Case 18
HISTORY AND PHYSICAL EXAMINATION
Electrodiagnostic (EDX) study was performed.
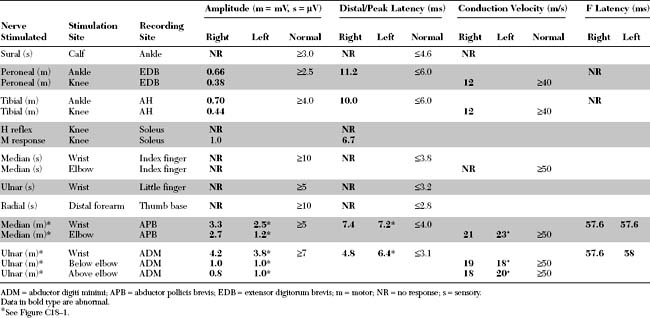
Please now review the Nerve Conduction Studies and Needle EMG tables.
QUESTIONS
EDX FINDINGS AND INTERPRETATION OF DATA
The abnormal EDX findings in this case include:
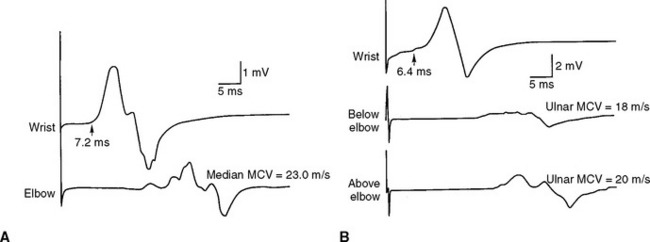
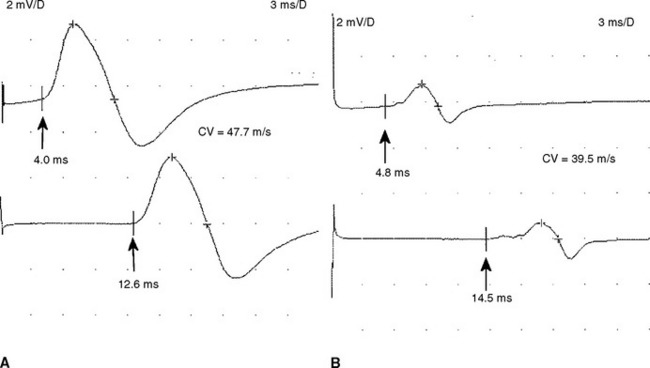
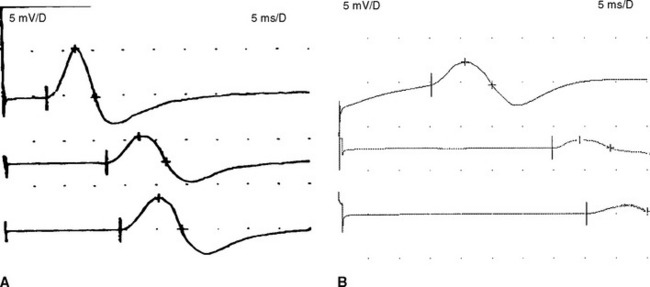
Figure C18-1 Motor nerve conduction studies of the left median nerve, recording abductor pollicis brevis (A) and the left ulnar nerve, recording abductor digiti minimi (B). Note the conduction blocks and dispersion between distal and proximal stimulations and the marked slowing of velocities and distal latencies. (See also the Nerve Conduction Studies table.)
DISCUSSION
Clinical Features
Peripheral Polyneuropathies
Peripheral polyneuropathy is a common presenting illness in neurologic practice with multiple, sometimes overwhelming, list of potential etiologies. Pattern recognition is a useful diagnostic approach but applies to a minority of patients who usually have advanced disease and often requires vast clinical experience such as by a seasoned neurologist. For an example, an asymmetrical polyneuropathy with predilection to cool skin areas (nipples, buttocks, and fingers) and skin ulcerations is highly suggestive of leprous neuropathy. Also, a distal sensory polyneuropathy with brisk reflexes, mild cognitive impairment, and a red tongue suggests combined system degeneration due to vitamin B12 deficiency. Another approach to the etiologic diagnosis of peripheral polyneuropathy is to order all available tests, including costly serology evaluations, on every patient with a polyneuropathy. Unfortunately, this irrational “shotgun” approach is quite common and often utilized by internists and some neurologists. It sometimes results in an incorrect diagnosis secondary to incidental abnormalities such as an elevated glucose on glucose tolerance test or antineuronal antiboby on serological testing.
A recommended and more rational approach may be initiated on every patient presenting with a peripheral polyneuropathy. This could be achieved by performing a thorough history and physical examination followed by EDX studies (see Figure C26–1, Case 26), and often results in limited and cost effective investigations (Table C18-1). Despite extensive investigations in specialized centers that includes EDX testing, antibody panels and genetic testing, up to 20% of patients with peripheral polyneuropathies will not have their exact causation identified. Of those with idiopathic etiology, it is estimated that a familial neuropathy accounts for about 40% if a meticulous family history is taken and relatives are carefully examined.
Table C18-1 Essential Facts Important in the Classification and Etiologic Diagnosis of Peripheral Polyneuropathy
|
If asymmetric, do the findings follow specific peripheral nerve distribution?
Distal – consider metabolic disturbance*, vitamin deficiency, toxins, drugs, critical illness, hereditary
|
CIDP = chronic inflammatory demyelinating polyradiculoneuropathy, HNPP = hereditary neuropathy with liability to pressure palsy, CMT = Chercot-Marie-Tooth disease, HIV = human immunodeficiency virus.
Chronic Demyelinating Polyneuropathies
In most peripheral polyneuropathies, it is often possible to define the predominant pathophysiologic mechanism, based on electrophysiologic and pathologic features, as being either primarily axonal or demyelinating. In demyelinating polyneuropathy, it is also useful to distinguish between neuropathies with segmental (multifocal) versus uniform slowing, based on electrophysiologic studies (see electrodiagnosis). Multifocal or segmental demyelinating polyneuropathies are almost always acquired, while uniform demyelinating polyneuropathies are typically hereditary.
The causes of chronic axonal neuropathies are abundant, while chronic demyelinating polyneuropathies have a fairly restrictive differential diagnosis (Table C18-2). Many acquired demyelinating polyneuropathies are immune in nature and respond to immunosupression or immunomodulation, while most axonal polyneuropathies are metabolic or toxic in nature. Since the differential diagnosis of chronic acquired demyelinating polyneuropathies is quite limited, the diagnostic work-up for patients with such entities is much less laborious and is quite different from that of patients with axonal neuropathies (Table C18-3).
Table C18-2 Common Causes of Chronic Demyelinating Peripheral Polyneuropathy
| Acquired (nonuniform multifocal slowing) |
CIDP = chronic inflammatory demyelinating polyradiculoneuropathy; HIV = human immunodeficiency virus; GM1 = ganglioside M1; MAG = myelin-associated glycoprotein; MGUS = monoclonal gammopathy of unknown significance; Ig = immunoglobulin; POEMS syndrome = polyneuropathy, organomegaly, endocrinopathy, monoclonal protein, and skin changes; HMSN = hereditary motor and sensory neuropathy; CMT = Charcot-Marie-Tooth disease; HNPP = hereditary neuropathy with liability to pressure palsy.
* May have multifocal slowing also, usually across common entrapment sites.
Table C18-3 Recommended Work-Up of Chronic Acquired Demyelinating Peripheral Polyneuropathy
BUN = blood urea nitrogen; CBC = complete blood count; CSF = cere-brospinal fluid; GM1 = ganglioside M1; HIV = human immunodeficiency virus; MAG = myelin-associated glycoprotein.
* Serum immunofixation is often necessary because routine serum protein electrophoresis may miss patients with a small amount of circulating paraprotein (M-protein).
Chronic Inflammatory Demyelinating Polyradiculoneuropathy
Patients with CIDP must be distinguished from patients with the more prevalent acquired axonal peripheral polyneuropathies. CIDP should also be separated from hereditary polyneuropathies, particularly those with demyelinating features such as CMT1, CMTX, and CMT3. CIDP should also be distinguished from other polyradiculopathies, such as meningeal carcinomatosus, Lyme disease, or sarcoidosis. When predominantly motor, CIDP may mimic neuromuscular junction disorders (such as the Lambert-Eaton myasthenic syndrome), motor neuron disorders, and myopathies.
Chronic inflammatory demyelinating polyradiculoneuropathy (CIDP) is a diagnosis of pattern recognition, based on clinical manifestations, EDX, cerebrospinal fluid examination, laboratory tests appropriate to the specific clinical situation, and, occasionally, results from nerve biopsy. The American Academy of Neurology defined criteria for the diagnosis of CIDP (Table C18-4). Four features are set as the basis of diagnosis: clinical, electrodiagnostic, pathologic, and cerebrospinal fluid studies. These are further divided into mandatory, supportive, and, where appropriate, exclusion. Mandatory features are those required for diagnosis and should be present in all definite cases. Supportive features are helpful in clinical diagnosis but by themselves do not make a diagnosis. Exclusion features strongly suggest alternative diagnoses.
Table C18-4 American Academy of Neurology Criteria for Diagnosis of Chronic Inflammatory Demyelinating Polyneuropathy
Diagnostic categories. Definite: Clinical A and C, Electrodiagnostic A, CSF A, and Pathology A and C. Probable: Clinical A and C, Electrodiagnostic A, and CSF A. Possible: Clinical A and C and Electrodiagnostic A.
* Criteria suggestive of partial conduction block: >20% drop in area or amplitude with <15% change in duration between proximal and distal sites.
† Criteria for abnormal temporal dispersion and possible conduction block: >20% drop in area or amplitude between proximal and distal sites with >15% change in duration between proximal and distal sites and. These criteria are only suggestive of partial conduction block as they are derived from studies of normal individuals. Additional studies, such as stimulation across short segments or recording of individual motor unit potentials, are required for confirmation.
‡ Demyelination by either electron microscopy (>5 fibers) or teased fiber studies (>12% of 50 teased fibers, minimum of four internodes each, demonstrating demyelination/remyelination).
Peripheral Polyneuropathy and Monoclonal Gammopathy of Undetermined Significance
The prevalence of monoclonal gammopathy of undetermined significance (MGUS) increases with age. It is present in 1% of patients older than 50 years of age and in 3% of patients older than 70 years of age. This entity must be distinguished from the more malignant myeloproliferative disorders, such as multiple myeloma, by obtaining complete blood count (CBC), calcium, blood urea nitrogen (BUN)/creatinine, a skeletal survey, and, at times, a bone marrow aspirate. Table C18-5 lists both the criteria needed to confirm the diagnosis of MGUS and its common characteristics. Although MGUS is relatively benign and is commonly asymptomatic, follow-up reveals that malignant myeloproliferative disorders will develop in up to one-third of these patients within 20 years. A good indication for this malignant transformation is a rising M-protein (paraprotein) value, especially one greater than 3 g/dL. Thus, a regular follow-up of the paraprotein value is warranted in all patients with MGUS.
Table C18-5 Monoclonal Gammopathy of Unknown Significance (MGUS)
| Criteria for Diagnosis | Characteristics |
|---|---|
There is continuous debate regarding the various acquired demyelinating polyneuropathies, namely CIDP, MGUS neuropathy, anti-MAG-associated neuropathy, and multifocal motor neuropathy with conduction block. Our current knowledge of the exact etiology and pathogenesis of these immune disorders is lacking. Figure C18-2 reveals a schematic representation of the significant overlap between all these disorders. Apart from the presence of a monoclonal protein, the clinical and EDX features of CIDP – with or without MGUS – are quite similar. However, as is shown in Table C18-6, there are certain features in the presentation and clinical course that tend to help differentiate between these disorders.
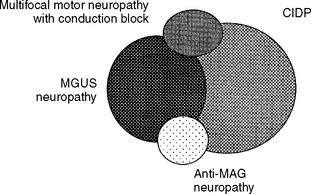
Table C18-6 Distinctive Features for Differentiating Between Chronic Inflammatory Demyelinating Polyradiculoneuropathy (CIDP) Without and With MGUS
| Feature | CIDP Without MGUS | CIDP With MGUS |
|---|---|---|
| Age | Relatively younger | Relatively older |
| Course | Progressive or relapsing | Frequently progressive |
| Neuropathy | Predominantly motor | Predominantly sensory with ataxia |
| Clinical deterioration | More rapid | Slow indolent |
| Functional impairment | Moderate to severe | Mild |
| Spontaneous improvement | Common | Rare |
| Response to therapy | Good | Less responsive |
Electrodiagnosis
In a patient with suspected peripheral polyneuropathy, the EDX study:
Thus, at the completion of an EDX test, the clinician should be able to better characterize the polyneuropathy and classify its pathophysiology. This helps establish a relatively short differential diagnosis and work-up aimed at identifying the cause of the neuropathy and planning its management (see Table C18-1).
Analyzing conduction times (velocities and latencies), as well as CMAP amplitude, area and duration, is an essential exercise in the EMG laboratory for establishing the primary pathologic process of a polyneuropathy. In most situations, the polyneuropathy falls in one of the following categories based on one of two primary nerve dysfunctions: the axon or its supporting myelin. Occasionally, such as in very mild polyneuropathies or in severe situations associated with absent responses, it may be difficult to establish the primary pathology based on EDX studies.
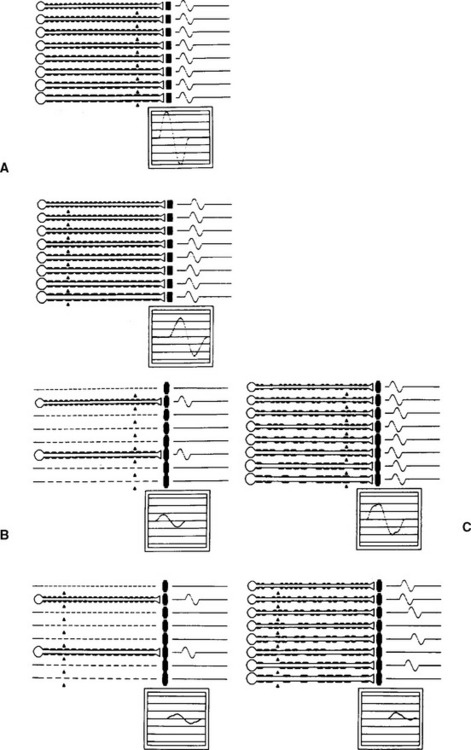
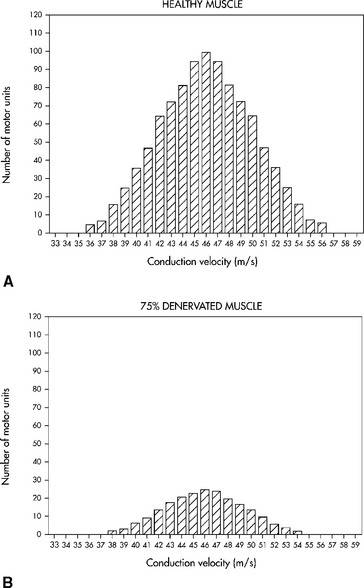
Multiple criteria have been set for the diagnosis of CIDP and are aimed at distinguishing the primary demyelinating polyneuropathy from the primary axonal polyneuropathy. Criteria proposed by Cornblath and Asbury were adopted by consensus to apply to patients with suspected CIDP (see Report from an Ad Hoc subcommittee, 1991). Table C18-7 reveals common nerve conduction criteria used to identify the acquired demyelinating polyneuropathies.
Table C18-7 Electrophysiologic Criteria for Acquired Demyelinating Polyneuropathy
| Criteria | Albers and Kelly | Asbury and Cornblath |
|---|---|---|
| Required |
MCV = motor conduction velocity; MDL = motor distal latency; ULN = upper limit of normal; LLN = lower limit of normal; CMAP = compound muscle action potential.
* Conduction block = >30% drop in CMAP amplitude between distal and proximal stimulations.
† Conduction block = >20% drop in CMAP area or amplitude between distal and proximal stimulations with <15% increase in CMAP duration; temporal dispersion = >20% drop in CMAP area or amplitude between distal and proximal stimulations with >15% increase in CMAP duration.
Adapted from Brown WF. Acute and chronic inflammatory demyelinating neuropathies. In: Brown WF, Bolton CF, eds. Clinical electromyography. Boston, MA: Butterworth-Heinemann, 1993, pp. 533–559; Albers JW, Kelly JJ. Acquired inflammatory demyelinating polyneuropathies: clinical and electrophysiologic features. Muscle Nerve 1989;12:435–451; Cornblath DR. Electrodiagnostic abnormalities in Guillain-Barré syndrome. Ann Neurol 1990;27(suppl):S17–S20.
Table C18-8 Electrodiagnostic Criteria of Conduction Block
All Amplitudes, areas and durations reflect negative-peak areas, amplitudes and durations.
* Caution should be taken in evaluating the tibial nerve, where stimulation at the knee can be submaximal, resulting in 50% or at times >50% drop in amplitude, especially in overweight patients.
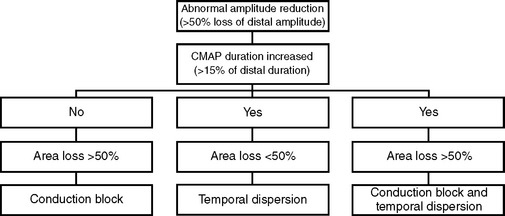
Patients with axonal polyneuropathy are sometimes erroneously diagnosed as demyelinating polyneuropathies and lead to wrong therapies that are potentially harmful. For example, a patient with alcoholic polyneuropathy misdiagnosed as CIDP may be treated with steroids, IVIG, or plasma exchange, all with potential adverse effects. The reasons for these misdiagnoses include the following:
FOLLOW-UP
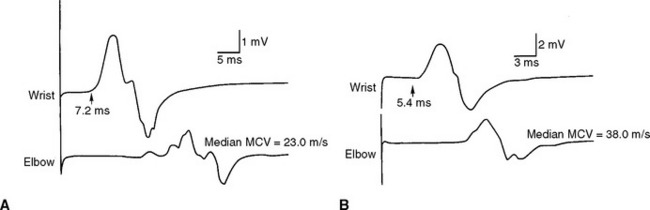
The patient was treated with weekly courses of plasma exchange, along with oral corticosteroids. She showed significant improvement in strength, pain level, and gait over the next 3 months. This was confirmed by repeat conduction studies, 6 months after treatment, which showed marked improvement, with resolution of most conduction blocks and improvement of both distal latencies and conduction velocities (Figure C18-8). Because of osteopenic vertebral fractures, steroids were discontinued, and the patient was maintained on bimonthly plasma exchange and azathioprine. Because of loss of appropriate venous access, she was then shifted to monthly intravenous immunoglobulin infusions (2 g/kg infused in 2 days). The patient strength was satisfactory on this regimen and a follow-up after 4 years showed no change in the paraprotein value
Ad hoc subcommittee of the American Academy of Neurology AIDS task force. Criteria for diagnosis of chronic inflammatory demyelinating polyneuropathy (CIDP). Neurology. 1991;41:617-618.
Albers JW, Kelly JJ. Acquired inflammatory demyelinating polyneuropathies: clinical and electrophysiologic features. Muscle Nerve. 1989;12:435-451.
Braun PE, Frail DE, Latov N. Myelin-associated glycoprotein is the antigen for monoclonal IgM polyneuropathy. J Neurochem. 1982;39:1261-1265.
Bromberg MB, Feldman EV, Albers JW. Chronic inflammatory demyelinating polyradiculoneuropathy: comparison of patients with and without an associated monoclonal gammopathy. Neurology. 1992;42:1157-1163.
Cook D, et al. High dose intravenous immunoglobulin in the treatment of demyelinating neuropathy associated with monoclonal gammopathy. Neurology. 1990;40:212-214.
Cornblath DR. Electrodiagnostic abnormalities in Guillain-Barré syndrome. Ann Neurol. 1990;27(suppl):S17-S20.
Cornblath DR, et al. Conduction block in clinical practice. Muscle Nerve. 1991;14:869-871.
Dyck PJ, et al. Plasma exchange in polyneuropathy associated with monoclonal gammopathy of undetermined significance. N Engl J Med. 1991;325:1482-1486.
Eurelings M, et al. Malignant transformation in polyneuropathy associated with monoclonal gammopathy. Neurology. 2005;64:2079-2084.
Gorson KC, Allan G, Ropper AH. Chronic inflammatory demyelinating polyneuropathy: clinical features and response to treatment in 67 consecutive patients with and without a monoclonal gammopathy. Neurology. 1997;48:321-328.
Katirji B. Chronic relapsing axonal neuropathy responsive to intravenous immunoglobulins. Neurology. 1997;48:1690-1694.
Kelly JJJr. The electrodiagnostic findings in polyneuropathies associated with monoclonal gammopathies. Muscle Nerve. 1990;13:1113-1117.
Kelly JJJr, et al. Prevalence of monoclonal proteins in peripheral neuropathy. Neurology. 1981;31:1480-1483.
Kelly JJJr, et al. Polyneuropathies associated with IgM monoclonal gammopathies. Arch Neurol. 1988;45:1355-1359.
Kyle RA. “Benign” monoclonal gammopathy: a misnomer? JAMA. 1984;251:1849-1854.
Kyle RA, Dyck PJ. Neuropathy associated with monoclonal gammopathies. In Dyck PJ, Thomas PK, editors: Peripheral neuropathy, 3rd ed., Philadelphia, PA: WB Saunders, 1993.
Latov N, et al. Plasma-cell dyscrasia and peripheral neuropathy with a monoclonal antibody to peripheral-nerve myelin. N Engl J Med. 1980;303:618-621.
Lewis RA, Sumner AJ. The electrodiagnostic distinctions between chronic familial and acquired demyelinative neuropathy. Neurology. 1982;32:592-596.
Notermans NC, et al. Polyneuropathy associated with monoclonal gammopathy of undetermined significance: a prospective study of the prognostic value of clinical and laboratory abnormalities. Brain. 1994;117:1385-1393.
Notermans NC, et al. Intermittent cyclophosphamide and prednisone treatment of polyneuropathy associated with monoclonal gammopathy of undetermined significance. Neurology. 1996;47:1227-1233.
Report from an ad hoc subcommittee of the American Academy of Neurology AIDS Task Force. Research criteria for the diagnosis of chronic inflammatory demyelinating polyneuropathy (CIDP). Neurology. 1991;41:617-618.
Rhee RK, England DR, Sumner AJ. Computer simulation of conduction block: effects produced by actual block versus interphase cancellation. Ann Neurol. 1990;28:146-159.
Simmons Z, et al. Presentation and initial clinical course in patients with chronic inflammatory demyelinating polyradiculoneuropathy: comparison of patients without and with monoclonal gammopathy. Neurology. 1993;43:2202-2209.
Simmons Z, et al. Long term follow-up of patients with chronic inflammatory demyelinating polyradiculoneuropathy, without and with monoclonal gammopathy. Brain. 1995;118:339-368.