Case 19
HISTORY AND PHYSICAL EXAMINATION
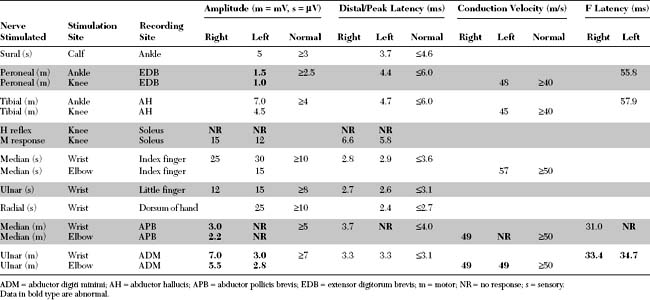
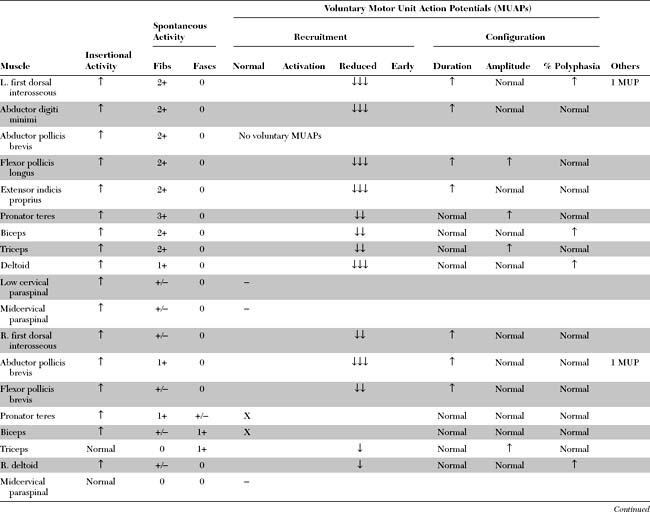
Please now review the Nerve Conduction Studies and Needle EMG tables.
QUESTIONS
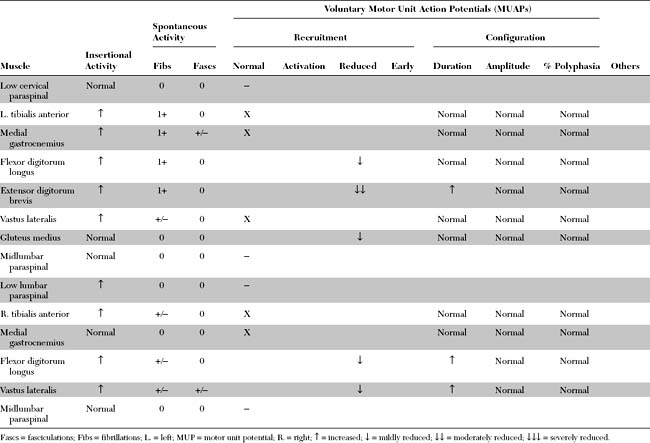
EDX FINDINGS AND INTERPRETATION OF DATA
Pertinent EDX findings in this patient include:
DISCUSSION
Pathology and Etiology
The pathology of sporadic ALS is represented by the selective loss of motor neurons in the spinal cord and brain stem, and cortical motor neurons (Betz cells). Classic findings on spinal cord sections include the loss of anterior horns, with degeneration of the pyramidal tracts (crossed and uncrossed) and dramatic preservation of the dorsal columns and spinocerebellar tracts. Although all motor neurons ultimately degenerate, there is relative sparing of the oculomotor nuclei in the brain stem and Onuff nucleus in the lumbosacral cord. Microscopically, there is, in addition to the loss of anterior horn motor neurons, frequent accumulation of neurofilaments in surviving neurons and dilatation of axons (“spheroids”). The pathologic findings in familial ALS are identical to those in the sporadic form, except that Lewy-like bodies frequently are identified in surviving motor neurons.
Clinical Features
The diagnosis of ALS is based on the presence of a progressive disorder with the characteristic combination of upper and lower motor neuron involvement. Many criteria have been proposed but most are inadequate, particularly those pertaining to early diagnosis and the definition of upper motor neuron involvement. Among them, the revised El Escorial diagnostic criteria currently are the most widely accepted for the diagnosis of ALS (Table C19-1).
Table C19-1 Revised El-Escorial Criteria for the Diagnosis of Amyotrophic Lateral Sclerosis
LMN = lower motor neuron; UMN = upper motor neuron.
Although lower motor neuron dysfunction dominates the clinical picture in many patients, there usually is evidence of upper motor neuron involvement as well. Extreme cases of “pure” lower motor neuron or “pure” upper motor neuron involvement exist, but they are less common than classic ALS. Because of this variability and the preponderance to lower or upper motor neurons, ALS variants commonly are separated from the classic form (Table C19-2).
Table C19-2 Amyotrophic Lateral Sclerosis (ALS) and Its Variants
| Disorder | Frequency (%) | Characteristics |
|---|---|---|
| Sporadic ALS | 90–95 | |
| Classic ALS | 82 | LMN and UMN dysfunctions |
| LMN-dominant ALS | LMN dysfunctions with subtle UMN signs | |
| UMN-dominant ALS | UMN dysfunctions with subtle or needle EMG signs of LMN dysfunctions | |
| Progressive bulbar palsy | 9 | Bulbar with or without pseudobulbar dysfunctions |
| Progressive muscular atrophy | 7 | Pure LMN dysfunctions |
| Primary lateral sclerosis | 2 | Pure UMN dysfunctions |
| Familial ALS | 5–10 | |
| Autosomal dominant ALS | ||
| SOD 1-linked | 20 (2% of ALS) | Linked to chromosome 21q22, associated with >50 mutations in gene for Cu, Zn SOD (Ala4 to valine) |
| Non-SOD 1-linked | Not linked to chromosome 21q22 | |
| Autosomal recessive ALS | Some are linked to chromosome 2q33 |
ALa4 = alanine4; Cu, Zn SOD = copper-zinc superoxide dismutase; LMN = lower motor neuron; SOD1 = superoxide dismustase; UMN = upper motor neuron.
Although ALS often can be readily diagnosed clinically, especially when both upper and lower motor neuron features are present, a definitive diagnosis sometimes may be difficult to attain, particularly during the early stages of the disease. Table C19-3 lists common disorders that may mimic ALS, thus posing difficulties in the diagnostic process.
Table C19-3 Differential Diagnosis of Amyotrophic Lateral Sclerosis
Electrodiagnosis
Nerve Conduction Studies
Although the major changes in ALS are seen on needle EMG, nerve conduction studies (NCS) should be done in all patients with suspected ALS to exclude other possible causes of weakness. Sensory NCSs are normal, although a subtle decrease in SNAP amplitudes has been reported in a few studies. Motor NCSs may show abnormalities that vary with the stage of disease. Normal study results are not uncommon early in the disease course. Later, low-amplitude CMAPs are frequently revealed; these may be regional (i.e., the result of motor conduction studies performed on weakened limb(s) because of anterior horn cell loss). In more advanced stages of the disease, diffusely low CMAP amplitudes with normal SNAP amplitudes, so called “low motor-normal sensory pattern,” is characteristic. This NCS pattern is not specific for the diagnosis and may be seen in spinal muscular atrophies, diffuse myelopathies or polyradiculopathies, axonal motor polyneuropathies, presynaptic neuromuscular junction disorders, and severe myopathies (see Figure C17–13). In contrast to CMAP amplitudes, motor conduction velocities, distal latencies, and F wave latencies are usually normal in ALS until significant degrees of axon loss have occurred, when mild slowing may be detected due to the loss of large and fast conducting axons. This slowing is proportional to the reduction in CMAP amplitude, and the conduction velocity does not decrease to less than 70 to 80% of the lower limit of normal. Motor conduction block, or significant CMAP temporal dispersion, should raise the suspicion of another disorder that may, at times, mimic motor neuron disease: multifocal motor neuropathy with conduction block.
Needle EMG
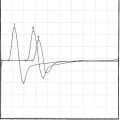
Fasciculation potentials are sporadic or quasi-rhythmic, spontaneous (involuntary) contractions of a group of muscle fibers that are innervated by a single motor unit. They can be of any shape and size, depending on the motor units from which they arise (Figure C19-1). They reflect irritability of the motor unit and usually originate from the distal nerve terminals, with spread by axon reflex to other parts of the unit. They frequently are visible on inspection. During needle EMG, fasciculation potentials are characterized by a random firing pattern. Fasciculations are particularly prominent in patients with ALS and have been closely linked to the disease since its first description by Charcot. Using simultaneous multichannel EMG recordings from different sites and muscles, it has been estimated that more than 90% of patients with ALS have fasciculations.
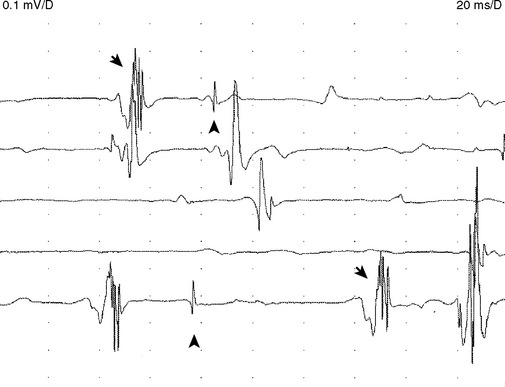
Although fasciculation potentials are extremely common in ALS, they also occur in other lower motor neuron disorders (radiculopathies and peripheral polyneuropathies), with the use of anticholinesterase medication, in hyperthyroidism and hypocalcemia, and in healthy muscles (particularly the calves). Thus, fasciculation potentials are nonspecific and may be benign, unless they are accompanied by fibrillation potentials or by MUAP changes.
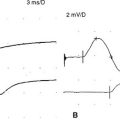
Reduced MUAP recruitment is caused by degeneration and loss of motor neurons in ALS, with the result that only a few can be activated voluntarily. The activated motor units fire more rapidly as anterior horn cells are lost (Figure C19-2). This EDX finding always is abnormal but, when isolated, does not mean automatically that axonal loss has occurred because it may happen if there is demyelination anywhere along the motor axon that results in the block of conduction transmission. In ALS patients, slow recruitment frequency (poor activation) of MUAPs in limbs where UMN loss predominates may also be evident but is a less frequent finding.
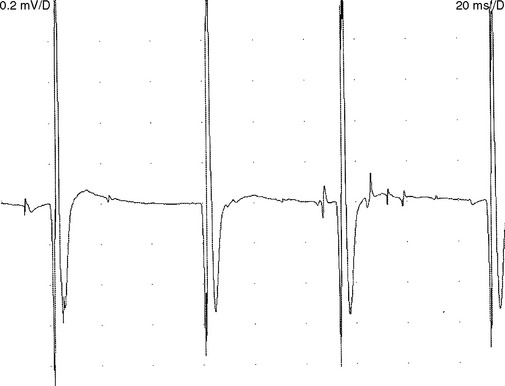
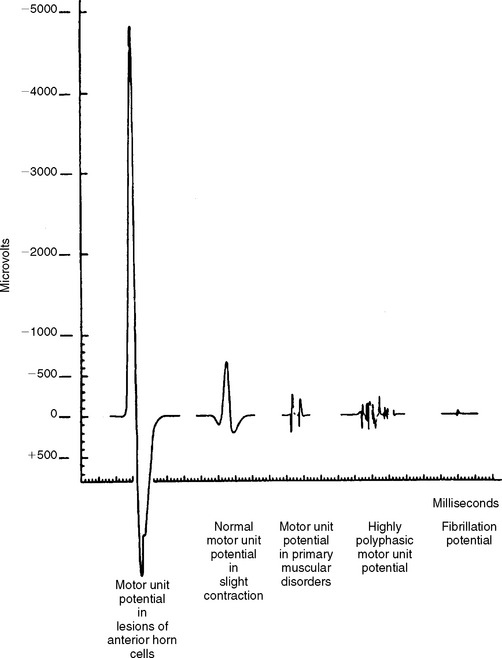
Reinnervated MUAPs dominate as collateral sprouting increases the number of muscle fibers per motor unit resulting in increased duration and amplitude MUAPs (Figure C19-2). Also, because of conduction slowing along the newly formed collateral sprouts, muscle fiber action potentials become asynchronous. This results in increased polyphasic MUAPs (more than four phases). Thus, a mixture of MUAPs often is seen on needle EMG, dependent on the stage of illness. Normal MUAPs are intermixed with polyphasic MUAPs, with or without satellite potentials (Figure C19-3), and with long-duration, high-amplitude MUAPs (Figure C19-4). Moment-to-moment MUAP amplitude variation, representing motor unit instability, may also be appreciated.

Figure C19-3 Polyphasic motor unit potential (MUAP) with satellite (linked) potentials, also called complex MUAP.
(From Daube J. AAEM minimonograph 11: needle electromyography in clinical electromyography. Muscle Nerve 1991;14:685–700, with permission.)
In summary, the findings on needle EMG are variable and depend on the stage of illness. At any one point in a patient’s illness, sampling many muscles in four limbs and the head often reveals a mixture of the following findings (listed in worsening severity):
Electrodiagnostic Criteria
Amyotrophic lateral sclerosis is a clinical disorder in which the EDX study plays a major role in supporting the diagnosis and in excluding entities that can mimic ALS. Electrophysiologic confirmation of ALS requires evidence of a widespread LMN degeneration, and hence the needle EMG examination should be performed on three or more regions of the neuraxis and should assess all the major segments in the limbs examined. When the bulbar region is assessed, changes must be observed in at least one muscle (including tongue, jaw muscles, and facial muscles). Needle EMG of the tongue is difficult due to failure to achieve adequate relaxation which results in inability to appreciate fibrillation or fasciculation potentials. Also, the tongue MUAPs normally are small and may appear similar to fibrillation potentials. The thoracic segment can only be assessed by needle EMG of the thoracic paraspinal muscles at or below the T6 level, and occasionally the abdominal muscles. Evaluation of higher thoracic segments may be misleading as denervation changes derived from lower cervical segments may manifest as far caudally as the T6 level.
The criteria proposed by Lambert in 1969 had been the most widely accepted for the diagnosis of ALS. These criteria require fibrillation with fasciculation potentials in three limbs, with the head counting as a “limb.” El Escorial criteria have adopted these standards with some revisions. Table C19-4 provides a summary of definitive EDX criteria for ALS.
Table C19-4 Electrodiagnostic Criteria for the Diagnosis of Amyotrophic Lateral Sclerosis (ALS)
* Regions are defined as follows: brain stem (bulbar), cervical (upper limbs), thoracic (back and abdomen), and lumbosacral (lower limbs). Involvement in a region is without regard to right or left side, but location is indicative of the level of neuraxis involved.
† Motor conduction velocities may be slowed but should not be lower than 70 to 80% of lower limits of normal values, in nerves with very low CMAP amplitudes (less than 50% of the lower limits of normal or less than 30% of normal mean).
The aforementioned criteria are fulfilled at the time of diagnosis in approximately two-thirds of patients with ALS. However, a significant proportion of patients with a clinical diagnosis of ALS fail to show these findings on EDX testing, particularly on initial studies. This is caused by the following limitations:
Behina M, Kelly J. Role of electromyography in amyotrophic lateral sclerosis. Muscle Nerve. 1991;14:1236-1241.
Bensimon G, et al. A controlled trial of riluzole in amyotrophic lateral sclerosis. N Engl J Med. 1994;330:585-591.
Bernstein LP, Antel JP. Motor neuron disease: decremental responses to repetitive stimulation. Neurology. 1981;31:204-205.
Bradley WG, et al. Morphometric and biochemical studies of peripheral nerves in amyotrophic lateral sclerosis. Ann Neurol. 1983;14:267-277.
Brooks BR, Miller RG, Swash M, et al. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord. 2000;1:293-299.
Brown RH. Superoxide dismutase and familial amyotrophic lateral sclerosis: new insights into mechanisms and treatments. Ann Neurol. 1996;39:145-146.
Brown RH, Swash M, Pasinelli P, editors. Amyotrophic lateral sclerosis, 2nd edn., Informa Healthcare, 2006.
Chancellor AM, Warlow CP. Adult onset motor neuron disease: worldwide mortality, incidence and distribution since 1950. J Neurol Neurosurg Psych. 1992;55:1106-1115.
Drachman DB, Kuncl RW. Amyotrophic lateral sclerosis: an unconventional autoimmune disease? Ann Neurol. 1989;26:269-274.
Gooch CL, Harati Y. Motor unit number estimation, ALS and clinical trials. Amyotroph Lateral Scler Other Motor Neuron Disord. 2000;1:71-82.
Gurney ME, et al. Benefit of vitamin E, Riluzole, and Gabapentin in a transgenic model of familial amyotrophic lateral sclerosis. Ann Neurol. 1996;39:147-157.
Hjorth RJ, Walsh JC, Willison RG. The distribution and frequency of spontaneous fasciculations in motor neuron disease. J Neurol Sci. 1973;18:469-474.
Killian JM, et al. Decremental motor responses to repetitive nerve stimulation in ALS. Muscle Nerve. 1994;17:747-754.
Kuncl RW, Cornblath DR, Griffin JW. Assessment of thoracic parapsinal muscles in the diagnosis of ALS. Muscle Nerve. 1988;11:484-492.
Lacomblez L, et al. Dose-ranging study of riluzole in amyotrophic lateral sclerosis. Lancet. 1996;347:1425-1431.
Lambert EH. Electromyography in amyotrophic lateral sclerosis. In: Kurland LT, Norris FH, editors. Motor neuron diseases. New York: Grune and Stratton, 1969.
Lambert EH, Mulder DW. Electromyographic studies in amyotrophic lateral sclerosis. Mayo Clin Proc. 1957;32:441-446.
Miller RG, Rosenberg JA, Gelinas DF, et al. Practice parameter: rhe care of the patient with amyotrophic lateral sclerosis (an evidence based review). Neurology. 1999;52:1311-1323.
Mitsumoto H, Chad D, Pioro EP, editors. Amyotrophic lateral sclerosis. Philadelphia, PA: FA Davis, 1998.
Moss AH, et al. Home ventilation for amyotrophic lateral sclerosis. Neurology. 1993;43:438-443.
Quality Standards Subcommittee of the American Academy of Neurology. Practice advisory on the treatment of amyotrophic lateral sclerosis with riluzole: report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 1997;49:657-659.
Rosenfeld J, Swash M. What’s in a name? Lumping or splitting ALS, PLS, PMA and the other motor neuron diseases. Neurology. 2006;66:6214-6254.
Roth G. The origin of fasciculation potentials. Ann Neurol. 1982;12:542-547.
Rothstein JD. Excitotoxic mechanisms in amyotrophic lateral sclerosis. In: Seratrice G, Munsat T, editors. Pathogenesis and therapy of amyotrophic lateral sclerosis, advances in neurology, vol. 68. Philadelphia, PA: Lippincott-Raven, 1995.
Rothstein JD, Martin LJ, Kuncl RK. Decreased glutamate transport by the brain and spinal cord in amyotrophic lateral sclerosis. N Engl J Med. 1992;326:1464-1468.
Shefner JM, Tyler HR, Krarup C. Abnormalities in the sensory action potential in patients with amyotrophic lateral sclerosis. Muscle Nerve. 1991;14:1242-1246.
Siddique T, et al. Linkage of a gene causing familial amyotrophic lateral sclerosis to chromosome 21 and evidence of genetic-locus heterogeneity. N Engl J Med. 1991;324:1381-1384.
Smith RG, et al. Serum antibodies to L-type calcium channels in patients with amyotrophic lateral sclerosis. N Engl J Med. 1992;327:1721-1728.
Sorenson EJ, Stalker AP, Kurland LT, et al. Amyotrophic lateral sclerosis in Olmsted County, Minnesota, 1925 to 1998. Neurology. 2002;59:280-282.
Urban PP, Vogt T, Hopf HC. Corticobulbar tract involvement in amyotrophic lateral sclerosis: a transcranial magnetic stimulation study. Brain. 1998;121:1099-1108.