CaV1.2 and β-Adrenergic Regulation of Cardiac Function
Overview of the β-Adrenergic Regulation of L-Type Calcium Channels
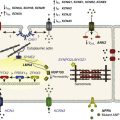
In cardiac cells, the L-type calcium channel (LTCC) current also called ICa,L underlies the plateau phase of the action potential (AP). Upon depolarization, ICa,L reflects calcium influx via the CaV1.2 channels. This current initiates cardiac contraction by gating the ryanodine receptor, thereby triggering the calcium release from the sarcoplasmic reticulum.1 Among several regulatory pathways of this current, the best described is the β-adrenergic stimulation, which contributes to the positive inotropic effects of catecholamines. To date, three β-adrenergic receptors (β-ARs), respectively β1-AR, β2-AR, and β3-AR, have been cloned,2 and this major achievement has led to the 2012 Nobel prize award to Robert Lefkowitz and Brian Kobilka for paving the road to the current understanding of their structures and functions. The classical pathway for β-AR receptor signaling is activation of adenylyl cyclases via Gαs, resulting in increased intracellular cyclic adenosine monophosphate (cAMP) levels. The primary target of cAMP is the cAMP-dependent protein kinase (PKA) that in turn phosphorylates the CaV1.2 channels among other key proteins of the excitation-contraction coupling (Figure 37-1). Although direct modulation by G proteins of LTCCs was first suggested to partially mediate the upregulation of ICa,L upon β-AR activation, it has been clearly established that a cAMP phosphorylation mediated by PKA is responsible for this increase.3,4
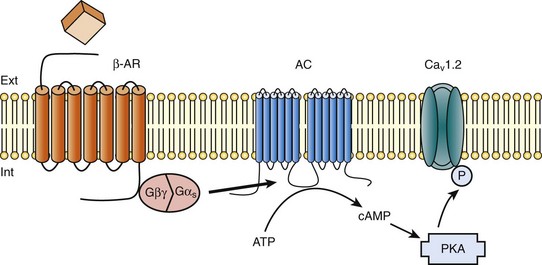
Figure 37-1 β-Adrenergic modulation of cardiac L-type calcium channels. Modulation of cardiac CaV1.2 channels by β-adrenergic receptor (β-AR) stimulation occurs via a G protein pathway. β-ARs couple to heterodimeric G proteins. Agonist catecholamine binding to β-AR activates the stimulatory heterotrimeric G protein by inducing the exchange of guanosine diphosphate (GDP) for guanosine-5′-triphosphate (GTP) on GαS. GαS dissociates from its Gβγ partner to stimulate the adenylyl cyclase (AC) present in the plasma membrane. The AC catalyses the conversion of adenosine triphosphate (ATP) in cyclic 3′,5′ monophosphate (cAMP) that in turn activates the cAMP dependent protein kinase (PKA) to phosphorylate CaV1.2.
This chapter reviews the literature on the β-AR regulation of LTCCs, with emphasis on recent information on the molecular mechanisms of PKA regulation of CaV1.2 channels and the local compartmentation of β-AR/cAMP/PKA signaling around these channels. We conclude with an overview of such modulation in a pathologic context. Additional details concerning LTCCs can be found in Chapter 2.
Molecular Mechanisms of Protein Kinase Regulation of L-Type Calcium Channels
LTCCs display three distinct gating modes upon depolarization: mode 1 corresponds to brief openings, mode 2 corresponds to long lasting openings, and mode 0 corresponds to a silent mode because of unavailability.5 PKA phosphorylation of CaV1.2 channels results in a shift of the channel from the gating mode 1 to mode 2.6 As a result, ICa,L density is increased twofold to threefold, and its voltage dependence shifted slightly toward hyperpolarized potentials (Figure 37-2, A). Voltage steady-state activation and inactivation in adult mouse ventricular myocytes are presented in Figure 37-2. Activation for ICa,L starts at –40 mV and is maximal around 5 mV while inactivation begins at –45 mV and is maximal at approximately 0 mV. The overlap of the two curves defines a “window current” between –40 and 0 mV (i.e., near the action potential plateau phase). The β-adrenergic stimulation leads to an increased window current because of the effect of PKA phosphorylation on channel activation. As presented in Figure 37-2, B, isoproterenol application at a maximal concentration of 100 nM shifts the activation by 5 mV toward negative potentials, whereas a minor shift of availability of approximately 2.5 mV is observed. During maintained depolarizations, ICa,L decreases with time, a phenomenon named inactivation, which depends on time, voltage, and intracellular calcium.7 Because β-AR activation enhances calcium entry, it also accelerates ICa,L inactivation,8 and calcium-dependent inactivation becomes the main mechanism by which the channel inactivates.9 Overall, β-AR stimulation promotes ICa,L and thus the calcium entry during action potential that is partially responsible for its positive inotropic effects.
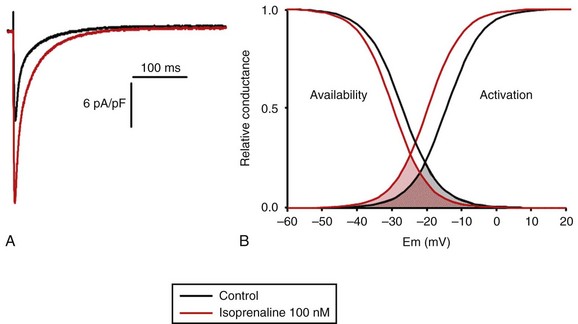
Figure 37-2 The effect of β-adrenergic stimulation on ICa,L in adult mice ventricular cardiomyocytes. A, Example of current traces of ICa,L recorded using the patch-clamp technique under whole-cell conditions at 0 mV (holding potential at –50 mV) obtained before and after activation of the β-adrenergic receptors with 100 nM isoproterenol. An approximately twofold increase of ICa,L amplitude is induced by the application of the agonist. B, Influence of β-adrenergic stimulation on cardiac calcium channel activation and availability: ICa,L availability and steady-state inactivation were measured by applying a 200-ms pulse from –60 to 60 mV, followed by a 3-ms repolarization to –50 mV, before a 200-ms test pulse to 0 mV. The present curves are mean data obtained from 16 different adult mice ventricular myocytes before and after application of 100 nM isoproterenol. Steady-state inactivation curves were obtained by normalizing the peak current at each test potential to the maximal current. Activation curves were derived from current–voltage relations and described by: G / Gmax = 1 / (1+exp[{V½,act − V} / k]). A and B were obtained in similar recording conditions as described by Leroy et al.91
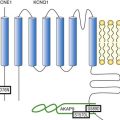
The LTCC current ICa,L in the working myocardium is mediated by the predominantly expressed CaV1.2 channels. These channels are multimeric proteins composed of a central subunit, α1C, the pore-forming subunit, which determines the main biophysical and pharmacologic properties of the channel. This subunit is a protein of approximately 240 kDa with 24 transmembrane segments according to its hydropathic profile, organized into four repeated domains (I to IV) of six transmembrane segments each (1 to 6), with intracellular N- and a large C-termini.4 Like other high-voltage–gated calcium channels, CaV1.2 associates with a largely extracellular disulfide-linked α2-δ subunit of 170 kDa.10 It also binds CaVβ subunits to its α interaction domain present in its intracellular I-II loop via their guanylate kinase–like domain.11 Four genes encode four CaVβ subunits (β1-4), but CaVβ2 is thought to be the main isoform expressed in the heart.12 Both α2-δ13, and CaVβ14 auxiliary subunits influence the biophysical properties and increase the trafficking of the channel at the plasma membrane. γ(4,6-8) subunits are also expressed in ventricular cells and, like γ1 for the skeletal muscle calcium channel CaV1.1, can interact with the CaV1.2 channel to modulate its function when coexpressed in HEK-293 cells; however, the exact role for these accessory proteins on CaV1.2 in native cardiac tissues remains to be determined.15
α-1 Subunit
If the main effects of β-AR stimulation on voltage dependence and amplitude of the ICa,L and their consequences for the fight-or-flight response are well documented in the literature, the molecular events that mediate the increase of CaV1.2 activity during a sympathetic stimulation remain elusive; this is due to the difficulty of reconstituting such modulation in heterologous overexpression systems. The α1C-subunit of CaV1.2 channels exhibits multiple potential PKA phosphorylation sites in the N- and the C-terminal regions (Figure 37-3).2 Despite the fact that α1C is a substrate for phosphorylation by PKA in vitro,16,17 several attempts to mimic the adrenergic stimulation of CaV1.2 channels in expression systems have failed.18–20 This failure led to the hypothesis that at least one missing link in heterologous systems would preclude the reconstitution of such modulation of overexpressed CaV1.2 channels. One of these links could be an A-kinase-anchoring protein (AKAP) that allows the cAMP modulation and the PKA phosphorylation of serine 1928 in the C-terminus of CaV1.2 channels in HEK293 cells.21 A conserved leucine zipper motif in the C-terminus of CaV1.2 identified in native cardiac cells directly anchors a low molecular weight AKAP15-PKA complex to ensure a fast and efficient β-adrenergic modulation of ICa,L (see Figure 37-3),22 allowing its phosphorylation at serine 1928 when β-ARs are stimulated.23 Surprisingly, the C-terminal part of rabbit CaV1.2 undergoes proteolytic processing by calpain at residue 1821, leaving two size forms of the α1C-subunit of these channels expressed in cardiac cells,17 whereas this distal 37 to 50-kDa peptide contains the serine 1928 phosphorylated by PKA and the binding site for AKAP15. In fact, this fragment constitutes a potent autoinhibitory domain that covalently associates with the proximal C-terminal part of α1C, reducing its open probability and shifting the voltage dependence of activation to depolarized potentials.24 The role of serine 1928 in ICa,L upregulation by β-AR has since been challenged. A first study showed that a DHP-resistant CaV1.2 channels mutated at position 1928 (S1928A) displays an unaltered response to the β-AR agonist isoproterenol when overexpressed in isolated ventricular myocytes.25 Moreover, the generation of a S1928A knock-in mouse model confirmed that the phosphorylation of this residue by PKA is not required for the functional effects of β-AR stimulation on CaV1.2 in cardiac cells, because these mice exhibit an essentially conserved response of ICa,L to isoproterenol and typical chronotropic and inotropic responses to β-AR stimulation.26 Therefore, although phosphorylation of serine 1928 within the C-terminus of CaV1.2 definitely occurs when β-ARs are stimulated,17,21,23 it does not correlate with the functional effects observed on ICa,L. As a result, PKA would phosphorylate other PKA sites within the channel or another binding partner to mediate the increase of its activity.
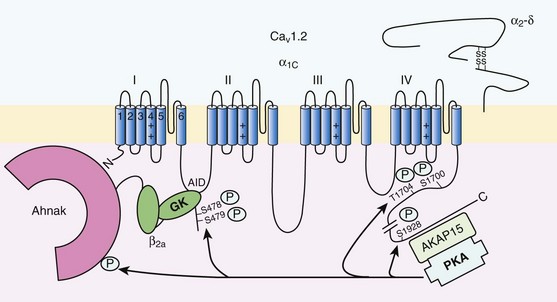
Figure 37-3 Subunit structure of cardiac L-type CaV1.2 channel and phosphorylation sites for PKA mediated upregulation of ICa,L. The α1C-subunit constitutes the pore-forming subunit of the cardiac CaV1.2 channel. It presents four domains (I to IV) composed of six transmembrane segments (S1 to S6), with S4 exhibiting positively charged residues that confer their sensitivity to potential and a pore loop between S5 and S6 of each domains. The intracellular I-II loop of the α1C-subunit associates with the β interaction domain (BID) in the guanylate kinase–like domain of the intracellular CaVβ2a auxiliary subunit via its α interaction domain (AID). CaV1.2 associates also with the largely extracellular glycosylphosphatidylinositol (GPI)-anchored α2-δ subunit. The C-terminal part of the α1C undergoes proteolytic cleavage by calpain at residue 1821. The cleaved distal fragment associates monocovalently with the proximal C-terminal part to inhibit the channel. This necessary association for β-AR regulation of CaV1.2 is relieved by the phosphorylation of serine 1700 by the PKA tethered in the distal C-terminus by AKAP15 to produce the upregulation of channel activity. PKA also phosphorylates serine 1704 to control its basal activity while phosphorylation of serine 1928 serves an undetermined regulatory function. PKA also phosphorylates CaVβ2a on the two serine residues 478 and 479. This subunit associates to the giant ahnak cytosqueletal protein that could also be an important factor for the cAMP/PKA regulation of CaV1.2 channels.
β-Subunit
Three serines (S459, S478, and S479) of the cardiac the CaVβ2a subunit were identified as putative PKA phosphorylation sites.27 Coexpression of this subunit with a CaV1.2 lacking the C-terminal part including the serine 1928 in TsA-201 cells allowed an upregulation of the current by cAMP/PKA pathway, whereas expression of mutated CaVβ2a at serine 478 and 479, but not at position 459, was unable to do so.28 The authors concluded that the CaVβ2a ancillary subunit was the main target for PKA to mediate the β–AR stimulation of ICa,L; however, this conclusion has been questioned by experiments realized in a more physiologic context. Overexpression of a CaVβ2a construct mutated for its PKA phosphorylation sites in ventricular cardiac cells did not prevent the cAMP/PKA modulation of ICa,L.14 Nonetheless, the CaVβ2 subunit can associate with the giant cytoskeletal protein of 700 kDa named ahnak, which emerged as an important player in the β-AR regulation of cardiac ICa,L (see Figure 37-3). Through its interaction with CaVβ2, ahnak would serve as a brake on ICa,L that would be relieved by PKA phosphorylation of the ancillary subunit and the cytoskeletal protein.29 Interestingly, ahnak polymorphism occurs, and the genetic variant generated interferes with the β-AR stimulation of ICa,L by reducing the CaVβ2 interaction with ahnak.29 However, if the involvement of the CaVβ or other binding partners such as ahnak cannot be ruled out completely, recent studies have reaffirmed the importance of the proteolytically cleaved distal C-terminus of CaV1.2 for its upregulation upon β-AR stimulation. In addition, a new model for the molecular basis of β-AR stimulation of CaV1.2 has been proposed.30 Overexpression in TsA-201 cells of a truncated CaV1.2 channel at position A1800 (the site of in vivo proteolytic cleavage previously determined for skeletal muscle CaV1.1 channel31) with α2δ1 and CaVβ1b and the distal C-terminal part of the channel (peptide from 1821-2171) produced a functional channel that is autoinhibited. Two presumed sites for PKA phosphorylation were identified upstream from the proteolytic site—a serine at position 1700 and a threonine 1704 at the interface of the distal and the proximal C-terminal parts. Although a mutation of S1928 confirmed that its phosphorylation is not required for the increase of the current, substitution of T1704 and S1700 for alanines revealed that phosphorylation of T1704 is required for basal CaV1.2 channel activity, whereas S1700 is crucial for its cAMP/PKA-induced upregulation. In this scheme, the AKAP15 is still required to coordinate the PKA phosphorylation of S1700 that disrupts the interaction of the noncovalently distal C-terminus, thus relieving the inhibition of the channel.30 This assumption is reinforced by the fact that mice expressing a CaV1.2 channel deleted for the distal C-terminus display altered cAMP/PKA regulation accompanied with reduced expression of AKAP15.32
Compartmentation of cAMP/PKA Regulation of L-Type Calcium Channels
In light of the knowledge accumulated over the years, it is evident that intracellular cAMP is not uniformly distributed, and nor is PKA uniformly activated within cardiomyocytes upon β-AR stimulation.33 On the contrary, a tight cAMP/PKA compartmentation is required for adequate processing and targeting of the information generated at the cell surface, conferring the specificity of the response to various hormones linked to Gαs-coupled receptors.34,35 In the case of β-ARs, several processes contribute to a localized cAMP/PKA response: catecholamines activate different β-AR subtypes located at different places of the cell surface (e.g., caveolae, T-tubules); Gαs activation of different adenylyl cyclase (AC) isoforms can lead to cAMP synthesis at different locations; cAMP diffusion may be restricted because of localized phosphodiesterase (PDE) activity; anchoring of PKA to AKAPs position PKA at different subcellular compartments to selectively phosphorylate a local pool of proteins for specific cellular processes.36,37 In addition, AKAPs also ensure that PKA is coupled to its upstream activators, including membrane β-ARs and ACs, and to signal termination enzymes such as PDEs and phosphatases.36
Receptor Specificity
Although only three types of β-adrenergic receptors (β-ARs) have been cloned (β1-, β2-, and β3-ARs), the effect of catecholamines in the human heart is generally attributed to β1– and β2-ARs. β1– and β2-ARs are highly homologous receptors and are both positively coupled to AC/cAMP/PKA cascade, ICa,L and cardiac performance33; however, they exert opposite effects on hypertrophy38,39 and apoptosis,40,41 and their respective contribution varies significantly depending on the cardiac tissue, pathophysiologic state, age, or developmental stage.42 Part of the difference is because β2-ARs couple not only to Gαs but also to Gαi proteins, and this confines the cAMP-dependent signal to the membrane compartment and to activation of the LTCCs.43 Another difference between the two β-AR subtypes is their location at the cell surface, with β2-ARs present in the caveolae/lipid rafts44,45 of the transverse-tubular structure46 and β1-ARs distributed throughout both caveolae–lipid rafts and nonlipid raft membrane domains44 and in both plasma and T-tubular membranes.46 Accordingly, the β2-AR downstream activation of ICa,L is sensitive to disruption of caveolae by cholesterol depletion, whereas the β1-AR stimulatory effect is not.47 Therefore, β2-AR stimulation exerts a local activation of LTCCs, whereas β1-AR stimulation leads to activation of LTCCs in the distance.43,48,49
The β3-AR differs from β1-and β2-AR subtypes in its molecular structure and pharmacologic functions.50 Expression of β3-AR was demonstrated in human myocardium at the mRNA51,52 and protein levels.52–55 Interestingly, β3-AR activation produces a negative inotropic effect in human endomyocardial biopsies from transplanted hearts50,51 and in left ventricular samples from failing and nonfailing explanted hearts50,54 that is due to activation of the NO/cGMP pathway, but increases ICa,L and contractility in human atrial tissue via the cAMP/PKA pathway (Figure 37-4).56 This effect is reminiscent of the contractile effects of the serotonin 5-HT4 receptors,57 which are also coupled to increases in force of contraction58 or ICa,L59 in atria but not in healthy ventricles.
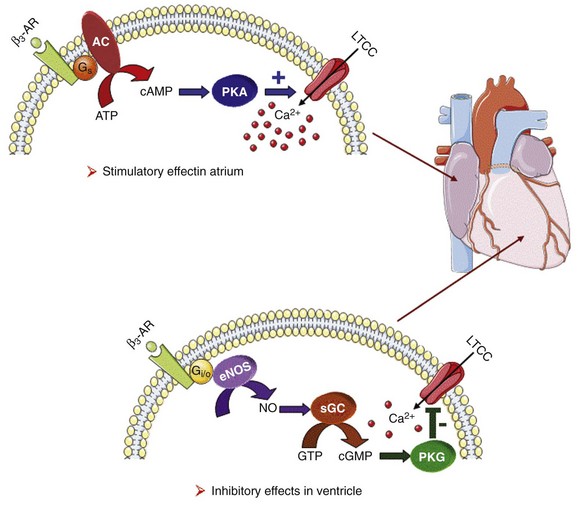
Figure 37-4 Opposite effect of β3-adrenergic receptor stimulation on L-type calcium channel (LTCC) activity in a human atrium and ventricle. In the atrium (top), the β3-adrenergic receptor (β3-AR) is positively coupled to the Gs-protein and to adenylyl cyclase (AC). Upon activation of the β3-AR, cAMP synthesis is increased, which leads to PKA phosphorylation of LTCCs and a stimulation of Ca2+ influx. In the human ventricle (bottom), the β3-AR is coupled to the endothelial nitric oxide synthase (eNOS), presumably via the Gi/o-protein. Activation of eNOS leads to NO production, which activates soluble guanylyl cyclase (sGC) and increases intracellular cGMP concentration. This leads to PKG-phosphorylation of LTCCs and an inhibition of Ca2+ influx.
Role of A-Kinase-Anchoring Proteins
Cyclic AMP signaling components are organized into multiprotein complexes, an arrangement that increases both efficiency and specificity of the transduction cascade. AKAPs have an essential role in these arrangements. AKAPs form a large family of proteins comprising more than 50 members whose primary function is to anchor PKA in the vicinity of its substrates, thus ensuring the preferential phosphorylation of a limited number of targets.60,61 As discussed earlier, AKAP15 (also called AKAP 7 or AKAP15/18) is the main AKAP controlling the PKA phosphorylation of LTCCs. However, in a recent study, CaV1.2 phosphorylation and β-AR stimulation of ICa,L was found to be unchanged in mice in which the gene encoding this protein was inactivated, suggesting that PKA is anchored by a different protein to the channel.62 Furthermore, AKAP5 was found to target ACs, PKA and phosphatases within caveolae to allow specific PKA phosphorylation of the subpopulation of channels present in this compartment upon β-AR stimulation.63 Although AKAPs share in common their ability to bind PKA, they are remarkably diverse scaffold proteins. Within each signalosome, AKAPs couple PKA to different substrates, enhancing the rate and fidelity of their phosphorylation by the kinase. Importantly, AKAPs not only bind PKA but act as scaffold proteins for other signaling components, including phosphatases 1 and 2,64,65 Epac,66 adenylyl cyclases,67,68 and PDEs.69 Recently, it has been demonstrated that the phosphoinositide 3-kinase p110γ, that was shown recently tether PKA,70 orchestrates multiprotein complexes including different PDEs to control cardiac CaV1.2 phosphorylation during β-AR stimulation.71 The combination of PDEs and phosphatases present in individual AKAP complexes will affect the duration, amplitude, and spatial extent of cAMP/PKA signaling. Thus, by bringing together different combinations of upstream and downstream signaling molecules, AKAPs provide the architectural infrastructure for specialization of the cAMP signaling network.61,66,72
Role of Phosphodiesterases
Localized cAMP signals can be generated by the interplay between discrete cAMP production sites and restricted diffusion within the cytoplasm. Restricted diffusion of cAMP can be achieved by several means. A first possibility is that physical barriers are created by specialized membrane structures within the cytoplasm. This method was initially proposed to explain the differences in cAMP concentration elicited by PGE1 at the plasma membrane and in the bulk cytosol of HEK293 cells, although an experimental proof that this actually occurs is still lacking.73 Another important mechanism that limits cAMP diffusion is cAMP degradation by PDEs, which appears to be critical to the formation of dynamic microdomains that confer specificity of the response.35,60
Cardiac cAMP PDEs degrading belong to five families (PDE1 to PDE4 and PDE8) that can be distinguished by distinct enzymatic properties and pharmacology.74 Among these families various enzymes were shown to degrade cAMP to allow a fine tuning of ICa,L regulation by PKA in the heart. Although PDE2 is not highly expressed in cardiomyocytes, it controls LTCC activity in various species, including human atrial myocytes.75–78 This enzyme is activated by cGMP, and stimulation of guanylyl cyclase strongly decreases local cAMP levels controlling ICa,L with only modest effects on its global concentration, suggesting the existence of a cAMP microdomain including β-AR and LTCCs under tight control of PDE2.79 Contrary to PDE2, PDE3 is inhibited by cGMP; this explains in part why cGMP at low concentration can also increase basal ICa,L as shown in human atrial myocytes.78,80 In rodents, PDE3 and PDE4 are the major contributors to the total cAMP-hydrolytic activity34,81 and PDE4 is dominant to modulate β-AR regulation of cAMP levels.49,82–84 Multiple PDE4 variants associate with β-ARs,85–87 RyR2,88 SERCA2,89,90 ICa,L,91 and IKs72 to exert local control of ECC. In larger mammals, PDE3 activity is dominant in microsomal fractions,92–94 and PDE3 inhibitors exert a potent positive inotropic effect.95 Selective inhibition of PDE3 with milrinone has been shown to improve cardiac contractility in patients with congestive heart failure.96 The role of PDE4 is less well defined, but evidence is emerging that PDE4 could also have an important role in these species. In the canine heart, a large PDE4 activity is found in the cytoplasm,92 but PDE4 is also present in microsomal fractions, where it accounts for approximately 20% of the activity.93 Recent studies have indicated that PDE4 is expressed in the human ventricle where, similar to rodents, it associates with β-ARs, RyR2, and phospholamban.81,88 Moreover, PDE4 is the main PDE modulating LTCC activity in rodent cardiomyocytes,77,84 and PDE4 was recently shown to control ICa,L, ECC and arrhythmias in the human atrium.97
Early evidence of the contribution of PDEs to intracellular cyclic nucleotide compartmentation was obtained by comparing the effects of the nonselective β-AR agonist Iso, or the nonselective PDE inhibitor, 3-isobutyl-1-methylxanthine (IBMX), or the PDE3 inhibitor milrinone in guinea pig perfused hearts. Whereas each of these treatments increased intracellular cAMP and produced positive inotropic and lusitropic effects, differences in the phosphorylation pattern of PLB, TnI, and MyBP-C by PKA were observed.98 These results were attributed to a functional cellular compartmentation of cAMP and PKA substrates owing to a different expression of PDEs at the membrane and in the cytosol.92 In canine ventricular myocytes, an increase in particulate but not total cAMP correlated to an increase in Ca2+ transient amplitude and decay kinetics.99 In response to β-AR stimulation, approximately 45% of the total cAMP was found in the particulate fraction, but this fraction declined to less than 20% when IBMX was added to Iso, although cAMP production was up to threefold to fourfold greater. These results show that cAMP-PDEs reside predominantly in the cytoplasm, where they prevent excessive cAMP accumulation upon β-AR activation. Thus, PDEs appear to be important to maintain the specificity of the β-AR response by limiting the amount of cAMP diffusing from membrane to cytoplasm. Similar results were obtained when studying the effect of PDE inhibition on ICa,L regulation by local application of Iso in frog ventricular myocytes.100 In the absence of IBMX, the application of Iso to half of the cell increased ICa,L half maximally, corresponding to activation of the channels located in the same part of the cell as the β-AR agonist. When IBMX was added with Iso on half of the cell, the effect of Iso was greatly potentiated because in this condition, LTCCs in the remote part of the cells could be recruited. These results suggest that PDE activity is important for the definition of local cAMP pools involved in the β-AR stimulation of LTCCs. Subsequent studies using ratiometric FRET biosensors to monitor cAMP directly have shown that the second messenger increases preferentially in discrete microdomains corresponding to the dyad region under β-AR stimulation, and that cAMP diffusion is limited by PDE activity.101,102 Other studies using recombinant cyclic nucleotide gated channels to measure cAMP generated at the plasma membrane identified specific functional coupling of individual PDE families, mainly PDE3 and PDE4, to β1-AR, β2-AR, PGE1-R, and Glu-R as a major mechanism enabling cardiac cells to generate heterogeneous cAMP signals in response to different hormones.84
Although PDE4 regulates ICa,L in cardiomyocytes,77,84 the molecular identity of the PDE4 regulating the LTCC was unveiled only recently.91 In mouse cardiomyocytes, PDE4B and PDE4D, but not PDE4A, were found to be part of a CaV1.2 signaling complex. However, in mice deficient for the Pde4d gene (Pde4d–/–), basal or β-AR stimulated ICa,L were not different from wild-type mice, whereas in Pde4b–/– mice the β-AR response of ICa,L was increased, together with an increase in cell contraction and Ca2+ transients.91 Upon β-AR stimulation in vivo, catheter-mediated burst pacing triggers ventricular tachycardia in Pde4b–/– mice but not in wild type.91 Thus, PDE4B is the main PDE4 isoform regulating cardiac LTCC activity and has a key role during β-AR stimulation of cardiac function. PDE4B, by limiting the amount of Ca2+ that enters the cell via the LTCCs, prevents Ca2+ overload and arrhythmias.
Role of Phosphatases
Formation of inside-out patches from rabbit ventricular myocytes causes run-down of LTCC activity that is blocked by okadaic acid, a serine/threonine phosphatase inhibitor.103 This observation provided early functional evidence that a phosphatase is anchored in close proximity to the channel that counteracts upregulation of CaV1.2 by phosphorylation. Later, two major cardiac serine–threonine phosphatases,104 phosphatase 2A (PP2A) and 2B (PP2B or calcineurin), were found to associate with cardiac CaV1.2.105,106 For PP2A, two attachment sites were identified within the C-terminus of the α1-subunit:107,108 one region spans residues 1795 to 1818 and the other spans residues 1965 to 1971. PP2B binds immediately downstream of residues 1965 to 1971 without competition between these two phosphatases for binding to this rather narrow region.108 Injection of a peptide that contains residues 1965 to 1971 and displaces PP2A but not PP2B from endogenous CaV1.2 increases basal and β-AR stimulated cardiac ICa,L.108 Similarly, inhibition of PP2B with cyclosporin A or the calcineurin autoinhibitory peptide increases cardiac ICa,L.109 This indicates that anchoring of PP2A and PP2B on cardiac Cav1.2 negatively regulates LTCC activity, most likely by counterbalancing basal and stimulated phosphorylation that is mediated by PKA and possibly other kinases.
β-Adrenergic Regulation of L-Type Calcium Channels in Pathologic Situations
CaV1.2 Involvement in Early and Delayed Afterdepolarizations
According to the Coumel’s triangle, the production of a clinical arrhythmia requires three ingredients: an arrhythmogenic substrate, a trigger factor, and modulation factors of which the most common is the autonomic nervous system.110 In pathologic conditions such as atrial fibrillation (AF), cardiac hypertrophy or heart failure (HF), these factors are united to trigger fatal cardiac arrhythmias. These pathologies are accompanied with structural changes, electrophysiological remodeling, and abnormal β-AR activation at tissue and cellular levels promoting aberrant electrical activities and modulation. Modified excitation-contraction coupling owing to altered calcium homeostasis in pathologic conditions is a major cause of arrhythmias.111 As described in the previous paragraph, a fine tuning of the LTCC (i.e., of calcium cycling) and its regulation in discrete subcellular compartments is required to achieve a normal cardiomyocyte function. In physiopathologic conditions, deregulation of this coupling leads to calcium waves and calcium alternans to promote reentrant arrhythmias and triggered activities.111,112 At the level of the cardiomyocyte in AF113 or in HF,114 calcium handling is perturbed, leading to afterdepolarizations. When action potential duration (APD) is excessively prolonged, early afterdepolarization (EAD) can occur during the repolarization phase, whereas arrhythmogenic delayed afterdepolarization (DAD) can occur when the cell is fully returned to its resting potential. Non-reentrant mechanisms involve triggered activities from either EADs or DADs. When APD is prolonged, owing to alteration of Na+ or K+ conductances (as in long QT syndromes or upon antiarrhythmic treatments), ICa,L recovery from inactivation occurs during the plateau phase of the AP which causes EADs.115 This is particularly true when ICa,L window current is increased upon β-AR activation, thus increasing calcium entry during the plateau phase and allowing its reactivation.115,116 EADs are also promoted by increased calmodulin-dependent protein kinase II (CaMKII) activity in hypertrophy and HF. CaMKII is activated by increased intracellular calcium levels upon β-AR activation via PKA-dependent and independent mechanisms.117,118 CaMKII in turn phosphorylates CaV1.2 on its CaVβ2a subunit, triggering afterdepolarizations.119,120 However, intracellular calcium overload, by activating the reverse mode of the Na+/Ca2+ exchange, generates a depolarizing current during the late AP phase 2 and phase 3, which is believed to act synergistically with ICa,L to induce EADs.121,122 Similarly, DADs occur at high intracellular calcium load, and their incidence is increased by both rapid pacing and β-AR stimulation.114,123 The main triggering mechanism for DADs is spontaneous calcium release activating a transient inward current (ITI) because of excess diastolic calcium handled by sarcolemmal Na+/Ca2+ exchange. Large enough DADs can eventually sufficiently depolarize the membrane potential to reach threshold and trigger an AP. Again, increased LTCC activity contributes to DAD occurrence and Ca2+-evoked arrhythmias. For example, mutations in the LTCC have been associated with a number of inherited arrhythmia syndromes.124 Increased LTCC owing to CaV1.2 mutations such as those depicted in the Timothy syndrome when reinforced upon β-AR stimulation is a source for intracellular calcium disorders triggering DADs and severe arrhythmias.125 These observations demonstrate that a fine tuning of the β-AR modulation of calcium entry via CaV1.2 channels in the myocytes is required to maintain proper calcium homeostasis and electrical activity of the heart.
Atrial Fibrillation
Atrial fibrillation (AF) is an extremely common cardiac arrhythmia, most prevalent in elderly people, that is profoundly influenced by the autonomic nervous system, especially by the adrenergic component.126 It is accompanied by APD shortening and intracellular calcium homeostasis disorders.113,127 Decreased depolarizing ICa,L contributes to such APD shortening by favoring repolarization thus reentry substrates. If reductions in ICa,L have been observed consistently in atrial myocytes from patients with permanent AF128–130 or from patients in sinus rhythm with a high risk of AF,131,132 the exact mechanism for such downregulation is not clearly established. This reduction might be primarily the result of transcriptional downregulation of the α1C-subunit of LTCCs via a Ca2+-dependent calmodulin-calcineurin-NFAT system at the cost of the APD reduction observed in AF.133 Besides, it has been also associated with diminished expression of the CaVβ and α2-δ accessory subunits,134 which are essential for α1C trafficking to the plasma membrane. Normal trafficking of CaV1.2 might as well be impaired by a zinc binding protein (ZnT-1) upregulated in patients with AF.135 Upregulation of a microRNA, miR-328, has also been correlated to AF producing downexpression of α1C and CaVβ1.136 Oxidative stress that occurs in AF could also participate to decrease LTCC activity by inducing S-nitrosylation of α1C137 and by promoting its β-AR stimulation to trigger arrhythmogenic EADs.138 ICa,L amplitude is clearly decreased in AF, but its β-AR regulation is not systematically modified.139 Although β-AR receptor density is not impaired in AF140, polymorphism of β1-AR occurs in patients with AF, leading to decreased β-AR cascade,141 that could partially explain the decreased LTCC activity in AF. Another plausible mechanism has been proposed as a decreased phosphorylation of the CaV1.2 channel because of suppress to the increased phosphatase 1 or phosphatase 2A activities observed in AF.130,142 This might be also influenced by the hyperphosphorylation by PKA of the peptide inhibitor 1 that is prominent in AF to increase its inhibitory effect on PP-1.142,143 Furthermore, depressed expression of the major PDE4 isoform detected in human atria, PDE4D, correlates with aging a well-known favoring factor for AF development.97 Although PDE3 is the main cAMP-degrading enzyme in human atria, PDE4 substantially contributes to the enzymatic control of β-AR stimulation of ICa,L in human atria, and its inhibition leads to arrhythmias because of dysregulated intracellular calcium homeostasis.97 All these observations suggest that altered β-AR modulation of CaV1.2 channels happens in AF to contribute to such arrhythmia.
Hypertrophy and Heart Failure
In human HF, a chronic activation of β-AR induced by the elevation of circulating catecholamine levels occurs and contributes to the progression of the disease,144,145 stressing the necessity for a strict control of β-AR signaling. This hypothesis is further demonstrated in animal models with exacerbated β-AR/cAMP/PKA signaling and that develop cardiac hypertrophy and heart failure.146–148 A hallmark of heart failure is β-AR desensitization with a loss of β-AR signaling compartmentation35,144 and intracellular calcium cycling perturbations,114 accompanied by profound alterations of the structure of the cardiomyocytes notably a loss of the T-tubules.149,150 Desensitization of β-AR is promoted by its phosphorylation by the G-protein–coupled receptor kinase 2 (GRK2), a serine/threonine kinase upregulated in hypertrophy and HF.151 The number of functional β-AR is reduced in HF, and their localization is changed, with a redistribution to cell crests of the β2-AR subtype normally localized in the T-tubules, whereas β1-R remain uniformly distributed at the membrane.46 Interestingly, 80% of the CaV1.2 channels are located in the T-tubules152 where they are targeted by the protein BIN-1.153 The number of T-tubular LTCCs is decreased in failing myocytes,154,155 a reduction associated with a down-regulation of BIN-1 in human failing cardiomyocytes.156 This must contribute to the decreased EC coupling157 that might be also affected by the architectural reorganization of the dyadic cleft.157,158 Nonetheless, there is no real consensus in the literature concerning a reduction of ICa,L amplitude in failing cardiomyocytes. If the reduction of CaV1.2 channels is well described in animals models for HF, it seems largely accepted that ICa,L amplitude is maintained especially in failing human cardiomyocytes.159,160 This apparent discrepancy might be explained by an increased open probability of single CaV1.2 channels because of either their increased phosphorylation161,162 to altered PKA and phosphatase activities155,163 or increased expression of CaVβ ancillary subunits.164,165 Furthermore, cardiac remodeling induced by chronic activation of β-AR signaling activates CaMKII,166,167 which is known to increase the CaV1.2 channel activity120 and participate in calcium influx remodeling in heart failure.168 Blunted β-AR stimulation of ICa,L in human failing cardiomyocytes has been consistently reported,162,169 probably because of more abundant heteromeric Gi/oα proteins,170–172 leading to an increased β2-R receptor coupling with Gi antagonizing the β1-AR stimulation of ICa,L.173 In addition, increased Gi/oα modifies PP1 and PP2A activities, which are two phosphatases that control LTCC phosphorylation.163 GRK2 could also contribute to the remodeling of the β-AR stimulation of ICa,L in HF, because knockout mice for this kinase appear to be more resistant to adverse remodeling following myocardial infarction, but they demonstrated an increased basal ICa,L amplitude and blunted β-AR stimulation.174 Furthermore, overexpression of βARKct, a peptide derived from the GRK2 C-terminus, is able to increase β-AR stimulation of ICa,L independently of PKA in normal and failing cardiomyocytes by sequestering Gβγ proteins.175 Moreover, expression of a PDE4 isoform—PDE4B, which modulates β-AR stimulation of ICa,L91—has been shown to be decreased in a rat model of compensated hypertrophy,176 suggesting that not only the production but the enzymatic degradation of the cAMP controlling ICa,L is affected in pathological conditions. Thus, in hypertrophy and HF, the β-AR regulation of CaV1.2 channels is altered, contributing to a dysregulation of calcium handling promoting calcium-induced arrhythmias. Accurate calcium influx is not only a determinant for normal cardiomyocyte function during the excitation-contraction coupling; recent evidence in the literature highlighted a possible role of CaV1.2 channels in gene transcription and prohypertrophic signaling.177 Few studies have stated that chronic increase calcium influx via CaV1.2 channels is deleterious by promoting prohypertrophic signaling pathways. For example, mice overexpressing the α1C-subunit of CaV1.2 channels exhibit cardiac growth and cardiomyopathy hypertrophy178 and similarly, overexpression of CaVβ2a leads to cardiac hypertrophy by increasing ICa,L, and activation of calcineurin/nuclear factor of activated T cells (NFAT) and CaMKII/HDAC signaling pathways.179 These assumptions are confirmed by the fact that LTCC blockers can prevent cardiac remodeling in animal subjected to pressure overload180 and by the attenuation of cardiac hypertrophy observed when the expression of CaVβ2 subunit is inhibited.181 Astonishingly, decreasing the expression of α1C in mice also produces cardiac hypertrophy and HF, but this deleterious effect appears not to be connected directly to the diminished ICa,L amplitude, but more possibly to the compensatory neuroendocrine stress induced to balance the decreased contractility.182 Recently, it has been proposed that PKA phosphorylation of the serine 1700 of α1C relieves its autoinhibition by inducing the dissociation of its proteolytically cleaved and covalently reassociated distal C-terminus from the proximal C-terminus.30 Interestingly, the C-terminus part of α1C can translocate to the nucleus to act as a transcription factor for its own expression and to activate hypertrophic signaling.183 A possible role of its C-terminus in hypertrophy and HF is reaffirmed by recent studies showing that deletion of the terminal part of the α1C-subunit results in HF in vivo.32,184 It is tempting to speculate that chronic β-AR stimulation promotes hypertrophy and HF by favoring a permanent dissociation of the C-terminus of the pore-forming subunit of CaV1.2 channels to induce cardiac remodeling.
References
1. Bers, DM. Cardiac excitation-contraction coupling. Nature. 2002; 415(6868):198–205.
2. van der Heyden, MA, Wijnhoven, TJ, Opthof, T. Molecular aspects of adrenergic modulation of cardiac L-type Ca2+ channels. Cardiovasc Res. 2005; 65(1):28–39.
3. Hartzell, HC, Méry, P-F, Fischmeister, R, et al. Sympathetic regulation of cardiac calcium current is due exclusively to cAMP-dependent phosphorylation. Nature. 1991; 351(6327):573–576.
4. Catterall, WA. Voltage-gated calcium channels. Cold Spring Harb Perspect Biol. 2011; 3(8):a003947.
5. Hess, P, Lansman, JB, Tsien, RW. Different modes of Ca channel gating behaviour favoured by dihydropyridine Ca agonists and antagonists. Nature. 1984; 311(5986):538–544.
6. Yue, DT, Herzig, S, Marban, E. β-adrenergic stimulation of calcium channels occurs by potentiation of high-activity gating modes. Proc Natl Acad Sci U S A. 1990; 87(2):753–757.
7. Cens, T, Rousset, M, Leyris, JP, et al. Voltage- and calcium-dependent inactivation in high voltage-gated Ca2+ channels. Prog Biophys Mol Biol. 2006; 90(1-3):104–117.
8. Argibay, JA, Fischmeister, R, Hartzell, HC. Inactivation, reactivation and pacing dependence of calcium current in frog cardiocytes: correlation with current density. J Physiol. 1988; 401:201–226.
9. Findlay, I. Physiological modulation of inactivation in L-type Ca2+ channels: one switch. J Physiol. 2004; 554(Pt 2):275–283.
10. Davies, A, Hendrich, J, Van Minh, AT, et al. Functional biology of the alpha(2)delta subunits of voltage-gated calcium channels. Trends Pharmacol Sci. 2007; 28(5):220–228.
11. Dolphin, AC. Beta subunits of voltage-gated calcium channels. J Bioenerg Biomembr. 2003; 35(6):599–620.
12. Colecraft, HM, Alseikhan, B, Takahashi, SX, et al. Novel functional properties of Ca2+ channel β subunits revealed by their expression in adult rat heart cells. J Physiol. 2002; 541(2):435–452.
13. Fuller-Bicer, GA, Varadi, G, Koch, SE, et al. Targeted disruption of the voltage-dependent calcium channel alpha2/delta-1-subunit. Am J Physiol Heart Circ Physiol. 2009; 297(1):H117–H124.
14. Miriyala, J, Nguyen, T, Yue, DT, et al. Role of Cavβ subunits, and lack of functional reserve, in protein kinase A modulation of cardiac Cav1. 2 channels. Circ Res. 2008; 102(7):e54–64.
15. Yang, L, Katchman, A, Morrow, JP, et al. Cardiac L-type calcium channel (Cav1. 2) associates with gamma subunits. FASEB J. 2011; 25(3):928–936.
16. Puri, TS, Gerhardstein, BL, Zhao, XL, et al. Differential effects of subunit interactions on protein kinase A- and C-mediated phosphorylation of L-type calcium channels. Biochemistry (Mosc). 1997; 36(31):9605–9615.
17. De Jongh, KS, Murphy, BJ, Colvin, AA, et al. Specific phosphorylation of a site in the full-length form of the alpha 1 subunit of the cardiac L-type calcium channel by adenosine 3’,5’-cyclic monophosphate-dependent protein kinase. Biochemistry (Mosc). 1996; 35(32):10392–10402.
18. Perez-Reyes, E, Yuan, WL, Wei, XY, et al. Regulation of the cloned L-type cardiac calcium channel by cyclic-AMP-dependent protein kinase. FEBS Lett. 1994; 342(2):119–123.
19. Zong, XG, Schreieck, J, Mehrke, G, et al. On the regulation of the expressed L-type calcium channel by cAMP-dependent phosphorylation. Pflügers Arch. 1995; 430(3):340–347.
20. Mikala, G, Klockner, U, Varadi, M, et al. cAMP-dependent phosphorylation sites and macroscopic activity of recombinant cardiac L-type calcium channels. Mol Cell Biochem. 1998; 185(1-2):95–109.
21. Gao, TY, Yatani, A, Dell’Acqua, ML, et al. cAMP-dependent regulation of cardiac L-type Ca2+ channels requires membrane targeting of PKA and phosphorylation of channel subunits. Neuron. 1997; 19(1):185–196.
22. Hulme, JT, Lin, TW, Westenbroek, RE, et al. β-adrenergic regulation requires direct anchoring of PKA to cardiac Cav1. 2 channels via a leucine zipper interaction with A kinase-anchoring protein 15. Proc Natl Acad Sci U S A. 2003; 100(22):13093–13098.
23. Hulme, JT, Westenbroek, RE, Scheuer, T, et al. Phosphorylation of serine 1928 in the distal C-terminal domain of cardiac Cav1. 2 channels during β1-adrenergic regulation. Proc Natl Acad Sci U S A. 2006; 103(44):16574–16579.
24. Hulme, JT, Yarov-Yarovoy, V, Lin, TW, et al. Autoinhibitory control of the Cav1. 2 channel by its proteolytically processed distal C-terminal domain. J Physiol. 2006; 576(Pt 1):87–102.
25. Ganesan, AN, Maack, C, Johns, DC, et al. β-Adrenergic stimulation of L-type Ca2+ channels in cardiac myocytes requires the distal carboxyl terminus of alpha1C but not serine 1928. Circ Res. 2006; 98(2):e11–8.
26. Lemke, T, Welling, A, Christel, CJ, et al. Unchanged β-adrenergic stimulation of cardiac L-type calcium channels in Cav1. 2 phosphorylation site S1928A mutant mice. J Biol Chem. 2008; 283(50):34738–34744.
27. Gerhardstein, BL, Puri, TS, Chien, AJ, et al. Identification of the sites phosphorylated by cyclic AMP-dependent protein kinase on the β2 subunit of L-type voltage-dependent calcium channels. Biochemistry (Mosc). 1999; 38(32):10361–10370.
28. Bunemann, M, Gerhardstein, BL, Gao, TY, et al. Functional regulation of L-type calcium channels via protein kinase A-mediated phosphorylation of the β2 subunit. J Biol Chem. 1999; 274(48):33851–33854.
29. Haase, H, Alvarez, J, Petzhold, D, et al. Ahnak is critical for cardiac CaV1. 2 calcium channel function and its β-adrenergic regulation. FASEB J. 2005; 19(14):1969–1977.
30. Fuller, MD, Emrick, MA, Sadilek, M, et al. Molecular mechanism of calcium channel regulation in the fight-or-flight response. Sci Signal. 2010; 3(141):ra70.
31. Hulme, JT, Konoki, K, Lin, TW, et al. Sites of proteolytic processing and noncovalent association of the distal C-terminal domain of Cav1. 1 channels in skeletal muscle. Proc Natl Acad Sci U S A. 2005; 102(14):5274–5279.
32. Fu, Y, Westenbroek, RE, Yu, FH, et al. Deletion of the distal C terminus of Cav1. 2 channels leads to loss of β-adrenergic regulation and heart failure in vivo. J Biol Chem. 2011; 286(14):12617–12626.
33. Xiang, YK. Compartmentalization of beta-adrenergic signals in cardiomyocytes. Circ Res. 2011; 109(2):231–244.
34. Rochais, F, Abi-Gerges, A, Horner, K, et al. A specific pattern of phosphodiesterases controls the cAMP signals generated by different Gs-coupled receptors in adult rat ventricular myocytes. Circ Res. 2006; 98(8):1081–1088.
35. Fischmeister, R, Castro, LRV, Abi-Gerges, A, et al. Compartmentation of cyclic nucleotide signaling in the heart: The role of cyclic nucleotide phosphodiesterases. Circ Res. 2006; 99(8):816–828.
36. Perino, A, Ghigo, A, Scott, JD, et al. Anchoring proteins as regulators of signaling pathways. Circ Res. 2012; 111(4):482–492.
37. Diviani, D, Maric, D, Lopez, IP, et al. A-kinase anchoring proteins: Molecular regulators of the cardiac stress response. Biochim Biophys Acta. 2012.
38. Morisco, C, Zebrowski, DC, Vatner, DE, et al. β-Adrenergic cardiac hypertrophy is mediated primarily by the β1-subtype in the rat heart. J Mol Cell Cardiol. 2001; 33(3):561–573.
39. Ahmet, I, Krawczyk, M, Heller, P, et al. Beneficial effects of chronic pharmacological manipulation of β-adrenoreceptor subtype signaling in rodent dilated ischemic cardiomyopathy. Circulation. 2004; 110(9):1083–1090.
40. Communal, C, Singh, K, Sawyer, DB, et al. Opposing effects of β1– and β2-adrenergic receptors on cardiac myocyte apoptosis – Role of a pertussis toxin-sensitive G proteins. Circulation. 1999; 100(22):2210–2212.
41. Zaugg, M, Xu, WM, Lucchinetti, E, et al. β-Adrenergic receptor subtypes differentially affect apoptosis in adult rat ventricular myocytes. Circulation. 2000; 102(3):344–350.
42. Brodde, OE, Bruck, H, Leineweber, K. Cardiac adrenoceptors: Physiological and pathophysiological relevance. J Pharmacol Sci. 2006; 100:323–337.
43. Chen-Izu, Y, Xiao, RP, Izu, LT, et al. Gi-dependent localization of β2-adrenergic receptor signaling to L-type Ca2+ channels. Biophys J. 2000; 79(5):2547–2556.
44. Xiang, Y, Rybin, VO, Steinberg, SF, et al. Caveolar localization dictates physiologic signaling of β2-adrenoceptors in neonatal cardiac myocytes. J Biol Chem. 2002; 277(37):34280–34286.
45. Calaghan, S, Kozera, L, White, E. Compartmentalisation of cAMP-dependent signalling by caveolae in the adult cardiac myocyte. J Mol Cell Cardiol. 2008; 45(1):88–92.
46. Nikolaev, VO, Moshkov, A, Lyon, AR, et al. β2-Adrenergic receptor redistribution in heart failure changes cAMP compartmentation. Science. 2010; 327(5973):1653–1657.
47. Balijepalli, RC, Foell, JD, Hall, DD, et al. Localization of cardiac L-type Ca2+ channels to a caveolar macromolecular signaling complex is required for β2-adrenergic regulation. Proc Natl Acad Sci U S A. 2006; 103(19):7500–7505.
48. Xiao, RP, Lakatta, EG. β1-Adrenoceptor stimulation and β2-adrenoceptor stimulation differ in their effects on contraction, cytosolic Ca2+, and Ca2+ current in single rat ventricular cells. Circ Res. 1993; 73:286–300.
49. Nikolaev, VO, Bunemann, M, Schmitteckert, E, et al. Cyclic AMP imaging in adult cardiac myocytes reveals far-reaching β1-adrenergic but locally confined β2-adrenergic receptor-mediated signaling. Circ Res. 2006; 99(10):1084–1091.
50. Rozec, B, Gauthier, C. β3-Adrenoceptors in the cardiovascular system: Putative roles in human pathologies. Pharmacol Ther. 2006; 111:652–673.
51. Gauthier, C, Tavernier, G, Charpentier, F, et al. Functional β3-adrenoceptor in the human heart. J Clin Invest. 1996; 98:556–562.
52. Moniotte, S, Vaerman, J, Kockx, MM, et al. Real-time RT-PCR for the detection of beta-adrenoceptor messenger RNAs in small human endomyocardial biopsies. J Mol Cell Cardiol. 2001; 33(12):2121–2133.
53. Chamberlain, PD, Jennings, KH, Paul, F, et al. The tissue distribution of the human β3-adrenoceptor studied using a monoclonal antibody: direct evidence of the β3-adrenoceptor in human adipose tissue, atrium and skeletal muscle. Int J Obes Relat Metab Disord. 1999; 23(10):1057–1065.
54. Moniotte, S, Kobzik, L, Feron, O, et al. Upregulation of β3-adrenoceptors and altered contractile response to inotropic amines in human failing myocardium. Circulation. 2001; 103(12):1649–1655.
55. De Matteis, R, Arch, JR, Petroni, ML, et al. Immunohistochemical identification of the β3-adrenoceptor in intact human adipocytes and ventricular myocardium: effect of obesity and treatment with ephedrine and caffeine. Int J Obes Relat Metab Disord. 2002; 26(11):1442–1450.
56. Skeberdis, VA, Gendvilienë, V, Zablockaitë, D, et al. β3-adrenergic receptor activation increases human atrial tissue contractility and stimulates the L-type Ca2+ current. J Clin Invest. 2008; 118:3219–3227.
57. Schoemaker, RG, Du, XY, Bax, WA, et al. 5-Hydroxytryptamine stimulates human isolated atrium but not ventricle. Eur J Pharmacol. 1993; 230:103–105.
58. Jahnel, U, Rupp, J, Ertl, R, et al. Positive inotropic response to 5-HT in human atrial but not in ventricular heart muscle. Naunyn Schmiedebergs Arch Pharmacol. 1992; 346:482–485.
59. Ouadid, H, Seguin, J, Dumuis, A, et al. Serotonin increases calcium current in human atrial myocytes via the newly described 5-hydroxytryptamine4 receptors. Mol Pharmacol. 1992; 41:346–351.
60. Scott, JD, Santana, LF. A-kinase anchoring proteins: getting to the heart of the matter. Circulation. 2010; 121(10):1264–1271.
61. Kritzer, MD, Li, J, Dodge-Kafka, K, et al. AKAPs: The architectural underpinnings of local cAMP signaling. J Mol Cell Cardiol. 2011.
62. Jones, BW, Brunet, S, Gilbert, ML, et al. Cardiomyocytes from AKAP7 knockout mice respond normally to adrenergic stimulation. Proc Natl Acad Sci U S A. 2012; 109(42):17099–17104.
63. Nichols, CB, Rossow, CF, Navedo, MF, et al. Sympathetic stimulation of adult cardiomyocytes requires association of AKAP5 with a subpopulation of L-type calcium channels. Circ Res. 2010; 107(6):747–756.
64. Marx, SO, Reiken, S, Hisamatsu, Y, et al. PKA phosphorylation dissociates FKBP12. 6 from the calcium release channel (Ryanodine receptor): Defective regulation in failing hearts. Cell. 2000; 101(4):365–376.
65. Marx, SO, Kurokawa, J, Reiken, S, et al. Requirement of a macromolecular signaling complex for β adrenergic receptor modulation of the KCNQ1-KCNE1 potassium channel. Science. 2002; 295(5554):496–499.
66. Dodge-Kafka, KL, Soughayer, J, Pare, GC, et al. The protein kinase A anchoring protein mAKAP co-ordinates two integrated cAMP effector pathways. Nature. 2005; 437:574–578.
67. Efendiev, R, Samelson, BK, Nguyen, BT, et al. AKAP79 interacts with multiple adenylyl cyclase (AC) isoforms and scaffolds AC 5 and 6 to AMPA receptors. J Biol Chem. 2010; 285(19):14450–14458.
68. Bauman, AL, Soughayer, J, Nguyen, BT, et al. Dynamic regulation of cAMP synthesis through anchored PKA-adenylyl cyclase V/VI complexes. Mol Cell. 2006; 23(6):925–931.
69. Dodge, KL, Khouangsathiene, S, Kapiloff, MS, et al. mAKAP assembles a protein kinase A/PDE4 phosphodiesterase cAMP signaling module. EMBO J. 2001; 20(8):1921–1930.
70. Perino, A, Ghigo, A, Ferrero, E, et al. Integrating cardiac PIP3 and cAMP signaling through a PKA anchoring function of p110gamma. Mol Cell. 2011; 42(1):84–95.
71. Ghigo, A, Perino, A, Mehel, H, et al. PI3Kγ Protects against catecholamine-induced ventricular arrhythmia through PKA-mediated regulation of distinct phosphodiesterases. Circulation. 2012. [PMID: 23008439 (in press)].
72. Terrenoire, C, Houslay, MD, Baillie, GS, et al. The cardiac IKs potassium channel macromolecular complex includes the phosphodiesterase PDE4D3. J Biol Chem. 2009; 284(14):9140–9146.
73. Rich, TC, Fagan, KA, Nakata, H, et al. Cyclic nucleotide-gated channels colocalize with adenylyl cyclase in regions of restricted cAMP diffusion. J Gen Physiol. 2000; 116(2):147–161.
74. Mika, D, Leroy, J, Vandecasteele, G, et al. PDEs create local domains of cAMP signaling. J Mol Cell Cardiol. 2012; 52:323–329.
75. Méry, P-F, Pavoine, C, Pecker, F, et al. Erythro-9-(2-hydroxy-3-nonyl)adenine inhibits cyclic GMP-stimulated phosphodiesterase in isolated cardiac myocytes. Mol Pharmacol. 1995; 48(1):121–130.
76. Rivet-Bastide, M, Vandecasteele, G, Hatem, S, et al. cGMP-stimulated cyclic nucleotide phosphodiesterase regulates the basal calcium current in human atrial myocytes. J Clin Invest. 1997; 99(11):2710–2718.
77. Verde, I, Vandecasteele, G, Lezoualc’h, F, et al. Characterization of the cyclic nucleotide phosphodiesterase subtypes involved in the regulation of the L-type Ca2+ current in rat ventricular myocytes. Br J Pharmacol. 1999; 127(1):65–74.
78. Vandecasteele, G, Verde, I, Rucker-Martin, C, et al. Cyclic GMP regulation of the L-type Ca2+ channel current in human atrial myocytes. J Physiol. 2001; 533(2):329–340.
79. Dittrich, M, Jurevicius, J, Georget, M, et al. Local response of L-type Ca2+ current to nitric oxide in frog ventricular myocytes. J Physiol. 2001; 534(1):109–121.
80. Kirstein, M, Rivet-Bastide, M, Hatem, S, et al. Nitric oxide regulates the calcium current in isolated human atrial myocytes. J Clin Invest. 1995; 95(2):794–802.
81. Richter, W, Xie, M, Scheitrum, C, et al. Conserved expression and functions of PDE4 in rodent and human heart. Basic Res Cardiol. 2011; 106(2):249–262.
82. Mongillo, M, McSorley, T, Evellin, S, et al. Fluorescence resonance energy transfer-based analysis of cAMP dynamics in live neonatal rat cardiac myocytes reveals distinct functions of compartmentalized phosphodiesterases. Circ Res. 2004; 95(1):65–75.
83. Rochais, F, Vandecasteele, G, Lefebvre, F, et al. Negative feedback exerted by PKA and cAMP phosphodiesterase on subsarcolemmal cAMP signals in intact cardiac myocytes. An in vivo study using adenovirus-mediated expression of CNG channels. J Biol Chem. 2004; 279(50):52095–52105.
84. Leroy, J, Abi-Gerges, A, Nikolaev, VO, et al. Spatiotemporal dynamics of β-adrenergic cAMP signals and L-type Ca2+ channel regulation in adult rat ventricular myocytes: Role of phosphodiesterases. Circ Res. 2008; 102(9):1091–1100.
85. Baillie, GS, Sood, A, McPhee, I, et al. β-Arrestin-mediated PDE4 cAMP phosphodiesterase recruitment regulates β-adrenoceptor switching from Gs to Gi. Proc Natl Acad Sci U S A. 2003; 100(3):941–945.
86. Richter, W, Day, P, Agraval, R, et al. Signaling from β1– and β2-adrenergic receptors is defined by differential interactions with PDE4. Embo J. 2008; 27(2):384–393.
87. De Arcangelis, V, Liu, R, Soto, D, et al. Differential association of phosphodiesterase 4D isoforms with β2-adrenoceptor in cardiac myocytes. J Biol Chem. 2009; 284(49):33824–33832.
88. Lehnart, SE, Wehrens, XHT, Reiken, S, et al. Phosphodiesterase 4D deficiency in the ryanodine receptor complex promotes heart failure and arrhythmias. Cell. 2005; 123(1):23–35.
89. Kerfant, BG, Zhao, D, Lorenzen-Schmidt, I, et al. PI3KY is required for PDE4, not PDE3, activity in subcellular microdomains containing the sarcoplasmic reticular calcium ATPase in cardiomyocytes. Circ Res. 2007; 101(4):400–408.
90. Beca, S, Helli, PB, Simpson, JA, et al. Phosphodiesterase 4D regulates baseline sarcoplasmic reticulum Ca2+ release and cardiac contractility, independently of L-type Ca2+ current. Circ Res. 2011; 109(9):1024–1030.
91. Leroy, J, Richter, W, Mika, D, et al. Phosphodiesterase 4B in the cardiac L-type Ca2+ channel complex regulates Ca2+ current and protects against ventricular arrhythmias. J Clin Invest. 2011; 121(7):2651–2661.
92. Weishaar, RE, Kobylarz-Singer, DC, Steffen, RP, et al. Subclasses of cyclic AMP-specific phosphodiesterase in left ventricular muscle and their involvement in regulating myocardial contractility. Circ Res. 1987; 61:539–547.
93. Lugnier, C, Muller, B, Lebec, A, et al. Characterization of indolidan-sensitive and rolipram-sensitive cyclic nucleotide phosphodiesterases in canine and human cardiac microsomal fractions. J Pharmacol Exp Ther. 1993; 265:1142–1151.
94. Smith, CJ, Huang, R, Sun, D, et al. Development of decompensated dilated cardiomyopathy is associated with decreased gene expression and activity of the milrinone-sensitive cAMP phosphodiesterase PDE3A. Circulation. 1997; 96(9):3116–3123.
95. Osadchii, OE. Myocardial phosphodiesterases and regulation of cardiac contractility in health and cardiac disease. Cardiovasc Drugs Ther. 2007; 21:171–194.
96. Monrad, ES, Baim, DS, Smith, HS, et al. Assessment of long-term therapy with milrinone and the effects of milrinone withdrawal. Circulation. 1986; 73(3 Pt 2):III205–12.
97. Molina, CE, Leroy, J, Xie, M, et al. Cyclic AMP phosphodiesterase type 4 protects against atrial arrhythmias. J Am Coll Cardiol. 2012; 59(24):2182–2190.
98. Rapundalo, ST, Solaro, RJ, Kranias, EG. Inotropic responses to isoproterenol and phosphodiesterase inhibitors in intact guinea pig hearts: comparison of cyclic AMP levels and phosphorylation of sarcoplasmic reticulum and myofibrillar proteins. Circ Res. 1989; 64:104–111.
99. Hohl, CM, Li, Q. Compartmentation of cAMP in adult canine ventricular myocytes—Relation to single-cell free Ca2+ transients. Circ Res. 1991; 69:1369–1379.
100. Jurevicius, J, Fischmeister, R. cAMP compartmentation is responsible for a local activation of cardiac Ca2+ channels by β-adrenergic agonists. Proc Natl Acad Sci U S A. 1996; 93(1):295–299.
101. Zaccolo, M, Pozzan, T. Discrete microdomains with high concentration of cAMP in stimulated rat neonatal cardiac myocytes. Science. 2002; 295(5560):1711–1715.
102. Nikolaev, VO, Lohse, MJ. Monitoring of cAMP synthesis and degradation in living cells. Physiology (Bethesda). 2006; 21:86–92.
103. Ono, K, Fozzard, HA. Phosphorylation restores activity of L-type calcium channels after rundown in inside-out patches from rabbit cardiac cells. J Physiol. 1992; 454:673–688.
104. Herzig, S, Neumann, J. Effects of serine/threonine protein phosphatases on ion channels in excitable membranes. Physiol Rev. 2000; 80(1):173–210.
105. Davare, MA, Horne, MC, Hell, JW. Protein phosphatase 2A is associated with class C L-type calcium channels (CaV1. 2) and antagonizes channel phosphorylation by cAMP-dependent protein kinase. J Biol Chem. 2000; 275(50):39710–39717.
106. Tandan, S, Wang, Y, Wang, TT, et al. Physical and functional interaction between calcineurin and the cardiac L-type Ca2+ channel. Circ Res. 2009; 105(1):51–60.
107. Hall, DD, Feekes, JA, Arachchige Don, AS, et al. Binding of protein phosphatase 2A to the L-type calcium channel Cav1. 2 next to Ser1928, its main PKA site, is critical for Ser1928 dephosphorylation. Biochemistry (Mosc). 2006; 45(10):3448–3459.
108. Xu, H, Ginsburg, KS, Hall, DD, et al. Targeting of protein phosphatases PP2A and PP2B to the C-terminus of the L-type calcium channel Ca v1. 2. Biochemistry (Mosc). 2010; 49(48):10298–10307.
109. Santana, LF, Chase, EG, Votaw, VS, et al. Functional coupling of calcineurin and protein kinase A in mouse ventricular myocytes. J Physiol. 2002; 544(Pt 1):57–69.
110. Coumel, P, Leenhardt, A. Mental activity, adrenergic modulation, and cardiac arrhythmias in patients with heart disease. Circulation. 1991; 83(4 Suppl):II58–70.
111. Weiss, JN, Nivala, M, Garfinkel, A, et al. Alternans and arrhythmias: from cell to heart. Circ Res. 2011; 108(1):98–112.
112. Eisner, DA, Li, Y, O’Neill, SC. Alternans of intracellular calcium: mechanism and significance. Heart Rhythm. 2006; 3(6):743–745.
113. Wakili, R, Voigt, N, Kaab, S, et al. Recent advances in the molecular pathophysiology of atrial fibrillation. J Clin Invest. 2011; 121(8):2955–2968.
114. Pogwizd, SM, Bers, DM. Cellular basis of triggered arrhythmias in heart failure. Trends Cardiovasc Med. 2004; 14(2):61–66.
115. January, CT, Riddle, JM. Early afterdepolarizations—Mechanism of induction and block—A role for L-type Ca2+ current. Circ Res. 1989; 64(5):977–990.
116. Volders, PGA, Kulcsar, A, Vos, MA, et al. Similarities between early and delayed afterdepolarizations induced by isoproterenol in canine ventricular myocytes. Cardiovasc Res. 1997; 34(2):348–359.
117. Pereira, L, Métrich, M, Fernández-Velasco, M, et al. The cAMP binding protein Epac modulates Ca2+ sparks by Ca2+/calmodulin kinase signalling pathway in rat cardiac myocytes. J Physiol. 2007; 583(Pt 2):685–694.
118. Mangmool, S, Shukla, AK, Rockman, HA. β-Arrestin-dependent activation of Ca2+/calmodulin kinase II after β1-adrenergic receptor stimulation. J Cell Biol. 2010; 189(3):573–587.
119. Wu, Y, Temple, J, Zhang, R, et al. Calmodulin kinase II and arrhythmias in a mouse model of cardiac hypertrophy. Circulation. 2002; 106(10):1288–1293.
120. Koval, OM, Guan, X, Wu, Y, et al. Cav1. 2 beta-subunit coordinates CaMKII-triggered cardiomyocyte death and afterdepolarizations. Proc Natl Acad Sci U S A. 2010; 107(11):4996–5000.
121. Sipido, KR, Volders, PGA, de Groot, SHM, et al. Enhanced Ca2+ release and Na/Ca exchange activity in hypertrophied canine ventricular myocytes—Potential link between contractile adaptation and arrhythmogenesis. Circulation. 2000; 102(17):2137–2144.
122. Spencer, CI, Sham, JS. Effects of Na+/Ca2+ exchange induced by SR Ca2+ release on action potentials and afterdepolarizations in guinea pig ventricular myocytes. Am J Physiol Heart Circ Physiol. 2003; 285(6):H2552–H2562.
123. Priori, SG, Chen, SR. Inherited dysfunction of sarcoplasmic reticulum Ca2+ handling and arrhythmogenesis. Circ Res. 2011; 108(7):871–883.
124. Napolitano, C, Antzelevitch, C. Phenotypical manifestations of mutations in the genes encoding subunits of the cardiac voltage-dependent L-type calcium channel. Circ Res. 2011; 108(5):607–618.
125. Sung, RJ, Wu, YH, Lai, NH, et al. β-adrenergic modulation of arrhythmogenesis and identification of targeted sites of antiarrhythmic therapy in Timothy (LQT8) syndrome: a theoretical study. Am J Physiol Heart Circ Physiol. 2010; 298(1):H33–H44.
126. Workman, AJ. Cardiac adrenergic control and atrial fibrillation. Naunyn Schmiedebergs Arch Pharmacol. 2010; 381(3):235–249.
127. Greiser, M, Lederer, WJ, Schotten, U. Alterations of atrial Ca2+ handling as cause and consequence of atrial fibrillation. Cardiovasc Res. 2011; 89(4):722–733.
128. Legrand, B, Hatem, S, Deroubaix, E, et al. Calcium current depression in isolated human atrial myocytes after cessation of chronic treatment with calcium antagonists. Circ Res. 1991; 69(2):292–300.
129. Van Wagoner, DR, Pond, AL, McCarthy, PM, et al. Outward K+ current densities and Kv1. 5 expression are reduced in chronic human atrial fibrillation. Circ Res. 1997; 80(6):772–781.
130. Christ, T, Boknik, P, Wohrl, S, et al. L-type Ca2+ current downregulation in chronic human atrial fibrillation is associated with increased activity of protein phosphatases. Circulation. 2004; 110(17):2651–2657.
131. Nattel, S, Maguy, A, Le Bouter, S, Yeh, YH. Arrhythmogenic ion-channel remodeling in the heart: heart failure, myocardial infarction, and atrial fibrillation. Physiol Rev. 2007; 87(2):425–456.
132. Dinanian, S, Boixel, C, Juin, C, et al. Downregulation of the calcium current in human right atrial myocytes from patients in sinus rhythm but with a high risk of atrial fibrillation. Eur Heart J. 2008; 29(9):1190–1197.
133. Qi, XY, Yeh, YH, Xiao, L, et al. Cellular signaling underlying atrial tachycardia remodeling of L-type calcium current. Circ Res. 2008; 103(8):845–854.
134. Bosch, RF, Scherer, CR, Rub, N, et al. Molecular mechanisms of early electrical remodeling: transcriptional downregulation of ion channel subunits reduces ICa,L and Ito in rapid atrial pacing in rabbits. J Am Coll Cardiol. 2003; 41(5):858–869.
135. Beharier, O, Etzion, Y, Levi, S, et al. The involvement of ZnT-1, a new modulator of cardiac L-type calcium channels, in. Ann N Y Acad Sci. 2010; Feb(1188):87–95.
136. Lu, Y, Zhang, Y, Wang, N, et al. MicroRNA-328 contributes to adverse electrical remodeling in atrial fibrillation. Circulation. 2010; 122(23):2378–2387.
137. Carnes, CA, Janssen, PM, Ruehr, ML, et al. Atrial glutathione content, calcium current, and contractility. J Biol Chem. 2007; 282(38):28063–28073.
138. Gaur, N, Rudy, Y, Hool, L. Contributions of ion channel currents to ventricular action potential changes and induction of early afterdepolarizations during acute hypoxia. Circ Res. 2009; 105(12):1196–1203.
139. Van Wagoner, DR, Pond, AL, Lamorgese, M, et al. Atrial L-type Ca2+ currents and human atrial fibrillation. Circ Res. 1999; 85(5):428–436.
140. Gaspo, R, Sun, H, Fareh, S, et al. Dihydropyridine and beta adrenergic receptor binding in dogs with tachycardia-induced atrial fibrillation. Cardiovasc Res. 1999; 42(2):434–442.
141. Parvez, B, Chopra, N, Rowan, S, et al. A common β1-adrenergic receptor polymorphism predicts favorable response to rate-control therapy in atrial fibrillation. J Am Coll Cardiol. 2012; 59(1):49–56.
142. El-Armouche, A, Boknik, P, Eschenhagen, T, et al. Molecular determinants of altered Ca2+ handling in human chronic atrial fibrillation. Circulation. 2006; 114(7):670–680.
143. Wittköpper, K, Fabritz, L, Neef, S, et al. Constitutively active phosphatase inhibitor-1 improves cardiac contractility in young mice but is deleterious after catecholaminergic stress and with aging. J Clin Invest. 2010; 120(2):617–626.
144. Lohse, MJ, Engelhardt, S, Eschenhagen, T. What is the role of β-adrenergic signaling in heart failure? Circ Res. 2003; 93(10):896–906.
145. Movsesian, MA, Bristow, MR. Alterations in cAMP-mediated signaling and their role in the pathophysiology of dilated cardiomyopathy. Curr Top Dev Biol. 2005; 68:25–48.
146. Engelhardt, S, Hein, L, Wiesmann, F, et al. Progressive hypertrophy and heart failure in beta1-adrenergic receptor transgenic mice. Proc Natl Acad Sci U S A. 1999; 96(12):7059–7064.
147. Antos, CL, Frey, N, Marx, SO, et al. Dilated cardiomyopathy and sudden death resulting from constitutive activation of protein kinase A. Circ Res. 2001; 89(11):997–1004.
148. Iwase, M, Bishop, SP, Uechi, M, et al. Adverse effects of chronic endogenous sympathetic drive induced by cardiac Gs alpha overexpression. Circ Res. 1996; 78(4):517–524.
149. Lyon, AR, Macleod, KT, Zhang, Y, et al. Loss of T-tubules and other changes to surface topography in ventricular myocytes from failing human and rat heart. Proc Natl Acad Sci U S A. 2009; 106(16):6854–6859.
150. Orchard, C, Brette, F. T-tubules and sarcoplasmic reticulum function in cardiac ventricular myocytes. Cardiovasc Res. 2008; 77(2):237–244.
151. Rengo, G, Lymperopoulos, A, Leosco, D, et al. GRK2 as a novel gene therapy target in heart failure. J Mol Cell Cardiol. 2011; 50(5):785–792.
152. Brette, F, Salle, L, Orchard, CH. Differential modulation of L-type Ca2+ current by SR Ca2+ release at the T-tubules and surface membrane of rat ventricular myocytes. Circ Res. 2004; 95(1):e1–7.
153. Hong, TT, Smyth, JW, Gao, D, et al. BIN1 localizes the L-type calcium channel to cardiac T-tubules. PLoS Biol. 8(2), 2010.
154. He, JQ, Conklin, MW, Foell, JD, et al. Reduction in density of transverse tubules and L-type Ca2+ channels in canine tachycardia-induced heart failure. Cardiovasc Res. 2001; 49(2):298–307.
155. Horiuchi-Hirose, M, Kashihara, T, Nakada, T, et al. Decrease in the density of t-tubular L-type Ca2+ channel currents in failing ventricular myocytes. Am J Physiol Heart Circ Physiol. 2011; 300(3):H978–H988.
156. Hong, TT, Cogswell, R, James, CA, et al. Plasma BIN1 correlates with heart failure and predicts arrhythmia in patients with arrhythmogenic right ventricular cardiomyopathy. Heart Rhythm. 2012; 9(6):961–967.
157. Gomez, AM, Valdivia, HH, Cheng, H, et al. Defective excitation-contraction coupling in experimental cardiac hypertrophy and heart failure. Science. 1997; 276(5313):800–806.
158. Crossman, DJ, Ruygrok, PN, Soeller, C, et al. Changes in the organization of excitation-contraction coupling structures in failing human heart. PLoS One. 6(3), 2011.
159. Pitt, GS, Dun, W, Boyden, PA. Remodeled cardiac calcium channels. J Mol Cell Cardiol. 2006; 41(3):373–388.
160. Benitah, JP, Alvarez, JL, Gomez, AM. L-type Ca2+ current in ventricular cardiomyocytes. J Mol Cell Cardiol. 2010; 48(1):26–36.
161. Handrock, R, Schroder, F, Hirt, S, et al. Single-channel properties of L-type calcium channels from failing human ventricle. Cardiovasc Res. 1998; 37(2):445–455.
162. Chen, X, Zhang, X, Harris, DM, et al. Reduced effects of BAY K 8644 on L-type Ca2+ current in failing human cardiac myocytes are related to abnormal adrenergic regulation. Am J Physiol Heart Circ Physiol. 2008; 294(5):H2257–H2267.
163. Kashihara, T, Nakada, T, Shimojo, H, et al. Chronic receptor-mediated activation of Gi/o proteins alters basal t-tubular and sarcolemmal L-type Ca2+ channel activity through phosphatases in heart failure. Am J Physiol Heart Circ Physiol. 2012; 302(8):H1645–H1654.
164. Hullin, R, Khan, IFY, Wirtz, S, et al. Cardiac L-type calcium channel beta-subunits expressed in human heart have differential effects on single channel characteristics. J Biol Chem. 2003; 278(24):21623–21630.
165. Hullin, R, Matthes, J, von Vietinghoff, S, et al. Increased expression of the auxiliary β2-subunit of ventricular L-type Ca2+ channels leads to single-channel activity characteristic of heart failure. PLoS One. 2007; 2(3):e292.
166. Hoch, B, Meyer, R, Hetzer, P, et al. Identification and expression of delta-isoforms of the multifunctional Ca2+/calmodulin-dependent protein kinase in failing and nonfailing human myocardium. Circ Res. 1999; 84(6):713–721.
167. Zhang, R, Khoo, MS, Wu, Y, et al. Calmodulin kinase II inhibition protects against structural heart disease. Nat Med. 2005; 11(4):409–417.
168. Wang, Y, Tandan, S, Cheng, J, et al. Ca2+/calmodulin-dependent protein kinase II-dependent remodeling of Ca2+ current in pressure overload heart failure. J Biol Chem. 2008; 283(37):25524–25532.
169. Chen, X, Piacentino, VI, Furukawa, S, et al. L-type Ca2+ channel density and regulation are altered in failing human ventricular myocytes and recover after support with mechanical assist devices. Circ Res. 2002; 91(6):517–524.
170. Neumann, J, Schmitz, W, Scholz, H, et al. Increase in myocardial Gi-proteins in heart failure. Lancet. 1988; 2(8617):936–937.
171. Xiao, RP, Zhang, SJ, Chakir, K, et al. Enhanced Gi signaling selectively negates β2-adrenergic receptor (AR)- but not β1-AR-mediated positive inotropic effect in myocytes from failing rat hearts. Circulation. 2003; 108(13):1633–1639.
172. El-Armouche, A, Zolk, O, Rau, T, et al. Inhibitory G-proteins and their role in desensitization of the adenylyl cyclase pathway in heart failure. Cardiovasc Res. 2003; 60(3):478–487.
173. He, JQ, Balijepalli, RC, Haworth, RA, et al. Crosstalk of beta-adrenergic receptor subtypes through Gi blunts beta-adrenergic stimulation of L-type Ca2+ channels in canine heart failure. Circ Res. 2005; 97(6):566–573.
174. Raake, PW, Vinge, LE, Gao, E, et al. G protein-coupled receptor kinase 2 ablation in cardiac myocytes before or after myocardial infarction prevents heart failure. Circ Res. 2008; 103(4):413–422.
175. Volkers, M, Weidenhammer, C, Herzog, N, et al. The inotropic peptide βARKct improves βAR responsiveness in normal and failing cardiomyocytes through GβY-mediated L-type calcium current disinhibition. Circ Res. 2011; 108(1):27–39.
176. Abi-Gerges, A, Richter, W, Lefebvre, F, et al. Decreased expression and activity of cAMP phosphodiesterases in cardiac hypertrophy and its impact on β-adrenergic cAMP signals. Circ Res. 2009; 105(8):784–792.
177. Best, JM, Kamp, TJ. Different subcellular populations of L-type Ca2+ channels exhibit unique regulation and functional roles in cardiomyocytes. J Mol Cell Cardiol. 2012; 52(2):376–387.
178. Wang, S, Ziman, B, Bodi, I, et al. Dilated cardiomyopathy with increased SR Ca2+ loading preceded by a hypercontractile state and diastolic failure in the alpha1CTG mouse. PLoS One. 2009; 4(1):e4133.
179. Chen, X, Nakayama, H, Zhang, X, et al. Calcium influx through Cav1. 2 is a proximal signal for pathological cardiomyocyte hypertrophy. J Mol Cell Cardiol. 2011; 50(3):460–470.
180. Semsarian, C, Ahmad, I, Giewat, M, et al. The L-type calcium channel inhibitor diltiazem prevents cardiomyopathy in a mouse model. J Clin Invest. 2002; 109(8):1013–1020.
181. Cingolani, E, Ramirez Correa, GA, et al. Gene therapy to inhibit the calcium channel β subunit: physiological consequences and pathophysiological effects in models of cardiac hypertrophy. Circ Res. 2007; 101(2):166–175.
182. Goonasekera, SA, Hammer, K, Auger-Messier, M, et al. Decreased cardiac L-type Ca2+ channel activity induces hypertrophy and heart failure in mice. J Clin Invest. 2012; 122(1):280–290.
183. Schroder, E, Byse, M, Satin, J. L-type calcium channel C terminus autoregulates transcription. Circ Res. 2009; 104(12):1373–1381.
184. Domes, K, Ding, J, Lemke, T, et al. Truncation of murine Cav1. 2 at Asp-1904 results in heart failure after birth. J Biol Chem. 2011; 286(39):33863–33871.