laboratory abnormalities typical of celiac disease despite adherence to a gluten-free diet for at least six months.118,173,174 Ten percent of patients with celiac disease are nonresponsive either immediately after the initial diagnosis (primary NRCD) or following a period of response to the gluten-free diet (secondary NRCD).173 An approach to the evaluation of NRCD that is based on the early identification and correction of common causes and that culminates in the diagnosis of refractory celiac disease (which affects 1% of patients with celiac disease) is outlined in Figure 104-7.118
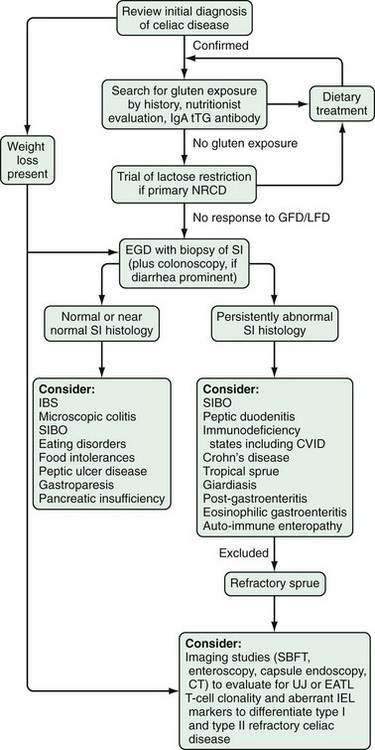
The single most common cause for NRCD is continued gluten ingestion, which is often inadvertent and occult. A persisting elevation of anti-tTG is strongly associated with ongoing gluten exposure.173 Intolerance to disaccharides (e.g., lactose, fructose) also is common, especially in primary NRCD. Thus, evaluation by an expert dietitian to seek ingestion of hidden gluten and intolerance of disaccharides is an essential next step.118
If no dietary causes can be identified, a small bowel biopsy should be repeated and the findings compared with the initial pretreatment biopsy. If the enteropathy has healed or is substantially improved, diagnostic considerations for ongoing symptoms and signs include irritable bowel syndrome (22%), small intestinal bacterial overgrowth (6%), other food allergies and intolerances (1%), and pancreatic insufficiency.173,174 If diarrhea is a prominent symptom, colonic biopsy also should be obtained and examined for microscopic colitis (6%). If repeat biopsies show ongoing changes consistent with active celiac disease, refractory celiac disease becomes likely. Other causes of a celiac-like enteropathy, however, should again be considered, including small bowel bacterial overgrowth, peptic duodenitis, hypogammaglobulinemia, tropical sprue, intestinal infections (e.g., giardiasis), Crohn’s disease, and autoimmune enteropathy.173,174 Patients with NRCD who show substantial weight loss are at a significantly greater risk for refractory celiac disease.173
REFRACTORY CELIAC DISEASE
Refractory celiac disease, also known as unclassified or intractable celiac disease, is defined as symptomatic, severe small intestinal villus atrophy that mimics celiac disease but does not respond to at least six months of a strict gluten-free diet and is not accounted for by other causes of villus atrophy or overt intestinal lymphoma.1,3,97 Refractory celiac disease is uncommon in adults, extremely rare in children, and largely a diagnosis of exclusion (Figs. 104-7 and 104-8). Symptoms can persist in treated celiac disease patients for a variety of reasons as described in the section on nonresponsive celiac disease.118,173–175
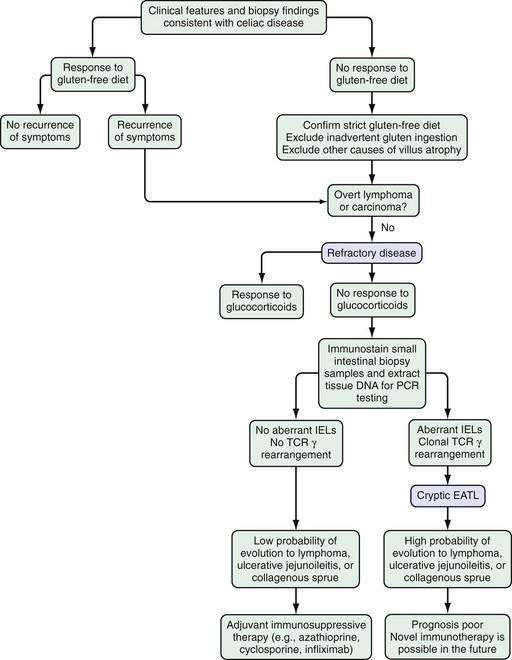
ULCERATIVE JEJUNOILEITIS
Ulcerative jejunoileitis, also known as chronic, nongranulomatous ulcerative enterocolitis or nongranulomatous jejunitis, is a rare but serious complication of celiac disease characterized by ulceration and strictures of the small intestine (see Chapter 115). Whether ulcerative jejunoileitis truly is a discrete entity has been questioned, because lymphoma ultimately is diagnosed in many of these patients.176 Indeed, ulcerative jejunoileitis in association with EATL previously was designated malignant histiocytosis.
Ulcerative jejunoileitis should be suspected in patients with celiac disease who present with weight loss, abdominal pain, and diarrhea that do not respond to a gluten-free diet. Typically, patients also experience recurrent episodes of intestinal ulceration and obstruction with gradual weight loss despite surgery and strict adherence to a gluten-free diet. Areas of intestinal ulceration and stricture formation typically cause hemorrhage and obstruction (Fig. 104-9); perforation with peritonitis also can occur. Diagnosis is made by enteroscopy, contrast studies of the small intestine, abdominal CT, capsule endoscopy, or laparotomy.
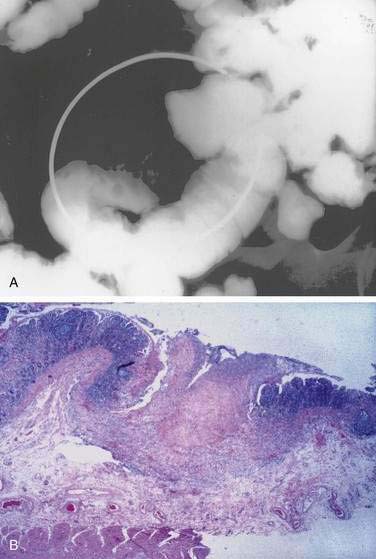
Some patients respond to a gluten-free diet, but surgical excision of the worst affected segments of small intestine so far has proved to be the most effective treatment. There is a high risk for transition to diffuse or multifocal EATL, but in a few patients with well-documented celiac disease and localized jejunoileitis, no evidence of malignant disease develops and there is a response to either surgical resection or therapy with glucocorticoids and azathioprine.177 Even in the absence of overt malignant transformation, however, the five-year survival rate for patients with ulcerative jejunoileitis is less than 50%.
COLLAGENOUS SPRUE
Collagenous disease is characterized by the development of a subepithelial collagen band thicker than 10 mm in the small intestine. Although collagenous disease has been regarded as an entity distinct from celiac disease,127 deposition of collagen under the intestinal epithelial cells has been noted in up to 36% of patients with classic celiac disease.178 Furthermore, there are several reports of patients with collagenous disease who have EMA179 and complications of refractory celiac disease, specifically ulcerative jejunoileitis180 and lymphoma.15
Although collagenous disease often is refractory to therapy, the presence of subepithelial collagen does not, a priori, preclude a successful response to gluten withdrawal.178,181 Collagenous disease should be distinguished from collagenous colitis, which rarely accompanies celiac disease and should be considered in the differential diagnosis of nonresponsive celiac disease.154 Compared with both celiac disease and collagenous colitis, the prognosis in collagenous disease is grim, with most reported patients dying from the disease.
TREATMENT
In patients with celiac disease and no demonstrable cause for lack of response to a gluten-free diet, a variety of treatments (based mostly on small, uncontrolled studies) have been described; these include glucocorticoids, immunosuppressive drugs, elimination diets, and dietary supplementation with zinc and copper.182–186
Evidence supporting the use of immunosuppressive therapy in the treatment of refractory celiac disease is based mainly on anecdotal reports; to date, no controlled trials have been performed.185 Glucocorticoid treatment also may be necessary in patients with refractory celiac disease (see later). Azathioprine or 6-mercaptopurine may be used as a glucocorticoid-sparing agent if a dose of 10 mg of prednisolone or more per day is required to keep the condition under control.182 In one open pilot study, 13 adult patients with refractory celiac disease were treated for two months with oral cyclosporine, in doses titrated to achieve serum levels of 100 to 200 ng/mL; small intestinal histology improved in eight patients (61%), and villi normalized in five (38%).186 Cyclosporine therapy has been reported to be lifesaving in occasional patients with refractory celiac disease–like disease, and it can result in reversal of glucocorticoid resistance, but its efficacy remains unproved.183 There also has been a report on the efficacy of infliximab, a chimeric antibody to TNF-α, in refractory celiac disease.184
In a study of patients with refractory celiac disease, 16 of 29 (55%) responded fully to treatment with oral enteric-release budesonide (Entocort, 9 mg daily), and an additional six (21%) showed a partial response.187 Improvements were evident in patients with coexisting microscopic colitis and in those without. Oral budesonide was well tolerated, and no serious side effects were reported. Because of its good side-effect profile, oral budesonide is fast becoming a drug of first choice for treatment of refractory celiac disease.
COMPLICATIONS
It has long been appreciated that patients with refractory celiac disease are at high risk for developing fatal complications such as ulcerative jejunoileitis and lymphoma. Until recently, the precise link between refractory celiac disease and these complications as well as between refractory celiac disease and celiac disease remained controversial. The spectrum of autoimmune enteropathy was implicated in a handful of adult refractory celiac disease patients by the presence of antienterocyte antibodies.188,189 It is now becoming clear, however, that refractory celiac disease, EATL, and ulcerative jejunoileitis represent a heterogeneous, but related, group of clinical conditions at the extreme end of the celiac disease spectrum. Moreover, there is now a growing realization that many of these patients have a cryptic intestinal T-cell lymphoma, characterized by phenotypically abnormal IELs that have monoclonal rearrangements of the TCR γ gene.15
Early immunophenotypical studies demonstrated that the normal cell counterpart of EATL was the IEL.190 It was not until 1995, however, that Murray and colleagues191 made the remarkable observation that in patients with overt EATL, lymphocytes from adjacent nonlymphomatous mucosa contained the identical monoclonal TCR gene rearrangement as the overt lymphoma and coined the term cryptic intestinal T-cell lymphoma. Ashton-Key and colleagues192 later confirmed this finding and showed that both the inflammatory ulcers and the intact (nonlymphomatous) mucosa in cases of ulcerative jejunoileitis harbored a monoclonal T-cell population and that the lymphomas that developed in these patients consisted of the identical T-cell clone.
Cellier and colleagues68 showed that the IELs in refractory celiac disease patients are abnormal in that they lack expression of CD8, which is consistently found on most normal or celiac disease IELs. Subsequent work confirmed this finding and showed that the abnormal IELs in ulcerative jejunoileitis and nonlymphomatous mucosa in EATL shared not only the genotype but also the immunophenotype of the lymphoma.193 Cellier and colleagues15 detected aberrant clonal IELs (similar to those in most cases of EATL) in 16 (84%) of 19 patients with refractory celiac disease (type II refractory celiac disease). Of these 19, 7 (37%) had collagenous disease, 6 (32%) had ulcerative jejunoileitis, 6 (32%) had mesenteric lymph node cavitation, and 3 (16%) developed overt EATL that was clonally identical to the IELs of the preexisting refractory celiac disease. The three patients (16%) without aberrant clonal IELs (type I refractory celiac disease) all made a complete clinical and histologic recovery with glucocorticoid therapy plus a gluten-free diet.
Thus, the cumulative evidence now points to type II refractory celiac disease being a manifestation of an aberrant clonal IEL-mediated neoplastic process. These cells have destructive properties, possible related to their cytotoxic phenotype,194 which leads to mucosal ulceration and lymph node cavitation, and they sometimes, but not always, undergo further molecular and clinical progression to lymphoma. As noted earlier, the proinflammatory cytokine IL-15 is massively increased in the intestine of patients with refractory celiac disease. IL-15 induces IEL secretion of IFN-γ and increases IEL cytotoxicity against epithelial cells, thereby favoring the severe enteropathy characteristic of type II refractory celiac disease.16 Increasing evidence suggests that IL-15, through its key role in modulating IEL homeostasis, ultimately might lead to lymphomatous transformation because IL-15 provides signals mandatory for the survival or expansion of the abnormal clonal IELs.
CELIAC DISEASE AND MALIGNANCY
In the past, patients with celiac disease or DH had been reported to have a 10-fold increased risk for certain gastrointestinal tract malignancies and a 40- to 70-fold increased risk for non-Hodgkin’s lymphoma (NHL).130,195 Recent studies, however, indicate that the increase in risk of malignancy, particularly lymphoma, is much less than initially believed. A large retrospective Swedish study followed almost 12,000 hospitalized patients who had either celiac disease or DH between 1964 and 1994, with a mean follow-up of 10 years.131 The overall cancer and malignancy lymphoma risks were increased only modestly (standardized incidence ratio [SIR], 1.3; 95% CI: 1.2-1.5; and SIR, 5.9; 95% CI: 4.3-7.9, respectively). In a prospective Italian study, patients with celiac disease had a 3.1-fold increased risk of NHL.196 Conversely, the prospective BioMed European Working Group on Celiac Disease and Malignancy, which reviewed data from 10 countries, reported that the prevalence of celiac disease was increased 2.6-fold in 1446 patients with NHL compared with 9659 control subjects.197 The European study suggested that the risk of NHL is even less evident in silent celiac disease.
Small intestinal lymphoma, often multifocal and diffuse, accounts for one half to two thirds of the malignancies complicating celiac disease and typically occurs after 20 to 40 years of disease (see Chapters 29 and 121).131,195 Whereas in the general population, most small intestinal lymphomas are of B-cell origin, intestinal lymphoma in celiac disease is typically of T-cell origin, and the term EATL (enteropathy-associated T-cell lymphoma) was coined to describe both the intestinal and extraintestinal lymphomas that complicate celiac disease. The European multicenter study indicated that intestinal T-cell lymphomas, chiefly EATL, are highly characteristic, occurring almost exclusively in those with celiac disease (odds ratio of 28 compared to population controls).
The clinical onset of EATL may be insidious, and its initial presentation and small bowel biopsy appearance can mimic those of untreated celiac disease. EATL commonly is accompanied by mucosal ulceration, as seen in ulcerative jejunoileitis, and these ulcers sometimes are the only endoscopic manifestation of lymphoma (see Chapters 29, 121). Although some patients with EATL have a partial or temporary response to a strict gluten-free diet, most are eventually unresponsive to gluten withdrawal. In patients whose disease was previously controlled on a gluten-free diet, recurrence of gastrointestinal symptoms (e.g., abdominal pain, weight loss, diarrhea) should raise the clinical suspicion of lymphoma. In some patients with lymphoma, mucosal histology adjacent to and distant from the lymphoma is indistinguishable from that of untreated celiac disease, yet the patient’s symptoms do not respond to gluten withdrawal.190 There is long-standing controversy whether such patients had latent celiac disease that became evident after lymphoma developed, refractory celiac disease complicated by lymphoma, or refractory enteropathy induced by primary intestinal T-cell lymphoma and indistinguishable by histologic criteria from celiac disease.15 Molecular and immunohistochemical studies that have advanced our understanding of the relationships among celiac disease, refractory celiac disease, and EATL are discussed in the section on refractory celiac disease.
Other features suggesting lymphoma include intestinal obstruction, intestinal bleeding, fever, hypoalbuminemia, lymphadenopathy, and erythrophagocytosis evident in bone marrow or the peripheral blood. Small intestinal radiology, enteroscopy with biopsy of the mucosa at multiple levels, capsule endoscopy, and CT or MR scanning may be helpful. Mesenteric lymphadenopathy with central cavitation has been described in celiac disease, both with198 and without199 lymphoma. If the index of suspicion is high and studies are not diagnostic, full-thickness biopsy specimens of the small intestine should be obtained at laparoscopy or laparotomy with careful examination of the entire length of the small intestine and examination of mesenteric lymph nodes. Even with such an aggressive approach, EATL can be extremely difficult to diagnose.
EATL commonly is fatal: Overall one-year and five-year survival rates of 31% and 11%, respectively, were reported in one small series, with long-term survival almost exclusively confined to those treated with chemotherapy.200
Carcinoma, particularly of the oropharynx, esophagus, and small intestine, account for one third of the remaining malignancies complicating celiac disease. The average patient so affected is older than 50 years. The Swedish study reported elevated risks for small intestinal cancer (SIR, 10), oropharyngeal cancer (SIR, 2.3), esophageal cancer (SIR, 4.2), and primary liver cancer (SIR, 2.7).131 Patients with DH also had a slightly increased overall cancer risk (SIR, 1.2) owing to excesses of lymphoma and leukemia, but they had no increases in gastrointestinal carcinomas.131
The mechanisms responsible for the increased prevalence of malignancy in celiac disease are unknown. Increased crypt mitotic activity, increased turnover of lymphoid cells within the mucosa, penetration of the damaged intestinal mucosa by carcinogens, infection with oncogenic viruses, and underlying abnormalities in the mucosal immune system and surface epithelium all are potential factors. In the Swedish study, the excess risk of malignancies, which was confined to adults, disappeared after a 10-year follow-up.131 This declining risk of malignancies with increased duration of follow-up, and thus with the length of gluten-free diet, supports the results of a previous study, which indicated that a strict gluten-free diet for five years reduced the risk of all malignancies, not just EATL, to that of the general population.130
PROGNOSIS
Celiac disease has an excellent prognosis if it is diagnosed early and the patient adheres to a lifelong gluten-free diet. Conversely, if it is not recognized and properly treated, patients can develop marked malnutrition and debilitation and can die of complications such as intercurrent infection, hemorrhage, or malignancy. Although earlier studies reported 1.9-fold201 and 3.4-fold202 increases in mortality, these studies included patients who were not adhering to a gluten-free diet as well as patients with refractory celiac disease and intestinal lymphoma. A study of 335 adults with celiac disease from Finland, at least 83% of whom adhered strictly to a gluten-free diet, showed that the five-year survival was comparable with that of the general population.142 Growth and development in infants and children with celiac disease proceed normally following gluten withdrawal. In adults, absorptive functions usually return, and many of the manifestations of disease disappear after a gluten-free diet is initiated. Certain complications, however, such as peripheral neuropathy, ataxia, or pathologic fractures secondary to severe osteopenic bone disease, particularly in the setting of secondary hyperparathyroidism, might not be completely reversible. The potential protective effect of gluten-free diet against the development of other autoimmune diseases and the functional consequences of a gluten-free diet in silent celiac disease remain unknown.
Several lines of evidence, gathered during recent years, suggest that celiac disease is not always a lifelong condition. First, the long-term follow-up of children with proven celiac disease shows that 10% to 20% develop latent celiac disease and become tolerant (defined on clinical, biological, and histologic grounds) to gluten during adolescence. Second, it also has been shown, in individual cases, that the mucosal lesions typical of the disease can appear de novo during adulthood.203 The factors leading to the appearance or disappearance of gluten-sensitive enteropathy, however, are still unknown. Although adolescent patients might stray from their gluten-free diet, often without apparent ill effects, their inability to tolerate gluten remains, and many asymptomatic adolescent patients can be shown to have persistent hematologic, biochemical, and morphologic abnormalities.22,85 If gluten ingestion continues into adult life, most patients with celiac disease eventually develop recurrent clinical evidence of celiac disease. Therefore, patients with unequivocal evidence of celiac disease in childhood should be encouraged to remain on a gluten-free diet indefinitely if recurrent clinical disease is to be avoided during adult life.
FUTURE THERAPIES
Improved knowledge of celiac disease epidemiology and pathogenesis has encouraged the search for alternatives to the gluten-free diet. Shan and colleagues204 showed that treating gliadin peptides with a combination of digestive enzymes in conditions mimicking the in vivo situation released a protease-resistant 33-amino-acid peptide (33-mer) encompassing a cluster of three immunodominant T-cell epitopes. The resistance of this peptide to human digestive enzymes was ascribed to its high proline content. Rapid hydrolysis, however, was achieved in the presence of a bacterial prolyl-endopeptidase.204 Oral glutenases from bacterial or cereal sources are now being studied for their potential therapeutic use in cleaving toxic gliadin peptides within the stomach and proximal small intestine, thereby potentially abrogating gluten toxicity.205
An increase in intestinal permeability is a well-documented feature of celiac disease and can facilitate the passage of gluten across the epithelial layer to be taken up by antigen-presenting cells and activate gliadin-specific T cells. Larazotide acetate is an octapeptide inhibitor of paracellular permeability whose structure is derived from a protein (zonula occludens toxin) secreted by Vibrio cholerae.206 In early clinical trials, larazotide acetate showed promise in preventing symptoms and signs of celiac disease activity during gluten challenge.206
Genetic modification of wheat to delete toxic peptides has been proposed to prevent the activation of celiac disease. This approach is complicated by the large number of T-cell epitopes and by the complexity of wheat genetics. Furthermore, nature has already provided a wealth of nontoxic cereal alternatives. An alternative strategy might be to develop peptide analogs capable of interfering with HLA-DQ binding and T-cell activation to redirect the immune response toward tolerance. In mice, a vaccine based on the intranasal administration of whole gliadin or of one of its isoforms partially inhibited the systemic T-cell response to parenteral challenge by whole gliadin.207 This approach is not easy to transpose into humans, however, because of the risk of enhancing immunization instead of promoting tolerance.
As mentioned earlier, tTG modification of gliadin peptides greatly increases their immunogenicity and toxicity in celiac disease.208 Thus, tTG inhibitors are also being evaluated for their ability to reduce gluten toxicity.
Another strategy, suggested by Maiuri and colleagues,38 is blockade of signals derived from the cytokine IL-15. This proposal may be premature for uncomplicated celiac disease in the absence of data concerning the consequence of IL-15 blockade in vivo in humans. Blocking IL-15 and its signals, however, is an attractive possibility in refractory celiac disease when patients have become unresponsive to the gluten-free diet and do not experience a response to conventional anti-inflammatory therapy.
Because celiac disease is, in most cases, a benign disease effectively treated by a safe, established diet, any alternative treatments that emerge must meet high standards of efficacy and safety. That stated, many patients with celiac disease would welcome the development of agents that could reduce the daily burden of strict gluten avoidance and also improve the control of their disease.209,210
Askling J, Linet M, Gridley G, et al. Cancer incidence in a population-based cohort of individuals hospitalized with celiac disease or dermatitis herpetiformis. Gastroenterology. 2002;123:1428. (Ref 131.)
Bernstein CN, Leslie WD, Leboff MS. AGA medical position statement: Guidelines on osteoporosis in gastrointestinal diseases. Gastroenterology. 2003;124:791. (Ref 168.)
Cellier C, Delabesse E, Helmer C, et al. Refractory sprue, coeliac disease, and enteropathy-associated T-cell lymphoma. French Coeliac Disease Study Group. Lancet. 2000;356:203. (Ref 15.)
Dicke W. Coeliac disease: Investigation of harmful effects of certain types of cereal on patients with coeliac disease. Utrecht: University of Utrecht; 1950. (Ref 6.)
Dieterich W, Ehnis T, Bauer M, et al. Identification of tissue transglutaminase as the autoantigen of celiac disease. Nat Med. 1997;3:797. (Ref 12.)
Fasano A, Berti I, Gerarduzzi T, et al. Prevalence of celiac disease in at-risk and not-at-risk groups in the United States: A large multicenter study. Arch Intern Med. 2003;163:286. (Ref 17.)
Green PH, Cellier C. Celiac disease. N Engl J Med. 2007;357:1731-43. (Ref 97.)
Leffler DA, Dennis M, Hyett B, et al. Etiologies and predictors of diagnosis in nonresponsive celiac disease. Clin Gastroenterol Hepatol. 2007;5:445-50. (Ref 173.)
Lundin KE, Scott H, Hansen T, et al. Gliadin-specific, HLA-DQ(α1*0501,β1*0201) restricted T cells isolated from the small intestinal mucosa of celiac disease patients. J Exp Med. 1993;178:187. (Ref 11.)
Molberg O, McAdam SN, Korner R, et al. Tissue transglutaminase selectively modifies gliadin peptides that are recognized by gut-derived T cells in celiac disease. Nat Med. 1998;4:713. (Ref 13.)
National Institutes of Health Consensus Development Conference Statement on Celiac Disease, June 28-30, 2004. Gastroenterology. 2005;128:S1-S9.
Schuppan D. Current concepts of celiac disease pathogenesis. Gastroenterology. 2000;119:234. (Ref 43.)
Shan L, Molberg O, Parrot I, et al. Structural basis for gluten intolerance in celiac sprue. Science. 2002;297:2218. (Ref 204.)
Sollid LM, Khosla C. Future therapeutic options for celiac disease. Nat Clin Pract Gastroenterol Hepatol. 2005;2(3):140-7. (Ref 18.)
Stern M. Comparative evaluation of serologic tests for celiac disease: A European initiative toward standardization. J Pediatr Gastroenterol Nutr. 2000;31:513. (Ref 98.)
1. Farrell RJ, Kelly CP. Celiac sprue. N Engl J Med. 2002;346:180.
2. Arranz E, Ferguson A. Intestinal antibody pattern of celiac disease: Occurrence in patients with normal jejunal biopsy histology. Gastroenterology. 1993;104:1263.
3. Trier JS. Diagnosis of celiac sprue. Gastroenterology. 1998;115:211.
4. Adams F. The extant works of Aretaeus of Cappodocian. London: Sydenham Society; 1856.
5. Gee S. On the coeliac affection. St Barth Hosp Rep. 1888;24:17.
6. Dicke W. Coeliac disease: Investigation of harmful effects of certain types of cereal on patients with coeliac disease. Utrecht: University of Utrecht; 1950.
7. van de Kamer JH, Weijers NA, Dicke WK. Coeliac disease: IV. An investigation into the injurious constituents of wheat in connection with their action in patients with coeliac disease. Acta Paediatr. 1953;42:223.
8. Paulley L. Observations on the aetiology of idiopathic steatorrhea. Br Med J. 1954;2:1318.
9. Rubin CE, Brandborg LL, Phelps PC, Taylor HCJr. Studies of celiac disease: I. The apparent identical and specific nature of the duodenal and proximal jejunal lesion in celiac disease and idiopathic sprue. Gastroenterology. 1960;38:28.
10. Howell MD, Austin RK, Kelleher D, et al. An HLA-D region restriction fragment length polymorphism associated with celiac disease. J Exp Med. 1986;164:333.
11. Lundin KE, Scott H, Hansen T, et al. Gliadin-specific, HLA-DQ(α1*0501,β1*0201) restricted T cells isolated from the small intestinal mucosa of celiac disease patients. J Exp Med. 1993;178:187.
12. Dieterich W, Ehnis T, Bauer M, et al. Identification of tissue transglutaminase as the autoantigen of celiac disease. Nat Med. 1997;3:797.
13. Molberg O, McAdam SN, Korner R, et al. Tissue transglutaminase selectively modifies gliadin peptides that are recognized by gut-derived T cells in celiac disease. Nat Med. 1998;4:713.
14. Anderson RP, Degano P, Godkin AJ, et al. In vivo antigen challenge in celiac disease identifies a single transglutaminase-modified peptide as the dominant α-gliadin T-cell epitope. Nat Med. 2000;6:337.
15. Cellier C, Delabesse E, Helmer C, et al. Refractory sprue, coeliac disease, and enteropathy-associated T-cell lymphoma. French Coeliac Disease Study Group. Lancet, 356, 2000:203
16. Mention JJ, Ben Ahmed M, Begue B, et al. Interleukin 15: A key to disrupted intraepithelial lymphocyte homeostasis and lymphomagenesis in celiac disease. Gastroenterology. 2003;125:730.
17. Fasano A, Berti I, Gerarduzzi T, et al. Prevalence of celiac disease in at-risk and not-at-risk groups in the United States: A large multicenter study. Arch Intern Med. 2003;163:286.
18. Sollid LM, Khosla C. Future therapeutic options for celiac disease. Nat Clin Pract Gastroenterol Hepatol. 2005;2(3):140-7.
19. Schuppan D, Junker Y. Turning swords into plowshares: Transglutaminase to detoxify gluten. Gastroenterology. 2007;133(3):1025-8.
20. Catassi C, Fabiani E, Ratsch IM, et al. The coeliac iceberg in Italy: A multicentre antigliadin antibodies screening for coeliac disease in school-age subjects. Acta Paediatr Suppl. 1996;412:29.
21. Maki M, Mustalahti K, Kokkonen J, et al. Prevalence of celiac disease among children in Finland. N Engl J Med. 2003;348:2517.
22. Johnston SD, Watson RG, McMillan SA, et al. Coeliac disease detected by screening is not silent—simply unrecognized. Q J Med. 1998;91:853.
23. Rossipal E. Incidence of coeliac disease in children in Austria. Z Kinderheilkd. 1975;119:143.
24. Cavell B, Stenhammar L, Ascher H, et al. Increasing incidence of childhood coeliac disease in Sweden: Results of a national study. Acta Paediatr. 1992;81:589.
25. Weile B, Cavell B, Nivenius K, Krasilnikoff PA. Striking differences in the incidence of childhood celiac disease between Denmark and Sweden: A plausible explanation. J Pediatr Gastroenterol Nutr. 1995;21:64.
26. Catassi C, Ratsch IM, Gandolfi L, et al. Why is coeliac disease endemic in the people of the Sahara? Lancet. 1999;354:647.
27. Madara JL, Trier JS. Structural abnormalities of jejunal epithelial cell membranes in celiac sprue. Lab Invest. 1980;43:254.
28. Wright NA, Watson AJ, Morley AR, et al. Cell production rate in mucosa of untreated coeliac disease. Gut. 1972;13:846.
29. Baklien K, Brandtzaeg P, Fausa O. Immunoglobulins in jejunal mucosa and serum from patients with adult coeliac disease. Scand J Gastroenterol. 1977;12:149.
30. Marsh MN. Gluten, major histocompatibility complex, and the small intestine: A molecular and immunobiologic approach to the spectrum of gluten sensitivity (“celiac sprue”). Gastroenterology. 1992;102:330.
31. Rubin CE, Brandborg LL, Flick AL, et al. Biopsy studies on the pathogenesis of celiac sprue. In: Wolstenholme GEW, Cameron MP, editors. Intestinal Biopsy. Boston: Little, Brown; 1962:67.
32. Grefte JM, Bouman JG, Grond J, et al. Slow and incomplete histological and functional recovery in adult gluten-sensitive enteropathy. J Clin Pathol. 1988;41:886.
33. De Vincenzi M, Luchetti R, Peruffo AD, et al. In vitro assessment of acetic acid–soluble proteins (glutenin) toxicity in celiac disease. J Biochem Toxicol. 1996;11:205.
34. Kasarda DD, Okita TW, Bernardin JE, et al. Nucleic acid (cDNA) and amino acid sequences of α-type gliadins from wheat (Triticum aestivum). Proc Natl Acad Sci U S A. 1984;81:4712.
35. Troncone R, Auricchio S, De Vincenzi M, et al. An analysis of cereals that react with serum antibodies in patients with coeliac disease. J Pediatr Gastroenterol Nutr. 1987;6:346.
36. Ciclitira PJ, Ellis HJ. Investigation of cereal toxicity in coeliac disease. Postgrad Med J. 1987;63:767.
37. Vader W, Kooy Y, Van Veelen P, et al. The gluten response in children with celiac disease is directed toward multiple gliadin and glutenin peptides. Gastroenterology. 2002;122:1729.
38. Maiuri L, Ciacci C, Ricciardelli I, et al. Association between innate response to gliadin and activation of pathogenic T cells in coeliac disease. Lancet. 2003;362:30.
39. Gianfrani C, Troncone R, Mugione P, et al. Celiac disease association with CD8+ T cell responses: Identification of a novel gliadin-derived HLA-A2-restricted epitope. J Immunol. 2003;170:2719.
40. Kagnoff MF, Paterson YJ, Kumar PJ, et al. Evidence for the role of a human intestinal adenovirus in the pathogenesis of coeliac disease. Gut. 1987;28:995.
41. Arato A, Kosnai I, Szonyi L, Toth M. Frequent past exposure to adenovirus 12 in coeliac disease. Acta Paediatr Scand. 1991;80:1101.
42. Shidrawi RG, Day P, Przemioslo R, et al. In vitro toxicity of gluten peptides in coeliac disease assessed by organ culture. Scand J Gastroenterol. 1995;30:758.
43. Schuppan D. Current concepts of celiac disease pathogenesis. Gastroenterology. 2000;119:234.
44. Schmitz J. Lack of oats toxicity in coeliac disease. BMJ. 1997;314:159.
45. Norris JM, Barriga K, Hoffenberg EJ, et al. Risk of celiac disease autoimmunity and timing of gluten introduction in the diet of infants at increased risk of disease. JAMA. 2005;293(19):2343-51.
46. Ellis A. Coeliac disease: Previous family studies. In: McConnel RB, editor. The Genetics of Coeliac Disease. Lancaster, England: MTP Press; 1981:197.
47. Sollid LM, Markussen G, Ek J, et al. Evidence for a primary association of celiac disease to a particular HLA-DQ alpha/beta heterodimer. J Exp Med. 1989;169:345.
48. Nilsen EM, Lundin KE, Krajci P, et al. Gluten-specific, HLA-DQ-restricted T cells from coeliac mucosa produce cytokines with Th1 or Th0 profile dominated by interferon-γ. Gut. 1995;37:766.
49. Smyth DJ, Plagnol V, Walker NM, et al. Shared and distinct genetic variants in type 1 diabetes and celiac disease. N Engl J Med. 2008;359:2767-77.
50. McManus R, Wilson AG, Mansfield J, et al. TNF-2, a polymorphism of the tumour necrosis-α gene promoter, is a component of the celiac disease major histocompatibility complex haplotype. Eur J Immunol. 1996;26:2113.
51. Djilali-Saiah I, Schmitz J, Harfouch-Hammoud E, et al. CTLA-4 gene polymorphism is associated with predisposition to celiac disease. Gut. 1998;43:187.
52. Sollid L. Celiac disease: Dissecting a complex inflammatory disorder. Nat Rev Immunol. 2002;9:647.
53. Kelly CP, Feighery CF, Gallagher RB, et al. Mucosal and systemic IgA anti-gliadin antibody in celiac disease: Contrasting patterns of response in serum, saliva, and intestinal secretions. Dig Dis Sci. 1991;36:743.
54. Leffler DA, Edwards George JB, et al. A prospective comparative study of five measures of gluten-free diet adherence in adults with coeliac disease. Aliment Pharmacol Ther. 2007;26:1227-35.
55. Niveloni S, Sugai E, Cabanne A, et al. Antibodies against synthetic deamidated gliadin peptides as predictors of celiac disease: Prospective assessment in an adult population with a high pretest probability of disease. Clin Chem. 2007;53:2186-92.
56. Volta U, Granito A, Fiorini E, et al. Usefulness of antibodies to deamidated gliadin peptides in celiac disease diagnosis and follow-up. Dig Dis Sci. 2008;53:1582-8.
57. Uibo O, Uibo R, Kleimola V, et al. Serum IgA anti-gliadin antibodies in an adult population sample: High prevalence without celiac disease. Dig Dis Sci. 1993;38:2034.
58. Hvatum M, Scott H, Brandtzaeg P. Serum IgG subclass antibodies to a variety of food antigens in patients with coeliac disease. Gut. 1992;33:632.
59. Chorzelski TP, Beutner EH, Sulej J, et al. IgA anti-endomysium antibody: A new immunological marker of dermatitis herpetiformis and coeliac disease. Br J Dermatol. 1984;111:395.
60. van de Wal Y, Kooy Y, van Veelen P, et al. Selective deamidation by tissue transglutaminase strongly enhances gliadin-specific T cell reactivity. J Immunol. 1998;161:1585.
61. Szabolcs M, Sipka S, Csorba S. In vitro cross-linking of gluten into high-molecular-weight polymers with transglutaminase. Acta Paediatr Hung. 1987;28:215.
62. Halttunen T, Maki M. Serum immunoglobulin A from patients with celiac disease inhibits human T84 intestinal crypt epithelial cell differentiation. Gastroenterology. 1999;116:566.
63. Halstensen TS, Scott H, Fausa O, Brandtzaeg P. Gluten stimulation of coeliac mucosa in vitro induces activation (CD25) of lamina propria CD4+ T cells and macrophages but no crypt-cell hyperplasia. Scand J Immunol. 1993;38:581.
64. Brandtzaeg P, Halstensen TS, Kett K, et al. Immunobiology and immunopathology of human gut mucosa: Humoral immunity and intraepithelial lymphocytes. Gastroenterology. 1989;97:1562.
65. Halstensen TS, Scott H, Brandtzaeg P. Human CD8+ intraepithelial T lymphocytes are mainly CD45RA-RB+ and show increased co-expression of CD45R0 in celiac disease. Eur J Immunol. 1990;20:1825-30.
66. Halstensen TS, Scott H, Brandtzaeg P. Intraepithelial T cells of the TcR gamma/delta+ CD8- and V delta 1/J delta 1+ phenotypes are increased in coeliac disease. Scand J Immunol. 1989;30:665.
67. Mak TW, Ferrick DA. The gamma/delta T-cell bridge: Linking innate and acquired immunity. Nat Med. 1998;4:764.
68. Cellier C, Patey N, Mauvieux L, et al. Abnormal intestinal intraepithelial lymphocytes in refractory sprue. Gastroenterology. 1998;114:471.
69. Maiuri L, Ciacci C, Ricciardelli I, et al. Association between innate response to gliadin and activation of pathogenic T cells in coeliac disease. Lancet. 2003;362:30.
70. Damen GM, Boersma B, Wit JM, Heymans HS. Catch-up growth in 60 children with celiac disease. J Pediatr Gastroenterol Nutr. 1994;19:394.
71. Ansaldi N, Tavassoli K, Dell’Olio D, et al. Clinical data on celiac disease with an early or late onset. Minerva Pediatr. 1991;43:377.
72. Ivarsson A, Hernell O, Stenlund H, Persson LA. Breast-feeding protects against celiac disease. Am J Clin Nutr. 2002;75:914-21.
73. Ascher H, Krantz I, Rydberg L, et al. Influence of infant feeding and gluten intake on coeliac disease. Arch Dis Child. 1997;76:113.
74. Norris JM, Barriga K, Hoffenberg EJ, et al. Risk of celiac disease autoimmunity and timing of gluten introduction in the diet of infants at increased risk of disease. JAMA. 2005;293(19):2343-51.
75. Beaumont DM, Mian MS. Coeliac disease in old age: “A catch in the rye.”. Age Ageing. 1998;27:535.
76. Vuoristo M, Miettinen TA. The role of fat and bile acid malabsorption in diarrhoea of coeliac disease. Scand J Gastroenterol. 1987;22:289.
77. Maton PN, Selden AC, Fitzpatrick ML, Chadwick VS. Defective gallbladder emptying and cholecystokinin release in celiac disease: Reversal by gluten-free diet. Gastroenterology. 1985;88:391.
78. Sanders DS, Carter MJ, Hurlstone DP, et al. Association of adult coeliac disease with irritable bowel syndrome: A case-control study in patients fulfilling ROME II criteria referred to secondary care. Lancet. 2001;358:1504.
79. O’Farrelly C, O’Mahony C, Graeme-Cook F, et al. Gliadin antibodies identify gluten-sensitive oral ulceration in the absence of villous atrophy. J Oral Pathol Med. 1991;20:476.
80. Trier JS. Celiac sprue and refractory sprue. In: Feldman M, Scharschmidt BF, Sleisenger MH, editors. Gastrointestinal and Liver Disease. 6th ed. Philadelphia: WB Saunders; 1997:1557.
81. O’Grady JG, Stevens FM, Harding B, et al. Hyposplenism and gluten-sensitive enteropathy: Natural history, incidence, and relationship to diet and small bowel morphology. Gastroenterology. 1984;87:1326.
82. Corazza GR, Di Sario A, Cecchetti L, et al. Bone mass and metabolism in patients with celiac disease. Gastroenterology. 1995;109:122.
83. Kemppainen T, Kroger H, Janatuinen E, et al. Osteoporosis in adult patients with celiac disease. Bone. 1999;24:249.
84. Vasquez H, Mazure R, Gonzalez D, et al. Risk of fractures in celiac disease patients: A cross-sectional, case-control study. Am J Gastroenterol. 2000;95:183.
85. Cellier C, Flobert C, Cormier C, et al. Severe osteopenia in symptom-free adults with a childhood diagnosis of coeliac disease. Lancet. 2000;355:806.
86. Thomason K, West J, Logan RF, et al. Fracture experience of patients with coeliac disease: A population-based survey. Gut. 2003;52:518.
87. Hadjivassiliou M, Grunewald RA, Chattopadhyay AK, et al. Clinical, radiological, neurophysiological, and neuropathological characteristics of gluten ataxia. Lancet. 1998;352:1582.
88. Lewis PD, Pallis CA. Neurological complications of coeliac disease and tropical sprue. In: Lewis PD, Pallis CA, editors. The neurology of gastrointestinal disease. London: WB Saunders; 1974:138.
89. Gobbi G, Bouquet F, Greco L, et al. Coeliac disease, epilepsy, and cerebral calcifications. The Italian Working Group on Coeliac Disease and Epilepsy. Lancet. 1992;340:439.
90. Ferroir JP, Fénelon G, Billy C, et al. Epilepsy, cerebral calcifications, and celiac disease. Rev Neurol (Paris). 1997;153:354.
91. Addolorato G, Capristo E, Ghittoni G, et al. Anxiety but not depression decreases in coeliac patients after one-year gluten-free diet: A longitudinal study. Scand J Gastroenterol. 2001;36:502.
92. Molteni N, Bardella MT, Bianchi PA. Obstetric and gynecological problems in women with untreated celiac sprue. J Clin Gastroenterol. 1990;12:37.
93. Collin P, Vilska S, Heinonen PK, et al. Infertility and coeliac disease. Gut. 1996;39:382.
94. Gasbarrini A, Torre ES, Trivellini C, et al. Recurrent spontaneous abortion and intrauterine fetal growth retardation as symptoms of coeliac disease. Lancet. 2000;356:399.
95. Farthing MJ, Rees LH, Dawson AM. Male gonadal function in coeliac disease: III. Pituitary regulation. Clin Endocrinol (Oxf). 1983;19:661.
96. Aine L, Maki M, Collin P, Keyrilainen O. Dental enamel defects in celiac disease. J Oral Pathol Med. 1990;19:241.
97. Green PH, Cellier C. Celiac disease. N Engl J Med. 2007;357:1731-43.
98. Stern M. Comparative evaluation of serologic tests for celiac disease: A European initiative toward standardization. J Pediatr Gastroenterol Nutr. 2000;31:513.
99. Dieterich W, Laag E, Schopper H, et al. Autoantibodies to tissue transglutaminase as predictors of celiac disease. Gastroenterology. 1998;115:1317.
100. Sulkanen S, Halttunen T, Laurila K, et al. Tissue transglutaminase autoantibody enzyme-linked immunosorbent assay in detecting celiac disease. Gastroenterology. 1998;115:1322.
101. Carroccio A, Vitale G, Di Prima L, et al. Comparison of anti-transglutaminase ELISAs and an anti-endomysial antibody assay in the diagnosis of celiac disease: A prospective study. Clin Chem. 2002;48:1546.
102. Gillett HR, Freeman HJ. Comparison of IgA endomysium antibody and IgA tissue transglutaminase antibody in celiac disease. Can J Gastroenterol. 2000;14:668.
103. Rostami K, Kerckhaert J, Tiemessen R, et al. Sensitivity of antiendomysium and antigliadin antibodies in untreated celiac disease: Disappointing in clinical practice. Am J Gastroenterol. 1999;94:888.
104. Volta U, Molinaro N, de Franceschi L, et al. IgA anti-endomysial antibodies on human umbilical cord tissue for celiac disease screening: Save both money and monkeys. Dig Dis Sci. 1995;40:1902.
105. Ferreira M, Davies SL, Butler M, et al. Endomysial antibody: Is it the best screening test for coeliac disease? Gut. 1992;33:1633.
106. Kapuscinska A, Zalewski T, Chorzelski TP, et al. Disease specificity and dynamics of changes in IgA class anti-endomysial antibodies in celiac disease. J Pediatr Gastroenterol Nutr. 1987;6:529.
107. Kilander AF, Dotevall G, Fallstrom SP, et al. Evaluation of gliadin antibodies for detection of coeliac disease. Scand J Gastroenterol. 1983;18:377.
108. Farrell RJ, Kelly CP. Diagnosis of celiac sprue. Am J Gastroenterol. 2001;96:3237.
109. Corrao G, Corazza GR, Andreani ML, et al. Serological screening of coeliac disease: Choosing the optimal procedure according to various prevalence values. Gut. 1994;35:771.
110. Grodzinsky E, Hed J, Skogh T. IgA antiendomysium antibodies have a high positive predictive value for celiac disease in asymptomatic patients. Allergy. 1994;49:593.
111. Tonutti E, Visentini D, Bizzaro N, et al. French-Italian Laboratory Study Group on Coeliac Disease. The role of antitissue transglutaminase assay for the diagnosis and monitoring of coeliac disease: A French-Italian multicentre study. J Clin Pathol. 2003;56:389.
112. Kumar PJ. European and North American populations should not be screened for celiac disease. Gut. 2002;52:170.
113. Fasano A. European and North American populations should be screened for celiac disease. Gut. 2002;52:168.
Ventura A, Magazzù G, Greco L. Duration of exposure to gluten and risk for autoimmune disorders in patients with celiac disease. SIGEP Study Group for Autoimmune Disorders in Celiac Disease. Gastroenterology. 1999;117:297-303.
114. Fabiani E, Taccari LM, Ratsch IM, et al. Compliance with gluten-free diet in adolescents with screening-detected celiac disease: A 5-year follow-up study. J Pediatr. 2000;136:841.
115. Hin H, Bird G, Fisher P, et al. Celiac disease in primary care: Case finding study. BMJ. 1999;318:164.
116. Setty M, Hormaza L, Guandalini S. Celiac disease: risk assessment, diagnosis, and monitoring. Mol Diagn Ther. 2008;12:289-98.
117. Wolters VM, Wijmenga C. Genetic background of celiac disease and its clinical implications. Am J Gastroenterol. 2008;103:190-5.
118. Abdallah H, Leffler D, Dennis M, Kelly CP. Refractory celiac disease. Curr Gastroenterol Rep. 2007;9:401-5.
119. Achkar E, Carey WD, Petras R, et al. Comparison of suction capsule and endoscopic biopsy of small bowel mucosa. Gastrointest Endosc. 1986;32:278.
120. Jabbari M, Wild G, Goresky CA, et al. Scalloped valvulae conniventes: An endoscopic marker of celiac sprue. Gastroenterology. 1988;95:1518.
121. Shah VH, Rotterdam H, Kotler DP, et al. All that scallops is not celiac disease. Gastrointest Endosc. 2000;51:717.
122. Kaukinen K, Turjanmaa K, Mäki M, et al. Intolerance to cereals is not specific for coeliac disease. Scand J Gastroenterol. 2000;35:942-6.
123. von Krainick HG, Debatin F, Gautier F, et al. Additional research on the injurious effect of wheat flour in coeliac disease: Acute gliadin reactions (gliadin shock). Helv Paediatr Acta. 1958;13:432.
124. Fine KD, Lee EL, Meyer RL. Colonic histopathology in untreated celiac sprue or refractory sprue: Is it lymphocytic colitis or colonic lymphocytosis? Hum Pathol. 1998;29:1433.
125. Walker-Smith J, Harrison M, Kilby A, et al. Cows’ milk-sensitive enteropathy. Arch Dis Child. 1978;53:375.
126. Ament ME, Rubin CE. Soy protein—another cause of the flat intestinal lesion. Gastroenterology. 1972;62:227.
127. Weinstein WM, Saunders DR, Tytgat GN, Rubin CE. Collagenous sprue—an unrecognized type of malabsorption. N Engl J Med. 1970;283:1297.
128. Mulder CJ, Tytgat GN. Coeliac disease and related disorders. Neth J Med. 1987;31:286.
129. Fry L. Dermatitis herpetiformis: Problems, progress and prospects. Eur J Dermatol. 2002;12:523.
130. Holmes GK, Prior P, Lane MR, et al. Malignancy in coeliac disease-effect of a gluten-free diet. Gut. 1989;30:333.
131. Askling J, Linet M, Gridley G, et al. Cancer incidence in a population-based cohort of individuals hospitalized with celiac disease or dermatitis herpetiformis. Gastroenterology. 2002;123:1428.
132. Lewis HM, Renaula TL, Garioch JJ, et al. Protective effect of gluten-free diet against development of lymphoma in dermatitis herpetiformis. Br J Dermatol. 1996;135:363.
133. Smith EP, Zone JJ. Dermatitis herpetiformis and linear IgA bullous dermatosis. Dermatol Clin. 1993;11:511.
134. Rose C, Dieterich W, Brocker EB, et al. Circulating autoantibodies to tissue transglutaminase differentiate patients with dermatitis herpetiformis from those with linear IgA disease. J Am Acad Dermatol. 1999;41:957.
135. Sardy M, Karpati S, Merkl B, et al. Epidermal transglutaminase (TGase3) is the autoantigen of dermatitis herpetiformis. J Exp Med. 2002;195:747.
136. Porter WM, Unsworth DJ, Lock RJ, et al. Tissue transglut-aminase antibodies in dermatitis herpetiformis. Gastroenterology. 1999;117:749.
137. Garioch JJ, Lewis HM, Sargent SA, et al. Twenty-five years’ experience of a gluten-free diet in the treatment of dermatitis herpetiformis. Br J Dermatol. 1994;131:541.
138. Hardman CM, Garioch JJ, Leonard JN, et al. Absence of toxicity of oats in patients with dermatitis herpetiformis. N Engl J Med. 1997;337:1884.
139. Sjoberg K, Eriksson KF, Bredberg A, et al. Screening for coeliac disease in adult insulin-dependent diabetes mellitus. J Intern Med. 1998;243:133.
140. Talal AH, Murray JA, Goeken JA, Sivitz WI. Celiac disease in an adult population with insulin-dependent diabetes mellitus: Use of endomysial antibody testing. Am J Gastroenterol. 1997;92:1280.
141. Cronin CC, Feighery A, Ferriss JB, et al. High prevalence of celiac disease among patients with insulin-dependent (type I) diabetes mellitus. Am J Gastroenterol. 1997;92:2210.
142. Collin P, Reunala T, Pukkala E, et al. Coeliac disease-associated disorders and survival. Gut. 1994;35:1215.
143. Counsell CE, Taha A, Ruddell WS. Coeliac disease and autoimmune thyroid disease. Gut. 1994;35:844.
144. Collin P, Maki M. Associated disorders in coeliac disease: Clinical aspects. Scand J Gastroenterol. 1994;29:769.
145. Freeman HJ. Hepatobiliary tract and pancreatic disorders in celiac disease. Can J Gastroenterol. 1997;11:77.
146. Ventura A, Magazzu G, Greco L. Duration of exposure to gluten and risk for autoimmune disorders in patients with celiac disease. Gastroenterology. 1999;117:297.
147. Sategna Guidetti C, Solerio E, Scaglione N, et al. Duration of gluten exposure in adult coeliac disease does not correlate with the risk for autoimmune disorders. Gut. 2001;49:502.
148. O’Donoghue DJ. Fatal pneumococcal septicaemia in coeliac disease. Postgrad Med J. 1986;62:229.
149. Shah A, Mayberry JF, Williams G, et al. Epidemiological survey of coeliac disease and inflammatory bowel disease in first-degree relatives of coeliac patients. Q J Med. 1990;74:283.
150. DuBois RN, Lazenby AJ, Yardley JH, et al. Lymphocytic enterocolitis in patients with “refractory sprue.”. JAMA. 1989;262:935.
151. Moayyedi P, O’Mahony S, Jackson P, et al. Small intestine in lymphocytic and collagenous colitis: Mucosal morphology, permeability, and secretory immunity to gliadin. J Clin Pathol. 1997;50:527.
152. Wolber R, Owen D, Freeman H. Colonic lymphocytosis in patients with celiac sprue. Hum Pathol. 1990;21:1092.
153. Loft DE, Marsh MN, Crowe PT. Rectal gluten challenge and diagnosis of coeliac disease. Lancet. 1990;335:1293.
154. McCashland TM, Donovan JP, Strobach RS, et al. Collagenous enterocolitis: A manifestation of gluten-sensitive enteropathy. J Clin Gastroenterol. 1992;15:45.
155. Fine KD, Do K, Schulte K, et al. High prevalence of celiac sprue-like HLA-DQ genes and enteropathy in patients with the microscopic colitis syndrome. Am J Gastroenterol. 2000;95:1974.
156. Miletic ID, Miletic VD, Sattely-Miller EA, Schiffman SS. Identification of gliadin presence in pharmaceutical products. J Pediatr Gastroenterol Nutr. 1994;19:27.
157. Dennis M, Case S. Going gluten free: A primer for clinicians. Pract Gastroenterol. 2004;28:86.
158. National Institutes of Health Consensus Development Conference Statement on Celiac Disease, June 28-30, 2004. Gastroenterology. 2005;128:S1-S9.
159. Janatuinen EK, Pikkarainen PH, Kemppainen TA, et al. A comparison of diets with and without oats in adults with celiac disease. N Engl J Med. 1995;333:1033.
160. Janatuinen EK, Kemppainen TA, Julkunen RJ, et al. No harm from five-year ingestion of oats in coeliac disease. Gut. 2002;50:332.
161. Hoffenberg EJ, Haas J, Drescher A, et al. A trial of oats in children with newly diagnosed celiac disease. J Pediatr. 2000;137:361.
162. Thompson T, Dennis M, Higgins LA, et al. Gluten-free diet survey: Are Americans with coeliac disease consuming recommended amounts of fibre, iron, calcium and grain foods? J Hum Nutr Diet. 2005;18:163-9.
163. Pink IJ, Creamer B. Response to a gluten-free diet of patients with the coeliac syndrome. Lancet. 1967;1:300.
164. MacDonald WC, Brandborg LL, Flick AL, et al. Studies of celiac sprue: IV. The response of the whole length of the small bowel to a gluten-free diet. Gastroenterology. 1964;47:573.
165. Valdimarsson T, Lofman O, Toss G, Strom M. Reversal of osteopenia with diet in adult coeliac disease. Gut. 1996;38:322.
166. Sategna-Guidetti C, Grosso SB, Grosso S, et al. The effects of one-year gluten withdrawal on bone mass, bone metabolism, and nutritional status in newly diagnosed adult coeliac disease patients. Aliment Pharmacol Ther. 2000;14:35.
167. Valdimarsson T, Toss G, Lofman O, Strom M. Three years’ follow-up of bone density in adult coeliac disease: Significance of secondary hyperparathyroidism. Scand J Gastroenterol. 2000;35:274.
168. Bernstein CN, Leslie WD, Leboff MS. AGA medical position statement: Guidelines on osteoporosis in gastrointestinal diseases. Gastroenterology. 2003;124:791.
169. Jones PE, Peters TJ. Oral zinc supplements in non-responsive coeliac syndrome: Effect on jejunal morphology, enterocyte production, and brush border disaccharidase activities. Gut. 1981;22:194.
170. Katz AJ, Falchuk ZM, Strober W, Shwachman H. Gluten-sensitive enteropathy: Inhibition by cortisol of the effect of gluten protein in vitro. N Engl J Med. 1976;295:131.
171. Mitchison HC, al Mardini H, Gillespie S, et al. A pilot study of fluticasone propionate in untreated coeliac disease. Gut. 1991;32:260.
172. Lloyd-Still JD, Grand RJ, Khaw KT, Shwachman H. The use of corticosteroids in celiac crisis. J Pediatr. 1972;81:1074.
173. Leffler DA, Dennis M, Hyett B, et al. Etiologies and predictors of diagnosis in nonresponsive celiac disease. Clin Gastroenterol Hepatol. 2007;5:445-50.
174. Abdulkarim AS, Burgart LJ, See J, Murray JA. Etiology of nonresponsive celiac disease: Results of a systematic approach. Am J Gastroenterol. 2002;97:2016.
175. Ciacci C, Mazzacca G. Unintentional gluten ingestion in celiac patients. Gastroenterology. 1998;115:243.
176. Baer AN, Bayless TM, Yardley JH. Intestinal ulceration and malabsorption syndromes. Gastroenterology. 1980;79:754.
177. Enns R, Lay T, Bridges R. Use of azathioprine for nongranulomatous ulcerative jejunoileitis. Can J Gastroenterol. 1997;11:503.
178. Bossart R, Henry K, Booth CC, Doe WF. Subepithelial collagen in intestinal malabsorption. Gut. 1975;16:18.
179. Freeman HJ. Hyposplenism, antiendomysial antibodies, and lymphocytic colitis in collagenous sprue. Can J Gastroenterol. 1999;13:347.
180. Guller R, Anabitarte M, Mayer M. Collagenous sprue and ulcerative jejuno-ileitis in a patient with gluten-induced enteropathy. Schweiz Med Wochenschr. 1986;116:1343.
181. Holtmann M, von Herbay A, Galle PR, Stremmel W. Long-term collagenous sprue: Remission with a gluten-free diet. Z Gastroenterol. 1999;37:1163.
182. Vaidya A, Bolanos J, Berkelhammer C. Azathioprine in refractory sprue. Am J Gastroenterol. 1999;94:1967.
183. Longstreth GF. Successful treatment of refractory sprue with cyclosporine. Ann Intern Med. 1993;119:1014.
184. Gillett HR, Arnott ID, McIntyre M, et al. Successful infliximab treatment for steroid-refractory celiac disease: A case report. Gastroenterology. 2002;122:800.
185. Ryan BM, Kelleher D. Refractory celiac disease. Gastroenterology. 2000;119:243.
186. Wahab PJ, Crusius JB, Meijer JW, et al. Cyclosporin in the treatment of adults with refractory coeliac disease: An open pilot study. Aliment Pharmacol Ther. 2000;14:767.
187. Brar P, Lee S, Lewis S, et al. Budesonide in the treatment of refractory celiac disease. Am J Gastroenterol. 2007;102(10):2265-9.
188. Corazza GR, Biagi F, Volta U, et al. Autoimmune enteropathy and villous atrophy in adults. Lancet. 1997;350:106.
189. Akram S, Murray JA, Pardi DS, et al. Adult autoimmune enteropathy: Mayo Clinic Rochester experience. Clin Gastroenterol Hepatol. 2007;5:1282-90.
190. Spencer J, Cerf-Bensussan N, Jarry A, et al. Enteropathy-associated T cell lymphoma (malignant histiocytosis of the intestine) is recognized by a monoclonal antibody (HML-1) that defines a membrane molecule on human mucosal lymphocytes. Am J Pathol. 1988;132:1.
191. Murray A, Cuevas EC, Jones DB, Wright DH. Study of the immunohistochemistry and T cell clonality of enteropathy-associated T cell lymphoma. Am J Pathol. 1995;146:509.
192. Ashton-Key M, Diss TC, Pan L, et al. Molecular analysis of T-cell clonality in ulcerative jejunitis and enteropathy-associated T-cell lymphoma. Am J Pathol. 1997;151:493.
193. Bagdi E, Diss TC, Munson P, Isaacson PG. Mucosal intra-epithelial lymphocytes in enteropathy-associated T-cell lymphoma, ulcerative jejunitis, and refractory celiac disease constitute a neoplastic population. Blood. 1999;94:260.
194. de Bruin PC, Connolly CE, Oudejans JJ, et al. Enteropathy-associated T-cell lymphomas have a cytotoxic T-cell phenotype. Histopathology. 1997;31:313.
195. Corrao G, Corazza GR, Bagnardi V, et al. Mortality in patients with coeliac disease and their relatives: A cohort study. Lancet. 2001;358:356.
196. Catassi C, Fabiani E, Corrao G, et al. Risk of non-Hodgkin lymphoma in celiac disease. JAMA. 2002;287:1413.
197. Cerf-Bensussan N, Cellier C, Heyman M, et al. Coeliac disease: An update on facts and questions based on the 10th International Symposium on Coeliac Disease. J Pediatr Gastroenterol Nutr. 2003;37:412.
198. Freeman HJ, Chiu BK. Small bowel malignant lymphoma complicating celiac sprue and the mesenteric lymph node cavitation syndrome. Gastroenterology. 1986;90:2008.
199. Matuchansky C, Colin R, Hemet J, et al. Cavitation of mesenteric lymph nodes, splenic atrophy, and a flat small intestinal mucosa: Report of six cases. Gastroenterology. 1984;87:606.
200. Egan LJ, Walsh SV, Stevens FM, et al. Celiac-associated lymphoma: A single-institution experience of 30 cases in the combination chemotherapy era. J Clin Gastroenterol. 1995;21:123.
201. Logan RF, Rifkind EA, Turner ID, Ferguson A. Mortality in celiac disease. Gastroenterology. 1989;97:265.
202. Nielsen OH, Jacobsen O, Pedersen ER, et al. Non-tropical sprue: Malignant diseases and mortality rate. Scand J Gastroenterol. 1985;20:13.
203. Schmitz J. Is celiac disease a lifelong disorder? Clin Invest Med. 1996;19:352.
204. Shan L, Molberg O, Parrot I, et al. Structural basis for gluten intolerance in celiac sprue. Science. 2002;297:2218.
205. Gass J, Bethune MT, Siegel M, et al. Combination enzyme therapy for gastric digestion of dietary gluten in patients with celiac sprue. Gastroenterol. 2007;133:472-80.
206. Paterson BM, Lammers KM, Arrieta MC, et al. The safety, tolerance, pharmacokinetic and pharmacodynamic effects of single doses of AT-1001 in coeliac disease subjects: A proof of concept study. Aliment Pharmacol Ther. 2007;26(5):757-66.
207. Maurano F, Siciliano RA, De Giulio B, et al. Intranasal administration of one α-gliadin can downregulate the immune response to whole gliadin in mice. Scand J Immunol. 2001;53:290.
208. Esposito C, Caputo I, Troncone R. New therapeutic strategies for coeliac disease: Tissue transglutaminase as a target. Curr Med Chem. 2007;14:2572-80.
209. Baldassarre M, Laneve AM, Grosso R, Laforgia N. Celiac disease: Pathogenesis and novel therapeutic strategies. Endocr Metab Immune Disord Drug Targets. 2008;8:152-8.
210. Troncone R, Ivarsson A, Szajewska H, Mearin ML. Review article: Future research on coeliac disease—a position report from the European multistakeholder platform on coeliac disease (CDEUSSA). Aliment Pharmacol Ther. 2008;27:1030-43.