Chapter 11 Cardiovascular adaptations to chronic exercise
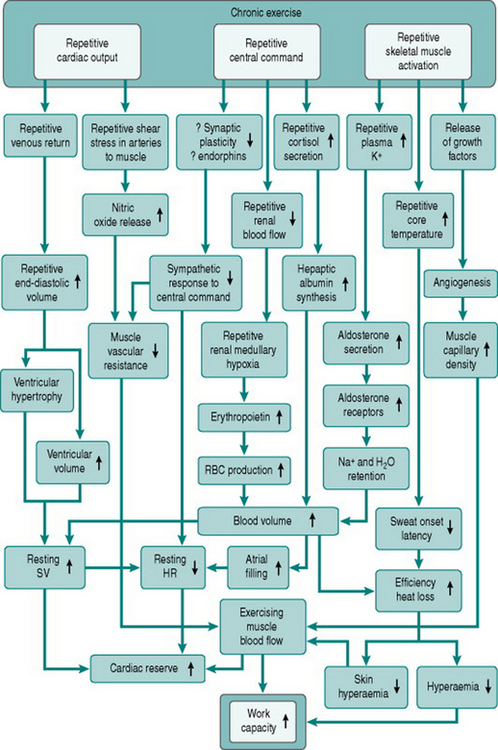
From the issues discussed in previous chapters it will be clear that one’s capacity to elevate cardiac output could be improved by increasing blood volume, by reducing resting heart rate (that is, increasing heart rate reserve) or by increasing stroke volume. It should also be remembered that cardiovascular capacity to service muscle metabolism is normally limited by the simultaneous thermoregulatory demands. Therefore, exercise capacity will be increased by minimizing hyperthermia and the associated encroachments on muscle blood flow and plasma volume that result from cutaneous vasodilation and sweating.
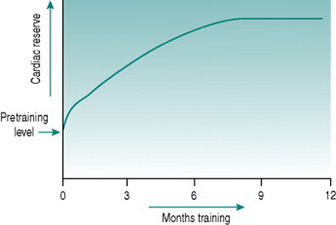
All of these adaptations occur, but they have very different timecourses. An initial increase in work capacity linked to increased cardiac reserve occurs over a matter of several weeks’ training, but the achievable cardiac output continues to rise over the succeeding months so that with any given training schedule, peak performance, as determined by cardiovascular efficiency, will not be reached until after around 9–12 months (Fig. 11.1).
BLOOD-VOLUME ADAPTATIONS
Red cell volume
The changes in plasma volume described above produce an initial fall in haematocrit, but with continued training there is, over the ensuing several weeks, increased erythropoiesis that restores the concentration of red cells to its previous level. The likely stimulus for increased red cell production is the recurrent renal medullary hypoxia associated with renal sympathetic activation, which results in release of erythropoietin from medullary tubule cells.
Collectively, the changes in blood volume constitute the major cause of the rise in cardiac reserve that is seen over the first 3–4 weeks of training (Fig. 11.1). As we shall see below, increased heart rate reserve also has a role during this period.
CARDIAC ADAPTATIONS
Heart rate reserve
Resting heart rate often begins to fall after the first few sessions of regular dynamic exercise and may be around 10 beats/min lower than the pretraining value after 2 weeks of training (Murray et al 2006). This confers an immediate advantage on exercise capacity, since it increases heart rate reserve. A 20 year old with a resting heart rate of 70 beats/min can theoretically increase cardiac output by (200–70/70) or 1.85-fold by tachycardia alone: with a resting heart rate of 60 beats/min this increase becomes (200–60/60) or 2.33-fold. Thus, in order to undertake any given submaximal workload, the trained individual needs only to increase heart rate (and, therefore, cardiac workload) by (1.85/2.33) or 80% of the amount needed before training.
In individuals who train intensively for prolonged periods, much greater degrees of bradycardia develop due to increased vagal tone, such that resting heart rate may be as low as 35 beats/min. This obviously confers a far larger cardiac reserve, with the 20-year-old athlete being theoretically able to increase his cardiac output by (200–35/35) or 4.7-fold using tachycardia alone, and reducing his cardiac workload for a given submaximal work intensity to (1.85/4.7) or 40% of that in an untrained age-matched individual. This more dramatic bradycardic effect of prolonged training is secondary to structural adaptation of the heart (see Cardiac hypertrophy, below).
During incremental dynamic exercise, it has been found that many trained athletes do not produce linear increases in heart rate up to their age-limited maximum. Instead, the slope of the heart rate/work curve flattens at around 85%  (Lepretre et al 2005). Since this workload corresponds approximately to the anaerobic threshold, the heart rate deflection point has been adopted in a number of centres as an easy, non-invasive monitor for setting training workloads. It has also been suggested that the deflection infers some advantage on athletes by allowing greater utilization of their capacity to increase cardiac output by increasing stroke volume, although no firm evidence base for this exists. Regardless of whether or not the phenomenon has a value, the mechanisms that underlie it are unknown, and its usefulness as a training aid is limited by its variable occurrence even in trained athletes.
(Lepretre et al 2005). Since this workload corresponds approximately to the anaerobic threshold, the heart rate deflection point has been adopted in a number of centres as an easy, non-invasive monitor for setting training workloads. It has also been suggested that the deflection infers some advantage on athletes by allowing greater utilization of their capacity to increase cardiac output by increasing stroke volume, although no firm evidence base for this exists. Regardless of whether or not the phenomenon has a value, the mechanisms that underlie it are unknown, and its usefulness as a training aid is limited by its variable occurrence even in trained athletes.
Cardiac hypertrophy
Like skeletal muscle, the myocardium responds to chronically increased workload by muscle cell growth, resulting in what is traditionally known as the ‘athlete’s heart’ (Iglesias Cubero et al 2000). As in skeletal muscle, the pattern of growth varies depending on whether the increased work is dynamic or resistive. Increased ventricular filling (increased preload), with no change in outflow resistance, results in moderately thickened muscle around an enlarged ventricular chamber (eccentric hypertrophy). By contrast, increased resistance to outflow (increased afterload) due, for instance, to increased peripheral resistance, leads to a markedly thickened ventricular wall with no alteration of chamber size (concentric hypertrophy) (Fig. 11.2).
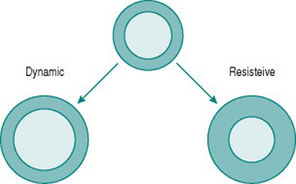
Chronic dynamic exercise exerts both of these effects on the heart; the increased preload, because of increased venous return, and the increased afterload, because of the pressor response to central command. Not surprisingly, therefore, the initial structural adaptations to dynamic training involve elements of both types of hypertrophy. In the right heart, little or no afterload increase takes place because the pulmonary vasculature is exempted from sympathetic vasoconstrictor influences and, in fact, pulmonary vascular resistance falls during exercise due to better ventilation/perfusion matching (see Chapter 8, p. 95). The right ventricle, therefore, shows uncomplicated eccentric hypertrophy. On the left side, however, there are increases in both preload and afterload, which induces some degree of concentric hypertrophy and limits the expansion of left ventricular chamber size until after around 6–9 months training. Thus, the full benefits of cardiac adaptation, in terms of increased stroke volume, are not evident until after this period.
VASCULAR ADAPTATIONS
The resting bradycardia associated with training is accompanied by reduced total peripheral resistance. In studies that have tracked previously sedentary subjects through a training programme, the fall in peripheral resistance has been seen to cause a fall in mean resting blood pressure of up to around 10 mmHg (Murray et al 2006). There is, however, considerable variation between individuals in the magnitude of the effect and the available data suggest that little or no further effect on pressure occurs in highly trained athletes than is seen with moderately improved fitness. The fall in peripheral resistance that follows training may involve several mechanisms.
Sympathetic drive
In view of the apparent reduction in sympathetic vasomotor tone following acute exercise (see Chapter 7, p. 79) and the likely baroreflex effects of expanded blood volume, it might be expected that resting sympathetic drive would be reduced following training. In fact, microneurographic studies of action potential frequency in sympathetic nerve filaments provide no support for this, although for technical reasons these studies use muscle nerves and so give no information on the behaviour of, for example, splanchnic vasomotor control (Alvarez et al 2005).
There is, by contrast, good evidence that training reduces pressor responses to stimuli that cause sympathetic activation. Trained individuals have been found to exhibit less blood pressure rise in response to some laboratory stressors such as mental arithmetic and also to have less pronounced pressor responses to given increments of exercise. These changes may be due to reduced gain in the hindbrain responses to central command and could involve endorphin release (see Chapter 7, p. 85). Desensitization of peripheral metaboreceptors and limb mechanoreceptors may also be involved. In terms of training-mediated effects on performance, damping of sympathetic drive may be beneficial in allowing greater increments of muscle blood flow in response to local dilator factors during muscle activation (see Chapter 6, p. 72).
Endothelial function
Training is associated with increased endothelium-dependent dilator capacity, as indicated by the magnitudes of responses to reactive hyperaemia (see Chapter 6, p. 66) and by resting plasma levels of nitric oxide (NO). One possible mechanism is that the increased endothelial turnover caused by repetitive exercise-induced shear stress results in a systemic endothelial lining with a younger mean cell age that, therefore, has greater secretory capacity. There is evidence that endothelial function is affected preferentially in the vascular beds of those muscles that have been repetitively exercised (Thijssen et al 2005), so the extent of peripheral resistance reduction is likely to correlate with the proportion of the whole-body musculature that is used. This is an issue that needs to be considered when designing training programmes for patients with limited mobility.
Angiogenesis
With prolonged dynamic training, local release of vascular endothelial growth factor and other growth factors leads to sprouting of new capillaries and arterioles in the active skeletal muscles and the myocardium. Appearance of these new vessels necessarily lowers regional vascular resistance, as well as reducing the distance for diffusion of nutrients and metabolites between blood stream and cells. Much less effect on vascular resistance is seen with static or intense resistive training, since the capillary density in type IIb glycolytic muscle is considerably sparser than in oxidative muscles.
THERMOREGULATORY ADAPTATIONS
This is a further rapid thermoregulatory adaptation to result from the increased aldosterone secretion that follows exercise-induced hyperkalaemia (see above, p. 130). As in the renal tubules, aldosterone acts on sweat ducts to reabsorb sodium. In the presence of greater numbers of aldosterone receptors, therefore, the sodium content of sweat falls dramatically, from around 50 mmol/L in a sedentary exercising subject to around 5 mmol/L in a trained subject. Thus, although total water loss in sweat may rise in the trained individual, due to the greater amount of muscle work undertaken, plasma and extracellular fluid osmolality are maintained far more efficiently.
Figure 11.3 summarizes the adaptive responses to dynamic training, as discussed above.
GENDER DIFFERENCES
Closely similar effects of training are seen in men and women with respect to fitness, although the absolute effect of training is limited in women by their smaller heart size, in a similar way to their lower limit to acute exercise. Some investigators have reported that women are less able than men to produce cardiac hypertrophy in response to increased preload and have suggested that peripheral vascular adaptations may play a greater part than in men. However, these conclusions may have been distorted by the smaller initial heart size in women: the consensus of opinion now is that equivalent cardiac changes proportionate to heart mass occur in both sexes (Whyte et al 2004).
 of short-term (of the order of weeks) training is, therefore, completely lost again within a matter of weeks, a fact that needs to be considered in relation to the potential long-term benefits of cardiac rehabilitation programmes.
of short-term (of the order of weeks) training is, therefore, completely lost again within a matter of weeks, a fact that needs to be considered in relation to the potential long-term benefits of cardiac rehabilitation programmes.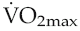 falls more gradually, as the reversals of muscle angiogenesis and cardiac hypertrophy take place over the order of months. Notwithstanding, there is some evidence that, in athletes who have trained for many years, some cardiac hypertrophy may persist permanently.
falls more gradually, as the reversals of muscle angiogenesis and cardiac hypertrophy take place over the order of months. Notwithstanding, there is some evidence that, in athletes who have trained for many years, some cardiac hypertrophy may persist permanently.ROLE OF EXERCISE IN CARDIOVASCULAR THERAPY
Common sites of atherosclerotic damage
The tissues most sensitive to this local deprivation of flow are those with the greatest continuous need for oxygen – the myocardium and the brain. So the characteristic results of atheromatous thrombus formation are myocardial infarctions or heart attacks, and cerebral infarctions or strokes. The effect of less severe restriction of blood flow due to atherosclerotic luminal narrowing is most usually seen as chest pain when acidic metabolites accumulate in an area of the myocardium where coronary perfusion is reduced, causing stimulation of chemosensitive nerve endings. This type of pain caused by inadequate local blood flow is known as angina.
Hypertension predisposes to atherosclerosis
The increased risk of cardiovascular events associated with hypertension is probably related to the rapid increase in vessel wall tension as transmural pressure rises, in line with Laplace’s law (see Chapter 5, p. 52), since increased tension will facilitate breakdown of the endothelial barrier at sites of pre-existing cell damage and so aid plaque formation. High intravascular pressure will also, by increasing blood flow velocity, increase marginal shear stress in large arteries and facilitate dislodgement of pre-existing plaques. In addition, the increased cardiac afterload due to hypertension will lead to concentric hypertrophy of the left ventricle. This lowers coronary capillary density and increases systolic endocardial compression, reducing functional cardiac reserve and predisposing the heart to hypoxic damage.
Chronic exercise has several anti-atherogenic effects
From consideration of these causal factors, chronic dynamic exercise can be seen to have several effects that potentially help prevent or reverse atherosclerosis-related cardiovascular changes. Improved endothelial cell function will reduce the susceptibility of the endothelium to traumatic or chemical damage. Increased endothelial dilator capacity and increased muscle capillary density will reduce peripheral resistance and, together with reduced sympathetic responses to arousal, will lower pressor responses to a variety of stimuli. This pressure reduction, the relative bradycardia induced by training and the conversion of concentric cardiac hypertrophy to eccentric hypertrophy will all reduce cardiac workload. Finally, the concomitant increase in coronary capillary density will help reduce any local myocardial ischaemia produced by atheromatous blockage. Figure 11.4 summarizes these therapeutic effects of training.
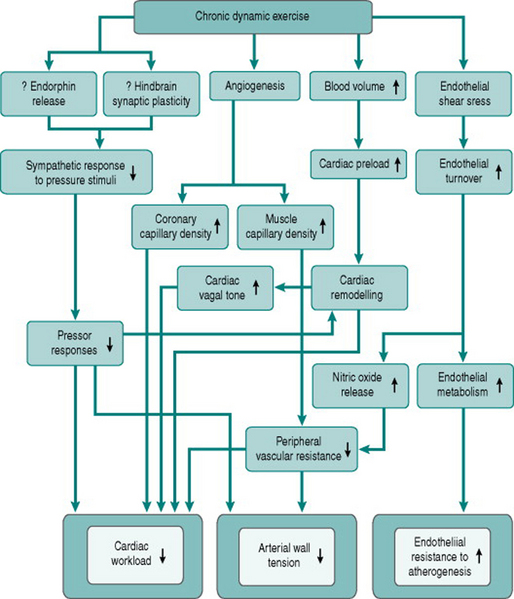
Exercise in cardiac rehabilitation
Following myocardial infarction the affected region of the myocardium may, depending on the degree of hypoxia, consist of dead muscle cells or contain at least some cells that can recover their contractile function. In either situation, chronic exercise provides a valuable stimulus to improvement of cardiac contractility. In the case of hypoxic but living myocardial tissue, exercise increases coronary perfusion by elevating blood pressure and by stimulating angiogenesis. Where an area of myocardium is dead, then the angiogenic effect of exercise helps to increase perfusion of the remaining myocardium. In both cases, the multiple consequences of training that reduce cardiac afterload (Fig. 11.4) will optimize resting cardiac function and enhance cardiac reserve.
Despite the clear theoretical benefits of exercise programmes in aiding recovery from cardiac infarcts, and general agreement that they are effective, very little objective data exist concerning the advantages and limitations of different exercise intensities or programme durations. Part of the problem is the difficulty in quantifying exercise performance in this patient group. At least initially, most are unable to work at more than around 20% of their calculated maximum work capacity because of their coronary impairment. In addition, almost all patients are routinely prescribed β-adrenoceptor antagonists to limit their cardiac workload. They are consequently not able to produce normal work-intensity-related tachycardia. Under these circumstances it is necessary to make a number of assumptions about the workloads undertaken, based primarily on the patients’ subjective perceptions of how hard they are working (American Association of Cardiovascular and Pulmonary Rehabilitation 2006). Well-designed research would be extremely helpful in ascertaining how these rehabilitation strategies can be optimized and how the benefits that accrue can be maintained after patients complete a supervised programme.
Alvarez GE, Halliwill JR, Ballard TP, Beske SD, Davy KP. Sympathetic neural regulation in endurance-trained humans: fitness vs. fatness. Journal of Applied Physiology. 2005;98:498-502.
American Association of Cardiovascular and Pulmonary Rehabilitation. AACVPR Cardiac Rehabilitation Resource Manual. Champaign, IL: Human Kinetics, 2006.
Iglesias Cubero G, Batalla A, Rodriguez Reguero JJ, et al. Left ventricular mass index and sports: the influence of different sports activities and arterial blood pressure. International Journal of Cardiology. 2000;75:261-265.
Lepretre P-M, Foster C, Koralsztein J-P, Billat VL. Heart rate deflection point as a strategy to defend stroke volume during incremental exercise. Journal of Applied Physiology. 2005;98:1660-1665.
Murray Á, Delaney T, Bell C. Rapid onset and offset of circulatory adaptations to exercise training in men. Journal of Human Hypertension. 2006;20:193-200.
Thijssen DH, Heesterbeek P, van Kuppevelt DJ, Duysens J, Hopman MT. Local vascular adaptations after hybrid training in spinal cord-injured subjects. Medicine and Science of Sports and Exercise. 2005;37:1112-1118.
Whyte GP, George K, Sharma S, et al. The upper limit of physiological cardiac hypertrophy in elite male and female athletes: the British experience. European Journal of Applied Physiology. 2004;92:592-597.