Case 24
HISTORY AND PHYSICAL EXAMINATION
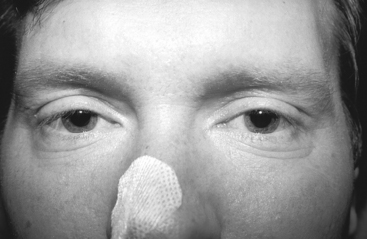
On examination on day 4, the patient was alert and intubated, and followed commands well. He had bilateral complete ophthalmoplegia to all gaze directions with bilateral ptosis (Figure C24-1). Pupils were dilated and unreactive to light or attempted accommodation. Corneal reflexes were depressed. The patient had bilateral peripheral facial weakness. His tongue was extremely weak, with no fasciculations. His palate did not move volitionally or to gagging. Neck flexors and extensors were weak (Medical Research Council [MRC] 3/5), as were proximal pelvic and shoulder girdle muscles (4/5). However, distal muscles were normal. Deep tendon reflexes were depressed (1/4). Sensation was normal. Cerebellar function also was normal.
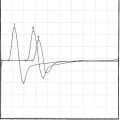
An electrodiagnostic (EDX) examination was requested.
Please now review the Nerve Conduction Studies and Needle EMG tables.
QUESTIONS
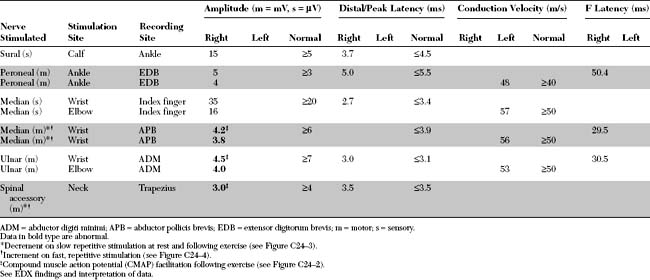
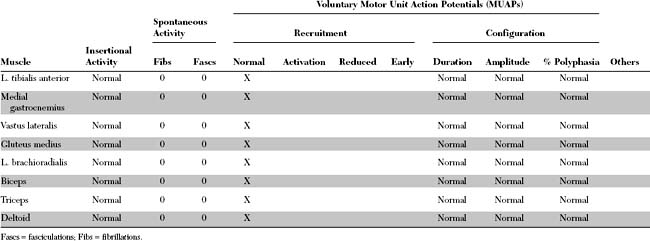
EDX FINDINGS AND INTERPRETATION OF DATA
The relevant EDX findings in this case are:
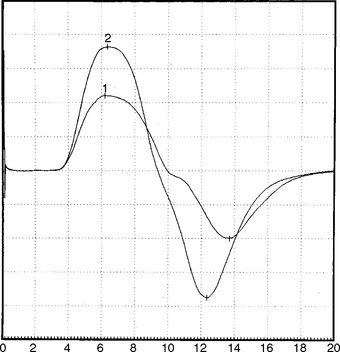
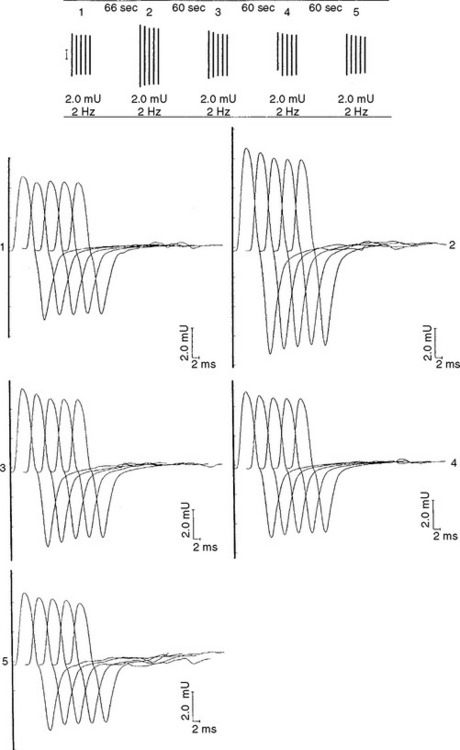

The EDX findings are consistent with a neuromuscular junction disorder of the presynaptic type, as supported by the borderline or low-amplitude baseline CMAPs, and the significant increment (>50%) of the CMAP amplitude after brief exercise and rapid repetitive stimulation of motor nerves. This case is consistent with botulism based on subacute progression of a descending muscle paralysis (ocular to bulbar to limbs), the muscarinic involvement (pupillary dilatation), and the EDX findings (presynaptic blockade). It is not consistent with Lambert-Eaton myasthenic syndrome because of the rapid evolution of symptoms, the prominent oculobulbar muscle weakness, and the relatively modest increment on rapid repetitive stimulation and after brief exercise (see electrodiagnosis).
DISCUSSION
Physiology and Pathophysiology
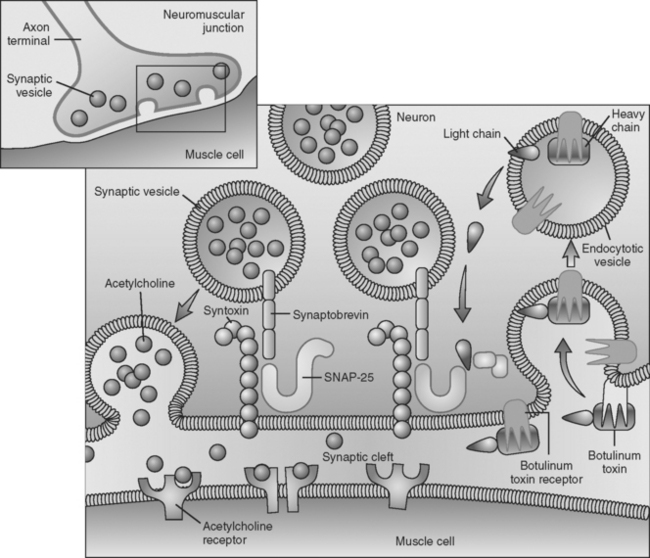
Botulinum toxin is an extremely potent toxin with doses as small as 0.05 to 0.1 μg causing death in humans. The toxin has significant affinity to both muscarinic and nicotinic cholinergic nerve terminals resulting in autonomic failure and skeletal muscle paralysis. The toxin results in failure of ACH release from the presynaptic terminal and ultimately leads to destruction of the presynaptic terminal. Botulinum toxin first attaches irreversibly to the axonal terminal, and enters via endocytosis without interfering with the calcium channel (calcium entry is not blocked by botulinum toxin). The toxin then interferes with the calcium-dependent intracellular cascade that is responsible for ACH release, by cleaving proteins essential for docking and fusion of the presynaptic vesicles at the presynaptic active zones. Electron microscopy of nerve endings exposed to the toxin reveal a “log jam” of vesicles in the presynaptic terminals. It is now known that various serotypes bind to different presynaptic proteins: botulinum toxin A and E hydrolyze synaptosomal-associated protein-25 (SNAP-25), a protein of the presynaptic membrane; botulinum toxins B, D, F, and G specifically cleave synaptobrevin, a membrane protein of the neurotransmitter-containing vesicles; botulinum toxin C cleaves both SNAP-25 and syntaxin, a nerve plasmalemma protein (Figure C24-5). Because the ultimate result of this intoxication is interference with neurotransmitter release (exocytosis of synaptic vesicles) and destruction of the nerve terminals, recovery of neurologic function is protracted since it is dependent on the regrowth of sprouts from the injured nerve terminal.
Clinical Features
The diagnosis of botulism may be difficult and requires a high index of suspicion. Many cases go unrecognized and are diagnosed with various neuromuscular, medical, and even psychiatric diagnoses (Table C24-1). The diagnosis is relatively easy in epidemics, or if two or more cases are identified simultaneously. Botulism should be suspected when there is:
Table C24-1 Diagnosis of 31 Previously Unrecognized Canadian Patients With Botulism After the Identification of Two Teenaged Sisters With Type B Botulism*
| Discharge Diagnosis | Number of Patients |
|---|---|
| Myasthenia gravis | 7 |
| Psychiatric illness† | 4 |
| Viral syndrome | 4 |
| Botulism‡ | 3 |
| Stroke | 3 |
| Guillain-Barré syndrome | 3 |
| Inflammatory myopathy | 2 |
| Diabetic complications | 1 |
| Hyperemesis gravidarum | 1 |
| Hypothyroidism | 1 |
| Laryngeal trauma | 1 |
| Overexertion | 1 |
* The outbreak was subsequently identified as spoiled commercial chopped garlic in soybean oil.
† Includes hysteria, agitated depression, separation reaction, and factitious weakness.
‡ All three patients were family members whose diagnosis represented the initial recognition of the outbreak.
Data from St. Louis ME et al. Botulism from chopped garlic: delayed recognition of a major outbreak. Ann Intern Med 1988;108:363–368.
The diagnosis of botulism is confirmed by:
Electrodiagnosis
The EDX studies provide a rapid evidence of botulism awaiting the bioassay and stool cultures. The latter two tests may also be negative. The EDX findings in botulism are compatible with a presynaptic defect of the neuromuscular junction (see electrodiagnosis in Cases 17 and 21). The findings are as follows:
Table C24-2 Electrophysiological Differences Between Two Common Presynaptic Neuromuscular Disorders (Botulism and Lambert-Eaton Myasthenic Syndrome)
| Electrophysiology | Botulism | Lambert-Eaton Myasthenic Syndrome |
|---|---|---|
| Baseline CMAPs | Low in amplitudes, particularly in proximal and weak muscles | Low in amplitudes in all muscles |
| CMAP increment | Present in clinically affected muscles | Present in all muscles |
| Degree of CMAP increment | Moderate (30–100%) | Marked (>200%) |
Although the clinical presentations of LEMS and botulism are quite different, their EDX findings are similar, but with certain distinctions, since both are due to a presynaptic defect of ACH release (see Table C24-2).
FOLLOW-UP
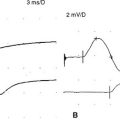
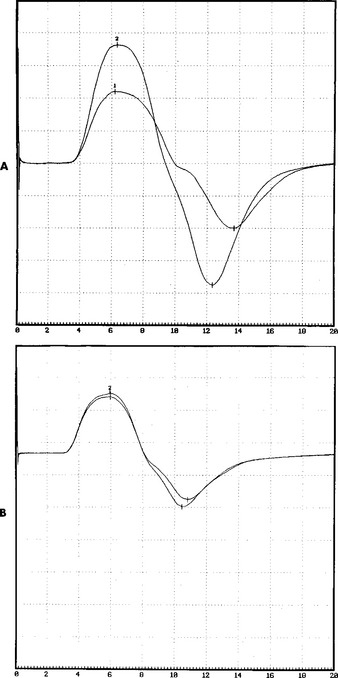
When the patient was seen 6 months after the onset of illness, he complained of easy fatigability and poor endurance. He had no appreciable muscle atrophy and minimal weakness of proximal and neck muscles (MRC 5-/5), and he had regained all deep tendon reflexes. Pupils were normal. The patient returned to work 1 month later. Examination 1 year later revealed no abnormality. Baseline CMAPs returned to normal, with absence of increment after rapid repetitive stimulation or postexercise facilitation (Figure C24-6).
Cherrington M. Botulism. Ten-year experience. Arch Neurol. 1974;30:432-437.
Cherrington M. Electrophysiologic methods as an aid in diagnosis of botulism: a review. Muscle Nerve. 1982;5:528-529.
Cherington M. Botulism: update and review. Semin Neurol. 2004;24:155-163.
Cornblath DR, Sladky JT, Sumner AJ. Clinical electrophysiology of infantile botulism. Muscle Nerve. 1983;6:448-652.
Hallet M. One man’s poison: clinical applications of botulinum toxin. N Engl J Med. 1999;341:118-120.
Hunter WB. Snappy exocytoxins. Nature. 1993;365:104-105.
Katirji B, Kaminski HJ. Electrodiagnostic approach to the patient with suspected neuromuscular junction disorder. Neurol Clin. 2002;20:557-586.
Pickett JB. Infant botulism: the first five years. Muscle Nerve. 1982;5:S26-S27.
Pickett J, et al. Syndrome of botulism in infancy: clinical and electrophysiologic study. N Engl J Med. 1976;295:770-772.
Robinson RF. Management of botulism. Ann Pharmacother. 2003;37:127-131.
Shapiro RL, Hatheway C, Swerdlow DL. Botulism in the United States: a clinical and epidemiologic review. Ann Intern Med. 1998;129:221-228.
St. Louis ME, et al. Botulism from chopped garlic: delayed recognition of a major outbreak. Ann Intern Med. 1988;108:363-368.