Cardiovascular System
Myocardial and Pericardial Diseases

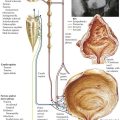
Congenital heart disease results from malformations of the heart and major vessels that develop during embryogenesis and are present at birth. A general classification of congenital malformations of the heart and major vessels is presented in Table 2-1. The ventricular septal defect (VSD) is the most common malformation presenting in infancy and childhood. Most VSDs result from defective closure of the membranous interventricular septum, although some are located in the muscular interventricular septum. As a result of the left-to-right shunt, patients present with systolic murmur, CHF, and progressive pulmonary HTN. If not surgically corrected, pulmonary arterial pressure reaches the systemic level, and the shunt becomes predominantly right to left, leading to late onset of cyanosis (Eisenmenger syndrome).
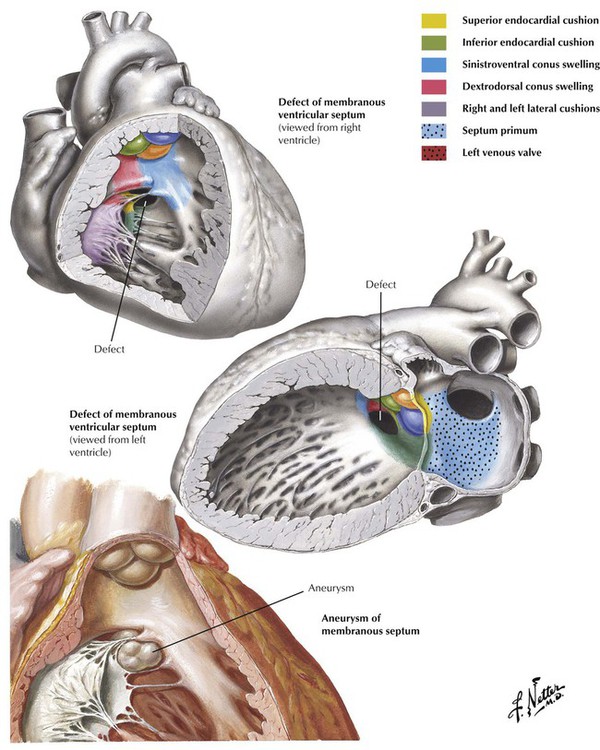
The ductus arteriosus is an arterial connection between the origin of the left pulmonary artery and the aorta that normally closes within hours after birth. Failure of this connection to close results in PDA. PDA is another type of high-pressure left-to-right shunt producing symptomatic disease in infants and children. Other anomalies of the aortic arch system, such as an aberrant right subclavian artery, give rise to anatomic variations of the normal pattern of origin of the great arteries. Some of these anomalies produce vascular rings that can compress the trachea and esophagus.
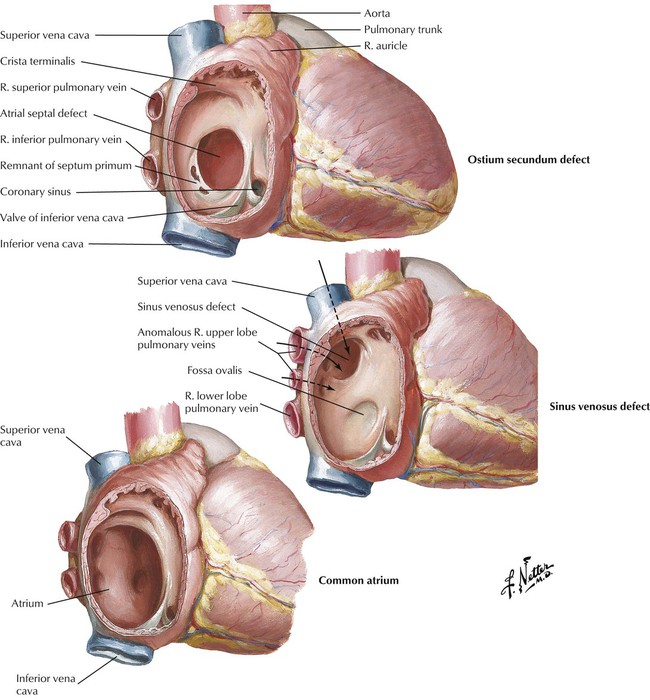
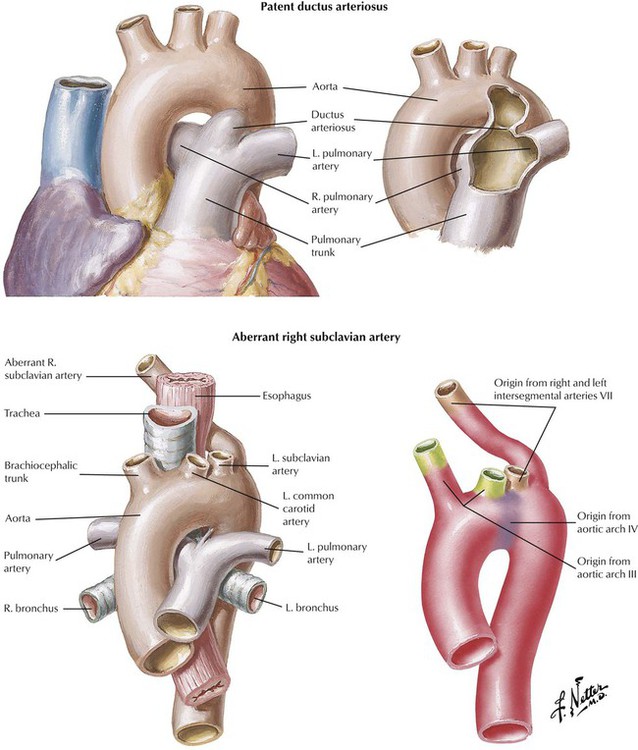
Ostium secundum defect, the most common atrial septal defect (ASD), is located in the middle portion of the interatrial septum in the region of the foramen ovale. This ASD occurs as a result of defective formation of septum primum and septum secundum tissue, which leads to failure of the ostium secundum to close. Sinus venosus defect, located high in the interatrial septum, is the result of defective incorporation of the sinus venosus into the RV. This ASD is associated with anomalous drainage of the right upper lobe pulmonary veins into the right atrium. Failure of formation of the septum primum and septum secundum results in a common atrium. Because left-to-right shunting occurs at low pressure, patients with ASDs tend to have pulmonary HTN and become symptomatic later in childhood or as adults in contrast to the usual course of patients with VSDs and PDAs.
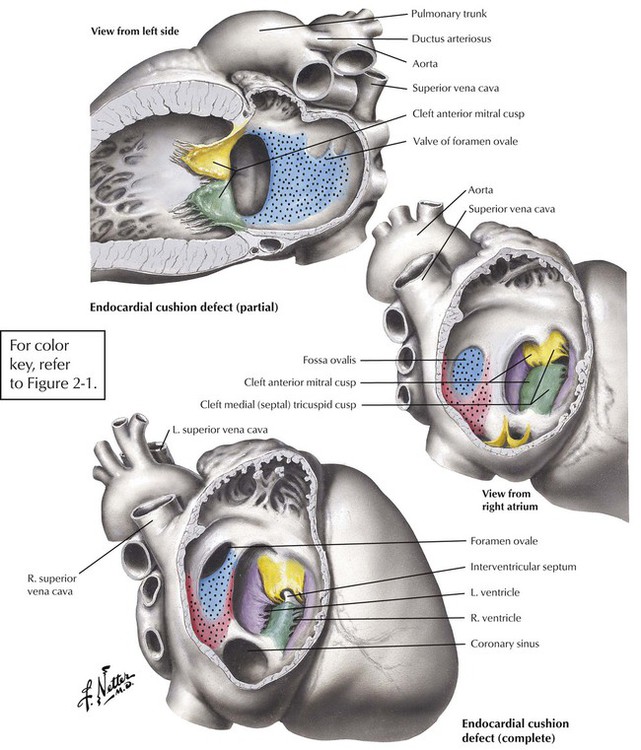
Atrioventricular septal defects (AVSDs) result from significantly defective formation of endocardial cushion tissue. The ASD component is low in the interatrial septum because of failure of closure of the ostium primum. The VSD component is in the region of the membranous interventricular septum. The partial endocardial-cushion defect is composed of an ostium primum type of ASD, a defective mitral valve with a cleft in the anterior leaflet, and subtle anomalies in the LV, but it is associated with a closed membranous interventricular septum. The complete endocardial-cushion defect, also called a persistent common atrioventricular canal, consists of a large ostium primum ASD, a membranous VSD, and an abnormal common atrioventricular valve straddling the AVSD.
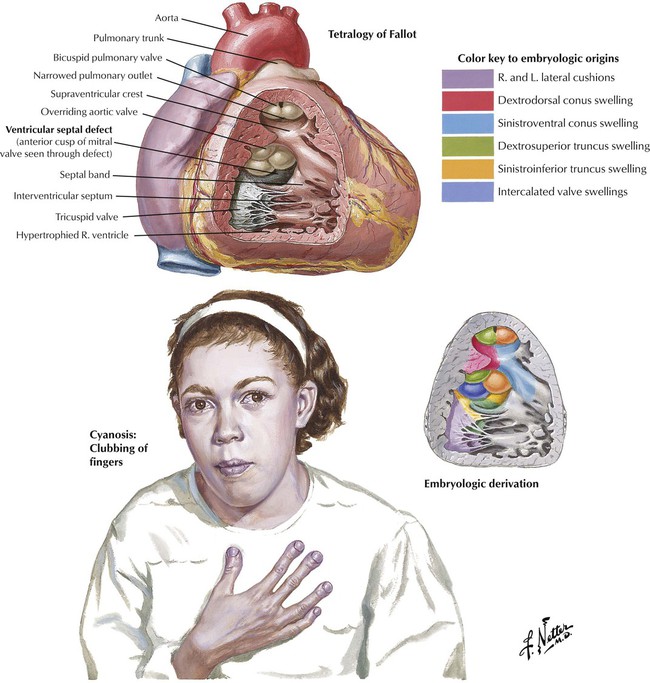
The tetralogy of Fallot is the most common form of cyanotic congenital heart disease, a state characterized by a right-to-left shunt with cyanosis at the time of presentation (i.e., cyanotic congenital heart disease). Depending on the severity of the defects, the presentation may occur in infancy (blue baby syndrome) but is not usually apparent until at least early childhood. The 4 components of the tetralogy of Fallot are (1) VSD; (2) obstruction of the right ventricular outflow tract, usually as a result of subpulmonic, infundibular stenosis; (3) an aorta that overrides the VSD; and (4) right ventricular hypertrophy. Complete surgical correction of the tetralogy of Fallot includes closure of the VSD and expansion of the right ventricular outflow tract.
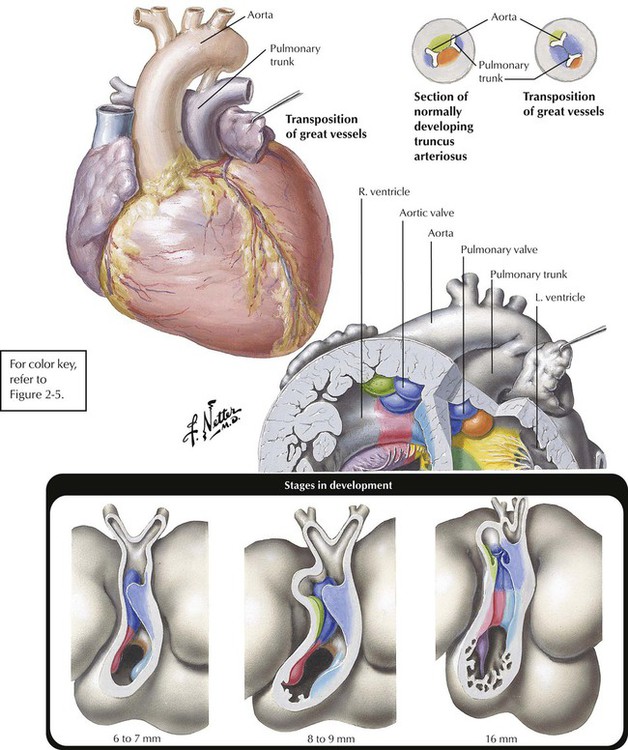
Transposition of the great vessels, or more specifically, congenitally complete transposition of the great vessels, is a condition in which the aorta takes origin anteriorly from the RV and the pulmonic trunk arises posteriorly from the LV. Transposition of the great vessels is compatible with postnatal life only when the anomaly occurs in association with one or more other defects, usually VSD, ASD, or PDA. The lower diagram shows the embryological development of transposition. Normally, two pairs of truncal swellings develop. In transposition, the wrong pair of truncal swellings becomes involved in partitioning the truncus, resulting in the abnormal position of the great vessels.
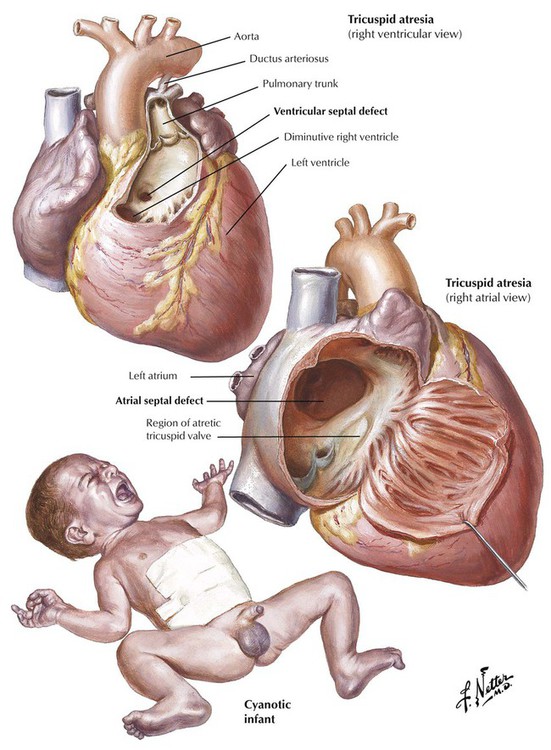
Tricuspid atresia, a severe complex anomaly of the right side of the heart with underdevelopment (hypoplasia) of the RV and a right-to-left shunt through an ASD, a VSD, or a PDA, results in severe cyanotic heart disease in infants. Next to transposition of the great vessels, it is the most common cause of severe cyanosis in the neonatal period, and the degree of cyanosis is usually more marked than in cases of transposition.
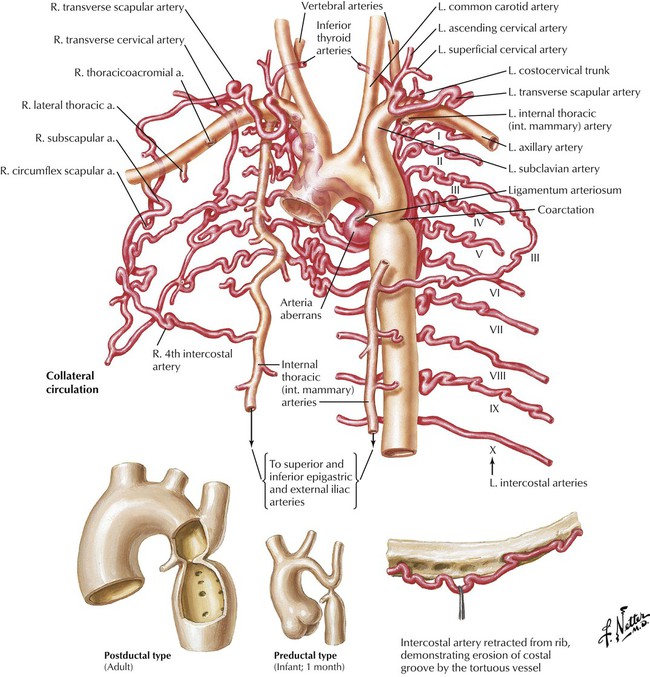
Coarctation of the aorta is a common obstructive congenital anomaly. There are 2 major types: (1) an infantile form, with tubular hypoplasia of the aortic arch proximal to a PDA, typically resulting in clinical problems in early childhood; and (2) an adult postductal form, in which there is a discrete ridgelike infolding of the aorta, just opposite the closed ductus arteriosus (the ligamentum arteriosum). The postductal type of coarctation leads to the development of an extensive collateral circulation (top image) to bypass the obstruction. The patient presents with HTN in the upper extremities and normal pressures in the lower extremities. Rib notching (produced by the enlarged collateral arteries) is seen on chest radiograph.
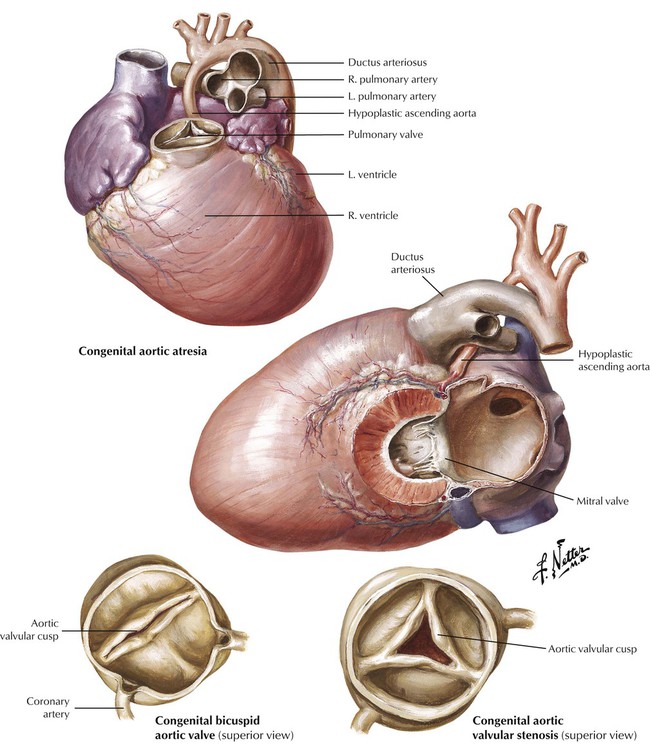
Left ventricular outflow tract (LVOT) obstruction can result from aortic stenosis or atresia. In severe congential aortic malformation, LVOT obstruction leads to underdevelopment (hypoplasia) of the LV and ascending aorta. There is a dense, porcelainlike endocardial fibroelastosis of the diminutive LV. The ductus arteriosus is open. This constellation of anomalies constitutes the hypoplastic left heart syndrome, a condition that is fatal in the first several days of postnatal life when the ductus closes, unless high-risk surgery is performed. Less severe congenital aortic stenoses are compatible with longer survival. The congenital bicuspid aortic valve occurs in approximately 1% to 2% of the population and can give rise to aortic stenosis in adulthood because of hemodynamic turbulence that leads to fibrosis and calcification.
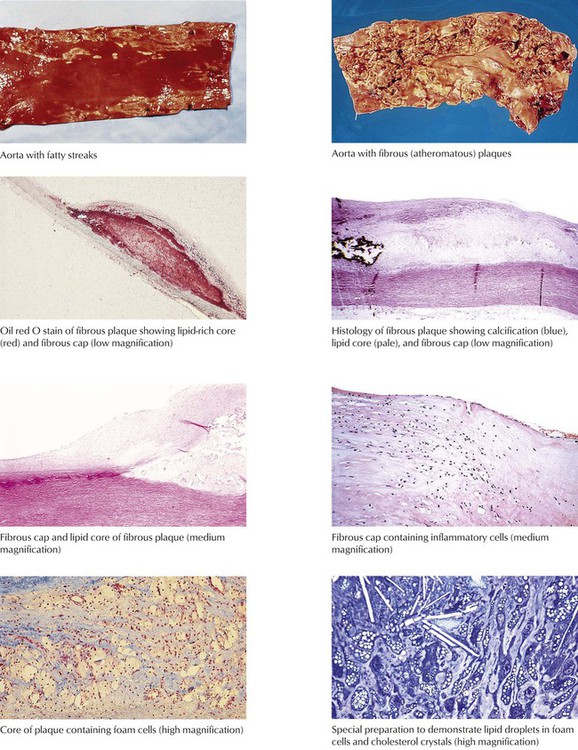
Atherosclerosis, the most prevalent and important form of arteriosclerosis, is a disease that typically affects the aorta and its major muscular distributing branches. The lesion of established atherosclerosis is the atherosclerotic (fibrous or atheromatous) plaque (gross photo, upper right). The fatty streak is the most obvious precursor lesion (gross photo, upper left). The photomicrographs show the features of atherosclerotic plaques, including fibrous capsule and lipid-rich core containing foam cells and cholesterol crystals.
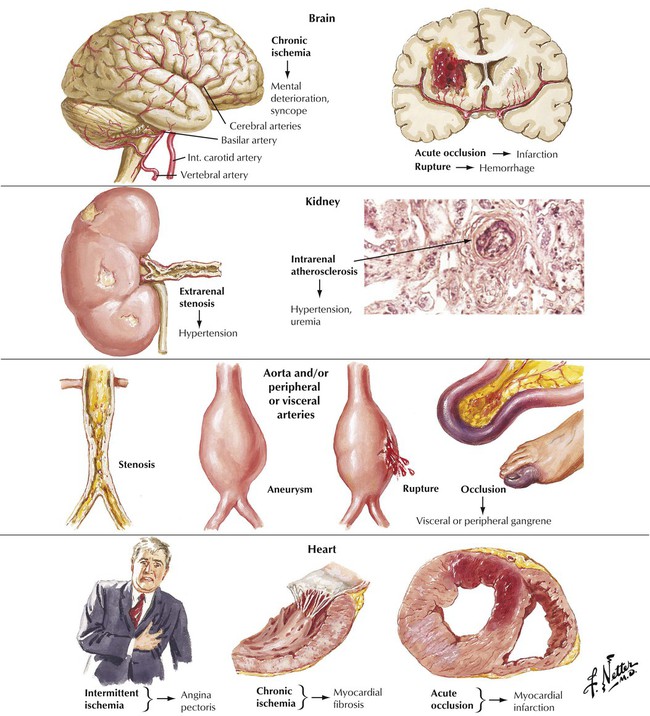
The major pathologic and clinical effects of atherosclerosis involve the brain, the kidneys, the aorta, the peripheral and visceral arteries, and the heart. According to the response to injury hypothesis, atherosclerosis develops as a response of the vessel wall to multifactorial and repetitive injury. Genetic susceptibility, environmental factors, and endogenous metabolic alterations are risk factors that participate in the pathogenesis of atherosclerosis and the formation of atherosclerotic plaques in vessels. Atherosclerosis progresses asymptomatically for years until a clinical threshold is reached. The onset of symptoms may be gradual or abrupt. The major treatable risk factors for development of atherosclerosis are a diet high in saturated fats and cholesterol, HTN, cigarette smoking, and diabetes mellitus.
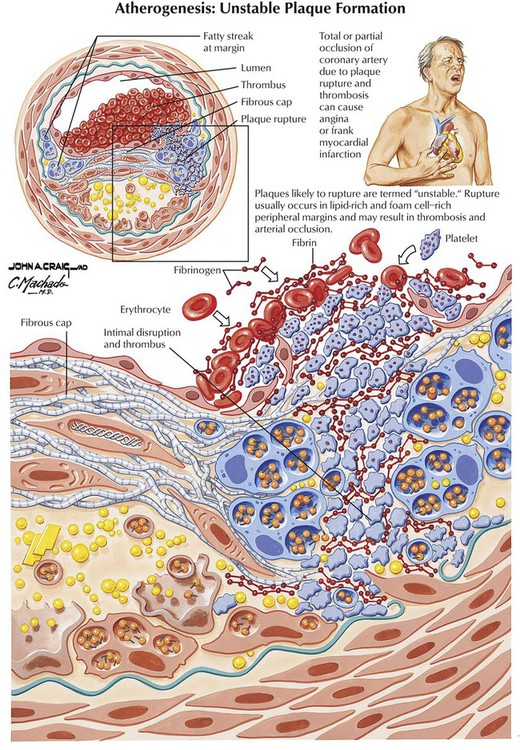
Coronary heart disease (atherosclerotic or ischemic heart disease), dysfunction and damage to the heart muscle resulting from coronary artery disease (CAD), is usually due to coronary atherosclerosis. Coronary atherosclerosis leads to progressive luminal narrowing of one or more of the coronary arteries by atherosclerotic plaques; these frequently have calcification. Coronary reserve is such that angina pectoris usually does not develop until there is at least 75% narrowing of the cross-sectional area. Most symptomatic disease is associated with the development of secondary changes in the plaques, especially surface ulceration, intraplaque hemorrhage, and thrombosis. Coronary thrombi can organize, leading to recanalization of the lumen. Nonatheromatous causes of myocardial ischemia include congenital coronary anomaly, coronary dissection, coronary vasculitis (Kawasaki disease, polyarteritis nodosa, and others), or a systemic hemodynamic problem, such as severe shock or profound anemia.
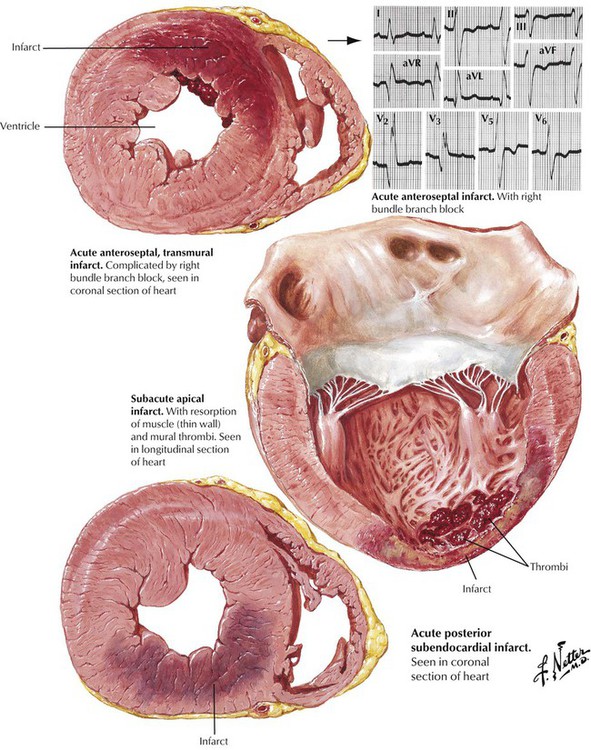
Angina pectoris, which is due to myocardial ischemia of short duration (approximately 15 minutes), results in reversible myocardial injury. Myocardial infarction, the death of heart muscle due to prolonged, severe ischemia, usually involves the LV. Myocardial necrosis generally begins after 45 minutes of severe ischemia and extends from the subendocardium into the subepicardium in a wave-front fashion over a period of approximately 3 to 4 hours. Subendocardial (intramural) myocardial infarcts are limited to the inner half of the wall. Transmural myocardial infarcts extend into the outer half of the wall. Lesions of the left anterior descending coronary system give rise to anterior and anteroseptal myocardial infarcts; lesions of the right coronary artery give rise to inferior (posteroapical) and posterior myocardial infarcts. Lesions of the left circumflex coronary artery give rise to lateral myocardial infarcts.
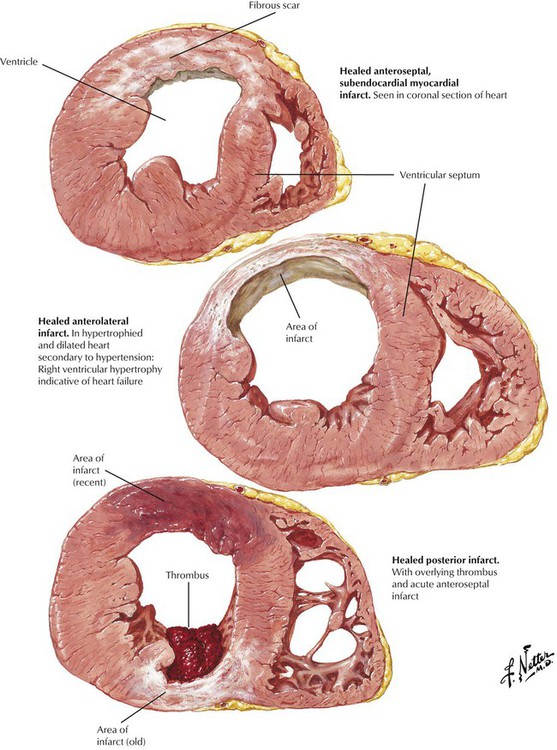
Acute myocardial infarction may result in death due to pump failure or ventricular fibrillation. If the patient survives, the infarct undergoes organization and healing. During the first 2 to 3 weeks, the necrotic myocardium is gradually replaced by granulation tissue; during the next 2 to 3 months, the granulation tissue is converted to fibrous scar. This illustration shows various patterns of healed myocardial infarcts. During healing, the thinned infarcted wall may expand to form a ventricular aneurysm. Mural thrombi can form over the infarct and give rise to systemic emboli.
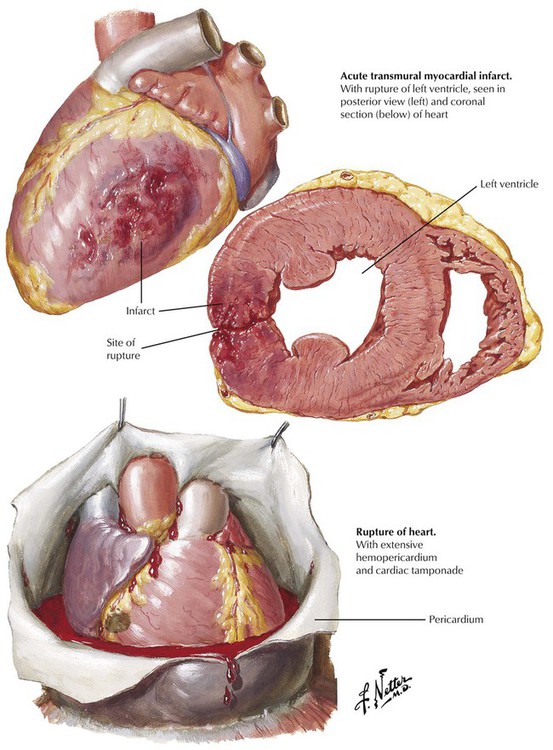
Less than 5% of acute myocardial infarcts rupture. These ruptures involve transmural myocardial infarcts that may rupture during the first 7 to 10 days after onset. Patients at highest risk are those with persistent HTN during their infarcts and those with infarcts in regions without fibrosis; typically, these are first infarcts. Over time, a dissection track develops from the left ventricular chamber through the necrotic myocardium, and the completed process results in abrupt development of hemopericardium, cardiac tamponade, and electromechanical dissociation (electrical rhythm on electrocardiogram [ECG] but no effective cardiac output). This is generally fatal. In some cases, the intramural dissection occurs slowly enough for a pericardial inflammatory reaction to occur and seal off a region of pericardium, containing the rupture. This gives rise to a wide-mouthed pseudoaneurysm that, unlike true aneurysms, is prone to late rupture. Other severe complications of acute myocardial infarction involve rupture of infarcted interventricular septum to produce a VSD and rupture of the head or entire trunk of an infarcted papillary muscle. These complications lead to systolic murmurs and cardiac failure.
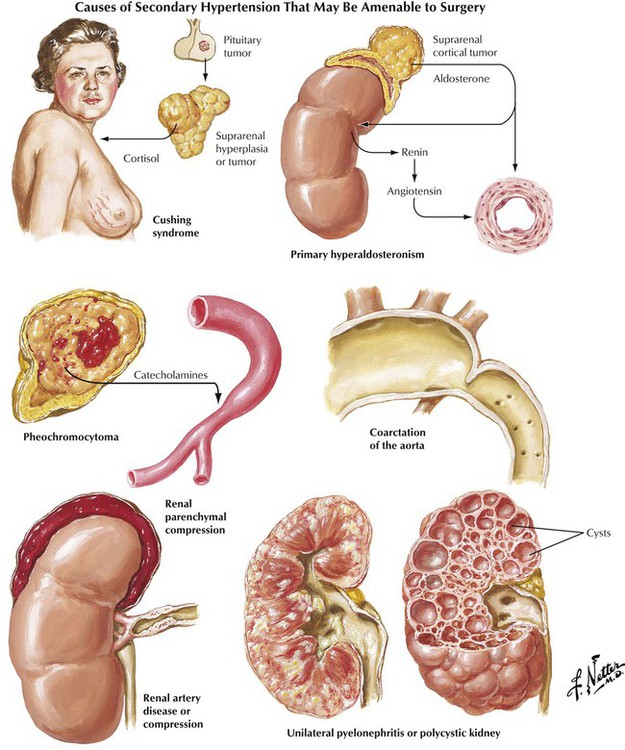
Increase of systemic arterial pressure greater than the normal values of 120 mm Hg systolic and 80 mm Hg diastolic leads to a constellation of changes known as hypertensive cardiovascular disease. The pathophysiologic basis of HTN is excessive arteriolar constriction leading to increased peripheral vascular resistance. This may be exacerbated by factors promoting increased cardiac output. The fundamental etiology of HTN is unknown in most patients, although genetic predisposition and certain environmental influences, particularly high sodium intake, are known to be important factors. This condition is known as essential, idiopathic, or primary HTN. In approximately 10% of patients, HTN is secondary to a recognizable lesion or disease. Parenchymal renal disease and renovascular disease are the most common of these entities that are amenable to surgical treatment. Endocrine disorders and coarctation of the aorta are less common.
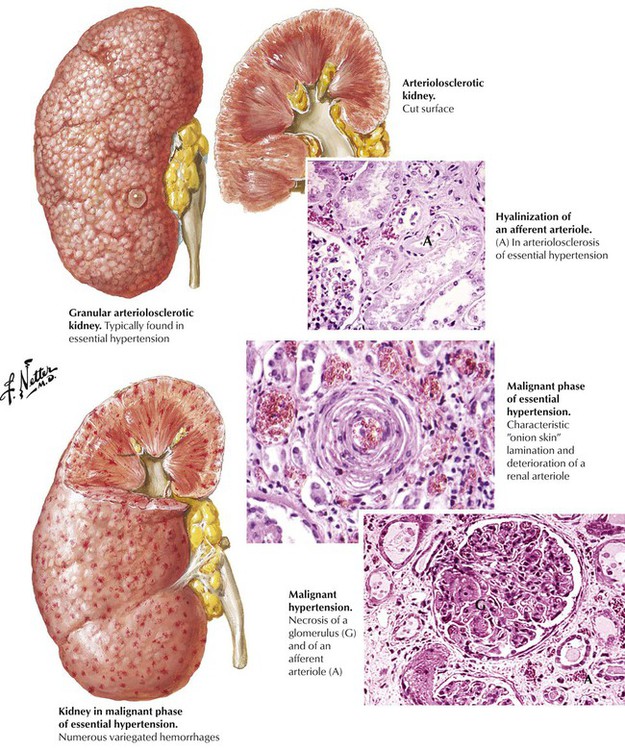
The natural history of HTN follows 2 general patterns. Benign HTN is characterized by mild to moderate increase of blood pressure and an asymptomatic period of several years before the inevitable onset of symptoms and end-organ damage (hence, the condition is not truly benign). Malignant HTN is characterized by marked increase of blood pressure and rapid progression over a few weeks to end-organ failure. Most patients with essential HTN follow the benign pattern, although it may accelerate to malignant HTN. The characteristic vascular lesion of benign essential HTN is widespread hyaline arteriolosclerosis manifest by thickening of the walls of the small arteries and arterioles by amorphous eosinophilic material composed of degenerated smooth cells and deposited plasma proteins. Hyaline arteriolosclerosis with associated small cortical scars (hyaline arteriolonephrosclerosis) is commonly seen in the kidneys. Hyperplastic arteriolosclerosis, marked luminal narrowing by cellular intimal proliferation in a lamellar, “onionskin” pattern, is the characteristic lesion of malignant HTN. In severe malignant HTN, fibrinoid necrosis of the glomerular arterioles occurs. An associated ischemic injury develops rapidly, leading to petechial hemorrhages in multiple organs, including the kidneys (hyperplastic arteriolonephrosclerosis).
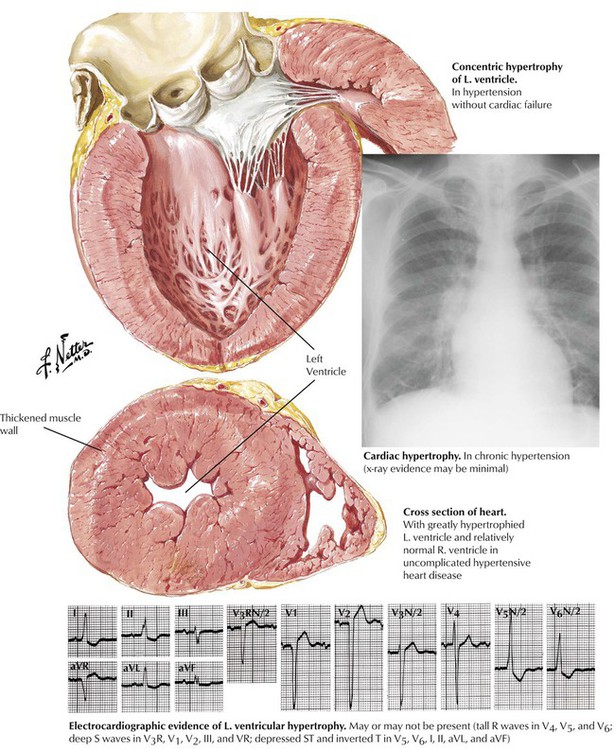
Hypertension, even of moderate degree, leads rapidly to cardiac hypertrophy, a compensatory increase of mass of the LV. The typical pattern of concentric hypertrophy of the LV, characterized by a thick wall and a relatively small chamber volume, is produced by a pressure load (afterload) on the ventricle. The heart size on cardiac silhouette is relatively normal, but the ECG shows increased voltage. When the limits of compensation are reached, the patient may have progressive cardiac decompensation accompanied by cardiac dilation. Cardiac hypertrophy is an independent risk factor for ventricular arrhythmias and sudden cardiac death.
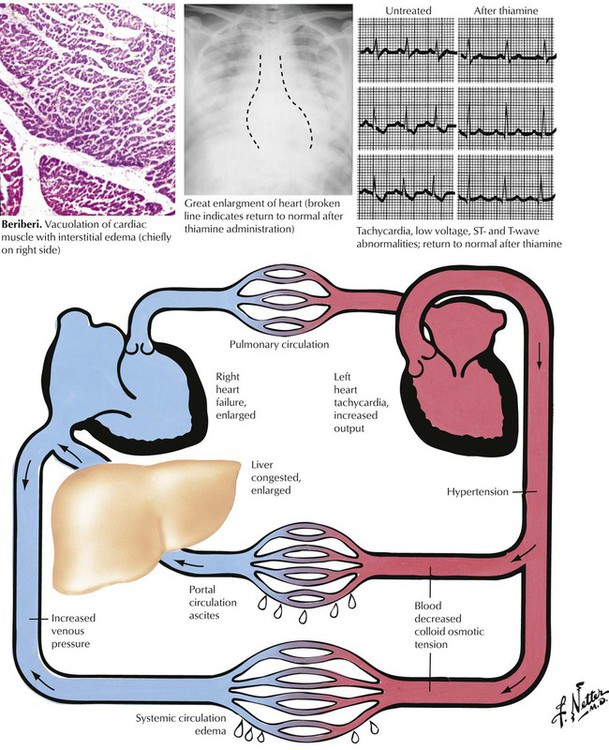
Heart failure is a state in which the heart fails as a pump to provide sufficient volume of circulating blood to meet the metabolic demands of the body. Because the dominant symptoms usually result from pulmonary or systemic venous congestion, the condition is termed congestive heart failure (CHF). Most commonly, heart failure is of the low cardiac output variety, but some conditions, including thiamine deficiency (beriberi), thyrotoxicosis, and severe anemia, produce cardiac failure with an increased circulating blood volume (high output cardiac failure), as shown here. The failure may be left-sided, right-sided, or combined left- and right-sided heart failure. This illustration shows the major manifestations of failure of the left and right ventricles. Cardiac transplantation or an artificial heart is the last therapeutic option. The most common conditions necessitating cardiac transplantation are end-stage ischemic heart disease (ischemic cardiomyopathy) and dilated (congestive) cardiomyopathy.
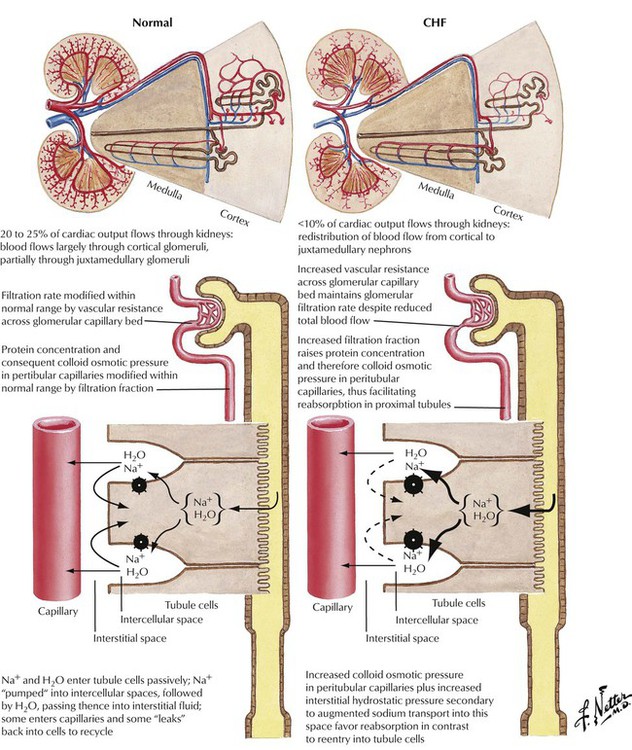
Abnormal renal function is important in the pathophysiology of CHF. In response to impaired left ventricular output, renal blood flow is decreased and redistributed from cortical to juxtamedullary nephrons. The subsequent increased glomerular vascular resistance produces increases in filtration fraction and colloid osmotic pressure in peritubular capillaries and also an increase in interstitial hydrostatic pressure secondary to augmented sodium transport, leading to increased sodium and water retention in the peritubular capillaries. The sodium and water retention contribute to the development of edema associated with CHF.
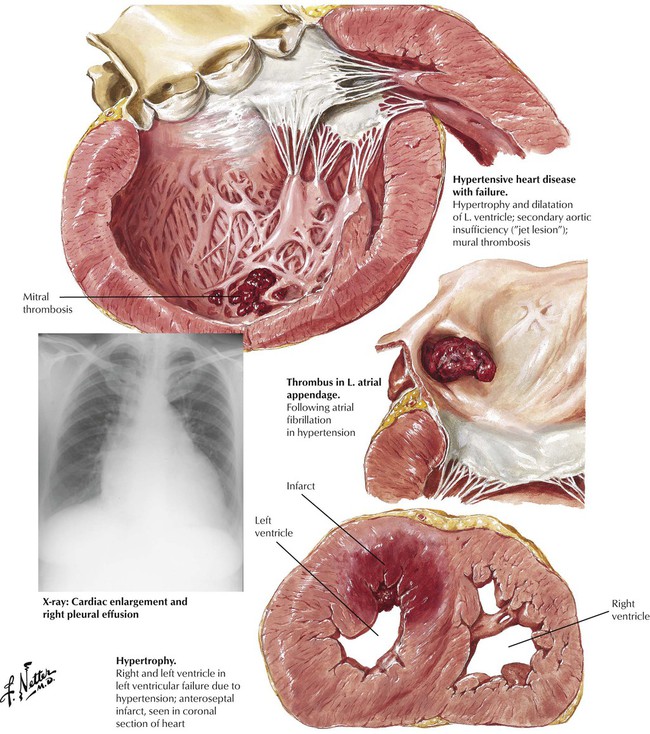
Most cases of CHF result from diseases that affect the LV initially or primarily, most commonly HTN and CAD. In response to chronic stress, the affected part of the heart undergoes compensatory hypertrophy. When the heart reaches a critical weight of 550 g, reserve is lost and progressive cardiac decompensation ensues. Heart failure results in progressive ventricular dilatation superimposed on the hypertrophy, which produces a pattern of so-called eccentric hypertrophy, as shown here. A severe acute load on the heart can produce failure and cardiac dilatation without previous hypertrophy. Stress of the atria can result in atrial fibrillation and formation of mural thrombi. The frequent coexistence of HTN and CAD can result in myocardial infarction of the hypertrophied LV.
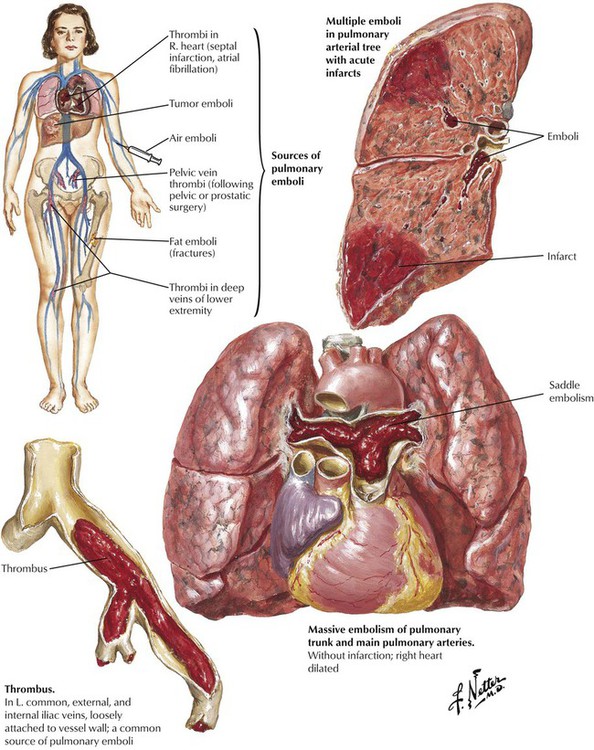
Cor pulmonale, the selective or primary impairment of the right heart (RV and right atrium) due to HTN in the pulmonary circulation, is caused by pulmonary vascular or parenchymal disease. Acute strain on the right heart is produced by a massive thromboembolus or by multiple segmental thromboemboli in the pulmonary trunk. A thromboembolus of sufficient magnitude may cause sudden death because the obstruction of the pulmonary vasculature produces pulmonary HTN and acute right-sided heart failure coupled with an impaired return of blood to the left heart with consequent decreased systemic and coronary perfusion and secondary left-sided heart failure. A thromboembolus usually does not result in pulmonary infarction. Because of the dual circulation from the pulmonary arteries and bronchial arteries, most segmental thromboemboli do not produce pulmonary infarcts. Pulmonary infarcts do occur in the presence of thromboemboli and impaired systemic circulation associated with preexistent CHF.
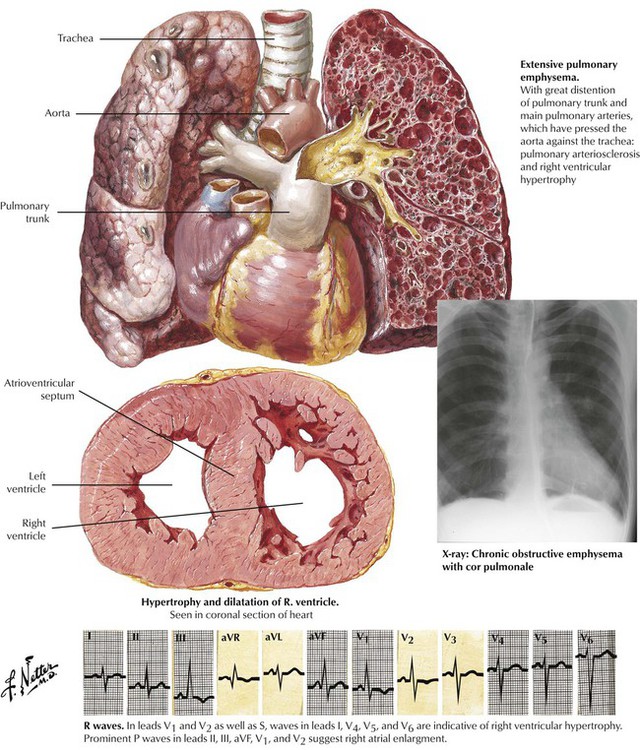
Chronic cor pulmonale typically develops in response to recurring pulmonary thromboembolic disease or chronic pulmonary parenchymal diseases, particularly chronic bronchitis and emphysema. The heart exhibits significant hypertrophy and dilatation of the RV with a normal-sized LV (unless the patient has other diseases, such as systemic HTN or CAD).
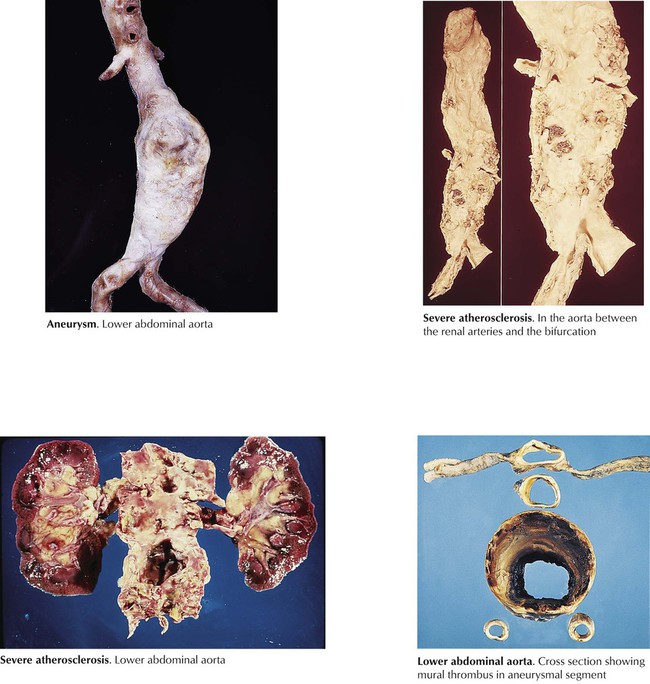
Atherosclerosis of the aorta is typically most severe in the lower abdominal aorta between the origin of the renal arteries and the aortic bifurcation. The frequent occurrence of abdominal atherosclerotic aortic aneurysms is due to the medial weakening that accompanies the severe atherosclerosis. Less frequently, the entire abdominal aorta and the descending thoracic aorta form a thoracoabdominal atherosclerotic aortic aneurysm. Aortic root and ascending aorta atherosclerotic aneurysms are secondary to end arteriolitis of the vasa vasorum produced years previously by systemic infection by Treponema pallidum (syphilitic or luetic aortitis), unless proven otherwise. Atherosclerotic aneurysms of the iliofemoral arteries also occur. The cavity of the atherosclerotic aneurysm frequently fills with unorganized mural thrombus, and the expanding aneurysms become increasingly susceptible to external rupture and life-threatening exsanguinations.
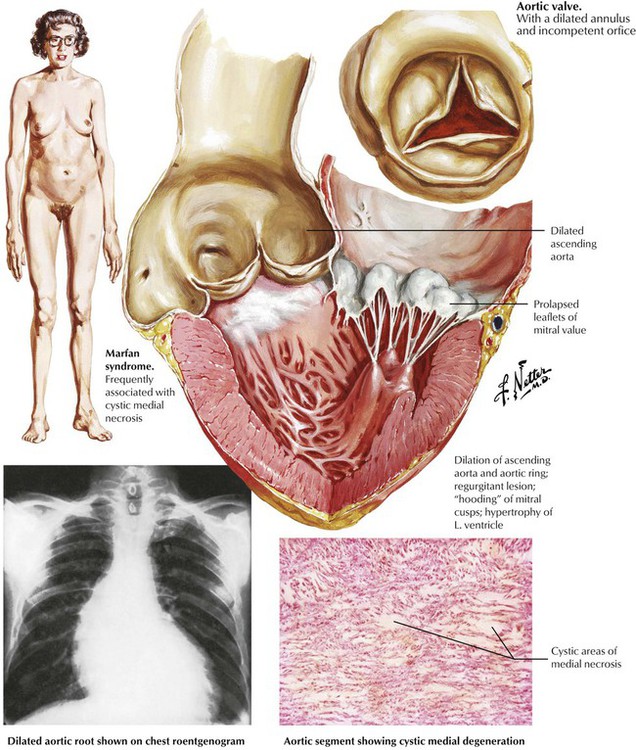
Primary degenerative disease of the aortic media manifests as cystic medial degeneration, also called cystic medial necrosis. The lesions, which consist of foci of an acid mucopolysaccharide (glycosaminoglycan)–rich ground substance, are devoid of smooth muscle cells and elastic fibers. Severe cystic medial degeneration develops as a component of genetic diseases of connective tissue, specifically, the Marfan syndrome and certain subtypes of the Ehlers-Danlos syndrome. Severe disease gives rise to annuloaortic ectasia, a progressive aneurysmal dilatation of the aortic root, with accompanying aortic valvular incompetence. Myxomatous degeneration of the mitral valve typically develops, which leads to mitral valvular prolapse and mitral incompetence. The aortic and mitral regurgitation place a volume load (preload) on the LV, which causes dilatation and hypertrophy (eccentric hypertrophy). The weakened and dilated aorta is prone to medial dissection or to focal perforation with external rupture and fatal exsanguination.
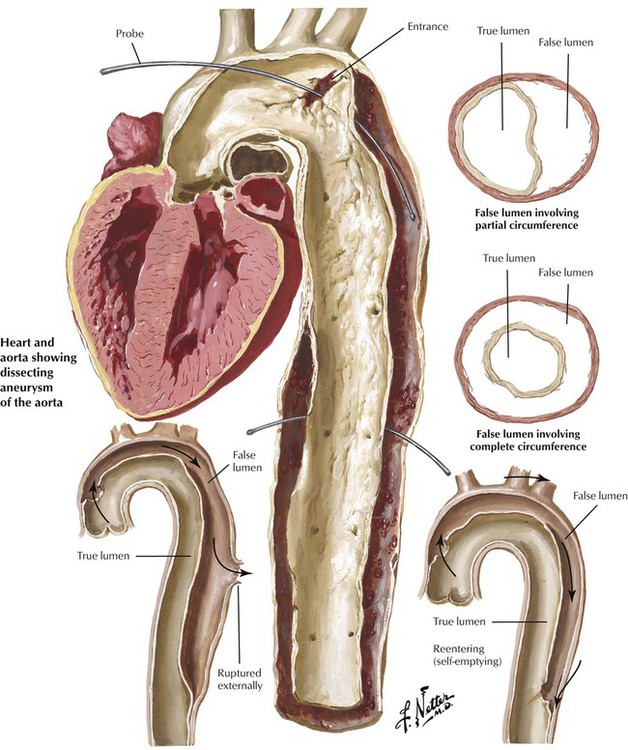
The effects of HTN with excessive hemodynamic trauma on a weakened aortic wall can lead to the formation of a hematoma in the media. The hematoma dissects longitudinally to split the media, which creates a dissecting hematoma or a dissecting aneurysm, a double-barreled aorta with true and false lumens. In most cases, a proximal intimal tear allows blood to enter the false lumen under systemic pressure. In type A dissections, the proximal intimal tear is in the ascending thoracic aorta, whereas in type B dissections, the proximal intimal tear is in the aortic arch or the descending thoracic aorta. Type A dissections, which are prone to external rupture into the mediastinum or pericardial cavity, necessitate surgical intervention. Some dissections develop distal tears and become chronic with the potential for late rupture. Blood pressure control is key in the treatment of any aortic dissection.
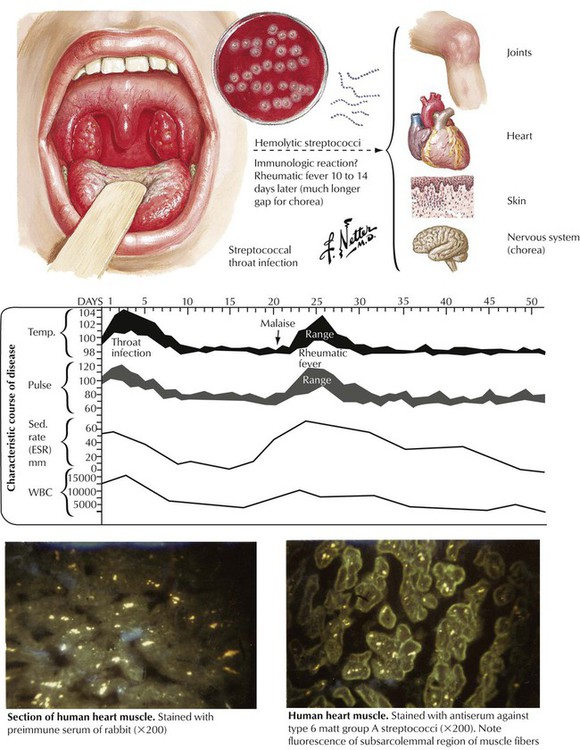
Acute RF is a multisystem immunologic illness often resulting in chronic rheumatic heart disease (RHD). RF generally affects children between the ages of 5 and 15 years. Ten to 14 days after infection with group A β-hemolytic streptococci, patients have multisystem manifestations, including skin rash (erythema annulare), subcutaneous nodules, migratory polyarthritis involving the larger joints of the extremities, and acute cardiac failure with mitral regurgitation. In some cases, central nervous system involvement manifests as spontaneous uncoordinated movements of the extremities (Sydenham chorea). The autoimmune attack of the target tissues of the host, which involves both humoral (antibody-mediated) and cellular (activated T lymphocytes) mechanisms, is a result of an immunologic reaction against the streptococci.
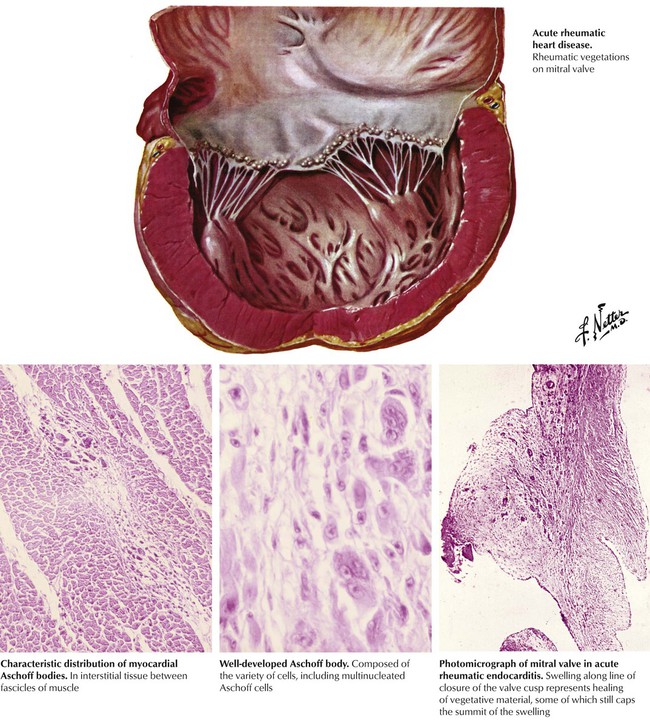
The basic tissue lesion of acute RF consists of fibrinoid necrosis of connective tissue accompanied by inflammatory cellular infiltrates composed of lymphocytes and macrophages. Acute RHD is produced by inflammation of all components of the heart (pancarditis) composed of fibrinous pericarditis, perivascular nodular foci of fibrinoid degeneration of collagen with surrounding granulomatous inflammation (Aschoff bodies), and similar inflammation of the mural endocardium and cardiac valves. The cardiac valvular lesions consist of small, nodular, wartlike fibrin thrombi (verrucae) along the line of closure of the valves, particularly the mitral and aortic valves. The cardiac inflammation leads to depressed myocardial contractile function and dilatation of the cardiac chambers, particularly the LV, and associated mitral valvular regurgitation.
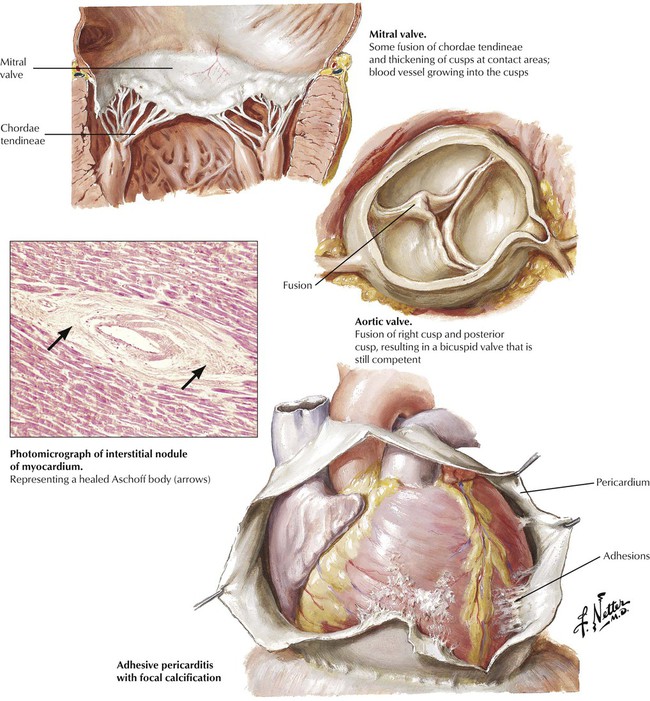
The heart manifests chronic residua at the sites of previous inflammation, including fibrous adhesions, which partially obliterate the pericardial space; perivascular scars in the myocardium; and alterations of the cardiac valves produced by the process of organization and healing. The inflammation of the cardiac valves elicits a granulation tissue response with ingrowth of small blood vessels (neovascularization) and fibroblasts, collagen production, diffuse fibrous thickening, and, later, dystrophic calcification. The organization and healing of the fibrinous verrucae lead to the partial or complete fusion of one or more of the commissures between adjacent leaflets. These changes distort the anatomy and function of the valves. A vicious cycle of increased hemodynamic turbulence and wear and tear ensues, leading to progressive distortion of valvular anatomy and function, until months to years after the acute RF, the patient becomes symptomatic with chronic RHD.
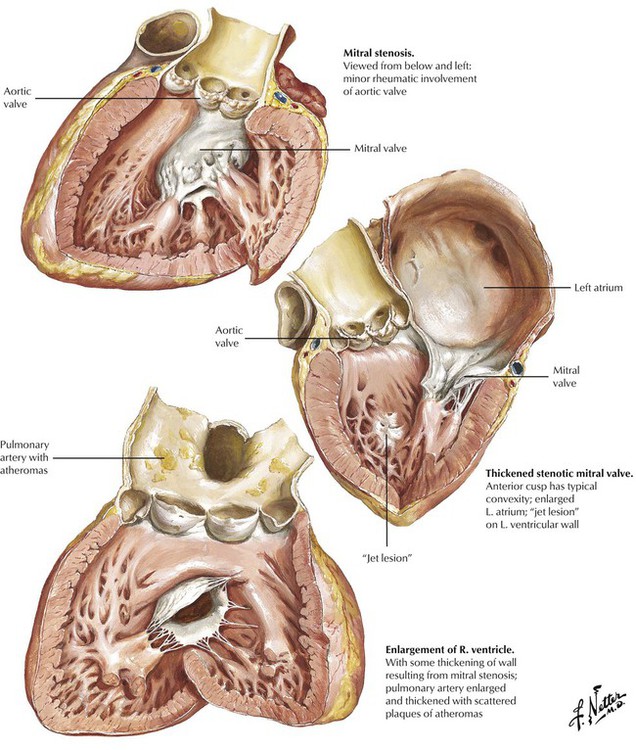
Chronic RHD accounts for nearly all cases of mitral stenosis.
Stenosis and incompetence of the mitral and aortic valve are produced by obstruction of the orifice and regurgitation of blood across the orifice, respectively. In rheumatic mitral stenosis, the shortening and thickening of the mitral leaflets, the fusion and thickening and the shortening of the chordae tendineae, and the fusion of the commissures results in a greatly reduced orifice. This “dam-in-the-stream” effect leads to increased left atrial pressure with subsequent atrial dilatation, formation of atrial mural thrombi, and atrial fibrillation. Increased pulmonary venous pressure, pulmonary congestion, increased pulmonary arterial pressure, and right heart strain leads to right ventricular hypertrophy and dilatation and functional tricuspid regurgitation. There is a characteristic opening snap and diastolic rumbling murmur at the cardiac apex.
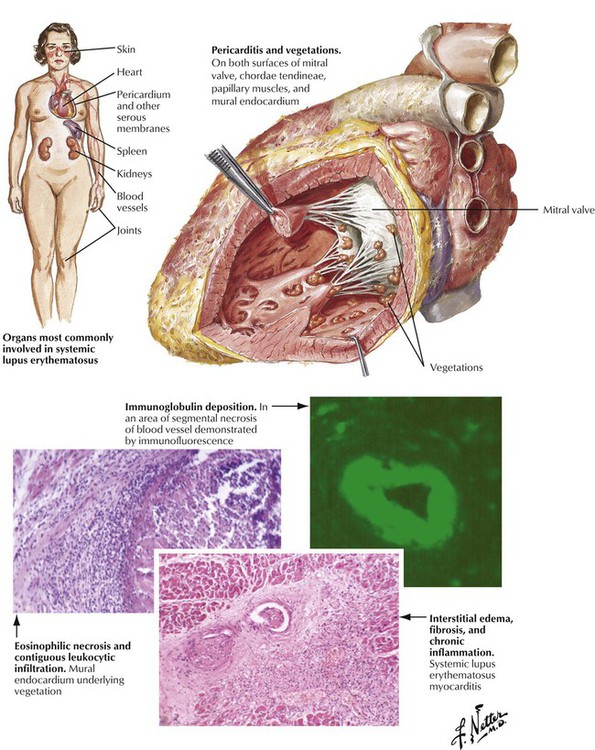
Systemic lupus erythematosus can produce pancarditis with fibrinous pericarditis and pericardial effusion, multifocal lymphohistiocytic myocarditis, and mural and valvular endocarditis. Valvular inflammation can be extensive, leading to fibrinous verrucae on the upper and lower surfaces of the valvular leaflets, particularly those of the mitral valve. This valvular pathology is known as the atypical verrucous endocarditis of Libman and Sacks. Healing of the inflammation leads to progressive valvular deformity, including fibrous adhesions of the posterior mitral leaflet to the adjacent left ventricular wall with resultant mitral stenosis or regurgitation or both.
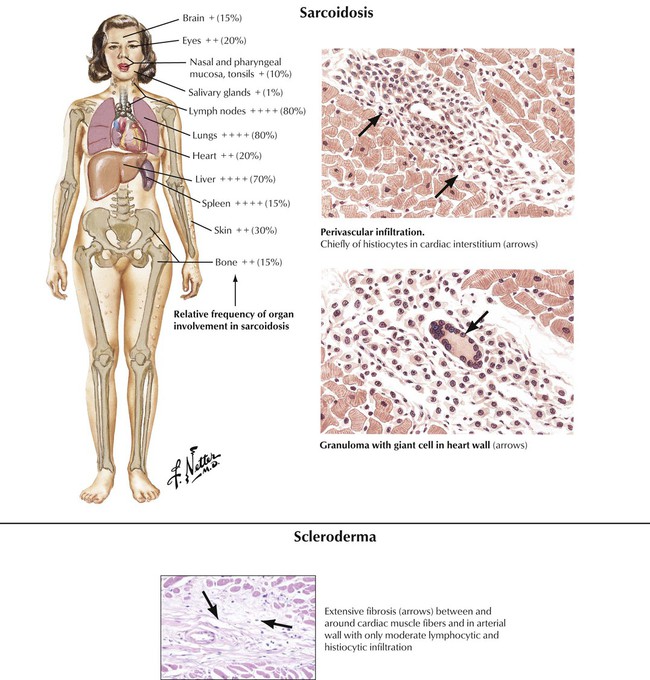
Progressive systemic sclerosis (scleroderma) can produce interstitial myocarditis and progressive myocardial fibrosis. Sarcoidosis can produce extensive replacement of the myocardium by multiple granulomas with multinucleated giant cells derived from macrophages and associated fibrosis. The process may involve the cardiac conduction system as well as the working myocardium.
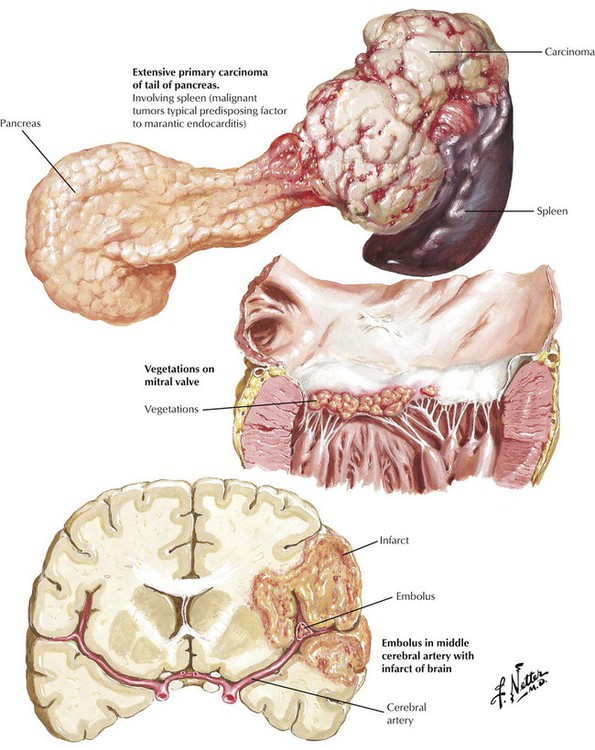
Nonbacterial thrombotic endocarditis (NBTE) consists of sterile thrombi that form as vegetations on the superior surfaces of the leaflets of the aortic, mitral, tricuspid, and pulmonic valves as a result of mild inflammation and associated surface endothelial damage. The lesions are frequently associated with disseminated intravascular coagulation. Predisposing illnesses are those that initiate a systemic reaction, including serious infections, shock of various causes, and extensive burns. The valvular lesions also develop with chronic wasting states, particularly in association with malignant tumors, leading to the pseudonym marantic endocarditis. The valvular lesions can be clinically silent or can give rise to serious symptoms due to embolization of the vegetations. After acute illness, the valvular lesions form fibrous tags along the line of closure of the valvular leaflets (Lambl excrescences).
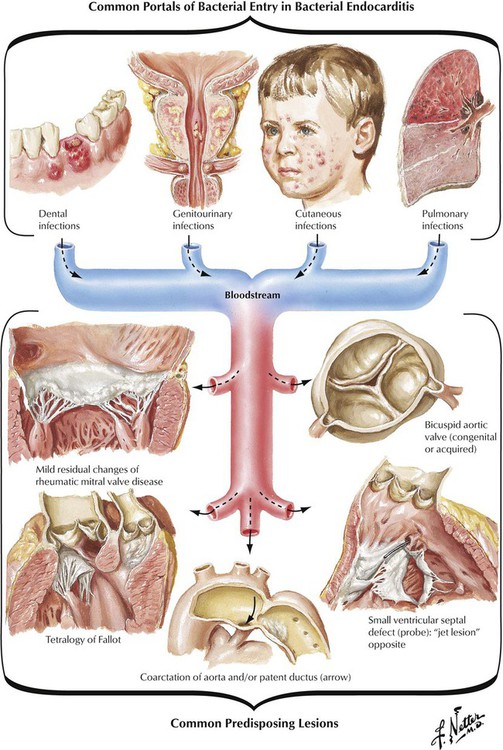
Infective (bacterial) endocarditis results from direct infection of the valvular or mural endocardium by bacteria or other microorganisms, including fungi and rickettsia. The bacteria or other microorganisms enter the bloodstream from the site of a local infection of the skin, the lungs, the genitourinary system, or the oral cavity. Sometimes there is no obvious site of infection. Some medical or dental procedures may lead to the seeding of the bloodstream with microorganisms. Whether IE follows an acute hectic course or a subtle subacute course depends on whether the virulence of the microorganism is high (Staphylococcus aureus, gram-negative bacteria, fungi, and others) or low (Streptococcus viridans and others), the presence or absence of a preexisting valvular or congenital defect, and the presence or absence of systemic conditions in the host (chronic alcoholism, intravenous drug abuse, immunosuppression).
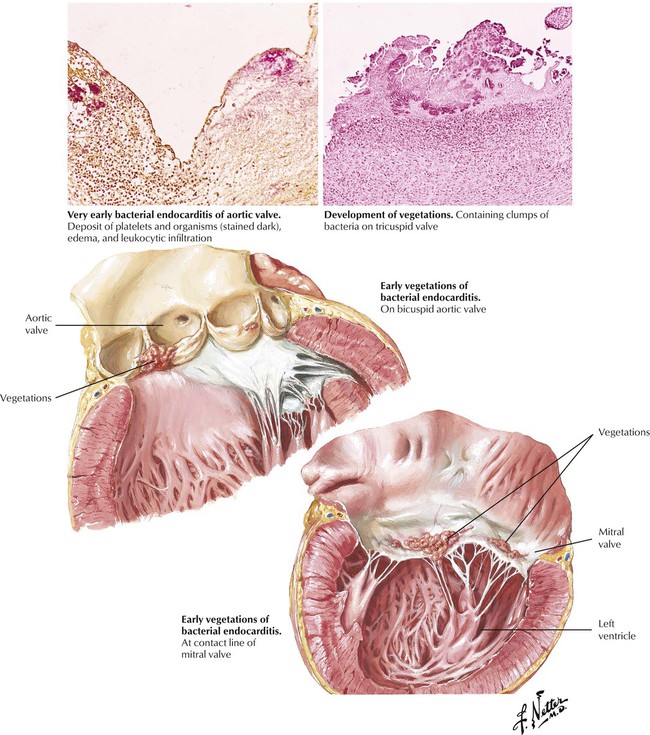
Infective endocarditis generally involves the cardiac valves, unless a congenital cardiac defect, which predisposes to mural endocarditis, is present at the site of a jet lesion. As part of a generalized inflammatory reaction to a bacteremia (or fungemia etc), small thrombi form over foci of endothelial damage on the endocardium, producing lesions similar to those of marantic endocarditis. These thrombi, which are sterile initially, become seeded with microorganisms rapidly, and the influx of neutrophils incites an accelerated inflammatory reaction. The surface thrombi grow to become vegetations. The toxic products of the bacteria and neutrophils produce necrosis of the valvular leaflets, which stimulates further suppurative inflammation.
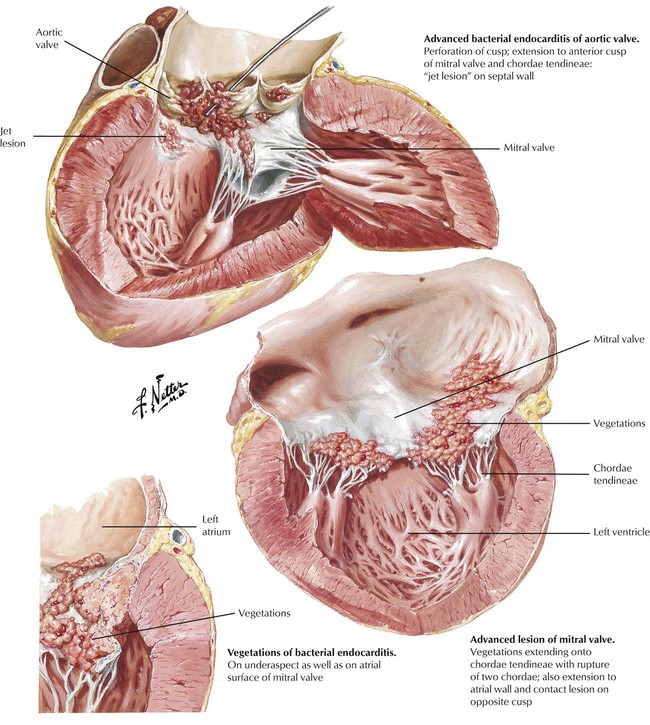
Progression of the inflammation can lead to the perforation of a valve leaflet or it can spread onto the chordae tendineae, leading to chordal rupture. The inflammation may also invade the valvular annulus, producing a valvular ring abscess. Generally, there is permanent damage to one or more of the cardiac valves, which leads to progressive valvular incompetence and cardiac failure. A major goal of clinical management is to make the diagnosis and institute high-dose intravenous antibiotic therapy to sterilize the vegetations and prevent the spread of the infection beyond the valve leaflets.
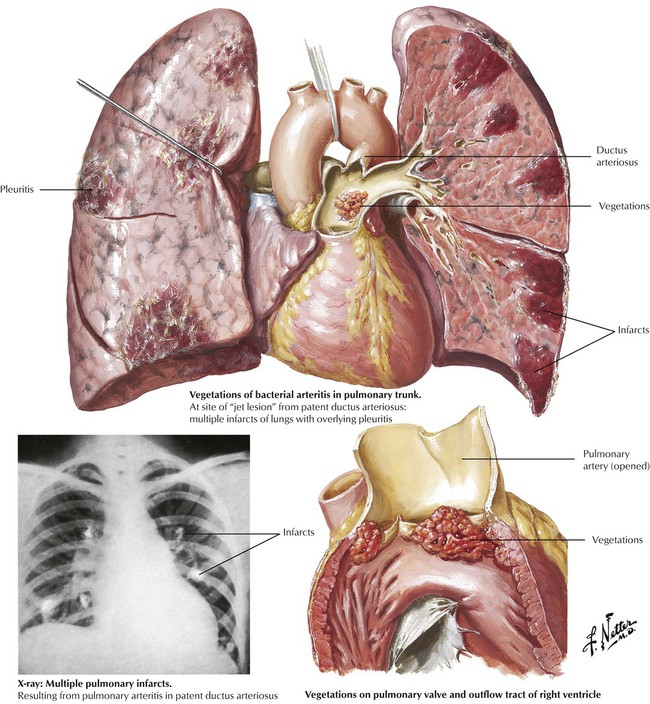
Infection of the right-sided mural endocardium can occur at the site of a ”jet lesion” produced by a ventricular septal defect with left-to-right shunting. Infection of the right-sided cardiac valves is also a complication of intravenous drug abuse with contaminated needles and foreign material. The patient may present with severe pneumonia due to seeding of the lungs with infected vegetations.
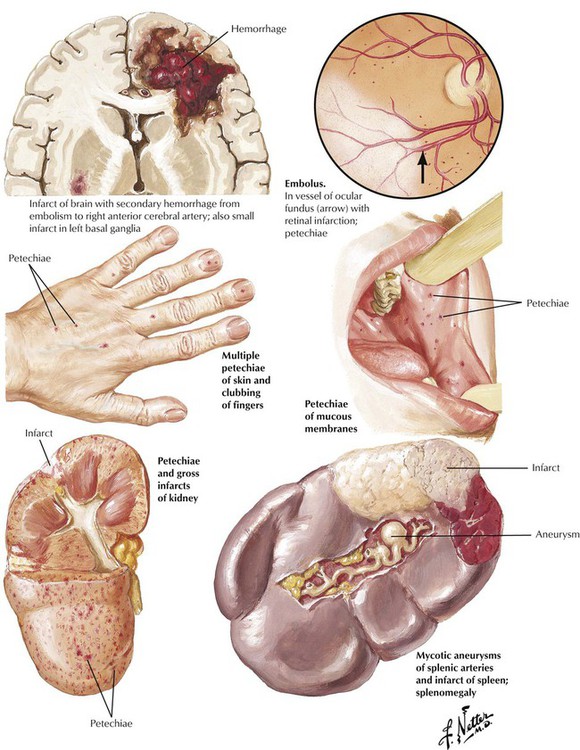
Embolization of infected vegetations is a serious complication of IE. Small thromboemboli lead to petechial hemorrhages in the skin and internal organs. Larger infected thomboemboli, which result from inflammatory damage to the vessel wall, produce mycotic (mushroomlike) aneurysms. Intraluminal obstruction produces infarcts of the tissue supplied by an end artery. With highly virulent organisms, the affected tissue develops an abscess (infected infarct). Infected emboli to one or more coronary arteries can lead to myocardial infarction or to the formation of myocardial abscesses.
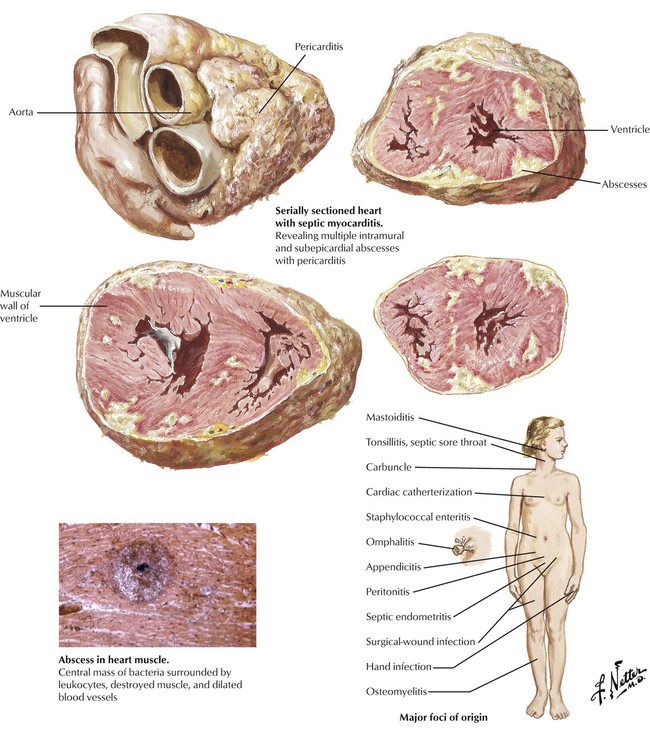
Some infections that originate in the skin or an internal organ can give rise to bacteremia or fungemia. Cardiac involvement, which includes fibrinopurulent pericarditis and multifocal suppurative myocarditis with abscess formation, can occur in the absence of valvular involvement, or the valves may show lesions of marantic endocarditis (with potential for the development of IE). Systemic fungal infections tend to occur in immunosuppressed patients, and the lesions show much diminished inflammatory cellular infiltrates. The heart may also be involved with various protozoal infections, such as Chagas disease.
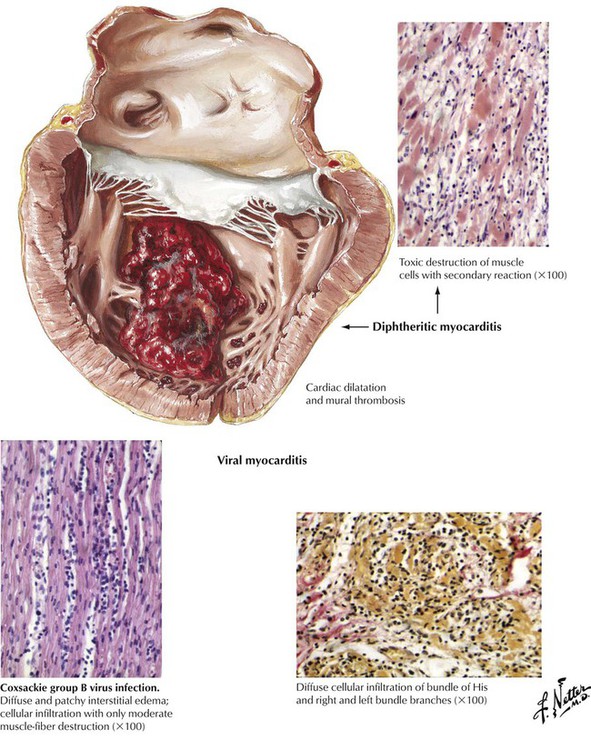
Microorganisms (viruses, rickettsiae, bacteria, fungi, and protozoa) or their toxins can produce a pattern of myocarditis or myopericarditis that is distinctive but not generally microorganism specific. Pericardial involvement consists of a fibrinous exudate often accompanied by a serous effusion. The type of inflammatory cellular infiltrate provides information about the underlying cause (neutrophils with bacterial infection, lymphocytes with viral infections, eosinophils with allergic reactions). Viral myocarditis is characterized by multifocal infiltration of the interstitium with lymphocytes and some macrophages (histiocytes) and by variable amounts of myocardial necrosis. The extent of inflammatory cellular infiltrate exceeds the amount of necrosis.
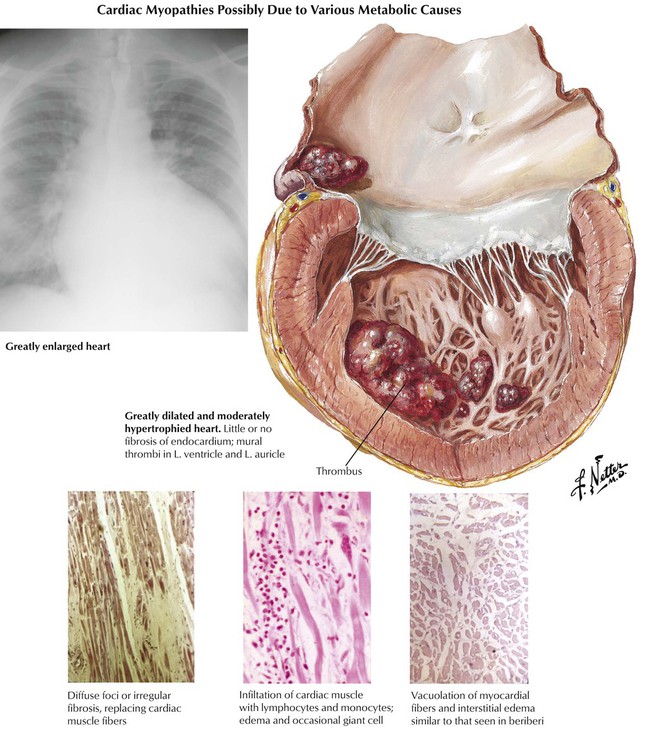
Primary (idiopathic) cardiomyopathies develop independently. Secondary cardiomyopathies occur as a component of cardiac disease (not originating in the myocardium) or systemic disease with cardiac involvement. Dilated (congestive) cardiomyopathy, the most common type, is characterized by the progressive development of cardiomegaly, CHF, and often arrhythmias. Pathologically, there is symmetrical hypertrophy and dilatation of the 4 cardiac chambers in the absence of significant coronary, valvular, or congenital cardiac lesions or prominent arteriolonephrosclerosis. Atrial or ventricular mural thrombi or both may form as a result of poor contractile function. The myocardium frequently shows nonspecific degeneration and fibrosis and, occasionally, some inflammatory infiltrates. The likely causes for this disease include previous myocarditis, chronic alcoholism, and genetic mutations often associated with familial disease.
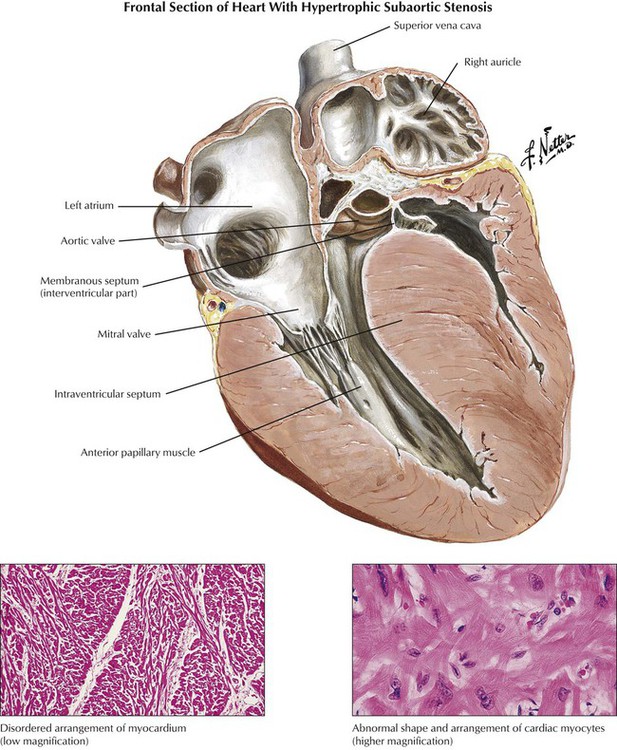
Hypertrophic cardiomyopathy includes the classic IHSS, a condition that produces obstruction of the LVOT at the subvalvular, valvular, or supravalvular level. IHSS is characterized pathologically by asymmetrical ventricular septal hypertrophy (the interventricular septum is at least 1.3 times the thickness of the left ventricular free wall, and, initially, left and right ventricular chambers are small). Myocytes in the affected myocardium have a disorganized, “herringbone” arrangement rather than the normal parallel pattern within muscle bundles. The abnormal pattern of contraction leads to the paradoxical systolic anterior motion of the anterior mitral leaflet toward the bulging interventricular septum, thereby producing functional LVOT obstruction (functional aortic stenosis) and mitral regurgitation. The IHSS phenotype is caused by a variety of genetic mutations of actin, myosin, and other contractile proteins.
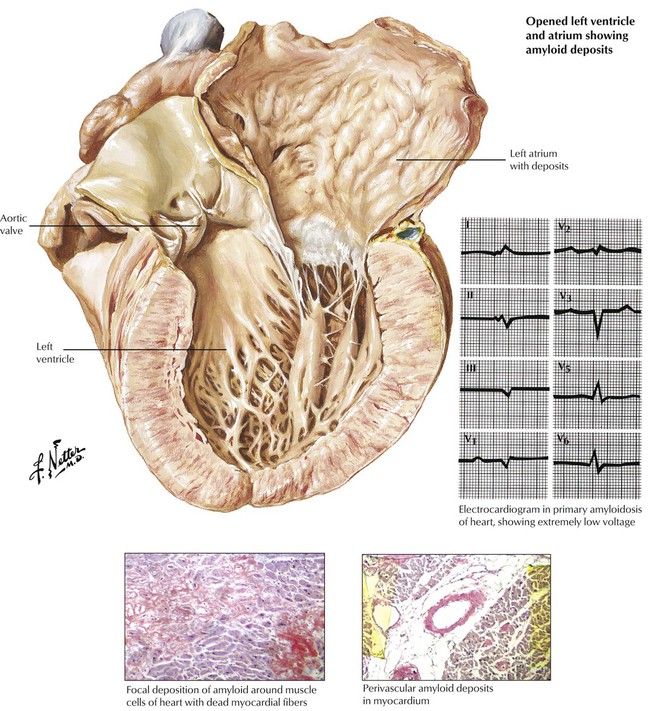
Cardiac involvement occurs most frequently in primary systemic amyloidosis and senile cardiac amyloidosis. Myocardial degeneration results from deposits of infiltrative amyloid, which surround the myocytes and cause the classic pattern of low voltage on the ECG. Cardiac amyloidosis and severe fibrosis (collagen deposition) of any cause can produce a restrictive cardiomyopathy. Endomyocardial biopsy is used to distinguish the two conditions and make the diagnosis of amyloidosis. Patients with restrictive cardiomyopathy typically present with symptoms of CHF dominated by those of right-sided failure and a normal-sized heart, which mimic constructive pericarditis on radiographic examination.
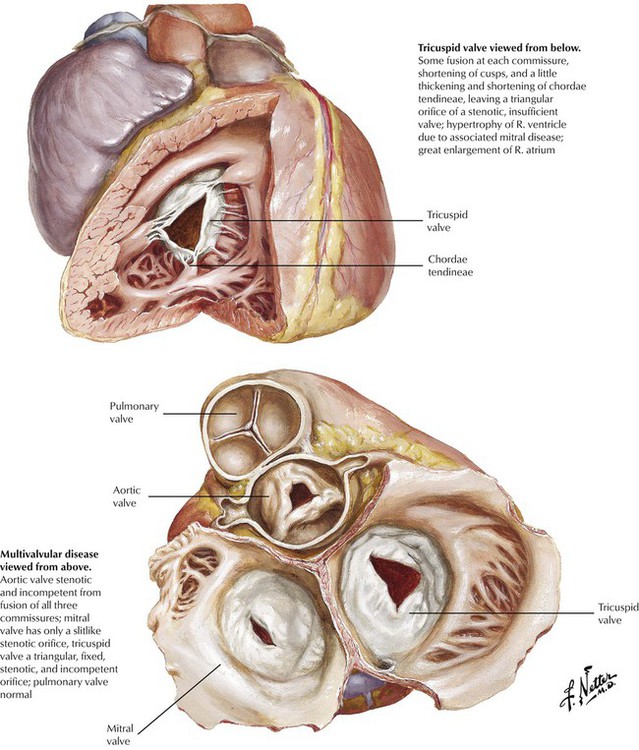
This illustration shows multivalvular disease involving the aortic, mitral, and tricuspid valves produced by chronic RHD. The diffuse fibrosis and variable commissural fusion result in dysfunction dominated by incompetence (insufficiency, regurgitation) or stenosis. Chronic RHD results in clinically significant pathology of the mitral valve alone in approximately 40% of cases, the mitral and aortic valves in another 40%, and the aortic valve alone in approximately 20%. Significant rheumatic lesions affect the tricuspid valve in a small percentage of cases, whereas the pulmonic valve is virtually never involved. RHD is only one of a number of diseases that produce significant valvular pathology, which results in incompetence, stenosis, or both of one or more of the valves.
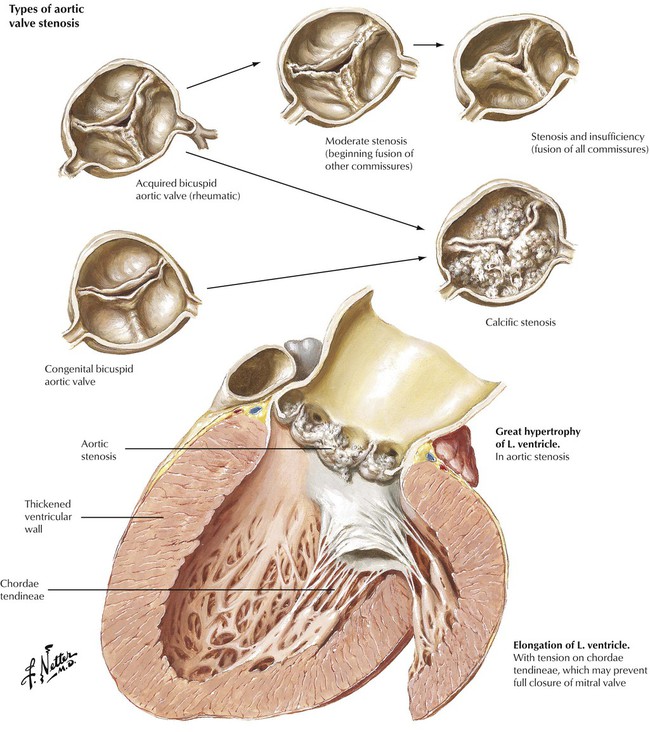
Valvular heart disease may result from congenital cardiac defects or from immunologic, inflammatory, infectious, or degenerative diseases of the heart. In the United States, nonrheumatic causes are responsible for most valvular heart disease necessitating surgical intervention. Stenosis of a congenital bicuspid aortic valve in a middle-aged individual and stenosis due to senile sclerosis (Mönckeberg) of a tricuspid aortic valve in an older individual are the conditions that lead most commonly to severe isolated aortic valve disease. RHD can produce aortic stenosis, which includes the formation of an acquired bicuspid valve due to fusion of one of the 3 commissures. Patients with valvular aortic stenosis experience left ventricular hypertrophy. They present with symptoms of fatigue and angina pectoris and exhibit a systolic ejection murmur along the right sternal border accompanied by diminished carotid pulsations.
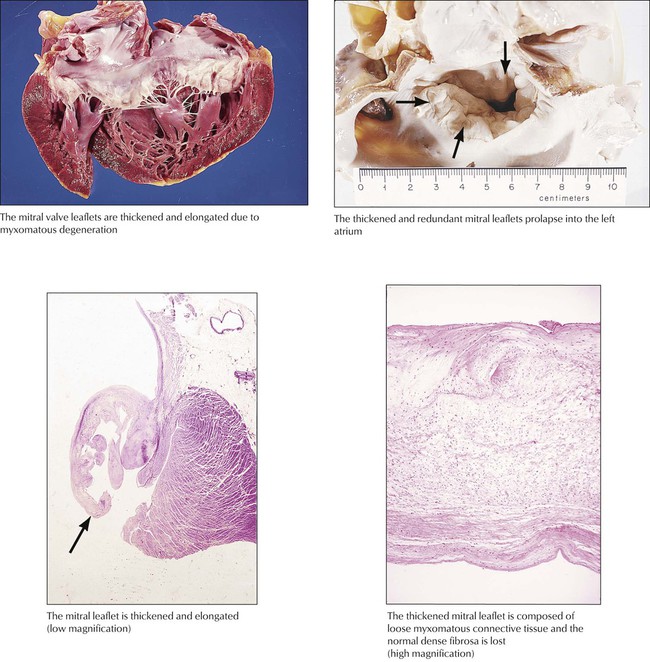
Myxomatous degeneration of the mitral valve gives rise to a redundant valve composed of white, glistening myxomatous tissue. Histologically, the normal fibrosa is replaced with myxomatous tissue. The redundant valve prolapses into the left atrium with each systole, which gives rise to mid to late systolic clicks and a short systolic murmur heard at the cardiac apex. The prolapse is readily detected by echocardiography. Some degree of mitral valve prolapse occurs in approximately 2% of the population, with higher frequency in females than in males. The cause is obscure, but there is some evidence that autonomic dysfunction gives rise to an abnormal pattern of cardiac contraction and secondary degenerative change of the mitral valve. A similar lesion is seen with Marfan syndrome. Progression of the mitral degeneration can give rise to progressive mitral regurgitation. Rupture of a chordae tendineae, either spontaneously or due to infection, can give rise to acute severe mitral regurgitation.
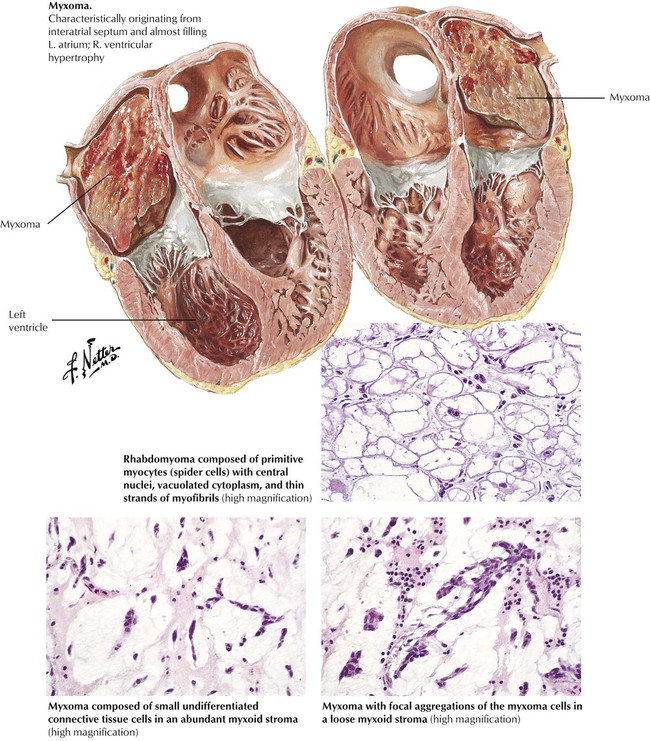
Primary tumors of the heart, most of which are benign, occur much less frequently than metastatic tumors of the heart but more often than the rare primary sarcomas of the heart. The most common primary tumor in adults is the myxoma, which consists of nondifferentiated small mesenchymal cells in an abundant myxoid stroma. It occurs most commonly in the left atrium and presents with symptoms mimicking mitral stenosis. In infants and children, the rhabdomyoma is most common. These tumors may be single or multiple and have a subendocardial, intramural, or subepicardial location. The lesions are composed of primitive myocytes (spider cells). The rhabdomyomas may occur as part of the tuberous sclerosis complex.
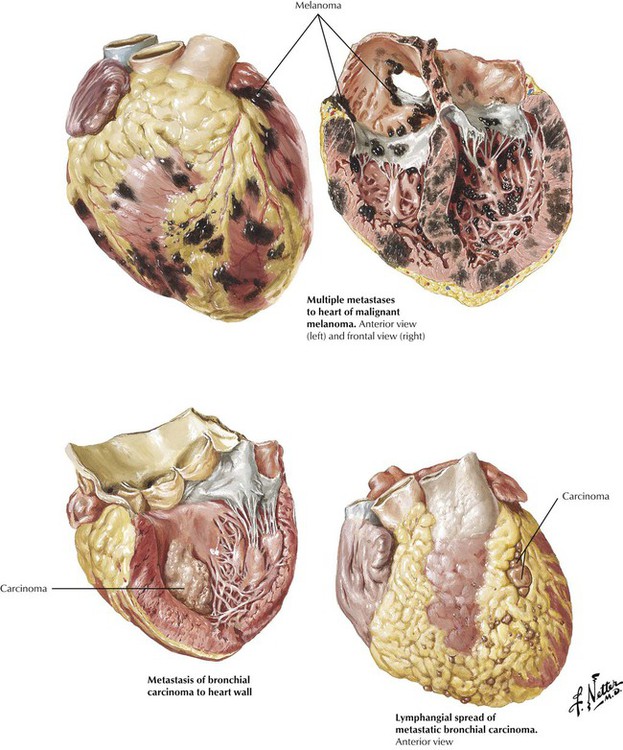
Metastatic cardiac tumors can arise from a number of different malignant neoplasms, including multiple myeloma, bronchogenic carcinoma, breast carcinoma, lymphomas, and leukemias. Various patterns of metastases, including large masses and lymphangitic spread of tumor, are illustrated.
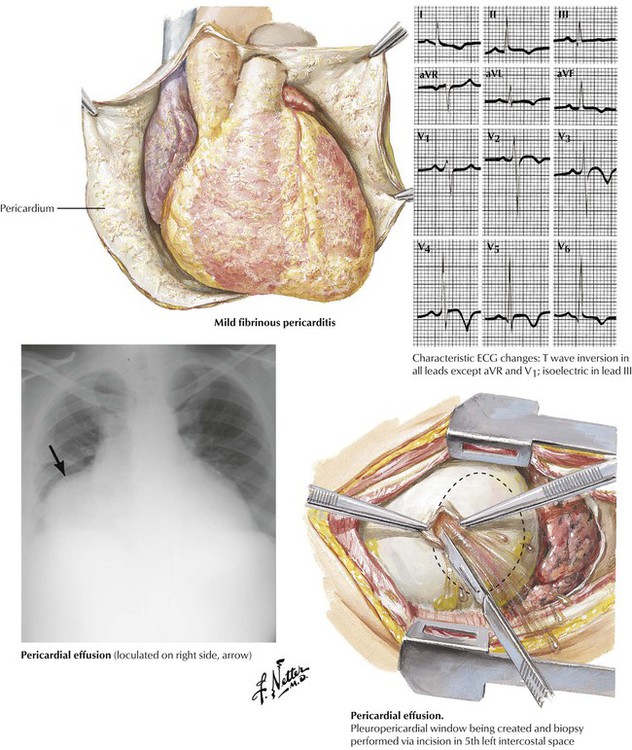
Congestive heart failure produces pericardial effusion consisting of clear fluid with low protein content (a transudate). Viral infections, renal failure (uremia), and noninfectious immunologic diseases result in fibrinous pericarditis that is often accompanied by pericardial effusion. Bacterial infections produce purulent pericarditis. Tuberculosis produces granulomatous pericarditis. Pericardial disease also can result from metastatic tumors. Fluid accumulation in the pericardium can result in impaired cardiac function depending on the rapidity and amount of fluid accumulation. Treatment may involve percutaneous pericardiocentesis or a surgically produced pericardial window.
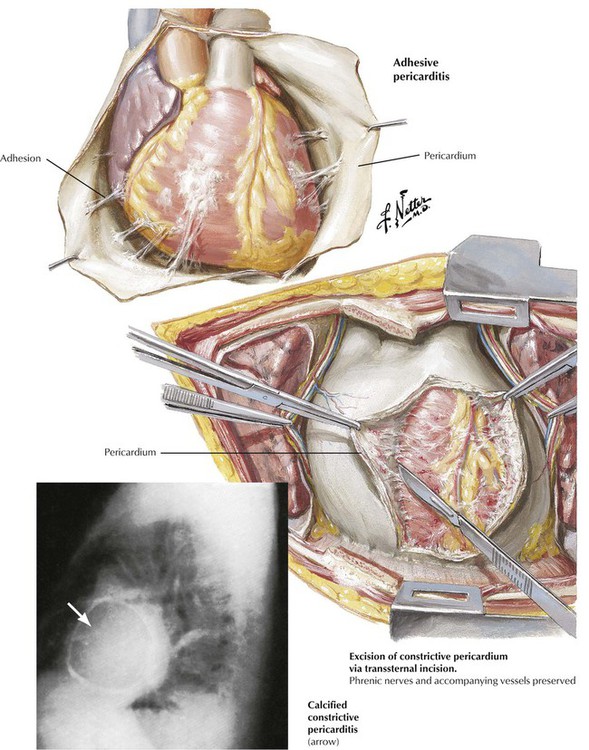
Healing of acute pericarditis, particularly with abundant fibrinous exudates or hemorrhage, gives rise to adhesive pericarditis. Adhesions may be severe and may be accompanied by calcification. Severe adhesive pericarditis can produce the clinical syndrome of constrictive pericarditis, which necessitates surgical relief. Previously, tuberculous pericarditis was the leading cause of constrictive pericarditis, but currently, most cases are idiopathic. In the age of hemodialysis, chronic renal disease is a relatively common cause of adhesive pericarditis.
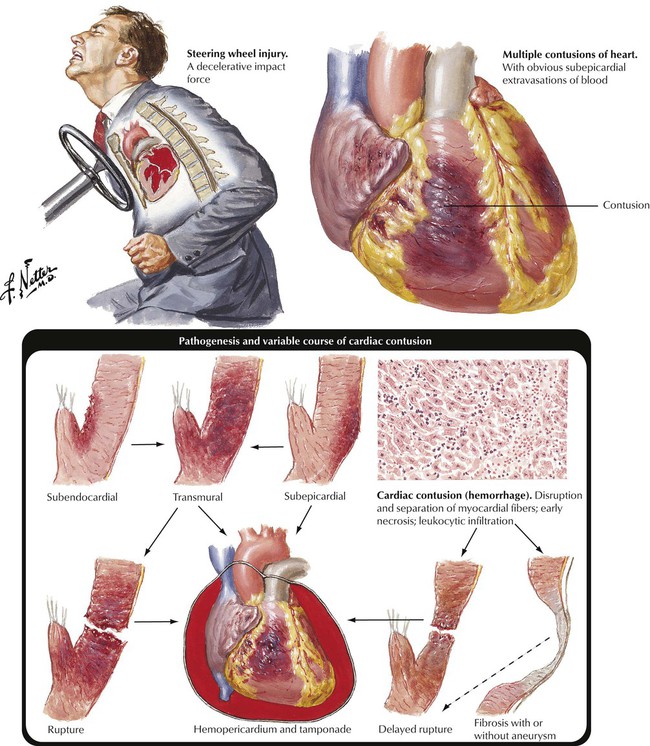
Severe crush injury of the chest can result in traumatic damage to the heart muscle known as a cardiac contusion. The lesions show hemorrhagic necrosis of the myocardium. The lesion can be transmural and result in cardiac rupture and cardiac tamponade due to hemopericardium. The lesions can heal with aneurysm formation. Other traumatic lesions include rupture of the interventricular septum, rupture of a papillary muscle, or rupture of a valve cusp. All of these lesions can produce acute cardiac decompensation. Gunshot or stab wounds of the chest can produce lacerations or perforations of various parts of the heart. This leads to severe bleeding into the pericardium and early or delayed cardiac tamponade due to hemopericardium. This life-threatening condition necessitates rapid evacuation of the hemopericardium.