Case 22
HISTORY AND PHYSICAL EXAMINATION
An electrodiagnostic (EDX) examination was performed.
Please now review the Nerve Conduction Studies and Needle EMG tables.
QUESTIONS
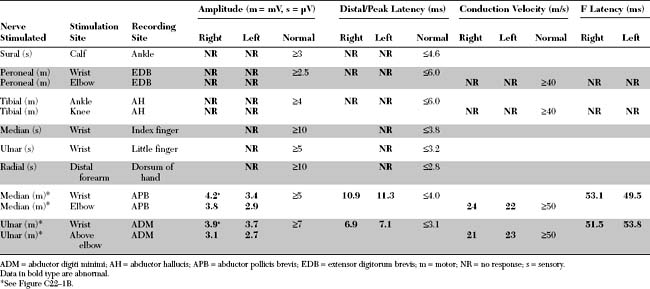
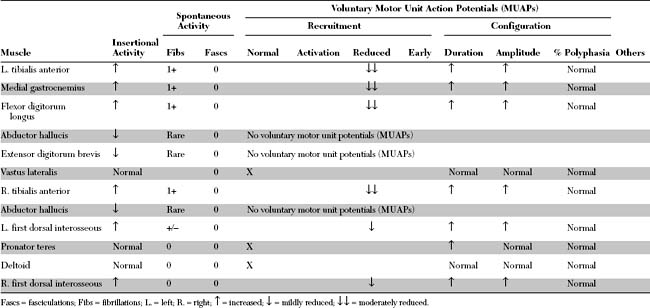
EDX FINDINGS AND INTERPRETATION OF DATA
Relevant EDX findings in this case include:
DISCUSSION
Classification
Hereditary neuropathies are a heterogeneous group of peripheral nerve disorders (Figure C22-2). Some have a known metabolic basis and potential therapies (Table C22-1). The hereditary neuropathies that are not based on known specific metabolic defect are classified into three clinical groups: (1) hereditary motor and sensory neuropathies (HMSNs); (2) hereditary sensory and autonomic neuropathies (HSANs); and (3) hereditary motor neuropathies (HMNs).
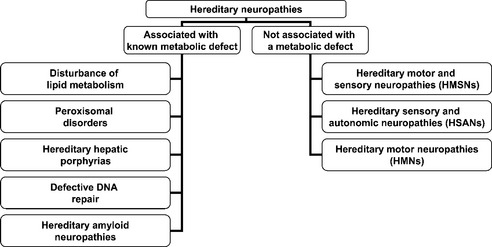
Figure C22-2 Classification of hereditary neuropathies.
(Adapted, with revisions, from Thomas PK. Classification and electrodiagnosis of hereditary neuropathies. In: Brown WF, Bolton CF, eds. Clinical electromyography, 2nd ed. Boston, MA: Butterworth-Heinemann, 1993, pp. 391–425.)

The hereditary motor and sensory neuropathies (HMSNs) were classified by Dyck and Lambert into three predominant types (1) HMSN I, a demyelinating type; (2) HMSN II, a neuronal (axonal) type; and (3) HMSN III (Dejerine-Sottas disease), a severe demyelinating neuropathy of infancy and early childhood. HMSN I and II are characterized by skeletal deformities (pes cavus, hammer toes, scoliosis), insidious onset of distal lower more than upper extremities weakness, atrophy and sensory loss, and reduced or absent deep tendon reflexes. Based on clinical examination, these disorders are difficult to distinguish from each other because of similar phenotypes. With the recent influence of chromosomal linkage and gene identification, the term Charcot-Marie-Tooth disease (CMT) reemerged which created some confusion in the nomenclature and classifications of these disorders.
Charcot-Marie-Tooth disease (CMT) is subdivided into six major types with some but not perfect correlation to the HMSN classification (Table C22-2). CMT1 and CMT2 are interchangeable with HMSN I and HMSN II. The name Dejerine-Sottas syndrome (DSS, Dejerine-Sottas disease) is preserved and is the same condition as HMSN III. The term CMT3 is not commonly used since the genes involved with DSS are the same as CMT1. CMT4 is a new designation for a group of autosomal recessive CMT and should not be confused with HMSN IV which is Refsum disease. CMTX is an X-linked disorder and hereditary neuropathy with liability to pressure palsy (HNPP) is a distinct disorder characterized by recurrent mononeuropathies.
Table C22-2 Charcot-Marie-Tooth Disease (CMT, Hereditary Motor and Sensory Neuropathy, HMSN) Subtypes, Its Variants and Their Genetic Causes
| Disorder | Locus/Gene | Protein |
|---|---|---|
| Charcot-Marie-Tooth Disease Type 1 (CMT1, HMSN I, Autosomal-Dominant, Demyelinating) | ||
| CMT1A | 17p11.2-12/PMP22* | Peripheral myelin protein 22 |
| CMT1B | 1q22-23/MPZ | Myelin protein zero |
| CMT1C | 16p13.1-12.3/LITAF | SIMPLE |
| CMT1D | 10q21.1-22.1/EGR2 | Early growth response protein 2 |
| Charcot-Marie-Tooth Disease Type 2 (CMT2, HMSN II, Autosomal-Dominant, Axonal) | ||
| CMT2A | 1p35-36/KFI1B | Kinesin-like protein Mitousin 2 |
| CMT2B | 3q13-22/RAB7 | Ras-related protein |
| CMT2C | 12q23-24/? | ? (unknown) |
| CMT2D | 7p15/GARS | Glycy-tRNA synthetase |
| CMT2E | 8P21/NEFL | Neurofilament triplet L protein |
| CMT2F | 7q11-21/HSP27 | Small heat shock protein |
| CMT2 | 1q22/MPZ | Myelin protein zero |
| Dejerine-Sottas Syndrome (DSS, HMSN III, CMT3, Autosomal-Dominant or Recessive, Demyelinating) | ||
| DSS A | 17p/PMP22 | Peripheral myelin protein 22 |
| DSS B | 1q/MPZ | Myelin protein zero |
| DSS C | 10q/EGR2 | Early growth response protein 2 |
| DSS D | 8q23 | Unknown |
| Charcot-Marie-Tooth Disease Type 4 (CMT4, Autosomal-Recessive, Axonal or Demyelinating) | ||
| CMT4A | 8q13-21/GDAP1 | Ganglioside-induced differentiation-associated protein-1 |
| CMT4B1 | 11q22/MTMR2 | Myotubularin-related protein-2 |
| CMT4B2 | 11p15/MTMR13 | Myotubularin-related protein-13 |
| CMT4C | 5q23-33/KIAA1985 | − |
| CMT4D | 8q24.3/NDRG1 | N-myc downstream-regulated gene-1 |
| CMT4E | 10q21.1-22.1/EGR2 | Early growth response protein 2 |
| CMT4F | 19q13.1-13.2/PRX | Periaxin gene |
| X-linked Charcot-Marie-Tooth Disease (CMTX, Axonal or Demyelinating) | ||
| CMTX (X-linked) | Xq13-q21/CX32(GJB1) | Connexin 32 (gap junction protein-β-1) |
| Hereditary Neuropathy with Liability to Pressure Palsy (HNPP, Demyelinating) | ||
| HNPP (autosomal dominant) | 17p11.22/PMP22† | Peripheral myelin protein 22 |
* Duplication (98%) and point mutation (2%).
† Deletion (80%) and point mutation (20%).
The hereditary sensory and autonomic neuropathies (HSANs) are very rare and familial neuropathies with selective involvement of the primary sensory, with or without the autonomic, fibers. They should be distinguished from inherited disorders that affect large primary afferent neurons (spinocerebellar degeneration). They are currently subdivided into five types, based on mode of inheritance, natural history, electrophysiologic characteristics and histopatologic findings (Table C22-3).
Table C22-3 Hereditary Sensory and Autonomic Neuropathy (HSAN)
| Disorder | Locus/Gene | Protein |
|---|---|---|
| HSAN I (hereditary sensory radicular neuropathy) | 9q22.1-q22.3/SPTLC1 | Serine-palitoyltransferase-1 |
| HSAN II (congenital sensory neuropathy) | 12p13-33 | |
| HSAN III (familial dysautonomia, Riley-Day syndrome) | 9q31-33/IKBKAP | Inhibitor of kappaB-kinase complex associated polypeptide |
| HSAN IV (congenital sensory neuropathy with anhidrosis) | 1q21-22/TRKA | |
| HSAN V | 1q21-22/NTRK1 |
The hereditary motor neuropathies (HMNs) are loosely subdivided into proximal and distal (Table C22-4). The proximal HMNs are better known as spinal muscular atrophies (SMAs). These are among the most common autosomal recessive disorders in childhood affecting 1/10 000 live births with carrier frequency of 1/50. Spinal muscular atrophy is caused by a deficiency of the ubiquitous survival motor neuron (SMN) protein, which is encoded by the SMN genes, SMN1 and SMN2, on chromosome 5q. The distal HMNs are a genetically and clinically heterogeneous group of disorders that are also known as spinal CMT because of their overlap with CMT. They are characterized by distal weakness with or without foot deformities, but without sensory or autonomic involvement. Sensory nerve action potentials are normal while the motor NCSs reveal low-amplitude CMAPs with normal or borderline velocities, consistent with motor axonopathy. The inheritance of HMNs is either dominant or recessive. Only few have been mapped to a chromosome or have a defined gene mutation.
Table C22-4 Hereditary Motor Neuropathy (HMN)
| Proximal HMN (Spinal Muscular Atrophy, SMA, Autosomal-Recessive, Mutations of the Survival Motor Neuron 1 (SMN 1) Gene on Chromosome 5q13) | |
| SMA I | Werdnig-Hoffmann disease. Onset before the age of 6 months, inability to sit or walk, and fatal before the age of 2 years |
| SMA II | Intermediate, arrested Werdnig-Hoffmann disease. Onset between 6 and 18 months of age, able to sit but not walk and survive beyond the age of 4 years |
| SMA III | Kugelberg-Welander disease |
| SMA IIIa | Onset between the age of 2 to 3 years, survive into adulthood and able to walk independently usually until age 20–40 years |
| SMA IIIb | Onset after the age of 3 years and able to walk independently till age 30–50 years |
| SMA IV | Adult SMA. Variable age of onset, but rarely before the age of 20 years and usually after the age of 30 years |
| Bulbospinal (Kennedy Disease, X-Linked CAG Repeat Expansion of the Androgen Receptor Gene on Chromosome Xq13.1) Distal HMN (Spinal Form of Charcot-Marie-Tooth Disease) | |
| HMN I | Juvenile onset, autosomal dominant |
| HMN II | Adult onset, autosomal dominant (12q24) |
| HMN III | Mild juvenile, autosomal recessive (11q13) |
| HMN IV | Severe juvenile, autosomal recessive |
| HMN V | Upper limb predominance, autosomal dominant (7p) |
| HMN VI | Severe infantile with respiratory distress, autosomal recessive |
| HMN VII | Vocal cord paralysis, autosomal dominant (9p21.1-p12) |
| Scapuloperoneal | |
| Type I | Autosomal dominant |
| Type II | Autosomal recessive |
| Bulbar | |
| Type I | Autosomal recessive (Vialetto-Van Laere syndrome) |
| Type II | Autosomal recessive (Fazio-Londe disease) |
Clinical Features and Genetics
Charcot-Marie-Tooth Disease 1 (CMT1, HMSN I)
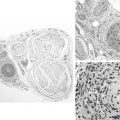
Common findings on examination include distal muscle weakness, atrophy, distal areflexia, pes cavus, and hammer toes (Figure C22-3). The atrophy is predominant in the foot but may extend into the distal legs, resulting in an “inverted champagne bottle” appearance to the leg, and into the hands, resulting in “claw hands.” Although most patients do not complain of positive sensory symptoms, there is distal loss of all sensory modalities. Pain, other than that related to foot deformity and callus formation, is rare. Scoliosis is present in a minority of patients. Enlarged and palpable peripheral nerves may be identified in some patients with HMSN I. Late in the disease, steppage gait and claw hands are common. Although the disorder is frequently disabling, the life expectancy of patients with the disease is normal.
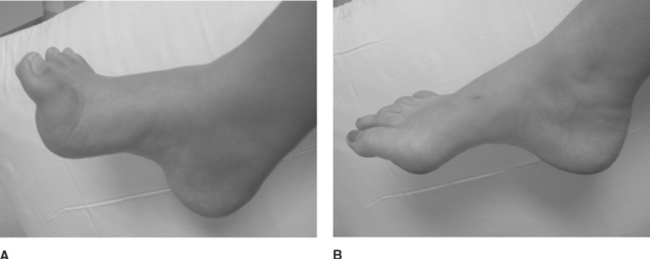
Molecular and genetic studies have further subdivided CMT1 into four subtypes, with no definitive phenotypic characteristics that could accurately distinguish among them. These are named 1A, 1B, 1C, and 1D (see Table C22-2). CMT1A is the most common inherited neuropathy. Most patients have a tandem duplication of a 1.5 Mb region, which contains the peripheral myelin protein-22 (PMP22) gene, on chromosome 17p11.2p12. Duplication of PMP22 gene leads to overexpression (increased dosage) of the peripheral myelin protein. Occasional patients have point mutations of the PMP22 gene complex. CMT1B is associated with mutations of the myelin protein zero (MPZ) gene located on chromosome 1q22-23. The exact function of PMP22 and MPZ is not well understood, but both proteins are integral parts that likely play a major role in myelin compaction. CMT1C and CMT1D have been mapped to chromosome 16p and 10q with gene loci, named LITAF and EGR2, respectively.
Charcot-Marie-Tooth Disease 2 (CMT2, HMSN II)
Hereditary motor and sensory neuropathy II (HMSN II, CMT2) is a heterogeneous group of inherited neuropathies that are due to primary axonal degeneration. They are not distinguishable from CMT1 except by having preserved conduction velocities (>38 m/s) and absence of onion bulb formation on nerve biopsy. In contrast to CMT1, CMT2 phenotypes do not have palpable or enlarged nerves, but tend to have a later age of onset, diffuse areflexia and less involvement of hand muscles. CMT2 is divided into several subtypes based on gene locus and product (see Table C22-2).
Charcot-Marie-Tooth Disease 4 (CMT4)
Autosomal-recessive forms of hereditary neuropathies are rare, usually present in small ethnic groups, and named collectively CMT4 (see Table C22-2). These disorders may present in infancy and childhood with delayed motor milestones, severe neuropathies, and areflexia. Some patients become wheelchair bound by adulthood.
Electrodiagnosis
Table C22-5 Electrophysiologic Characteristics of Demyelinating Polyneuropathy
| Inherited (e.g., CMT1) | Acquired (e.g., CIDP) |
|---|---|
| Diffuse slowing | Multifocal slowing |
| Symmetrical slowing | Asymmetrical slowing |
| No conduction block | Frequent conduction blocks |
| Slight temporal dispersion | Prominent temporal dispersion |
CIDP = chronic inflammatory demyelinating polyneuropathy; CMT1 = Charcot-Marie-Tooth disease 1.
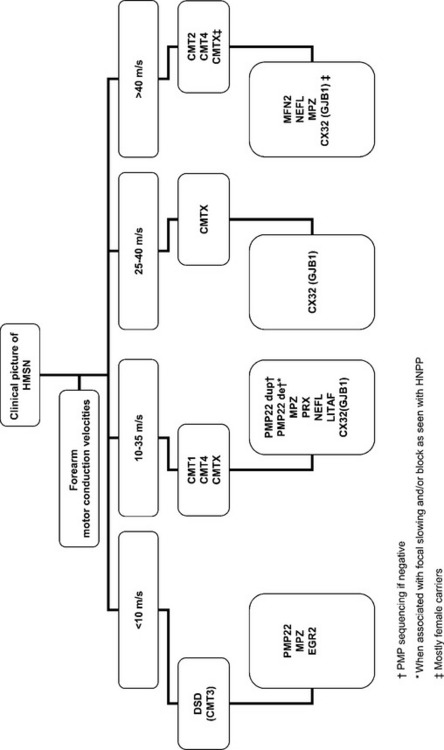
In summary, hereditary demyelinating neuropathy are characterized by diffuse, uniform, and symmetrical slowing, without conduction block or temporal dispersion. Common causes of hereditary demyelinating peripheral neuropathy with uniform slowing are shown in Table C22-6.
Table C22-6 Hereditary Demyelinating Peripheral Neuropathy Associated With Prominent and Uniform Slowing of Conduction Velocities
* Often associated with multifocal slowing and/or conduction block at common entrapment sites.
Buchthal F, Behse F. Peroneal muscular atrophy and related disorders: I. Clinical manifestations as related to biopsy findings, nerve conduction and electromyography. Brain. 1977;100:41-66.
Garcia CA, et al. Clinical variability in two pairs of identical twins with the Charcot-Marie-Tooth disease type 1A duplication. Neurology. 1995;45:2090-2093.
Harding AE. Inherited neuronal atrophy and degeneration predominantly of lower motor neurons. In: Dyck PJ, et al, editors. Peripheral neuropathy. Philadelphia, PA: WB Saunders, 1993.
Harding AE, Thomas PK. The clinical features of hereditary motor and sensory neuropathy, types I and II. Brain. 1980;103:259-280.
Lewis RA, Sumner AJ. The electrodiagnostic distinctions between chronic familial and acquired demyelinative neuropathies. Neurology. 1982;32:592-596.
Scherer SS. Findings the causes of inherited neuropathies. Arch Neurol. 2006;63:812-816.
Shy ME, Lupski JR, Chance PF, et al. Hereditary motor and sensory neuropathies: an overview of clinical, genetic, electrophysiologic, and pathologic features. In: Dyck PJ, Thomas PK, editors. Peripheral neuropathy. 4th ed. Philadelphia, PA: WB Saunders; 2005:1623-1658.