[level-membership-for-internal-medicine-category]
12 Cardiovascular disease
Cardiac arrhythmias
Approach to the patient
History
Examination
Investigations
Twelve-lead electrocardiogram (ECG)
This test provides a three-dimensional snapshot of the electrical activity of the heart and is a useful screening tool. The sum of the depolarization (activation) and repolarization (recovery) potentials of the atrial and ventricular myocardium gives rise to the ECG waveform (Fig. 12.1).
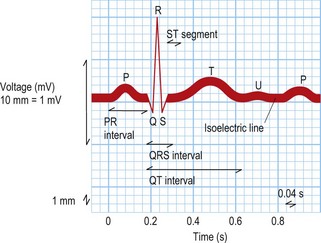
Atrial depolarization spreads from the sinus node inferiorly and to the left, producing a P wave that is usually positive in lead II and negative in aVR. Ventricular activation begins with depolarization of the interventricular septum from left to right, producing a small, positive R wave in lead V1 and a small, negative Q wave in lead V6. Depolarization of the remaining ventricular mass is usually dominated by the more massive left ventricle and therefore directed to the left and posteriorly. This results in a negative S wave in lead V1 and a positive R wave in lead V6. The normal transition from leads V1 to V6 is marked by a progressive increase in R wave amplitude and a simultaneous decrease in S wave amplitude. QRS duration is usually 100 ms or less. Ventricular repolarization usually proceeds in the reverse direction to depolarization, i.e. apex to base and epicardium to endocardium, producing a deflection of similar polarity called the T wave. See Fig. 12.1 and Box 12.1.
Box 12.1 Normal ECG intervals
| P wave duration | 120 ms or less |
| PR interval | 120–200 ms |
| QRS duration | 100 ms or less |
| Corrected QT interval | 440 ms or less in males |
| 460 ms or less in females |
Electrophysiology study (EPS)
| Action | Measurements | Note |
|---|---|---|
| Sinus rhythm | Basic intervals (PA, AH, His duration, HV, QRS and corrected QT) | Is there pre-excitation or evidence for slowed conduction? |
| Single ventricular extra-stimulus testing | Retrograde AV node effective refractory period (AVNERP) | Is there VA conduction? If so, is it via the AV node and decremental, or an accessory pathway? |
| Incremental ventricular pacing | Retrograde Wenckebach cycle length (WCL) | |
| Single atrial extra-stimulus testing | Anterograde AVNERP | Is there AV conduction? If so, is it via the AV node and decremental, or an accessory pathway? Is there evidence for dual AV node physiology? |
| Incremental atrial pacing | Anterograde WCL | |
| Arrhythmia induction pacing from the atrium | Atrial effective refractory period (AERP) Coupling interval(s) for arrhythmia induction |
Extra-stimulus testing with 2 or more extra-stimuli, burst pacing and 2 or more premature beats during sinus rhythm |
| Wellen’s protocol | Ventricular effective refractory period (VERP) Coupling interval(s) for arrhythmia induction |
Used to induce ventricular tachycardia. Ventricular extra-stimulus testing with up to 3 extra-stimuli |
Pathophysiology of arrhythmias
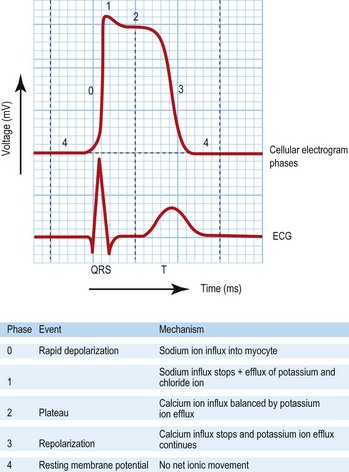
Mechanisms and diagnosis of bradyarrhythmias
Sinus node-dependent arrhythmias
These include sinus bradycardia and sinus pauses with or without an escape rhythm.
Sinus bradycardia
Treatment of sinus bradycardia involves identifying and excluding specific causes, if possible. Specific measures are summarized in Box 12.2.
Atrio-ventricular node-dependent arrhythmias
Second-degree block
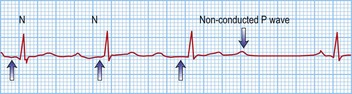
Third-degree or complete block (AV dissociation)
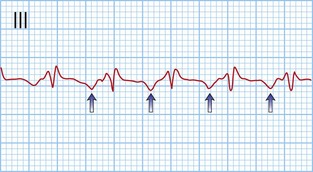
This is when no atrial electrical impulses are conducted to the ventricles. Hence, there is a continuously changing relationship between the P waves and QRS complexes (Fig. 12.4). Usually, there is a regular escape rhythm that arises below the level of block. A narrow QRS complex (< 125 ms) escape rhythm is from the His bundle and more stable than a broad QRS complex (> 125 ms), ventricular escape rhythm.

Bundle branch blocks and hemiblocks
These are due to delayed conduction within the His–Purkinje system, producing QRS complex durations > 120 ms. Causes include ischaemic heart disease, idiopathic fibrosis of the conduction system, cardiomyopathies, aortic stenosis, hypertensive heart disease, recurrent pulmonary emboli, cor pulmonale, congenital lesions, infective endocarditis and myotonic dystrophy.
Mechanisms and diagnosis of tachyarrhythmias
Atrial origin
Focal tachycardias
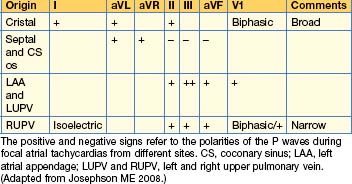
The morphology of the P waves during tachycardia is highly variable, depending on the site of the tachycardia focus. If the focus is close to the sinus node, typical P waves may be seen. P wave morphologies may give some indication as to the site of the tachycardia focus (Table 12.2).
Flutters
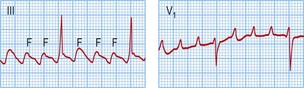
Typical atrial flutter usually, but not always, has negative sawtooth-like flutter waves in the inferior ECG leads and positive flutter waves in lead V1 (Fig. 12.6). Frequently, typical atrial flutter is conducted with a 2-to-1 AV block, giving a heart rate of 150 bpm. The re-entry circuit is well characterized, and the zone of slow conduction critical to sustaining the circuit is the cavo-tricuspid isthmus.
Atrial fibrillation (AF)
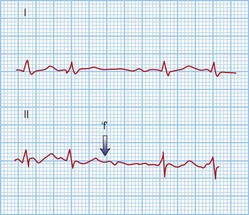
AF is the commonest arrhythmia globally, with a prevalence of 0.5–1% of the general population and 5–10% of the population over 65 years. It is characterized by rapid (300–600 bpm), irregularly irregular contractions of the atria. The 12-lead ECG shows no regular atrial activity (Fig. 12.7) and the ventricles beat in an irregularly irregular fashion at rates of up to 150–200 bpm. The mechanisms underlying AF are thought to be a combination of triggering atrial ectopics usually originating within the pulmonary veins, and progressive electrical changes within the atrial myocardium (remodelling) that allow it to be sustained. AF is a consequence of a wide range of pathologies but it can exist independently in a structurally normal heart (lone AF).
• Acute AF
• Chronic AF
Atrio-ventricular junction origin
Atrio-ventricular node re-entry tachycardia (AVNRT)
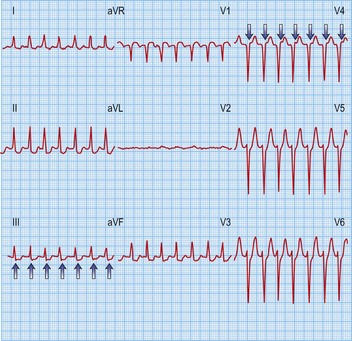
Accessory pathway-dependent tachycardias
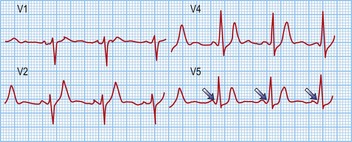
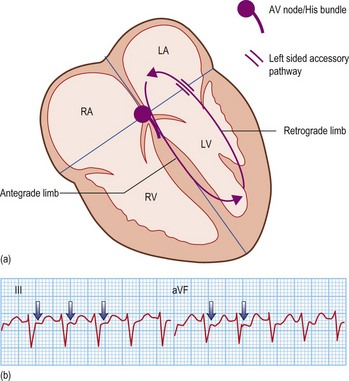
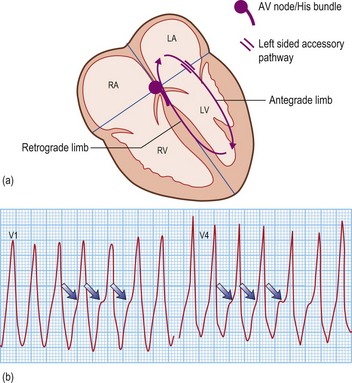
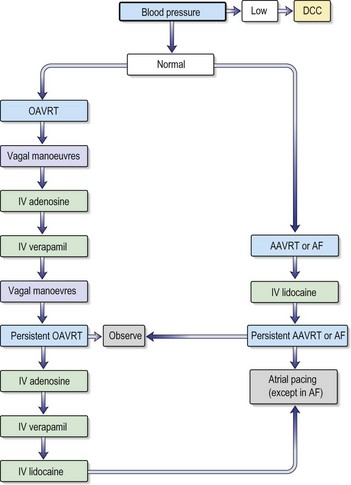
Long RP tachycardias
This is a special group of supraventricular tachycardias whose RP interval is > 50% of the RR interval (Fig. 12.14). The possible causes include focal atrial tachycardia, atypical AVNRT and orthodromic AVRT.
Ventricular origin
Ventricular tachycardia (VT)
Table 12.3 ECG features distinguishing ventricular tachycardia from supraventricular tachycardia with aberrant conduction
| ECG criterion | VT | SVT with aberrant conduction |
|---|---|---|
| AV relationship | Usually dissociated | Usually associated |
| Capture and fusion beats | Present | Absent |
| QRS duration | > 140 msec with RBBB morphology > 160 msec with LBBB morphology |
≤ 140 msec ≤ 160 msec |
| Mean frontal plane axis | −90 to ±180° | Similar to sinus rhythm |
| QRS transition across chest leads | Usually positive or negative concordance, i.e. no transition | Similar to sinus rhythm |
LBBB/RBBB, left/right bundle branch block.
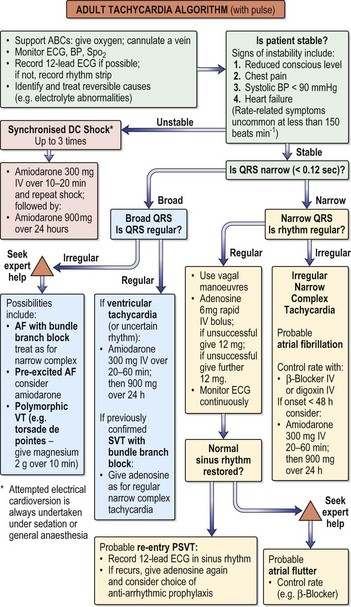
Fig. 12.15 Adult tachycardia algorithm (with pulse).
(Reproduced with permission from the Resuscitation Council (http://www.resus.org.uk/siteindex.htm.)
•Investigations
Ventricular fibrillation (VF)
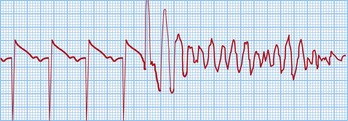
This is a cardiac arrest rhythm requiring immediate electrical defibrillation. There are advanced life support treatment algorithms that should be applied (p. 705). The underlying causes are similar to those of VT; the commonest is ischaemic heart disease with previous or acute myocardial infarction. The ECG shows disorganized electrical activity (Fig. 12.16) that is usually sustained. Unless there is an obvious reversible cause, such as acute myocardial ischaemia, all patients require ICD implantation.
Sudden cardiac death
Ion channel disorders
Long QT syndrome (LQTS)
This is another cause of potentially lethal ventricular arrhythmias in structurally normal hearts and presents with sudden death or syncope. It is characterized by prolongation of the QT interval > 440 msec due to abnormal myocardial repolarization. The underlying causes may be ion channel mutations, certain drugs and electrolyte disturbances. The prevalence is around 1 in 4000, although it is probably under-diagnosed.
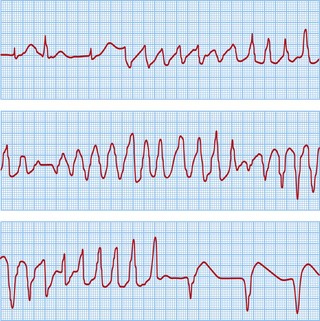
The classical VT in LQTS is torsades de pointes. This is a polymorphic VT in which the amplitude of the QRS complexes constantly changes around the isoelectric line (Fig. 12.17). This causes palpitations or syncope. Most episodes are self-terminating but SCD results from degeneration into VF.
Drug therapy
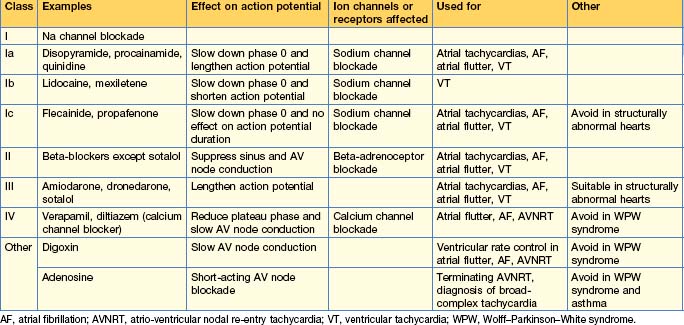
Anti-arrhythmic drugs are commonly used and work by affecting different components of the cardiac action potential. Consequently, they may precipitate arrhythmias and depress cardiac contractility. The Vaughan Williams classification describes them according to the effect on the action potential (Table 12.4). The use of individual drugs has been outlined in previous sections.
Device therapy
Pacing
| I Chamber paced | II Chamber sensed | III Response to sensing |
|---|---|---|
| O: None | O: None | O: None |
| A: Atrium | A: Atrium | T: Triggered |
| V: Ventricle | V: Ventricle | I: Inhibited |
| D: Dual (A and V) | D: Dual (A and V) | D: Dual (T and I) |
DDD and VVI are most commonly used.
Box 12.5 Indications for pacing
• Complications
Biventricular (cardiac resynchronization) pacing
Implantable cardioverter-defibrillators (ICDs)
These are specialized pacing devices that retain all of the functions of a standard pacemaker but, in addition, have the capacity to recognize and treat ventricular arrhythmias (VT, VF). The treatment modalities include ventricular burst pacing at rates exceeding the VT rate (anti-tachycardia pacing, ATP) and shocks (up to 41 J).
ICDs are implanted for both primary and secondary prevention of SCD and are the most effective treatment available at present. The indications are summarized in Box 12.6.
Box 12.6 Permanent indications for ICD implantation (based on NICE guidelines)
Primary prevention
Inappropriate therapies and device failure
In some patients, the ICD over-senses the ventricular activity, giving an erroneously fast ventricular rate that may cause inappropriate therapies. This can be due to high T wave voltages, atrial activity, skeletal muscle activity (myopotentials) and noise from electrical appliances, e.g. lead fractures or a loose set screw securing the shocking lead to the generator. Over-sensing of the T wave or atrial activity may be resolved by decreasing the ventricular lead’s sensitivity without compromising VF detection. Alternatively, the shocking lead would require repositioning or replacement if fractured.
Electrical storm
Typical precipitants are listed in Box 12.7. Not infrequently, there is no obvious cause. The ICD is studied to confirm VT/VF and exclude atrial tachycardias. The treatment of storms is to identify and correct the precipitant and to provide symptomatic relief. If patients are receiving multiple shocks, they should be sedated and given opiates. If the VT can be treated by ATP rather than shocks, the ICD should be reprogrammed appropriately. Patients with a monomorphic VT should also be considered for catheter ablation. The mainstay of drug therapy is β-blockade, which may be IV initially, together with amiodarone or mexiletene. In Brugada syndrome, these drugs may exacerbate the storm and should be avoided if possible. Instead, patients should be started on an isoprenaline infusion, and class Ia drugs such as quinidine have been shown to be effective. If electrical storms fail to abate despite all treatment, general anaesthesia and intra-aortic balloon counterpulsation therapy may be effective.
Appropriate therapies
If patients are repeatedly receiving therapy from the ICD, particularly shocks, the device programming and medications should be reviewed. Recently, there have been reports demonstrating that ATP is very effective for termination of fast VT, where previously a shock would have been delivered. All patients should be assessed for acute heart failure and treated appropriately. As the heart failure progresses, many patients develop slower VTs that may fall outside the VT therapy zone. These are difficult problems to treat, since patients may already be on optimal medical therapy and ICD reprogramming may lead to inappropriate therapy for sinus tachycardia. Catheter ablation of VT may be considered but remains a high-risk procedure in these patients.
Heart failure
Causes
Systolic and diastolic ventricular dysfunction usually coexist to cause decreased cardiac output.
Pathophysiology
Clinical syndromes of heart failure

Signs of heart failure
Objective evidence, e.g. an echo, is required for diagnosis and management of the cause.
Diagnosis
Management
Lifestyle
Pharmacological (Tables 12.6 and 12.7)
Table 12.7 Drugs available for the treatment of heart failure
| Starting dose (daily) | Maximum dose (daily) | |
|---|---|---|
| Loop diuretics | ||
| Furosemide | 20–40 mg | 250–500 mg |
| Bumetanide | 0.5–1 mg | 5–10 mg |
| Torasemide | 5.0 mg | 40 mg |
| Thiazide diuretics | ||
| Bendroflumethiazide | 2.5 mg | 10 mg |
| Chlortalidone | 25 mg | 200 mg |
| Metolazone | 2.5 mg | 10 mg |
| Angiotensin-converting enzyme (ACE) inhibitors | ||
| Captopril | 6.25 mg 3 times daily | 50 mg 3 times daily |
| Enalapril | 2.5 mg | 10–20 mg twice daily |
| Lisinopril | 2.5–5.0 mg | 30–35 mg |
| Perindopril | 2 mg | 4 mg |
| Ramipril | 1.25 mg | 10 mg |
| Angiotensin II receptor antagonists (ARBs) | ||
| Candesartan | 4 mg | 32 mg |
| Valsartan | 20 mg twice daily | 160 mg twice daily |
| Beta-adrenoreceptor blocking drugs | ||
| Carvedilol | 3.125 mg twice daily | 25-50 mg twice daily |
| Bisoprolol | 1.25 mg | 10 mg |
| Nebivolol | 1.25 mg | 10 mg |
| Aldosterone antagonists | ||
| Spironolactone | 12.5–25 mg | 50–200 mg |
| Eplerenone | 25 mg | 50 mg |
| Cardiac glycoside | ||
| Digoxin | Loading dose: 375–1500 mcg in divided doses over 24 hours | Maintenance dose: 62.5–500 mcg once daily with serum concentration ≥ 6 hrs post-dose of ≤ 2 ng/mL |
Devices
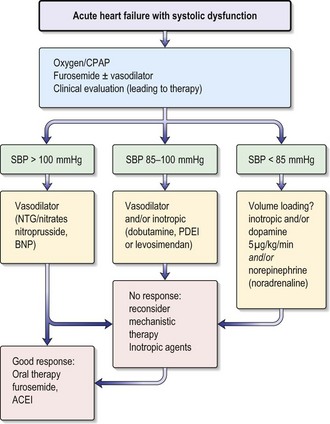
Acute heart failure
Acute heart failure (AHF) occurs with the rapid onset of symptoms and signs of heart failure secondary to abnormal cardiac function, causing elevated cardiac filling pressures. This leads to severe dyspnoea as fluid accumulates in the interstitial and alveolar spaces of the lung (pulmonary oedema). AHF has a poor prognosis, with a 60-day mortality rate of nearly 10% and a rate of death or rehospitalization of 35% within 60 days. In patients with acute pulmonary oedema the in-hospital mortality rate is 12% and by 12 months this rises to 30%. Poor prognostic indicators include a high (≥ 16 mmHg) pulmonary capillary wedge pressure (PCWP), low serum sodium concentration, increased LV end-diastolic dimension on echo and low oxygen consumption.
The aetiology of AHF is similar to chronic heart failure.
Several clinical syndromes of AHF can be defined (Table 12.8). In a clinical environment both the Killip score (based on a cardio-respiratory clinical assessment) and the Forrester classification (based on right heart catheterization findings) are used to provide therapeutic and prognostic information.
| Range of presentations | Signs and symptoms | Criteria |
|---|---|---|
| Mild acute decompensated heart failure | Mild signs and symptoms of AHF | Does not fulfil criteria for cardiogenic shock, pulmonary oedema or hypertensive crisis |
| Hypertensive acute heart failure | Signs and symptoms of heart failure are accompanied by high BP | High BP, CXR evidence of pulmonary oedema, relatively preserved LV function |
| Pulmonary oedema | Severe respiratory distress, with basal lung crackles and orthopnoea | O2 saturation < 90% on room air prior to treatment and CXR evidence of pulmonary oedema |
| Cardiogenic shock | Low BP, tachycardia, cool peripheries, pulmonary oedema, oliguria and/or altered mental state | Systolic BP < 90 mmHg, reduction of MAP (> 30 mmHg) ± oliguria (< 0.5 mL/kg/hour), pulse rate > 90 bpm |
| High-output failure | High heart rate with warm peripheries, pulmonary congestion, and sometimes low BP as in septic shock | High cardiac output, warm peripheries, tachycardia and pulmonary congestion |
| Right heart failure | Raised JVP, hepatomegaly, peripheral oedema, ascites and hypotension |
MAP, mean arterial pressure.
(Adapted from Nieminen et al 2005)
Diagnosis
Treatment
The goals of treatment in a patient with AHF include:
Box 12.8 Treatment of acute heart failure
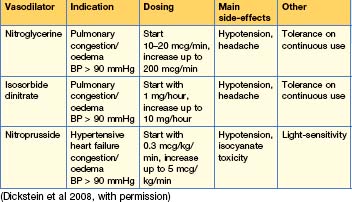
Table 12.10 Dosing of positive inotropic agents in acute heart failure
| Bolus | Infusion rate | |
|---|---|---|
| Dobutamine | No | 2–20 mcg/kg/min (β+) |
| Dopamine | No | < 3 mcg/kg/min: renal effect (δ+) 3–5 mcg/kg/min: inotropic (β+) > 5 mcg/kg/min: (β+), vasopressor (α+) |
| Milrinone | 25–75 mcg/kg over 10–20 mins | 0.375–0.75 mcg/kg/min |
| Enoximone | 0.25–0.75 mg/kg | 1.25–7.5 mcg/kg/min |
| Levosimendan* | 12 mcg/kg over 10 mins (optional)† | 0.1 mcg/kg/min, which can be decreased to 0.05 or increased to 0.2 mcg/kg/min |
| Noradrenaline (norepinephrine) | No | 0.2–1.0 mcg/kg/min |
| Adrenaline (epinephrine) | Bolus: 1 mg can be given IV during resuscitation, repeated every 3–5 mins | 0.05–0.5 mcg/kg/min |
* This agent also has vasodilator properties.
† In hypotensive patients (systolic BP < 100 mmHg) initiation of therapy without a bolus is recommended.
(Dickstein et al 2008, with permission)
Ventricular assist devices (VADs)
VADs are mechanical devices that replace or help the failing ventricles in delivering blood around the body. The left ventricular assist device (LVAD) receives blood from the left ventricle and delivers it to the aorta; the right ventricular assist device (RVAD) receives blood from the right ventricle and delivers it to the pulmonary artery. The devices can be extracorporeal (suitable for short-term support) or intracorporeal (suitable for long-term support as a bridge to transplantation or as destination therapy in patients with end-stage heart failure who are not candidates for transplantation). The main problems with VADs include thromboembolism, bleeding, infection and device malfunction.
Coronary Artery Disease
Risk factors
Definitions and spectrum of disease
Clinical features
Investigations
Twelve-lead ECG
Biochemical serum markers of myocardial damage
Imaging
Management of acute coronary syndromes
Classification of ACS
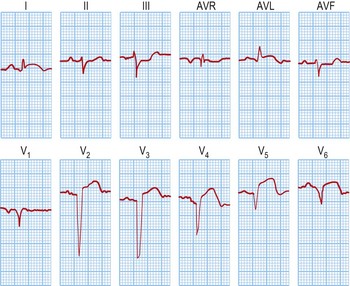
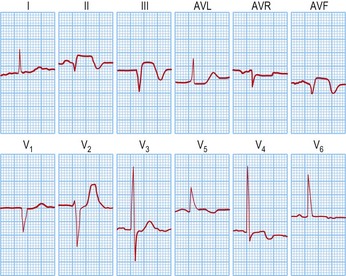
Immediate medical management in A&E (Table 12.11)
Table 12.11 Pharmacological therapy in acute coronary syndromes
| Drug | Dose | Notes |
|---|---|---|
| Antiplatelet | ||
| Aspirin | 150–300 mg chewable or soluble aspirin, then 75–100 mg oral daily | Caution if active peptic ulceration |
| Clopidogrel | 300 mg oral loading dose, then 75 mg oral daily | Caution — increased risk of bleeding, avoid if CABG planned |
| Prasugrel | 60 mg loading dose then 10 mg if >60 kg or 5 mg if <60 kg daily | Used in combination with aspirin |
| Anti-thrombin | ||
| Heparin | 5000 U IV bolus, then 0.25 U/kg/hour | Measure anticoagulant effect with APTT at 6 hours |
| Low molecular weight heparins | e.g. Enoxaparin 1 mg/kg SC twice daily | |
| Fondaparinux | 2.5 mg SC once daily | |
| Bivalirudin | 750 mcg/kg IV bolus, then 0.75 mg/kg/hour | |
| Glycoprotein IIB/IIIA inhibitors | ||
| Abciximab | 0.25 mg/kg IV bolus, then 0.125 mcg/kg/min up to 10 mcg/min IV × 12 hours | Indicated if coronary intervention likely within 24 hours |
| Eptifibatide | 180 mcg/kg IV bolus, then 2 mcg/kg/min IV × 72 hours | Indicated in high-risk patients managed without coronary intervention or during PCI |
| Tirofiban | 0.4 mcg/kg/min for 30 mins, then 0.1 mcg/kg/min × 48–108 hours | Indicated in high-risk patients managed without coronary intervention or during PCI |
| Analgesia | ||
| Diamorphine or morphine | 2.5–5.0 mg IV10 mg IV | Prescribe with antiemetic, e.g. metoclopramide 10 mg IV |
| Myocardial energy consumption reduction | ||
| Atenolol | 5 mg IV, repeated after 15 mins, then 25–50 mg oral daily | Avoid in asthma, heart failure, hypotension, bradyarrhythmias |
| Metoprolol | 5 mg IV, repeated to a maximum of 15 mg, then 25–50 mg orally twice daily. In STEMI, wait 48 hours | Avoid in asthma, heart failure, hypotension, bradyarrhythmias |
| Coronary vasodilatation | ||
| Glyceryl trinitrate | 2–10 mg/hour IV/buccal/sublingual | Maintain systolic BP > 90 mmHg |
| Plaque stabilization/ventricular remodelling | ||
| HMG-CoA reductase inhibitors (statins) | ||
| Simvastatin Pravastatin Atorvastatin |
20–40 mg oral 20–40 mg oral 80 mg oral |
Combine with dietary advice and modification |
| ACE inhibitors | ||
| Ramipril Lisinopril |
2.5–10 mg oral 5–10 mg oral |
Monitor renal function |
APTT, activated partial thromboplastin time; CABG, coronary artery bypass graft; PCI, percutaneous coronary intervention; STEMI, ST elevation myocardial infarction.
Reperfusion in STEMI ACS
Percutaneous coronary intervention (PCI)
Thrombolytic therapy
Thrombolytic agents (Table 12.12)
Table 12.12 Fibrinolytic regimes for ST elevation myocardial infarction (STEMI)
| Initial treatment (route is IV for all) | Need for heparin | |
|---|---|---|
| Streptokinase | 1.5 million U in 100 mL of 5% dextrose or 0.9% saline infusion over 30–60 mins | No |
| Alteplase (tPA) | 0.15 mg bolus 0.75 mg/kg infusion over 30 mins then 0.5 mg/kg infusion over 60 mins Total dose must be < 100 mg |
24–48 hours (or 4–8 days with LMWH), e.g. enoxaparin 1 mg/kg SC twice daily |
| Reteplase (rPA) | 10 U bolus + 10 U bolus (30 mins apart) | 24–48 hours (or LMWH as above) |
| Tenecteplase (TNK-tPA) | 30 mg single bolus if < 60 kg 35 mg single bolus if 60 to < 70 kg 40 mg single bolus if 70 to < 80 kg 55 mg single bolus if 80 to < 90 kg 50 mg single bolus if ≥ 90 kg |
24–48 hours (or LMWH as above) |
LMWH, low molecular weight heparin.
Patients can be transferred to CCU and given fibrinolysis, provided there is no delay. Otherwise, fibrinolysis should be started in A&E in a monitored bed. The transfer of a patient should never delay initiation of thrombolytic therapy.
NSTEMI ACS
Risk stratification, GP IIb/IIIa antagonists and revascularization in NSTEMI ACS
All other patients must be ‘risk-stratified’ using the results of their serum troponin level 12 hours post admission. There is a clear increase in risk of mortality with increasing troponin levels. Either the TIMI (Table 12.13) or the GRACE risk score can be used. A TIMI score of > 4 (or a GRACE risk score of > 140) is classified as high-risk.
Table 12.13a TIMI risk score in acute coronary syndrome (NSTEMI/UA)
| Risk factor | Score |
|---|---|
| Age > 65 | 1 |
| ≥ 3 risk factors for CAD | 1 |
| Known CAD (stenosis > 50%) | 1 |
| Aspirin use in last 7 days | 1 |
| Recent (≤ 24 hours) severe angina | 1 |
| Raised cardiac markers | 1 |
| ST deviation ≥ 0.5 mm | 1 |
| Elevated cardiac markers (CK-MB or troponin) | 1 |
CAD, coronary artery disease; CK-MB, creatine kinase; NSTEMI, non-ST elevation myocardial infarction; UA, unstable angina.
Table 12.13b Risk of cardiac events (%) by 14 days in TIMI IIb
| Total score | Death or MI (%) | Death, MI or urgent revascularization (%) |
|---|---|---|
| 0/1 | 3 | 4.75 |
| 2 | 3 | 8.3 |
| 3 | 5 | 13.2 |
| 4 | 7 | 19.9 |
| 5 | 12 | 26.2 |
| 6/7 | 19 | 40.9 |
•Intermediate- and high-risk patients
Other therapies used in ACS
Secondary prevention and medical treatment
Pharmacological treatment
All ACS patients should be prescribed (and be discharged on) all of the following medications:
Full fasting lipid profiles should be obtained in all patients with CAD, to include LDL-C, HDL-C and triglycerides. Diet must be modified (p. 617), as this will improve the lipid profile in addition to medical therapy, e.g. bezafibrate 200 mg 3 times daily, used in addition to a statin in patients with a low HDL-C. Patients who have any of the following will need further assessment, treatment and family screening, if necessary, in the lipid clinic:
The following may also be of use in ACS patients:
Pre-discharge risk stratification and follow-up
All STEMI patients who have received successful fibrinolysis should proceed to a coronary angiogram. Those with a low risk, or if coronary angiography is not immediately available, should have a submaximal (modified Bruce) pre-discharge exercise test for risk stratification or an early outpatient (usually 6 weeks post discharge) symptom-limited (full Bruce) exercise test. Alternative non-invasive imaging (e.g. dobutamine stress echocardiography) should be used when exercise testing is contraindicated. Patients who have had successful PCI do not need routine exercise tests prior to discharge. Exercise testing is now less often performed, being replaced by a functional imaging study.
Management of complications of ACS
Cardiogenic shock
Mechanical complications post MI
Heart failure
This is a poor prognostic feature post MI. Acute heart failure should be followed up and treated by the heart failure team (p. 419). An echocardiogram allows assessment of ventricular function and identification of other potentially treatable contributing causes (e.g. VSD). Any reversible causes, such as arrhythmia, ischaemia and drug therapy, need treatment. Any patients with an ejection fraction of < 35% is at high risk of SCD and must be discussed with a cardiologist (ideally, an electrophysiologist) prior to discharge, as they will benefit from insertion of an ICD, which can detect and treat dangerous ventricular arrhythmias.
Arrhythmias and bradycardias
• Ventricular tachycardia (VT)
Pericarditis and Dressler’s syndrome
Management of chronic stable angina
Risk stratification and investigation
These patients are usually referred to a rapid-access chest pain clinic or an outpatient clinic; they may occasionally present in A&E. Based on age, sex, history and a 12 lead ECG, a diagnosis of angina can be made, and the patient treated for angina. An exercise ECG is now not recommended (NICE guidelines) if the likelihood of coronary artery disease (CAD) is more than 90%. If there is doubt, CT calcium scoring (p. 432) can be helpful. If the score is 1-400, CT coronary angiography should be performed. If the angiogram shows CAD, treat as angina; if there is doubt perform functional tests. These tests include myocardial perfusion scintigraphy with single photon emission computed tomography (MAS with SPECT), stress echocardiography or contrast enhanced MR, depending on availability of the tests. Exercise ECGs are still being performed.
ECG exercise test
Coronary angiography
Revascularization
Secondary prevention and medical treatment
All patients should follow lifestyle advice (p. 441) and should be on aspirin 75 mg, a statin, an ACE inhibitor and a β-blocker (p. 441). All should be given a GTN spray to use as required. In addition, the following may be used as second-line anti-anginal treatments:
Valvular heart disease
Mitral regurgitation
Mitral regurgitation is common and may be a benign condition. It is secondary to intrinsic mitral valve disease or LV dilatation of any cause. Causes therefore include mitral valve prolapse, rheumatic heart disease, infective endocarditis and collagen vascular diseases, which primarily affect the valve, and ischaemic heart disease and dilated cardiomyopathy, which allow the mitral valve annulus to dilate. In addition to these chronic conditions, the mitral valve is susceptible to acute ischaemic, traumatic or infective injury with papillary muscle dysfunction, chordal rupture or valve leaflet destruction causing sudden valve failure.
Pathophysiology
Clinical features
Diagnosis
Management
Mitral stenosis
Mitral stenosis is almost always rheumatic in origin.
Diagnosis
Management
Aortic stenosis
Pathophysiology
Diagnosis
Management
Aortic regurgitation
Aortic regurgitation can present acutely or follow a chronic course. The relative paucity of symptoms early in the disease continues until there is advanced disease, in which case LV dysfunction may be irreversibly impaired. Exercise tolerance and serial estimations of valvular and ventricular function should be made by echo.
Pathophysiology
Clinical features
Diagnosis
Tricuspid regurgitation
There is almost always a minor degree of tricuspid regurgitation on echocardiography.
Special considerations for valvular heart disease
Pregnancy
Cardiomyopathy
Dilated cardiomyopathy
Clinical features
Hypertrophic cardiomyopathy
Hypertrophic cardiomyopathy (HCM) is common in the general population, with an incidence of 1 : 500, and represents a common cause of SCD in young adults, although the annual risk of SCD is low (~1%). Increased surveillance and use of genetic screening have revealed a wide heterogeneity in the cardiac morphological characteristics and prognosis of this condition, even with similar genetic mutations. Thus some patients may have the disease-causing mutation without obvious hypertrophy but their histology shows the classical features of HCM.
Pathophysiology
Clinical features
[/level-membership-for-internal-medicine-category][not-level-membership-for-internal-medicine-category]
12 Cardiovascular disease
Cardiac arrhythmias
Approach to the patient
History
Examination
Investigations
Twelve-lead electrocardiogram (ECG)
This test provides a three-dimensional snapshot of the electrical activity of the heart and is a useful screening tool. The sum of the depolarization (activation) and repolarization (recovery) potentials of the atrial and ventricular myocardium gives rise to the ECG waveform (Fig. 12.1).
Atrial depolarization spreads from the sinus node inferiorly and to the left, producing a P wave that is usually positive in lead II and negative in aVR. Ventricular activation begins with depolarization of the interventricular septum from left to right, producing a small, positive R wave in lead V1 and a small, negative Q wave in lead V6. Depolarization of the remaining ventricular mass is usually dominated by the more massive left ventricle and therefore directed to the left and posteriorly. This results in a negative S wave in lead V1 and a positive R wave in lead V6. The normal transition from leads V1 to V6 is marked by a progressive increase in R wave amplitude and a simultaneous decrease in S wave amplitude. QRS duration is usually 100 ms or less. Ventricular repolarization usually proceeds in the reverse direction to depolarization, i.e. apex to base and epicardium to endocardium, producing a deflection of similar polarity called the T wave. See Fig. 12.1 and Box 12.1.
Box 12.1 Normal ECG intervals
| P wave duration | 120 ms or less |
| PR interval | 120–200 ms |
| QRS duration | 100 ms or less |
| Corrected QT interval | 440 ms or less in males |
| 460 ms or less in females |
Electrophysiology study (EPS)
| Action | Measurements | Note |
|---|---|---|
| Sinus rhythm | Basic intervals (PA, AH, His duration, HV, QRS and corrected QT) | Is there pre-excitation or evidence for slowed conduction? |
| Single ventricular extra-stimulus testing | Retrograde AV node effective refractory period (AVNERP) | Is there VA conduction? If so, is it via the AV node and decremental, or an accessory pathway? |
| Incremental ventricular pacing | Retrograde Wenckebach cycle length (WCL) | |
| Single atrial extra-stimulus testing | Anterograde AVNERP | Is there AV conduction? If so, is it via the AV node and decremental, or an accessory pathway? Is there evidence for dual AV node physiology? |
| Incremental atrial pacing | Anterograde WCL | |
| Arrhythmia induction pacing from the atrium | Atrial effective refractory period (AERP) Coupling interval(s) for arrhythmia induction |
Extra-stimulus testing with 2 or more extra-stimuli, burst pacing and 2 or more premature beats during sinus rhythm |
| Wellen’s protocol | Ventricular effective refractory period (VERP) Coupling interval(s) for arrhythmia induction |
Used to induce ventricular tachycardia. Ventricular extra-stimulus testing with up to 3 extra-stimuli |
Pathophysiology of arrhythmias
Mechanisms and diagnosis of bradyarrhythmias
Sinus node-dependent arrhythmias
These include sinus bradycardia and sinus pauses with or without an escape rhythm.
Sinus bradycardia
Treatment of sinus bradycardia involves identifying and excluding specific causes, if possible. Specific measures are summarized in Box 12.2.
Atrio-ventricular node-dependent arrhythmias
Second-degree block
Third-degree or complete block (AV dissociation)
This is when no atrial electrical impulses are conducted to the ventricles. Hence, there is a continuously changing relationship between the P waves and QRS complexes (Fig. 12.4). Usually, there is a regular escape rhythm that arises below the level of block. A narrow QRS complex (< 125 ms) escape rhythm is from the His bundle and more stable than a broad QRS complex (> 125 ms), ventricular escape rhythm.
Bundle branch blocks and hemiblocks
These are due to delayed conduction within the His–Purkinje system, producing QRS complex durations > 120 ms. Causes include ischaemic heart disease, idiopathic fibrosis of the conduction system, cardiomyopathies, aortic stenosis, hypertensive heart disease, recurrent pulmonary emboli, cor pulmonale, congenital lesions, infective endocarditis and myotonic dystrophy.
Mechanisms and diagnosis of tachyarrhythmias
Atrial origin
Focal tachycardias
The morphology of the P waves during tachycardia is highly variable, depending on the site of the tachycardia focus. If the focus is close to the sinus node, typical P waves may be seen. P wave morphologies may give some indication as to the site of the tachycardia focus (Table 12.2).
Flutters
Typical atrial flutter usually, but not always, has negative sawtooth-like flutter waves in the inferior ECG leads and positive flutter waves in lead V1 (Fig. 12.6). Frequently, typical atrial flutter is conducted with a 2-to-1 AV block, giving a heart rate of 150 bpm. The re-entry circuit is well characterized, and the zone of slow conduction critical to sustaining the circuit is the cavo-tricuspid isthmus.
Atrial fibrillation (AF)
AF is the commonest arrhythmia globally, with a prevalence of 0.5–1% of the general population and 5–10% of the population over 65 years. It is characterized by rapid (300–600 bpm), irregularly irregular contractions of the atria. The 12-lead ECG shows no regular atrial activity (Fig. 12.7) and the ventricles beat in an irregularly irregular fashion at rates of up to 150–200 bpm. The mechanisms underlying AF are thought to be a combination of triggering atrial ectopics usually originating within the pulmonary veins, and progressive electrical changes within the atrial myocardium (remodelling) that allow it to be sustained. AF is a consequence of a wide range of pathologies but it can exist independently in a structurally normal heart (lone AF).
• Acute AF
• Chronic AF
Atrio-ventricular junction origin
Atrio-ventricular node re-entry tachycardia (AVNRT)
Accessory pathway-dependent tachycardias
Long RP tachycardias
This is a special group of supraventricular tachycardias whose RP interval is > 50% of the RR interval (Fig. 12.14). The possible causes include focal atrial tachycardia, atypical AVNRT and orthodromic AVRT.
Ventricular origin
Ventricular tachycardia (VT)
Table 12.3 ECG features distinguishing ventricular tachycardia from supraventricular tachycardia with aberrant conduction
| ECG criterion | VT | SVT with aberrant conduction |
|---|---|---|
| AV relationship | Usually dissociated | Usually associated |
| Capture and fusion beats | Present | Absent |
| QRS duration | > 140 msec with RBBB morphology > 160 msec with LBBB morphology |
≤ 140 msec ≤ 160 msec |
| Mean frontal plane axis | −90 to ±180° | Similar to sinus rhythm |
| QRS transition across chest leads | Usually positive or negative concordance, i.e. no transition | Similar to sinus rhythm |
LBBB/RBBB, left/right bundle branch block.
Fig. 12.15 Adult tachycardia algorithm (with pulse).
(Reproduced with permission from the Resuscitation Council (http://www.resus.org.uk/siteindex.htm.)
•Investigations
Ventricular fibrillation (VF)
This is a cardiac arrest rhythm requiring immediate electrical defibrillation. There are advanced life support treatment algorithms that should be applied (p. 705). The underlying causes are similar to those of VT; the commonest is ischaemic heart disease with previous or acute myocardial infarction. The ECG shows disorganized electrical activity (Fig. 12.16) that is usually sustained. Unless there is an obvious reversible cause, such as acute myocardial ischaemia, all patients require ICD implantation.
Sudden cardiac death
Ion channel disorders
Long QT syndrome (LQTS)
This is another cause of potentially lethal ventricular arrhythmias in structurally normal hearts and presents with sudden death or syncope. It is characterized by prolongation of the QT interval > 440 msec due to abnormal myocardial repolarization. The underlying causes may be ion channel mutations, certain drugs and electrolyte disturbances. The prevalence is around 1 in 4000, although it is probably under-diagnosed.
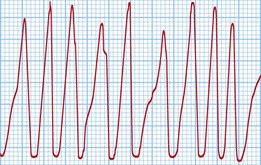
The classical VT in LQTS is torsades de pointes. This is a polymorphic VT in which the amplitude of the QRS complexes constantly changes around the isoelectric line (Fig. 12.17). This causes palpitations or syncope. Most episodes are self-terminating but SCD results from degeneration into VF.
Drug therapy
Anti-arrhythmic drugs are commonly used and work by affecting different components of the cardiac action potential. Consequently, they may precipitate arrhythmias and depress cardiac contractility. The Vaughan Williams classification describes them according to the effect on the action potential (Table 12.4). The use of individual drugs has been outlined in previous sections.
Device therapy
Pacing
| I Chamber paced | II Chamber sensed | III Response to sensing |
|---|---|---|
| O: None | O: None | O: None |
| A: Atrium | A: Atrium | T: Triggered |
| V: Ventricle | V: Ventricle | I: Inhibited |
| D: Dual (A and V) | D: Dual (A and V) | D: Dual (T and I) |
DDD and VVI are most commonly used.
