Case 20
HISTORY AND PHYSICAL EXAMINATION
| Right | Left | |
|---|---|---|
| Deltoid | 4/5 | 4/5 |
| Triceps | 4/5 | 4/5 |
| Biceps | 4/5 | 4/5 |
| Hand grip | 4+/5 | 4+/5 |
| Iliopsoas | 4−/5 | 4−/5 |
| Quadriceps | 4−/5 | 4−/5 |
| Hamstrings | 4−/5 | 4−/5 |
| Ankle dorsiflexion | 4+/5 | 4+/5 |
| Plantar dorsiflexion | 4+/5 | 4+/5 |
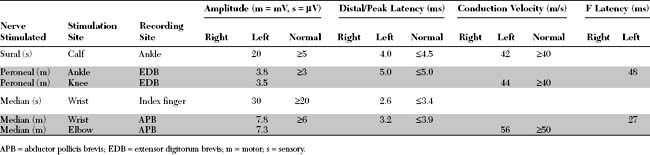
An electrodiagnostic (EDX) examination was performed.
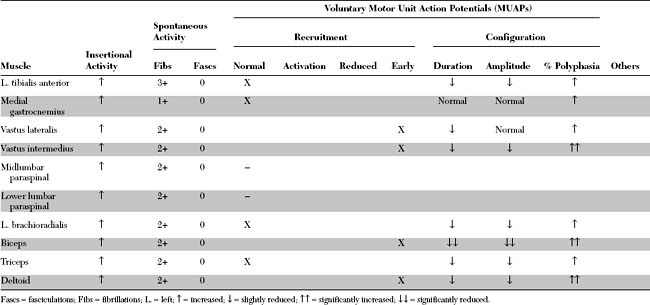
Please now review the Nerve Conduction Studies and Needle EMG tables.
QUESTIONS
EDX FINDINGS AND INTERPRETATION OF DATA
Pertinent EDX findings in this case include the following:
DISCUSSION
Classification and Pathology
The inflammatory myopathies are a heterogeneous group of disorders that share a common pathologic feature: inflammatory cells in muscles. They comprise three major categories of muscle disease, polymyositis (PM), dermatomyositis (DM), and inclusion body myositis (IBM), conditions that are clinically, histologically, and pathogenetically distinct. Inflammatory myopathies are often classified into two major types (Table C20-1), a primary and secondary (associated with systemic or other identifiable disorders).
Table C20-1 General Classification of Inflammatory Myopathies
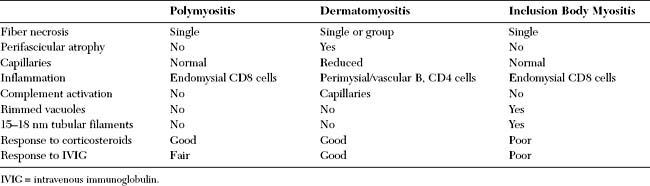
Pathologically, the inflammatory myopathies are characterized by mononuclear cell inflammation, segmental muscle fiber necrosis, and muscle fiber regeneration. The pathologic findings in PM and DM have both common and diverse features. Both also are different from the findings in inclusion body myositis. Table C20-2 shows the major differences among these three primary inflammatory myopathies.
Clinical Features
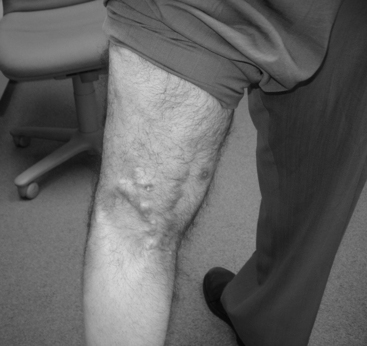
In DM only, there is an associated skin rash that can be the presenting symptom. Two classic rashes are characteristic. The first is a “heliotrope rash,” an erythematous, violaceous (hence the name) rash over the malar and periorbital areas that may extend to involve other sun-exposed areas, such as the dorsum of the hands, knees, elbows, or forehead. The second rash is Gottron papules, an erythematous papular rash over the knuckles of the fingers. Subcutaneous calcinosis complicates up to half of children with juvenile DM and may be present in chronic DM. These lesions are most often seen in the buttocks, thighs, knuckles, and elbows (Figure C20-1). They may be painful, ulcerate through the skin, or get infected.
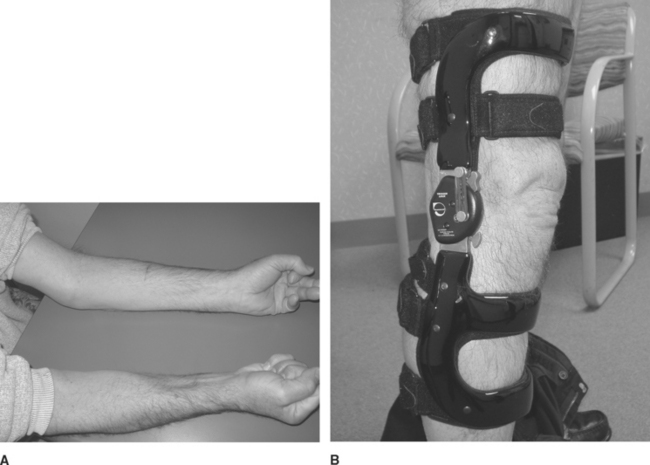
In contrast to PM and DM, IBM affects patients over the age of 50, has a male predominance, and is more common in the white than black population. The onset is insidious and it progresses slowly, evolving over years. It is the most common inflammatory myopathy in patients over the age of 50. Clinically, IBM has a unique muscle involvement that easily distinguishes it from the other inflammatory myopathies. There is usually asymmetrical involvement of the finger flexors and wrist flexors in the upper extremities, and the knee extensors and ankle dorsiflexors in the lower extremities (Figure C20-2). Dysphagia is common and afflicts up to 60% of patients as the disease progresses.
The diagnoses of DM and PM are confirmed based on the combination of clinical, laboratory, electrophysiologic, and pathologic findings. In 1975, Bohan and Peter proposed a classification that has been used since, with some revisions, in confirming the diagnoses of PM and DM. Based on these criteria, the confidence limits in the diagnosis of PM or DM range from definite to probable to possible (Table C20-3). The differential diagnoses of PM include IBM, polymyalgia rheumatica, metabolic myopathies (such as acid maltase deficiency), and limb girdle muscular dystrophy. In dermatomyositis, the presence of typical skin rash combined with muscle weakness often is diagnostic.
Table C20-3 Criteria and Confidence Limits in the Diagnosis of Polymyositis and Dermatomyositis
| Criteria | |
| Confidence Limits | ||
|---|---|---|
| Diagnosis | Polymyositis | Dermatomyositis |
| Definite | 4 criteria without rash | 3 or 4 criteria plus rash |
| Probable | 3 criteria without rash | 2 criteria plus rash |
| Possible | 2 criteria without rash | 1 criteria plus rash |
* See electrodiagnosis discussion for details.
† See Table C20-2 for differences between PM, DM, and inclusion body myositis.
Electrodiagnosis
Electrodiagnostic Findings of Myopathies in General
Sensory nerve conduction studies (NCS) in myopathy are normal, except in certain myopathies in which an associated peripheral polyneuropathy may occur (such as myotonic dystrophy or Kearns-Sayre syndrome). Similarly, motor NCSs usually are normal; however, motor studies may reveal low-amplitude compound muscle action potentials (CMAPs) when recording severely affected muscles. Examples include median and ulnar motor NCSs in adults with myotonic dystrophy, advanced Duchenne muscular dystrophy or critical illness myopathy. Proximal motor NCSs, such as musculocutaneous and femoral studies, may be low in amplitudes if performed on children with Duchenne muscular dystrophy. F waves and H reflexes are universally normal except when there is an associated polyneuropathy.
The changes seen on needle EMG in myopathy include one or more of the following.

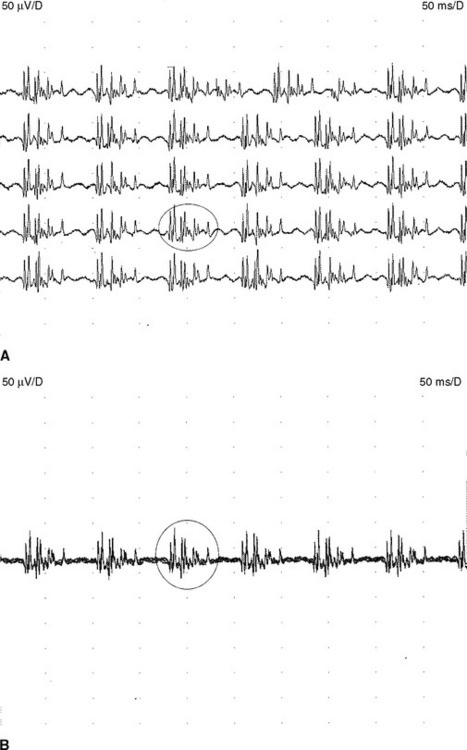

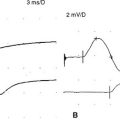
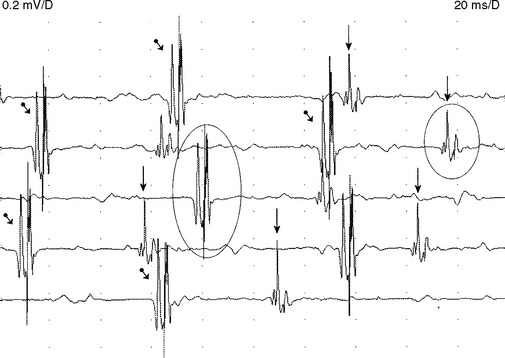
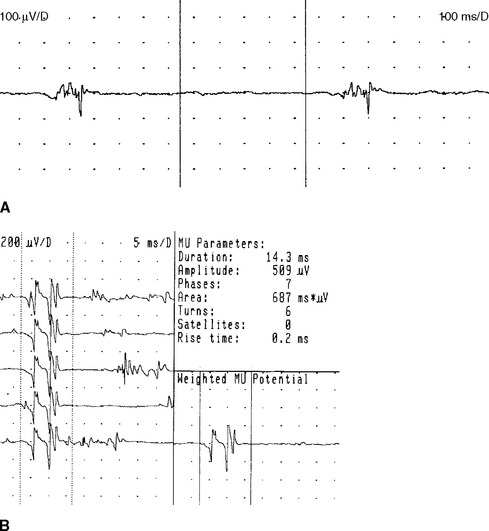
Although MUAP changes were suspected to exist in myopathy, Kugelberg (1949) is credited for a major contribution to our current understanding of the characteristic MUAP changes that occur in myopathy. These MUAP changes reflect the disintegration of motor unit structure in myopathy caused by muscle fiber loss, the variation in muscle fiber diameter, and an increased amount of connective tissue, along with the presence of regenerated and reinnervated muscle fibers. In general, MUAPs in myopathy are short in duration, low in amplitude and polyphasics (Figure C20-6). These units are very small compared with both normal MUAPs and sprouted (“neurogenic”) MUAPs (Figure C20-7). Typical MUAP changes seen in myopathy include:
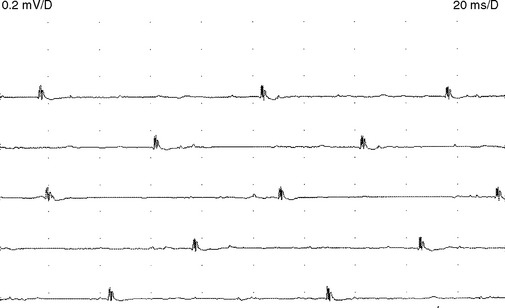
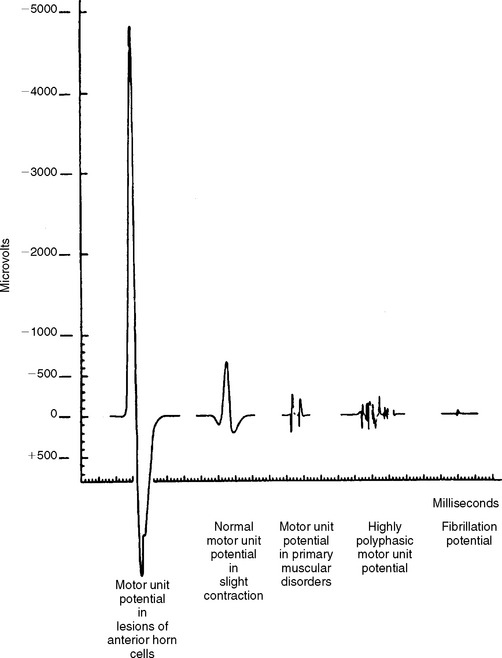
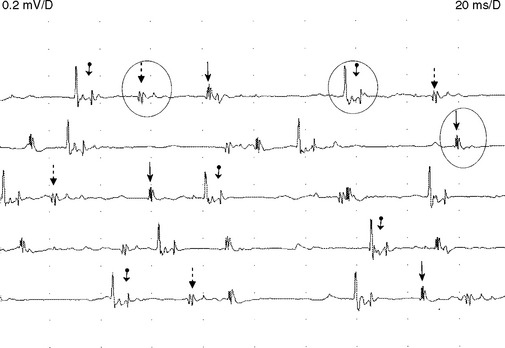
Figure C20-8 Several MUAPs recorded from the biceps muscle, recorded in a raster form, of the same patient as Figure C20-5. Note the short-duration, low-amplitude, and polyphasic MUAPs (solid arrow and dashed-line arrow) and the MUAP with satellite (linked) potentials (bulleted arrow).
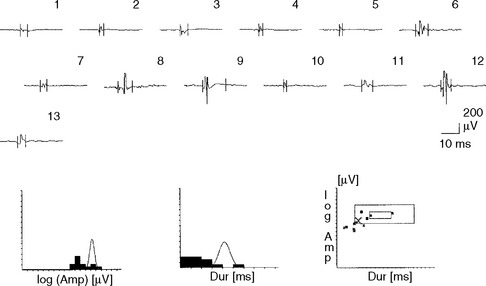
The alterations in MUAP configuration that occur in myopathy are not absolute. Instead, the MUAP changes form a continuum that ranges from normal to grossly abnormal MUAPs, with many MUAPs falling at points on the continuum between these two. Hence, identifying a myopathic process by needle EMG is one of the most difficult tasks for electromyographers. Evaluation of a patient with suspected myopathy requires meticulous analysis of the morphology of many MUAPs, within different areas of the muscle, in many muscles and in multiple extremities including the paraspinal muscles. Automated analysis of MUAPs, using computer-assisted EDX equipment that incorporates triggered and delayed sweeps, has allowed better determination of MUAP changes, especially those associated with myopathy (Figure C20-10).
The EDX assessment of recruitment is the most subjective parameter studied in the EMG laboratory, and attainment of accurate results is highly dependent on experience. Interpretation of recruitment is a particularly difficult task in myopathy. Interference pattern computer-assisted analysis, which typically judges the “turns and amplitudes,” has gained some popularity but has not been used extensively in routine EDX studies.
The recruitment of MUAPs in various myopathies may take one of these forms:
The EDX examination, however, has two relevant limitations in the diagnosis of myopathy:
Certain needle EMG features may accompany the MUAP changes and are helpful in the differential diagnosis of myopathies. Two features are instrumental in the accurate diagnosis of myopathy: fibrillation potentials and myotonic discharges. Based on these electrical potentials and the MUAP changes associated with myopathy, the EDX findings in myopathy may be easily divided into six general categories (Table C20-4):
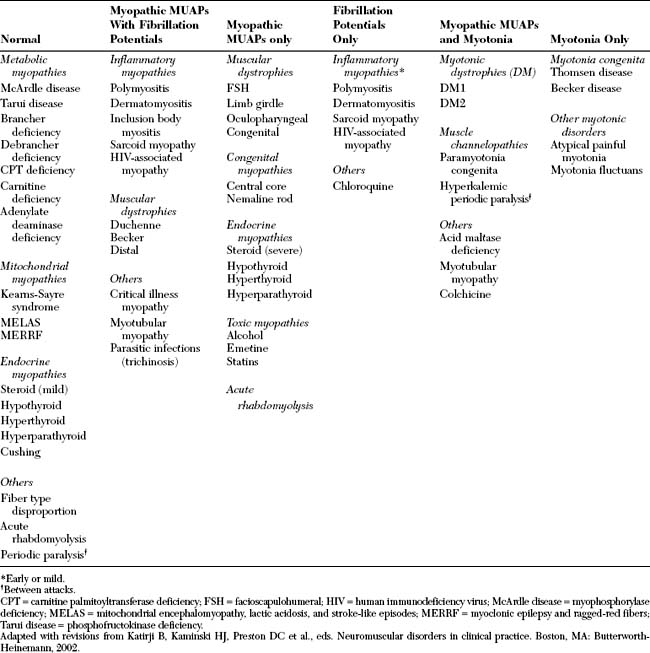
Electrodiagnostic Findings in Polymyositis/Dermatomyositis
Needle EMG findings during the active phases of PM/DM consist of the following.
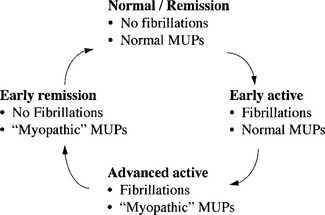
The EDX findings in polymyositis and dermatomyositis follow a cyclic pattern. Fibrillation potentials appear first at relapse and disappear early during remission, but abnormal MUAPs become evident later in relapse and last longer before resolution (Figure C20-12). This changing pattern must be recognized after treatment, when serial studies are performed on patients with PM/DM. For example, when fibrillations are not detected in a patient with PM/DM who is experiencing worsening weakness while taking corticosteroids, the diagnosis of iatrogenic “steroid myopathy” becomes more likely because the latter is not associated with fibrillation potentials.
FOLLOW-UP
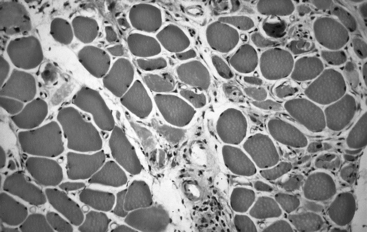
A muscle biopsy was obtained from the quadriceps muscle. The findings were diagnostic of inflammatory myopathy, particularly PM (Figure C20-13). No rimmed vacuoles were seen. The patient was started on prednisone 80 mg/day. She showed a dramatic improvement in strength, accompanied by a decline in CK. Prednisone was tapered slowly with no evidence of recurrence. One year later, she displayed normal strength and CK while taking prednisone 10 mg every other day.
Bohan A, Peter JB. Polymyositis and dermatomyositis. N Engl J Med. 1975;292:344-347. 403–407
Buchthal F. Electromyography in the evaluation of muscle diseases. Neurol Clin. 1985;3(3):573-598.
Chad D. Inflammatory myopathies. In: Katirji B, Kaminski HJ, Preston DC, Ruff RL, Shapiro EB, editors. Neuromuscular disorders in clinical practice. Boston, MA: Butterworth-Heinemann; 2002:1169-1180.
Dalakas MC. Polymyositis, dermatomyositis, and inclusion body myositis. N Engl J Med. 1991;325:1487-1498.
Dalakas MC. Inflammatory, immune, and viral aspects of inclusion-body myositis. Neurology. 2006;66(2 Suppl 1):S33-38.
Dalakas MC, Hohlfeld R. Polymyositis and dermatomyositis. Lancet. 2003;362(9388):971-982.
Dalakas MC, et al. A controlled trial of high-dose intravenous immune globulin infusions as treatment for dermatomyositis. N Engl J Med. 1993;329:1993-1998.
Daube JR. Application of quantitative methods in neuromuscular disorders. In: Halliday AM, Butler SR, Paul R, editors. A textbook of clinical neurophysiology. New York: John Wiley, 1987.
Fellows LK, Foster BJ, Chalk CH. Clinical significance of complex repetitive discharges. A case-control study. Muscle Nerve. 2003;28:504-507.
Katirji B, Al-Jaberi M. Creatine kinase revisited. J Clin Neuromusc Dis. 2001;2:158-163.
Kugelberg E. Electromyogram in muscular disorders. J Neurol Neurosurg Psychiatry. 1947;10:122-133.
Lacomis D. Electrodiagnostic approach to the patient with suspected myopathy. Neurol Clin N Am. 2002;20:587-603.
Lambert EH, Sayre GP, Eaton LM. Electrical activity in muscle in polymyositis. Trans Am Neurol Assoc. 1954;79:64-69.
Mastaglia FL, Ojeda VJ. Inflammatory myopathies: parts 1 and 2. Ann Neurol. 1985;7:215-227. 317–323
Mitz M, et al. Electromyographic and histologic paraspinal abnormalities in polymyositis/dermatomyositis. Arch Phys Med Rehabil. 1981;62:118-121.
Sandstedt PER, Henriksson KG, Larsson LE. Quantitative electromyography in polymyositis and dermatomyositis. Acta Neurol Scand. 1982;65:110-121.
Sigurgeirsson B, et al. Risk of cancer in patients with dermatomyositis or polymyositis. N Engl J Med. 1992;326:363-367.
Streib E. Differential diagnosis of myotonic disorders. Muscle Nerve. 1987;10:603-615.
Streib E, Daube JR. Electromyography of paraspinal muscles. Neurology. 1975;25:386.
Streib EW, Wilbourn AJ, Mitsumoto H. Spontaneous electrical muscle fiber activity in polymyositis and dermatomyositis. Muscle Nerve. 1979;2:14-18.
Trojaborg W. Quantitative electromyography in polymyositis: a reappraisal. Muscle Nerve. 1990;13:964-971.
van der Meulen MFG, Bronner IM, Hoogendijk JE, et al. Polymyositis. An overdiagnosed entity. Neurology. 2003;61:316-321.
Wilbourn AJ. Electrodiagnostic examination with myopathies. J Clin Neurophysiol. 1993;10:132-148.