[level-membership-for-anesthesiology-category]
39 Cardiopulmonary Resuscitation
Epidemiology and Outcome of In-hospital Cardiopulmonary Arrest
Mechanics of Cardiopulmonary Resuscitation
Defibrillation and Cardioversion
Vascular Access and Monitoring during Cardiopulmonary Resuscitation
Medications Used during Cardiopulmonary Resuscitation
Special Cardiac Arrest Situations
Historical Background
In 1814, a description in poetical form of the Rules of the Humane Society for recovering drowned persons included the following description of mouth-to-mouth resuscitation1:
Let one the mouth, and either nostril close
While through the other the bellows gently blows.
Thus the pure air with steady force convey,
To put the flaccid lungs again in play.
Should bellows not be found, or found too late,
Let some kind soul with willing mouth inflate;
External cardiac massage was successfully conducted more than 100 years ago in two children (ages 8 and 13 years) after circulatory arrest precipitated by chloroform anesthesia during a surgical procedure.2 In 1904, Crile described the effectiveness of external cardiac compressions in maintaining the circulation of dogs.3
After multiple reports that attested to the effectiveness of mouth-to-mouth resuscitation,4–6 in 1958 the National Academy of Sciences National Research Council recommended mouth-to-mouth resuscitation with maximum backward tilt of the head as the preferred technique for all individuals requiring emergency artificial ventilation. In 1960, external cardiac compression was revived as a resuscitation technique when Kouwenhoven, Jude, and Knickerbocker7 demonstrated its effectiveness when combined with artificial respirations. Many of their patients, including the first, were in cardiac arrest as a result of anesthesia. Before this study, internal cardiac compression was the accepted technique, with its effectiveness demonstrated by experience in cardiac bypass surgery. In 1947, Beck and associates8 successfully internally defibrillated the human heart; and in 1956, Zoll and colleagues9 performed the first successful external defibrillation of a human heart.
Epidemiology and Outcome of In-Hospital Cardiopulmonary Arrest
A 2009 review of cardiac arrest events submitted to the National Registry of Cardiopulmonary Circulation included 3342 pediatric events, excluding events in a delivery room or neonatal intensive care unit (NICU).10 Seventy-three percent of the inpatient cardiac arrests reported occurred in an ICU, 7% in a general inpatient area, 11% in an emergency department, and 3% in an operating room or postanesthesia care unit. Return of spontaneous circulation (ROSC) was achieved in 65%, 24-hour survival occurred in 47%, and 30% of children survived until hospital discharge. Other large series of in-hospital pediatric cardiac arrest report survival until hospital discharge ranging from 14% to 44%,11–14 with the 44% survival representing cardiac arrests that occurred in a pediatric cardiac ICU. In another multicenter cohort study of in-hospital pediatric cardiac arrest,15 48.7% of the 353 children survived until hospital discharge. Survivors had greater body temperatures, greater pH values, and reduced serum lactate concentrations compared with nonsurvivors. Nonsurvivors were more likely to have a tracheal tube before the arrest, and to receive sodium bicarbonate, calcium, and vasopressin during the arrest. In this study, postoperative cardiopulmonary resuscitation (CPR) was associated with decreased mortality.
Mechanics of Cardiopulmonary Resuscitation
Airway
In the child without an artificial airway, the use of BVM devices may result in a significant risk of gastric inflation, followed by pulmonary aspiration of gastric contents. Abdominal distention (gastric and bowel) can significantly compromise oxygenation; therefore the stomach should be vented when excessive gastric inflation occurs. One study found a 28% incidence of pulmonary aspiration in a series of failed resuscitations.16 For this reason as well as for the risk of barotrauma and volutrauma, excessive inflation pressures should be avoided. However, effective bilateral ventilation is best judged by visualizing bilateral chest excursions and listening to the quality of the breath sounds rather than setting a preset maximal inflation pressure.
Breathing
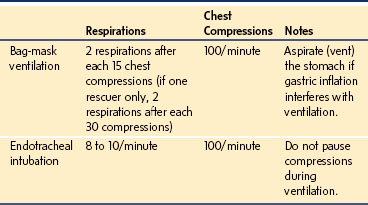
Overventilation is common during CPR, resulting in greater mean intrathoracic pressures than required, which decreases venous return and reduces cardiac output.17 In cardiopulmonary arrest, a less than normal minute ventilation may be appropriate, because cardiac output and delivery of CO2 to the lungs are diminished. If an artificial airway is not in place for single person rescue, two breaths should be given for each 30 chest compressions. If an artificial airway is not in place for two person rescue, two breaths should be given after each 15 chest compressions. Once an artificial airway is in place, a ventilator rate of 8 to 10 per minute without pausing during rapid chest compressions should be used (Table 39-1).
Circulation
During cardiac arrest, chest compressions provide the sole perfusion to a child’s vital organs; therefore optimal performance of CPR is critical. Key elements to providing quality chest compressions include (1) ensuring an adequate rate (100 compressions per minute), (2) ensuring adequate chest wall depression (one third to half of the anteroposterior chest diameter), (3) releasing completely between compressions to allow full chest wall recoil, (4) minimizing interruptions in chest compressions, and (5) ensuring that the child is on a sufficiently hard surface to allow effective chest compressions.18 In short, push hard and push fast, release completely, and do not interrupt compressions unnecessarily. Incomplete recoil during CPR is associated with higher intrathoracic pressures and significantly decreased venous return, and coronary and cerebral perfusion.19
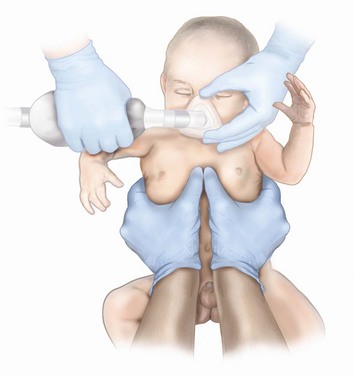
If a child is small enough (e.g., younger than 6 months) that the person providing chest compressions can comfortably encircle the chest with his or her hands, chest compressions should be performed using the circumferential technique, with thumbs depressing the sternum and the fingers supporting the infant’s back and circumferentially squeezing the thorax (Fig. 39-1). In larger infants, the sternum can be compressed using two fingers; and in the child, either one or two hands can be used, depending on the size of the child and of the rescuer.19 Whichever method is used, focused attention must remain on delivering effective compressions with minimal interruptions.20 In all cases other than circumferential CPR, a backboard must be used. Properly delivered chest compressions are tiring to the provider, and providers should rotate approximately every 2 minutes to prevent compressor fatigue and deterioration in the quality and rate of chest compressions.19
Mechanisms of Blood Flow
External chest compressions provide cardiac output through two mechanisms: the cardiac pump mechanism and the thoracic pump mechanism. By the cardiac pump mechanism of blood flow, blood is squeezed from the heart by compression of the heart between the sternum and the vertebral column, exiting the heart only anterograde because of closure of the atrioventricular valves. Between compressions, ventricular pressure decreases below atrial pressure, allowing the atrioventricular valves to open and the ventricles to fill. This sequence of events resembles the normal cardiac cycle. Although the cardiac pump is likely not the dominant blood flow mechanism during most closed-chest CPR, specific clinical situations have been identified in which the cardiac pump mechanism is more prominent. For example, a smaller, more compliant chest may allow for more direct cardiac compression (Fig. 39-2). Increasing, the applied force during chest compressions also increases the likelihood of direct cardiac compression.
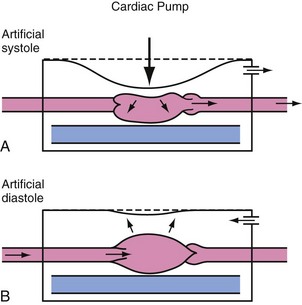
Several observations do not support the cardiac pump as the primary mechanism of blood flow during CPR. Angiographic studies show that blood passes from the vena cava through the right heart into the pulmonary artery and from the pulmonary veins through the left heart into the aorta during a single chest compression.21,22 Echocardiographic studies show that the atrioventricular valves are open during blood ejection.21,23,24 Without closure of atrioventricular valves during chest compression, the cardiac pump mechanisms cannot account for forward movement of blood during CPR.
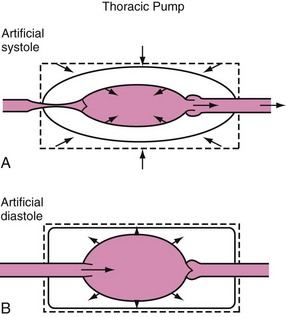
In 1976, Criley and colleagues25 made the dramatic observation that several patients who developed ventricular fibrillation during cardiac catheterization produced enough blood flow to maintain consciousness by repetitive coughing.25 The production of blood flow by increasing thoracic pressure without direct cardiac compression describes the thoracic pump mechanism, in which the heart is a passive conduit for blood flow. The intrathoracic pressure is greater than the extrathoracic pressure during the compression phase of CPR, at which time blood flows out of the thorax, with venous valves preventing excessive retrograde blood flow (Fig. 39-3). Experimental and clinical data support both mechanisms of blood flow during CPR in human infants.
Rate and Duty Cycle
The recommended rate of chest compressions for all patients is 100 per minute, with great care taken to minimize interruptions in chest compressions and to ensure adequate compression depth.20 This rate represents a compromise that attempts to maximize contributions from both the thoracic pump and cardiac pump mechanism of blood flow.
Duty cycle is defined as the percent of the compression–relaxation cycle that is devoted to compression. If blood flow is generated by direct cardiac compression, then primarily the force of compression determines the stroke volume. Prolonging the compression (increasing the duty cycle) beyond the time necessary for full ventricular ejection should have no additional effect on stroke volume. Increasing the rate of compressions should increase cardiac output, because a fixed volume of blood is ejected with each cardiac compression. In contrast, if blood flow is produced by the thoracic pump mechanism, the volume of blood that is ejected comes from a large reservoir of blood contained within the capacitance vessels in the chest. With the thoracic pump mechanism, flow is enhanced by increasing either the force of compression or the duty cycle but is not affected by changes in compression rate over a wide range of rates, given a set duty cycle.26
Different animal models yield conflicting results as to the optimal compression rate and duty cycle. However, a rate of compression during conventional CPR of 100 per minute satisfies both those who prefer the faster rates and those who support a longer duty cycle. This is true because it is easier to produce a longer duty cycle when compressions are administered at a faster rate.27,28
Defibrillation and Cardioversion
Electric Countershock
Electric countershock, or defibrillation, is the treatment of choice for ventricular fibrillation and pulseless ventricular tachycardia. Defibrillation should not be delayed to secure an airway, because the likelihood of restoring an organized rhythm decreases with increased duration of fibrillation. Ventricular fibrillation is terminated by simultaneous depolarization and sustained contraction of a critical mass of myocardium,29 allowing return of spontaneous, coordinated cardiac contractions, assuming the myocardium is well oxygenated and the acid-base status is relatively normal. Drug treatment may be required as an adjunct to defibrillation, but by itself cannot be relied on to terminate ventricular fibrillation.
An older generation of defibrillators that is still present in many hospitals delivers energy in a monophasic damped sinusoidal waveform (Fig. 39-4, A). This type of instrument delivers a single, unidirectional current with a gradual decrease to zero current. By contrast, the newer generation of biphasic defibrillators delivers a current in a positive direction for a set period, followed by a reversal in current (see Fig. 39-4, B). Biphasic defibrillators are more effective than monophasic defibrillators in terminating ventricular fibrillation in adults; therefore their use is recommended where possible.
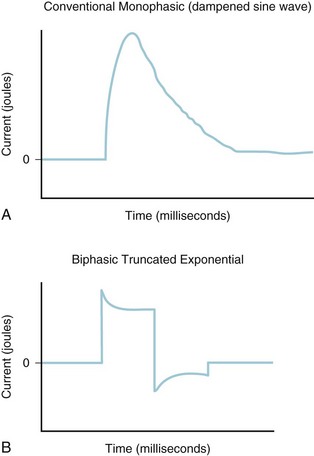
FIGURE 39-4 Energy delivery during conventional monophasic (A) and biphasic truncated exponential (B) defibrillation.
In the majority of adult cases, energy levels of 100 to 200 joules are successful when shocks are delivered with minimal delay.30,31 The goal of defibrillation is to deliver a minimum of electrical energy to a critical mass of ventricular muscle while avoiding excessive current that could further damage the heart. The most reliable predictor of success of defibrillation is the duration of fibrillation before the first countershock.32 Acidosis and hypoxemia also decrease the success of defibrillation.32
Practical Aspects of Defibrillation in Children
For children with in-hospital ventricular fibrillation or pulseless ventricular tachycardia, defibrillation should be attempted as soon as possible, with optimal CPR until the defibrillator is ready to deliver a shock. For the first defibrillation attempt, 2 joules/kg of delivered energy should be administered (Fig. 39-5). After shock delivery, CPR should resume immediately with chest compressions for five duty cycles (2 minutes). If one shock fails to eliminate ventricular fibrillation, the incremental benefit of another immediate shock is small. Resumption of CPR is likely to confer a greater benefit than another shock. CPR may provide coronary perfusion, increasing the likelihood of defibrillation with a subsequent shock. It is important to minimize the time between chest compressions and shock delivery and between shock delivery and resumption of postshock compressions.20 Approximately 2 minutes of CPR should be delivered before a second attempt at defibrillation at twice the original energy level (4 joules/kg).20
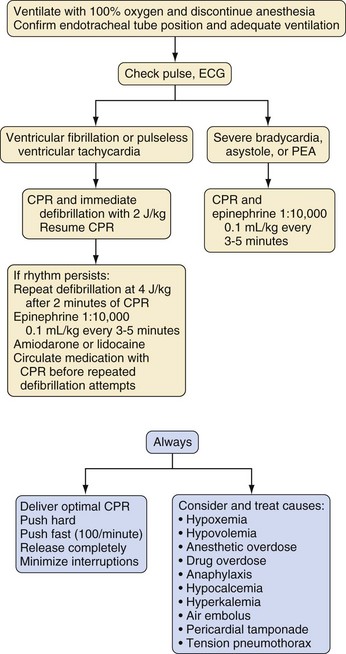
If ventricular fibrillation or pulseless ventricular tachycardia persists beyond the second defibrillation attempt, standard doses of epinephrine should be administered (with subsequent doses every 3 to 5 minutes during persistent cardiac arrest). After 2 minutes of chest compressions, defibrillation should be attempted again, followed by administration of amiodarone (5 mg/kg) or lidocaine (1 mg/kg) with subsequent defibrillation attempts. It is not necessary to increase the energy level on each successive shock during defibrillation after the second dose. However, successful defibrillation has been reported with currents in excess of 4 joules/kg without adverse sequelae, up to a maximum dose not exceeding 10 joules/kg or the adult level, whichever is less.20 This sometimes occurs when a fixed energy level, adult automated external defibrillator (AED) is used in a small child.
Automated External Defibrillation
Use of AEDs is now standard therapy in out-of-hospital resuscitation of adults.19,30 AEDs are now deemed appropriate for use in children older than 1 year. If available, use of pediatric attenuator pads or a pediatric mode on the AED should be used in children 1 to 8 years of age, but if unavailable (and a standard defibrillator is similarly unavailable), an unmodified AED should be used.
Vascular Access and Monitoring during Cardiopulmonary Resuscitation
Vascular Access and Fluid Administration
One of the key aspects of successful CPR is early establishment of a route for administration of fluids and medications. If intravenous access cannot be established rapidly, the intraosseous or endotracheal route should be used (see Chapter 48).
Intraosseous Access
The intraosseous route can be used to administer all medications and fluids used during CPR, including whole blood. An intraosseous needle also may be used to obtain initial blood samples, although acid-base analysis will be inaccurate after administration of sodium bicarbonate via the intraosseous needle. Intraosseous access should be considered a temporary measure during emergencies when other access is not available. The placement of an intraosseous needle in the older child (older than 10 years) and adult, although possible, is difficult owing to the thick bony cortex; however, a 50% success rate has been reported in these age-groups.33
The technique of placing an intraosseous line is straightforward. A specialized intraosseous needle or, if not available, a standard 16- or 18-gauge needle, a spinal needle with stylet, or bone marrow needle is inserted into the anterior surface of the tibia 1 to 2 cm below and 1 cm medial to the tibial tuberosity (avoiding the epiphyseal plate). The needle is directed at 90 degrees to the anteromedial surface of the tibia, just distal to the tuberosity (see Fig. 48-6 and E-Fig. 48-1). When the needle passes through the cortex into the marrow, a sudden loss of resistance is sensed. Successful placement has been achieved if the needle is in the marrow cavity, as evidenced by the needle standing upright without support. If the needle has slipped into the subcutaneous tissue, its upright position cannot be maintained without support. Free flow of the infusate without significant subcutaneous infiltration should also be demonstrated. The technique has a small complication rate,34 although possible complications include osteomyelitis, fat and bone marrow embolisms, and compartment syndrome. To avoid these potential complications, intravenous access should replace intraosseous access as soon as possible. The onset of action and concentration of most drugs after intraosseous administration are comparable with venous administration.35 A relatively new device, the EZ-IO (Vidacare, Shavano Park, Tex.) provides the most rapid means for intraosseous access (see E-Fig. 48-1).


Endotracheal Medication Administration
In the absence of other vascular access, medications including lidocaine, atropine, naloxone, and epinephrine (mnemonic LANE) can be administered via the endotracheal tube.36,37 The use of ionized medications such as sodium bicarbonate or calcium chloride is not recommended by this route. The peak concentration of epinephrine or lidocaine administered via the endotracheal route may be less compared with the intraosseous route. For example, the peak drug concentration of epinephrine after endotracheal administration was only 10% of that after intravenous administration in anesthetized dogs. The recommended dose for epinephrine via the endotracheal tube is 10 times the intravenous or intraosseous dose or 0.1 mg/kg for bradycardia or pulseless arrest.
The volume and the diluent in which the medications are administered through an endotracheal tube may be important. When large volumes of fluid are used, pulmonary surfactant may be altered or destroyed, resulting in atelectasis. The total volume of fluid delivered into the trachea with each drug administered should not exceed 10 mL in children and 5 mL in infants and neonates.38 However, administering an adequate volume of a drug is important to reach a large area of mucosal surface beyond the tip of the endotracheal tube for absorption. Absorption into the systemic circulation may be further enhanced by deep intrapulmonary administration by passing a catheter beyond the tip of the tracheal tube deep into the bronchial tree. The risk associated with the endotracheal route of drug administration is the formation of an intrapulmonary depot of drug, which may prolong the drugs’ effect. This could theoretically result in postresuscitation hypertension and tachycardia or the recurrence of fibrillation after normal circulation is restored.
Monitoring During Cardiopulmonary Resuscitation
The etco2 monitor provides important information during the course of resuscitation. Because the generation of exhaled CO2 depends on pulmonary blood flow, it can provide a useful indicator of the adequacy of cardiac output generated by chest compressions. As the cardiac output increases, the ETco2 increases and the difference between end-tidal and arterial CO2 becomes smaller.39 In animal models, etco2 during CPR correlates with coronary perfusion pressure and with ROSC.40,41 In adults during cardiac arrest, an etco2 greater than 10 mm Hg is positively associated with ROSC and hospital survival.42,43 When the etco2 is less than 10 mm Hg, efforts should be taken to enhance the quality of chest compressions (push hard, push fast, release completely, minimize interruptions, and optimize hand position). A reduced etco2 may occur transiently in the presence of adequate chest compressions after administration of epinephrine owing to an increase in intrapulmonary shunting.
Temperature should be monitored during and after CPR. The resuscitation of the child with hypothermia as the cause of cardiac arrest must be continued until the child’s core temperature exceeds 95° F (35° C). A glass bulb thermometer measures the temperature to very low values. Repeated measurements of core body temperature should be made at several sites (rectal, bladder, esophageal, axillary, or tympanic membrane) where possible, to avoid misleading temperature readings from a single site, because local body temperature may vary with changes in regional blood flow during CPR. Hyperthermia should be aggressively treated in the periarrest period, because postarrest hyperthermia is associated with worse outcomes in children.44 Evidence suggests a benefit to induced hypothermia after resuscitation from cardiac arrest in adults45,46 and after perinatal hypoxic or ischemic injury.47 The data available to support the use of hypothermia in infants and children after cardiac arrest is from case series and retrospective studies. Pending results of a randomized controlled trial (Therapeutic Hypothermia After Pediatric Cardiac Arrest), clinicians may choose to control a child’s temperature in the range of 91.4° to 95° F (33° to 35° C) for 12 to 48 hours after resuscitation with slow subsequent rewarming (see section on Postresuscitation Stabilization).
Medications Used during Cardiopulmonary Resuscitation
α- and β-Adrenergic Agonists
In 1963, only 3 years after the original description of closed-chest CPR, Redding and Pearson48 demonstrated that early administration of epinephrine in a canine model of cardiac arrest improved the success rate of CPR. They also demonstrated that the increase in aortic diastolic pressure with the administration of α-adrenergic agonists was responsible for the improved success of resuscitation. They theorized that vasopressors such as epinephrine were of value because the drug increased peripheral vascular tone and, hence, coronary perfusion pressure. The relative importance of α- and β-adrenergic agonist actions during resuscitation has been widely investigated. In a canine model of cardiac arrest, only 27% of dogs that received a pure β-adrenergic receptor agonist along with an α-adrenergic antagonist were resuscitated successfully, compared with 100% of dogs that received a pure α-adrenergic agonist and a β-adrenergic antagonist. Other investigators have demonstrated that the α-adrenergic effects of epinephrine resulted in intense vasoconstriction of the resistance vessels of all organs of the body, except those supplying the heart and brain.49 Because of the widespread vasoconstriction in nonvital organs, adequate perfusion pressure and thus blood flow to the heart and brain can be achieved despite the fact that cardiac output is very low during CPR.49–51
The increase in aortic diastolic pressure associated with epinephrine administration during CPR is critical for maintaining coronary blood flow and enhancing the success of resuscitation.52,53 Even though the contractile state of the myocardium is increased by the use of β-adrenergic agonists in the spontaneously beating heart, β-adrenergic agonists may actually decrease myocardial blood flow by increasing intramyocardial wall pressure and vascular resistance during CPR.54 By its inotropic and chronotropic effects, β-adrenergic stimulation increases myocardial O2 demand, which, when superimposed on low coronary blood flow, increases the risk of ischemic injury.
Epinephrine
Epinephrine (adrenaline) is an endogenous catecholamine with potent α- and β-adrenergic stimulating properties. The α-adrenergic action increases systemic and pulmonary vascular resistance, increasing both systolic and diastolic blood pressure. The increase in diastolic blood pressure directly increases coronary perfusion pressure, thereby increasing coronary blood flow and increasing the likelihood of ROSC.52,53 The β-adrenergic effect increases myocardial contractility and heart rate and relaxes smooth muscle in the skeletal muscle vascular bed and bronchi. Epinephrine also increases the vigor and intensity of ventricular fibrillation, increasing the likelihood of successful defibrillation.55
Larger than necessary doses of epinephrine may be deleterious. Epinephrine may worsen myocardial ischemic injury secondary to increased O2 demand and may result in postresuscitative tachyarrhythmias, hypertension, and pulmonary edema. Epinephrine causes hypoxemia and an increase in alveolar dead space ventilation by redistributing pulmonary blood flow.39,56 Prolonged peripheral vasoconstriction by excessive doses of epinephrine may delay or impair reperfusion of systemic organs, particularly the kidneys and gastrointestinal tract.
Routine use of large-dose epinephrine in in-hospital pediatric cardiac arrest should be avoided. A randomized, controlled trial in 2003 compared high-dose with standard-dose epinephrine for children with in-hospital cardiac arrest refractory to initial standard-dose epinephrine. Survival was reduced at 24 hours, with a trend toward decreased survival to hospital discharge in the children who received large doses of epinephrine.57 Despite these data, large doses of epinephrine may be considered in special cases (e.g., β-blocker overdose), particularly when diastolic blood pressure remains low despite excellent chest compression and several standard doses of epinephrine.
Vasopressin
Vasopressin is a long-acting endogenous hormone that causes vasoconstriction (V1 receptor) and reabsorption of water in the renal tubule (V2 receptor). In experimental models of cardiac arrest, vasopressin increases blood flow to the heart and brain and improves long-term survival compared with epinephrine.58,59 In a randomized trial comparing the efficacy of epinephrine to vasopressin in shock-resistant out-of-hospital ventricular fibrillation in adults, vasopressin produced a greater rate of ROSC.60 In a study of in-hospital adult cardiac arrest, vasopressin produced a rate of survival to hospital discharge similar to that of epinephrine.61
In a pediatric porcine model of prolonged ventricular fibrillation, the use of vasopressin and epinephrine in combination resulted in greater left ventricular blood flow than either vasopressor alone, and both vasopressin alone and vasopressin plus epinephrine resulted in superior cerebral blood flow than epinephrine alone.62 By contrast, in a pediatric porcine model of asphyxial cardiac arrest, ROSC was more likely in piglets treated with epinephrine than in those treated with vasopressin.63 Pediatric64–66 case series and reports suggested that vasopressin64 or its long-acting analog, terlipressin,65,66 may be effective in refractory cardiac arrest. In a 2009 National Registry of Cardiopulmonary Resuscitation (NRCPR) review, vasopressin was associated with reduced ROSC and a trend toward reduced 24-hour and discharge survival. There is insufficient evidence to make a recommendation for its routine use during cardiac arrest.20
Atropine
Atropine, a parasympatholytic agent, blocks cholinergic stimulation of the muscarinic receptors in the heart, increasing the sinus rate and shortening atrioventricular node conduction time. Atropine may activate latent ectopic pacemakers. Atropine has little effect on systemic vascular resistance, myocardial perfusion pressure, or contractility.67
The recommended pediatric dose of atropine is 0.02 mg/kg, with a maximum dose of 2 mg. The increase in heart rate after intravenous atropine (20 µg/kg) in infants and children may be attenuated compared with that in adults.68 Although a minimum dose of 0.1 mg has been entrenched in the literature, it is not evidence-based.68,69 Atropine may be given by any route, including intravenous, intraosseous, endotracheal, intramuscular, and subcutaneous. After intravenous administration, its onset of action is within 30 seconds and its peak effect occurs in 1 to 2 minutes. The recommended adult dose is 0.5 mg every 3 to 5 minutes until the desired heart rate is obtained, up to a maximum of 3 mg.
Sodium Bicarbonate
The routine use of sodium bicarbonate during CPR remains controversial, and it remains American Heart Association Class Indeterminate. Acidosis may depress myocardial function, prolong diastolic depolarization, depress spontaneous cardiac activity, decrease the electrical threshold for ventricular fibrillation, and reduce the cardiac response to catecholamines.70–72 Acidosis also vasodilates systemic vessels and attenuates the vasoconstrictive response of peripheral vessels to catecholamines,73 which is the opposite of the desired vascular effect during CPR. In children with a reactive pulmonary vascular bed, acidosis causes pulmonary hypertension. Therefore correction of even mild acidosis may be helpful in resuscitating children with increased pulmonary vascular resistance. Additionally, the presence of severe acidosis may increase the threshold for myocardial stimulation in a child with an artificial cardiac pacemaker.74 Other situations in which administration of bicarbonate is indicated include tricyclic antidepressant overdose, hyperkalemia, hypermagnesemia, or sodium channel blocker poisoning.
Potentially deleterious effects of bicarbonate administration include metabolic alkalosis, hypercapnia, hypernatremia, and hyperosmolality. In a 2004 multicenter cohort study of in-hospital pediatric cardiac arrest, the use of sodium bicarbonate was associated with increased mortality.15 Alkalosis causes a leftward shift of the oxyhemoglobin dissociation curve and thus impairs release of O2 from hemoglobin to tissues at a time when O2 delivery may already be reduced.75 Alkalosis also can result in hypokalemia by enhancing potassium influx into cells and in ionic hypocalcemia by increasing protein binding of ionized calcium. The marked hypercapnic acidosis that occurs during CPR in the venous circulation, including the coronary sinus, may be exacerbated by the administration of bicarbonate.76 Myocardial acidosis during cardiac arrest is associated with decreased myocardial contractility.72 Hypernatremia and hyperosmolality may decrease tissue perfusion by increasing interstitial edema in microvascular beds.
Paradoxical intracellular acidosis after bicarbonate administration can occur with the rapid entry of CO2 into cells with a slow egress of hydrogen ions out of cells; however, in neonatal rabbits recovering from hypoxic acidosis, bicarbonate administration increased both arterial pH and intracellular brain pH as measured by nuclear magnetic resonance spectroscopy.77,78 Likewise, in rats, intracellular brain adenosine triphosphate concentration did not change during severe intracellular acidosis in the brain produced by extreme hypercapnia.78 In a separate animal study, bicarbonate slowed the rate of decrease of both arterial and cerebral pH during prolonged CPR, suggesting that the blood-brain pH gradient is maintained during CPR.79 Given the potentially deleterious effects of bicarbonate administration, its use should be limited to cases in which there is a specific indication, as discussed earlier.
Calcium
Calcium administration during CPR should be restricted to cases with a specific indication for calcium (e.g., hypocalcemia, hyperkalemia, hypermagnesemia, and calcium channel blocker overdose). These restrictions are based on the possibility that exogenously administered calcium may worsen ischemia-reperfusion injury. Intracellular calcium overload occurs during cerebral ischemia by the influx of calcium through voltage-dependent and agonist-dependent (e.g., N-methyl-d-aspartate [NMDA]) calcium channels. Calcium plays an important role in the process of cell death in many organs, possibly by activation of intracellular enzymes such as nitric oxide synthase, phospholipase A and C, and others.80
The calcium ion is essential in myocardial excitation-contraction coupling, in increasing ventricular contractility, and in enhancing ventricular automaticity during asystole. Ionized hypocalcemia is associated with decreased ventricular performance and the peripheral blunting of the hemodynamic response to catecholamines.81,82 Severe ionized hypocalcemia has been documented in adults suffering from out-of-hospital cardiac arrest82 and in animals during prolonged CPR.83 Thus children at risk for ionized hypocalcemia should be identified and treated as expeditiously as possible. Both total and ionized hypocalcemia may occur in children with either chronic or acute disease. Ionized hypocalcemia also occurs during massive or rapid transfusion of blood products (particularly whole blood and fresh frozen plasma) because citrate and other preservatives in stored blood products rapidly bind calcium. Because of this effect, ionized hypocalcemia is a known cause of cardiac arrest in the OR and should be treated immediately with calcium chloride or calcium gluconate (see Chapter 10). The magnitude of hypocalcemia in this setting depends on the rate and volume of blood products administered and the hepatic and renal function of the child. Administration of fresh frozen plasma at a rate in excess of 1 mL/kg/min significantly decreases the ionized calcium concentration in anesthetized children.84
The pediatric dose of calcium chloride for resuscitation is 20 mg/kg with a maximum dose of 2 g. Calcium gluconate is as effective as calcium chloride in increasing the ionized calcium concentration.85,86 The dose of calcium gluconate should be three times that of calcium chloride (milligram per kilogram,) (i.e., 20 mg/kg calcium chloride is equivalent to 60 mg/kg calcium gluconate), with a maximum dose of 2 g in children. Calcium should be given slowly through a large-bore, free-flowing intravenous cannula, or preferably a central venous line. When administered too rapidly, calcium may cause bradycardia, heart block, or ventricular standstill. Severe tissue necrosis occurs when calcium infiltrates into subcutaneous tissue. Calcium administration is not recommended for pediatric cardiopulmonary arrest in the absence of documented hypocalcemia, calcium channel blocker overdose, hypermagnesemia, or hyperkalemia (Class III, level of evidence [LOE] B). Routine calcium administration in cardiac arrest provides no benefit and may be harmful.74,87
Glucose
The administration of glucose during CPR should be restricted to children with documented hypoglycemia because of the possible detrimental effects of hyperglycemia on the brain during or after ischemia. The mechanism by which hyperglycemia exacerbates ischemic neurologic injury may be due to an increased production of lactic acid in the brain by anaerobic metabolism. During ischemia under normoglycemic conditions, brain lactate concentration reaches a plateau. In a hyperglycemic milieu, however, brain lactate concentration continues to increase for the duration of the ischemic period.88
Clinical studies have shown a direct correlation between the initial post–cardiac arrest serum glucose concentration and poor neurologic outcome,89–92 although the greater glucose concentration may be a marker rather than a cause of more severe brain injury.90 However, given the likelihood of additional ischemic and hypoxic events in the postresuscitation period, it seems prudent to maintain serum glucose concentrations within the normal range. Additional studies are needed to determine if the benefit from tight control of serum glucose after cardiac arrest outweighs the risk of iatrogenic hypoglycemia. Some groups of children, including preterm infants and debilitated children with small endogenous glycogen stores, are more prone to developing hypoglycemia during and after a physiologic stress such as surgery. Bedside monitoring of the serum glucose concentration is critical during and after a cardiac arrest and allows for the opportunity to administer glucose before the critical point of small substrate delivery has been reached. The dose of glucose generally needed to correct hypoglycemia is 0.5 g/kg given as 5 mL/kg of 10% dextrose in infants or 1 mL/kg of 50% dextrose in an older child. The osmolarity of 50% dextrose is approximately 2700 mOsm/L and has been associated with intraventricular hemorrhage in neonates and infants; therefore the more dilute concentration is recommended in infants.
Amiodarone
Amiodarone has now supplanted lidocaine as the first drug of choice for medical management of shock-resistant ventricular tachycardia and fibrillation. The role of amiodarone was established for cardiac arrest after a series of studies showed it to be more effective than lidocaine in the management of refractory tachyarrhythmias in adults. Compared with lidocaine, amiodarone results in an increased rate of survival to hospital admission in patients with shock-resistant out-of-hospital ventricular fibrillation.93
Early reports on the use of oral amiodarone in children were favorable.94–96 Recent data on amiodarone use in children are limited to case reports and descriptive case series. Nevertheless, it is now used widely for serious pediatric arrhythmias in the nonresuscitation environment and appears to be effective and have an acceptable short-term safety profile.
Hypotension is commonly reported with intravenous administration and may limit the rate at which the drug can be given; however, the development of hypotension is less common with the newer, aqueous formulation.97 The overall hemodynamic impact of intravenous administration will depend on the balance of its effect on rate control, myocardial performance, and vasodilation. Dosage recommendations for children are based on limited clinical studies. The dose is extrapolated from data on adults; 5 mg/kg intravenously for life-threatening arrhythmias. This dose can be repeated if necessary to control the arrhythmia. Intravenous loading doses are followed by a continuous infusion of 10 to 20 mg/kg/day if there is a risk of arrhythmia recurrence. The ideal rate of bolus administration is unclear; in adults, once diluted, it is given as an intravenous push. It is best administered over 20 to 60 minutes to avoid profound vasodilation. We recommend slow intravenous push (2 to 3 minutes) for pulseless ventricular tachycardia or ventricular fibrillation until the arrhythmia is controlled and then a slower bolus (up to 10 minutes) for the remainder of the dose. An alternative dosing regimen for children is 1 mg/kg intravenous push every 5 minutes up to 5 mg/kg. The use of the small aliquot bolus technique may be particularly appropriate for infants younger than 12 months of age.
Amiodarone-induced torsades de pointes has been described.98 The use of amiodarone should be avoided in combination with other drugs that prolong the QT interval, as well as in the setting of hypomagnesemia and other electrolyte abnormalities that predispose to torsades de pointes. Severe bradycardia and heart block have also been described, especially in the postoperative period, and ventricular pacing wires are recommended in this setting. Both amiodarone and inhalation anesthetic agents prolong the QT interval; however, no specific data exist to evaluate the use of amiodarone for ventricular arrhythmias in children receiving inhalation anesthetics. It would seem prudent to be especially vigilant for this adverse effect in this circumstance.
Noncardiac adverse effects are often seen, especially with chronic dosing.99 The most serious of these has been the development of interstitial pneumonitis seen most commonly in patients with preexisting lung disease.100 The incidence in children is unknown. Rarely, an acute illness similar to acute respiratory distress syndrome illness has been reported in both infants and adults at the initiation of treatment.101 The lung disease may remit with early discontinuation of the drug. Hypothyroidism, hepatotoxicity, photosensitivity, and corneal opacities are also common side effects with chronic use.99
Lidocaine
Toxic effects of lidocaine occur when the serum concentration exceeds 7 to 8 µg/mL and include seizures, psychosis, drowsiness, paresthesias, disorientation, agitation, tinnitus, muscle spasms, and respiratory arrest. The treatment of choice for lidocaine-induced seizures is a benzodiazepine (midazolam or lorazepam) or a barbiturate (e.g., phenobarbital; chronic therapy also increases the hepatic metabolism of lidocaine).102 Conversion of second-degree heart block to complete heart block has been described,103 as has severe sinus bradycardia. Lidocaine is not as effective as amiodarone for improving ROSC or survival to hospital admission among adults with ventricular fibrillation refractory to ventricular fibrillation and shock.104
Special Cardiac Arrest Situations
Perioperative Cardiac Arrest
The incidence, causes, and risk factors associated with anesthesia- and operative-related cardiac arrest have been evaluated by the Pediatric Perioperative Cardiac Arrest registry.105,106 Cardiovascular causes of cardiac arrest were the most common (41% of all arrests), with hypovolemia from blood loss and hyperkalemia from transfusion of stored blood the most common identifiable cardiovascular causes. Among respiratory causes of arrest (27%), airway obstruction from laryngospasm was the most common cause. Vascular injury incurred during placement of central venous catheters was the most common equipment-related cause of arrest. The cause of arrest varied by phase of anesthesia care.
Cardiac arrest in the OR should have the greatest potential for a successful outcome, because it is a witnessed arrest with virtually instantaneous availability of skilled personnel, monitoring equipment, resuscitative equipment, and drugs. Whenever a cardiac arrest occurs in the OR, the circumstances causing the arrest should be rapidly determined. The circumstances of the arrest may provide a clue as to the cause, such as hyperkalemia after succinylcholine administration or rapid blood transfusion, hypocalcemia during a rapid infusion of fresh frozen plasma or large blood transfusion, or a sudden fall in etco2 indicating air, blood clot, or tumor embolism. A bradyarrhythmia always must be assumed to be first resulting from hypoxemia; second, caused by anesthetic overdose (real or relative); and third, possibly related to a vagal reflex caused by surgical or airway manipulation. Administering 100% O2 and ensuring adequate ventilation is always the first maneuver, regardless of the cause of the bradycardia. In reflex-induced bradycardia, atropine may be the first drug of choice, but in extreme cases of bradycardia, whatever the mechanism, epinephrine should be used. Hypotension and a low cardiac output state must be rapidly corrected by appropriate administration of intravenous fluids, vasopressors, and adequate chest compressions to circulate drugs to have the needed clinical effect. Once chest compressions are required, the standard American Heart Association recommendations for CPR generally apply and this includes the frequent administration of epinephrine. Figure 39-5 presents an algorithm for the differential diagnosis and treatment of the more common causes of acute OR-associated cardiac dysfunction.
Supraventricular Tachycardia
Adenosine is the medical treatment of choice for SVT. The underlying mechanism in children is usually a reentry circuit involving the atrioventricular node. Adenosine causes a temporary block in the atrioventricular node and interrupts this reentry circuit. The initial dose is 0.1 mg/kg given as a rapid intravenous bolus. Central venous administration is preferable because the drug is rapidly metabolized by red blood cell adenosine deaminase and therefore has a half-life of only 10 seconds. When the drug is given peripherally, the intravenous line should be immediately and rapidly flushed with 10 mL of saline. If there is no interruption in the reentry circuit, successive doses of 0.2 and 0.4 mg/kg should be given. In neonates, a smaller initial dose of 0.05 mg/kg is given and increased by 0.05 mg/kg/dose until termination of the arrhythmia up to a maximum dose of 0.3 mg/kg.107 When SVT appears without any circulatory compromise, conversion of the arrhythmia may first be attempted with a vagal maneuver such as ice to the face. If this is ineffective, then adenosine should be utilized.
Other medications used to treat SVT have a greater incidence of adverse effects than adenosine. Digoxin is often ineffective and causes frequent arrhythmias. Verapamil should be avoided in infants because of its association with congestive heart failure and cardiac arrest because of its negative inotropic effects.108 Flecainide is effective in treating SVT but has many cardiac and noncardiac adverse effects109; its role for hemodynamically unstable SVT remains to be established. Other therapies include β-adrenergic blockers, edrophonium, and α-agonists. If SVT persists despite medical therapy and the child progresses to circulatory instability, electrical cardioversion should proceed immediately.
Adjunctive Cardiopulmonary Resuscitation Techniques
Open-Chest Cardiopulmonary Resuscitation
The use of open-chest cardiac massage, although generally replaced by closed-chest CPR, still has an active role in the OR and ICU especially during and after thoracic surgery. Compared with closed-chest CPR, open-chest CPR generates greater cardiac output and vital organ blood flow. During open-chest CPR, less elevation occurs in intrathoracic, right atrial, and intracranial pressure, resulting in greater coronary and cerebral perfusion pressure and greater myocardial and cerebral blood flow.110–112
Typically, in the OR and ICU, open-chest CPR is preferable to closed-chest CPR in the child who has had a recent sternotomy. Open-chest CPR is also indicated for selected children when closed-chest CPR has failed, although exactly which children should receive this method of resuscitation under this condition is controversial. When initiated early after failure of closed-chest CPR, open-chest CPR may improve outcome.113–115 When performed after 15 minutes of closed-chest CPR, open-chest CPR significantly improves coronary perfusion pressure and the rate of successful resuscitation.116
Extracorporeal Membrane Oxygenation
In institutions with the ability to rapidly mobilize an extracorporeal circuit, extracorporeal cardiopulmonary bypass (CPB) should be considered for refractory pediatric cardiac arrest when the condition leading to arrest is reversible and when the period of no flow (cardiac arrest without CPR) was brief. Survival with a good neurologic outcome is possible after more than 50 minutes of CPR in selected children who were resuscitated via extracorporeal CPB.117,118 CPB requires major technical support and sophistication but can be rapidly implemented in hospitals set up to do so. However, absence of a formal rapid deployment extracorporeal membrane oxygenation (ECMO) team does not preclude resuscitation ECMO in pediatric cardiac patients with good results.119 Extracorporeal CPB should be reserved for children who have effective CPR initiated immediately after cardiac arrest.
Active Compression-Decompression
Active compression-decompression CPR uses a negative-pressure “pull” on the thorax during the release phase of chest compression using a handheld suction device. This technique has been shown to improve vascular pressures and minute ventilation during CPR in animals and humans.120–124 The hemodynamic benefit of this technique is attributed to enhancement of venous return by the negative intrathoracic pressure generated during the decompression phase. Thus, when this technique was used with a device adding impedance to inspiration, vascular pressures and flow increased further.125 Its effectiveness in adults shows promise, with increased survival and a trend toward neurologic improvement in prehospital victims.126–128 However, two recent, larger trials did not demonstrate improved survival in in-hospital or prehospital victims of cardiac arrest, nor did any subgroup demonstrate benefit from active compression-decompression CPR.129–131 The complication rate, including fatal rib and sternal fractures, may be greater with this technique.132
Postresuscitation Stabilization (Post–Cardiac Arrest Care)
Mitigation of neurologic injury after cardiac arrest has been a goal of many investigator groups. In adult patients with out-of-hospital ventricular fibrillation and in asphyxiated newborns,47 therapeutic hypothermia has been shown to be of benefit. In a retrospective study involving five hospitals, the effectiveness of hypothermia therapy was neither supported nor refuted.133 Fink and colleagues134 studied the feasibility of achieving mild hypothermia after pediatric cardiac arrest and found that they were reliably able to achieve a target temperature of 89.6° to 93.2° F (32° to 34° C) in less than 3 hours. A multicenter, randomized, controlled trial of systemic hypothermia for 48 hours after nontraumatic cardiac arrest (Therapeutic Hypothermia After Pediatric Cardiac Arrest [THAPCA]) is currently ongoing.
1 Scherlis L. Poetical version of the Rules of the Humane Society for recovering drowned persons. Crit Care Med. 1981;9:430–431.
2 Pearson J. Historical and experimental approaches to modern resuscitation. Springfield, Ill.: Charles C Thomas; 1965.
3 Crile G. The resuscitation of the apparently dead and a demonstration of the pneumatic rubber suit as a means of controlling blood pressure. Trans South Surg Gynecol Assoc. 1904;16:362.
4 Elam J, Brown E, Elder J. Artificial respiration by mouth-to-mask method. N Engl J Med. 1954;250:749–754.
5 Gordon A, Frye C, Gittelson L, et al. Mouth-to-mouth versus manual artificial respiration for children and adults. JAMA. 1958;167:320–328.
6 Safar P. Ventilatory efficiency of mouth-to-mouth respiration. JAMA. 1958;167:335–341.
7 Kouwenhoven W, Jude J, Knickerbocker G. Closed-chest cardiac massage. JAMA. 1960;173:1064–1067.
8 Beck C, Pritchard W, Feil H. Ventricular fibrillation of long duration abolished by electric shock. JAMA. 1947;135:985–986.
9 Zoll P, Linenthal A, Norman L, et al. Treatment of unexpected cardiac arrest by external electric stimulation of the heart. N Engl J Med. 1956;254:541–546.
10 Donoghue A, Berg RA, Hazinski MF, et al. Cardiopulmonary resuscitation for bradycardia with poor perfusion versus pulseless cardiac arrest. Pediatric. 2009;124:1541–1548.
11 Nadkarni VM, Larkin GL, Peberdy MA, et al. First documented rhythm and clinical outcome from in-hospital cardiac arrest among children and adults. JAMA. 2006;295:50–57.
12 Reis AG, Nadkarni V, Perondi MB, et al. A prospective investigation into the epidemiology of in-hospital pediatric cardiopulmonary resuscitation using the international Utstein reporting style. Pediatrics. 2002;109:200–209.
13 Parra DA, Totapally BR, Zahn E, et al. Outcome of cardiopulmonary resuscitation in a pediatric cardiac intensive care unit. Crit Care Med. 2000;28:3296–3300.
14 Slonim AD, Patel KM, Ruttimann UE, Pollack MM. Cardiopulmonary resuscitation in pediatric intensive care units. Crit Care Med. 1997;25:1951–1955.
15 Meert KL, Donaldson A, Nadkarni V, et al. Multicenter cohort study of in-hospital pediatric cardiac arrest. Pediatr Crit Care Med. 2009;10(5):544–553.
16 Lawes E, Baskett P. Pulmonary aspiration during unsuccessful cardiopulmonary resuscitation. Intensive Care Med. 1987;13:379–382.
17 Aufderheide T, Sigurdsson G, Pirrallo R, et al. Hyperventilation-induced hypotension during cardiopulmonary resuscitation. Circulation. 2004;109:1960–1965.
18 American Heart Association. 2005 American Heart Association Guidelines for Cardiopulmonary Resuscitation and Emergency Cardiovascular Care. Circulation. 2005;112(Suppl 1):IV-1–203.
19 Berg MD, Schexnayder SM, Chameides L, et al. Part 13: pediatric basic life support: 2010 American Heart Association Guidelines for Cardiopulmonary Resuscitation and Emergency Cardiovascular Care. Circulation. 2010;122(suppl 3):S862–S875.
20 Kleinman ME, Chameides L, Schexnayder SM, et al. Part 14: Pediatric advanced life support: 2010 American Heart Association Guidelines for Cardiopulmonary Resuscitation and Emergency Cardiovascular Care. Circulation. 2010;122(suppl 3):S876–S908.
21 Niemann J, Rosborough J, Hausknecht M, et al. Pressure-synchronized cineangiography during experimental cardiopulmonary resuscitation. Circulation. 1981;64:985–991.
22 Cohen J, Chandra N, Alderson P, et al. Timing of pulmonary and systemic blood flow during intermittent high intrathoracic pressure cardiopulmonary resuscitation in the dog. Am J Cardiol. 1982;49:1883–1889.
23 Werner J, Greene H, Janko C, et al. Visualization of cardiac valve motion in man during external chest compression using two-dimensional echocardiography: implications regarding the mechanism of blood flow. Circulation. 1981;63:1417–1421.
24 Rich S, Wix H, Shapiro E. Clinical assessment of heart chamber size and valve motion during cardiopulmonary resuscitation by two-dimensional echocardiography. Am Heart J. 1981;102:368–373.
25 Criley J, Blaufuss A, Kissel G. Cough-induced cardiac compression: self-administered form of cardiopulmonary resuscitation. JAMA. 1976;236:1246–1250.
26 Chandra N, Tsitlik J, Halperin H, et al. Observations of hemodynamics during human cardiopulmonary resuscitation. Crit Care Med. 1990;18:929–934.
27 Maier G, Tyson G, Olsen C, et al. The physiology of external cardiac massage: high-impulse cardiopulmonary resuscitation. Circulation. 1984;70:86–101.
28 Ornato J, Gonzalez E, Garnett A, et al. Effect of cardiopulmonary resuscitation compression rate on end-tidal carbon dioxide concentration and arterial pressure in man. Crit Care Med. 1988;16:241–245.
29 Zipes D, Fischer J, King R, et al. Termination of ventricular fibrillation in dogs by depolarizing a critical amount of myocardium. Am J Cardiol. 1975;36:37–44.
30 Weaver W, Cobb L, Copass M, et al. Ventricular defibrillation: a comparative trial using 175-J and 320-J shocks. N Engl J Med. 1982;307:1101–1106.
31 Campbell N, Webb S, Adgey A, et al. Transthoracic ventricular defibrillation in adults. BMJ (Clin Res Ed). 1977;2:1379–1381.
32 Kerber R, Sarnat W. Factors influencing the success of ventricular defibrillation in man. Circulation. 1979;60:226–230.
33 Glaeser PW, Hellmich TR, Szewczuga D, et al. Five-year experience in prehospital intraosseous infusions in children and adults. Ann Emerg Med. 1993;22:1119–1124.
34 Meola F. Bone marrow infusions as routine procedure in children. J Pediatr. 1944;25:13–16.
35 Andropoulos DB, Soifer SJ, Schreiber MD. Plasma epinephrine concentrations after intraosseous and central venous injection during cardiopulmonary resuscitation in the lamb. J Pediatr. 1990;116:312–315.
36 Ward JTJ. Endotracheal drug therapy. Am J Emerg Med. 1983;1:71–82.
37 Johnston C. Endotracheal drug delivery. Pediatr Emerg Care. 1992;8:94–97.
38 Greenberg M, Roberts J, Baskin S. Use of endotracheally administered epinephrine in a pediatric patient. Am J Dis Child. 1981;135:767–768.
39 Tang W, Weil M, Gazmuri R, et al. Pulmonary ventilation/perfusion defects induced by epinephrine during cardiopulmonary resuscitation. Circulation. 1991;84:2101–2107.
40 Sanders A, Kern K, Otto C, et al. End-tidal carbon dioxide monitoring during cardiopulmonary resuscitation: a prognostic indicator for survival. JAMA. 1989;262:1347–1351.
41 von Planta M, von Planta I, Weil MH, et al. End tidal carbon dioxide as an haemodynamic determinant of cardiopulmonary resuscitation in the rat. Cardiovasc Res. 1989;23:364–368.
42 Sanders AB, Ewy GA, Bragg S, et al. Expired Pco2 as a prognostic indicator of successful resuscitation from cardiac arrest. Ann Emerg Med. 1985;14:948–952.
43 Cantineau JP, Lambert Y, Merckx P, et al. End-tidal carbon dioxide during cardiopulmonary resuscitation in humans presenting mostly with asystole: a predictor of outcome. Crit Care Med. 1996;24:791–796.
44 Hickey RW, Kochanek PM, Ferimer H, et al. Hypothermia and hyperthermia in children after resuscitation from cardiac arrest. Pediatrics. 2000;106(1 pt 1):118–122.
45 The Hypothermia after Cardiac Arrest Study Group. Mild therapeutic hypothermia to improve the neurologic outcome after cardiac arrest. N Engl J Med. 2002;346:549–556.
46 Bernard S, Gray T, Buist M, et al. Treatment of comatose survivors of out-of-hospital cardiac arrest with induced hypothermia. N Engl J Med. 2002;346:564–569.
47 Shankaran S, Laptook A, Wright L, et al. Whole-body hypothermia for neonatal encephalopathy: animal observations as a basis for a randomized, controlled pilot study in term infants. Pediatrics. 2002;110:377–385.
48 Redding JS, Pearson JW. Evaluation of Drugs for Cardiac Resuscitation. Anesthesiology. 1963;24:203–207.
49 Michael J, Guerci A, Koehler R, et al. Mechanisms by which epinephrine augments cerebral and myocardial perfusion during cardiopulmonary resuscitation in dogs. Circulation. 1984;69:822–835.
50 Koehler R, Michael J. Cardiopulmonary resuscitation, brain blood flow, and neurologic recovery. Crit Care Clin. 1985;1:205–222.
51 Schleien CL, Dean JM, Koehler RC, et al. Effect of epinephrine on cerebral and myocardial perfusion in an infant animal preparation of cardiopulmonary resuscitation. Circulation. 1986;73:809–817.
52 Niemann JT, Criley JM, Rosborough JP, Niskanen RA, Alferness C. Predictive indices of successful cardiac resuscitation after prolonged arrest and experimental cardiopulmonary resuscitation. Ann Emerg Med. 1985;14:521–528.
53 Sanders A, Ewy G, Taft T. Prognostic and therapeutic importance of the aortic diastolic pressure in resuscitation from cardiac arrest. Crit Care Med. 1984;12:871–873.
54 Downey J, Chagrasulis R, Hemphill V. Quantitative study of intramyocardial compression in the fibrillating heart. Am J Physiol Cell Physiol. 1979;237:H191–H196.
55 Otto C, Yakaitis R. The role of epinephrine in CPR: a reappraisal. Annals of Emergency Medicine. 1984;13(2):840–843.
56 von Planta I, Wagner O, von Planta M, et al. Coronary perfusion pressure, end-tidal CO2 and adrenergic agents in haemodynamic stable rats. Resuscitation. 1993;25:203–217.
57 Perondi M, Reis A, Paiva E, et al. A comparison of high-dose and standard-dose epinephrine in children with cardiac arrest. N Engl J Med. 2004;350:1722–1730.
58 Lindner K, Prengel A, Pfenninger E, et al. Vasopressin improves vital organ blood flow during closed-chest cardiopulmonary resuscitation in pigs. Circulation. 1995;91:215–221.
59 Prengel AW, Lindner KH, Keller A. Cerebral oxygenation during cardiopulmonary resuscitation with epinephrine and vasopressin in pigs. Stroke. 1996;27:1241–1248.
60 Lindner KH, Dirks B, Strohmenger HU, et al. Randomised comparison of epinephrine and vasopressin in patients with out-of-hospital ventricular fibrillation. Lancet. 1997;349:535–537.
61 Babbs CF, Berg RA, Kette F, et al. Use of pressors in the treatment of cardiac arrest. Ann Emerg Med. 2001;37(4 Suppl):S152–S162.
62 Wenzel V, Krismer AC, Voelckel WG, et al. The use of arginine vasopressin during cardiopulmonary resuscitation: an analysis of experimental and clinical experience and a view of the future. Anaesthetist. 2002;51:191–202.
63 Voelckel WG, Lurie KG, McKnite S, et al. Comparison of epinephrine and vasopressin in a pediatric porcine model of asphyxial cardiac arrest. Crit Care Med. 2000;28:3777–3783.
64 Mann K, Berg RA, Nadkarni V. Beneficial effects of vasopressin in prolonged pediatric cardiac arrest: a case series. Resuscitation. 2002;52:149–156.
65 Matok I, Vardi A, Augarten A, et al. Beneficial effects of terlipressin in prolonged pediatric cardiopulmonary resuscitation: a case series. Crit Care Med. 2007;35:1161–1164.
66 Gil-Anton J, Lopez-Herce J, Morteruel E, Carrillo A, Rodriguez-Nunez A. Pediatric cardiac arrest refractory to advanced life support: is there a role for terlipressin? Pediatr Crit Care Med. 2010;11:139–141.
67 Brown JH, Laiken N. Muscarinic receptor agonists and antagonists. In Brunton LL, Chabner BA, Knollmann BC, eds.: Goodman & Gilman’s the pharmacological basis of therapeutics, 12th ed, New York: McGraw-Hill, 2011. Chapter 9 http://www.accessmedicine.com/content.aspx?aID=16660596 Accessed September 20, 2012
68 Dauchot P, Gravenstein JS. Effects of atropine on the electrocardiogram in different age groups. Clin Pharm Ther. 1971;12:274–280.
69 Kottmeier C, Gravenstein J. The parasympathomimetic activity of atropine and atropine methylbromide. Anesthesiology. 1968;29:1125–1133.
70 Huang YG, Wong KC, Yip WH, et al. Cardiovascular responses to graded doses of three catecholamines during lactic and hydrochloric acidosis in dogs. Br J Anaesth. 1995;74:83–590.
71 Burchfield D, Preziosi M, Lucas V, et al. Effects of graded doses of epinephrine during asphyxia-induced bradycardia in newborn lambs. Resuscitation. 1993;25:235–244.
72 Pannier J, Leusen I. Contraction characteristics of papillary muscle during changes in acid-base composition of the bathing-fluid. Arch Int Physiol Biochim. 1968;76:624–634.
73 Wood W, Manley EJ, Woodbury R. The effects of CO2-induced respiratory acidosis on the depressor and pressor components of the dog’s blood pressure to epinephrine. J Pharmacol Exp Ther. 1963;139:238.
74 Dohrmann ML, Goldschlager NF. Myocardial stimulation threshold in patients with cardiac pacemakers: effect of physiologic variables, pharmacologic agents, and lead electrodes. Cardiol Clin. 1985;3:527–537.
75 Bishop R, Weisfeldt M. Sodium bicarbonate administration during cardiac arrest: effect on arterial pH, Pco2, and osmolality. JAMA. 1976;235:506–509.
76 Grundler W, Weil M, Rackow E. Arteriovenous carbon dioxide and pH gradients during cardiac arrest. Circulation. 1986;74:1071–1074.
77 Sessler D, Mills P, Gregory G, et al. Effects of bicarbonate on arterial and brain intracellular pH in neonatal rabbits recovering from hypoxic lactic acidosis. J Pediatr. 1987;111:617–623.
78 Cohen Y, Chang L, Litt L, et al. Stability of brain intracellular lactate and 31P-metabolite levels at reduced intracellular pH during prolonged hypercapnia in rats. J Cereb Blood Flow Metab. 1990;10:277–284.
79 Eleff S, Sugimoto H, Shaffner D, et al. Acidemia and brain pH during prolonged cardiopulmonary resuscitation in dogs. Stroke. 1995;26:1028–1034.
80 Morley P, Hogan M, Hakim A. Calcium-mediated mechanisms of ischemic injury and protection. Brain Pathol. 1994;4:37–47.
81 Bristow M, Schwartz H, Binetti G, et al. Ionized calcium and the heart: elucidation of in vivo concentration-response relationships in the open-chest dog. Circ Res. 1977;41:565–574.
82 Urban P, Scheidegger D, Buchmann B, et al. Cardiac arrest and blood ionized calcium levels. Ann Intern Med. 1988;109:110–113.
83 Cairns C, Niemann J, Pelikan P, et al. Ionized hypocalcemia during prolonged cardiac arrest and closed-chest CPR in a canine model. Ann Emerg Med. 1991;20:1178–1182.
84 Coté C, Drop L, Hoaglin D, et al. Ionized hypocalcemia after fresh frozen plasma administration to thermally injured children: effects of infusion rate, duration, and treatment with calcium chloride. Anesth Analg. 1988;67:152–160.
85 Heining M, Band D, Linton R. Choice of calcium salt: a comparison of the effects of calcium chloride and gluconate on plasma ionized calcium. Anaesthesia. 1984;39:1079–1082.
86 Coté C, Drop L, Daniels A, et al. Calcium chloride versus calcium gluconate: comparison of ionization and cardiovascular effects in children and dogs. Anesthesiology. 1987;66:465–470.
87 Srinivasan V, Morris MC, Helfaer MA, et al. Calcium use during in-hospital pediatric cardiopulmonary resuscitation: a report from the National Registry of Cardiopulmonary Resuscitation. Pediatrics. 2008;121:e1144–e1151.
88 Siesjo BK. Cerebral circulation and metabolism. J Neurosurg. 1984;60:883–908.
89 Pulsinelli WA, Levy DE, Sigsbee B, et al. Increased damage after ischemic stroke in patients with hyperglycemia with or without established diabetes mellitus. Am J Med. 1983;74:540–544.
90 Longstreth WJ, Inui T. High blood glucose level on hospital admission and poor neurological recovery after cardiac arrest. Ann Neurol. 1984;15:59–63.
91 Ashwal S, Schneider S, Tomasi L, et al. Prognostic implications of hyperglycemia and reduced cerebral blood flow in childhood near-drowning. Neurology. 1990;40:820–823.
92 Longstreth WJ, Diehr P, Cobb L, et al. Neurologic outcome and blood glucose levels during out-of-hospital cardiopulmonary resuscitation. Neurology. 1986;36:1186–1191.
93 Sarkozy A, Dorian P. Strategies for reversing shock-resistant ventricular fibrillation. Curr Opin Crit Care. 2003;9:189–193.
94 Coumel P, Lucet V, Do Ngoc D. The use of amiodarone in children. Pacing Clin Electrophysiol. 1983;6(5 pt 1):930–939.
95 Pickoff AS, Singh S, Flinn CJ, et al. Use of amiodarone in the therapy of primary ventricular arrhythmias in children. Dev Pharmacol Ther. 1983;6:73–82.
96 Coumel P, Fidelle J. Amiodarone in the treatment of cardiac arrhythmias in children: one hundred thirty-five cases. Am Heart J. 1980;100(6 pt 2):1063–1069.
97 Somberg JC, Bailin SJ, Haffajee CI, et al. Intravenous lidocaine versus intravenous amiodarone (in a new aqueous formulation) for incessant ventricular tachycardia. Am J Cardiol. 2002;90:853–859.
98 Silvetti MS, Drago F, Bevilacqua M, Ragonese P. Amiodarone-induced torsades de pointes in a child with dilated cardiomyopathy. Ital Heart J. 2001;2:231–236.
99 Jafari-Fesharaki M, Scheinman MM. Adverse effects of amiodarone. Pacing Clin Electrophysiol. 1998;21(1 pt 1):108–120.
100 Jessurun GA, Boersma WG, Crijns HJ. Amiodarone-induced pulmonary toxicity: predisposing factors, clinical symptoms and treatment. Drug Saf. 1998;18:339–344.
101 Birmingham WP. More on an infant with acute pulmonary toxicity during amiodarone therapy. Am J Cardiol. 1998;81:1171.
102 Greenblatt D, Gross P, Bolognini V. Pharmacotherapy of cardiopulmonary arrest. Am J Hosp Pharm. 1976;33:579–583.
103 Lichstein E, Chadda K, Gupta P. Atrioventricular block with lidocaine therapy. Am J Cardiol. 1973;31:277–281.
104 Dorian P, Cass D, Schwartz B, et al. Amiodarone as compared with lidocaine for shock-resistant ventricular fibrillation. N Engl J Med. 2002;346:884–890.
105 Bhananker SM. anesthesia-related cardiac arrest in children: update from the Pediatric Perioperative Cardiac Arrest Registry. Anesth. Analg. 2007;105:344–350.
106 Geiduschek JM. Registry offers insight on preventing cardiac arrest in children. Soc Am Anesth. 1998;62:6–18.
107 Green A, Giattina K. Adenosine administration for neonatal SVT. Neonatal Netw. 1993;12:15–18.
108 Epstein M, Kiel E, Victorica B. Cardiac decompensation following verapamil therapy in infants with supraventricular tachycardia. Pediatrics. 1985;75:737–740.
109 Hopson J, Buxton A, Rinkenberger R, et al. Safety and utility of flecainide acetate in the routine care of patients with supraventricular tachyarrhythmias: results of a multicenter trial. The Flecainide Supraventricular Tachycardia Study Group. Am J Cardiol. 1996;77:72A–82A.
110 DelGurecio L, Feins N, Cohn J, et al. Comparison of blood flow during external and internal cardiac message in man. Circulation. 1965;3(Suppl 1):I171.
111 Bircher N, Safar P, Stewart R. A comparison of standard, “MAST”-augmented, and open-chest CPR in dogs: a preliminary investigation. Crit Care Med. 1980;8:147–152.
112 Weiser F, Adler L, Kuhn L. Hemodynamic effects of closed and open-chest cardiac resuscitation in normal dogs and those with acute myocardial infarction. Am J Cardiol. 1962;10:555.
113 Kern K, Sanders A, Ewy G. Open-chest cardiac massage after closed-chest compression in a canine model: when to intervene. Resuscitation. 1987;15:51–57.
114 Sanders A, Kern K, Atlas M, et al. Importance of the duration of inadequate coronary perfusion pressure on resuscitation from cardiac arrest. J Am Coll Cardiol. 1985;6:113–118.
115 Hachimi-Idrissi S, Leeman J, Hubloue Y, et al. Open chest cardiopulmonary resuscitation in out-of-hospital cardiac arrest. Resuscitation. 1997;35:151–156.
116 Sanders A, Kern K, Ewy G, et al. Improved resuscitation from cardiac arrest with open-chest massage. Ann Emerg Med. 1984;13:672–675.
117 Morris M, Wernovsky G, Helfaer M, Nadkarni V. Survival outcomes following extracorporeal cardiopulmonary resuscitation from in-hospital pediatric cardiac arrest. Pediatric. Crit Care Med. 2004;5:440–446.
118 Duncan B, Ibrahim A, Hraska V, et al. Use of rapid-deployment extracorporeal membrane oxygenation for the resuscitation of pediatric patients with heart disease after cardiac arrest. J Thorac Cardiovasc Surg. 1998;116:305–311.
119 Ghez O, Fouilloux V, Charpentier A, Fesquet P, et al. Absence of rapid deployment extracorporeal membrane oxygenation (ECMO) team does not preclude resuscitation ECMO in pediatric cardiac patients with good results. ASAIO J. 2007;53:692–695.
120 Cohen T, Tucker K, Redberg R, et al. Active compression-decompression resuscitation: a novel method of cardiopulmonary resuscitation. Am Heart J. 1992;124:1145–1150.
121 Tucker K, Khan J, Savitt M. Active compression-decompression resuscitation: effects on pulmonary ventilation. Resuscitation. 1993;26:125–131.
122 Cohen T, Tucker K, Lurie K, et al. Active compression-decompression: a new method of cardiopulmonary resuscitation. Cardiopulmonary Resuscitation Working Group. JAMA. 1992;267:2916–2923.
123 Lindner K, Pfenninger E, Lurie K, et al. Effects of active compression-decompression resuscitation on myocardial and cerebral blood flow in pigs. Circulation. 1993;88:1254–1263.
124 Shultz J, Coffeen P, Sweeney M, et al. Evaluation of standard and active compression-decompression CPR in an acute human model of ventricular fibrillation. Circulation. 1994;89:684–693.
125 Lurie K, Coffeen P, Shultz J, et al. Improving active compression-decompression cardiopulmonary resuscitation with an inspiratory impedance valve. Circulation. 1995;91:1629–1632.
126 Lurie K, Shultz J, Callaham M, et al. Evaluation of active compression-decompression CPR in victims of out-of-hospital cardiac arrest. JAMA. 1994;271:1405–1411.
127 Cohen T, Goldner B, Maccaro P, et al. A comparison of active compression-decompression cardiopulmonary resuscitation with standard cardiopulmonary resuscitation for cardiac arrests occurring in the hospital. N Engl J Med. 1993;329:1918–1921.
128 Plaisance P, Adnet F, Vicaut E, et al. Benefit of active compression-decompression cardiopulmonary resuscitation as a prehospital advanced cardiac life support: a randomized multicenter study. Circulation. 1997;95:955–961.
129 Stiell I, Hebert P, Wells G, et al. The Ontario trial of active compression-decompression cardiopulmonary resuscitation for in-hospital and prehospital cardiac arrest. JAMA. 1996;275:1417–1423.
130 Schwab T, Callaham M, Madsen CD, et al. A randomized clinical trial of active compression-decompression CPR vs. standard CPR in out-of-hospital cardiac arrest in two cities. JAMA. 1995;273:1261–1268.
131 Mauer D, Schneider T, Dick W, et al. Active compression-decompression resuscitation: a prospective, randomized study in a two-tiered EMS system with physicians in the field. Resuscitation. 1996;33:125–134.
132 Rabl W, Baubin M, Broinger G, et al. Serious complications from active compression-decompression cardiopulmonary resuscitation. Int J Legal Med. 1996;109:84–89.
133 Doherty DR, Parshuram CS, Gaboury I, et al. Hypothermia therapy after pediatric cardiac arrest. Circulation. 2009;119:1492–1500.
134 Fink EL, Clark RS, Kochanek PM, Bell MJ, Watson RS. A tertiary care center’s experience with therapeutic hypothermia after pediatric cardiac arrest. Pediatr Crit Care Med. 2010;11:66–74.
American Heart Association. American Heart Association guidelines for cardiopulmonary resuscitation and emergency cardiovascular care. Circulation. 2010;122:S640–S933.
Nadkarni VM, Larkin GL, Peberdy MA, et al. First documented rhythm and clinical outcome from in-hospital cardiac arrest among children and adults. JAMA. 2006;295:50–57.
Perondi M, Reis A, Paiva E, et al. A comparison of high-dose and standard-dose epinephrine in children with cardiac arrest. N Engl J Med. 2004;350:1722–1730.
[/level-membership-for-anesthesiology-category][not-level-membership-for-anesthesiology-category]
39 Cardiopulmonary Resuscitation
Epidemiology and Outcome of In-hospital Cardiopulmonary Arrest
Mechanics of Cardiopulmonary Resuscitation
Defibrillation and Cardioversion
Vascular Access and Monitoring during Cardiopulmonary Resuscitation
Medications Used during Cardiopulmonary Resuscitation
Special Cardiac Arrest Situations
Historical Background
In 1814, a description in poetical form of the Rules of the Humane Society for recovering drowned persons included the following description of mouth-to-mouth resuscitation1:
Let one the mouth, and either nostril close
While through the other the bellows gently blows.
Thus the pure air with steady force convey,
To put the flaccid lungs again in play.
Should bellows not be found, or found too late,
Let some kind soul with willing mouth inflate;
External cardiac massage was successfully conducted more than 100 years ago in two children (ages 8 and 13 years) after circulatory arrest precipitated by chloroform anesthesia during a surgical procedure.2 In 1904, Crile described the effectiveness of external cardiac compressions in maintaining the circulation of dogs.3
After multiple reports that attested to the effectiveness of mouth-to-mouth resuscitation,4–6 in 1958 the National Academy of Sciences National Research Council recommended mouth-to-mouth resuscitation with maximum backward tilt of the head as the preferred technique for all individuals requiring emergency artificial ventilation. In 1960, external cardiac compression was revived as a resuscitation technique when Kouwenhoven, Jude, and Knickerbocker7 demonstrated its effectiveness when combined with artificial respirations. Many of their patients, including the first, were in cardiac arrest as a result of anesthesia. Before this study, internal cardiac compression was the accepted technique, with its effectiveness demonstrated by experience in cardiac bypass surgery. In 1947, Beck and associates8 successfully internally defibrillated the human heart; and in 1956, Zoll and colleagues9 performed the first successful external defibrillation of a human heart.
Epidemiology and Outcome of In-Hospital Cardiopulmonary Arrest
A 2009 review of cardiac arrest events submitted to the National Registry of Cardiopulmonary Circulation included 3342 pediatric events, excluding events in a delivery room or neonatal intensive care unit (NICU).10 Seventy-three percent of the inpatient cardiac arrests reported occurred in an ICU, 7% in a general inpatient area, 11% in an emergency department, and 3% in an operating room or postanesthesia care unit. Return of spontaneous circulation (ROSC) was achieved in 65%, 24-hour survival occurred in 47%, and 30% of children survived until hospital discharge. Other large series of in-hospital pediatric cardiac arrest report survival until hospital discharge ranging from 14% to 44%,11–14 with the 44% survival representing cardiac arrests that occurred in a pediatric cardiac ICU. In another multicenter cohort study of in-hospital pediatric cardiac arrest,15 48.7% of the 353 children survived until hospital discharge. Survivors had greater body temperatures, greater pH values, and reduced serum lactate concentrations compared with nonsurvivors. Nonsurvivors were more likely to have a tracheal tube before the arrest, and to receive sodium bicarbonate, calcium, and vasopressin during the arrest. In this study, postoperative cardiopulmonary resuscitation (CPR) was associated with decreased mortality.
Mechanics of Cardiopulmonary Resuscitation
Airway
In the child without an artificial airway, the use of BVM devices may result in a significant risk of gastric inflation, followed by pulmonary aspiration of gastric contents. Abdominal distention (gastric and bowel) can significantly compromise oxygenation; therefore the stomach should be vented when excessive gastric inflation occurs. One study found a 28% incidence of pulmonary aspiration in a series of failed resuscitations.16 For this reason as well as for the risk of barotrauma and volutrauma, excessive inflation pressures should be avoided. However, effective bilateral ventilation is best judged by visualizing bilateral chest excursions and listening to the quality of the breath sounds rather than setting a preset maximal inflation pressure.
Breathing
Overventilation is common during CPR, resulting in greater mean intrathoracic pressures than required, which decreases venous return and reduces cardiac output.17 In cardiopulmonary arrest, a less than normal minute ventilation may be appropriate, because cardiac output and delivery of CO2 to the lungs are diminished. If an artificial airway is not in place for single person rescue, two breaths should be given for each 30 chest compressions. If an artificial airway is not in place for two person rescue, two breaths should be given after each 15 chest compressions. Once an artificial airway is in place, a ventilator rate of 8 to 10 per minute without pausing during rapid chest compressions should be used (Table 39-1).
Circulation
During cardiac arrest, chest compressions provide the sole perfusion to a child’s vital organs; therefore optimal performance of CPR is critical. Key elements to providing quality chest compressions include (1) ensuring an adequate rate (100 compressions per minute), (2) ensuring adequate chest wall depression (one third to half of the anteroposterior chest diameter), (3) releasing completely between compressions to allow full chest wall recoil, (4) minimizing interruptions in chest compressions, and (5) ensuring that the child is on a sufficiently hard surface to allow effective chest compressions.18 In short, push hard and push fast, release completely, and do not interrupt compressions unnecessarily. Incomplete recoil during CPR is associated with higher intrathoracic pressures and significantly decreased venous return, and coronary and cerebral perfusion.19
If a child is small enough (e.g., younger than 6 months) that the person providing chest compressions can comfortably encircle the chest with his or her hands, chest compressions should be performed using the circumferential technique, with thumbs depressing the sternum and the fingers supporting the infant’s back and circumferentially squeezing the thorax (Fig. 39-1). In larger infants, the sternum can be compressed using two fingers; and in the child, either one or two hands can be used, depending on the size of the child and of the rescuer.19 Whichever method is used, focused attention must remain on delivering effective compressions with minimal interruptions.20 In all cases other than circumferential CPR, a backboard must be used. Properly delivered chest compressions are tiring to the provider, and providers should rotate approximately every 2 minutes to prevent compressor fatigue and deterioration in the quality and rate of chest compressions.19
Mechanisms of Blood Flow
External chest compressions provide cardiac output through two mechanisms: the cardiac pump mechanism and the thoracic pump mechanism. By the cardiac pump mechanism of blood flow, blood is squeezed from the heart by compression of the heart between the sternum and the vertebral column, exiting the heart only anterograde because of closure of the atrioventricular valves. Between compressions, ventricular pressure decreases below atrial pressure, allowing the atrioventricular valves to open and the ventricles to fill. This sequence of events resembles the normal cardiac cycle. Although the cardiac pump is likely not the dominant blood flow mechanism during most closed-chest CPR, specific clinical situations have been identified in which the cardiac pump mechanism is more prominent. For example, a smaller, more compliant chest may allow for more direct cardiac compression (Fig. 39-2). Increasing, the applied force during chest compressions also increases the likelihood of direct cardiac compression.
Several observations do not support the cardiac pump as the primary mechanism of blood flow during CPR. Angiographic studies show that blood passes from the vena cava through the right heart into the pulmonary artery and from the pulmonary veins through the left heart into the aorta during a single chest compression.21,22 Echocardiographic studies show that the atrioventricular valves are open during blood ejection.21,23,24 Without closure of atrioventricular valves during chest compression, the cardiac pump mechanisms cannot account for forward movement of blood during CPR.
In 1976, Criley and colleagues25 made the dramatic observation that several patients who developed ventricular fibrillation during cardiac catheterization produced enough blood flow to maintain consciousness by repetitive coughing.25 The production of blood flow by increasing thoracic pressure without direct cardiac compression describes the thoracic pump mechanism, in which the heart is a passive conduit for blood flow. The intrathoracic pressure is greater than the extrathoracic pressure during the compression phase of CPR, at which time blood flows out of the thorax, with venous valves preventing excessive retrograde blood flow (Fig. 39-3). Experimental and clinical data support both mechanisms of blood flow during CPR in human infants.
Rate and Duty Cycle
The recommended rate of chest compressions for all patients is 100 per minute, with great care taken to minimize interruptions in chest compressions and to ensure adequate compression depth.20 This rate represents a compromise that attempts to maximize contributions from both the thoracic pump and cardiac pump mechanism of blood flow.
Duty cycle is defined as the percent of the compression–relaxation cycle that is devoted to compression. If blood flow is generated by direct cardiac compression, then primarily the force of compression determines the stroke volume. Prolonging the compression (increasing the duty cycle) beyond the time necessary for full ventricular ejection should have no additional effect on stroke volume. Increasing the rate of compressions should increase cardiac output, because a fixed volume of blood is ejected with each cardiac compression. In contrast, if blood flow is produced by the thoracic pump mechanism, the volume of blood that is ejected comes from a large reservoir of blood contained within the capacitance vessels in the chest. With the thoracic pump mechanism, flow is enhanced by increasing either the force of compression or the duty cycle but is not affected by changes in compression rate over a wide range of rates, given a set duty cycle.26
Different animal models yield conflicting results as to the optimal compression rate and duty cycle. However, a rate of compression during conventional CPR of 100 per minute satisfies both those who prefer the faster rates and those who support a longer duty cycle. This is true because it is easier to produce a longer duty cycle when compressions are administered at a faster rate.27,28
Defibrillation and Cardioversion
Electric Countershock
Electric countershock, or defibrillation, is the treatment of choice for ventricular fibrillation and pulseless ventricular tachycardia. Defibrillation should not be delayed to secure an airway, because the likelihood of restoring an organized rhythm decreases with increased duration of fibrillation. Ventricular fibrillation is terminated by simultaneous depolarization and sustained contraction of a critical mass of myocardium,29 allowing return of spontaneous, coordinated cardiac contractions, assuming the myocardium is well oxygenated and the acid-base status is relatively normal. Drug treatment may be required as an adjunct to defibrillation, but by itself cannot be relied on to terminate ventricular fibrillation.
An older generation of defibrillators that is still present in many hospitals delivers energy in a monophasic damped sinusoidal waveform (Fig. 39-4, A). This type of instrument delivers a single, unidirectional current with a gradual decrease to zero current. By contrast, the newer generation of biphasic defibrillators delivers a current in a positive direction for a set period, followed by a reversal in current (see Fig. 39-4, B). Biphasic defibrillators are more effective than monophasic defibrillators in terminating ventricular fibrillation in adults; therefore their use is recommended where possible.
FIGURE 39-4 Energy delivery during conventional monophasic (A) and biphasic truncated exponential (B) defibrillation.
In the majority of adult cases, energy levels of 100 to 200 joules are successful when shocks are delivered with minimal delay.30,31 The goal of defibrillation is to deliver a minimum of electrical energy to a critical mass of ventricular muscle while avoiding excessive current that could further damage the heart. The most reliable predictor of success of defibrillation is the duration of fibrillation before the first countershock.32 Acidosis and hypoxemia also decrease the success of defibrillation.32
Practical Aspects of Defibrillation in Children
For children with in-hospital ventricular fibrillation or pulseless ventricular tachycardia, defibrillation should be attempted as soon as possible, with optimal CPR until the defibrillator is ready to deliver a shock. For the first defibrillation attempt, 2 joules/kg of delivered energy should be administered (Fig. 39-5). After shock delivery, CPR should resume immediately with chest compressions for five duty cycles (2 minutes). If one shock fails to eliminate ventricular fibrillation, the incremental benefit of another immediate shock is small. Resumption of CPR is likely to confer a greater benefit than another shock. CPR may provide coronary perfusion, increasing the likelihood of defibrillation with a subsequent shock. It is important to minimize the time between chest compressions and shock delivery and between shock delivery and resumption of postshock compressions.20 Approximately 2 minutes of CPR should be delivered before a second attempt at defibrillation at twice the original energy level (4 joules/kg).20
If ventricular fibrillation or pulseless ventricular tachycardia persists beyond the second defibrillation attempt, standard doses of epinephrine should be administered (with subsequent doses every 3 to 5 minutes during persistent cardiac arrest). After 2 minutes of chest compressions, defibrillation should be attempted again, followed by administration of amiodarone (5 mg/kg) or lidocaine (1 mg/kg) with subsequent defibrillation attempts. It is not necessary to increase the energy level on each successive shock during defibrillation after the second dose. However, successful defibrillation has been reported with currents in excess of 4 joules/kg without adverse sequelae, up to a maximum dose not exceeding 10 joules/kg or the adult level, whichever is less.20 This sometimes occurs when a fixed energy level, adult automated external defibrillator (AED) is used in a small child.
Automated External Defibrillation
Use of AEDs is now standard therapy in out-of-hospital resuscitation of adults.19,30 AEDs are now deemed appropriate for use in children older than 1 year. If available, use of pediatric attenuator pads or a pediatric mode on the AED should be used in children 1 to 8 years of age, but if unavailable (and a standard defibrillator is similarly unavailable), an unmodified AED should be used.
Vascular Access and Monitoring during Cardiopulmonary Resuscitation
Vascular Access and Fluid Administration
One of the key aspects of successful CPR is early establishment of a route for administration of fluids and medications. If intravenous access cannot be established rapidly, the intraosseous or endotracheal route should be used (see Chapter 48).
Intraosseous Access
The intraosseous route can be used to administer all medications and fluids used during CPR, including whole blood. An intraosseous needle also may be used to obtain initial blood samples, although acid-base analysis will be inaccurate after administration of sodium bicarbonate via the intraosseous needle. Intraosseous access should be considered a temporary measure during emergencies when other access is not available. The placement of an intraosseous needle in the older child (older than 10 years) and adult, although possible, is difficult owing to the thick bony cortex; however, a 50% success rate has been reported in these age-groups.33
The technique of placing an intraosseous line is straightforward. A specialized intraosseous needle or, if not available, a standard 16- or 18-gauge needle, a spinal needle with stylet, or bone marrow needle is inserted into the anterior surface of the tibia 1 to 2 cm below and 1 cm medial to the tibial tuberosity (avoiding the epiphyseal plate). The needle is directed at 90 degrees to the anteromedial surface of the tibia, just distal to the tuberosity (see Fig. 48-6 and E-Fig. 48-1). When the needle passes through the cortex into the marrow, a sudden loss of resistance is sensed. Successful placement has been achieved if the needle is in the marrow cavity, as evidenced by the needle standing upright without support. If the needle has slipped into the subcutaneous tissue, its upright position cannot be maintained without support. Free flow of the infusate without significant subcutaneous infiltration should also be demonstrated. The technique has a small complication rate,34 although possible complications include osteomyelitis, fat and bone marrow embolisms, and compartment syndrome. To avoid these potential complications, intravenous access should replace intraosseous access as soon as possible. The onset of action and concentration of most drugs after intraosseous administration are comparable with venous administration.35 A relatively new device, the EZ-IO (Vidacare, Shavano Park, Tex.) provides the most rapid means for intraosseous access (see E-Fig. 48-1).
Endotracheal Medication Administration
In the absence of other vascular access, medications including lidocaine, atropine, naloxone, and epinephrine (mnemonic LANE) can be administered via the endotracheal tube.36,
[/not-level-membership-for-anesthesiology-category]
