[level-membership-for-neurology-category]
Case 21
HISTORY AND PHYSICAL EXAMINATION
Please now review the Nerve Conduction Studies and Needle EMG tables.
QUESTIONS
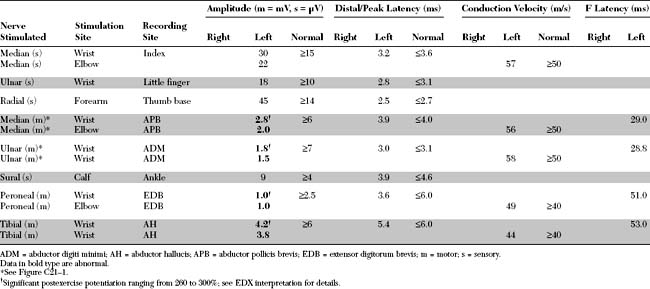
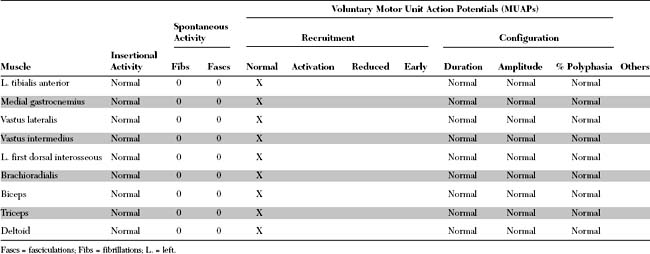
EDX FINDINGS AND INTERPRETATION OF DATA
Relevant EDX findings in this case include:
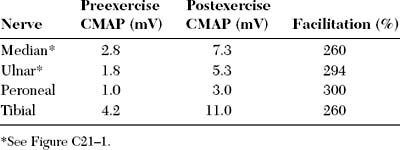
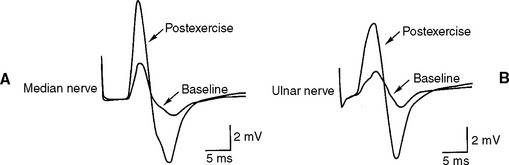

DISCUSSION
Clinical Features
Lambert-Eaton myasthenic syndrome, also referred to as the myasthenic syndrome, affects primarily adults older than 40 years of age, with a slight predilection to men. Patients present with proximal muscle weakness (especially of the lower extremities) and minimal ocular and bulbar weakness, and are susceptible to fatigue. Deep tendon reflexes are characteristically absent or reduced. Autonomic complaints (especially dry mouth) and transient paresthesias may also occur. A helpful and distinctive clinical finding is muscle facilitation: after a brief period (∼10 seconds) of intensive exercise of a muscle, muscle power is much transiently stronger and the deep tendon reflex to that muscle is enhanced. Unfortunately, this sign cannot always be confirmed during bedside evaluation. Figures C21-3 and C21-4 show the common signs and symptoms of LEMS patients, based on series of 50 patients.
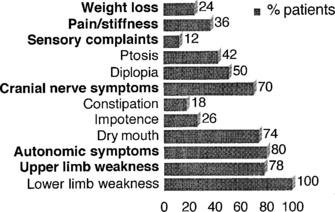
Figure C21-3 Lambert-Eaton myasthenic syndrome. Symptoms during the course of illness in 50 cases.
(Adapted from O’Neil JH, Murray NMF, Newsom-Davis J. The Lambert-Eaton myasthenic syndrome: a review of 50 cases. Brain 1988:11:577–596.)
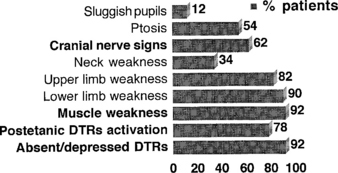
Figure C21-4 Lambert-Eaton myasthenic syndrome. Signs during the course of illness in 50 cases.
(Adapted from O’Neil JH, Murray NMF, Newsom-Davis J. The Lambert-Eaton myasthenic syndrome: a review of 50 cases. Brain 1988;11:577–596.)
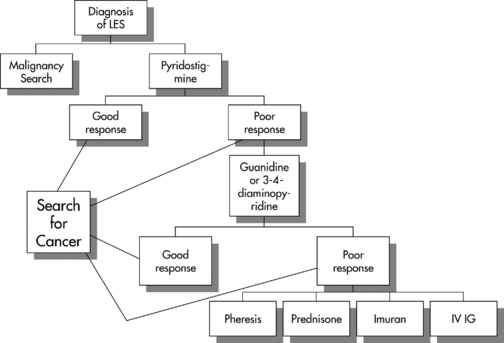
Treatment of LEMS is difficult. Treatment of the primary cancer is essential but seldom results in improvement of the neurologic symptoms. Figure C21-5 outlines a practical algorithmic treatment plan of weakness for patients with LEMS. Measures to combat weakness include:
Electrodiagnosis
Nerve Conduction Studies
Nerve conduction studies (NCSs) in LEMS reveal normal sensory responses. However, motor NCSs disclose low or borderline-low CMAP amplitude in all motor nerves (discussed in a later section). These are usually associated with normal distal latencies, conduction velocities, and F wave latencies.
In general, low-amplitude CMAPs with normal SNAPs are infrequent findings in the EMG laboratory, especially when the findings are diffuse (i.e., every motor NCS has low CMAP, and every sensory NCS reveals normal SNAP). Figure C21-6 outlines the common site of pathology in patients manifesting low-amplitude CMAP responses in all or most motor nerves. These disorders are distinguished by a detailed needle EMG and repetitive nerve stimulation. Table C21-1 lists the common causes of such findings, as seen in the EMG laboratory.
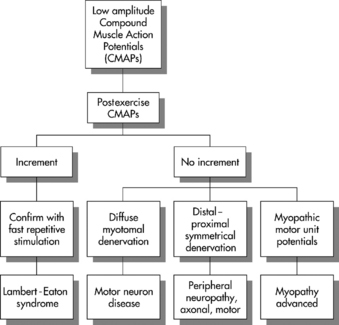
Table C21-1 Causes of Diffuse Low-Amplitude CMAPs (Compound Muscle Action Potentials) and Normal SNAPs (Sensory Nerve Action Potentials)
Repetitive Nerve Stimulation
RNS in Healthy Individuals
Slow or fast rates of motor nerve stimulation do not abolish any endplate potential (EPP); all remain above threshold because of the presence of a “safety factor” (many more quanta (vesicles) are released with a single stimulus than are needed to generate an EPP). Thus, the CMAP (= summated muscle fiber action potentials, MFAPs) does not change (no decrement). After rapid RNS or following brief exercise, there usually is a slight physiologic increment of the CMAP, which does not exceed 25% of the baseline CMAP. This physiologic post-tetanic facilitation is believed to be caused by increased synchrony of MFAPs after tetanic stimulation (Figures C21-7 and C21-8).
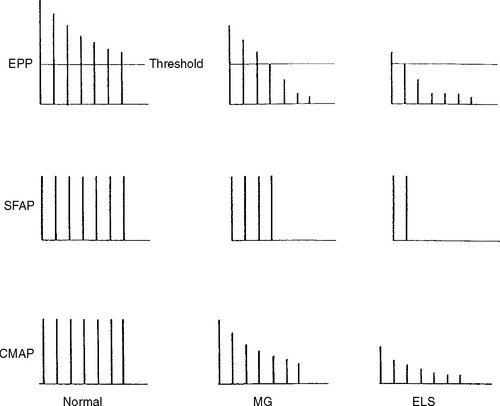
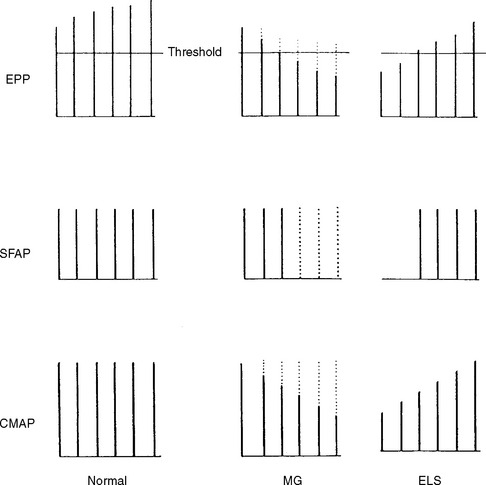
RNS in Patients With Lambert-Eaton Myasthenic Syndrome
RNS in Patients With Myasthenia Gravis (MG)
Single-Fiber EMG
Single-fiber jitter analysis is abnormal with frequent blocking in LEMS, as well as in myasthenia gravis. It is difficult to distinguish MG from LEMS using recruited (voluntary) single-fiber EMG because both disorders lead to a prolonged jitter, with or without blocking (for details, refer to Case 17). However, with stimulation jitter techniques, one can differentiate LEMS from myasthenia. Using a rapid rate of stimulation (>10 Hz), block or jitter or both improve significantly in LEMS owing to enhancement of ACH release by the influx of Ca2+ into the presynaptic terminal (Figure C21-9). However, at this rate of stimulation, the jitter does not change or worsen in myasthenia gravis.
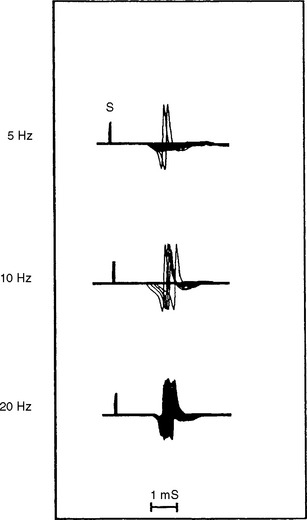
Conclusion
The EDX findings in LEMS include low-amplitude CMAP, increment of CMAP after brief exercise, and increment of CMAP after rapid RNS. Table C21-2 summarizes the EDX findings in LEMS and myasthenia gravis.

The diagnosis of LEMS must be considered and excluded in all patients whose nerve conduction studies show low or borderline-low baseline CMAP amplitudes at rest, with normal sensory responses. Low or borderline-low CMAP amplitudes at rest should be followed by a repeat distal nerve stimulation after 10 seconds of exercise to exclude the possibility of LEMS. Assessing CMAP after brief exercise in patients with suspected LEMS is as accurate as results obtained after rapid RNS. It has the advantage of being much less painful and thus can be done on many motor nerves. It is recommended that postexercise CMAP is performed on several motor nerves, as a screening, in patients with suspected LEMS. If postexercise facilitation is present, then one motor nerve (such as a median or ulnar nerve) is stimulated with a rapid train (20–50 Hz) to verify the diagnosis.
Patients with LEMS are often misdiagnosed as MG. This occurs when slow RNSs (2–3 Hz) are only performed and result in CMAP decrements that are frequent and common finding in MG and LEMS. Most of these patients have low amplitude or borderline CMAPs on NCSs that are overlooked. Repeating distal nerve stimulation after 10 seconds of exercise on all nerves with low amplitude or borderline CMAPs, which is often done prior to slow RNS, should exclude or confirm the diagnosis of LEMS. If postexercise facilitation is present, a rapid RNS would be then done for confirmation. Table C21-3 lists the common differentiating clinical and electrophysiologic features of LEMS and MG.
Table C21-3 Differential Diagnosis Between Generalized Myasthenia Gravis and Lambert-Eaton Myasthenic Syndrome
| Myasthenia Gravis | Lambert-Eaton Myasthenic Syndrome | |
|---|---|---|
| Ocular involvement | Common and prominent | Uncommon and subtle |
| Bulbar involvement | Common and prominent | Uncommon and subtle |
| Deep tendon reflexes | Normal | Absent or depressed |
| Sensory symptoms | None | Paresthesias are common |
| Autonomic involvement | None | Dry mouth, impotence and gastroparesis |
| Tensilon test | Frequently positive | May be positive |
| Serum antibodies directed against | Postsynaptic Ach receptors or MuSK | Presynaptic voltage-gated calcium channels |
| Baseline CMAPs | Normal | Low in amplitude |
| Postexercise CMAPs | No change | Significant facilitation (>50–100%)* |
| Slow repetitive stimulation | Decrement | Decrement |
| Rapid repetitive stimulation | No change or decrement | Increment† |
| Single-fiber EMG | Increased jitter with blocking | Increased jitter with blocking |
| Rapid-rate stimulation jitter | Does not change or worsens jitter | Improves jitter‡ |
Ach = acetylcholine; CMAPs = compound muscle action potentials; EMG = electromyography; MuSK = muscle-specific kinase.
† See Figure C21-2 (bottom tracing).
FOLLOW-UP
A review of the CT scan performed earlier revealed a small mass in the azygoesophageal recess. A barium swallow confirmed the presence of an extrinsic indentation of the lower esophagus. After an unrevealing bronchoscopy, a right thoracotomy was performed. The mass was consistent with SCLC, with 9 of 27 positive lymph nodes. An extensive search for distant metastases was negative. The patient underwent radiation therapy and a 6-month course of chemotherapy. Her muscle weakness did not respond despite 3 months of plasmapheresis (twice per week) and pyridostigmine, 120 mg every 3 hours. After the completion of chemotherapy, she was placed on guanidine, and the dose was increased to 500 mg qid, with no effect. Prednisone, 80 mg daily, was added, also with no beneficial result.
Chalk CH, et al. Response of the Lambert-Eaton myasthenic syndrome to treatment of associated small-cell lung carcinoma. Neurology. 1990;40(10):1552-1556.
Eaton LM, Lambert EH. Electromyography and electric stimulation of nerves in diseases of motor unit: observations on myasthenic syndrome associated with malignant tumors. JAMA. 1957;163:1117-1124.
Hughes R, Katirji MB. The Eaton-Lambert (myasthenic) syndrome in association with systemic lupus erythematosus. Arch Neurol. 1986;43:1186-1187.
Jablecki C. Lambert-Eaton myasthenic syndrome. Muscle Nerve. 1984;7:250-257.
Katirji B. Lambert-Eaton myasthenic syndrome: a harbinger to transitional cell carcinoma of the urinary bladder. J Clin Neuromusc Dis. 2000;1:134-136.
Lambert EH, Eaton LM, Rooke ED. Defect of neuromuscular conduction associated with malignant neoplasms. Am J Physiol. 1956;187:612-613.
Lennon VA, et al. Calcium-channel antibodies in the Lambert-Eaton syndrome and other paraneoplastic syndromes. N Engl J Med. 1995;332:1467-1474.
Leys K, et al. Calcium channel autoantibodies in the Lambert-Eaton myasthenic syndrome. Ann Neurol. 1991;29(3):307-314.
Maddison P, Newsom-Davis J. The Lambert-Eaton myasthenic syndrome. In: Katirji B, Kaminski HJ, Preston DC, Ruff RL, Shapiro EB, editors. Neuromuscular disorders in clinical practice. Boston, MA: Butterworth-Heinemann; 2002:931-941.
Maddison P, Newsom-Davis J, Mills KR. Distribution of electrophysiological abnormality in Lambert-Eaton myasthenic syndrome. J Neurol Neurosurg Psychiatry. 1998;65:213-217.
Maddison P, Newsom-Davis J, Mills KR, et al. Favourable prognosis in Lambert-Eaton myasthenic syndrome and small-cell lung carcinoma. Lancet. 1999;353:117-118.
McEvoy KM, et al. 3,4-Diaminopyridine in the treatment of Lambert-Eaton syndrome. N Engl J Med. 1989;321:1567-1571.
O’Neil JH, Murray NMF, Newsom-Davis J. The Lambert-Eaton myasthenic syndrome: a review of 50 cases. Brain. 1988;111:577-596.
Saunders DB. Lambert-Eaton myasthenic syndrome: clinical diagnosis, immune-mediated mechanisms and update on therapies. Ann Neurol. 1995;37(S1):S63-S73.
Tim RW, Saunders DB. Repetitive nerve stimulation studies in Lambert-Eaton myasthenic syndrome. Muscle Nerve. 1994;17:995-1001.
Ueno S, Hara Y. Lambert-Eaton myasthenic syndrome without anti-calcium channel antibody: adverse effect of calcium antagonist, diltiazem. J Neurol Neurosurg Psychiatry. 1992;55(5):409-410.
[/level-membership-for-neurology-category][not-level-membership-for-neurology-category]
Case 21
HISTORY AND PHYSICAL EXAMINATION
Please now review the Nerve Conduction Studies and Needle EMG tables.
QUESTIONS
EDX FINDINGS AND INTERPRETATION OF DATA
Relevant EDX findings in this case include:
DISCUSSION
Clinical Features
Lambert-Eaton myasthenic syndrome, also referred to as the myasthenic syndrome, affects primarily adults older than 40 years of age, with a slight predilection to men. Patients present with proximal muscle weakness (especially of the lower extremities) and minimal ocular and bulbar weakness, and are susceptible to fatigue. Deep tendon reflexes are characteristically absent or reduced. Autonomic complaints (especially dry mouth) and transient paresthesias may also occur. A helpful and distinctive clinical finding is muscle facilitation: after a brief period (∼10 seconds) of intensive exercise of a muscle, muscle power is much transiently stronger and the deep tendon reflex to that muscle is enhanced. Unfortunately, this sign cannot always be confirmed during bedside evaluation. Figures C21-3 and C21-4 show the common signs and symptoms of LEMS patients, based on series of 50 patients.
Figure C21-3 Lambert-Eaton myasthenic syndrome. Symptoms during the course of illness in 50 cases.
(Adapted from O’Neil JH, Murray NMF, Newsom-Davis J. The Lambert-Eaton myasthenic syndrome: a review of 50 cases. Brain 1988:11:577–596.)
Figure C21-4 Lambert-Eaton myasthenic syndrome. Signs during the course of illness in 50 cases.
(Adapted from O’Neil JH, Murray NMF, Newsom-Davis J. The Lambert-Eaton myasthenic syndrome: a review of 50 cases. Brain 1988;11:577–596.)
The disorder may be mistaken for myasthenia gravis or myopathy. However, the diagnosis
[/not-level-membership-for-neurology-category]
