[level-membership-for-neurology-category]
Case 25
HISTORY AND PHYSICAL EXAMINATION
| Right | Left | |
|---|---|---|
| Shoulder abduction | 2/5 | 5/5 |
| Elbow flexion | 3/5 | 5/5 |
| Elbow extension | 4−/5 | 5/5 |
| Pronation | 0/5 | 3/5 |
| Fingers flexion | 0/5 | 3/5 |
| Wrist flexion | 1/5 | 1/5 |
| Wrist extension | 2/5 | 5/5 |
| Finger extension | 3/5 | 4−/5 |
| Finger abduction | 4−/5 | 3/5 |
| Right | Left | |
|---|---|---|
| Hip flexion | 5/5 | 5/5 |
| Hip extension | 5/5 | 5/5 |
| Knee extension | 5/5 | 5/5 |
| Knee flexion | 5/5 | 5/5 |
| Foot dorsiflexion | 5/5 | 1/5 |
| Toe dorsiflexion | 5/5 | 0/5 |
| Plantar flexion | 5/5 | 5/5 |
| Ankle inversion | 5/5 | 5/5 |
| Ankle eversion | 5/5 | 1/5 |
Please now review the Nerve Conduction Studies and Needle EMG tables.
QUESTIONS
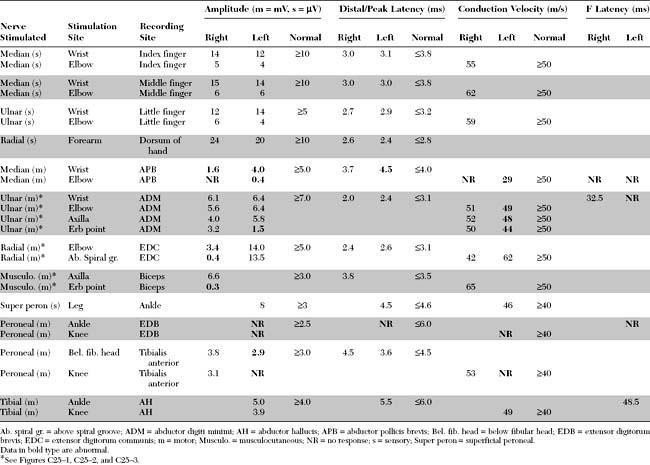
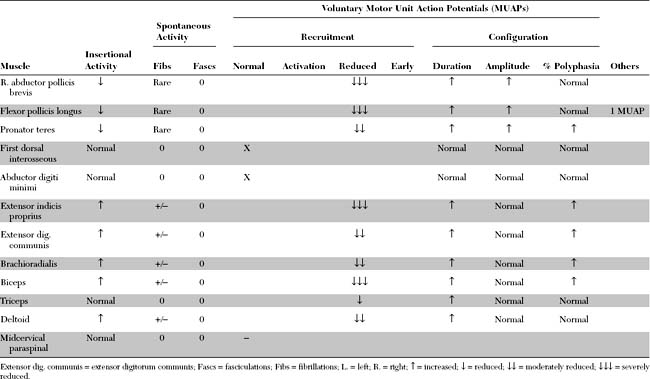
EDX FINDINGS AND INTERPRETATION OF DATA
Relevant EDX findings in this case include:
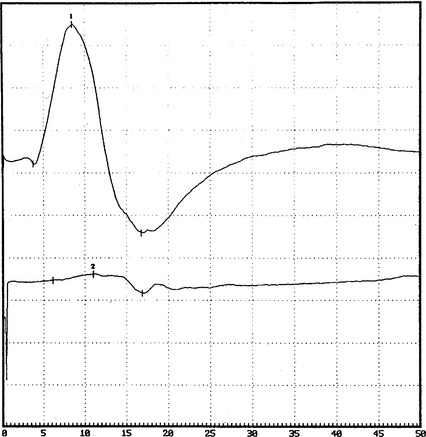
DISCUSSION
Definition and Pathogenesis
Multifocal motor neuropathy (MMN), described in the mid-1980s, is a rare disorder with a prevalence of 1 to 2 individuals per 100 000. It is characterized by specific EDX finding, i.e., motor conduction blocks, which is the gold standard for diagnosis. The disorder is important to recognize since it is treatable and responsive to immunomodulating therapies, and may mimic amyotrophic lateral sclerosis (ALS) which has a poor prognosis for survival.
CLINICAL FEATURES
Multifocal motor neuropathy presents insidiously with asymmetrical weakness often in the distribution of individual nerves. The age of onset of first symptoms is between 20 and 50 years of age in about 80% of patients, and the disorder is more common in men than women (ratio of 2.6/1). In more than 80% of patients, the weakness starts in the upper limbs, usually hand and forearm muscles. Other than the hypoglossal nerve, cranial nerve involvement is rare. Unilateral or bilateral phrenic nerve palsy causing respiratory failure may occur and is occasionally the presenting symptom. The disorder is slowly progressive, usually for more than 6 months and often years. Sometimes, the history is one of a stepwise progression with episodes of rapid worsening followed by prolonged periods of stabilization. The deep tendon reflexes are variable; they are usually depressed or absent diffusely or in weak limbs only. They may be normal or even brisk in one-third of patients, leading to confusion with ALS. Muscle atrophy is not prominent in weak muscles, despite the degree and chronicity of weakness; it may be present over the long term in the distribution of one or more affected nerves, implicating motor axon loss and predicting poor response to therapy. Mild sensory complaints may be present, but the sensory examination is usually normal except for minor vibration sense abnormalities in the lower extremities. A high titer of anti-GM1 antibody is present in approximately 50% of patients, although this varies between 30 and 80%, probably due to the different methodology utilized for antibody measurement. The cerebrospinal fluid protein is usually normal, but may be elevated in one-third of patients without exceeding 100 mg/dL.
Multifocal motor neuropathy should be distinguished from amyotrophic lateral sclerosis, particularly in patients with predominant or exclusive lower motor neuron findings. Clues on clinical examination of patients with MMN include the distribution of weakness, which follows peripheral nerves rather than spinal segments, the insidious course over many years, and the lack of pyramidal signs. It should be cautioned that preserved or brisk reflexes may be present in one-third of patients with MMN. Also, other forms of anterior horn cell disorders, such the spinal muscular atrophies, brachial amyotrophic diplegia (the flail arm syndrome) and monomelic amyotrophy (Hirayama disease) should be excluded. The flail arm syndrome (brachial amyotrophic diplegia), a variant of the progressive muscular atrophy form of ALS, is characterized by progressive proximal and distal upper limb weakness and ultimate variable involvement of the lower limbs. Monomelic amyotrophy (Hirayama disease) affects young men between the age of 15 and 22 and presents with an asymmetrical wasting and weakness of distal upper limb muscles. The disorder is benign, initially progressive over several years and then becoming static. Finally, MMN should be distinguished from other chronic acquired demyelinating peripheral polyneuropathies that may be associated with conduction block or predominantly motor including chronic inflammatory demyelinating polyradiculoneuropathy (CIDP) and its variant the Lewis-Sumner syndrome (multifocal acquired demyelinating sensory and motor neuropathy, MADSAM), osteosclerotic myeloma (POEMS syndrome), and MGUS neuropathy.
Diagnostic criteria for MMN were proposed. The aim of these criteria is to strengthen the diagnosis of MMN and exclude other disorders that may mimic it. They mostly emphasize the mononeuropathy multiplex-like distribution of weakness, presence of multifocal motor conduction block, lack of sensory loss and lack of pyramidal signs. Table C25-1 shows recently accepted criteria for accurate diagnosis of MMN.
Table C25-1 Criteria for the Diagnosis of Multifocal Motor Neuropathy
| Core Criteria |
Definite multifocal motor neuropathy. Core and exclusion criteria with definite conduction block in two or more motor nerves. Probable multifocal motor neuropathy. Core and exclusion criteria with (1) probable conduction block in two or more motor nerves, or (2) definite conduction block in one motor nerve and probable conduction block in another motor nerve, or (3) definite conduction block in one motor nerve and at least one of the “other supportive criteria.”
Adopted with revisions from European Federation of Neurological Societies/Peripheral Nerve Society. Guideline on management of multifocal motor neuropathy. J Periph Nerv Syst 2006:11;1–8; Olney RK, Lewis RA, Putnam TD et al. Consensus criteria for the diagnosis of multifocal motor neuropathy. Muscle Nerve 2003;27:117–121.
Electrodiagnosis
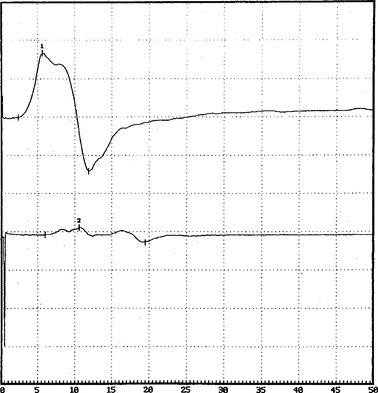
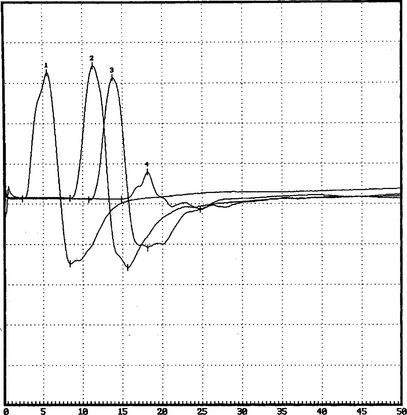
There are no uniformly accepted criteria for the identification of conduction block. Conduction block is defined as a decrease in the compound muscle action potential (CMAP) amplitude and area on proximal versus distal nerve stimulation, without evidence of significant temporal dispersion (i.e., prolongation of the CMAP duration; Figure C25-4). Table C25-2 lists recommended practical criteria for the diagnosis of conduction block, particularly in patients with suspected MMN. A detailed and meticulous nerve conduction study of multiple nerves and along many segments of these nerves are essential for the diagnosis of conduction block, which is a prerequisite for establishing the diagnosis of MMN. In general, CMAP amplitude and area decay should be less stringent when evaluating short nerve segments such as with the inching technique (Figure C25-5). This technique may allow precise localization of conduction block by finding an abrupt and focal reduction of CMAP area and amplitude over a very short segment of the nerve. It also helps excluding pseudoconduction block that may be associated with axonal loss and phase cancellation.

Table C25-2 Electrodiagnostic Criteria for Partial Conduction Block in Multifocal Motor Neuropathy*
* All amplitudes, areas, and durations reflect negative-peak areas, amplitudes, and durations comparing responses of proximal to distal stimulations.
† Conduction blocks at common entrapment sites are excluded.
‡ These requirements should be more stringent and sometimes cannot be included in nerves with very low distal CMAP amplitudes (<20% of the lower limit of normal or <1 mV).
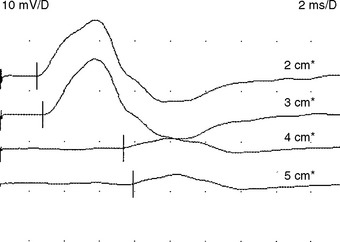
It is also important to emphasize avoiding over diagnosing conduction block. Table C25-3 reveals some common errors that are made in the EDX laboratory when attempting to diagnose conduction block. Two situations remain the most challenging and controversial. (1) Differentiating conduction block from abnormal temporal dispersion causes the most difficulty since temporal dispersion may result in CMAP amplitude and area reduction, due to the effects of phase cancellation. Computer analysis studies had suggested that a reduction of CMAP area of greater than 50% is always caused by a degree of conduction block. (2) Evaluating for conduction block in the context of axonal loss, such as in peripheral nerves with very low distal CMAPs (<20% of the lower limit of normal or <1 mV) is subject to error. Most proposed criteria intentionally restrict including these nerves to avoid confusion between conduction block and phase cancellation associated with axonal loss.
Table C25-3 Common False-Positive Results in the Diagnosis of Conduction Block
A finding that is unique to MMN is that sensory conductions across segments with motor conduction block are normal (Figure C25-6). Also, despite severe conduction block of the motor nerves, sensory conduction studies, which typically are performed in distal nerve segments (hand or foot), are normal. This distinctive characteristic helps to differentiate MMN from CIDP (and other acquired demyelinating neuropathies), in which the disruption of impulses usually affects both sensory and motor fibers.

Unsettled Issues
Are Antiglycolipid Antibodies Essential for Diagnosis?
High titers of anti-GM1 antibodies are often associated with MMN and decrease with successful treatment. However, the role of these antibodies in the pathogenesis of MMN remains unclear for several reasons. First, only about 50% of the patients with definite MMN have elevated antibodies. Second, elevated anti-GM1 antibodies are not specific for MMN since they may be seen in a variety of neuromuscular disorders, including Guillain-Barré syndrome, amyotrophic lateral sclerosis, and lower motor neuron disease. Third, sera from patients with MMN with or without anti-GM1 antibodies produce conduction block in animals. Fourth, most seronegative patients respond to IVIG and immune therapies in a fashion similar to GM1-positive patients. Hence, elevated titers of anti-GM1 antibodies are not considered a core component of the diagnostic criteria of MMN (see Table C25-1).
CIDP and Its Variants
Though MMN is a purely motor disorder, sensory symptoms are sometimes reported by patients and objective sensory loss may be seen in about 20% of patients. Also, subtle abnormalities in sensory NCSs and sural nerve biopsies may be noted in a few patients. These sensory abnormalities may suggest alternative diagnosis particularly the Lewis-Sumner syndrome (MADSAM), a variant of CIDP. In general, if the sensory nerve action potentials are unelicitable or are grossly abnormal, other demyelinating neuropathies, particularly the Lewis-Sumner syndrome and CIDP, must be interpreted (Table C25-4). The distinction between MMN and CIDP and the Lewis-Sumner syndrome is important since plasma exchange and corticosteroids are usually effective in CIDP and the Lewis-Sumner syndrome, while ineffective and sometimes harmful in MMN.
FOLLOW-UP
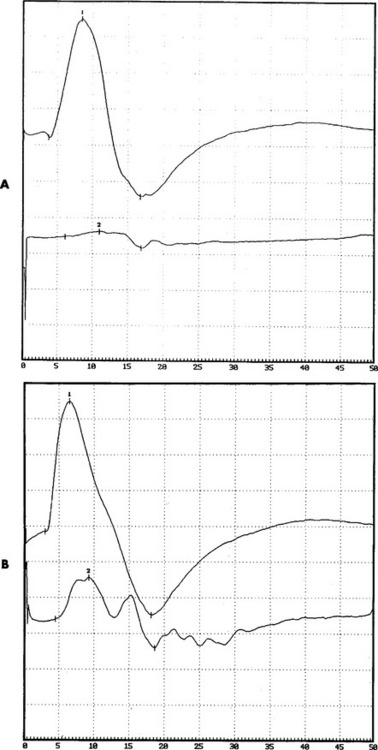
Antibody to GM1 was elevated moderately in the serum, with a titer of 1/1600 (normal = <1/800). Serum protein electrophoresis and immunofixation were normal. Cerebrospinal fluid examination revealed normal protein. The patient was infused with IVIG (total 2 g/kg), which resulted in dramatic improvement of the right shoulder and wrist and the left hand. This effect was maximal in 10 days and stabilized for 2 weeks, but the patient’s strength worsened again. She was given periodic IVIG every 4 weeks, with good results. Neurologic examination 6 months after the institution of therapy revealed significant improvement of the right upper extremity, particularly the biceps and wrist extensors, and of the left median innervated muscles. Repeat motor conduction showed significant improvement in conduction blocks (Figure C25-7). The patient has been maintained for the last 10 years on periodic IVIG every 6 to 8 weeks with no loss of effectiveness.
American Association of Electrodiagnostic Medicine. Consensus criteria for the diagnosis of partial conduction block. Muscle Nerve. 1999;22:S225-S229.
Boonyapisit K, Katirji B. Multifocal motor neuropathy with conduction block presenting with respiratory failure. Muscle Nerve. 2000;23:1887-1890.
Chaudhry V, et al. Multifocal motor neuropathy: response to human immune globulin. Ann Neurol. 1993;33:237-242.
Chaudhry V, Corse AM, Cornblath DR, et al. Multifocal motor neuropathy: electrodiagnostic features. Muscle Nerve. 1994;17:198-205.
Delmont E, Azulay JP, Girogi, et al. Multifocal motor neuropathy with or without conduction block: a single entity? Neurology. 2006;67:592-596.
European Federation of Neurological Societies/Peripheral Nerve Society. Guideline on management of multifocal motor neuropathy. J Periph Nerv Syst. 2006;11:1-8.
Kaji R, et al. Pathological findings at the site of conduction block in multifocal motor neuropathy. Ann Neurol. 1993;33:152-158.
Katirji B. Chronic relapsing axonal neuropathy responsive to intravenous immunoglobulins. Neurology. 1997;48:1690-1694.
Katz JS, Wolfe GI, Bryan WW, et al. Electrophysiologic findings in multifocal motor neuropathy. Neurology. 1997;48:700-707.
Kornberg AJ, Pestronk A. Chronic motor neuropathies: diagnosis, therapy, and pathogenesis. Ann Neurol. 1995;37(S1):S43-S50.
Krarup C, et al. A syndrome of asymmetric limb weakness with motor conduction block. Neurology. 1990;40:118-127.
Lewis RA. Multifocal motor neuropathy and Lewis-Sumner syndrome. Two distinct entities. Muscle Nerve 199;22: 1738–1739.
Lewis RA, et al. Multifocal demyelinating neuropathy with persistent conduction block. Neurology. 1982;32:958-964.
Nobile-Orazio E, Cappellari A, Priori A. Multifocal motor neuropathy: current concept and controversies. Muscle Nerve. 2005;31:663-680.
Oh SJ, et al. Multifocal demyelinating motor neuropathy: pathologic evidence of inflammatory demyelinating polyradiculoneuropathy. Neurology. 1995;45:1828-1832.
Olney RK, Lewis RA, Putnam TD, et al. Consensus criteria for the diagnosis of multifocal motor neuropathy. Muscle Nerve. 2003;27:117-121.
Parry GJ. Antiganglioside antibodies do not necessarily play a role in multifocal motor neuropathy. Muscle Nerve. 1994;17:97-99.
Parry GJ, Clarke S. Multifocal acquired demyelinating neuropathy masquerading as motor neuron disease. Muscle Nerve. 1988;11:103-107.
Pestronk A. Motor neuropathies, motor neuron disorders and antiglycolipid antibodies. Muscle Nerve. 1991;14:927-936.
Robers M, et al. Multifocal motor neuropathy human sera block distal motor nerve conduction in mice. Ann Neurol. 1995;38:111-118.
Terenghi F, Cappellari A, Bersano A, et al. How long is IVIG effective in multifocal motor neuropathy? Neurology. 2004;62:666-668.
Van Schaik IN, Bossuyt PM, Brand A, et al. Diagnostic value of GM1 antibodies in motor neuron disorders and neuropathies: a meta-analysis. Neurology. 1995;45:1570-1577.
Van Schaik IN, Van den Berg LH, de Haan R, et al. Intravenous immunoglobulin for multifocal motor neuropathy. Cochrane Database of Systematic Reviews. 2005:2.
[/level-membership-for-neurology-category][not-level-membership-for-neurology-category]
Case 25
HISTORY AND PHYSICAL EXAMINATION
| Right | Left | |
|---|---|---|
| Shoulder abduction | 2/5 | 5/5 |
| Elbow flexion | 3/5 | 5/5 |
| Elbow extension | 4−/5 | 5/5 |
| Pronation | 0/5 | 3/5 |
| Fingers flexion | 0/5 | 3/5 |
| Wrist flexion | 1/5 | 1/5 |
| Wrist extension | 2/5 | 5/5 |
| Finger extension | 3/5 | 4−/5 |
| Finger abduction | 4−/5 | 3/5 |
| Right | Left | |
|---|---|---|
| Hip flexion | 5/5 | 5/5 |
| Hip extension | 5/5 | 5/5 |
| Knee extension | 5/5 | 5/5 |
| Knee flexion | 5/5 | 5/5 |
| Foot dorsiflexion | 5/5 | 1/5 |
| Toe dorsiflexion | 5/5 | 0/5 |
| Plantar flexion | 5/5 | 5/5 |
| Ankle inversion | 5/5 | 5/5 |
| Ankle eversion | 5/5 | 1/5 |
Please now review the Nerve Conduction Studies and Needle EMG tables.
QUESTIONS
EDX FINDINGS AND INTERPRETATION OF DATA
Relevant EDX findings in this case include:
DISCUSSION
Definition and Pathogenesis
Multifocal motor neuropathy (MMN), described in the mid-1980s, is a rare disorder with a prevalence of 1 to 2 individuals per 100 000. It is characterized by specific EDX finding, i.e., motor conduction blocks, which is the gold standard for diagnosis. The disorder is important to recognize since it is treatable and responsive to immunomodulating therapies, and may mimic amyotrophic lateral sclerosis (ALS) which has a poor prognosis for survival.
