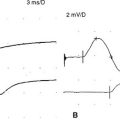
Case 1
HISTORY AND PHYSICAL EXAMINATION
EMG examination was performed 4 weeks after the onset of acute footdrop.
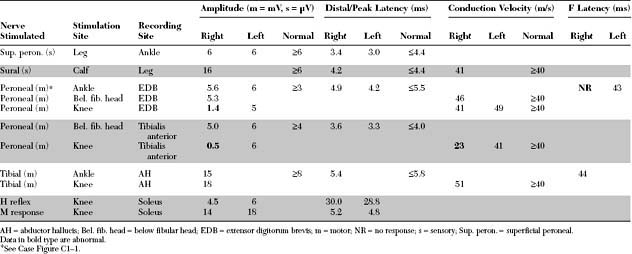
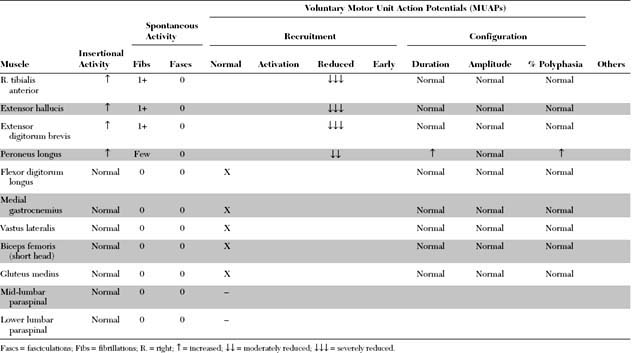
Please now review the Nerve Conduction Studies and Needle EMG tables.
QUESTIONS
EDX FINDINGS AND INTERPRETATION OF DATA
The pertinent electrodiagnostic EDX features in this case include the following:
DISCUSSION
Structure of Peripheral Nerve
The peripheral nerve consists of both unmyelinated and myelinated fibers and their supporting elements. The unmyelinated axons are surrounded only by the plasma membrane of a Schwann cell. The myelinated axons are engulfed by a Schwann cell that wraps around the axons multiple times, thereby insulating the axon with multiple layers of cell membrane, which is rich in lipid sphingomyelin. The myelinated axon is surrounded completely by myelin and Schwann cells, except at certain gaps. In adults, the gaps between myelin segments, called the nodes of Ranvier, measure approximately 1 μm, while myelinated segments between nodes, called the internodal segments, measure approximately 1 mm each.
Pathology of Peripheral Nerve Injury
Nerve injuries that are associated with focal interruption of the continuity of the axons cause significant changes in the structure of the peripheral nerve distal to the lesion (Table C1-1). The distal axons undergo a degenerative process, known as wallerian degeneration. This occurs since all the necessary building blocks needed for maintaining the axon are made in the cell body (peikaryon) and cannot reach the distal stump. The rate at which wallerian degeneration proceeds varies depending on the nerve injured, axon diameter, and the length of distal stump (the larger and the longer the distal stump the more time is needed for wallerian degeneration to be completed). Within hours of most nerve injuries, myelin begins to retract from the axons at the nodes of Ranvier. This is followed by swelling of the distal nerve segment, leakage of axoplasm, and subsequently the disappearance of neurofibrils. Within days, the axon and myelin fragment, and digestion of nerve components starts. By the end of the first week, the axon and myelin become fully digested and Schwann cells start to bridge the gap between the two nerve segments. In chronic nerve lesions, the endoneurial tubes in the distal stump shrink, the nerve fascicles atrophy distal to the lesion, and, in complete nerve transection, the severed ends retract away from each other.
Table C1-1 Consequences of Focal Axonal Injury Distal to the Lesion
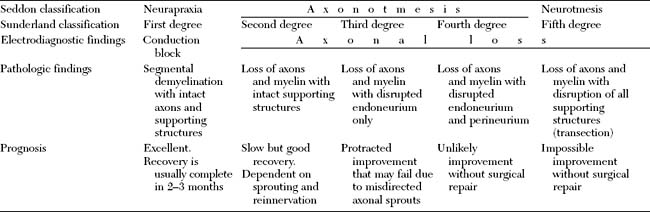
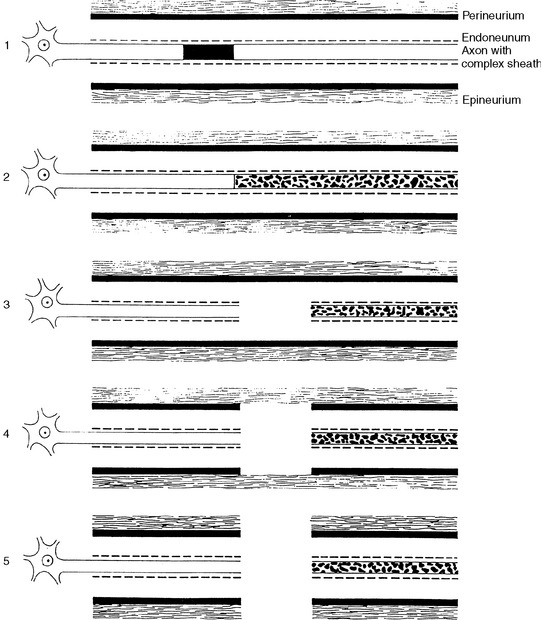
Classification of Peripheral Nerve Injury
Many classifications of peripheral nerve injury have been suggested, but Seddon’s and Sunderland’s classifications are the most widely used in clinical practice. These are based on the functional status of the nerve and on histologic findings. They are shown in Table C1-2 and in Figure C1-2, with their corresponding electrophysiologic findings.
Electrodiagnosis of Peripheral Nerve Injury
The EDX studies are the cornerstone in the diagnosis and management of nerve injuries by providing valuable information as to the location of the lesion, and its severity, pathophysiology, and prognosis (Table C1-3). Intraoperatively, the EDX studies guide the surgeon during the procedure and help assess the status of the regenerating axons within the injured nerve segment. During the recovery stage of peripheral nerve injury that may occur spontaneously or after surgical repair, the EDX studies are also essential in the evaluation of remyelination, regeneration, and reinnervation.
Table C1-3 Role of Electrodiagnostic Studies in Peripheral Nerve Injury
Localization of Nerve Lesions Using Nerve Conduction Studies
Focal Slowing
Focal slowing of conduction usually is caused by widening of the nodes of Ranvier (paranodal demyelination) and, sometimes, focal axonal narrowing. It is evident on NCSs by slowing of conduction of a specific nerve segment, while other segments of the same nerve as well as neighboring nerves remain normal. When the large myelinated fibers are slowed to essentially the same extent, focal slowing across the involved nerve segment is synchronized. This is manifested by either a prolongation of distal latencies (in distal lesions) or slowing in conduction velocities (in proximal lesions), while the CMAP amplitude, duration, and area are not affected and do not change when the nerve is stimulated proximal to the lesion. When variable number of the medium or small nerve fibers (average or slower conducting axons) are affected only, desynchronized (differential) slowing of conduction across the nerve segment is evident. In this situation, the CMAP is dispersed on stimulation proximal to the lesion and has prolonged duration, with normal (nondispersed) response on distal stimulation. If this finding is isolated, the distal latency or conduction velocity, which represent the speed of the largest (fastest) axons, are normal. However, in most clinical situations, the large fibers are often involved also, desynchronized slowing is usually accompanied by slowing at the involved segment, resulting in concomitant slowing of distal latency or conduction velocity.
Conduction Block
Temporal dispersion and phase cancellation are more prominent in sensory nerve conduction studies due to (1) the disparity of sensory fiber conduction velocities which are almost double that of the motor axons (25 m/s) and (2) the surface recorded nerve action potentials are triphasic. The SNAP may normally decrease in amplitude and area by 50+ or more and its duration can increase by 100+ or more with proximal stimulation in antidromic studies (or with proximal recording with orthodromic studies). Hence, it is a common practice not to rely on sensory studies in the diagnosis of conduction block. Figures C1-3 and C1-4 depict the concept of temporal dispersion and phase cancellation using computer modeling.


Impeding transmission of action potentials is the basis of conduction block. This usually results from internodal demyelination, but can occur in axonal loss before wallerian degeneration (“axonal” conduction block). Blockage of the transmission of electrical impulses anywhere throughout the course of motor axons results in motor weakness that is often indistinguishable from weakness that results from loss of motor neurons or motor axons. Experimental evidence on tourniquet paralysis on baboon hind limb showed that conduction block is reversible and the distal nerve remains normal and excitable (Figure C1-5).
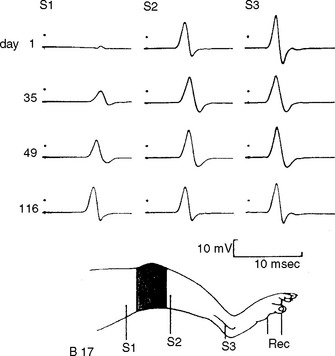
In practice, conduction block is defined as a relative decrease in the CMAP amplitude and area with proximal stimulation, when compared with the CMAP on distal stimulation, without significant prolongation of CMAP duration. Conduction block should be distinguished from physiologic or abnormal temporal dispersion. Based on experimental studies, differential slowing along medium and thinly myelinated fibers may result in temporal dispersion and phase cancellation manifesting as significant drop of amplitude that may occasionally reach up to 80+. This is often associated with obvious and marked prolongation of CMAP duration. In contrast to amplitude decay, differential slowing does not drop the area beyond 50+. Hence, in true conduction block a significant drop in amplitude should always be corroborated by a similar drop in CMAP area.
There are no uniformly accepted criteria for the identification of conduction block. Table C1-4 reveals some of the common errors made in the EMG laboratory in the diagnosis of true conduction block. Table C1-5 lists practical criteria for the diagnosis of conduction block. In general, an amplitude change should be always supported by area change, since a significant drop in amplitude up to 50+ or more may occasionally be due solely to abnormal temporal dispersion while an area drop of more than 50+ is always due to conduction block. In clinical practice, the identification of demyelinative conduction block is an excellent tool for precisely localizing peripheral nerve lesions. Conduction block is often caused by acute nerve compression such as peroneal mononeuropathy at the fibular neck or radial mononeuropathy across the spiral groove. It is also a common finding in immune-mediated peripheral neuropathies such as acute inflammatory demyelinating polyneuropathy, chronic inflammatory demyelinating polyneuropathy, or multifocal motor neuropathy. Finally, conduction block usually is reversible and amenable to treatment, by removing the offending compression factor from the injured nerve or immunotherapy.
Table C1-4 Common Errors in the Diagnosis of Conduction Block
Table C1-5 Electrodiagnosis of Conduction Block*
CMAP = compound muscle action potential.
* All amplitudes, areas, and durations reflect negative-peak areas, amplitudes, and durations.
† Caution should be used in evaluating the tibial nerve since stimulation at the knee may result in more than 50% decrease in amplitude, especially in obese patients.
Axonal Loss
NCSs, done on patients who harbor axonal damage before the completion of wallerian degeneration, require special attention since they can be a source of error in localizing, characterizing, or prognosticating nerve lesions. However, these early studies are useful since they often help localizing lesions better than if NCSs are done after the completion of wallerian degeneration.
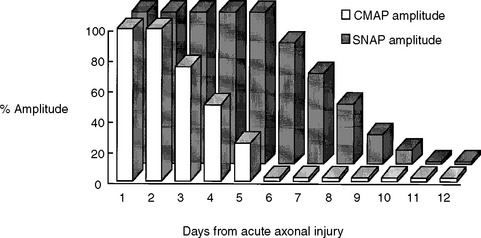
Early after axonal damage, the distal stump remains excitable for a variable period with some differences between the motor and sensory responses. The distal CMAP remains normal for 1 to 2 days after injury, giving rise to a pattern of conduction block on NCS that mimics the one seen with segmental demyelination. This pattern is sometimes referred to as “axonal noncontinuity, early axon loss, and axon discontinuity” conduction block. It is important to recognize this pattern since it carries poor prognosis, in contrast to the conduction block that is caused by segmental demyelination which usually recovers rapidly and completely. As wallerian degeneration progresses following axon injury, the distal CMAP then falls precipitously to reach its nadir by 5–6 days postinjury. After this time, the conduction block pattern is replaced by unelicitable CMAPs in complete lesions or low-amplitude CMAPs in partial lesions that are independent of the stimulation sites. In contrast to the motor studies, the distal sensory nerve remains excitable for a slightly longer period. The distal SNAP remains normal for 5–6 days and then decreases rapidly to reach its nadir in 10–11 days (Figure C1-6). Thus, repeat studies performed after the completion of wallerian degeneration prove that the lesion is due to axonal loss, by revealing a decrease in distal CMAP to values very similar to proximal CMAP values, along with low or absent SNAP.
Localization of Nerve Lesions Using Needle Electromyography
Axon loss lesions studied by NCSs prior to wallerian degeneration, as well as demyelinating (neurapraxic) lesions, are often precisely localized to a short segment of the nerve due to the presence of conduction block across that segment. Hence, localization of lesions by needle EMG is most important in axon loss lesions that are first studied following the completion of wallerian degeneration of motor axons (more than 5–6 days postinjury). These lesions are associated with nonlocalizable NCSs that are characterized by low-amplitude or unelicitable CMAPs from all nerve simulation sites.
Timing of Electrodiagnostic Studies in Peripheral Nerve Injury
In patients with closed nerve trauma or severe limb trauma at several sites, where the exact site of injury may not be clear, early NCSs are very useful in attempting to identify conduction block across the site of the lesion. This should be done, if possible, very early and before 3–5 days from injury since the distal CMAP reaches its nadir after that time. Detecting conduction block with this early study is extremely useful in precise localization of the site of the lesion, though finding a conduction block cannot distinguish whether the lesion is due to axon loss, demyelination, or a mixture of both. A repeat study after allowing time for the completion of motor and sensory wallerian degeneration (i.e., after 10–11 days from injury) helps establish the pathophysiologic diagnosis and estimate the degree of injury and prognosis.
Electrodiagnostic Studies During the Recovery Phase
Reinnervation by Collateral Sprouting
Collateral sprouting in partial axon loss lesions starts as early as 1–2 days after injury. However, the early signs of reinnervation first become evident on needle EMG by one month, and are usually definite by 2–3 months postinjury. Immediately following nerve injury, there is a decrease in MUAP recruitment in affected muscles that is appropriate to the number of lost axons. In the first few weeks after injury, MUAPs of surviving axons retain their normal morphology. As collateral sprouting proceeds, muscle fibers become progressively incorporated to the territory of the motor unit.
Early on, the collateral axons (sprouts) have thin or incomplete myelin. Hence, action potentials along collateral sprouts conduct slowly. This is often reflected on needle EMG by MUAPs with satellite potentials (linked or parasite potentials), late spikes of the MUAP that are distinct and time-locked with the main potentials. The satellite potential trails the main MUAP because the newly formed nerve terminal may be long, or small and thinly myelinated, or both, resulting in slower conduction. When a satellite potential is suspected on needle EMG, it is useful to use a trigger line to demonstrate that this potential is time-locked to the main potential (Figure C1-7).
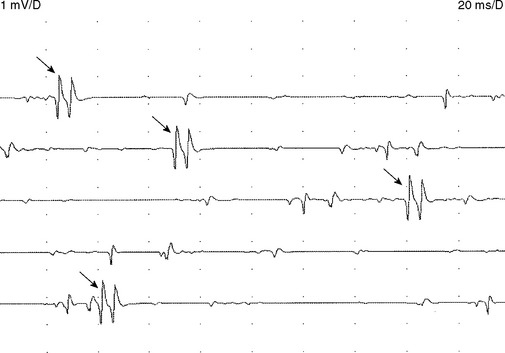
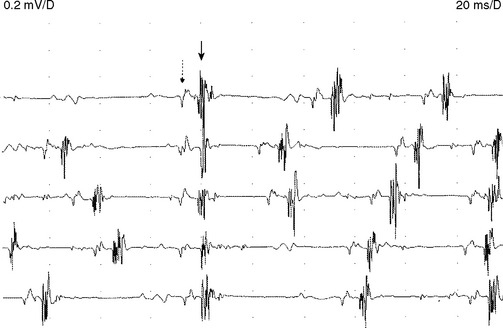
Reinnervation MUAPs, including satellite potentials, may be unstable (Figure C1-8). The MUAPs may show evidence of intermittent nerve conduction blocking or neuromuscular junction blocking due insecure action potential transmission at the sprout or endplate, respectively. This results in individual muscle fibers being either blocked or come to action potential at varying intervals, leading to a MUAP that changes in configuration from impulse to impulse (amplitude or number of phases or both). Over time, the sprout matures and the conduction velocity increases and the satellite potential then fires more closely to the main potential, and ultimately fuses to become an additional phase or serration within the main MUAP complex (Figure C1-9). In general, MUAPs become more stable, more polyphasic, and longer in duration as collateral sprouting continues. In very chronic lesions, MUAPs are typically stable with long duration and high amplitude and little or no polyphasia, reflecting the maturity of all the nerve sprouts (Figure C1-10). Also, as reinnervation proceeds, there is a decline in the number of fibrillation potentials, since reinnervated muscle fibers will cease to generate this spontaneous activity.
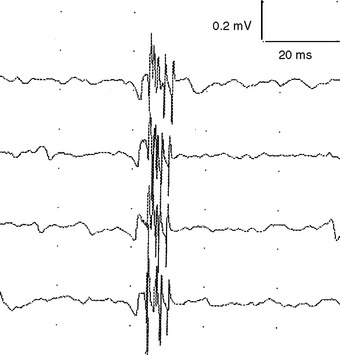
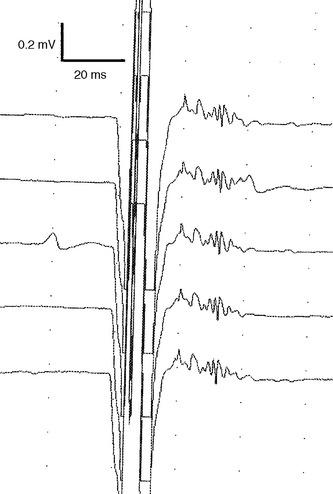
Reinnervation by Axon Regeneration
In complete or very severe axon loss peripheral nerve lesions, improvement is dependent solely or primarily on axonal regeneration that may occur spontaneously or following surgical repair. Unfortunately, in most cases of nerve injury, regeneration is slow and incomplete. In more severe axon loss lesions, the regenerating axons may not find intact endoneurial tubes and sometimes form a neuroma with tangled axons at the site of injury. In such lesions as well as lesions with complete transection, surgical repair is often needed.
On needle EMG, the early signs of regeneration can be confirmed by the appearance of small, complex, unstable MUAPs, sometimes referred to as “nascent” MUAPs, that precedes the onset of visible voluntary contraction. These units appear first in muscles nearest to the site of the injury and progress distally, and hence are useful in assessing the advancement of this proximodistal regeneration. Nascent MUAPs are very low in amplitude and extremely polyphasic, with normal or increased duration. These small nascent MUAPs mimic the MUAPs seen with necrotizing myopathies. Nascent MUAPs are often unstable due to conduction or neuromuscular junction blocking and are associated with decreased MUAP recruitment (Figure C1-11). As reinnervation proceeds, nascent MUAPs that are unstable become transformed into stable, long-duration, and polyphasic MUAPs, reflecting increased numbers of muscle fibers per motor unit, full myelination of the regenerating axons, and the maturity of the neuromuscular junctions. Similar to what is seen in reinnervation by collateral sprouting, there is also a decline in the number of fibrillation potentials and the progressive improvement of the SNAP and CMAP on NCS. However, in these severe or complete nerve lesions, it is common that the CMAP and SNAP never return to baseline values and there is often permanent slowing and dispersion of the CMAPs due to the extreme variability in the diameter and myelination of the regenerated axons that results in significant differential slowing of conduction velocities.
Aberrant Regeneration
The most common neurologic sequelae of aberrant reinnervation occur after facial nerve injury including after idiopathic Bell’s palsy. Aberrant regeneration between motor axons results in facial synkinesis, mainly contraction of the lower facial muscles on the affected side whenever there is an eye blink or vice versa. Other much less common, yet more publicized, examples of abnormal regeneration patterns are the “crocodile tears,” manifested as lacrimation of the ipsilateral eye during chewing, and the Marin-Amat syndrome, or “jaw-winking,” manifested as closure of the ipsilateral eyelid when the jaw opens.
FOLLOW-UP
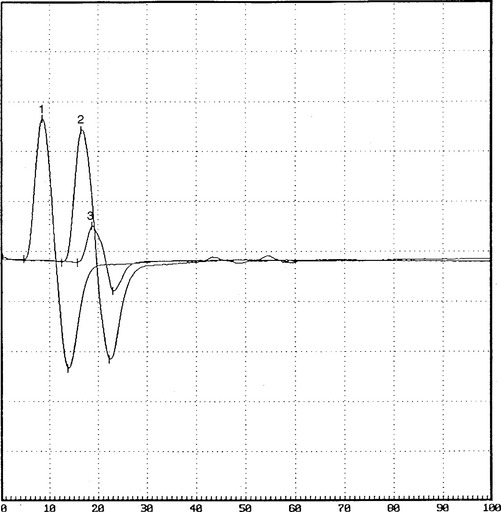
On further questioning, it was determined that the patient was involved in a long meeting that lasted approximately 12 hours, during most of which he was seated around a table. He also admitted to frequent leg crossing. More importantly, he had been on an intensive weight loss program for 2 to 3 weeks and had lost 30 pounds (see Case 8 for further discussions). When he was seen 2 months later, he had no residual weakness and minimal numbness on the dorsum of the foot. Repeat nerve conduction studies showed significant improvement of proximal peroneal (knee) CMAP amplitude and area (Figure C1-12).
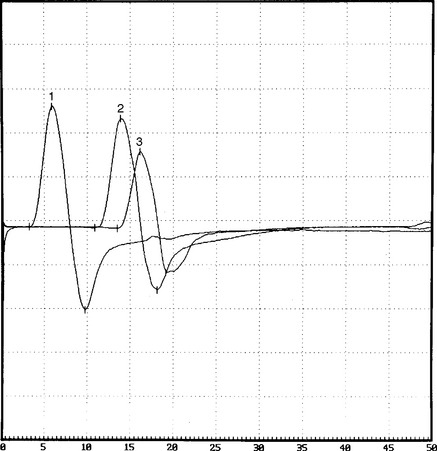
Figure C1-12 Follow-up peroneal motor nerve conduction studies recording extensor digitorum brevis stimulating ankle (1), below fibular head (2), and knee (3). Note the significant improvement of CMAP amplitude and area stimulating at the knee (waveform 3) compared with the initial study done at presentation (compare with Figure C1-1). (Sensitivity: vertical scale = 2 mV.)
Brushart TME. Central course of digital axons within the median nerve. J Comp Neurol. 1991;311:197-209.
Chaudhry V, Cornblath DR. Wallerian degeneration in human nerves: serial electrophysiologic studies. Muscle Nerve. 1992;15:687-693.
Cornblath DR, et al. Conduction block in clinical practice. Muscle Nerve. 1991;14:869-871.
Fowler CJ, Danta G, Gilliatt RW. Recovery of nerve conduction after a pneumatic tourniquet: observation on the hind-limb of the baboon. J Neurol Neurosurg Psychiatry. 1972;35:638-647.
Gilliatt RW, Taylor JC. Electrical changes following section of the facial nerve. Proc R Soc Med. 1959;52:1080.
Jabaley ME, Wallace WH, Heckler FR. Internal topography of major nerves of the forearm and hand: a current view. J Hand Surg [Am]. 1980;5:1-18.
Katirji MB, Wilbourn AJ. Common peroneal mononeuropathy: a clinical and electrophysiologic study of 116 lesions. Neurology. 1988;38:1723-1728.
Kimura J, et al. Relation between size of compound sensory or muscle action potentials, and length of nerve segment. Neurology. 1986;36:647-652.
Kline DG. Surgical repair of peripheral nerve injury. Muscle Nerve. 1990;13:843-852.
Miller RG. Injury to peripheral motor nerves. Muscle Nerve. 1987;10:698-710.
Oh SJ, Kim DE, Kuruoglu HR. What is the best diagnostic index of conduction block and temporal dispersion? Muscle Nerve. 1994;17:489-493.
Rhee RK, England JD, Sumner AJ. Computer simulation of conduction block: effects produced by actual block versus interphase cancellation. Ann Neurol. 1990;28:146-159.
Robinson LR. Traumatic injury to peripheral nerves. Muscle Nerve. 2000;23:863-873.
Seddon H. Three types of nerve injury. Brain. 1943;66:237-288.
Spinner RJ, Kline DG. Surgery for peripheral nerve and brachial plexus injuries or other nerve lesions. Muscle Nerve. 2000;23:680-695.
Stewart JD. Peripheral nerve fascicles: anatomy and clinical relevance. Muscle Nerve. 2003;28:525-541.
Sunderland S. The anatomy and physiology of nerve injury. Muscle Nerve. 1990;13:771-784.
Sunderland S. Nerve injuries and their repair: a critical appraisal. Edinburgh: Churchill Livingstone, 1991.
Taylor PK. CMAP dispersion, amplitude decay and area decay in a normal population. Muscle Nerve. 1993;16:1181-1187.
Wilbourn AJ. Nerve conduction studies: types, components, abnormalities and value in localization. Neurol Clin N Am. 2002;20:305-338.