[level-membership-for-critical-care-medicine-category]
10 Cardiovascular Alterations and Management
After reading this chapter, you should be able to:
• explain the pathophysiology of coronary artery disease, clinical manifestations of acute coronary syndromes and management of events
• discuss the collaborative care for a patient with chest pain
• list the diagnostic tests used to assess myocardial ischaemia
• outline the actions and contraindications of thrombolytic drugs
• outline the clinical manifestations of right and left ventricular failure
• discuss the goals of heart failure treatment
• discuss the pathophysiology of the four different types of cardiomyopathy and how it affects cardiac function
• outline the actions of angiotensin converting enzyme inhibitors, beta-blockers, loop diuretics and spironolactone and how they relate to the pathophysiology of heart failure
Coronary Heart Disease
Coronary heart disease (CHD) is the term used to describe the effects of a reduction or complete obstruction of blood flow through the coronary arteries due to narrowing from atherosclerosis and/or thrombus. Although some patients may be asymptomatic, the commonest manifestations of CHD are chest pain due to angina, acute coronary syndrome (ACS, a term used to collectively describe acute myocardial infarction [AMI] and unstable angina) and sudden death. CHD may also cause arrhythmias and heart failure.1
CHD is the leading cause of death, premature death and disability in Australia and New Zealand.2,3 In 2007, more than 22,000 people died of CHD in Australia, more than 5000 in New Zealand in 2004 and 7.2 million worldwide.2–4 Death rates have fallen by about 76% since the 1960s, primarily due to improvements in risk factors and health care for those at risk. However, the burden of CHD remains high, with 1.5% of Australians reporting CHD symptoms.2 Furthermore, CHD is the single leading health problem worldwide because of a rising incidence in developing countries.4
Myocardial Ischaemia
When coronary blood flow is insufficient to meet myocardial tissue demand for oxygen, myocardial ischaemia occurs. Critical restriction to blood flow occurs when the diameter of the lumen of the blood vessel is reduced by more than half. Coronary blood flow is also determined by perfusion pressure, which can be adversely affected by abnormalities in blood flow (valvular disease), vessel wall (coronary spasm) and the blood (anaemia, polycythaemia).5 Myocardial oxygen demand is influenced by heart rate, strength of myocardial contraction and left ventricular wall tension. As the myocardium receives most of its blood supply during diastole, a rise in heart rate will decrease the duration of diastole and therefore coronary perfusion. Sympathetic stimulation increases the force of contraction and therefore oxygen demand. Left ventricular wall tension increases with the changes in preload associated with filling and afterload associated with systemic vascular resistance. During activity, pyrexia and arrhythmias, these effects may compound due to sympathetic stimulation, causing an increased oxygen demand and reduced coronary perfusion.
Angina
A fixed coronary artery lesion, causing limitation of oxygen supply at times of increased demand, results in stable angina. Therefore, symptoms arise during periods of physical and emotional stress and resolve within 2–10 minutes of rest. Symptoms tend to be worse in the morning (coinciding with a peak in blood pressure), after heavy meals and in cold weather. The severity of symptoms has little correlation with the progress of the disease. However, a patient with a typical history of angina has a high probability of CHD and a higher risk of AMI and sudden death in the following year.6
Unstable Angina and Acute Myocardial Infarction
Unstable angina and AMI form a continuum on the basis of reduction in coronary blood flow and subsequent damage to myocardial cells. Unstable angina may indicate transient ischaemia, whereas AMI indicates myocardial tissue death. The term ‘acute coronary syndrome’ (ACS) is now used to represent this continuum.7 ACS results from the rupture or erosion of an atherosclerotic plaque, leading to release of vasoconstrictor substances and potentially triggering coagulation activity (see Figure 10.1). Formation of thrombi results in intermittent and/or prolonged obstruction of the coronary artery. Therefore, ACS typically presents as a recent history of angina (within the past 4–6 weeks); a change in symptoms including increased frequency, more easily provoked or occurring in the absence of physical or emotional stress, more severe or prolonged and/or less responsive to nitrate therapy. ACS is a medical emergency, with up to a third of ACS patients at risk of AMI and death within 3 months.7 There is a high risk of death if the patient experiences more than 20 minutes of pain at rest (pain at rest is associated with changes in ST segment of 1 mm or more on a 12-lead ECG), if there was MI within the previous two weeks, or if pulmonary oedema or mitral regurgitation is present.7
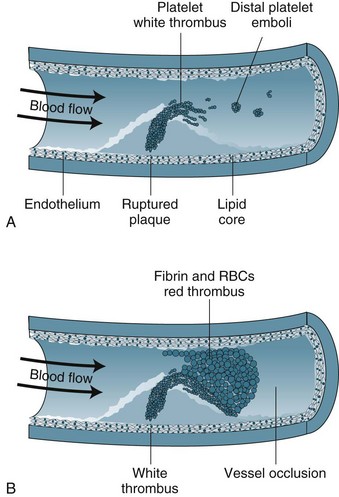
Myocardial Infarction
Myocardial infarction (MI) occurs when blood flow to the myocardium is severely impaired for more than 20 minutes as myocardial cell necrosis begins. Coronary artery thrombus arising from an atherosclerotic plaque is found in the majority of patients dying of AMI.8 Cellular death begins in the subendocardial layer and progresses through the full muscle thickness, so that by 2 hours with total occlusion a full ‘transmural’ infarction will result. However, the full extent of tissue death may occur as a single incident or evolve over several days, depending on the degree of obstruction to blood flow.
The location and impact of the infarction will depend on which coronary artery has been obstructed:
• Left anterior descending (LAD) affects the function of the left ventricle and interventricular septum, including ventricular conduction tissue. Patients with anteroseptal MI are at high risk of heart failure, cardiogenic shock and mortality due to pump deficits.
• Circumflex (CX) affects the left ventricle lateral and posterior walls and the SA node in 50% of people.5 The impact on pump efficiency of lateral and posterior wall necrosis is not as severe as anteroseptal infarcts, although patients are at more risk of arrhythmias.
• Right coronary artery (RCA) affects the inferior wall of the left ventricle and the right ventricle, as well as the AV node in most patients and the SA node in 50% of people. There is potentially severe impact on ventricular function if both the inferior wall and the right ventricle are affected, as well as a high risk of arrhythmias due to SA and AV node involvement.
Patient Assessment and Diagnostic Features
A key feature of assessment of the patient with chest pain is the use of protocols and guidelines to promote rapid assessment so that revascularisation procedures such as thrombolysis and percutaneous coronary intervention (PCI) can be implemented as soon as possible. This means that assessment may begin as early as in the ambulance, with ECG transmission to hospital ED where rapid, early triage models of care are in place.9 Additionally assessment also needs to determine whether there are any contraindications for thrombolysis.
The assessment method used depends on the condition of the patient but should occur within 10 minutes of arrival.7 This initial history will focus on the nature of symptoms such as pain. Pain assessment is complex, and the use of an acronym such as PQRST (see Table 10.1) is useful to incorporate precipitating and palliative factors, qualitative descriptors, location, radiation and length of time. A pain scale is included to help rate the intensity of pain. Asking patients for descriptive words is useful in assessment as many patients will deny pain and instead use words such as pressure, tightness or constriction. It is essential not to ignore other presentations, as patients with atypical symptoms, such as women, often have a delayed diagnosis and treatment and a higher mortality (50%) than with typical symptoms (18%).7 Differentiating this pain from any previous pain is also useful. The brief history should include a short cardiovascular risk profile: (a) previous cardiac history such as angina, MI, revascularisation; and (b) family history, smoking, hypertension, diabetes.
| P | Precipitating | Exercise and activity Stress and anxiety Cold weather |
| Palliating | Stop activity Rest Nitroglycerin |
|
| Q | Quality | Heavy, tight, choking, vice-like, constricting |
| R | Region, Radiation | Left side of chest, shoulder, arm and jaw Retrosternal and radiating to the neck |
| S | Severity | Rate pain on scale of 1 (no pain) to 10 (worst pain possible) |
| T | Time | Pain lasts longer than 10 minutes despite nitroglycerin Pain comes and goes but lasts longer than 20 minutes |
Hudak CM, Gallo BM, Morton PG. Critical care nursing, A holistic approach. (7th Ed) Philadelphia: Lippincott 1998.
Practice tip
Because of changes in neuroreceptors, older patients and diabetic patients may not describe the typical anginal pain. Women also may not describe classic angina symptoms and may use different descriptors from men.4 Be alert for prodromal symptoms, such as increased shortness of breath, weakness and fainting.
Electrocardiographic examination
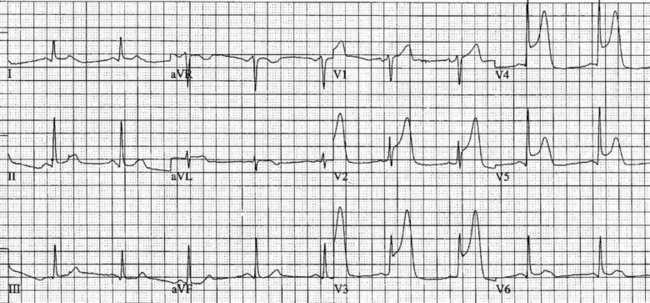
Patients with chest discomfort should be assessed by an appropriately qualified person and have an ECG recorded within 5 minutes of arrival at a healthcare facility to determine the presence and extent of myocardial ischaemia, the risk of adverse events and to provide a baseline for subsequent changes.7 Most importantly, the ECG is essential to determine whether emergency reperfusion is required, and is recommended as the sole test for selecting patients for PCI or thrombolysis. Where ST segment monitoring is available, this should be continuous. Alternatively, if chest discomfort persists, ECGs should be repeated every 15 minutes. Even when chest pain resolves it is important to record a series of 12-lead ECGs during admission to determine changes over time. (The normal ECG is covered in Chapter 9, whereas this section addresses ischaemic changes in the ECG.)
Myocardial ischaemia, injury or infarction cause cellular alterations and affect depolarisation and repolarisation.10 Myocardial ischaemia may be a transient finding on the ECG. Ischaemia results in T wave inversion or ST segment depression in the leads facing the ischaemic area.11 Ischaemic T waves are usually symmetrical, narrower and more pointed. ST segment depression of 1 mm for 0.08 seconds is indicative of ischaemia, especially when forming a sharp angle with an upright T wave.12 These changes are reversible with reduction in demand (e.g. by rest, nitrates).
On acute presentation, myocardial injury (infarction) is most commonly associated with ST segment elevation on the ECG, although this is not universal. In addition, a typical pattern of ECG changes over time (evolution of the ST segments, Q wave development and T wave inversion) are often seen (described below), but these changes too are not universal. The distinction between the various acute coronary syndromes, including ST elevation acute coronary syndrome (STEACS), ST elevation myocardial infarction (STEMI) and non-ST elevation myocardial infarction (non-STEMI), is important for ensuring appropriate assessment and protocol-based treatment13 for the various presentations.
• anteroseptal wall of left ventricle, V1–V4;
• anterior wall of the left ventricle, V1-V6, I and aVL;
Additional leads are needed to view the right ventricle and posterior wall. Chest electrodes can be placed on the right chest wall using the same landmarks as the left chest to view the right ventricle (see Chapter 9). Further electrodes, V7–V9, may be placed over the posterior of the left chest to view the posterior wall. Other indicative signs of posterior wall damage are a small r wave in V1 and/or ST depression in V3 and V4 as these may be reciprocal changes. The endocardial surface of the posterior wall faces the praecordial leads of the ECG so the signs of ischaemia and infarction are reversed or reciprocal such as ST depraession or a small r wave. If these signs are present a left-sided ECG, V7–V9, should be done to confirm or rule out a posterior infarction.
Typical ECG evolution pattern
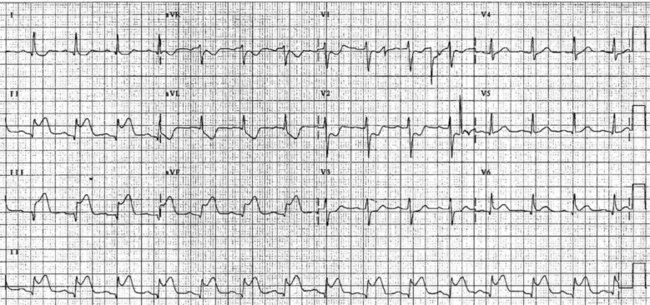
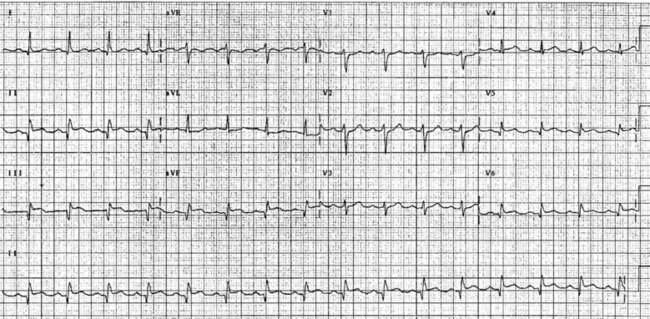
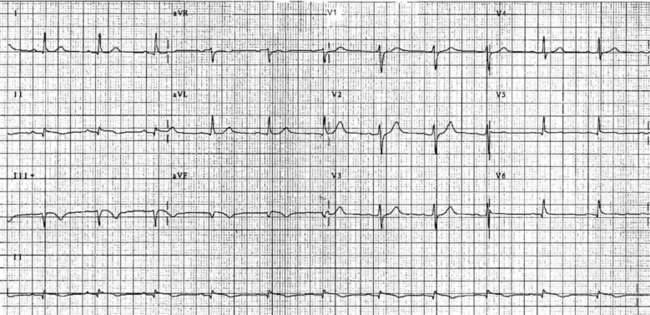
The initial ECG features of myocardial infarction are ST segment elevation with tall T-waves recorded in leads overlying the area of damaged myocardium. These changes gradually change, or evolve, over time, with ST segments returning to baseline (within hours), while Q waves develop (hours to days) and T waves become inverted (days to weeks). The time course for the evolutionary changes is accelerated by reperfusion, e.g. PCI, thrombolysis or surgery. Thus an almost fully-evolved pattern may be seen within hours if successful reperfusion has been undertaken (see Figures 10.2–10.4 for an example). Given the expected time course for evolution, it is possible to approximate how recently infarction has occurred, which is essential in determining management:
• acute (or hyperacute): there is ST elevation but Q waves or T inversion have not yet developed (see Figure 10.5).
• recent: Q waves have developed. ST segment elevation may still be present. Evolution is underway. The infarction is more than 24 hours old.
• old (fully evolved): Q waves and T inversion are present. ST segments are no longer elevated. Infarction occurred anything from a few days to years ago.
Biochemical markers
Intracellular cardiac enzymes enter the blood as ischaemic cells die, and elevated levels are used to confirm myocardial infarction and estimate the extent of cell death. The cardiac troponins T and I (cTnT and cTnI) have been found to be both sensitive and specific measures of cardiac muscle damage.14 Troponin I is rapidly released into the bloodstream, so it is especially useful for the diagnosis and subsequent risk stratification of patients presenting with chest pain in the early stages. Troponin I is also a more appropriate marker to use in postoperative and trauma patients than creatine kinase–MB (CK-MB), as CK-MB levels will be affected by muscle damage. However, CK-MB is less costly and more readily available, and so is still often used, particularly in the presence of a non-diagnostic ECG. C-reactive protein assays may prove to be useful, as baseline and discharge levels are predictive of subsequent cardiac events. However, the laboratory facilities are not readily available.
Coronary angiography and left heart catheterisation
Coronary angiography gives a detailed record of coronary artery anatomy and pathophysiology. Specially designed catheters are advanced with the assistance of a guidewire into the ascending aorta via the femoral or brachial arteries and manoeuvred into the ostium of each coronary artery. Contrast media is then injected and images are taken from several views to provide detailed information on the extent, site and severity of coronary artery lesions and the blood flow into each artery. This flow is graded using the Thrombolysis in Myocardial Infarction (TIMI) studies system (see Table 10.2).15 Typically, a left ventricular angiogram is performed during the same procedure to assess the appearance and function of the left ventricle, mitral and aortic valves. If CHD is present, treatment is determined as appropriate according to the severity (PCI, coronary artery bypass grafting or medical therapy). The nursing care for coronary angiography is similar to PCI, and is covered under that section.
TABLE 10.2 Thrombolysis in Myocardial Infarction (TIMI) flow grades in coronary arteries15
| TIMI 0 | No perfusion and no antegrade flow beyond the occlusion. |
| TIMI 1 | Penetration with minimal perfusion, and contrast does not opacify the entire bed distal to the stenosis during the picture run. |
| TIMI 2 | Partial perfusion and contrast opacifies the entire coronary bed distal to the stenosis, although entry to this area is slower than with unaffected coronary beds. |
| TIMI 3 | Complete perfusion and filling and clearance of contrast is rapid and comparable to other coronary beds. |
Exercise test
Exercise testing with ECG monitoring forms part of the diagnostic screen for patients suspected of stable angina. The Bruce protocol is used most often and considered positive for CHD if there is 1 mm or more of reversible ST segment depression.16 False-positive tests are more common in populations with a lower incidence of CHD, including women.17
Collaborative Management of Angina and Acute Coronary Syndrome
The management of stable angina patients is aimed at: (a) secondary prevention of cardiac events; (b) symptom control with medication; (c) revascularisation; and (d) rehabilitation (see Figure 10.6). (Revascularisation by coronary artery bypass graft is reviewed in Chapter 12; revascularisation by percutaneous coronary angioplasty is reviewed in the next section.)

Treatment of acute coronary syndrome aims at rapid diagnosis and prompt re-establishment of flow through the occluded artery to ensure myocardial perfusion and reduce size of infarction. In addition, treatment aims to:18
• minimise the area of myocardial ischaemia by increasing coronary perfusion and decreasing myocardial workload
• maximise oxygen delivery to tissues
• control pain and sympathetic stimulation
• counter detrimental effects of reperfusion
The ideal place to manage ACS or MI patients is in the coronary care unit, where continuous, specialised nursing care is available and there is rapid access to treatments.19 Secondary prevention of cardiac events includes the provision of medications, such as antiplatelet therapy and lipid-lowering therapy.
Reperfusion therapy
Thrombolytic therapy
Thrombolytic therapy has been demonstrated to show a significant reduction in mortality in the high-risk group described above.20 The greatest reduction in mortality occurs if the reperfusion occurs within the first ‘golden’ hour of presentation.20 Thrombolysis can be delivered effectively in many settings where other methods of reperfusion are not available.
Streptokinase and tenecteplase are the most commonly prescribed thrombolytic agents. Streptokinase is prepared from beta-haemolytic streptococci and is a potent plasminogen activator.21 Streptokinase is not thrombus-specific, so plasmin is released into the general circulation that may break down any recent clot formed as a result of surgery, injection or healing, leading to a potential increase in haemorrhagic episodes. Streptokinase is bacterial in origin, so it is antigenic. Most individuals have been exposed to beta-haemolytic streptococci so antibodies are often present, which means a higher dose may be required owing to the destruction of some of the enzyme when administered. Occasionally an escalated allergic response will occur and will need urgent treatment. This is more likely if streptokinase has been administered in the previous 6 months. Streptokinase is given intravenously over 60–90 minutes, because it has a short half-life.
The drug tissue-type plasminogen activator (tPA) is available as alteplase, tenecteplase and reteplase. These agents are of human origin, made by recombinant DNA techniques.22 The drug activates only plasminogen present in blood clots, so the risk of haemorrhage is decreased. Unlike streptokinase, tPA can be given repeatedly without risk of anaphylactic reaction. However, tPA costs about 10 times as much as streptokinase, so it is occasionally still reserved for patients who have recently received streptokinase or are at risk of allergic reaction. Often patients with anterior ischaemic changes are treated with tPA (alteplase) based on the GUSTO-1 trial that showed improved outcomes in terms of reduction of ischaemia.23 Alteplase is usually given by infusion, whereas reteplase, which has a longer half-life, can be given in two bolus injections.
• Observations. Assess neurological state including orientation, any IV sites and urinalysis for the presence of bleeding. Along with vital signs, these are attended every 15 minutes for the first hour, half-hourly for an hour, and then hourly according to the patient’s condition, however, patients are advised to report any bleeding postdischarge as well.
• ECG monitoring. This includes 12-lead ECG on return and ongoing ECG monitoring and chest pain assessment to detect reocclusion. Patients need to be requested to inform nursing staff of any chest pain or discomfort.
• IV anticoagulants such as heparin and/or oral antiplatelet drugs, such as clopidogrel or ticlopidine, may be given following thrombolysis to prevent reocclusion in the stent. Assess International Normalised Ratio (INR), prothrombin (PT) and partial thromboplastin time (PTT), as bleeding is more likely to occur if anticoagulants are above the therapeutic range.
Coronary angioplasty
Coronary angioplasty (PTCA) procedures are being used about twice as frequently as coronary artery bypass graft surgery, with 155 PTCA procedures performed for every 100,000 population in Australia in 2008–09.2 PTCA rates have grown dramatically in patients aged over 75 years. In this procedure, a catheter is introduced by the brachial or femoral artery into the coronary arteries and advanced into the area of occlusion or stenosis under the guidance of imagery and specifically designed catheters. A balloon attached to the end of the catheter is then inflated to widen the lumen of the artery by stretching the vessel wall, rupturing the atheromatous plaque and cracking the intima and media of the artery (see Figure 10.7).
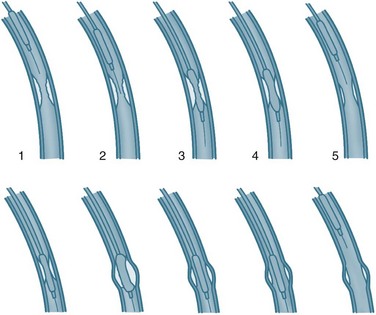
PTCA tends to be reserved for patients with single- or double-vessel disease as assessed on coronary artery angiograms. Angioplasty provides better symptom relief than medication alone, but there is no evidence of survival benefits.24 Primary angioplasty results in a higher rate of patency of the affected artery in AMI (>90%), lower rates of CVA and reinfarction and higher short-term survival than thrombolysis.25 PTCA is recommended in all patients presenting with chest pain who meet the indications for reperfusion when: (a) facilities are available and can be achieved within 60 minutes; (b) there are contraindications to fibrinolytic therapy described above; (c) ischaemia would result in large anterior AMI within 4 hours; or (d) haemodynamic instability or cardiogenic shock are present.
A stent is usually inserted to prevent abrupt closure and maintain patency for longer.26 The structure of the stent within the vessel enlarges the lumen and prevents vessel stricture. Restenosis due to intimal hyperplasia is a relatively common complication, occurring 10–12 weeks postimplantation. In response to this problem, drug-eluting stents have been developed. The drug coatings include sirolimus, a macrolide antibiotic that has been demonstrated to effectively decrease hyperplasia and prevent reduction of flow.27 Paclitaxel has also shown promise in a series of studies.28 In addition to dactinomycin, these drugs are undergoing approval processes.
Nursing management of patients post-PTCA includes care of the puncture site to prevent bleeding and detect arterial changes (including clot and aneurysm).29 The process used to create and maintain access for insertion of the catheters can damage the blood vessel(s) and alter perfusion to the limb. The sheath used to aid insertion and maintain access is usually maintained for 1–2 hours post-procedure for emergency access. Care is as follows:
• Observations. Observe access site for haemorrhage and haematoma, assess perfusion to the lower limb, including colour, warmth and pulses. This monitoring needs to be done often in the first few hours, when complications are most likely to occur.
• ECG monitoring. This includes 12-lead ECG on return and ongoing ECG monitoring and chest pain assessment to detect reocclusion. Patients need to be requested to inform nursing staff of any chest pain or discomfort.
• Vital signs. These are recorded every 15 minutes for the first hour, half-hourly for one hour, and then hourly according to the patient’s condition.
• Removal of sheath. This is usually performed by medical or specially trained nursing staff.
• Achievement of haemostasis. Use either application of pressure for at least 5 minutes or vascular sealing.29
• Assess International Normalised Ratio (INR), prothrombin (PT) and partial thromboplastin time (PTT), as bleeding is more likely to occur if anticoagulants are above the therapeutic range. Weight-adjusted heparin (100 units/kg) is usually used during PTCA to prevent thrombus formation, and glycoprotein IIb/IIIa inhibitors such as abciximab may be used to prevent platelet aggregation and thrombus formation for patients at high risk of occlusion.
• Bedrest (2–6 hours) is used to discourage the patient from moving the joint of the insertion site to prevent clot displacement and haematoma formation. Initially the patient should lie relatively flat if femoral artery access has been used, then progress to sitting. The period of rest has been demonstrated to be safely reduced to 1 hour in low-risk patients (normotensive and normal platelet count).29
• Pain relief is used primarily to promote comfort for patients who find bedrest to cause pain and discomfort.
• Urine output. Adequate urine output is essential as radiographic IV contrast is cleared by the kidneys, so it is vital that nurses ensure good hydration and monitor initial urine output.
• Oral antiplatelet drugs, such as clopidogrel or ticlopidine, may be given prior to the procedure to prevent later reocclusion in the stent. Usually patients will be discharged on this medication to continue for up to 3 months while endothelium lines the stent/injured area. Unless contraindicated, all patients will take aspirin for the rest of their lives.30,31
Many patients find the PTCA procedure and confirmation of CHD diagnosis stressful.32 It is an important nursing role to provide patients with preparatory information about the procedure and care required during recovery. As family members provide valuable support and reminders about recovery, these people should be included in any information sessions. The patient and family need to be provided with information about the possibility of restenosis, mobility restrictions at home and the lifestyle changes needed to reduce the risk of worsening CHD.
Nursing management of ACS and MI
Symptom relief should be provided, including analgesia for pain. Analgesia management should be conducted by nurses because of their continued contact and thus more accurate assessment and treatment of pain.18 It is essential to treat pain, not only for the distress it causes patients but also because pain causes stimulation of the sympathetic nervous system (SNS). SNS responses include elevated heart rate and potential for arrhythmias, peripheral vasoconstriction and increased myocardial contractility and, therefore, an overall increase in myocardial oxygen demand. Effective treatments for pain include IV morphine and nitrates. The IV route is preferable, as absorption is predictable and additional punctures in thrombolysed patients are not required. Morphine has the additional benefit of reducing anxiety in a distressing situation and should be initially provided at a dose of 2.5–5 mg at 1 mg/min, followed by 2.5 mg doses as indicated. While there is little randomised controlled trial evidence to support this particular practice, it is generally accepted to be appropriate. A standardised method of pain evaluation and charting should be used to ensure consistent assessment and treatment. An antiemetic such as metoclopramide should be given concurrently to lessen and prevent nausea. Other drugs, such as beta-blockers and nitrates, decrease myocardial workload, contributing to pain reduction.
Nursing care for thrombolysis
Patients receiving thrombolytics require constant observation, regular non-invasive blood pressure measurement for hypotension, and monitoring for allergic reactions to streptokinase. Continuous ECG monitoring for arrhythmias and ST segment changes is essential. Some arrhythmias, particularly idioventricular arrhythmias, are associated with reperfusion and tend to be benign. ST segment monitoring and assessment of pain help evaluate the effectiveness of the thrombolysis. Thrombolysis is considered to have failed if the patient is still in pain and the ST segment has not resolved within 60–90 minutes.18 If thrombolysis fails, patients are at high risk for other interventions, so repeat thrombolysis is often the only treatment option. Salvage or rescue angioplasty may be undertaken if available at the site.
Medications
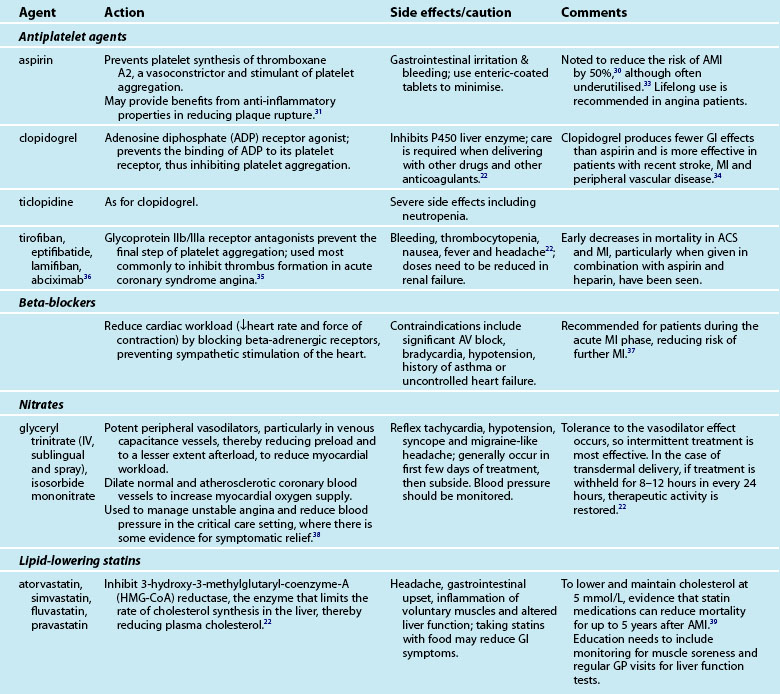
Provision of medications and assessment of the effectiveness of treatment is a major component of the nurse’s role in caring for the cardiac patient. Many of the medications are accompanied by side effects and interactions with other drugs, which the nurse must monitor. An array of medications is used to treat AMI patients, including aspirin, lipid-lowering agents, beta-blockers and organic nitrates (see Table 10.3).
Symptom control
Control of anginal symptoms with medication usually includes sublingual glyceryl trinitrate (GTN) for immediate symptom control and one or more antianginal medications for sustained symptom management.18 Beta-blockers are usually commenced unless contraindicated. Calcium channel blockers may be used in patients who do not have cardiac failure or heart block. (These medications are described in the next section.) The choice of medication may depend on how acceptable the patient finds the reduction in symptoms and the presence of side effects. Patients need to take antianginal agents continuously, regardless of symptoms. Patients should also be encouraged to take sublingual GTN prophylactically.
Angina may also be managed by avoiding situations that trigger angina. Education needs to be directed at awareness of symptoms and management of unstable angina and AMI symptoms, and the need for emergency care. Although these patients are at low risk of further cardiovascular events in the short term, in the medium to long term, risk may accumulate. Patients with angina are encouraged to attend cardiac rehabilitation programs to learn how to deal with symptom management.41
Angiotensin-converting enzyme (ACE) inhibitors have been recommended for all post-AMI patients while in hospital, with review of prescription at 4–6 weeks postdischarge. Patients with left ventricular failure should be maintained on ACE inhibitors. Similarly, diuretics provide the mainstay of the management of left ventricular failure if it is present (see Chapter 19). Diabetic patients have a higher mortality after AMI in both acute and long-term phases. Provision of an insulin-glucose infusion for BSL >11 mmol/L during the acute phase, followed by subcutaneous injections for at least 3 months, has been demonstrated to significantly reduce mortality up to 3 years post-AMI.42
Transfer to a step-down unit or general ward usually occurs when the patient is pain-free and is haemodynamically stable. Stability means that patients are not dependent on IV inotropic or vasoactive support and have no arrhythmias. Discharge home after AMI varies, but usually occurs at day 3 for low-risk patients.18
Independent Practice
Emotional responses and patient and family support
ACS or AMI is usually accompanied by feelings of acute anxiety and fear, as most patients are aware of the significant threat posed to their health.18 For many patients it may also be the first experience of acute illness and associated aspects such as ambulance transport, emergency care and hospitalisation, so they may experience shock and disbelief as well. Fast-track processes require patients and their families to process a large amount of information and make decisions quickly, and this, added to an alien environment, full of unfamiliar technology and personnel, can be quite distressing. However, the environment can also promote a feeling of security for patients and their families. Patients’ perceptions of the CCU environment have been linked to recovery.43
Anxiety is a common response to the stress of an acute cardiac event and leads to important physiological and psychological changes.44 The sympathetic nervous system is stimulated, resulting in increased heart rate, respiration and blood pressure. These responses increase the workload of the heart and therefore myocardial oxygen demand. In an acute cardiac event, these demands occur when perfusion is already poor and may lead to worse outcomes, including ventricular arrhythmias and increased myocardial ischaemia. Therefore, staff working in emergency and coronary care should employ strategies to reduce a patient’s anxiety.
Increasing a patient’s sense of control, calm and confidence in care reduces the patient’s sense of vulnerability, whether it is realistic or not.44 This can be achieved by:
• providing order and predictability in routines, allowing the patient to make choices, providing information and explanations, and including the patient in decision making
• using a calm, confident approach
• communicating with patients and families, while reducing conversation demands as excessive conversation by patients may unnecessarily raise heart rate45
• restricting the number and type of visitors in the acute phase is customary, but many patients feel safer if a family member is present
• provision of comprehensive information to families, with more concise information in understandable language for patients.
Cardiac rehabilitation
Coronary heart disease is a chronic disease process, which often presents with acute events such as ACS or AMI. Like all chronic illnesses, it has implications for patients in terms of lifestyle change, uncertainty of long-term outcomes, functional changes and social and economic alterations. Cardiac rehabilitation aims to address these issues. The World Health Organization describes cardiac rehabilitation as ‘the sum of activities required to influence favourably the underlying cause of the disease, as well as to ensure the patients the best possible physical, mental and social conditions so that they may, by their own efforts, preserve, or resume when lost, as normal a place as possible in the life of the community’.46Systematic, individualised rehabilitation and secondary prevention need to be offered to all AMI patients. Participation in well-structured, multidisciplinary programs has been demonstrated to reduce mortality by up to 30%.47 Additional benefits have been shown for improvements in exercise tolerance, symptoms, serum lipids, psychological wellbeing and cessation of smoking.48–50
Cardiac rehabilitation is structured around four phases, beginning with phase I, during admission.50 The components of phase I include:
• information regarding the disease process, the prognosis, and an optimal approach to recovery, early mobilisation and discharge planning
• assessment of patients’ understanding of their diagnosis and treatment as a foundation for self-management
• discharge planning which incorporates discussions on adaptation to the functional and lifestyle changes needed for secondary prevention – dietary intake of lipids, exercise, smoking cessation, stress management and symptom monitoring, and management of acute symptoms
• early mobilisation as an inpatient to encourage a positive approach to recovery with monitoring of the response to activity in heart rate, shortness of breath and chest pain to determine the rate of progress. (Most hospital units use an activity progress chart for this purpose based on metabolic equivalents [METs]).
The phases that follow, from II to IV, are managed in the outpatient setting and begin with assessment, liaison with multidisciplinary professionals and health education. Phase II occurs in the immediate postdischarge period and includes liaison with community-based carers and services and further assessment. In phase III, tailored, supervised exercise programs are usually conducted and there is a range of psychosocial interventions, such as support sessions and stress management. Finally, in phase IV the focus is on chronic disease management and maintaining risk modification behaviours. All phases require incorporation of the principles of adult learning to maximise learning and behaviour change. These principles include recognition of ‘readiness to learn’.50 Adults are ready to learn most effectively when they are physically and emotionally stable and are aware of the problem or need to learn. Nurses, because of their expertise and continual presence, are best placed to assess and provide education at optimal times.
Complications of Myocardial Infarction
Cardiogenic shock
Cardiogenic shock occurs as a complication of MI in about 5–10% of patients and is the most common cause of death in hospitals.50 It arises from loss of contractile force, and generally occurs when ventricular damage is more than 40% and ejection fraction less than 35%. Cardiogenic shock and the related management are described in more detail in Chapter 12.
Arrhythmias
Arrhythmias often occur in ACS and AMI and are often the cause of death in the prehospital phase. Management of the prehospital phase centres on community education and an effective, rapidly responsive ambulance service, as exemplified in Seattle in the USA.51 Arrhythmias may be generated by poorly perfused tissue and electrolyte alterations, and increased sympathetic tone during infarction, but are more often due to a failing left ventricle. They may also complicate reperfusion after successful revascularisation.52 It is essential to rapidly and effectively treat arrhythmias in the ACS and AMI context. The goal of treatment is to maintain cardiac output while reducing workload. Arrhythmias and management are described in Chapter 11.
Pericarditis
Pericarditis is an inflammation of the visceral and parietal layers of the pericardium that cover the heart. This inflammation occurs in approximately 20% of AMI patients within the following 2–3 days.10 The patient experiences chest pain, which may be confused with ischaemic pain. This confusion with an ischaemic event may be compounded by the additional presence of ST segment elevation on the ECG. However, pericardial pain increases with deep inspiration and a pericardial rub is often present. Electrocardiographically, the elevated ST segments of pericarditis are typically concave upwards (saddle-shaped) and often widespread, contrasting with convex ST segment elevation limited to the distribution of a single coronary artery in infarction.53 Pericarditis normally responds to anti-inflammatory treatment by aspirin, indomethacin and/or corticosteroids. Approximately 1–5% of AMI patients develop pericarditis as a late complication, 2 weeks to a few months post-AMI.18 Usually this late-onset pericarditis is associated with Dressler’s syndrome and may be an autoimmune response to myocardial injury. This is a chronic condition requiring systemic corticosteroid treatment.
Structural defects
Papillary muscle rupture most often occurs 2–7 days after MI. Patients experience a sudden onset of pulmonary oedema secondary to pulmonary hypertension and cardiogenic shock. Additional heart sounds and a systolic murmur will be heard. Urgent surgery is required, as the mortality rate for papillary muscle rupture is 95%.54 Cardiac rupture most often occurs within 5 days of MI and is commonest in older women. The patient experiences continuous chest pain, dyspnoea and hypotension as tamponade develops. Symptoms may worsen rapidly and result in pulseless electrical activity (PEA) unless surgery is undertaken immediately.
Heart Failure
In normal circumstances, the heart is a very effective, efficient pump with reserve mechanisms available to allow output to meet changing demands. These mechanisms include (a) increasing heart rate to increase total cardiac output, (b) dilation to create muscle stretch and more effective contraction, (c) hypertrophy of myocytes over time to generate more force, and (d) increasing stroke volume by increasing venous return and increased contractility. Heart failure is a complex clinical condition that is characterised by an underlying structural abnormality or dysfunction that results in the inability of the ventricle to fill with or eject blood.55 The condition is also known as congestive cardiac failure, a term commonly used in the USA but not in Australia. Chronic heart failure (CHF) describes the long-term inability of the heart to meet metabolic demands.
The burden of disease associated with heart failure is on the rise due to our ageing population, the prevalence of coronary heart disease and hypertension, the decrease in fatality from acute coronary syndrome and improved methods of diagnosis.55 Survival rates and prognosis for heart failure patients are extremely poor. Approximately 50% of patients diagnosed with heart failure will die within five years of diagnosis.56–58 When compared with those patients with cancer, heart failure patients have the poorest five-year survival rate, with the exception of lung cancer.59 In Australia during 2001–2002, 41,874 patients were hospitalised with a primary diagnosis of CHF (0.7% of all hospitalisations).60 Internationally, heart failure is the most common cause of hospitalisation in patients aged over 70 years.55 Approximately 40% of patients admitted to hospital with heart failure will be readmitted or die within one year.61
Over 50% of patients newly diagnosed with heart failure have concurrent ischaemic heart disease, hypertension is present in 65% and idiopathic dilated cardiomyopathy (5–10% of cases).55 The causes of heart failure can be categorised according to (a) myocardial disease, (b) arrhythmias, (c) valve disease, (d) pericardial disease and (e) congenital heart disease.62 Myocardial disease may be caused by myocardial infarction and fibrosis from prolonged ischaemic heart disease which accounts for approximately two-thirds of systolic heart failure causing systolic dysfunction and a reduced ejection fraction.
Arrhythmias, including both brady- and tachyarrhythmias, may cause heart failure due to changes in filling time affecting preload and resultant cardiac output. Myocardial oxygen demand is increased and if the heart is poorly perfused, muscle contraction will be affected. Frequent premature contractions and atrial fibrillation disturb mechanical coordination so that the ventricles may not be adequately filled for efficient contraction. Heart failure patients are also at high risk of sudden cardiac death due to ventricular fibrillation or tachycardia. Valvular disease causing heart failure usually involves valves on the left side of the heart (mitral and/or aortic valves). Aortic stenosis results in an increase in afterload and ventricular hypertrophy develops with reduced diastolic compliance resulting in a reduced ejection fraction. Mitral stenosis is usually due to rheumatic heart disease. Valvular incompetence results in a dilated ventricle to accommodate the regurgitant volume. Stroke volume increases in an attempt to empty its contents and ventricular muscle mass increases. However, over time the ventricle is unable to maintain the increased workload and heart failure develops. Valvular heart disease and treatment is described in more detail in Chapter 12.
• Backward failure: refers to the systemic and pulmonary congestion that occurs as a result of failure of the ventricle to expel its volume.
• Forward failure: is due to an inadequate cardiac output and leads to decrease in vital organ perfusion.
• Acute heart failure: includes the initial hospitalisation for the diagnosis of heart failure and exacerbations of chronic heart failure.
• Chronic heart failure: develops over time as a result of the inability of compensatory mechanisms to maintain an adequate cardiac output to meet metabolic demands.
• Systolic heart failure: refers to the inability of the ventricle to contract adequately during systole resulting in a reduced ejection fraction and an increased end-diastolic volume. This is the most common form of heart failure.
• Diastolic heart failure (or heart failure with preserved systolic function [HFSF]): indicates normal systolic function with a normal ejection fraction but impaired relaxation so there is a resistance to filling with increased filling pressures. Diastolic dysfunction usually occurs in conjunction with systolic dysfunction and is more common in the elderly.
• Low cardiac output syndrome: this occurs in response to hypovolaemia and/or hypertension. Severe vasoconstriction further reduces the cardiac output.
• High cardiac output syndrome is the result of an increase in metabolic demands causing a decrease in SVR leading to an increase in stroke volume and cardiac output. Burns and sepsis are the main causes.
• Left sided heart failure: occurs when there is a reduced left ventricular stroke volume resulting in accumulation of blood in the pulmonary system.
• Right sided heart failure: is the congestion of blood in the systemic system due to the inability of the right ventricle to expel its blood volume.
Responses to Heart Failure
When heart failure occurs, several adaptive responses are initiated by the body in an attempt to maintain normal perfusion (see Figure 10.8). These mechanisms are successful in the normal heart, but contribute to decreased effectiveness in the failing heart. The compensatory mechanisms include:
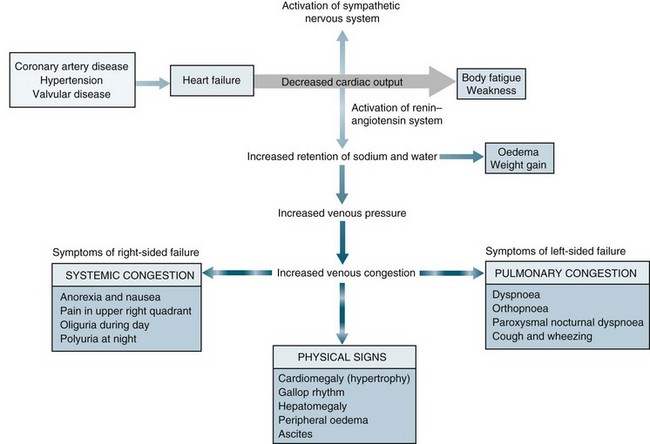
The sympathetic nervous system is the first response to be stimulated in heart failure. It occurs within seconds of a reduction in cardiac output and the parasympathetic system becomes inhibited. The baroreceptor reflexes are activated in response to a reduced arterial pressure. The beta-adrenergic receptors located in the heart are activated resulting in an increase in heart rate and contractility to increase stroke volume and cardiac output. Sympathetic nervous system response in the peripheral vascular system results in vasoconstriction which increase systemic venous return (SVR) and mean systemic filling pressures. This results in an increase in venous return, preload and afterload (see Figure 10.8). The consequence of this activation is increased myocardial oxygen demand. Although blood flow to essential organs is maintained, perfusion to the kidneys, gastrointestinal system and skin is reduced and peripheral resistance increased. Chronic activation of vasoconstrictors contributes to the progression of cardiac failure through increased resistance and effects on cardiac structure, causing hypertrophy and fibrosis and downregulation of beta-adrenergic receptors and endothelial dysfunction. Chronic poor perfusion to skeletal muscles may contribute to changes in muscle metabolism, resulting in further reductions in exercise tolerance.
The next compensatory mechanism to be activated is the RAAS. This is stimulated within minutes, in response to a decrease in kidney perfusion resulting in a decrease in glomerular filtration rate. Activation of this response results in an increase in SVR and sodium and water reabsorption which then increases the circulating blood volume, systemic filling pressures and venous return enhancing preload and afterload (see Chapter 9).
The Frank-Starling response is also activated. As the end-diastolic volume increases (preload) in response to sympathetic nervous system stimulation ventricular dilatation occurs stimulating the Frank-Starling response. As the myocardial fibres are stretched during diastole the force of contractility also increases to expel the increasing preload. This is a major mechanism of the heart to maintain a normal cardiac output. Optimal contractility occurs when the diastolic volume is 12–18 mmHg.63 However, when the ventricle is damaged, such as in MI, the sympathetic nervous system increases heart rate and contractility further increasing cardiac workload and exacerbating myocardial dysfunction which increases end-diastolic volume (preload) and ventricular dilatation further and heart failure progresses. As ventricular dilatation continues, ventricular hypertrophy results. The myocardium also increases its muscle mass in an attempt to increase contractility called ventricular remodelling. However, overtime ventricular hypertrophy results in changes to end-diastolic compliance and contractility due to the thickened ventricular wall, impaired muscle function and growth of collagen. These result in further impairment of ventricular function (see Figure 10.9). Ventricular hypertrophy also has a depressant effect of ventricular compliance, heart rate and contractility resulting in an increase in end-diastolic pressure with no associated increase in contractility. As the pulmonary artery pressures increase, pulmonary oedema and cardiogenic shock develop.
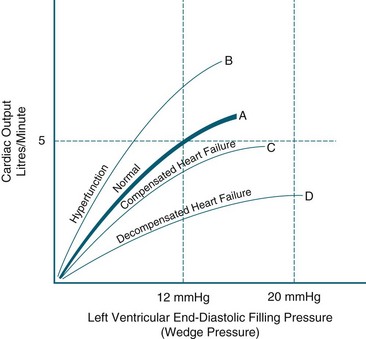
Left Ventricular Failure
Left ventricular failure (LVF), compared with other forms of heart failure, is characterised by breathlessness, orthopnoea and paroxysmal nocturnal dyspnoea, irritating cough and fatigue (see Table 10.4). Left ventricular failure exists when the ventricle has an ejection fraction of less than 40%, resulting in increased end-diastolic volume and increased intraventricular pressure.64 The left atrium is unable to empty into the left ventricle adequately and pressure in the left atrium rises. This pressure is reflected in the pulmonary veins and causes pulmonary congestion. When pulmonary venous congestion exceeds 20 mmHg, fluid moves into the pulmonary interstitium. Raised pulmonary interstitial pressure reduces pulmonary compliance, increases the work of breathing and is experienced by the patient as shortness of breath. Increased blood volume in the lung also initiates shallow, rapid breathing and the sensation of breathlessness. Patients also experience orthopnoea (dyspnoea while lying flat) and paroxysmal nocturnal dyspnoea (PND), because when lying, blood is redistributed from gravity-dependent areas of the body to the lung. Sitting upright or standing, and sleeping with additional pillows, relieves breathlessness at night.64

Acute pulmonary oedema results when pulmonary capillary pressure exceeds approximately 30 mmHg, and then fluid from the vessels begins to leak into the alveoli (see Figure 10.10).63 This fluid leak decreases the area available for normal gas exchange and severe shortness of breath results, often accompanied by pink, frothy sputum and noisy respirations. This causes patients to experience severe anxiety and decreased oxygen levels. Pulmonary oedema is a medical emergency and requires urgent treatment.
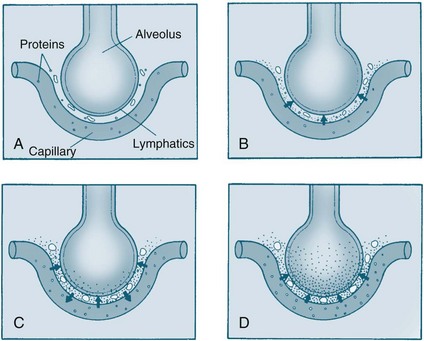
FIGURE 10.10 Pathophysiology of pulmonary oedema. As pulmonary oedema progresses, it inhibits oxygen and carbon dioxide exchange at the alveolar–capillary interface. (A) Normal relationship. (B) Increased pulmonary capillary hydrostatic pressure causes fluid to move from the vascular space into the pulmonary interstitial space. (C) Lymphatic flow increases and pulls fluid back into the vascular or lymphatic space. (D) Failure of lymphatic flow and worsening of left-sided heart failure causes further movement of fluid into the interstitial space and then into the alveoli.106
Right Ventricular Failure
Right ventricular failure (RVF) does not usually occur in isolation, except in the presence of severe lung disease, such as chronic obstructive pulmonary disease, pulmonary hypertension or a massive pulmonary embolus.60 In this case, right ventricular failure is due to resistance to outflow. The right ventricle can adapt to fairly large changes in volume; however, when cardiac output decreases, end-diastolic volume increases, and the right atrium is unable to empty adequately. Right atrial pressure rises and is reflected into the venous system. Jugular vein distension occurs, and the veins are usually visible above the clavicle. Symptoms of right heart failure are not as specific as left ventricular failure, and are mostly related to low cardiac output and raised venous pressure (see Table 10.4). Ascites and oedema tend to progress insidiously, and dependent oedema in the feet and ankles is often most prominent. Weight gain is an important sign as one kilogram of weight gain equals one litre of excess fluid. Liver congestion may result in tenderness, ascites and jaundice. Nausea and anorexia may be present and are a result of an increased intra-abdominal pressure. Many signs are not readily distinguishable from left ventricular failure, including extra heart sounds.
Patient Assessment, Diagnostic Procedures and Classification
Assessment and diagnosis are summarised in a diagnostic algorithm (see Figure 10.11). A full assessment and history is essential to determine the cause(s) of CHF and to assess the severity of the disease. A careful physical assessment is important for initial diagnosis and to evaluate the effectiveness of treatments and progress of the disease. The depth and time taken to conduct the assessment depend on the severity of symptoms. The physical examination of the patient focuses on cardiovascular and pulmonary assessment.
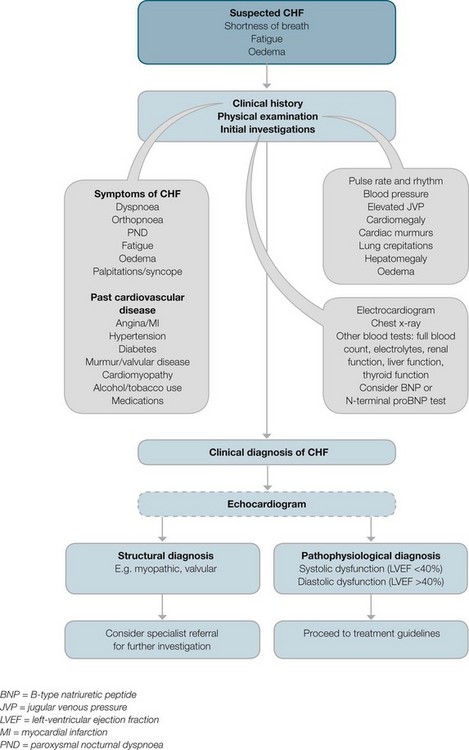
FIGURE 10.11 Diagnostic algorithm for CHF.
Courtesy National Heart Foundation of Australia and the Cardiac Society of Australia and New Zealand.55
Cardiovascular assessment includes:
• pulse rate and rhythm: The pulse rate is generally elevated due to a low cardiac output. However, if the patient is prescribed beta-adrenergic blocking agents and/or angiotensin converting enzyme (ACE) inhibitors, the pulse rate may be low.
• palpation of the praecordium and apical impulse: This may be displaced laterally and downward to the left due to an increased heart size.
• auscultation of a third heart sound (S3 gallop): This occurs due to a low ejection fraction and diastolic dysfunction. A fourth heart sound may also be present due to a decrease in ventricular compliance.
• assessment of jugular venous pressure (JVP): This is to estimate the degree of venous volume. If raised it reflects hypervolaemia, right ventricular failure, and reduced right ventricular compliance. It can also be raised in the presence of tricuspid valve disease. The hepatojugular reflex is also assessed by pressing on the liver and observing an increase in JVP. This results in an increase in blood flow to the right atrium.
• blood pressure: Lying and standing blood pressure are measured to assess postural hypotension due to a low cardiac output and also the prescribing of beta-adrenergic blocking agents and ACE inhibitors.
• peripheries: Look for the presence of cyanosis which may be due to vasoconstriction. Assess the fingers for clubbing which indicates long-term cyanosis usually as a consequence of congenital heart disease. Also assess the patient for ankle oedema. Peripheral oedema up to the midcalves indicates a moderate amount of excess fluid and the patient may require a bolus dose of diuretic medication.
Heart failure is usually classified according to the severity of symptoms. In chronic heart failure, the New York Heart Association (NYHA) Functional Classification is commonly used to classify patients on the basis of the activity level that initiates symptoms (see Table 10.5).
TABLE 10.5 New York Heart Association functional classification of heart failure64
| Class | Definition |
|---|---|
| I | Normal daily activity does not initiate symptoms. There are no limitations on activity |
| II | Ordinary activities initiate onset of symptoms, but symptoms subside with rest. Slight limitation of daily activities. |
| III | A small amount of activity initiates symptoms; patients are usually symptom-free at rest. Marked limitation of activity. |
| IV | Any type of activity initiates symptoms, and symptoms are present at rest. |
Diagnostic Tests
Tests used to diagnose heart failure include:
• trans-thoracic echocardiography is the most useful investigation to confirm diagnosis. This is the gold standard diagnostic test for heart failure and should always be undertaken when possible. This test is vital, as it can distinguish systolic dysfunction (left ventricular ejection fraction [LVEF] <40%) from diastolic dysfunction, and therefore help determine treatment.55 Information on left and right ventricular size, volumes, left ventricular thrombus and ventricular wall thickness and motion can be provided. Assessment of valve structure and function as well as intracardiac and pulmonary pressures can be determined, without the need for invasive techniques. Pulsed-wave Doppler and tissue Doppler studies can be used to determine diastolic dysfunction.
• assessment of cardiac function can also be done by invasive techniques (e.g. coronary angiography) and nuclear cardiology tests (e.g. gated radionuclide angiocardiography).
• ECG should be done as an initial investigation. Most common abnormalities include ST-T wave changes, left bundle branch block, left anterior hemiblock, left ventricular hypertrophy, atrial fibrillation and sinus tachycardia.
• chest X-ray for cardiomegaly and pulmonary markings, including evidence of interstitial oedema: perihilar pulmonary vessels, small basal pleural effusions obscuring the costophrenic angles, Kerley B lines (indicating raised left atrial pressure).
• full blood count for anaemia and mild thrombocytopenia. Any signs of anaemia should be further investigated.
• urea, creatinine and electrolytes for dilutional hyponatraemia, hypokalaemia, hyperkalaemia, low magnesium, and glomerular filtration rate. These should be closely monitored if there are any changes in clinical status and/or drug therapy such as ACEIs and diuretics.
• liver function tests for elevated levels of AST, ALT, LDH and serum bilirubin.
• thyroid function tests particularly in patients with no history of coronary artery disease and who develop atrial fibrillation.
• urinalysis for specific gravity and proteinuria.
• myocardial ischemia and viability need to be assessed in patients with heart failure and coronary artery disease. These can be assessed by a stress ECG, stress echocardiography or a stress nuclear study. Coronary angiography is useful to determine the contribution of coronary artery disease in these patients.
• natriuretic peptides includes plasma ANP and B-type natriuretic peptide (BNP). BNP or N-terminal proBNP is not recommended to be used to diagnose chronic heart failure as an elevated BNP may be due to other causes.55 However, it is useful to differentiate between dyspnoea due to chronic heart failure and dyspnoea due to chronic obstructive pulmonary disease.
• endomyocardial biopsy should be conducted if there is a suspicion of cardiomyopathy.
Nursing Management
Treatment of CHF is lifelong and multifactorial, requiring a well-coordinated, multidisciplinary approach. The goals of heart failure treatment are to identify and eliminate the precipitating cause, promote optimal cardiac function, enhance patient comfort by relieving signs and symptoms, and help the patient and family cope with any lifestyle changes. Clinical practice guidelines have been developed to guide the treatment of heart failure on the basis of ventricular dysfunction and grade of symptoms (see Figures 10.12–10.14).55
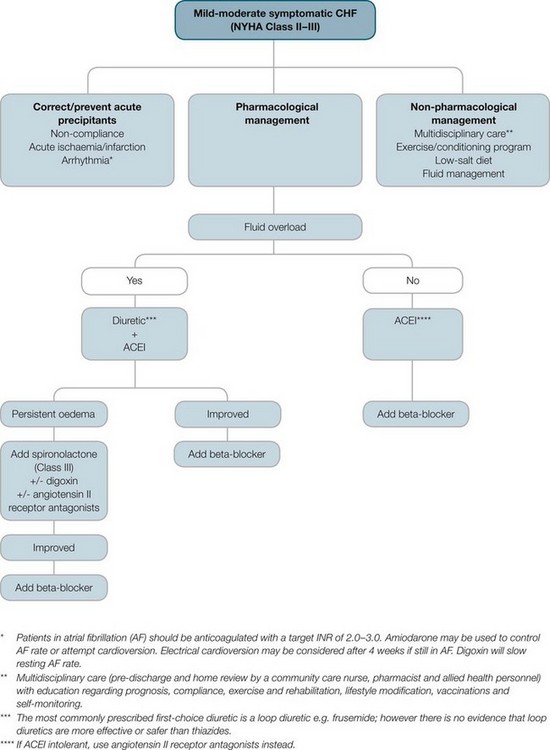
FIGURE 10.12 Pharmacological treatment of systolic heart failure.
Courtesy National Heart Foundation of Australia and the Cardiac Society of Australia and New Zealand.55
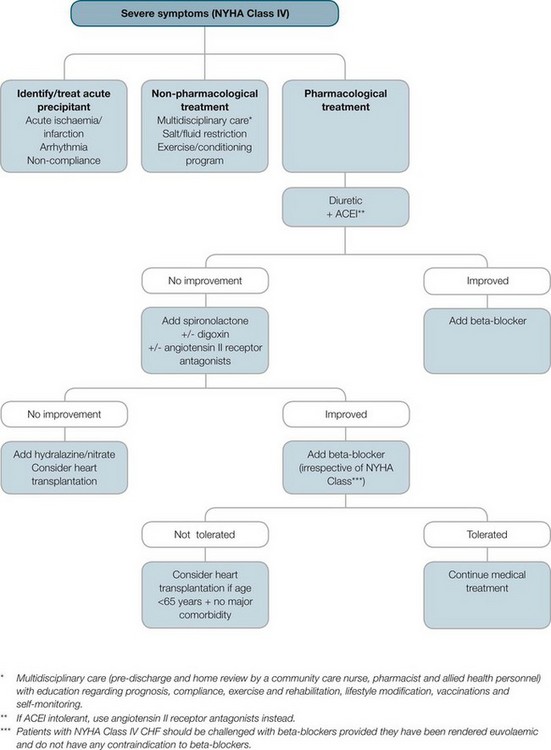
FIGURE 10.13 Pharmacological treatment of refractory systolic heart failure.
Courtesy National Heart Foundation of Australia and the Cardiac Society of Australia and New Zealand.55
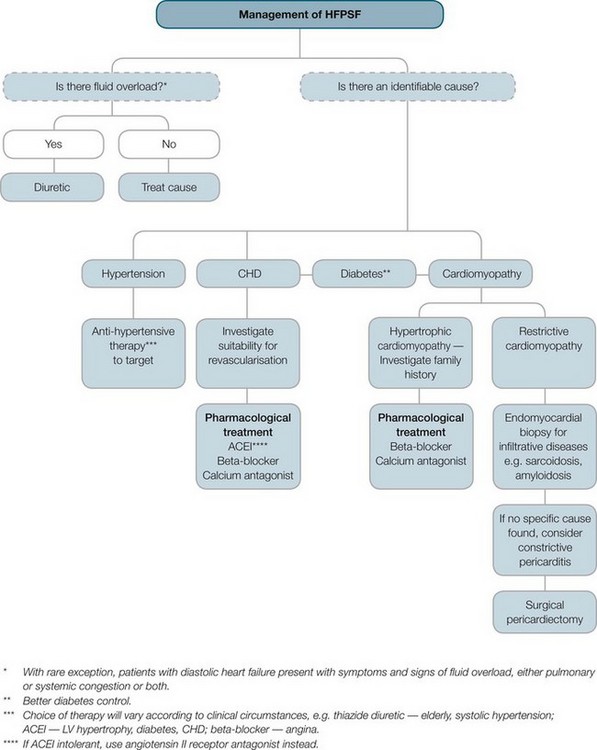
FIGURE 10.14 Management of HFPSF.
Courtesy National Heart Foundation of Australia and the Cardiac Society of Australia and New Zealand.55
Planning for hospital discharge begins early in the admission and aims to promote quality of life for the patient and prevent unnecessary admissions. Several health care services have been implemented to support the transition from hospital to home as it is during the first 30 days post-discharge that nearly 20% of heart failure patients are readmitted to hospital.65 There are currently over 70 outreach heart failure programs throughout Australia that support heart failure patients post-discharge.66 The main goals of these programs are to reduce symptom burden, improve functional capacity and minimise hospital readmissions. These programs range from in-hospital visits to facilitate discharge planning, nurse-led heart failure outpatient clinics, home visit programs and heart failure specific exercise programs. Several meta-analyses of home visit programs have shown a reduction in hospital admissions and mortality67,68 and these programs are now standard care for heart failure patients.55 Home visit heart failure programs involve a heart failure nurse visiting the patient at home and providing education to the patient and carer, assessing their symptoms and educating the patients and their carers about self-management strategies. Nurse-led outpatient clinics also reduce hospital admissions and mortality69,70 and play an important role in the management of heart failure patients post-discharge.
Management of heart failure post the acute phase is based on three principles: self-care management, long-term lifestyle changes and adherence to pharmacotherapy. Management of self-care is the key to non-pharmacological management of heart failure. Self-care refers to the decision-making process of patients concerning their choice of healthy behaviour and response to worsening symptoms when they occur. It involves cognitive decision making, requiring the recognition of signs and symptoms that indicate a change in condition, which is based on knowledge and prior experiences of deterioration.71–73
Lifestyle Modification and Self-care Management
• the disease process. This involves discussing what heart failure is, signs and symptoms and why they occur, and strategies to improve their symptoms
• medications and side effects
• self-monitoring and acute symptoms
• the importance of adherence to their medications and management plan.
Restriction of fluid to 1–1.5 L/day is one of the most important strategies that patients can adhere to in order to improve their symptoms. Patients are encouraged to weigh themselves daily and to identify any increase in weight as an increase of 1 kg equals 1 litre of excess fluid. National guidelines stipulate that if their weight increases by 2 kg over 2 days they need to see their local doctor as soon as possible.55 Patients that adhere to their management plan and closely monitor their daily weight may self-manage their volume status by using a flexible diuretic action plan as developed by their cardiologist. In addition, patients should be advised of early warning signs of excess fluid volume and decompensation, such as increasing dyspnoea, fatigue and peripheral oedema.
Sleep apnoea also occurs commonly in CHF patients. There are two types: obstructive sleep apnoea and central sleep apnoea. Obstructive sleep apnoea occurs due to airway collapse and is associated with obesity. It can be treated with weight reduction and night-time continuous positive airway pressure (CPAP). The use of CPAP for obstructive sleep apnoea results in an improvement in LVEF due to an increase in left ventricular filling and emptying rates, and a decrease in systolic blood pressure and left ventricular chamber size.74 Central sleep apnoea (Cheyne-Stokes respirations) occurs due to pulmonary congestion and high sympathetic stimulation in patients with severe heart failure and may be treated with CPAP. However, the benefits of oxygen therapy have not been proven. However, exercise is equally important, to prevent the deconditioning of skeletal muscle that occurs in CHF. Exercise training – including walking, exercise bicycle and light resistance – has been shown to improve functional capacity, symptoms, neurohormonal abnormalities, quality of life and mood in CHF.64 The Heart Foundation of Australia recommends that all stable CHF patients, regardless of age, should be considered for referral to a tailored exercise program (preferably a heart failure specific exercise program) or modified cardiac rehabilitation program.55 Heart failure exercise programs comprise resistance training and have been shown to improve functional capacity, heart failure symptoms and survival and reduced hospitalisations.75 In patients with symptomatic heart failure physical activity should be undertaken under the supervision of trained heart failure specialists, e.g. physiotherapist or exercise physiologist, who can tailor the level of exercise to the degree of severity of symptoms. Many CHF patients have co-morbidities such as arthritis, which make exercise programs difficult, but maintaining general activity should be encouraged.
Dietary sodium intake should be reduced to 2 g/day for patients with moderate to severe heart failure and to 3 g/day for mild heart failure.55 Reduction in sodium intake helps reduce fluid retention, diuretic requirements and potassium excretion. A large proportion of an individual’s sodium intake can come from processed foods, so patients are encouraged to read nutrition labels and reduce the intake of these foods. Salt intake can also be reduced by avoiding adding salt in cooking or to meals. As CHF patients who are overweight increase demands on their heart, weight loss by lowering dietary fat intake may improve symptoms and quality of life. These patients may require referral to a dietician for weight loss management. In patients with moderate to severe heart failure, cardiac cachexia and anaemia are common which further exacerbate weakness and fatigue. These patients will require a referral to a dietician for nutritional support. Other lifestyle changes are: smoking cessation, ideally no alcohol otherwise limit alcohol to less than 2 standard drinks/day (alcohol is a myocardial toxin and reduces contractility), limit caffeinated drinks to 1–2 drinks/day (to decrease risk of arrhythmias), control diabetes, annual vaccinations for influenza and regular pneumococcal disease vaccinations.55
Medications
Pharmacological management relies on the following categories of drugs: ACE inhibitors, beta-adrenergic blocking agents, angiotension receptor blocking agents (ARBs), diuretics, digoxin and antiarrhythmic drugs. (Beta-adrenergic blocking agents and antiarrhythmic drugs are reviewed on page 266). The main actions and adverse effects of these drugs in heart failure are summarised in Table 10.6.
TABLE 10.6 Common medications for the treatment of heart failure55,64,107,108
| Drug/example | Action | Major adverse effects |
|---|---|---|
| First line pharmacotherapy | ||
Angiotensin-converting enzyme inhibitors
ACE inhibitors are the cornerstone of CHF treatment, as they have been demonstrated to prolong survival, improve patient symptoms and exercise tolerance, prevent hospitalisation and improve ejection fraction in CHF patients.76,77 All patients with symptomatic systolic LV dysfunction should be prescribed ACE inhibitors.55,61 Drugs in this group (captopril, enalapril, lisinopril) act on the renin–angiotensin system by specifically preventing the conversion of angiotensin I into angiotensin II.78 As a result, systemic vascular resistance (afterload) is decreased. This is particularly important in preventing the progression of CHF, because blockade of the renin–angiotensin system prevents further development of systolic dysfunction. In addition, because angiotensin II also stimulates the release of aldosterone, sodium and water retention are decreased (preload). This may also be beneficial when ACE inhibitors are prescribed with diuretics, as potassium loss is limited. Further, ACE inhibitors inhibit the breakdown of bradykinin (a vasodilator), which also contributes to decreasing vascular resistance. The total reduction of systemic vascular resistance reduces the workload of the heart without affecting heart rate or cardiac output.
Common adverse effects of ACE inhibitors primarily result from hypotension, including dizziness and headache. Other side effects include hyperkalaemia, deterioration of renal function, and an unproductive cough, which may respond to asthma prophylactic medications. Initial doses of ACE inhibitors should be low, as severe – though transient – symptomatic hypotension can occur, worsening of renal function and hyperkalaemia. The dose of ACE inhibitors needs to be gradually increased to maximum dose over 2–3 months to optimise the survival and functional capacity benefits. This group of drugs is contraindicated in patients with bilateral renal artery stenosis due to the danger of developing renal failure. One important adverse effect of ACEIs is that it cannot be taken in conjunction with NSAIDs as NSAIDs reduce the action of ACE inhibitors.79
Beta-adrenergic blocking agents
All patients with symptomatic systolic left ventricular dysfunction should be prescribed a beta-adrenergic blocking agent. Beta-adrenergic blocking agents (carvedilol, metoprolol, bisoprolol) are used in CHF to inhibit the adverse effects of chronic activation of the sympathetic nervous system and improve ventricular function (see Chapter 10). In heart failure beta-2 receptors predominate with beta-1 receptors being downregulated. In heart failure beta-adrenergic blocking agents reduce this neurohormonal activity. The addition of a beta-adrenergic blocker has been demonstrated to reduce symptoms, reduce hospitalisations and prolong survival in patients.80,81 Similar to ACE inhibitors the dose of beta-adrenergic blocking agents needs to be gradually increased. Once the patient is eurovolaemic they should be commenced on low dose and gradually increased to maximal dose over several months.
Angiotensin receptor blocking agents
The primary use of angiotensin receptor blocking agents (ARBs) is in patients who are intolerant of ACE inhibitors such as ACEI cough. They have a similar action as ACE inhibitors, however, ARBs block the angiotensin II receptor that responds to angiotensin II stimulation. ACE inhibitors on the other hand act on the enzyme that produces angiotensin II.78 They have similar benefits as ACE inhibitors, improving survival, LVEF and heart failure symptoms and a reduction in hospitalisations.82,83 Similar to ACE inhibitors, ARBs are commenced on a low dose and gradually up-titrated to optimal dose over two months. Adverse effects are: deterioration in renal function, hyperkalaemia and symptomatic hypotension.61
Diuretics
• Loop diuretics: These drugs (frusemide, ethacrynic acid and bumetanide) act on the ascending limb of the loop of Henle of the nephron. They prevent the reabsorption of chloride and sodium ions from the loop, so that increased concentrations are present in the loop, attracting more water and increasing urine volume. Intravenous administration of frusemide is often used to manage preload in acute exacerbations. In fluid-overloaded patients, the aim is to achieve increased urine output and a weight reduction of 0.5–1 kg daily, until clinical euvolaemia is achieved. Hypokalaemia is a common adverse effect, and patients on long-term diuretics need regular monitoring and may require potassium supplements. Hyponatraemia may also occur at high doses, and needs careful management in heart failure patients. Ototoxicity, presenting as tinnitus, vertigo and deafness, can occur at high doses, so IV delivery of frusemide should be no faster than 4 mg/min.
• Thiazide and thiazide-like diuretics: These drugs (chlorothiazide, hydrochlorothiazide, chlorthalidone) act on the ascending loop of the nephron and decrease sodium reabsorption. As a result, the fluid in the collecting ducts is more concentrated and attracts more water. Thiazides also cause peripheral arteriole vasodilation, which may be beneficial in hypertensive patients. Adverse effects are similar to loop diuretics due to potassium and sodium loss, and supplementation may be necessary. When ACE inhibitors are prescribed concurrently, there is less potassium loss (details below). Hyperglycaemia can occur, so diabetics need monitoring. Impotence may also occur, as well as sensitivity due to the presence of sulphonamide in the drug structure.
• Aldosterone antagonists: These are potassium-sparing diuretics and include spironolactone.55 Aldosterone acts on the distal convoluted tubule of the nephron to cause sodium retention and thus water retention, although potassium is lost. Antagonists stop this action, so potassium is not lost and not as much sodium retained, thus there is minor diuresis. Spironolactone is particularly useful in chronic heart failure because there is excessive aldosterone production, causing oedema. There is the potential that spironolactone, by blocking aldosterone systemically, may prevent the negative effects of aldosterone on the heart, such as fibrosis, hypertrophy and arrhythmogenesis. Adverse effects include hyperkalaemia, which may occur more readily in CHF patients because of renal failure, and because of its potentially lethal effects requires regular monitoring. Other effects include hyponatraemia and feminisation effects such as gynaecomastia. Spironolactone is recommended for use in patients with severe symptomatic (NYHA class III–IV) systolic heart failure in addition other pharmacotherapy such as ACE inhibitors. Aldosterone antagonists have additional survival benefits and reduce hospital readmission.84,85
Cardiac glycosides
Arrhythmias are common in heart failure and need to be treated. The agent must be carefully selected, as chronic heart failure patients often have complex medication regimens and interactions may occur. Also, some ventricular antiarrhythmics, like class 1 agents (e.g. flecainide), are associated with sudden death in CHF. Implantable cardioverter-defibrillator (ICD) therapy may be more effective in treating ventricular arrhythmias. ICDs reduce mortality by 20–30%86 and are first-line therapy in patients with a history of VF or sustained VT, LVEF ≤30% at least one month post myocardial infarction or three months post CABGs and symptomatic heart failure and LVEF ≤35%.55 Cardiac resynchronisation therapy (CRT) (also known as biventricular pacing) is also indicated in patients with symptomatic heart failure to reduce asynchronous pacing of the left ventricle (QRS duration > 150 ms). Systolic function is improved when the left and right ventricles are paced simultaneously. Often patients with a prolonged QRS will have a combination of an ICD with CRT therapy. ICDs and CRT are discussed in more detail in Chapter 11.
Acute Exacerbations of Heart Failure
Acute exacerbations of CHF usually occur as episodes of decompensation due to progression of the disease or non-adherence to their management plan.87 Acute episodes usually present as congestive heart failure with associated pulmonary oedema, cardiogenic shock (see Chapter 21) or decompensated CHF.55 Patients with severe dyspnoea due to pulmonary congestion should be administered oxygen therapy. If their hypoxaemia does not improve then they may benefit from bilevel positive airway pressure (BiPAP) to support ventilation and gas exchange. The use of continuous positive airway pressure ventilation (CPAP) or BiPAP in acute pulmonary oedema will reduce the need for intubation and mechanical ventilation.
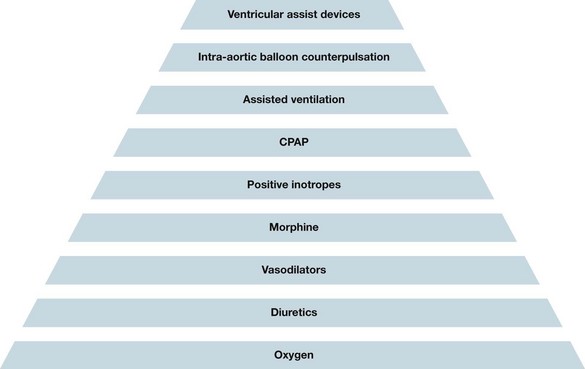
The mainstay of treatment of an acute exacerbation is pharmacological, so a combination of the medications is given, usually comprising diuretics, morphine and nitrates. The nitrates and morphine cause vasodilatation. Morphine also reduces the respiratory drive and respiratory workload. Nitrates also cause epicardial artery dilatation and reduce preload which also helps to relieve symptoms of pulmonary congestion particularly at night when filling pressures are increased due to the recumbent position of sleeping.55 Diuretics should be administered intravenously to optimise the excretion of intra and extravascular cellular fluid to reduce circulating blood volume to reduce cardiac workload. Fluid restriction, usually to 1–1.5 L in 24 hours, is begun. A urinary catheter may need to be inserted so that accurate, continuous measures of urine output can be gained and an accurate fluid balance calculated. This is necessary, along with consistent daily weighing, to determine the effectiveness of diuretic therapy and renal status. Various positive inotropes may be administered (e.g. IV dobutamine causes vasodilatation; IV dopamine to improve renal function) to improve contractility and reduce systemic venous return. Various mechanical devices are also available, e.g. intra-aortic balloon pump, LVAD (discussed in Chapter 12). CRT with or without an ICD may be implanted. CRT is recommended in NYHA class III–IV patients on optimal pharmacological therapy, LVEF ≤35%, QRS duration > 120 ms, and sinus rhythm.55 All of these criteria must be fulfilled. Criteria for implantation of an ICD include: symptomatic patients (NYHA class II–IV) and LVEF ≤35%, LVEF <30% one month post AMI or three months post CABGs, spontaneous VT with structural CHD, or survived a cardiac arrest due to VT or VF which was not due to a reversible cause. If a patient is to have an ICD implanted then extensive counselling pre- and post-implantation must be undertaken with the patient and carer to ensure they are aware of the painful and unexpected shocks that may be delivered.55 Figure 10.15 provides an overview of the escalation of treatment for acute heart failure.55
Selected Cases
Cardiomyopathy
Dilated Cardiomyopathy
Dilated cardiomyopathy (DCM) is the most common form of cardiomyopathy and is characterised by ventricular and atrial dilation and systolic dysfunction.88 All four chambers become enlarged which is not in proportion to the degree of hypertrophy. It presents as heart failure of variable severity, sometimes complicated by thromboembolism, at least partly due to atrial fibrillation, which is common. Conduction abnormalities are common in DCM further exacerbating AV dyssynchrony and left ventricular dysfunction. DCM is the most common cause of sudden cardiac death due to ventricular arrhythmias. Annual mortality from DCM ranges from 10–50%.89 Idiopathic DCM is the most common cause of heart failure in young people. Aetiology of DCM includes coronary heart disease, myocarditis, cardiotoxins, genetics and alcohol.
Diagnosis
Many of the features of DCM are non-specific. Heart failure, as mentioned, is present with typical symptoms of dyspnoea, fatigue, peripheral oedema and cardiomegaly. S3 and S4 heart sounds may be present on auscultation. Atrial and ventricular arrhythmias are common, particularly atrial fibrillation, ventricular tachycardia, ventricular fibrillation and torsades de pointes. Left bundle branch block (LBBB) is often present, which worsens systolic performance and shortens survival, especially when the QRS is markedly prolonged.88 Echocardiography demonstrates the defining abnormalities and may be useful in revealing atrial thrombus. Occasionally, endocardial biopsy is undertaken to differentiate from myocarditis or rarer causes of cardiomyopathy.
Management
Treatment for DCM is similar to that of heart failure and includes beta-adrenergic blocker therapy, ACEIs, diuretics and antiarrhythmic therapy where indicated or, if necessary, an ICD for recurrent haemodynamically significant ventricular arrhythmias.88 The use of cardiac resynchronisation therapy (CRT) has produced significant clinical improvements and is recommended for DCM patients with NYHA functional class III–IV, optimal medical therapy, LVEF ≤35%, and sinus rhythm with QRS greater than 120 msec.61,90 Cardiac transplantation is considered when standard therapies fail to influence clinical progression and left ventricular assist devices and ICDs may be used as a bridge to transplantation.
Hypertrophic Cardiomyopathy
Hypertrophic cardiomyopathy (HCM) is a genetic abnormality that gives rise to inappropriate hypertrophy especially in the intraventricular septum with preserved or hyperdynamic systolic function. The main abnormality with HCM is diastolic rather than systolic as in DCM. The hypertrophy is not a compensatory response to excessive load, such as in aortic stenosis or hypertension. Left ventricular hypertrophy of variable patterns is seen, occasionally with disproportionate septal hypertrophy, which causes left ventricular outflow tract obstruction (LVOTO) in which HCM progresses to hypertrophic obstructive cardiomyopathy, or HOCM. In HCM the muscle mass is large and hypercontractile, but the left ventricular cavity is small. The increase in left ventricular systolic pressure and the altered relaxation cause diastolic dysfunction and impaired ventricular filling. Mitral regurgitation is common. These abnormalities combine to produce pulmonary congestion and dyspnoea due to a raised end-diastolic pressure. Sudden cardiac death, often after exertion or other increases in contractility, is sometimes seen in HCM and is thought to be partly attributable to outflow obstruction.91 It is the most common cause of death in athletes.63
Diagnosis
Echocardiography will confirm the presence and pattern of hypertrophy and the presence (or absence) of an outflow tract gradient. Examination findings include cardiomegaly and pulmonary congestion. An S4 heart sound is common, and the ECG shows left ventricular hypertrophy and often ventricular arrhythmias. When the obstructive form (HOCM) is present, a systolic murmur, mitral regurgitation murmur and deep narrow Q waves on ECG, may be present.88 The majority of patients are asymptomatic and when they present to hospital it will be in severe symptoms of dyspnoea, angina and syncope. Angina is the result of an imbalance between oxygen supply and demand due to the increased myocardial mass and not due to atherosclerosis.
Management
Treatment for HCM is aimed at the prevention of sudden cardiac death and pharmacotherapy to increase diastolic filling and to reduce the LVOTO. Pharmacotherapy includes beta-adrenergic blocker or calcium channel blocker therapy, as these decrease contractility and lessen outflow tract obstruction. Care is necessary with medication selection, as vasodilation may worsen obstruction, causing haemodynamics to suffer.88 The impact of atrial fibrillation, by worsening the ventricular filling defect, can be dramatic in HCM patients and will require antiarrhythmics and anticoagulation. If ventricular arrhythmias are present, or there is a family history of sudden cardiac death, treatment with an ICD should be considered.92 For severely symptomatic patients or those worsening despite maximal drug treatment, surgical myectomy to reduce the size of the septum and lessen obstruction may be necessary and can result in a marked improvement of symptoms.92 Septal ablation with alcohol injected into the first septal branch of the left anterior descending artery is a less invasive alternative, a procedure that is usually undertaken with pacemaker insertion as AV block is produced. Although surgical myectomy remains the gold standard, both treatments provide effective symptom relief and improvement in heart failure severity.92 If the patient with HCM deteriorates and is hospitalised, positive inotropes, chronotropes and nitrates worsen LVOTO and should be avoided. However, beta-adrenergic blockers, amiodarone and calcium antagonists such as verapramil are indicated.88 Due to the familial nature of HCM, relatives aged 12–18 years also need to be screened for HCM.
Restrictive Cardiomyopathy
Restrictive cardiomyopathies (RCMs) limit diastolic distensibility or compliance of the ventricles. The stiff ventricular walls feature diastolic dysfunction and there is impaired ventricular filling. Infiltrates into the interstitium and the replacement of normal myocardium with abnormal tissue hamper this relaxation.88 Initially, systolic function and wall thickness are normal. However, as the disease progresses systolic dysfunction occurs. RCM is commonly caused by myocardial infiltration, as in amyloidosis, sarcoidosis, fibrosis or cardiac metastases, or may be idiopathic.88 Endomyocardial disease is more common in tropical countries, but in the Western world, RCMs are the least common form of cardiomyopathy.88
Diagnosis
Clinically there is heart failure (increase in JVP, dyspnoea, S3 and S4 heart sounds, and oedema), particularly right ventricular, and infiltration of the conduction system may cause conduction defects and heart block. Low-voltage ECGs are commonly seen. Patients commonly present with decreased exercise tolerance due to the impaired ability to increase heart rate and cardiac output because of reduced ventricular filling. Restrictive cardiomyopathy must be distinguished from constrictive pericarditis (which it may closely resemble), as pericarditis may be easily managed.88 If echocardiography demonstrates a restrictive pattern then a myocardial biopsy may be undertaken to determine its aetiology, especially in the case of systemic infiltrative disease.88
Management
There is no treatment for RCM so the aim of therapy is to relieve symptoms. This includes diuretics, corticosteroids and pacing. The use of nitrates should be done with caution as the filling defect can be worsened by decreased venous return or hypovolaemia. Generally, prognosis is poor with many dying within 1–2 years of diagnosis.92
Hypertensive Emergencies
Acute, uncontrolled hypertension is often divided into two categories: hypertensive emergencies and hypertensive urgencies. In hypertensive emergencies blood pressure needs to be reduced within one hour to prevent end-organ damage, such as hypertensive encephalopathy, papilloedema or aortic dissection.93 Immediate blood pressure reduction with IV agents under critical care monitoring is needed. By contrast, hypertensive urgencies are those in which end-organ damage is not occurring, and although prompt management is required, this can be approached more gradually with oral antihypertensive agents under close supervision, without necessarily requiring admission to a critical care unit.93 Previous hypertension is not always present, but because of chronic adaptive vascular changes may provide some level of protection against acute tissue injury. Symptoms may not develop until the blood pressure exceeds 220/110 mmHg, whereas in patients without previous hypertension, hypertensive emergencies may occur at levels of even 160/100 mmHg.94 When the diastolic pressure is persistently above 130 mmHg, there is risk of vascular damage and must be treated.
Diagnosis
A thorough history is taken, including any hypertension management, known renal or cerebrovascular disease, eclampsia in previous pregnancies if gravid, or use of stimulants or illicit drugs such as cocaine. Patient assessment should include evidence of end-organ damage, such as back pain (aortic dissection), neurological damage: headache, altered consciousness, confusion, visual loss, stupor or seizure activity (encephalopathy); cardiac damage: chest pain, ST segment changes, cardiac enlargement, or the development of heart failure or pulmonary oedema; and renal damage: oliguria and azotaemia.93 Serum urea, creatinine, electrolytes, urinalysis, ECG and chest X-ray should be performed.
Management
More severe, or malignant, hypertension may cause retinal haemorrhage or papilloedema, and emergency treatment should immediately be instituted. Other contexts in which there is a need for rapid treatment of severe hypertension include intracranial bleeding, acute myocardial infarction, phaeochromocytoma, recovery from cardiac surgery, and bleeding from vascular procedure sites. Hypertensive emergencies in pregnancy threaten both the mother and the fetus.95
The aim of treatment is to acutely lower the blood pressure, but neither too quickly nor too dramatically. Recommendations vary, but an initial aim of 150/110–160/100 mmHg within 2–6 hours, or a 25% reduction in mean arterial pressure within 2 hours, has been described.96,97 Continuous direct arterial pressure monitoring should be in place during treatment. Intravenous sodium nitroprusside, a rapidly acting arterial and venous dilator, is most frequently used, at doses of 0.25–10 µg/kg/min.97 Weaning of nitroprusside is undertaken after the later introduction of oral antihypertensives. Care is required to avoid hypotension during treatment, as well as rebound hypertension as nitroprusside is withdrawn. Rapidly acting beta-adrenergic blocking agents with short half-lives such as IV esmolol may be used at doses of 50–100 µg/kg/min (or higher) in patients without standard contraindications to beta-adrenergic blockers (asthma, heart failure).97 Glyceryl trinitrate infusions at 10–100 µg/min or higher are used for combined venous and arterial dilation, especially if there is angina.97 Intravenous frusemide may be introduced during the acute phase. After intravenous therapies have been established and progress towards target pressures is made, oral agents are introduced. These include oral beta-adrenergic blockers, calcium channel blockers, ACE inhibitors and diuretics.
Infective Endocarditis
Infective endocarditis remains a potentially life-threatening disorder, with mortality remaining as high as 20–25%98 even in this era of relative rheumatic fever control. This same era, however, sees other means of developing endocarditis, with factors such as longer life, IV drug use, prosthetic valves, greater rates of cannulation during hospitalisation, cardiac surgery, resistant organisms, and increased numbers of immunocompromised patients from immunosuppressant drugs and HIV/AIDS.99,100
Infection of the endocardium, often with involvement of the cardiac valves, occurs most commonly due to staphylococcal, streptococcal and enterococcal bacteraemia.99,100 The definition of infective endocarditis now also includes an infection of any structure within the heart such as prosthetic valves, implanted devices and chordae tendineae.101 Infective endocarditis can be acute or subacute. Acute infective endocarditis progresses over days to weeks with destruction of valves and metastatic infection. Subacute infective endocarditis occurs over weeks to months and is milder than acute infective endocarditis. Endothelial damage occurs in the endocardium. Platelet-fibrin deposits form and a lesion develops. Bacterial colonisation then occurs and vegetation adheres to the endocardial lesion. Many of the signs and symptoms of infective endocarditis are due to the immune response to the microorganism. The patient presents with fever, and general features of febrile illness, which may include septic shock. Joint pain is common and septic arthritis is sometimes seen. Cardiac symptoms develop when there is valvular involvement, which may manifest as erosion through valve leaflets producing regurgitation, fusing of valve leaflets or vegetations (outgrowths from valve structures), producing valvular stenosis or regurgitation.100 The mitral valve is more commonly affected, but aortic valve involvement carries a worse prognosis.98 Conduction system involvement manifests as arrhythmias and conduction defects. Embolic complications are relatively common and multifactorial. Septic emboli, embolisation of atrial thrombi when atrial fibrillation is present, and fragmentation of vegetations may all give rise to pulmonary and systemic emboli. These most often present as splenic infarction, stroke, peripheral vascular occlusion and renal failure.98
Diagnosis
• evidence of endocardial involvement (positive echocardiography, abscess, partial dehiscence of a prosthetic valve, or new valvular vegetation)
Minor clinical criteria include:
• fever with body temperature ≥38°C
• predisposing heart condition or intravenous drug use
• vascular signs: arterial emboli, intracranial haemorrhage, Janeway lesions (erythematous spots on the palms and feet) or conjunctival haemorrhages
• immunological signs: Osler nodes (painful, reddened nodules on the fingers and the feet), or glomerulonephritis98
Management
Prosthetic valve endocarditis must be aggressively managed, as mortality may be as high as 65%.100 Impaired valvular opening, even obstruction, may occur or the prosthetic valve may become unseated.100 Reoperation to replace the affected valve should be undertaken when valvular dysfunction is present. Antibiotic therapy is provided empirically until blood culture and sensitivities are established. Cardiac failure, if present, is managed along standard lines (see section on Nursing management of acute heart failure). Observations during endocarditis should be directed at detecting embolic complications involving the brain, kidneys, or spleen; development and progress of heart failure; progress of the febrile illness, including hydration and dietary status.
Prophylactic antibiotic coverage should be undertaken for at-risk patients 1 hour before dental procedures are to be performed, in particular for those with previous rheumatic fever or endocarditis, or prosthetic valves.101 Antibiotic prophylaxis for genitourinary and gastrointestinal procedures is no longer recommended.101
Aortic Aneurysm
The aorta is the major blood vessel leaving the heart. An aneurysm is a local dilation or outpouching of a vessel wall and comes in several forms (see Figure 10.16). Most aortic aneurysms are fusiform and saccular, and occur in the abdominal aorta. A fusiform aneurysm is uniform in shape with symmetrical dilation that involves the whole circumference of the aorta.102 A saccular aneurysm has dilation of part of the aortic wall so the dilation is very localised.102 A dissecting aneurysm occurs when the layers of the wall of the aorta continue to separate and fill with blood, resulting in obstructed blood flow. The aorta is particularly susceptible to aneurysm formation because of constant stress on the vessel wall and the absence of penetrating vasa vasorum that normally provide perfusion to the adventitia. As the blood flows through the aneurysm it becomes turbulent and some blood may stagnate along the walls allowing a thrombus to form. This thrombus in addition to atherosclerotic debris may embolise into the distal arteries compromising their circulation. Atherosclerosis is the commonest cause of aneurysm, because plaque formation erodes the vessel wall. Other causes include syphilis, infection, inflammatory diseases and trauma. Aneurysms occur most often in men and in people with the risk factors of hypertension or smoking. Approximately 80% of aortic aneurysms rupture into the left retroperitoneum which may contain the rupture. However, the other 20% rupture into the peritoneal cavity and uncontrolled haemorrhage results.102
FIGURE 10.16 Aneurysm. Major types of aneurysm: (A) fusiform aneurysm has an entire section of an artery dilated, occurring most often in the abdominal aorta due to atherosclerosis; (B) sacculated aneurysm affects one side of an artery, usually in the ascending aorta; (C) dissecting aneurysm results from a tear in the intima, causing blood to shunt between the intima and media; (D) pseudoaneurysm usually results from arterial trauma, such as intra-aortic balloon pump catheter or an arterial introducer; the opening does not heal properly and is covered by a clot that can burst at any time.106
Management
Management of asymptomatic aneurysms is conservative, unless the size of the aneurysm is >1.5 times the normal size of the aortic segment102 or the situation is acute. The primary aim is to lower hypertension and prevent increases in thrombus size and emboli through the administration of aspirin. Usually the patient has regular monitoring to assess the aneurysm and to determine the timing and need for surgical repair.
Nursing management of dissecting aortic aneurysm involves the following:
Ventricular Aneurysm
Less than 5% of patients post-STEMI, particularly a transmural anterior infarction, develop a left ventricular aneurysm.103 Post-STEMI, dyskinetic or akinetic areas of the left ventricle are common and known as regional wall motion abnormalities. It is in these areas that there is a risk of an aneurysm developing. Ventricular aneurysms are more likely to develop post anterior STEMI with a totally occluded LAD with poor collateral circulation.
Aneurysms form when the intraventricular tension stretches the dyskinetic area and a thin weak layer of necrotic muscle and fibrous tissue develops and bulges with each contraction of the ventricle resulting in a reduction in stroke volume. Aneurysms range from 1–8 cm in diameter and are four times more likely to occur at the apex and anterior wall rather than the inferoposterior wall.103 Large ventricular aneurysms may result in a reduction in stroke volume causing an increase in myocardial oxygen demand (MvO2) resulting in angina and heart failure. The mortality rate in people with ventricular aneurysms is four times higher than those with no aneurysm due to a higher risk of tachyarrhythmias and sudden cardiac death. Unlike aortic aneurysms these aneurysms rarely rupture so their management is usually conservative. Diagnosis of a ventricular aneurysm is by echocardiography. Ventricular aneurysm should be considered when ST segment elevation persists beyond 1 week after myocardial infarction.
Summary
• May affect either the left, right or both ventricles, resulting in different symptoms being displayed by the patient.
• Diagnosis is usually made on the basis of echocardiography, ECG, chest X-ray, full blood count, electrolytes, liver function tests and urinalysis.
• In acute heart failure, CPAP or BiPAP may be necessary to improve hypoxaemia
• Pharmacological therapy of acute heart failure consists of: morphine, nitrates and diuretics. Positive inotropes may also be used such as IV dopamine and dobutamine to improve renal perfusion and contractility
• Many patients with heart failure will also have a pacemaker with cardiac resynchronisation therapy and/or a defibrillator to improve cardiac function and reduce the incidence of sudden death
• Patient care must be lifelong and coordinated between all members of the healthcare team. Broad interventions, including medications, diet and lifestyle modification, may be appropriate for some patients, while palliative care might be more appropriate for other patients.
Learning activities
1. Discuss the various categories included under the umbrella ‘acute coronary syndrome’, including their identifying and differentiating features.
2. Differentiate common versus prognostic symptoms of acute coronary syndrome.
3. Describe the nursing care for a patient undergoing primary PTCA.
4. Identify the common complications of myocardial infarction in the early recovery period.
5. Why is silent ischaemia more common in patients with CHD and diabetes?
6. In the case study, what is the significance of an S3 heart sound in the clinical setting of heart failure?
7. Discuss the compensatory mechanisms that are activated in heart failure and their effect on the cardiovascular system.
8. Why did the patient in the case study meet the criteria for implantation of an ICD with CRT?
9. In the case study, when the patient was discharged from hospital, why were they prescribed perindopril, bisoprolol, spironolactone and frusemide? Include in your answer the reason for prescribing these medications and their actions and major side effect.
10. Observe an echocardiograph and ask the sonographer to explain what is visualised on the screen, particularly in Doppler mode. Ask them to identify areas of hypokinesis or akinesis and any evidence of dyssynchrony between the ventricles.
1 Badellino K. Pathogenesis of atherosclerosis. In: Moser D, Riegel B. Cardiac nursing: A companion to Braunwald’s heart disease. St Louis: Saunders Elsevier, 2008.
2 Australian Institute of Health and Welfare (AIHW). Australia’s health 2010 update [AIHW Cat. No. 12]. AIHW: Canberra, 2010.
3 Hay DR. Cardiovascular disease in New Zealand, 2004: technical report 82. Auckland: National Heart Foundation of New Zealand; 2004.
4 World Health Organization (WHO). World health report 2008 – Primary health care now more than ever. Geneva: WHO; 2008.
5 Guyton A. Textbook of medical physiology, 8th edn. Philadelphia: WB Saunders; 1991.
6 Galandi MM, Lampe FC, Wood DA. Incidence, clinical characteristics and short-term prognosis of angina pectoris. Br Heart J. 1995;73:193–198.
7 NHFA and Cardiac Society of Australia and New Zealand. Guidelines for the management of acute coronary syndrome 2006. Med J Aust. 2006;184:S1–32.
8 De Wood MA, Spores J, Notske R, Mouser LT, Burroughs R, et al. Prevalence of total coronary occlusion during the early hours of transmural myocardial infarction. New Engl J Med. 1980;303:897–902.
9 Boden WE, Eagle K, Granger CB. Reperfusion strategies in acute ST-segment elevation myocardial infarction: a comprehensive review of contemporary management options. J Am Coll Cardiol. 2007;50:917–929.
10 McCance K. Structure and function of the cardiovascular and lymphatic systems. In: Huether S, McCance K. Understanding pathophysiology. St Louis: Mosby, 2002.
11 Adams J, Trent Rawles J. Earliest electrocardiographic evidence of myocardial infarction: implications for thrombolytic treatment. BMJ. 1993;307:409–413.
12 Molliterno DJ, Sgarbosso EB, Armstrong PW, et al. ST depression versus T-wave inversion: GUSTO II results. J Am Coll Cardiol. 1996;27(Suppl. A):182A.
13 Pollack CV, Jr., Diercks DB, Roe MT, Peterson ED. American College of Cardiology/American Heart Association guidelines for the management of patients with ST-elevation myocardial infarction: implications for emergency department practice. Ann Emerg Med. 2005;45(4):363–376.
14 Murphy MJ, Berding CB. Use of measurements of myoglobin and cardiac troponin in the diagnosis of acute myocardial infarction. Crit Care Nurse. 1999;19(1):58–66.
15 Belenkie I, Knudtson ML, Roth DL, Hansen JL, Traboulsi M, et al. Relation between flow grade after thrombolytic therapy and the effect of angioplasty on left ventricular function: a prospective randomized trial. Am Heart J. 1991;121(2,1):407–416.
16 Froelicher VF, Jr., Thompson AJ, Jr., Davis G, Stewart AJ, Triebwasser JH. Prediction of maximal oxygen consumption: comparison of the Bruce and Balke treadmill protocols. Chest. 1975;68(3):331–336.
17 Melin JA, Wijns W, Vanbutsele RJ, Robert A, DeCostes P, et al. Alternative strategies for coronary artery disease in women: demonstration of the usefulness and efficiency of probability analysis. Circulation. 1985;71(3):535–552.
18 Moser D, Riegel B. Care of patients with acute coronary syndrome: ST-segment elevation myocardial infarction. In: Moser D, Riegel B. Cardiac nursing: A companion to Braunwald’s heart disease. St Louis: Saunders Elsevier, 2008.
19 Lawrence-Mathew PJ, Wilson AT, Woodmansey PA, Channer KS. Unsatisfactory management of patients with myocardial infarction admitted to general medical wards. J R Coll Phys Lond. 1994;28(1):49–51.
20 Fibrinolytic Therapy Trialists’ (FTT) Collaborative Group. Indications for fibrinolytic therapy in suspected acute myocardial infarction: collaborative overview of early mortality and major morbidity results from all randomised trials of more than 1000 patients. Lancet. 1994;343:311–322.
21 Boersma E, Maas AC, Deckers JW, Simoons ML. Early thrombolytic treatment in acute myocardial infarction. J Adv Nurs. 1996;12:677–682.
22 Galbraith A, Bullock S, Manias E. Fundamentals of pharmacology: a text for nurses and allied health professionals. Sydney: Prentice Hall Health; 2004.
23 The Guston authors. GUSTO-1 Global utilisation of streptokinase and t-PA for occluded coronary arteries. An international randomised trial comparing four thrombolytic strategies for acute myocardial infarction. New Engl J Med. 1993;329:673–682.
24 RITA-2 Trial Participants. Coronary angioplasty versus medical therapy: the second Randomised Intervention Treatment of Angina (RITA-2) trial. Lancet. 1997;350:461–468.
25 Zijlstra F, Hoorntje JC, De Boer MJ, Reiffers S, Miedema K, et al. Long-term benefit of primary angioplasty as compared with thrombolytic therapy for acute myocardial infarction. New Engl J Med. 1999;341:1413–1419.
26 Fischman Dl, Leon MB, Baim DS. A randomised comparison of coronary stent placement and balloon angioplasty in the treatment of coronary artery disease: stent restenosis study investigators. New Engl J Med. 1994;331(8):496–501.
27 van Hout BA, Serruys PW, Lemos PA, van den Brand MJ, van Es GA, et al. One year cost effectiveness of sirolimus eluting stents compared with bare metal stents in the treatment of single native de novo coronary lesions: an analysis from the RAVEL trial. Heart. 2005;91(4):507–512.
28 Halkin A, Stone GW. Polymer-based paclitaxel-eluting stents in percutaneous coronary intervention: a review of the TAXUS trials. J Intervent Cardiol. 2004;17(5):271–282.
29 Schiks IE, Schoonhoven L, Aengevaeren WR, Nogarede-Hoekstra C, van Achterberg T, Verheugt FW. Ambulation after femoral sheath removal in percutaneous coronary intervention: a prospective comparison of early vs. late ambulation. J Clin Nurs. 2009;18:1862–1870.
30 Antiplatelet Trialists’ Collaboration. Collaborative overview of randomised trials of antiplatelet therapy: Prevention of death, myocardial infarction and stroke by prolonged antiplatelet therapy in various categories of patients. BMJ. 1994;308:81–106.
31 Lewis HD, Jr., Davis JW, Archibald DG, Steinke WE, Smitherman TC, et al. Protective effects of aspirin against acute myocardial infarction and death in men with unstable angina: results of a Veterans Administration Cooperative Study. New Engl J Med. 1983;309:396–403.
32 Trotter R, Gallagher R, Donoghue J. Anxiety in patients undergoing percutaneous coronary interventions. Heart Lung. July 2010. DOI: doi:10.1016/j.hrtlng.2010.05.054 Epub ahead of print
33 Cannon CP, Molterno DJ, Every N, et al. Implementation of AHCPR guidelines for unstable angina in 1996. Unfortunate differences between men and women: results from the GUARANTEE registry. J Am Coll Cardiol. 1997;29(Suppl):217A.
34 CAPRIE Steering Committee. A randomised controlled trial of clopidogrel versus aspirin in patients at risk of ischaemic events (CAPRIE). Lancet. 1996;348:1329–1339.
35 PRISM-PLUS Study Investigators. Inhibition of the platelet glycoprotein IIb/IIIa receptor with tirofiban in unstable angina and non-Q-wave myocardial infarction. Platelet Receptor Inhibition in Ischemic Syndrome Management in Patients Limited by Unstable Signs and Symptoms (PRISM-PLUS). New Engl J Med. 1998;338:1488–1497.
36 Neumann FJ, Zohlnhofer D, Fakhoury L, Ott I, Gawaz M, et al. Effect of glycoprotein IIb/IIIa receptor blockade on platelet-leukocyte interaction and surface expression of the leukocyte integrin Mac-1 in acute myocardial infarction. J Am Coll Cardiol. 1999;34:1420–1426.
37 Yusuf S, Wittes J, Friedman L. Overview of results of randomised clinical trials in heart disease II. Unstable angina, heart failure, primary prevention with aspiring, and risk factor modification. JAMA. 1988;260:2259–2263.
38 Kaplan K, Davison R, Parker M, Przybylek J, Teagarden JR, et al. Intravenous nitroglycerin for the treatment of angina at rest unresponsive to standard nitrate therapy. Am J Cardiol. 1983;51:694–698.
39 Sacks FM, Pieffer MA, Moye LA, Rouleau JL, Rutherford JD, et al. The effect of pravastatin on coronary events after myocardial infarction in patients with average cholesterol levels. N Engl J Med. 1996;335:1001–1009.
40 Quinn T, Webster R, Hatchett R. Coronary heart disease: angina and acute myocardial infarction. In: Hatchett R, Thompson D. Cardiac nursing: a comprehensive guide. Philadelphia: Churchill Livingston Elsevier, 2002.
41 Thompson Dr, Bowman GS. Evidence for the effectiveness of cardiac rehabilitation. Clin Effect Nurs. 1997;1:64–75.
42 Malmberg K, Norhammer A, Wedel H, Ryden L. Glycometabolic state at admission: important risk marker of mortality in conventionally treated patients with diabetes mellitus and acute myocardial infarction: long term results from the Diabetes and Insulin-Glucose Infusion in Acute Myocardial Infarction (DIGAMI) study. Circulation. 1999;99:2626–2632.
43 Proctor T, Yarcheski A, Oriscello RG. The relationship of hospital process variables to patient outcome post myocardial infarction. Int J Nurs Stud. 1996;33(2):121–130.
44 De Jong M. Impact of anxiety on cardiac disease. In: Moser D, Riegel B. Cardiac nursing: A companion to Braunwald’s heart disease. St Louis: Saunders Elsevier, 2008.
45 Baker CF, Garvin BJ, Kennedy CW, Polivka BJ. The effect of environmental sound and communication on CCU patients’ heart rate and blood pressure. Res Nurs Health. 1993;16:415–421.
46 World Health Organization. Needs and action priorities in cardiac rehabilitation and secondary prevention in patients with CHD. Copenhagen: WHO Regional Office for Europe; 1993.
47 Taylor RS, Brown A, Ebrahim S, et al. Exercise-based rehabilitation for patients with coronary heart disease: systematic review and meta-analysis of randomised controlled trials. Am J Med. 2004;116:682–692.
48 Oldridge N, Guyatt G, Jones N, Crowe J, Singer J. Effects on quality of life with comprehensive cardiac rehabilitation after acute myocardial infarction. Am J Cardiol. 1991;74:1240–1244.
49 Gulanick M, Berra K. Cardiac rehabilitation. In: Moser D, Riegel B. Cardiac nursing: A companion to Braunwald’s heart disease. St Louis: Saunders Elsevier, 2008.
50 Santoro GM, Buonamici P. Reperfusion therapy in cardiogenic shock complicating acute myocardial infarction. Am Heart J. 1999;138(2:2):S126–S138.
51 Cobb LA, Weaver WD, Fahrenbruch CE, Hallstrom AP, Copass MK, Eberhardt F, Bode F. Community-based interventions for sudden cardiac death: impact limitations and changes. Circulation. 1992;85(Suppl 1):98–102.
52 Bonnemeier H, Ortak J, Wiegand UK, Eberhardt F, Bode F, et al. Accelerated idioventricular rhythm in the post-thrombolytic era: incidence, prognostic implications, and modulating mechanisms after direct percutaneous coronary intervention. Ann Noninvas Electrocardiol. 2005;10(2):179–187.
53 Wagner GS, Marriott HJL. Marriott’s practical electrocardiography, 10th edn. Baltimore: Lippincott, Williams & Wilkins; 2000.
54 Appel S. Care of patients with complications of acute myocardial infarction. In: Moser D, Riegel B. Cardiac nursing: A companion to Braunwald’s heart disease. St Louis: Saunders Elsevier, 2008.
55 National Heart Foundation of Australia and the Cardiac Society of Australia and New Zealand (Chronic Heart Failure Guidelines Expert Writing Panel). Guidelines for the prevention, detection and management of people with chronic heart failure in Australia 2006. Melbourne: National heart Foundation of Australia; 2006.
56 Najafi F, Dobson AJ, Jamrozik K. Recent changes in heart failure hospitalizations in Australia. Eur J Heart Fail. 2007;9:228–233.
57 Levy D, Kenchaiah S, Larson M, Benjamin EJ, Kupka MJ, et al. Long-term trends in the incidence of and survival with heart failure. N Engl J Med. 2002;347:1397–1402.
58 Roger VL, Weston SA, Redfeild MM, Hellermann-Homan JP, Killian J, et al. Trends in heart failure incidence and survival in a community-based population. JAMA. 2004;292:344–350.
59 Stewart S, MacIntyre K, Hole DA, Capewell S, McMurray JJV. More malignant than cancer? Five-year survival following a first admission for heart failure in Scotland? Eur J Heart Fail. 2001;3:315–322.
60 Australian Institute of Health and Welfare. Heart, stroke and vascular diseases: Australian facts [AIHW cat. no. CVD 27]. AIHW and National Heart Foundation of Australia: Canberra, 2004.
61 Dickstein K, Cohen-Solal A, Filippatos G, McMurray JJV, Ponikowski P, et al. Task force for the diagnosis and treatment of chronic heart failure of the European Society of Cardiology. Guidelines for the diagnosis and treatment of acute and chronic heart failure 2008 of the European Society of Cardiology. Eur Heart J. 2008;29:2388–2442.
62 Abraham WT, Krum H. Heart failure: a practical approach to treatment. New York: McGraw Hill Medical; 2007.
63 Soine L. Heart failure and cardiogenic shock. Woods SL, Froelicher ESS, Motzer SU, Bridges EJ. Cardiac nursing, 6th edn, Baltimore: Lippincott: Williams & Wilkins, 2010.
64 Gould M. Chronic heart failure. In: Hatchett R, Thompson D. Cardiac nursing: a comprehensive guide. Edinburgh: Churchill Livingstone Elsevier, 2002.
65 McMurray JJV, Stewart S. Epidemiology, aetiology, and prognosis of heart failure. Heart. 2000;83:596–602.
66 Driscoll A, Worrall-Carter L, Hare DL, Davidson PM, Riegel B, et al. Evidence-based chronic heart failure management programs: myth or reality. Qual Safe Health Care. 2009;18(6):450–455.
67 McAlister FA, Stewart S, Ferrua S, McMurray JJV. Multidisciplinary strategies for the management of heart failure patients at high risk for readmission: A systematic review of randomised trials. J Am Coll Cardiol. 2004;44(4):810–819.
68 Whellan DJ, Hasselblad V, Peterson E, O’Connor CM, Schulman KA. Meta-analysis and review of heart failure disease management randomised controlled clinical trials. Am Heart J. 2005;149:722–729.
69 Phillips CO, Singa RM, Rubin HR, Jaarsma T. Complexity of program and clinical outcomes of heart failure disease management incorporating specialist nurse-led heart failure clinics. A meta-regression analysis. Eur J Heart Fail. 2005;7:333–341.
70 Driscoll A, Toia D, Gibcus J, Srivastava PM, Hare DL. Heart Failure Nurse Practitioner clinic: an innovative approach for optimisation of beta-blockers. Heart Lung Circulation. 2008;17(1):S13.
71 Driscoll A, Davidson P, Clark R, Huang N, Aho Z on behalf of National Heart Foundation consumer resource working group. Tailoring consumer resources to enhance self-care in chronic heart failure. ACC. 2009;22(3):133–140.
72 Bennett SJ, Cordes DK, Westmoreland G, Castro R, Donnelly E. Self-care strategies for symptom management in patients with chronic heart failure. Nurse Res. 2000;49(3):139–145.
73 Riegel B, Carlson VV. A situation-specific theory of heart failure self-care. J Cardiovasc Nurs. 2008;23(3):190–196.
74 Kaneko Y, Floras JS, Usui K, Plante J, Tkacova R, et al. Cardiovascular effects of continuous positive airway pressure in patients with chronic heart failure and obstructive sleep apnoea. N Engl J Med. 2003;348:1233–1241.
75 Piepoli MF, Davos C, Francis DP. Coats AJ for the ExTraMATCH Collaborative. Exercise training meta-analysis of trials in patients with chronic heart failure (ExTraMATCH). BMJ. 2004;328(7443):189–200.
76 CONSENSUS Trial Study Group. Effects of enalapril on mortality in severe congestive cardiac failure. Results of the Cooperative North Scandinavian Enalapril Survival Study (CONSENSUS). N Engl J Med. 1987;316:1429–1435.
77 SOLVD Investigators. Effect of enalapril on survival in patients with reduced left ventricular ejection fractions and congestive heart failure. N Engl J Med. 1991;325:293–302.
78 Opie LH, Pfeffer MA. Inhibitors of angiotensin-converting enzyme, angiotesin II receptor, aldosterone and renin. Opie LH, Gersh BJ. Drugs for the heart, 7th edn, Philadelphia: Saunders, 2009.
79 Ailabouni W, Eknoyan G. Nonsteroidal anti-inflammatory drugs and acute renal failure in the elderly.A risk-benefit assessment. Drugs Aging. 1996;9(5):341–351.
80 Hjalmarson A, Goldstein S, Fagerberg B, Wedel H, Waagstein F, et al. Effects of controlled-release metoprolol on total mortality, hospitalizations, and well-being in patients with heart failure: the Metoprolol CR/XL Randomized Intervention Trial in congestive heart failure (MERIT-HF). MERIT-HF Study Group. JAMA. 2000;283:1295–1302.
81 Packer M, Fowler MB, Roecker EB, Coats AJ, Katus HA, et al. Effect of carvedilol on the morbidity of patients with severe chronic heart failure: results of the carvedilol prospective randomized cumulative survival (COPERNICUS) study. Circulation. 2002;106:2194–2199.
82 McMurray JJ, Ostergren J, Swedberg K, Granger CB, Held P, et al. Effects of candesartan in patients with chronic heart failure and reduced left-ventricular systolic function taking angiotensin converting-enzyme inhibitors: the CHARM-Added trial. Lancet. 2003;362(9386):767–771.
83 Cohn JN, Tognoni G, for the Valsartan Heart Failure Trial Investigators. A randomized trial of the angiotensin receptor blocker valsartan in chronic heart failure. N Engl J Med. 2001;345(23):1667–1675.
84 Pitt B, Zannad F, Remme WJ, Cody R, Castaigne A, et al. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. Randomized Aldactone Evaluation Study Investigators. N Engl J Med. 1999;341:709–717.
85 Pitt B, Remme W, Zannad F, Neaton J, Martinez F, et al. Eplerenone, a selective aldosterone blocker, in patients with left ventricular dysfunction after myocardial infarction. N Engl J Med. 2003;348:1309–1321.
86 Bokhari F, Newman D, Greene M, Korley V, Mangat I, Dorian P. Long-term comparison of the implantable cardioverter defibrillator versus amiodarone: eleven-year follow-up of a subset of patients in the Canadian Implantable Defibrillator Study (CIDS). Circulation. 2004;110:112–116.
87 Moser D, Mann D. Improving outcomes in heart failure: it’s not unusual beyond usual care (Editorial). Circulation. 2002;105:2810–2812.
88 Wynne JA, Braunwald E. The cardiomyopathies and myocarditides. In: Zipes DP, Libby P, Bonow RO, Braunwald E. Braunwald’s heart disease: a textbook of cardiovascular medicine. 7th edn. Philadelphia: Elsevier Saunders; 2008:1404.
89 Deedwania PC. The key to unravelling the mystery of mortality in heart failure: An integrated approach. Circulation. 2003;107:1719.
90 Hunt SA, Abraham WT, Chin MH, Feldman AM, Francis GS, et al. ACC/AHA 2005 Guideline Update for the Diagnosis and Management of Chronic Heart Failure in the Adult: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Update the 2001 Guidelines for the Evaluation and Management of Heart Failure): developed in collaboration with the American College of Chest Physicians and the International Society for Heart and Lung Transplantation: endorsed by the Heart Rhythm Society. Circulation. 2005;112:e154–e235.
91 Finkelmeier BA. Cardiomyopathies. In: Finkelmeier BA, ed. Cardiothoracic surgical nursing. 2nd edn. Philadelphia:, Williams & Wilkins; 2000:59.
92 Ralph-Edwards A, Woo A, McCrindle BW, Shapero JL, Schwartz L, et al. Hypertrophic obstructive cardiomyopathy: comparison of outcomes after myectomy or alcohol ablation adjusted by propensity score. J Thorac Cardiovasc Surg. 2005;129(2):351–358.
93 Kaplan NM. Systemic hypertension: mechanisms and diagnosis. In: Zipes DP, Libby P, Bonow RO, Braunwald E. Braunwald’s heart disease: a textbook of cardiovascular medicine. 7th edn. Philadelphia: Elsevier Saunders; 2008:807.
94 Vaughan CJ, Delanty N. Hypertensive emergencies. Lancet. 2000;356(9227):411–417.
95 Vidaeff AC, Carroll MA, Ramin SM. Acute hypertensive emergencies in pregnancy. Crit Care Med. 2005;33(Suppl 10):S307–S312.
96 Shapiro S. Cardiac problems in critical care. Bongard FS, Sue DY. Current critical care: diagnosis and treatment, 2nd edn, New York: Lange Medical Books/McGraw-Hill, 2002.
97 Baas LS. Hypertensive emergencies. Baird MS, Keen JH, Swearingen PL. Manual of critical care nursing: nursing interventions and collaborative management, 5th edition, St Louis: Elsevier Mosby, 2005.
98 Karchmer AW. Infective endocarditis. In: Zipes DP, Libby P, Bonow RO, Braunwald E. Braunwald’s heart disease: a textbook of cardiovascular medicine. 7th edn. Philadelphia: Elsevier Saunders; 2008:1077.
99 Baas LS. Acute infective endocarditis. Baird MS, Keen JH, Swearingen PL. Manual of critical care nursing: nursing interventions and collaborative management, 5th edn, St Louis: Elsevier Mosby, 2005.
100 Crawford MH, Durack DT. Clinical presentation of infective endocarditis. Cardiol Clin. 2003;21:159–166.
101 Bashore T, Cabell CH, Fowler V. Update on infective endocarditis. Current Prob Cardio. 2006;31:274–352.
102 Isselbacher EM, Eagle KA, Descanctis RW. Diseases of the aorta. In: Zipes DP, Libby P, Bonow RO, Braunwald E. Braunwald’s heart disease: a textbook of cardiovascular medicine. 7th edn. Philadelphia: Elsevier Saunders; 2008:1546.
103 Antman EM. ST-Elevation myocardial infarction: Management. In: Zipes DP, Libby P, Bonow RO, Braunwald E. Braunwald’s heart disease: a textbook of cardiovascular medicine. 7th edn. Philadelphia: Elsevier Saunders; 2008:1215.
104 Bryant B, Knights K, Saterno E. Pharmacology for Health Professionals. Sydney: Mosby; 2003.
105 Bersten AD, Soni N, Oh TE. Oh’s intensive care manual, 5th edn. Oxford: Butterworth-Heinemann; 2003.
106 Urden L, Stacy K, Logh M. Thelan’s critical care nursing: diagnosis and management, 5th edn. St Louis: Mosby; 2006.
107 Holland R, Battersby J, Harvey I, Lenaghan E, Smith J, Hay L. Systematic review of multidisciplinary interventions in heart failure. Heart. 2005;91(7):899–906.
108 Davies MK, Gibbs CR, Lip GYH. ABC of heart failure. Management: diuretics, ACE inhibitors, and nitrates. BMJ. 2000;320(7232):428–431.
109 Michaelson CR. Congestive heart failure. St Louis: Mosby; 1983.
110 Hudak CM, Gallo BM, Morton PG. Critical care nursing: A holistic approach, 7th edn. Philadelphia: Lippincott & Williams; 1998.
[/level-membership-for-critical-care-medicine-category][not-level-membership-for-critical-care-medicine-category]
10 Cardiovascular Alterations and Management
After reading this chapter, you should be able to:
• explain the pathophysiology of coronary artery disease, clinical manifestations of acute coronary syndromes and management of events
• discuss the collaborative care for a patient with chest pain
• list the diagnostic tests used to assess myocardial ischaemia
• outline the actions and contraindications of thrombolytic drugs
• outline the clinical manifestations of right and left ventricular failure
• discuss the goals of heart failure treatment
• discuss the pathophysiology of the four different types of cardiomyopathy and how it affects cardiac function
• outline the actions of angiotensin converting enzyme inhibitors, beta-blockers, loop diuretics and spironolactone and how they relate to the pathophysiology of heart failure
Coronary Heart Disease
Coronary heart disease (CHD) is the term used to describe the effects of a reduction or complete obstruction of blood flow through the coronary arteries due to narrowing from atherosclerosis and/or thrombus. Although some patients may be asymptomatic, the commonest manifestations of CHD are chest pain due to angina, acute coronary syndrome (ACS, a term used to collectively describe acute myocardial infarction [AMI] and unstable angina) and sudden death. CHD may also cause arrhythmias and heart failure.1
CHD is the leading cause of death, premature death and disability in Australia and New Zealand.2,3 In 2007, more than 22,000 people died of CHD in Australia, more than 5000 in New Zealand in 2004 and 7.2 million worldwide.2–4 Death rates have fallen by about 76% since the 1960s, primarily due to improvements in risk factors and health care for those at risk. However, the burden of CHD remains high, with 1.5% of Australians reporting CHD symptoms.2 Furthermore, CHD is the single leading health problem worldwide because of a rising incidence in developing countries.4
Myocardial Ischaemia
When coronary blood flow is insufficient to meet myocardial tissue demand for oxygen, myocardial ischaemia occurs. Critical restriction to blood flow occurs when the diameter of the lumen of the blood vessel is reduced by more than half. Coronary blood flow is also determined by perfusion pressure, which can be adversely affected by abnormalities in blood flow (valvular disease), vessel wall (coronary spasm) and the blood (anaemia, polycythaemia).5 Myocardial oxygen demand is influenced by heart rate, strength of myocardial contraction and left ventricular wall tension. As the myocardium receives most of its blood supply during diastole, a rise in heart rate will decrease the duration of diastole and therefore coronary perfusion. Sympathetic stimulation increases the force of contraction and therefore oxygen demand. Left ventricular wall tension increases with the changes in preload associated with filling and afterload associated with systemic vascular resistance. During activity, pyrexia and arrhythmias, these effects may compound due to sympathetic stimulation, causing an increased oxygen demand and reduced coronary perfusion.
Angina
A fixed coronary artery lesion, causing limitation of oxygen supply at times of increased demand, results in stable angina. Therefore, symptoms arise during periods of physical and emotional stress and resolve within 2–10 minutes of rest. Symptoms tend to be worse in the morning (coinciding with a peak in blood pressure), after heavy meals and in cold weather. The severity of symptoms has little correlation with the progress of the disease. However, a patient with a typical history of angina has a high probability of CHD and a higher risk of AMI and sudden death in the following year.6
Unstable Angina and Acute Myocardial Infarction
Unstable angina and AMI form a continuum on the basis of reduction in coronary blood flow and subsequent damage to myocardial cells. Unstable angina may indicate transient ischaemia, whereas AMI indicates myocardial tissue death. The term ‘acute coronary syndrome’ (ACS) is now used to represent this continuum.7 ACS results from the rupture or erosion of an atherosclerotic plaque, leading to release of vasoconstrictor substances and potentially triggering coagulation activity (see Figure 10.1). Formation of thrombi results in intermittent and/or prolonged obstruction of the coronary artery. Therefore, ACS typically presents as a recent history of angina (within the past 4–6 weeks); a change in symptoms including increased frequency, more easily provoked or occurring in the absence of physical or emotional stress, more severe or prolonged and/or less responsive to nitrate therapy. ACS is a medical emergency, with up to a third of ACS patients at risk of AMI and death within 3 months.7 There is a high risk of death if the patient experiences more than 20 minutes of pain at rest (pain at rest is associated with changes in ST segment of 1 mm or more on a 12-lead ECG), if there was MI within the previous two weeks, or if pulmonary oedema or mitral regurgitation is present.7
Myocardial Infarction
Myocardial infarction (MI) occurs when blood flow to the myocardium is severely impaired for more than 20 minutes as myocardial cell necrosis begins. Coronary artery thrombus arising from an atherosclerotic plaque is found in the majority of patients dying of AMI.8 Cellular death begins in the subendocardial layer and progresses through the full muscle thickness, so that by 2 hours with total occlusion a full ‘transmural’ infarction will result. However, the full extent of tissue death may occur as a single incident or evolve over several days, depending on the degree of obstruction to blood flow.
The location and impact of the infarction will depend on which coronary artery has been obstructed:
• Left anterior descending (LAD) affects the function of the left ventricle and interventricular septum, including ventricular conduction tissue. Patients with anteroseptal MI are at high risk of heart failure, cardiogenic shock and mortality due to pump deficits.
• Circumflex (CX) affects the left ventricle lateral and posterior walls and the SA node in 50% of people.5 The impact on pump efficiency of lateral and posterior wall necrosis is not as severe as anteroseptal infarcts, although patients are at more risk of arrhythmias.
• Right coronary artery (RCA) affects the inferior wall of the left ventricle and the right ventricle, as well as the AV node in most patients and the SA node in 50% of people. There is potentially severe impact on ventricular function if both the inferior wall and the right ventricle are affected, as well as a high risk of arrhythmias due to SA and AV node involvement.
Patient Assessment and Diagnostic Features
A key feature of assessment of the patient with chest pain is the use of protocols and guidelines to promote rapid assessment so that revascularisation procedures such as thrombolysis and percutaneous coronary intervention (PCI) can be implemented as soon as possible. This means that assessment may begin as early as in the ambulance, with ECG transmission to hospital ED where rapid, early triage models of care are in place.9 Additionally assessment also needs to determine whether there are any contraindications for thrombolysis.
The assessment method used depends on the condition of the patient but should occur within 10 minutes of arrival.7 This initial history will focus on the nature of symptoms such as pain. Pain assessment is complex, and the use of an acronym such as PQRST (see Table 10.1) is useful to incorporate precipitating and palliative factors, qualitative descriptors, location, radiation and length of time. A pain scale is included to help rate the intensity of pain. Asking patients for descriptive words is useful in assessment as many patients will deny pain and instead use words such as pressure, tightness or constriction. It is essential not to ignore other presentations, as patients with atypical symptoms, such as women, often have a delayed diagnosis and treatment and a higher mortality (50%) than with typical symptoms (18%).7 Differentiating this pain from any previous pain is also useful. The brief history should include a short cardiovascular risk profile: (a) previous cardiac history such as angina, MI, revascularisation; and (b) family history, smoking, hypertension, diabetes.
| P | Precipitating | Exercise and activity Stress and anxiety Cold weather |
| Palliating | Stop activity Rest Nitroglycerin |
|
| Q | Quality | Heavy, tight, choking, vice-like, constricting |
| R | Region, Radiation | Left side of chest, shoulder, arm and jaw Retrosternal and radiating to the neck |
| S | Severity | Rate pain on scale of 1 (no pain) to 10 (worst pain possible) |
| T | Time | Pain lasts longer than 10 minutes despite nitroglycerin Pain comes and goes but lasts longer than 20 minutes |
Hudak CM, Gallo BM, Morton PG. Critical care nursing, A holistic approach. (7th Ed) Philadelphia: Lippincott 1998.
Practice tip
Because of changes in neuroreceptors, older patients and diabetic patients may not describe the typical anginal pain. Women also may not describe classic angina symptoms and may use different descriptors from men.4 Be alert for prodromal symptoms, such as increased shortness of breath, weakness and fainting.
Electrocardiographic examination
Patients with chest discomfort should be assessed by an appropriately qualified person and have an ECG recorded within 5 minutes of arrival at a healthcare facility to determine the presence and extent of myocardial ischaemia, the risk of adverse events and to provide a baseline for subsequent changes.7 Most importantly, the ECG is essential to determine whether emergency reperfusion is required, and is recommended as the sole test for selecting patients for PCI or thrombolysis. Where ST segment monitoring is available, this should be continuous. Alternatively, if chest discomfort persists, ECGs should be repeated every 15 minutes. Even when chest pain resolves it is important to record a series of 12-lead ECGs during admission to determine changes over time. (The normal ECG is covered in Chapter 9, whereas this section addresses ischaemic changes in the ECG.)
Myocardial ischaemia, injury or infarction cause cellular alterations and affect depolarisation and repolarisation.10 Myocardial ischaemia may be a transient finding on the ECG. Ischaemia results in T wave inversion or ST segment depression in the leads facing the ischaemic area.11 Ischaemic T waves are usually symmetrical, narrower and more pointed. ST segment depression of 1 mm for 0.08 seconds is indicative of ischaemia, especially when forming a sharp angle with an upright T wave.12 These changes are reversible with reduction in demand (e.g. by rest, nitrates).
On acute presentation, myocardial injury (infarction) is most commonly associated with ST segment elevation on the ECG, although this is not universal. In addition, a typical pattern of ECG changes over time (evolution of the ST segments, Q wave development and T wave inversion) are often seen (described below), but these changes too are not universal. The distinction between the various acute coronary syndromes, including ST elevation acute coronary syndrome (STEACS), ST elevation myocardial infarction (STEMI) and non-ST elevation myocardial infarction (non-STEMI), is important for ensuring appropriate assessment and protocol-based treatment13 for the various presentations.
• anteroseptal wall of left ventricle, V1–V4;
• anterior wall of the left ventricle, V1-V6, I and aVL;
Additional leads are needed to view the right ventricle and posterior wall. Chest electrodes can be placed on the right chest wall using the same landmarks as the left chest to view the right ventricle (see Chapter 9). Further electrodes, V7–V9, may be placed over the posterior of the left chest to view the posterior wall. Other indicative signs of posterior wall damage are a small r wave in V1 and/or ST depression in V3 and V4 as these may be reciprocal changes. The endocardial surface of the posterior wall faces the praecordial leads of the ECG so the signs of ischaemia and infarction are reversed or reciprocal such as ST depraession or a small r wave. If these signs are present a left-sided ECG, V7–V9, should be done to confirm or rule out a posterior infarction.
Typical ECG evolution pattern
The initial ECG features of myocardial infarction are ST segment elevation with tall T-waves recorded in leads overlying the area of damaged myocardium. These changes gradually change, or evolve, over time, with ST segments returning to baseline (within hours), while Q waves develop (hours to days) and T waves become inverted (days to weeks). The time course for the evolutionary changes is accelerated by reperfusion, e.g. PCI, thrombolysis or surgery. Thus an almost fully-evolved pattern may be seen within hours if successful reperfusion has been undertaken (see Figures 10.2–10.4 for an example). Given the expected time course for evolution, it is possible to approximate how recently infarction has occurred, which is essential in determining management:
• acute (or hyperacute): there is ST elevation but Q waves or T inversion have not yet developed (see Figure 10.5).
• recent: Q waves have developed. ST segment elevation may still be present. Evolution is underway. The infarction is more than 24 hours old.
• old (fully evolved): Q waves and T inversion are present. ST segments are no longer elevated. Infarction occurred anything from a few days to years ago.
Biochemical markers
Intracellular cardiac enzymes enter the blood as ischaemic cells die, and elevated levels are used to confirm myocardial infarction and estimate the extent of cell death. The cardiac troponins T and I (cTnT and cTnI) have been found to be both sensitive and specific measures of cardiac muscle damage.14 Troponin I is rapidly released into the bloodstream, so it is especially useful for the diagnosis and subsequent risk stratification of patients presenting with chest pain in the early stages. Troponin I is also a more appropriate marker to use in postoperative and trauma patients than creatine kinase–MB (CK-MB), as CK-MB levels will be affected by muscle damage. However, CK-MB is less costly and more readily available, and so is still often used, particularly in the presence of a non-diagnostic ECG. C-reactive protein assays may prove to be useful, as baseline and discharge levels are predictive of subsequent cardiac events. However, the laboratory facilities are not readily available.
Coronary angiography and left heart catheterisation
Coronary angiography gives a detailed record of coronary artery anatomy and pathophysiology. Specially designed catheters are advanced with the assistance of a guidewire into the ascending aorta via the femoral or brachial arteries and manoeuvred into the ostium of each coronary artery. Contrast media is then injected and images are taken from several views to provide detailed information on the extent, site and severity of coronary artery lesions and the blood flow into each artery. This flow is graded using the Thrombolysis in Myocardial Infarction (TIMI) studies system (see Table 10.2).15 Typically, a left ventricular angiogram is performed during the same procedure to assess the appearance and function of the left ventricle, mitral and aortic valves. If CHD is present, treatment is determined as appropriate according to the severity (PCI, coronary artery bypass grafting or medical therapy). The nursing care for coronary angiography is similar to PCI, and is covered under that section.
TABLE 10.2 Thrombolysis in Myocardial Infarction (TIMI) flow grades in coronary arteries15
| TIMI 0 | No perfusion and no antegrade flow beyond the occlusion. |
| TIMI 1 | Penetration with minimal perfusion, and contrast does not opacify the entire bed distal to the stenosis during the picture run. |
| TIMI 2 | Partial perfusion and contrast opacifies the entire coronary bed distal to the stenosis, although entry to this area is slower than with unaffected coronary beds. |
| TIMI 3 | Complete perfusion and filling and clearance of contrast is rapid and comparable to other coronary beds. |
Exercise test
Exercise testing with ECG monitoring forms part of the diagnostic screen for patients suspected of stable angina. The Bruce protocol is used most often and considered positive for CHD if there is 1 mm or more of reversible ST segment depression.16 False-positive tests are more common in populations with a lower incidence of CHD, including women.17
Collaborative Management of Angina and Acute Coronary Syndrome
The management of stable angina patients is aimed at: (a) secondary prevention of cardiac events; (b) symptom control with medication; (c) revascularisation; and (d) rehabilitation (see Figure 10.6). (Revascularisation by coronary artery bypass graft is reviewed in Chapter 12; revascularisation by percutaneous coronary angioplasty is reviewed in the next section.)
Treatment of acute coronary syndrome aims at rapid diagnosis and prompt re-establishment of flow through the occluded artery to ensure myocardial perfusion and reduce size of infarction. In addition, treatment aims to:18
• minimise the area of myocardial ischaemia by increasing coronary perfusion and decreasing myocardial workload
• maximise oxygen delivery to tissues
• control pain and sympathetic stimulation
• counter detrimental effects of reperfusion
The ideal place to manage ACS or MI patients is in the coronary care unit, where continuous, specialised nursing care is available and there is rapid access to treatments.19 Secondary prevention of cardiac events includes the provision of medications, such as antiplatelet therapy and lipid-lowering therapy.
Reperfusion therapy
Thrombolytic therapy
Thrombolytic therapy has been demonstrated to show a significant reduction in mortality in the high-risk group described above.20 The greatest reduction in mortality occurs if the reperfusion occurs within the first ‘golden’ hour of presentation.20 Thrombolysis can be delivered effectively in many settings where other methods of reperfusion are not available.
Streptokinase and tenecteplase are the most commonly prescribed thrombolytic agents. Streptokinase is prepared from beta-haemolytic streptococci and is a potent plasminogen activator.21 Streptokinase is not thrombus-specific, so plasmin is released into the general circulation that may break down any recent clot formed as a result of surgery, injection or healing, leading to a potential increase in haemorrhagic episodes. Streptokinase is bacterial in origin, so it is antigenic. Most individuals have been exposed to beta-haemolytic streptococci so antibodies are often present, which means a higher dose may be required owing to the destruction of some of the enzyme when administered. Occasionally an escalated allergic response will occur and will need urgent treatment. This is more likely if streptokinase has been administered in the previous 6 months. Streptokinase is given intravenously over 60–90 minutes, because it has a short half-life.
The drug tissue-type plasminogen activator (tPA) is available as alteplase, tenecteplase and reteplase. These agents are of human origin, made by recombinant DNA techniques.22 The drug activates only plasminogen present in blood clots, so the risk of haemorrhage is decreased. Unlike streptokinase, tPA can be given repeatedly without risk of anaphylactic reaction. However, tPA costs about 10 times as much as streptokinase, so it is occasionally still reserved for patients who have recently received streptokinase or are at risk of allergic reaction. Often patients with anterior ischaemic changes are treated with tPA (alteplase) based on the GUSTO-1 trial that showed improved outcomes in terms of reduction of ischaemia.23 Alteplase is usually given by infusion, whereas reteplase, which has a longer half-life, can be given in two bolus injections.
• Observations. Assess neurological state including orientation, any IV sites and urinalysis for the presence of bleeding. Along with vital signs, these are attended every 15 minutes for the first hour, half-hourly for an hour, and then hourly according to the patient’s condition, however, patients are advised to report any bleeding postdischarge as well.
• ECG monitoring. This includes 12-lead ECG on return and ongoing ECG monitoring and chest pain assessment to detect reocclusion. Patients need to be requested to inform nursing staff of any chest pain or discomfort.
• IV anticoagulants such as heparin and/or oral antiplatelet drugs, such as clopidogrel or ticlopidine, may be given following thrombolysis to prevent reocclusion in the stent. Assess International Normalised Ratio (INR), prothrombin (PT) and partial thromboplastin time (PTT), as bleeding is more likely to occur if anticoagulants are above the therapeutic range.
Coronary angioplasty
Coronary angioplasty (PTCA) procedures are being used about twice as frequently as coronary artery bypass graft surgery, with 155 PTCA procedures performed for every 100,000 population in Australia in 2008–09.2 PTCA rates have grown dramatically in patients aged over 75 years. In this procedure, a catheter is introduced by the brachial or femoral artery into the coronary arteries and advanced into the area of occlusion or stenosis under the guidance of imagery and specifically designed catheters. A balloon attached to the end of the catheter is then inflated to widen the lumen of the artery by stretching the vessel wall, rupturing the atheromatous plaque and cracking the intima and media of the artery (see Figure 10.7).
PTCA tends to be reserved for patients with single- or double-vessel disease as assessed on coronary artery angiograms. Angioplasty provides better symptom relief than medication alone, but there is no evidence of survival benefits.24 Primary angioplasty results in a higher rate of patency of the affected artery in AMI (>90%), lower rates of CVA and reinfarction and higher short-term survival than thrombolysis.25 PTCA is recommended in all patients presenting with chest pain who meet the indications for reperfusion when: (a) facilities are available and can be achieved within 60 minutes; (b) there are contraindications to fibrinolytic therapy described above; (c) ischaemia would result in large anterior AMI within 4 hours; or (d) haemodynamic instability or cardiogenic shock are present.
A stent is usually inserted to prevent abrupt closure and maintain patency for longer.26 The structure of the stent within the vessel enlarges the lumen and prevents vessel stricture. Restenosis due to intimal hyperplasia is a relatively common complication, occurring 10–12 weeks postimplantation. In response to this problem, drug-eluting stents have been developed. The drug coatings include sirolimus, a macrolide antibiotic that has been demonstrated to effectively decrease hyperplasia and prevent reduction of flow.27 Paclitaxel has also shown promise in a series of studies.28 In addition to dactinomycin, these drugs are undergoing approval processes.
Nursing management of patients post-PTCA includes care of the puncture site to prevent bleeding and detect arterial changes (including clot and aneurysm).29 The process used to create and maintain access for insertion of the catheters can damage the blood vessel(s) and alter perfusion to the limb. The sheath used to aid insertion and maintain access is usually maintained for 1–2 hours post-procedure for emergency access. Care is as follows:
• Observations. Observe access site for haemorrhage and haematoma, assess perfusion to the lower limb, including colour, warmth and pulses. This monitoring needs to be done often in the first few hours, when complications are most likely to occur.
• ECG monitoring. This includes 12-lead ECG on return and ongoing ECG monitoring and chest pain assessment to detect reocclusion. Patients need to be requested to inform nursing staff of any chest pain or discomfort.
• Vital signs. These are recorded every 15 minutes for the first hour, half-hourly for one hour, and then hourly according to the patient’s condition.
• Removal of sheath. This is usually performed by medical or specially trained nursing staff.
• Achievement of haemostasis. Use either application of pressure for at least 5 minutes or vascular sealing.29
• Assess International Normalised Ratio (INR), prothrombin (PT) and partial thromboplastin time (PTT), as bleeding is more likely to occur if anticoagulants are above the therapeutic range. Weight-adjusted heparin (100 units/kg) is usually used during PTCA to prevent thrombus formation, and glycoprotein IIb/IIIa inhibitors such as abciximab may be used to prevent platelet aggregation and thrombus formation for patients at high risk of occlusion.
• Bedrest (2–6 hours) is used to discourage the patient from moving the joint of the insertion site to prevent clot displacement and haematoma formation. Initially the patient should lie relatively flat if femoral artery access has been used, then progress to sitting. The period of rest has been demonstrated to be safely reduced to 1 hour in low-risk patients (normotensive and normal platelet count).29
• Pain relief is used primarily to promote comfort for patients who find bedrest to cause pain and discomfort.
• Urine output. Adequate urine output is essential as radiographic IV contrast is cleared by the kidneys, so it is vital that nurses ensure good hydration and monitor initial urine output.
• Oral antiplatelet drugs, such as clopidogrel or ticlopidine, may be given prior to the procedure to prevent later reocclusion in the stent. Usually patients will be discharged on this medication to continue for up to 3 months while endothelium lines the stent/injured area. Unless contraindicated, all patients will take aspirin for the rest of their lives.30,31
Many patients find the PTCA procedure and confirmation of CHD diagnosis stressful.32 It is an important nursing role to provide patients with preparatory information about the procedure and care required during recovery. As family members provide valuable support and reminders about recovery, these people should be included in any information sessions. The patient and family need to be provided with information about the possibility of restenosis, mobility restrictions at home and the lifestyle changes needed to reduce the risk of worsening CHD.
Nursing management of ACS and MI
Symptom relief should be provided, including analgesia for pain. Analgesia management should be conducted by nurses because of their continued contact and thus more accurate assessment and treatment of pain.18 It is essential to treat pain, not only for the distress it causes patients but also because pain causes stimulation of the sympathetic nervous system (SNS). SNS responses include elevated heart rate and potential for arrhythmias, peripheral vasoconstriction and increased myocardial contractility and, therefore, an overall increase in myocardial oxygen demand. Effective treatments for pain include IV morphine and nitrates. The IV route is preferable, as absorption is predictable and additional punctures in thrombolysed patients are not required. Morphine has the additional benefit of reducing anxiety in a distressing situation and should be initially provided at a dose of 2.5–5 mg at 1 mg/min, followed by 2.5 mg doses as indicated. While there is little randomised controlled trial evidence to support this particular practice, it is generally accepted to be appropriate. A standardised method of pain evaluation and charting should be used to ensure consistent assessment and treatment. An antiemetic such as metoclopramide should be given concurrently to lessen and prevent nausea. Other drugs, such as beta-blockers and nitrates, decrease myocardial workload, contributing to pain reduction.
Nursing care for thrombolysis
Patients receiving thrombolytics require constant observation, regular non-invasive blood pressure measurement for hypotension, and monitoring for allergic reactions to streptokinase. Continuous ECG monitoring for arrhythmias and ST segment changes is essential. Some arrhythmias, particularly idioventricular arrhythmias, are associated with reperfusion and tend to be benign. ST segment monitoring and assessment of pain help evaluate the effectiveness of the thrombolysis. Thrombolysis is considered to have failed if the patient is still in pain and the ST segment has not resolved within 60–90 minutes.18 If thrombolysis fails, patients are at high risk for other interventions, so repeat thrombolysis is often the only treatment option. Salvage or rescue angioplasty may be undertaken if available at the site.
Medications
Provision of medications and assessment of the effectiveness of treatment is a major component of the nurse’s role in caring for the cardiac patient. Many of the medications are accompanied by side effects and interactions with other drugs, which the nurse must monitor. An array of medications is used to treat AMI patients, including aspirin, lipid-lowering agents, beta-blockers and organic nitrates (see Table 10.3).
Symptom control
Control of anginal symptoms with medication usually includes sublingual glyceryl trinitrate (GTN) for immediate symptom control and one or more antianginal medications for sustained symptom management.18 Beta-blockers are usually commenced unless contraindicated. Calcium channel blockers may be used in patients who do not have cardiac failure or heart block. (These medications are described in the next section.) The choice of medication may depend on how acceptable the patient finds the reduction in symptoms and the presence of side effects. Patients need to take antianginal agents continuously, regardless of symptoms. Patients should also be encouraged to take sublingual GTN prophylactically.
Angina may also be managed by avoiding situations that trigger angina. Education needs to be directed at awareness of symptoms and management of unstable angina and AMI symptoms, and the need for emergency care. Although these patients are at low risk of further cardiovascular events in the short term, in the medium to long term, risk may accumulate. Patients with angina are encouraged to attend cardiac rehabilitation programs to learn how to deal with symptom management.41
Angiotensin-converting enzyme (ACE) inhibitors have been recommended for all post-AMI patients while in hospital, with review of prescription at 4–6 weeks postdischarge. Patients with left ventricular failure should be maintained on ACE inhibitors. Similarly, diuretics provide the mainstay of the management of left ventricular failure if it is present (see Chapter 19). Diabetic patients have a higher mortality after AMI in both acute and long-term phases. Provision of an insulin-glucose infusion for BSL >11 mmol/L during the acute phase, followed by subcutaneous injections for at least 3 months, has been demonstrated to significantly reduce mortality up to 3 years post-AMI.42
Transfer to a step-down unit or general ward usually occurs when the patient is pain-free and is haemodynamically stable. Stability means that patients are not dependent on IV inotropic or vasoactive support and have no arrhythmias. Discharge home after AMI varies, but usually occurs at day 3 for low-risk patients.18
Independent Practice
Emotional responses and patient and family support
ACS or AMI is usually accompanied by feelings of acute anxiety and fear, as most patients are aware of the significant threat posed to their health.18 For many patients it may also be the first experience of acute illness and associated aspects such as ambulance transport, emergency care and hospitalisation, so they may experience shock and disbelief as well. Fast-track processes require patients and their families to process a large amount of information and make decisions quickly, and this, added to an alien environment, full of unfamiliar technology and personnel, can be quite distressing. However, the environment can also promote a feeling of security for patients and their families. Patients’ perceptions of the CCU environment have been linked to recovery.43
Anxiety is a common response to the stress of an acute cardiac event and leads to important physiological and psychological changes.44 The sympathetic nervous system is stimulated, resulting in increased heart rate, respiration and blood pressure. These responses increase the workload of the heart and therefore myocardial oxygen demand. In an acute cardiac event, these demands occur when perfusion is already poor and may lead to worse outcomes, including ventricular arrhythmias and increased myocardial ischaemia. Therefore, staff working in emergency and coronary care should employ strategies to reduce a patient’s anxiety.
Increasing a patient’s sense of control, calm and confidence in care reduces the patient’s sense of vulnerability, whether it is realistic or not.44 This can be achieved by:
• providing order and predictability in routines, allowing the patient to make choices, providing information and explanations, and including the patient in decision making
• using a calm, confident approach
• communicating with patients and families, while reducing conversation demands as excessive conversation by patients may unnecessarily raise heart rate45
• restricting the number and type of visitors in the acute phase is customary, but many patients feel safer if a family member is present
• provision of comprehensive information to families, with more concise information in understandable language for patients.
Cardiac rehabilitation
Coronary heart disease is a chronic disease process, which often presents with acute events such as ACS or AMI. Like all chronic illnesses, it has implications for patients in terms of lifestyle change, uncertainty of long-term outcomes, functional changes and social and economic alterations. Cardiac rehabilitation aims to address these issues. The World Health Organization describes cardiac rehabilitation as ‘the sum of activities required to influence favourably the underlying cause of the disease, as well as to ensure the patients the best possible physical, mental and social conditions so that they may, by their own efforts, preserve, or resume when lost, as normal a place as possible in the life of the community’.46Systematic, individualised rehabilitation and secondary prevention need to be offered to all AMI patients. Participation in well-structured, multidisciplinary programs has been demonstrated to reduce mortality by up to 30%.47 Additional benefits have been shown for improvements in exercise tolerance, symptoms, serum lipids, psychological wellbeing and cessation of smoking.48–50
Cardiac rehabilitation is structured around four phases, beginning with phase I, during admission.50 The components of phase I include:
• information regarding the disease process, the prognosis, and an optimal approach to recovery, early mobilisation and discharge planning
• assessment of patients’ understanding of their diagnosis and treatment as a foundation for self-management
• discharge planning which incorporates discussions on adaptation to the functional and lifestyle changes needed for secondary prevention – dietary intake of lipids, exercise, smoking cessation, stress management and symptom monitoring, and management of acute symptoms
• early mobilisation as an inpatient to encourage a positive approach to recovery with monitoring of the response to activity in heart rate, shortness of breath and chest pain to determine the rate of progress. (Most hospital units use an activity progress chart for this purpose based on metabolic equivalents [METs]).
The phases that follow, from II to IV, are managed in the outpatient setting and begin with assessment, liaison with multidisciplinary professionals and health education. Phase II occurs in the immediate postdischarge period and includes liaison with community-based carers and services and further assessment. In phase III, tailored, supervised exercise programs are usually conducted and there is a range of psychosocial interventions, such as support sessions and stress management. Finally, in phase IV the focus is on chronic disease management and maintaining risk modification behaviours. All phases require incorporation of the principles of adult learning to maximise learning and behaviour change. These principles include recognition of ‘readiness to learn’.50 Adults are ready to learn most effectively when they are physically and emotionally stable and are aware of the problem or need to learn. Nurses, because of their expertise and continual presence, are best placed to assess and provide education at optimal times.
Complications of Myocardial Infarction
Cardiogenic shock
Cardiogenic shock occurs as a complication of MI in about 5–10% of patients and is the most common cause of death in hospitals.50 It arises from loss of contractile force, and generally occurs when ventricular damage is more than 40% and ejection fraction less than 35%. Cardiogenic shock and the related management are described in more detail in Chapter 12.
Arrhythmias
Arrhythmias often occur in ACS and AMI and are often the cause of death in the prehospital phase. Management of the prehospital phase centres on community education and an effective, rapidly responsive ambulance service, as exemplified in Seattle in the USA.51 Arrhythmias may be generated by poorly perfused tissue and electrolyte alterations, and increased sympathetic tone during infarction, but are more often due to a failing left ventricle. They may also complicate reperfusion after successful revascularisation.52 It is essential to rapidly and effectively treat arrhythmias in the ACS and AMI context. The goal of treatment is to maintain cardiac output while reducing workload. Arrhythmias and management are described in Chapter 11.
Pericarditis
Pericarditis is an inflammation of the visceral and parietal layers of the pericardium that cover the heart. This inflammation occurs in approximately 20% of AMI patients within the following 2–3 days.10 The patient experiences chest pain, which may be confused with ischaemic pain. This confusion with an ischaemic event may be compounded by the additional presence of ST segment elevation on the ECG. However, pericardial pain increases with deep inspiration and a pericardial rub is often present. Electrocardiographically, the elevated ST segments of pericarditis are typically concave upwards (saddle-shaped) and often widespread, contrasting with convex ST segment elevation limited to the distribution of a single coronary artery in infarction.53 Pericarditis normally responds to anti-inflammatory treatment by aspirin, indomethacin and/or corticosteroids. Approximately 1–5% of AMI patients develop pericarditis as a late complication, 2 weeks to a few months post-AMI.18 Usually this late-onset pericarditis is associated with Dressler’s syndrome and may be an autoimmune response to myocardial injury. This is a chronic condition requiring systemic corticosteroid treatment.
Structural defects
Papillary muscle rupture most often occurs 2–7 days after MI. Patients experience a sudden onset of pulmonary oedema secondary to pulmonary hypertension and cardiogenic shock. Additional heart sounds and a systolic murmur will be heard. Urgent surgery is required, as the mortality rate for papillary muscle rupture is 95%.54 Cardiac rupture most often occurs within 5 days of MI and is commonest in older women. The patient experiences continuous chest pain, dyspnoea and hypotension as tamponade develops. Symptoms may worsen rapidly and result in pulseless electrical activity (PEA) unless surgery is undertaken immediately.
Heart Failure
In normal circumstances, the heart is a very effective, efficient pump with reserve mechanisms available to allow output to meet changing demands. These mechanisms include (a) increasing heart rate to increase total cardiac output, (b) dilation to create muscle stretch and more effective contraction, (c) hypertrophy of myocytes over time to generate more force, and (d) increasing stroke volume by increasing venous return and increased contractility. Heart failure is a complex clinical condition that is characterised by an underlying structural abnormality or dysfunction that results in the inability of the ventricle to fill with or eject blood.55 The condition is also known as congestive cardiac failure, a term commonly used in the USA but not in Australia. Chronic heart failure (CHF) describes the long-term inability of the heart to meet metabolic demands.
The burden of disease associated with heart failure is on the rise due to our ageing population, the prevalence of coronary heart disease and hypertension, the decrease in fatality from acute coronary syndrome and improved methods of diagnosis.55 Survival rates and prognosis for heart failure patients are extremely poor. Approximately 50% of patients diagnosed with heart failure will die within five years of diagnosis.56–58 When compared with those patients with cancer, heart failure patients have the poorest five-year survival rate, with the exception of lung cancer.59 In Australia during 2001–2002, 41,874 patients were hospitalised with a primary diagnosis of CHF (0.7% of all hospitalisations).60 Internationally, heart failure is the most common cause of hospitalisation in patients aged over 70 years.55 Approximately 40% of patients admitted to hospital with heart failure will be readmitted or die within one year.61
Over 50% of patients newly diagnosed with heart failure have concurrent ischaemic heart disease, hypertension is present in 65% and idiopathic dilated cardiomyopathy (5–10% of cases).55
[/not-level-membership-for-critical-care-medicine-category]
