33 Cardiopulmonary Cerebral Resuscitation
Between 1960—when closed-chest compressions were first introduced—and 2000, there was little or no change in long-term survival after cardiac arrest.1,2 However, regional efforts to improve resuscitation practices at multiple levels, including the emergency response and the post–cardiac arrest care, have resulted in significant improvements in meaningful survival over time.3,4 There is accumulating evidence about which aspects of post–cardiac arrest management influence final outcomes,5,6 and specific patterns of physiologic changes after cardiac arrest have been described.7 Improving outcome will require an integrated approach to immediate resuscitation and subsequent intensive care management. This chapter will review the epidemiology of cardiac arrest, the initial approach for reversing cardiopulmonary arrest, modifications of this approach appropriate for specific disease states, and post–cardiac arrest care designed to minimize brain injury.
 Epidemiology
Epidemiology
In industrialized countries, heart disease is the overall leading cause of death. Estimates of the incidence of cardiopulmonary arrest outside the hospital vary from 55 to 100 to 120 events per 100,000 people per year,8,9,10 with one large sample estimating a median incidence of 52.1 per 100,000 people per year.11 Likewise, median survival after out-of-hospital cardiac arrest is estimated at 8.4%,11 but the range varies from several large U.S. cities that reported survival rates less than 2%12,13 to exemplary systems with survival over 16%.11 The incidence of cardiac arrest in the hospital is about 0.17 events per hospital bed per year.14 For inpatients experiencing cardiac arrest, survival to hospital discharge is estimated at 17%. Less than half of cardiac arrests occur in an intensive care unit (ICU) setting, and survival does not appear to be related to the location of collapse.15 As many as 17% of episodes of respiratory compromise in the hospital may progress to cardiac arrest.16
Demographic features of sudden cardiac death are similar to the characteristics of cardiovascular disease. Sudden cardiac death is more common in males than females both outside the hospital10 and in the hospital.14 However, the incidence of cardiac arrest is higher in women (6%) than in men (4.4%) who are admitted to the hospital for acute myocardial infarction (MI).17 Cardiac arrest outside the hospital affects blacks more than whites or Asians.10,12 In addition, cardiac arrest is more common in areas with lower socioeconomic status,18 and survival may be worse for individuals in regions with lower property values.19 While sudden death can affect patients of all ages, the mean age for sudden cardiac arrest is between 65 and 70 years in most studies.10,11,14
Two temporally and mechanistically separate processes contribute to mortality: cardiopulmonary collapse and neurologic injury. In evidence of the first process, only one-third of patients who collapse outside of the hospital have restoration of circulation long enough to be admitted to the hospital. Likewise, only 44% of patients who collapse in the hospital have return of circulation.14 In evidence of the second process, two-thirds of patients admitted to the hospital after out-of-hospital collapse20 and 60% of patients resuscitated from cardiac arrest in the hospital14 die prior to discharge from the hospital. The most common reason for death among patients admitted to the ICU after out-of-hospital cardiac arrest is postischemic brain injury, whereas multiple organ failure is more common for patients after in-hospital cardiac arrest.21 Failure to awaken contributes to withdrawal of care and in-hospital death for as many as 44% to 68% of patients after initial restoration of circulation.14,22
 Restoring Circulation
Restoring Circulation
Acute treatment of cardiac arrest consists of two concurrent, goal-directed activities: (1) artificial circulation (usually chest compressions augmented by peripheral vasoconstrictors) to circulate oxygenated blood to heart and brain and (2) electric shock to terminate ventricular fibrillation (VF) and unstable tachyarrhythmias. There is increasing recognition that continuous, uninterrupted chest compressions are critically important for restoring circulation.23 Electrical rescue shocks are used only when appropriate. Rescue shock is the only procedure for which interruption of artificial circulation is absolutely necessary and justified.
Continuous reassessment of the patient can be reduced to constant awareness of two parameters (Figure 33-1). The organization of the electrocardiogram (ECG) and the presence of pulses will prompt appropriate selection of therapy. The recommended division of time and prioritization of activities to accomplish these goals is depicted in Figure 33-2. All other activities, including medications and advanced airway maneuvers, are designed to supplement these two core activities. Optimization of resuscitation requires that any interruption in the two core activities, especially artificial circulation, be minimized.
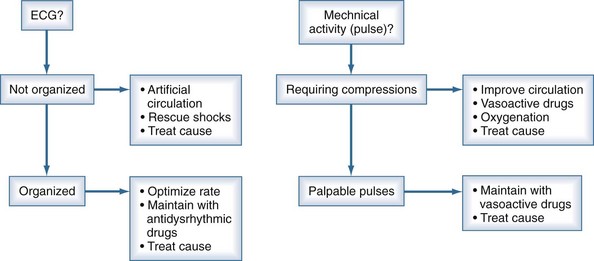
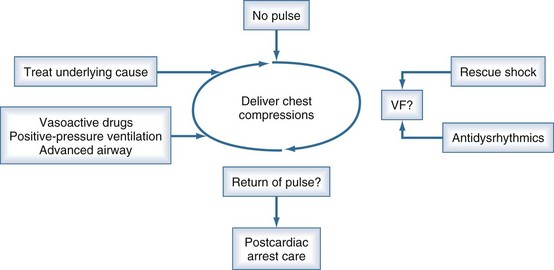
The American Heart Association and European Resuscitation Council provide consensus scientific statements about the acute management of cardiac arrest.24 Those guidelines have detailed review of specific drugs and procedures. The following section provides an overview of airway management, circulation support, rescue shock for defibrillation, and drug therapy during cardiac arrest.
Airway and Ventilation
Obstruction of the airway can occur in any patient with impaired consciousness including cardiac arrest.25 If uncorrected, this obstruction prevents oxygenation and ventilation, leading to or perpetuating cardiopulmonary collapse. In patients who are comatose because of primary cardiac arrest, the airway usually is not patent. Animal models typically do not mimic ventilation through the human airway, because the most common research animals (dogs and swine) have straight oral-tracheal passages.
Agonal respirations occur after acute cardiac arrest for an additional 1 to 2 minutes.26 These respirations may confuse lay people, delaying recognition of cardiac arrest. It is unclear whether agonal respirations can generate sufficient ventilation to support life. The presence of gasping is associated with survival, but it also may be a surrogate marker for brief collapse-to-resuscitation intervals.27 Regardless, the amplitude and frequency of agonal respirations declines over 1 to 2 minutes, necessitating artificial ventilation for all patients requiring more than momentary resuscitation efforts.
Simple maneuvers can open the human airway. Extension of the neck (head tilt) and forward displacement of the mandible (chin lift) straightens and opens the pharynx. The tongue can be displaced from the posterior pharynx by insertion of an oropharyngeal airway. With these steps, positive-pressure ventilation can be provided using mouth-to-mouth or bag-valve-mask ventilation. A positive-pressure breath of as little as 400 mL in adults (6-7 mL/kg) delivered over 2 to 3 seconds will cause the chest to rise.28 Recent studies have indicated that hyperventilation or hyperexpansion of the chest can impair venous return and decrease circulation during resuscitation.29 In addition, minute ventilations that are smaller than those required for long-term support probably provide adequate gas exchange during cardiac arrest.
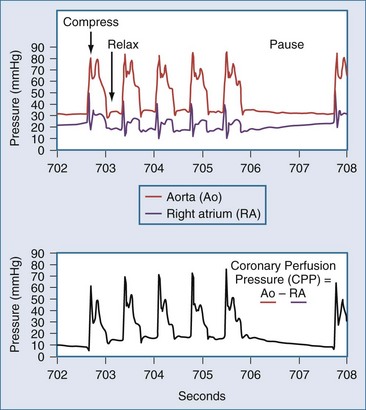
The optimal rate and timing for positive-pressure breathes during resuscitation in humans has not been established. The need for gas exchange must be balanced against the fact that interrupting chest compressions even for a few seconds can reduce coronary perfusion pressure (Figure 33-3).30 In swine, comparison of different ratios of chest compressions to ventilation suggests that 2 breaths per 50 chest compressions or more may be optimal for resuscitation.31 An extreme point of view is that chest compressions without any artificial ventilation may be sufficient to accomplish resuscitation in certain individuals.32 This position is contrary to early work demonstrating that the human airway collapses in most unconscious subjects, and that compressions alone are unable to provide ventilation.25 Recent studies in humans indicate that chest compressions generate tidal volumes that are likely less than the physiologic dead space.33 Nevertheless, passive insufflation of oxygen without positive-pressure ventilation has been reported to be beneficial in humans.34 As a compromise, some medical systems employ continuous chest compressions without pauses for positive-pressure ventilation. In these systems, positive-pressure breathes are delivered asynchronously by bag-mask during chest compressions, and the actual minute ventilation achieved is unknown. Therefore, the most recent guidelines recommend a ratio of 30 chest compressions to 2 ventilations, but even higher ratios or asynchronous ventilation may be optimal. Certainly, the duration of any pauses to deliver breaths must be minimized.
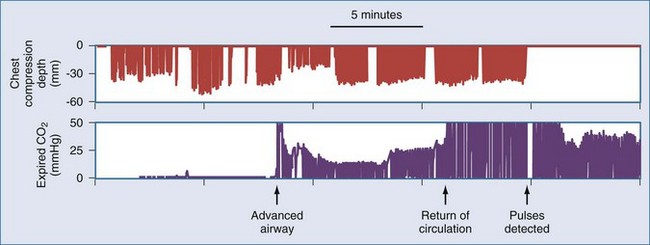
Waveform capnography is an extremely useful monitor during resuscitation, both for confirming ventilation and for monitoring adequacy of circulation. During cardiac arrest, end-tidal CO2 measurement is related to cardiac output and pulmonary blood flow.35 Therefore, CO2 levels may be very low (<10 mm Hg) at the onset of resuscitation. Adequate artificial circulation will cause CO2 levels to increase, and these levels may be used as a feedback to improve or modify chest compressions. Data from emergency department patients suggest that an end-tidal CO2 level greater than 15 to 16 mm Hg is associated with successful cardiac resuscitation.36,37 Conversely, end-tidal CO2 less than 10 mm Hg after 20 minutes of resuscitative efforts appears to confirm failure of resuscitation.38 However, drugs commonly used during resuscitation can disrupt the association between capnography readings and pulmonary blood flow. For example, epinephrine infusion reduces CO2 levels, and sodium bicarbonate infusion produces a transient but profound elevation of CO2 levels. An abrupt increase in end-tidal CO2 levels, usually to levels over 35 mm Hg, accompanies the return of spontaneous circulation. This finding may be useful for recognizing return of circulation, thereby minimizing any interruptions of chest compressions for pulse checks (Figure 33-4).
Airway Devices
A second difficulty with BVM ventilation is insufflation of the stomach.39 Excessive air in the stomach can promote emesis, and the abdominal distension may impair venous return and lung compliance.40 The esophagus prevents air entry into the stomach unless upper airway pressures exceed 15 to 20 cm H2O.41 However, esophageal muscle tone declines during cardiac arrest, allowing air to enter the stomach with upper airway pressures over 5 to 8 cm H2O.42 If the upper airway is not patent, providers may try to ventilate with increased pressure to achieve chest rise. Furthermore, rapid squeezing of the bag during the excitement of the situation results in too-high upper airway pressures. To avoid these problems, rescuers should emphasize gentle and well-paced inflation of the lungs during resuscitation.
Tracheal intubation can secure the airway definitively. A cuffed tracheal tube protects from emesis and maintains airway patency. However, laryngoscopy requires an interruption in chest compressions, and the tracheal tube by itself does not correct cardiac arrest. Observations of paramedics have documented extremely long interruption of chest compressions during “uncomplicated” tracheal intubation.43 Therefore, consideration should be made to delay tracheal intubation during initial resuscitation to reduce delays or interruptions of other life-saving interventions. After restoration of circulation, patients with coma or continued respiratory failure will require tracheal intubation.
Alternative supraglottic airway adjuncts such as double-lumen combination tracheal-esophageal tubes (e.g., Combi-tube), laryngeal tubes (e.g., King-LT), or laryngeal mask airways (LMA) can be used to temporarily manage the airway during resuscitation.44,45 These devices have the advantage that they can be inserted blindly in seconds without laryngoscopy and without interruption of chest compressions.39 The degree to which these devices can protect from aspiration is debated, but it is clear that they can allow adequate control of the airway to achieve resuscitation during cardiac arrest. Clinicians should strongly consider using these supraglottic airways as the first advanced airway during resuscitation because of the advantage of these devices for reducing interruptions in chest compressions.
Artificial Circulation
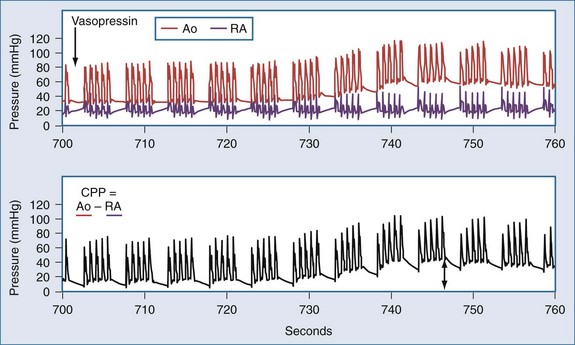
In the patient without pulses, circulation of blood can be accomplished by mechanical compression of the heart and chest. The critical parameter for restoring spontaneous circulation is the development of adequate coronary perfusion pressure (CPP). CPP is quantified by the pressure gradient between the aorta and the inside of the ventricles (usually approximated by the pressure in the right atrium or the central venous pressure [CVP]). Measurement of CPP in clinical practice is difficult unless the patient has invasive monitoring prior to cardiopulmonary collapse. CVP can be estimated from a central line, and peripheral arterial pressures developed approximate aortic pressures. In the spontaneously beating heart, most blood flows through the ventricular walls during diastole, when the ventricular pressure is lowest. With mechanical compression of the heart and chest during resuscitation, the primary perfusion of the heart occurs during the relaxation phase (see Figure 33-3). Therefore, CPP is usually measured at the end of the relaxation phase. CPP is highly correlated with myocardial perfusion, and consequently with the likelihood of resuscitation.46 In humans, return of circulation requires that the developed CPP exceed 15 to 20 mm Hg. It is likely that with CPP less than 15 mm Hg, perfusion is inadequate to replete the energy state of the myocardium during cardiac arrest.
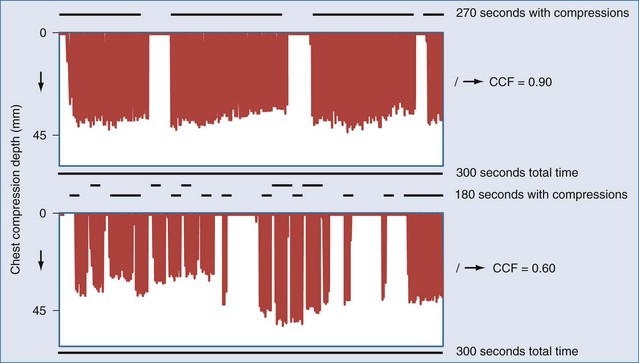
Even brief interruptions in chest compressions can result in decreased CPP. This fact has clinical importance, as evidenced by the fact that more uninterrupted chest compression are highly associated with restoring circulation and survival.23 When chest compressions are measured during actual resuscitations by paramedics or hospital providers, interruptions and pauses are frequent.47,48 These observations have prompted the development of monitor-defibrillators with features to measure and record chest compressions, and even to provide real-time feedback to providers.49 These devices often employ an accelerometer in the defibrillation pads or in a separate attachment that is placed on the sternum of the patient. It is unknown if real-time feedback can improve resuscitation success, but it is clear that the continuity and quality of chest compressions are important parameters to maximize for resuscitation to succeed. Recent literature has used the chest-compression fraction to quantify this parameter (Figure 33-5).
Direct cardiac compression via a thoracotomy is more effective than external chest compressions, producing roughly threefold increases in CPP.50,51 This approach also allows recognition of cardiac tamponade and treatment by pericardiotomy. Mechanical activity and fibrillation are immediately visible, and electrical rescue shocks or pacing can be applied directly to the heart. In the setting of cardiopulmonary collapse due to exsanguination, thoracotomy also allows aortic compression to shunt blood to heart and brain, as well as direct control of intrathoracic bleeding. Until the 1960s, thoracotomy was the standard approach for treatment of sudden cardiac arrest, but this procedure has now been supplanted by closed-chest compressions. Cases series describe how this technique continues to be successful, and its use should be considered when closed-chest compressions are ineffective.50 Open-chest cardiac massage is most likely to succeed if initiated early during resuscitation.52
Delivery of chest compressions is often inadequate, and uninterrupted chest compressions are critical for restoration of circulation.23,30,53,54 A variety of mechanical devices have been developed to provide more consistent and continuous chest compressions.55 Some of these devices exploit circumferential compression or active compression/decompression of the chest to increase the efficiency of artificial circulation. Clinical trials comparing these devices have not demonstrated any benefit versus manual chest compressions, and one trial found a trend towards worse neurologic outcome.56 While no current device is poised to solve this challenge, the constant attention of industry to this area illustrates the need for strategies to improve delivery of chest compressions.
Extracorporeal perfusion for restoration of circulation is highly effective and can be used to resuscitate subjects for whom chest compressions have failed.57,58,59 With extracorporeal support, more time becomes available to address the primary cause for cardiac arrest. However, this approach requires specialized technical skill and has increased cost and risk. Logistical issues include limited availability of perfusion equipment, setup time for circuit priming, and delays in establishing adequate venous and arterial access. Development of portable cardiopulmonary bypass devices that can be primed quickly, along with improved techniques for rapid vascular access, could broaden the use of this technology. At present, this approach is used only in specialized centers that have devoted institutional resources to set up a dedicated program.
ECG Monitoring
VF and asystole lie along a continuum of not-organized ECG. Arbitrary peak-to-peak amplitude of the ECG is usually used to distinguish asystole (amplitude < 0.1-0.2 mV) from VF (amplitude > 0.2 mV).60 However, VF also exhibits temporal structure that may be absent in asystole.61 VF is a chaotic electrical activity formed by multiple interacting waves of activation within the heart.62 VF emerges from broken wavefronts that result from an area of ischemia (as in MI), an area of prolonged refractoriness (as in drug-induced or inherited prolonged QT intervals), or too-rapid succession of activation potentials (as in tachycardia or an “R on T” premature beat). As the organization and amplitude of these waves decline, because of ischemia or hypoxemia, the amplitude of the ECG also declines. Reperfusion of the heart in asystole may restore VF. Furthermore, the amplitude and organization of the VF increase with reperfusion, providing a marker of adequate artificial perfusion.
Rational Use of Rescue Shocks for Defibrillation
Delivery of immediate transthoracic electric (rescue) shocks to patients in VF can convert VF into an organized cardiac rhythm. Rescue shocks are highly effective when VF is of very brief duration (<1-2 minutes). These shocks may work by depolarizing the heart, canceling the original wavefronts, or by prolonging the refractory periods.62 Although rescue shocks can successfully restore an organized rhythm, repeated shocks may directly damage the myocardium. The precise magnitude of this damage is still unclear.63 Nevertheless, optimal therapy should provide rescue shocks at the lowest effective energy while minimizing the number of unsuccessful rescue shocks.
Rescue shocks are more likely to fail when cardiac arrest has lasted more than a few minutes. In the out-of-hospital setting when the collapse is not witnessed by the paramedic, only 9% to 12% of rescue shocks restore an organized ECG.64,65 Furthermore, resuscitation is less likely after rescue shocks that convert VF into asystole.66 In one model for defibrillation, a “critical mass” of the heart must be depolarized by a rescue shock to ensure that VF activation potentials are extinguished.62 If a critical mass is not defibrillated, chaotic activity in the remaining regions will spread throughout the heart, rekindling VF. However, even when the entire heart is depolarized by the shock, VF may recur, perhaps because of heterogeneous areas of refractoriness or persistent foci.67 Regardless, shocks must be of sufficient intensity to deliver depolarizing current to the majority of the heart.
Several maneuvers can facilitate electrical defibrillation. Multiphasic shock waveforms that produce more effective depolarization of individual myocytes (biphasic waveforms, for example) tend to accomplish defibrillation with less energy than monophasic waveforms.62 Consequently, most defibrillators available now deliver biphasic waveforms. Increased pressure of paddles from 0.5 to 8 kg on the chest will decrease transthoracic impedance by as much as 14%, increasing delivery of current to the heart.68,69 This advantage of paddles must be weighed against the increased safety and convenience afforded by hands-free self-adhesive defibrillation pads, and most settings now have adopted the hands-free pads. In the past, multiple shocks would be delivered in rapid succession to decrease chest impedance. However, repetitive shocks decrease chest impedance only about 8% or less in actual patients,68,70,71 which does not justify the interruption of artificial circulation to deliver “stacked” shocks. Minimal interruption of chest compressions to deliver a single shock, followed by immediate resumption of chest compressions, is now recommended. Reducing the delay from the last chest compression to the delivery of the rescue shock has also been associated with greater resuscitation success.72 Therefore, chest compressions should continue while the defibrillator is charging and only stop at the last moment prior to shock. Rescue shocks of sufficient energy with multiphasic waveforms should be delivered singly to the patient in VF using firm paddle pressure on the chest or hands-free self-adherent pads.
For VF that has lasted more than a 3 to 4 minutes, preclinical data suggest that delaying rescue shocks until after a few minutes of chest compressions will improve rescue shock success. In animals, reperfusion prior to rescue shocks appears to be preferable to immediate rescue shock after more than 5 minutes of untreated VF.73–76 To date, two clinical studies in out-of-hospital cardiac arrest have found that either 90 seconds or 3 minutes of chest compressions prior to delivery of the initial rescue shock improved resuscitation rates for subjects with VF outside the hospital, particularly when rescuer response intervals are longer than 4 minutes.2,77 However, a third study found no difference in outcomes with 5 minutes of chest compressions prior to shock,78 while a fourth study found no difference in outcomes with 3 minutes of chest compressions prior to shock.79 Finally, a large multicenter trial comparing immediate rescue shock to 3 minutes of chest compressions prior to rescue shock recently stopped enrollment, reportedly finding no difference between groups.80 Taken together, the clinical data suggest that the first rescue shock for VF should be delivered as soon as possible within 3 to 5 minutes as long as chest compressions are started immediately, but that there is no reason to intentionally delay the rescue shock.
Quantitative analysis of the VF waveform can distinguish early VF from late VF and may be useful for estimating the likelihood of rescue shock success.81 Larger amplitude of VF suggests early VF and is associated with more successful resuscitation.82 However, amplitude can be affected by body habitus and other recording conditions. Frequency-based measures, as well as nonlinear dynamical measures, also can be used to quantify VF and estimate the probability of rescue shock success and are less dependent on recording conditions.83–86 All these measures, or some combination of these measures, are likely to be implemented in future generations of defibrillators. These devices may provide real-time, semiquantitative estimates of the probability that a rescue shock will succeed in restoring an organized rhythm. Using this information, the clinician will be able to choose to shock VF when the probability of shock success is high, or to concentrate on improving the situation with artificial perfusion when the probability of shock success is low.
Drug Therapy
All drug therapy in cardiac arrest can be divided into three categories: pressors, antidysrhythmics, and metabolic drugs. There is good evidence that pressors improve artificial circulation, making restoration of pulses more likely. However, recent data in out-of-hospital cardiac arrest accentuate the point that no drug therapy has been demonstrated to improve long-term survival.87 Antidysrhythmic drugs are effective for preventing dysrhythmias and therefore have a role in stabilizing the heart once circulation is restored. The value of antidysrhythmic drugs for terminating VF or reversing asystole is less clear. Metabolic drugs, primarily bicarbonate, can be used to reverse acidosis or other electrolyte problems when they are recognized. However, there are no data to support routine use of these drugs for all patients.
Pressors used during resuscitation include epinephrine and vasopressin. Both of these drugs can increase CPP via actions on α-adrenergic (epinephrine) or vasopressin receptors (Figure 33-6).88,89 Epinephrine is usually administered in 1-mg (~0.015 mg/kg) increments. In laboratory studies, the pressor effects of epinephrine during cardiac arrest are brief (~5 minute). Vasopressin has been administered as 40-unit boluses (~0.5 units/kg) and produces a longer-lasting increase in CPP (~10 minutes). Both drugs should be titrated to improvement in clinical indicators (ECG waveform, mechanical activity, changes in end-tidal CO2 or CPP). Direct comparisons between vasopressin and epinephrine have failed to demonstrate a clear superiority of one drug over the other or of the combination of the two over either alone.90–93
The 1-mg dose of epinephrine may be too small to restore pulses after circulatory arrest lasting more than a few minutes. However, trials in out-of-hospital patients comparing higher initial boluses of epinephrine (15 mg versus 1 mg) found a higher rate of restoration of pulses (13% versus 8%) and admission to the hospital (18% versus 10%),94 but overall survival was not different. Comparison of a lower dose (7 mg versus 1 mg) of epinephrine in both in-hospital and out-of-hospital cardiac arrest found no change in restoration of pulses or survival.95 Likewise, comparison of 0.02 mg/kg versus 0.2 mg/kg epinephrine found no change in restoration of pulses, or survival.96
It is possible that the β-adrenergic effect of these higher doses of epinephrine produces toxicity that limits long-term survival. Postarrest impairment of cardiac index and oxygen delivery has been related to epinephrine dose.97 Likewise, neurologic impairment has been related to epinephrine dose.98 No trial has completed a direct comparison of epinephrine with more selective α-adrenergic agents such as phenylephrine. However, one trial found no advantage from administration of 11 mg of norepinephrine.94
Vasopressin can increase CPP without complicating β-adrenergic effects. Resuscitation rates and survival are identical for patients resuscitated with vasopressin and standard doses of epinephrine after in-hospital90 or out-of-hospital cardiac arrest.91 However, post hoc analyses suggest vasopressin may be superior for resuscitation and survival of patients whose first ECG rhythm is asystole and for those subjects requiring multiple doses of vasopressors.91 Subsequent studies examined the combination of epinephrine with vasopressin versus epinephrine alone for treatment of cardiac arrest.92,93 These studies found no difference in outcomes with the combination of drugs. Therefore, use of either vasopressin or epinephrine is justified in the setting of cardiac arrest. None of the studies on these pressors standardized post–cardiac arrest care, and therefore all are limited for assessing drug effects on neurologic recovery.
Although preclinical and existing clinical data support the conclusion that vasoactive drugs during CPR can increase the probability of restoring spontaneous circulation, it is unclear whether these drugs actually improve overall survival. A recent trial in out-of-hospital cardiac arrest compared resuscitation without intravenous (IV) drugs to resuscitation with IV drugs.87 In this study, which accounted for many aspects of post–cardiac arrest care, IV drugs clearly increased the rate of restoration of pulses (32% versus 21%), but the rate of neurologically good survival did not differ (9.8% versus 8.1%). It is possible the study was underpowered to detect differences in long-term outcomes. However, the data raise the worrisome possibilities that when IV drugs are required to restore cardiac activity, severe brain injury has already occurred, or even that the drugs used add to brain injury.98
The role of antidysrhythmic drugs during cardiac arrest is equivocal.99,100 Atropine may relieve bradycardia when it is vagally mediated. However, nervous system influences on the heart are largely eliminated after more than 1 to 2 minutes of circulatory arrest. Therefore, there is little expectation that atropine will improve resuscitation from asystole or PEA. Lidocaine, procainamide, and bretylium have a long history of use in the treatment of VF. The basis for this use is principally the observation that these drugs can suppress dysrhythmias prior to cardiac arrest. Once VF is established, lidocaine can actually increase the electrical energy required to defibrillate by more than 50%.101 This effect is not true for agents that are less potent, as sodium channel antagonists. For example, administration of amiodarone (5 mg/kg) is superior to placebo102 and to lidocaine103 for restoration of pulses to out-of-hospital patients with VF that is not terminated by three rescue shocks. These studies did not control subsequent critical care and are thus not designed to determine any effect on long-term survival. In summary, antidysrhythmic drugs are commonly used during resuscitation, but only amiodarone use has supporting human data.
Empirical treatments of metabolic disturbances during cardiac arrest are not supported by prospective human data. Bicarbonate or other buffers may improve acidemia resulting from ischemia, and systems with increased use of bicarbonate report higher rates of successful resuscitation.104 However, no trial has demonstrated that sodium bicarbonate administration improves outcome.105,106 Aminophylline has been proposed as an antagonist of adenosine released during ischemia. Adenosine is hypothesized to suppress cardiac electrical activity. Two prospective studies of aminophylline administration to subjects with PEA or asystole failed to demonstrate any improvement in resuscitation.107,108 Use of dextrose-containing fluids versus dextrose-free fluids did not alter outcome for out-of-hospital cardiac arrest.109 Other metabolic therapies including calcium and magnesium also lack supporting data.110,111 However, it is appropriate to consider specific use of these agents to correct known abnormalities that are contributing to cardiac arrest, such as known hyperkalemia, calcium channel blocker overdose, torsades, or hypomagnesemia.
 Aspects of Cardiac Arrest in Specific Situations
Aspects of Cardiac Arrest in Specific Situations
The original etiology of cardiac arrest may not be known during acute resuscitation. However, if this information is available, treatment and prognosis can be individualized to the specific patient. Among out-of-hospital patients, as many as 66% have primary cardiac disturbances.112 For in-hospital patients experiencing cardiac arrest, dysrhythmia and cardiac ischemia account for 59% of events.14 This section reviews unique features of cardiac arrest resulting from both cardiac and noncardiac causes.
Primary Cardiac Events
Cardiac arrest is most commonly attributable to cardiac disease. A primary dysrhythmia or cardiogenic shock is the most common proximate cause of cardiac arrest.112,113 Patients undergoing angioplasty have 1.3% incidence of cardiac arrest, and survival in these patients resembles survival in other populations.114 Among patients admitted to the hospital with acute MI, cardiac arrest occurs in 4.8%.17 Dysrhythmias are common during the hours after reperfusion therapy,114 although reperfusion therapy reduces the overall risk of cardiac arrest.115 During acute MI, cardiac arrest is most likely in patients with lower serum potassium levels, more than 20 mm of total ST elevation, and a prolonged QTc interval during the first 2 hours of their event.115 With a mean follow up of 43 months, 3.3% of subjects surviving acute MI suffered sudden cardiac death.116 Abnormalities of the heart are present in most cases of cardiac arrest, with coronary artery disease present in at least 65% of autopsies.117 Taken together, these data suggest that most patients with cardiac arrest will have contributing cardiovascular disease.
An acute coronary syndrome is present in more than half of patients presenting with primary cardiac arrest outside of the hospital. When angiography was performed on consecutive patients resuscitated from cardiac arrest, coronary artery occlusion was identified in 48%.118 Similarly, 51% of initially resuscitated outpatients exhibited cardiac enzyme elevation or ECG evidence of acute MI.119 In one series, troponin T was elevated in 40% of out-of-hospital patients undergoing CPR, regardless of whether circulation was restored.120 The direct myocardial injury from defibrillation and CPR may cause spurious elevations of creatine kinase that are unrelated to cardiovascular disease.121 However, cardiac troponin elevations are believed to reflect acute MI rather than injury from electric shocks.122 Thus the 40% of subjects undergoing CPR with elevated troponin probably suffered myocardial injury prior to collapse.
The high likelihood of an acute coronary syndrome in the patient suffering cardiac arrest should prompt consideration of antiplatelet therapy, anticoagulation, beta-blockade, and nitrates during the post–cardiac arrest care. Unless a clearly noncardiac etiology for cardiac arrest is evident, acute coronary angiography may reveal an indication for angioplasty, thrombolysis, or other reperfusion therapy. Early angioplasty or reperfusion therapy is associated with improved survival and outcome.17,118,123,124 Primary angioplasty is safe in comatose patients undergoing hypothermia treatment, and good outcomes have been reported.125,126 Therefore, coma and its treatment should not delay emergent treatment of acute coronary syndromes if they are suspected.
Primary ventricular tachyarrhythmias are rapidly reversible and may be more likely than PEA or asystole for patients with a primary cardiac cause of collapse. Ventricular tachyarrhythmias are the initially recorded rhythm in 23% to 41% of out-of-hospital cardiac arrests10,11,127 and in 25% of in-hospital cardiac arrests.14 Because VF is rapidly reversible, patients with this rhythm comprise the majority of survivors of cardiac arrest. Data collected over 3 decades in one city noted that the prevalence of VF in out-of-hospital cardiac arrest has declined since 1978.10 This trend may reflect a change in preventive medicine or in the epidemiology of cardiovascular disease over time. Once defibrillation is accomplished, prophylactic treatment of ventricular dysrhythmias is usually not required and may be harmful, given the side effects of most drugs. However, recurrent malignant dysrhythmias may be treated with infusions of lidocaine, amiodarone, procainamide, or other antidysrhythmics while searching for and treating the underlying etiology. Current practice most often employs amiodarone or lidocaine.
Long-term antidysrhythmia treatment should be considered for patients who survive sudden cardiac arrest. At a minimum, treatment should be considered for patients with decreased left ventricular function or primary dysrhythmia without a reversible cause.128 Importantly, subjects surviving a life-threatening ventricular dysrhythmia had a 15% to 20% risk of death during a mean of 16 months of follow-up, even when a reversible cause of the dysrhythmia such as electrolyte disturbance or hypoxia can be identified.129 Implantable defibrillators have been found to be superior to antidysrhythmic drugs for reducing the risk of subsequent death.130 This benefit is primarily in subjects with a left ventricular ejection fraction (LVEF) less than 0.35.131 Implantable defibrillators were not better than antidysrhythmic drugs in a European trial that enrolled subjects resuscitated from cardiac arrest secondary to ventricular dysrhythmia without regard to LVEF.132 Nevertheless, these devices offer significant hope of preventing sudden cardiac death, and identification of patients they may benefit is an active area of research. At present, implantable defibrillators should be discussed for patients who recover from coma with LVEF less than 0.35 or survive a ventricular arrhythmia in the absence of clearly reversible causes.
Asphyxia
Asphyxia-induced cardiac arrest can result from drowning, choking, asthma, progressive respiratory failure with hypoxemia, or traumatic coma with hypoventilation. Acute asphyxia causes transient tachycardia and hypertension, followed by bradycardia and hypotension, progressing to PEA or asystole. This period of blood flow with severe hypoxemia prior to cardiac arrest may make asphyxiation a more severe injury than VF or other rapid causes of circulatory arrest.133 Brain edema is more common on CT scans after resuscitation when cardiac arrest was caused by pulmonary rather than cardiac etiologies.134
During cardiac arrest, pulmonary edema develops from redistribution of blood into the pulmonary vasculature.135 Thus, oxygenation is only worsened in the asphyxiated patient. Attention to the primary cause of asphyxia, as well as to maneuvers that will increase oxygenation (e.g., increased end-expiratory pressure or increased inspiration to expiration time ratios), may be necessary.
Pulmonary Embolism
Pulmonary emboli may occur in the postsurgical patient, as well as in medical patients with impaired mobility.136 In one series, pulmonary emboli were present in 10% of in-hospital deaths,137 and the prevalence among out-of-hospital deaths was similar.138 Pulmonary emboli can result in rapid cardiopulmonary collapse and should be considered as a possible etiology of cardiac arrest in the proper clinical setting or when collapse is preceded by sudden shortness of breath, hypoxemia and/or pleuritic chest pain.
Administration of bolus fibrinolytic drugs (tissue plasminogen activator, streptokinase, or urokinase) has been speculated to help acutely during resuscitation of a patient with a suspected pulmonary embolism. Smaller pulmonary emboli can lead to cardiac arrest due to hypoxemia, and resuscitation may be possible prior to fibrinolysis if adequate oxygen exchange can be restored. Tenecteplase has been used with reported success in non-randomized trials during resuscitation of undifferentiated patients,139 but it failed to demonstrate benefit in a larger randomized trial of undifferentiated patients in cardiac arrest.140 Likewise, a randomized trial of tissue plasminogen activator to patients with out-of-hospital cardiac arrest and an initial rhythm of PEA failed to demonstrate any benefit, although drug administration was late during resuscitation.141 There are few data about treatment of cardiac arrest resulting from massive pulmonary emboli, and certainly the overall prognosis in this setting is dismal. Individual decisions to use fibrinolytics in this setting must be tempered by the low probability of success.
Electrolyte Disturbances
Potassium disturbances are the most likely electrolyte disturbance to result in cardiac arrest. In cardiac patients, hypokalemia has been linked to the incidence of VF after MI.115,142 Hypokalemia also may account for the increased incidence of sudden death in patients taking large doses of diuretics. VF is rare in patients where serum potassium is maintained over 4.5 mEq/L. Conversely, hyperkalemia can prolong repolarization increasing the likelihood of VF initiation. Hyperkalemia may also suppress automaticity in the myocardial electrical system, leading to bradycardic PEA or asystole. Interestingly, cardiac arrest occurring during hemodialysis is not associated with high or low potassium levels but is more common when patients are dialyzed against a low (0 or 1 mEq/L) potassium dialysate.143 These data suggest that rapid changes in potassium rather than the absolute value are important triggers of cardiac arrest in this population. Derangements of calcium and magnesium may produce similar or synergistic changes in cardiac conduction.
Poisoning
Cardiac arrest can result from drug overdose. Therapy does not change except when specific antidotes or countermeasures to the poison are available. For example, calcium channel blocker overdose may be countered by administration of IV calcium.144 Beta-blocker toxicity may require large doses of inotropic agents145 or may respond to glucagon.146 Digoxin overdose may respond to digoxin-binding antibodies.147 In the case of narcotic-induced respiratory depression, subsequent cardiac arrest usually is a specific case of asphyxia rather than specific cardiotoxic effects. Case reports have suggested treating local anesthetic toxicity with 1 to 3 mL/kg of Intralipid.148 This intervention is likely to be explored for other lipid-soluble poisonings.149 One principle of poisonings is that the patient was often healthy prior to the event, and may recover well once the poison is eliminated. This potential for a better outcome may justify longer and more aggressive efforts at resuscitation.
Sepsis
Cardiac arrest can develop from sepsis for several reasons. Direct myocardial depression occurs, probably due to humoral factors.150,151 Vasodilation results in apparent hypovolemia. Finally, impaired oxygen extraction, shunting, and mitochondrial depression can produce cellular hypoxia. Because pump and vascular failure are the principle physiologic derangements, the most common initial ECG rhythm would be expected to be a rapid PEA that slows to asystole with ischemia. When these processes have progressed to cardiac arrest, large doses of inotropes, vasoconstrictors, and volume may be needed to restore circulation. Acute volume resuscitation may require 100 mL/kg of isotonic fluids or more and must be titrated to physiologic endpoints (for example, central venous oxygen saturation, CVP, or urine output) rather than according to recipe. Because the underlying sepsis physiology will still be present if pulses are restored, these patients may prove exceedingly unstable during the subacute recovery period and have a reduced chance for survival.27,152–154
Trauma/Hemorrhage
Hypovolemic cardiac arrest occurs after severe trauma, gastrointestinal hemorrhage, or other blood loss. Absence of venous return results in an empty heart that cannot produce cardiac output despite normal inotropic state and normal vascular tone. As with sepsis, this situation would most likely present with a rapid PEA that slows to asystole, but VF can develop in response to the global ischemia. Because cardiac function and vascular function are initially normal, inotropes and vasoconstrictors are unlikely to benefit hypovolemic cardiac arrest. Rapid replacement of volume with crystalloid infusion is indicated. Colloid or blood infusion should correct the situation more rapidly.155 After restoration of circulation, patients with hemorrhagic cardiac arrest are likely to develop multisystem organ failure.155
During hypovolemic cardiac arrest, the empty cardiac ventricles render external chest compressions ineffective. If blood loss is ongoing or if massive volume replacement cannot be instituted rapidly, thoracotomy allows clamping or compression of the aorta, perhaps retaining sufficient blood in the proximal aorta to perfuse the coronary and cerebral arteries. This procedure has reported success in the treatment of penetrating traumatic injuries,156 but not in blunt trauma.157 Survival is better if thoracotomy occurs in the operating room after brief loss of pulses, and best if the penetrating injury has created cardiac tamponade that is directly relieved by pericardotomy. Clearly, restoration of circulation must be accompanied by repair of the site of hemorrhage.
Hypothermia
Hypothermia represents an important situation where prolonged resuscitative efforts are justified. If hypothermia develops prior to circulatory arrest, the tolerance of the heart and brain to ischemia is greatly prolonged. Survival with favorable neurologic recovery has been reported after cold-water submersion or exposure with cardiac arrest and resuscitation efforts lasting several hours.158,159 Although all data are retrospective, subjects in whom circulatory arrest occurs because of hypothermia appear to be more salvageable than subject who asphyxiate or have circulatory arrest prior to becoming cold.160
Treatment should be based upon the initial temperature of the patient. Between 32°C and 37°C, no change in drug or electrical treatment is required, and this level of hypothermia may be beneficial for resuscitation of both brain and heart.161,162 Between 29°C and 32°C, cardiac activity may be preserved, and external warming (warm air, heating lights, warm blankets) and warm IV fluids should accompany usual resuscitation efforts. The likelihood of generating sufficient perfusion to rewarm the body declines as temperature decreases from 32°C to 29°C, and more invasive warming should be considered if there is not a rapid response with external warming. More invasive and aggressive treatment will almost certainly be required for cooler patients, because both mechanical and electrical activity of the heart is disrupted at temperatures below 28°C. Patients below this temperature may exhibit PEA, VF that is refractory to defibrillation attempts, or asystole. Repetitive rescue shocks in such patients are not justified and may be detrimental. Efficacy of most resuscitation drugs may be impaired.
Several techniques for active rewarming during resuscitation of victims of severe hypothermia are available. Given the potentially prolonged tolerance of the cold patient to ischemia, there may be sufficient time to establish arterial and venous access for partial or complete cardiopulmonary bypass. Extracorporeal circulation is particularly useful in these subjects because it can provide artificial circulation at the same time as rewarming.160,163,164 In the absence of extracorporeal circulation, placement of thoracostomy tubes and lavage of the chest with warm fluids is an option.165 Thoracostomy is intuitively preferable to peritoneal lavage because the heart will be directly warmed. Warm air forced over the body surface can rewarm a patient, although this technique may provide the least heat exchange.166 In any case, it is difficult to determine whether or not circulation can be reestablished in the profoundly hypothermic patient until near physiologic core temperatures (33°C-37°C) are restored.
Other Medical Conditions
Comorbidities have a tremendous influence on the outcome from cardiac arrest.113,167,168 In some cases, cardiac arrest may be an expected progression of the patient’s disease, but guidelines about limiting resuscitation were not set out. For example, no survivors were reported among cancer patients with expected cardiac arrest.168 Therefore, it may be appropriate to set limits on resuscitation efforts in certain medical conditions prior to cardiopulmonary collapse. Ideally, discussion about the expectations for resuscitative efforts should be held with the patient, their family, or the patient’s representatives prior to cardiac arrest. If those discussions did not occur prior to the first cardiac arrest, they should follow promptly any initially successful resuscitation.
 Post–Cardiac Arrest Care to Minimize Brain Injury
Post–Cardiac Arrest Care to Minimize Brain Injury
Management of the patient after restoration of circulation affects ultimate outcome. For example, long-term survival differed for comparable patients treated by a single ambulance service but delivered to separate hospitals.5,6 Institutional differences in in-hospital management were identified, particularly the permitted frequency of hyperthermia and hyperglycemia, that may have accounted for these differences. Despite the apparent importance of post–cardiac arrest critical care, there are few guidelines for treatment.
Brain injury appears not to be acute neuronal necrosis during ischemia, but instead to be an active process that develops over hours to days after resuscitation. Multiple cellular and molecular mechanisms contribute to neurologic injury after global brain ischemia.169 During brain ischemia and immediately after reperfusion, studies have detected increased release of excitatory amino acids, free radicals, and energy failure. Protein synthesis is inhibited at the level of translation initiation for several hours.170 There are focal disturbances of cerebral blood flow.171 Specific intracellular and extracellular signaling pathways are activated for several hours after brain ischemia,172,173 which may lead to specific changes in gene transcription. Finally, activation of specific proteases between 24 and 72 hours after reperfusion is associated with appearance of histologic signs of neuronal death.174 The relative contribution of each of these processes to neuronal injury is unknown, and all may contribute synergistically to brain injury. All represent potential targets for therapeutic intervention.
Despite detailed knowledge of the mechanisms involved with brain ischemia, drugs that target specific pathways provide modest effects in laboratory studies, and no drug to date has demonstrated clear benefit in human trials. Randomized trials have examined thiopental, the calcium channel blocker lidoflazine, magnesium, and diazepam.175–177 One explanation for this failure is that multiple mechanisms contribute simultaneously to the process of ischemic neuronal death. Antagonizing one pathway leading to neuronal death may leave other backup mechanisms unaffected. Less-specific therapies or multifaceted therapies that affect multiple pathways may prove more effective.
In support of this idea, prospective randomized clinical trials confirm that induction of mild hypothermia (32°C-34°C) for 12 to 24 hours after resuscitation improves survival and neurologic recovery.161,178 Observational data also support avoidance of fever, hypotension, and hyperglycemia.5,179–181 Therefore, systematic brain-oriented intensive care rather than a single therapeutic drug or intervention is required to improve outcome (Table 33-1).
| Temperature | Avoid fever for 48 hours. |
| Induced mild hypothermia 32°C-34°C for 12-24 hours | |
| Rewarm slowly (<0.25°C/h). | |
| Cardiovascular | Mean arterial pressure 80-100 mm Hg for first day (inotropic and vasopressor support as needed; invasive monitoring as needed). |
| Suppress dysrhythmias. | |
| Reperfusion therapy for acute myocardial infarction, regardless of concurrent coma or treatment with hypothermia. | |
| Medical management for acute coronary syndromes (antiplatelet drugs, anticoagulation). | |
| Pulmonary | Usual care. |
| Avoid hyperventilation. | |
| Avoid hypoxia or hyperoxia. | |
| Pneumonitis is common. | |
| Gastrointestinal | Usual care. |
| Consider early refeeding (after hypothermia) to reduce translocation. | |
| Fluids/Electrolytes | Monitor CVP and urine output with hypothermia/rewarming. |
| Monitor potassium/electrolytes during temperature changes. | |
| Keep potassium ≥ 4.5 mEq/L. | |
| Monitor glucose frequently, avoid hyperglycemia > 180 mg/dL. | |
| Infection | Bacteremia and pneumonia are common. |
| Prophylactic antibiotics are of unproven benefit. | |
| Antipyretics are reasonable. | |
| Neurologic | CT scan to exclude intracranial lesions. |
| Sedation and muscle relaxation as needed for hypothermia induction. | |
| Monitor for seizures with EEG if available. | |
| Serial clinical examinations for prognosis. | |
| Examinations may change dramatically over first 72 hours (or longer with hypothermia treatment). | |
| EEG, SSEP, and MRI with DWI may supplement clinical examination for prognosis in selected patients. |
CT, computed tomography; CVP, central venous pressure; DWI, diffusion-weighted imaging; EEG, electroencephalogram; MRI, magnetic resonance imaging; SSEP, somatosensory evoked potential.
Temperature Control
Meticulous avoidance of fever is important during the first 24 to 48 hours after ischemic brain injury. Temperature control after cardiac arrest may be confounded by the fact that bacteremia and spontaneous fever is common in the resuscitated patient.182,183 The benefit of lower temperatures for injured brain has been demonstrated after traumatic brain injury, stroke, and cardiac arrest.179,184,185 Mechanistically, temperature probably affects more than brain metabolic rate. For example, manipulations of temperature that improve neurologic recovery in laboratory studies produce no effect on jugular venous lactate or oxygen uptake.186 Recent laboratory investigations suggest that a variety of signaling pathways and cellular responses are sensitive to relatively small (1°C-2°C) changes in brain temperature.172,173
Induction of mild hypothermia for resuscitated patients produces a 24% to 30% relative risk reduction for death or poor neurologic outcome.187 Mild hypothermia (32°C-34°C) maintained for 12 or 24 hours significantly improved the odds of survival and good neurologic outcome for subjects resuscitated from VF cardiac arrest.161,178 There is no biological basis to believe that this neurologic benefit of induced hypothermia is specific to patients with one type of cardiac rhythm, and use in all patients who remain comatose after cardiac arrest seems reasonable. Multiple case series report successful application of therapeutic hypothermia for patients after out-of-hospital and in-hospital cardiac arrest with all initial rhythms.3,188,189 At the time of resuscitation, many patients are already mildly hypothermic, with core temperatures between 35°C and 35.5°C.161,178,190 This spontaneous cooling may result from equilibration of core and peripheral blood compartments during circulatory arrest. Subsequent to restoration of circulation, patients will rewarm within a few hours unless specific interventions are instituted.179,191
After cardiac arrest, mild hypothermia can be induced and maintained by a variety of techniques including surface cooling and endovascular devices.162 Surface cooling with ice packs and cooling blankets is tolerated by the comatose patient.178,192 Initial studies using surface cooling alone suggested that it is slow and may require 4 to 6 hours to reach 34°C.161,184,193 However, neuromuscular blockade and sedation can help prevent shivering or other compensatory responses and thereby greatly facilitate surface cooling. Recent reports indicated that core temperature can be reduced below 34°C for the majority of patients within 4 hours with surface cooling.194 There are few direct comparisons of surface cooling and endovascular cooling. What data exist suggest that endovascular devices are no faster than water-circulating surface devices, but that endovascular catheters provide more stable control of temperature over time.195,196 Local cooling of the head is unlikely to produce brain hypothermia when there is adequate perfusion by warm core blood,190 although the head can be an effective site for removing heat from the body.197
Additional maneuvers are available to accelerate cooling after cardiac arrest. For example, rapid infusion of 30 mL/kg cold (4°C) crystalloid produces a rapid decrease in core temperature and is tolerated by the post–cardiac arrest patient.191,198–200 Effective use of cold fluid boluses requires that they are administered quickly into the central circulation (via central line or under pressure infusion via peripheral line). The volume required may limit this intervention to those patients without renal failure or pulmonary edema. Gastric or bladder lavage are labor intensive and less effective than cold fluid administration. It is also critical to appreciate that cold IV fluids only produce a transient decrease in core temperature, requiring that a maintenance technique (endovascular or surface cooling device) be in place after the infusion.191,200 Taken together, these data support the rapid induction of mild hypothermia by bolus infusion of cold IV fluids (unless contraindicated), followed by maintenance of hypothermia by surface or endovascular cooling, neuromuscular paralysis, and sedation. One additional precaution that must be considered if neuromuscular blockade is required is the high incidence of seizures after cardiac arrest. Neurophysiologic monitoring should be in place if the patient is paralyzed.
Fluid and electrolyte shifts are the primary management concerns during induction of hypothermia. Induction of cooling can result in peripheral vasoconstriction, with an apparent reduction in vascular volume.201 CVP will rise, followed by diuresis. Conversely, at the time of rewarming, vessels will dilate, CVP will fall, and the patient may appear relatively hypovolemic. Volume status should be followed closely, and a need for additional volume infusion to maintain blood pressure and urine output should be anticipated at the time of rewarming. Inattention to this fluid shift was cited as a pitfall in trials of therapeutic hypothermia for traumatic brain injury.201 The initial diuresis, along with shifts between intracellular and extracellular compartments, can result in hypokalemia, hypophosphatemia, and hypomagnesemia at cooling, followed by hyperkalemia at rewarming.202,203 Frequent monitoring and correction of electrolytes during these transitions is warranted.
When indicated, primary angioplasty can be conducted in patients undergoing hypothermia treatement.125,126 Conscious patients undergoing angioplasty for acute MI tolerate mild hypothermia.162 In these patients, cooling did not interfere with defibrillation when it was required. Cardiovascular complications of hypothermia are rare with temperatures greater than 30°C. Cooling from 37°C to 31°C actually has a positive inotropic effect, increasing stroke volume to a greater extent than it decreases heart rate.204 Systemic vascular resistance does not appear to change greatly. Clinical data report a transient 18% decline in cardiac index with cooling to 33°C.205
Other complications of mild hypothermia are few when the cooling period lasts less than 24 hours. Infections become more common if cooling is prolonged for more than 24 hours. There is a suggestion that infections were slightly more common in post–cardiac arrest patients cooled for 24 hours,161 but not in those cooled for 12 hours.178 Although mild hypothermia can inhibit platelet function and coagulation,206 these changes are of small magnitude, leading to few bleeding complications in studies to date. These studies included subjects with concurrent trauma or administration of heparinoids and glycoprotein IIb/IIIa inhibitors.162,184 There are reports of infrequent but increased bleeding in post–cardiac arrest patients treated with hypothermia and cardiac catheterization (6.2% of patients).207 Elevations of pancreatic enzymes have been reported in cooled patients, but these changes resolve with rewarming.161,205 Creatinine clearance and platelet count may fall during cooling, but both parameters normalize with rewarming.205
Blood Pressure and Cerebral Blood Flow
After cardiac arrest, the heart experiences a reversible period of decreased mechanical function.208 The biochemical basis for this dysfunction is an active area of study. Moreover, reperfusion at the time of resuscitation includes oxidative stress or other triggers that can lead to myocyte death over time.209 From a clinical standpoint, some vasoactive drug support is necessary in a large proportion of patients resuscitated from cardiac arrest lasting more than 3 or 4 minutes. This dependence on vasoactive drugs should decline over the subsequent 24 to 48 hours.
Autoregulation of cerebral blood flow is disturbed after cardiac arrest. Although rarely used clinically, measurement of oxygen saturation in the jugular bulb venous blood allows calculation of brain oxygen extraction. Furthermore, cerebral blood flow can be estimated using transcranial Doppler ultrasound or nuclear imaging. During the first day after resuscitation from cardiac arrest, patients exhibit increased cerebral vascular resistance210 and impaired cerebral autoregulation.211,212 When autoregulation is present, it is right shifted such that brain perfusion declines when mean arterial pressure (MAP) declines below 80 to 120 mm Hg. When blood pressure is maintained, clinical positron emission tomography (PET) studies suggest that regional perfusion remains matched to metabolic activity after cardiac arrest.213,214 Thus, available data indicate that normal perfusion of the brain requires a higher MAP than normal during the first few hours after cardiac arrest. Periods of hypotension after restoration of pulses may add significant secondary ischemic brain injury.
If tolerated by the heart, relative hypertension (MAP of 80–100 mm Hg) should be maintained to prevent brain hypoperfusion. Maintaining this level of hypertension often requires infusions of inotropes and/or pressors. In support of this recommendation, hypotension during the first 2 hours after resuscitation is associated with poor neurologic recovery in patients admitted to the hospital after cardiac arrest.180 No specific choice of pressors has been demonstrated to be superior. Dopamine 5 to 20 µg/kg/min, norepinephrine (0.01-1 µg/kg/min) and/or epinephrine (0.01-1 µg/min) are all potential agents. In addition, the doses of these agents must be titrated, and patient requirements may be very dynamic in the early period after cardiac arrest.
Glucose Control
Elevated serum glucose is associated with poor outcome after cardiac arrest109 and may be a marker of prolonged or difficult resuscitation. Both epinephrine and physiologic stress can elevate serum glucose. However, multivariate models that accounted for resuscitation time and medication usage still show an effect of serum glucose on admission and during the first 48 hours of intensive care on long-term outcome.5,215 Despite this association, both studies noted that monitoring of glucose in nondiabetic patients was infrequent.
It has not been established whether any specific strategy to control hyperglycemia will improve outcome after cardiac arrest. Intensive glycemic control with a low target range (72-108 mg/dL, 4-6 mmol/L) is quite controversial outside of the surgical population where it was first studied216 and has not proven beneficial and may be harmful in medical intensive care.217,218 After cardiac arrest, there was no difference in outcome when a moderate glucose was targeted (108-144 mg/dL; 6-8 mmol/L) versus a strict lower range (72-108 mg/dL; 4-6 mmol/L).219 However, the incidence of hypoglycemic events was higher in the strict versus moderate control group (18% versus 2% of patients). Given the available data, control of glucose levels above 180 mg/dL (10 mmol/L) seems reasonable, and 144 to 180 mg/dL (8-10 mmol/L) would be a safe target range. However, lower ranges (72-108 mg/dL; 4-6 mmol/L) have risk of complication without benefit and should be avoided.
Hematologic Changes
Cardiac arrest is associated with activation of coagulation that is not balanced by fibrinolysis. This hematologic profile is reminiscent of disseminated intravascular coagulation and may contribute to subsequent end-organ dysfunction. Markers of thrombogenesis that have been reported include increased thrombin-antithrombin complexes and fibrinopeptide A.220,221 These increases are not balanced by fibrinolytic factors for at least 24 hours. The etiology of these changes is unknown and may be related to ischemic injury to the endothelium.
At present, use of anticoagulation is variable, and there are no prospective trials evaluating the effect of anticoagulation after resuscitation. Anticoagulation and even fibrinolytic drugs are safe after cardiopulmonary resuscitation.140,221–223 A retrospective series noted a univariate relationship between anticoagulation and 6-month survival that was not significant in a multivariate model.215 Given the hematologic evidence of active thrombogenesis, these data suggest that at least anticoagulation should be considered immediately after resuscitation.
Infection
The physiology of the post–cardiac arrest patient resembles the systemic inflammatory response syndrome, and post–cardiac arrest infections are common. Bacteremia has been noted in 39% of patients during the first 12 hours after resuscitation.182 Fever is common in patients within 48 hours after resuscitation from cardiac arrest if active temperature control is not in place. Potential causes include contamination during emergent line placement, aspiration or transient bacteremia during airway management, and mesenteric ischemia contributing to bacterial translocation from the gut. Endotoxin and various cytokines are elevated in serum after resuscitation.224 While an intestinal origin of endotoxin was suspected, pulmonary infections were more common than bacteremia. Pneumonitis is evident in 33% to 41% of patients after cardiac arrest,161,207 although microbiological confirmation of pneumonia is often lacking. Severe infections may contribute to overall mortality.183 Despite these observations, the role of routine antibiotics and antipyretics has not been examined.
 Predicting Neurologic Recovery
Predicting Neurologic Recovery
The goal of clinical practice always is to restore the patient to full consciousness and function.225 All subjects with circulatory arrest of more than 1 or 2 minutes will be comatose at initial presentation, but some of these same patients can recover and awaken. Therefore, signs of neurologic activity immediately after restoration of circulation are good, but their absence does not preclude eventual recovery. Unfortunately, many cardiac arrest survivors fail to completely awaken and may meet criteria for a persistent vegetative state.226,227 The status of patients who do not quite meet these criteria but are not awake has been described as a minimally conscious state.228
Determining the neurologic prognosis of patients resuscitated from cardiac arrest has been the subject of multiple reviews.229,230 At this time, there are limited data about whether the traditional findings associated with universally poor outcome231–233 are still valid in the setting of modern ICU care and treatment with therapeutic hypothermia (targeted temperature management). In fact, good survival has been reported after some situations previously believed to have universally poor outcome such as post–cardiac arrest status myoclonus and seizures234 or the absence of cortical responses on evoked potentials.235 In general, there is agreement that the neurologic examination continues to evolve over a longer time period than the 1 to 3 days recommended in historical publications,229,230 suggesting that longer periods of support and observation may be appropriate for some patients. There is also belief that a multimodal approach to determining prognosis that includes imaging and neurophysiologic studies may be useful to sort out difficult cases in which the physical examination is equivocal.234 Unfortunately, the published case series from modern ICU care are too small to provide tight confidence intervals for determining when further support is futile. The intensivist can assist families and proxy decision makers by interpreting available data in terms of the probabilities of meaningful survival, and by revising the likelihood of meaningful survival based on the progress of the patient from day to day.
Several clinical signs have been used to assess and predict neurologic recovery after cardiac arrest. A classic case series found that pupillary reaction to light, corneal reflexes, and motor activity can change over the first 72 hours after resuscitation.233 By 72 hours, absence of eye reflexes and failure to have a localizing response to pain were highly predictive of permanent coma. A systematic review of literature since that initial report confirms the value of these clinical findings.231 These classic series show how specific clinical signs can provide quantitative estimates of the probability of awakening, but the actual benchmarks and timing of examinations proposed in these studies were derived from data in the 1980s.
Only a few modern case series have reported the sensitivity and specificity of clinical signs for determining poor outcome.234,236,237 These studies tend to support the finding that persistent absence of one or more brainstem reflexes for more than 3 days portends poor outcome, but the prediction is not perfect, and the small number of patients observed with absent brainstem reflexes results in wide confidence intervals. A significant difference in these studies is the less robust predictive value of motor examination after 3 days. Specifically, motor response less than flexion at 3 days was reported to have a false-positive rate ([FPR] percentage where test predicts poor outcome, but patients have good outcome) of 14% (95% CI 3%-44%)236 or 8% (FPR 8%, 95% CI 2%-25%).234 Therefore, the modern case series suggest that improvement in the clinical examination can occur over a more protracted time course than previously believed.
Neurophysiology
It is common for an electroencephalogram (EEG) to be obtained for prognostic purposes. However, the prognostic value of EEG after resuscitation from cardiac arrest is limited by its nonspecific nature and by the dynamic changes in EEG over time.238 Perhaps the greatest utility of the EEG is to diagnose seizures and exclude nonconvulsive seizures as an etiology of unresponsiveness. Seizures are diagnosed clinically in 5% to 20% of comatose patients after cardiac arrest,161,239 and the true incidence of nonconvulsive electrographic seizures may be higher. Termination of seizures, if possible, is essential to allow untainted assessment of the neurologic examination. Aside from seizures, certain malignant EEG patterns have a strong albeit imperfect association with poor outcome. Specifically, generalized suppression (<20 µV), burst-suppression pattern associated with generalized epileptic activity, or diffuse periodic complexes on a flat background during the first week after resuscitation are associated with poor neurologic outcome.231 Thus EEG cannot be used by itself to determine prognosis, but the information provided by EEG can exclude confounders (seizures) and can be integrated into the total clinical picture used to assess prognosis.
Electrophysiologic response to stimuli also can be used to assess whether the patient is neurologically intact. Recovery of longer-latency event-related potentials is associated with awakening.238,240,241 Conversely, absence of a cortical response to somatosensory evoked potentials (SSEPs) is very specific for poor neurologic outcome.230,231 Cortical response is usually assessed as the N20 response to electrical stimulation of the median nerve. Like EEG, SSEP responses vary with the elapsed time since resuscitation.238 Recent data suggest that the use of therapeutic hypothermia may increase the time-dependent changes in SSEP. One case series reported two patients treated with hypothermia who had absent N20 responses at 3 days after cardiac arrest but recovered cognition.235 Therefore, it may be reasonable to repeat SSEPs that show absent N20s several days apart in order to avoid false-negative tests.
Blood Markers
Several neuronal peptides appear in the blood after injury to the brain, including neuron-specific enolase (NSE) and the glia-derived protein, S-100B. After cardiac arrest, NSE reaches a maximum level in serum at 72 hours. High NSE levels at 48 to 72 hours after resuscitation are associated with poor outcome.241–243 Serial NSE levels that continue to rise over the first 72 hours also are associated with poor outcome.243 In contrast to NSE, peak levels of S-100B in serum occur during the first 24 hours after resuscitation.242 Higher S-100B levels are also associated with poor neurologic outcome.242,244 Hypothermia treatment appears to alter serum NSE levels.245 Use of NSE or S-100B to determine prognosis is limited by the absence of a clear cutoff value that is unsurvivable. In addition, laboratory assays are not sufficiently standardized to be certain that cutoff values derived in one location are relevant to another, and at least NSE can be released by non-brain injury.246 These neuronal markers might be considered a tool for following brain injury, analogous to troponin levels for following myocardial injury, but lack the specificity or clarity required to guide therapeutic decisions.
Imaging Studies
Imaging of the brain is important to exclude injury incurred at the time of collapse and to exclude intracranial causes of collapse. A noncontrast cranial computed tomography (CT) scan to exclude hemorrhage may be prudent in the comatose patient after cardiac arrest prior to anticoagulation or fibrinolytic therapy. Several series have reported a reasonable yield of clinical information from CT scans after cardiac arrest.247,248 In general, noncontrast CT scan is insufficiently sensitive to determine prognosis after cardiac arrest unless severe changes are present. For example, severe generalized edema is often associated with loss of brainstem reflexes and may progress to herniation and brain death (Figure 33-7).
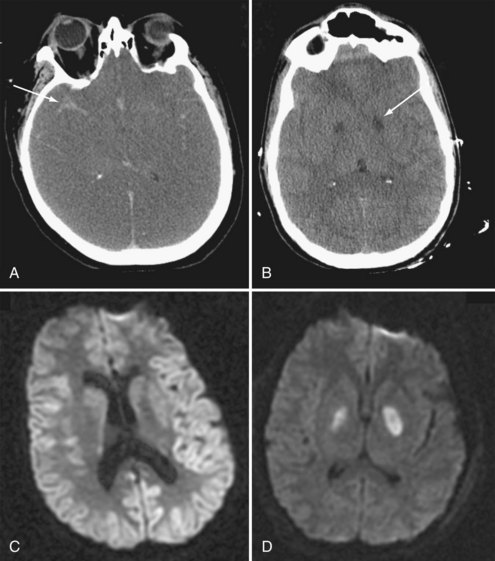
Magnetic resonance imaging (MRI) has a capacity to visualize more subtle changes in brain after cardiac arrest. For example, increased cortical signals on diffusion-weighted images (DWI) or fluid-attenuated inversion recovery (FLAIR) are associated with poor neurologic outcome.249 For patients who remain comatose for several days and in whom clinical or electrophysiologic testing is indeterminate, MRI may be considered as an adjunct to assess the extent of brain injury. Expectations and enthusiasm for long-term support may be reduced if extensive cortical lesions are present, whereas persistence may be justified if injury is limited. In the long term, cognitive deficits are associated with global brain volume loss after cardiac arrest.250 An important caveat with interpretation of all brain imaging after global ischemia is the differing clinical impact of lesions in different brain regions. The anatomic complexity of the brain precludes any simple quantitative relationship between the number or size of lesions and outcome. At present, the role of MRI as an adjunct for assessing post–cardiac arrest brain damage is evolving, and its interpretation should be considered in combination with neurologic or neuroradiologic expertise.
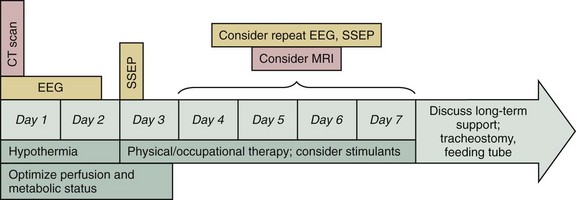
In summary, the determination of neurologic prognosis after cardiac arrest varies from patient to patient. Changes in the clinical examination are the cornerstone of prognostication. While the patient is recovering, hypothermia and supportive care may increase the likelihood of recovery. However, electrophysiologic and imaging techniques can add additional useful information to help guide clinicians and families. The approximate timing for each of these studies is depicted in Figure 33-8.
 Rehabilitation
Rehabilitation
The role of rehabilitation or other therapy in recovery from neurologic impairment after cardiac arrest is relatively unstudied. It is clear that both patients and their caregivers have complex needs if neurologic injury is severe.251 Older data suggest that long-term improvement is less common when neurologic devastation follows a medical cause like cardiac arrest than when it results from traumatic brain injury.227 More recent work suggests that rehabilitation can produce similar improvements after global brain ischemia.252,253 Nevertheless, early consideration of rehabilitative services including physical therapy and occupational therapy may help promote recovery, just as in acute stroke.254 Physical stimulation and maintenance of muscle tone could conceivably promote awakening. When arousal or level of consciousness is impaired after traumatic brain injury, stimulants such as methylphenidate or amantadine have been employed with reduction in total ICU stay or improved final status.255,256 While these data are few and indirect, addition of stimulants for post–cardiac arrest patients who linger in intermediate coma might be considered if medically tolerated.
 Withdrawal of Support
Withdrawal of Support
For adults who are neurologically devastated after cardiac arrest in North America, it is more common to die in the hospital than to receive long-term care. An estimated 44% of patients who are initially resuscitated from cardiac arrest in the hospital have withdrawal of care later during their hospitalization.14 For patients resuscitated from out-of-hospital cardiac arrest, 68% have do-not-resuscitate (DNR) status established in the hospital, perhaps representing a comparable outcome.22 These decisions are often based on the neurologic prognosis of the patient,21 and such decisions limit the number of neurologically impaired individuals who are discharged from the hospital. Consequently, quality of life for those patients who do leave the hospital is generally high.20,257,258 Popular reports of awakening after long coma may cause inappropriate optimism for families of patients or surrogate decision makers. Partial awakening of the patient into a persistent vegetative state or minimally conscious state can further confuse their expectations. Decision makers should receive information about these syndromes, realistic expectations of recovery, and any specific considerations for the individual patient. Religious, cultural, and personal beliefs will contribute to decisions, and appropriate social service and pastoral support should be provided.
 Summary
SummaryKey Points
Aufderheide TP, Sigurdsson G, Pirrallo RG, Yannopoulos D, McKnite S, von Briesen C, et al. Hyperventilation-induced hypotension during cardiopulmonary resuscitation. Circulation. 2004;109:1960-1965.
Bobrow BJ, Ewy GA, Clark L, Chikani V, Berg RA, Sanders AB, et al. Passive oxygen insufflation is superior to bag-valve-mask ventilation for witnessed ventricular fibrillation out-of-hospital cardiac arrest. Ann Emerg Med. 2009;54:656-662.
Christenson J, Andrusiek D, Everson-Stewart S, Kudenchuk P, Hostler D, Powell J, et al. Resuscitation Outcomes Consortium Investigators. Chest compression fraction determines survival in patients with out-of-hospital ventricular fibrillation. Circulation. 2009;120:1241-1247.
Don CW, Longstreth WTJr, Maynard C, Olsufka M, Nichol G, Ray T, et al. Active surface cooling protocol to induce mild therapeutic hypothermia after out-of-hospital cardiac arrest: a retrospective before-and-after comparison in a single hospital. Crit Care Med. 2009;37:3062-3069.
Kim F, Olsufka M, Longstreth WTJr, Maynard C, Carlbom D, Deem S, et al. Pilot randomized clinical trial of prehospital induction of mild hypothermia in out-of-hospital cardiac arrest patients with a rapid infusion of 4°C normal saline. Circulation. 2007;115:3064-3070.
Olasveengen TM, Sunde K, Brunborg C, Thowsen J, Steen PA, Wik L. Intravenous drug administration during out-of-hospital cardiac arrest: a randomized trial. JAMA. 2009;302:2222-2229.
Sunde K, Pytte M, Jacobsen D, Mangschau A, Jensen LP, Smedsrud C, et al. Implementation of a standardised treatment protocol for post resuscitation care after out-of-hospital cardiac arrest. Resuscitation. 2007;73:29-39.
1 Stratton S, Niemann JT. Effects of adding links to “the chain of survival” for prehospital cardiac arrest: a contrast in outcomes in 1975 and 1995 at a single institution. Ann Emerg Med. 1998;31:471-477.
2 Cobb LA, Fahrenbruch CE, Walsh TR, et al. Influence of cardiopulmonary resuscitation prior to defibrillation in patients with out-of-hospital ventricular fibrillation. JAMA. 1999;281:1182-1188.
3 Sunde K, Pytte M, Jacobsen D, Mangschau A, Jensen LP, Smedsrud C, et al. Implementation of a standardised treatment protocol for post resuscitation care after out-of-hospital cardiac arrest. Resuscitation. 2007;73:29-39.
4 Rea TD, Helbock M, Perry S, Garcia M, Cloyd D, Becker L, et al. Increasing use of cardiopulmonary resuscitation during out-of-hospital ventricular fibrillation arrest: survival implications of guideline changes. Circulation. 2006;114:2760-2765.
5 Langhelle A, Tyvold SS, Lexow K, Hapnes SA, Sunde K, Steen PA. In-hospital factors associated with improved outcome after out-of-hospital cardiac arrest. A comparison between four regions in Norway. Resuscitation. 2003;56:247-263.
6 Engdahl J, Abrahamsson P, Bang A, Lindquist J, Karlsson T, Herlitz J. Is hospital care of major importance for outcome after out-of-hospital cardiac arrest? Resuscitation. 2000;43:201-211.
7 Neumar RW, Nolan JP, Adrie C, Aibiki M, Berg RA, Böttiger BW, et al. Post-cardiac arrest syndrome: epidemiology, pathophysiology, treatment, and prognostication. A consensus statement from the International Liaison Committee on Resuscitation (American Heart Association, Australian and New Zealand Council on Resuscitation, European Resuscitation Council, Heart and Stroke Foundation of Canada, InterAmerican Heart Foundation, Resuscitation Council of Asia, and the Resuscitation Council of Southern Africa); the American Heart Association Emergency Cardiovascular Care Committee; the Council on Cardiovascular Surgery and Anesthesia; the Council on Cardiopulmonary, Perioperative, and Critical Care; the Council on Clinical Cardiology; and the Stroke Council. Circulation. 2008 Dec 2;118(23):2452-2483.
8 Lloyd-Jones D, Adams RJ, Brown TM, Carnethon M, Dai S, De Simone G, et alAmerican Heart Association Statistics Committee and Stroke Statistics Subcommittee. Executive summary: heart disease and stroke statistics–2010 update: a report from the American Heart Association. Circulation. 2010 Feb 23;121(7):948-954. No abstract available. Erratum in: Circulation. 2010 Mar 30;121(12):e259
9 Zheng ZJ, Croft JB, Giles WH, Mensah GA. Sudden cardiac death in the United States, 1989-1998. Circulation. 2001;104:2158-2163.
10 Cobb LA, Fahrenbruch CE, Olsufka M, Copass MK. Changing incidence of out-of-hospital ventricular fibrillation 1980-2000. JAMA. 2002;288:3008-3013.
11 Nichol G, Thomas E, Callaway CW, Hedges J, Powell JL, Aufderheide TP, et alResuscitation Outcomes Consortium Investigators. Regional variation in out-of-hospital cardiac arrest incidence and outcome. JAMA. 2008;300:1423-1431.
12 Becker LB, Ostrander MP, Barrett J, Kondos GT. Outcome of CPR in a large metropolitan area: Where are the survivors? Ann Emerg Med. 1991;20:48-54.
13 Lombardi G, Gallagher J, Gennis P. Outcome of out-of-hospital cardiac arrest in New York City. JAMA. 1994;271:678-683.
14 Peberdy MA, Kaye W, Ornato JP, Larkin GL, Nadkarni V, Mancini ME, et al. Cardiopulmonary resuscitation of adults in the hospital: a report of 14 720 cardiac arrests from the National registry of Cardiopulmonary Resuscitation. Resuscitation. 2003;58:297-308.
15 Schultz SC, Cullinane DC, Pasquale MD, Magnant C, Evans SRT. Predicting in-hospital mortality during cardiopulmonary resuscitation. Resuscitation. 1996;33:13-17.
16 Wang HE, Abella BS, CW Callaway. American Heart Association National Registry of Cardiopulmonary Resuscitation Investigators. Risk of cardiopulmonary arrest after acute respiratory compromise in hospitalized patients. Resuscitation. 2008;79:234-240.
17 Ornato JP, Peberdy MA, Tadler SC, Strobos NC. Factors associated with the occurrence of cardiac arrest during hospitalization for acute myocardial infarction in the second national registry of myocardial infarction in the US. Resuscitation. 2001;48:117-123.
18 Reinier K, Stecker EC, Vickers C, Gunson K, Jui J, Chugh SS. Incidence of sudden cardiac arrest is higher in areas of low socioeconomic status: a prospective two year study in a large United States community. Resuscitation. 2006;70:186-192.
19 Vaillancourt C, Lui A, De Maio VJ, Wells GA, Stiell IG. Socioeconomic status influences bystander CPR and survival rates for out-of-hospital cardiac arrest victims. Resuscitation. 2008;79:417-423.
20 De Vos R, de Haes HC, Koster RW, de Haan RJ. Quality of survival after cardiopulmonary resuscitation. Arch Intern Med. 1999;159:249-254.
21 Laver S, Farrow C, Turner D, Nolan J. Mode of death after admission to an intensive care unit following cardiac arrest. Intensive Care Med. 2004;30:2126-2128.
22 Niemann JT, Stratton SJ. The Utstein template and the effect of in-hospital decisions: the impact of do-not-attempt resuscitation status on survival to discharge statistics. Resuscitation. 2001;51:233-237.
23 Christenson J, Andrusiek D, Everson-Stewart S, Kudenchuk P, Hostler D, Powell J, et alResuscitation Outcomes Consortium Investigators. Chest compression fraction determines survival in patients with out-of-hospital ventricular fibrillation. Circulation. 2009;120:1241-1247.
24 Morrison LJ, Deakin CD, Morley PT, et alAdvanced Life Support Chapter Collaborators. Part 8: Advanced life support: 2010 International consensus on Cardiopulmonary Resuscitation and Emergency Cardiovascular Care Science with Treatment Recommendations. Circulation. 2010;122(16 Suppl 2):S345-S421. 19
25 Safar P, Brown TC, Holtey WJ, Wilder RJ. Ventilation and circulation with closed-chest cardiac massage in man. JAMA. 1961;176:574-576.
26 Clark JJ, Larsen MP, Culley LL, Graves JR, Eisenberg MS. Incidence of agonal respirations in cardiac arrest. Ann Emerg Med. 1992;21:1464-1467.
27 Mullie A, Lewi P, Van Hoeyweghen R. Pre-CPR conditions and the final outcome of CPR. The Cerebral Resuscitation Study Group. Resuscitation. 1989;17(Suppl):S11-S21.
28 Baskett P, Nolan J, Parr M. Tidal volumes which are perceived to be adequate for resuscitation. Resuscitation. 1996;31:231-234.
29 Aufderheide TP, Sigurdsson G, Pirrallo RG, Yannopoulos D, McKnite S, von Briesen C, et al. Hyperventilation-induced hypotension during cardiopulmonary resuscitation. Circulation. 2004;109:1960-1965.
30 Kern KN, Hilwig RW, Berg RA, Sanders AB, Ewy GA. Importance of continuous chest compressions during cardiopulmonary resuscitation: improved outcome during a simulated single lay-rescuer scenario. Circulation. 2002;105:645-649.
31 Sanders AB, Kern KB, Berg RA, Hilwig RW, Heidenrich J, Ewy GA. Survival and neurologic outcome after cardiopulmonary resuscitation with four different chest compression-ventilation ratios. Ann Emerg Med. 2002;40:553-562.
32 Berg RA, Kern KB, Sanders AB, Otto CW, Hilwig RW, Exy GA. Bystander cardiopulmonary resuscitation: is ventilation necessary? Circulation. 1993;88:1907-1915.
33 Deakin CD, O’Neill JF, Tabor T. Does compression-only cardiopulmonary resuscitation generate adequate passive ventilation during cardiac arrest? Resuscitation. 2007;75:53-59.
34 Bobrow BJ, Ewy GA, Clark L, Chikani V, Berg RA, Sanders AB, et al. Passive oxygen insufflation is superior to bag-valve-mask ventilation for witnessed ventricular fibrillation out-of-hospital cardiac arrest. Ann Emerg Med. 2009;54:656-662.
35 Weil MH, Bisera J, Trevino RP, Rackow EC. Cardiac output and end-tidal carbon dioxide. Crit Care Med. 1985;13:907-909.
36 Salen P, O’Connor R, Sierzenski P, Passarello B, Pancu D, Melanson S, et al. Can cardiac sonography and capnography be used independently and in combination to predict resuscitation outcomes? Acad Emerg Med. 2001;8:610-615.
37 Callaham M, Barton C. Prediction of outcome from cardiopulmonary resuscitation from end-tidal carbon dioxide concentration. Crit Care Med. 1990;18:358-362.
38 Levine RL, Wayne MA, Miller CC. End-tidal carbon dioxide and outcome of out-of-hospital cardiac arrest. New Engl. J Med. 1997;337:301-306.
39 Gabrielli A, Layon AJ, Wenzel V, Dorges V, Idris AH. Alternative ventilation strategies in cardiopulmonary resuscitation. Curr Opin Crit Care. 2002;8:199-211.
40 Wenzel V, Idris AH, Banner MJ, Kubilis PS, Band R, Williams JLJr, Lindner KH. Respiratory system compliance decreases after cardiopulmonary resuscitation and stomach inflation: impact of large and small tidal volumes on calculated peak airway pressure. Resuscitation. 1998;38:113-118.
41 Weiler N, Heinrichs W, Dick W. Assessment of pulmonary mechanics and gastric inflation pressure during mask ventilation. Prehosp Disaster Med. 1995;10:101-105.
42 Bowman FP, Menegazzi JJ, Check BD, Duckett TM. Lower esophageal sphincter pressure during prolonged cardiac arrest and resuscitation. Ann Emerg Med. 1995;26:216-219.
43 Wang HE, Simeone SJ, Weaver MD, Callaway CW. Interruptions in cardiopulmonary resuscitation from paramedic endotracheal intubation. Ann Emerg Med. 2009;54:645-652.
44 Rumball CJ, MacDonald D. The PTL, Combitube, laryngeal mask, and oral airway: a randomized prehospital comparative study of ventilatory device effectiveness and cost-effectiveness in 470 cases of cardiorespiratory arrest. Prehosp Emerg Care. 1997;1:1-10.
45 Staudinger T, Brugger S, Watschinger B, Roggla M, Dielacher C, Lobl T, et al. Emergency intubation with the Combitube: comparison with the endotracheal airway. Ann Emerg Med. 1993;22:1573-1575.
46 Paradis NA, Martin GB, Rivers EP, Goetting MG, Appleton TJ, Feingold M, et al. Coronary perfusion pressure and the return of spontaneous circulation in human cardiopulmonary resuscitation. JAMA. 1990;263:1106-1113.
47 Abella BS, Alvarado JP, Myklebust H, Edelson DP, Barry A, O’Hearn N, et al. Quality of cardiopulmonary resuscitation during in-hospital cardiac arrest. JAMA. 2005;293:305-310.
48 Wik L, Kramer-Johansen J, Myklebust H, Sørebø H, Svensson L, Fellows B, et al. Quality of cardiopulmonary resuscitation during out-of-hospital cardiac arrest. JAMA. 2005;293:299-304.
49 Abella BS, Edelson DP, Kim S, Retzer E, Myklebust H, Barry AM, et al. CPR quality improvement during in-hospital cardiac arrest using a real-time audiovisual feedback system. Resuscitation. 2007;73:54-61.
50 Boczar ME, Howard MA, Rivers EP, Martin GB, Horst EM, Lewandowski C, et al. A technique revisited: hemodynamic comparison of close and open-chest cardiac massage during human cardiopulmonary resuscitation. Crit Care Med. 1995;23:498-503.
51 Alzaga-Fernandez AG, Varon J. Open-chest cardiopulmonary resuscitation: past, present and future. Resuscitation. 2005;64:149-156.
52 Takino M, Okada Y. The optimum timing of resuscitative thoracotomy for non-traumatic out-of-hospital cardiac arrest. Resuscitation. 1993;26:69-74.
53 Berg RA, Sanders AB, Kern KB, Hilwig RW, Heidenreich JW, Porter ME, et al. Adverse hemodynamic effects of interrupting chest compressions for rescue breathing during cardiopulmonary resuscitation for ventricular fibrillation cardiac arrest. Circulation. 2001;13(104):2465-2470.
54 Eftestol T, Sunde K, Steen PA. Effects of interrupting precordial compressions on the calculated probability of defibrillation success during out-of-hospital cardiac arrest. Circulation. 2002;105:2270-2273.
55 Lurie K, Plaisance P, Sukhum P, Soleil C. Mechanical advances in cardiopulmonary resuscitation. Curr Opin Crit Care. 2001;7:170-175.
56 Hallstrom A, Rea TD, Sayre MR, Christenson J, Anton AR, Mosesso VNJr, et al. Manual chest compression vs use of an automated chest compression device during resuscitation following out-of-hospital cardiac arrest: a randomized trial. JAMA. 2006;295:2620-2628.
57 Bartlett RH, Roloff DW, Custer JR, Younger JG, Hirschl RB. Extracorporeal life support: the University of Michigan experience. JAMA. 2000;283:904-908.
58 Nagao K, Hayashi N, Kanmatsuse K, Arima K, Ohtsuki J, Kikushima K, et al. Cardiopulmonary cerebral resuscitation using emergency cardiopulmonary bypass, coronary reperfusion therapy and mild hypothermia in patients with cardiac arrest outside the hospital. J Am Coll Cardiol. 2000;36:776-783.
59 Nichol G, Karmy-Jones R, Salerno C, Cantore L, Becker L. Systematic review of percutaneous cardiopulmonary bypass for cardiac arrest or cardiogenic shock states. Resuscitation. 2006;70:381-394.
60 Gliner BE, White RD. Electrocardiographic evaluation of defibrillation shocks delivered to out-of-hospital sudden cardiac arrest patients. Resuscitation. 1999;41:133-144.
61 Sherman L, Callaway C, Menegazzi J. Ventricular fibrillation exhibits dynamical properties and self-similarity. Resuscitation. 2000;47:163-173.
62 White RD. New concepts in transthoracic defibrillation. Emerg Med Clin N Am. 2002;20:785-807.
63 Walcott GP, Killingsworth CR, Ideker RE. Do clinically relevant transthoracic defibrillation energies cause myocardial damage and dysfunction? Resuscitation. 2003;59:59-70.
64 Hargarten KM, Stueven HA, Waite EM, Olson DW, Mateer JR, Aufderheide TP, et al. Prehospital experience with defibrillation of coarse ventricular fibrillation: a ten year review. Ann Emerg Med. 1990;19:157-162.
65 Lo BM, Quinn SM, Hostler D, Callaway CW. Rescue shock outcomes during out-of-hospital cardiac arrest. Resuscitation. 2007;75:469-475.
66 Niemann JT, Strattion SJ, Cruz B, Lewis RJ. Outcome of out-of-hospital postcountershock asystole and pulseless electrical activity. Crit Care Med. 2001;29:2366-2370.
67 Chattipakorn N, Fotuhi PC, Chattipakorn SC, Ideker RE. Three-dimensional mapping of earliest activation after near-threshold ventricular defibrillation shocks. J Cardiovasc Electrophysiol. 2003;24:65-69.
68 Kerber RE, Grayzel J, Hoyt R, Marcus M, Kennedy J. Transthoracic resistance in human defibrillation: influence of body weight, chest size, serial shocks, paddle contact size, and paddle contact pressure. Circulation. 1981;63:676-682.
69 Deakin CD, Sado DM, Petley GW, Clewlow F. Determining the optimal paddle force for external defibrillation. Am J Cardiol. 2002;90:812-813.
70 Deakin CD, Ambler JJ, Shaw S. Changes in transthoracic impedance during sequential biphasic defibrillation. Resuscitation. 2008;78:141-145.
71 Walker RG, Koster RW, Sun C, Moffat G, Barger J, Dodson PP, et al. Defibrillation probability and impedance change between shocks during resuscitation from out-of-hospital cardiac arrest. Resuscitation. 2009;80:773-777.
72 Edelson DP, Abella BS, Kramer-Johansen J, Wik L, Myklebust H, Barry AM, et al. Effects of compression depth and pre-shock pauses predict defibrillation failure during cardiac arrest. Resuscitation. 2006;71:137-145.
73 Menegazzi JJ, Callaway CW, Sherman LD, Hostler DP, Wang HE, Fertig KC, et al. The ventricular fibrillation scaling exponent can guide timing of defibrillation and other therapies. Circulation. 2004;109:926-931.
74 Niemann J, Cairns C, Sharma J, Lewis R. Treatment of prolonged ventricular fibrillation. Immediate countershock versus high-dose epinephrine and CPR preceding countershock. Circulation. 1992;85:281-287.
75 Yakaitis RW, Ewy GA, Otto CW, Taren DL, Moon TE. Influence of time and therapy on ventricular defibrillation in dogs. Crit Care Med. 1980;8:157-163.
76 Menegazzi JJ, Seaberg DC, Yealy DM, Davis EA, MacLeod BA. Combination pharmacotherapy with delayed countershock vs standard advanced cardiac life support after prolonged ventricular fibrillation. Prehosp Emerg Care. 2000;4:31-37.
77 Wik L, Hansen TB, Fylling F, Vaagnes P, Auestad BH, Steen PA. Delaying defibrillation to give basic cardiopulmonary resuscitation to patients with out-of-hospital ventricular fibrillation. JAMA. 2003;289:1389-1395.
78 Jacobs IG, Finn JC, Oxer HF, Jelinek GA. CPR before defibrillation in out-of-hospital cardiac arrest: a randomized trial. Emerg Med Australas. 2005;17:39-45.
79 Baker PW, Conway J, Cotton C, Ashby DT, Smyth J, Woodman RJ, et al. Clinical Investigators. Defibrillation or cardiopulmonary resuscitation first for patients with out-of-hospital cardiac arrests found by paramedics to be in ventricular fibrillation? A randomised control trial. Resuscitation. 2008;79:424-431.
80 Stiell IG, Callaway C, Davis D, Terndrup T, Powell J, Cook A, et al. ROC Investigators. Resuscitation Outcomes Consortium (ROC) PRIMED cardiac arrest trial methods part 2: rationale and methodology for “Analyze Later vs. Analyze Early” protocol. Resuscitation. 2008;78:186-195.
81 Reed MJ, Clegg GR, Robertson CE. Analysing the ventricular fibrillation waveform. Resuscitation. 2003;57:11-20.
82 Weaver WD, Cobb LA, Dennis D, Ray R, Hallstrom AP, Copass MK. Amplitude of ventricular fibrillation waveform and outcome after cardiac arrest. Ann Intern Med. 1986;102:53-55.
83 Brown C, Dzworczyk R. Signal analysis of the human electrocardiogram during ventricular fibrillation: frequency and amplitude parameters as predictors of successful countershock. Ann Emerg Med. 1996;27:184-188.
84 Strohmenger HU, Linder KH, Lurie KG, Welz A, Georgieff M. Frequency of ventricular fibrillation as a predictor of defibrillation success during cardiac surgery. Anesth Analg. 1994;79:434-438.
85 Eftestol T, Sunde K, Aase S, Husoy J, Steen P. Predicting outcome of defibrillation by spectral characterization and nonparametric classification of ventricular fibrillation in patients with out-of-hospital cardiac arrest. Circulation. 2000;102:1523-1529.
86 Callaway C, Sherman L, Mosesso V, Dietrich T, Holt E, Clarkson M. Scaling exponent predicts defibrillation success for out-of-hospital ventricular fibrillation cardiac arrest. Circulation. 2001;103:1656-1661.
87 Olasveengen TM, Sunde K, Brunborg C, Thowsen J, Steen PA, Wik L. Intravenous drug administration during out-of-hospital cardiac arrest: a randomized trial. JAMA. 2009;302:2222-2229.
88 Paradis NA, Martin GB, Rosenberg J, River EP, Goetting MG, Appleton TJ, et al. The effect of standard- and high-dose epinephrine on coronary perfusion pressure during prolonged cardiopulmonary resuscitation. JAMA. 1991;265:1139-1144.
89 Wenzel V, Lindner KH. Arginine vasopressin during cardiopulmonary resuscitation: laboratory evidence, clinical experience and a view to the future. Crit Care Med. 2002;30:S157-S161.
90 Stiell IG, Herbert PC, Wells GA, Vandemheen KL, Tang AS, Higginson LA, et al. Vasopressin versus epinephrine for in-hospital cardiac arrest: a randomized controlled trial. Lancet. 2001;358:105-109.
91 Wenzel V, Krismer AC, Arntz HR, Sitter H, Stadlbauer KH, Lindner KH. A comparison of vasopressin and epinephrine for out-of-hospital cardiopulmonary resuscitation. New Engl J Med. 2004;350:105-113.
92 Callaway CW, Hostler D, Doshi AA, Pinchalk M, Roth RN, Lubin J, et al. Usefulness of vasopressin administered with epinephrine during out-of-hospital cardiac arrest. Am J Cardiol. 2006;98:1316-1321.
93 Gueugniaud PY, David JS, Chanzy E, Hubert H, Dubien PY, Mauriaucourt P, et al. Vasopressin and epinephrine vs. epinephrine alone in cardiopulmonary resuscitation. N Engl J Med. 2008;359:21-30.
94 Callaham M, Madsen C, Barton C, Saunders C, Daley M, Pointer J. A randomized clinical trial of high-dose epinephrine and norepinephrine versus standard-dose epinephrine in prehospital cardiac arrest. JAMA. 1992;268:2667-2672.
95 Stiell IG, Hebert PC, Weitzman BN, Wells GA, Raman S, Stark RM, et al. High-dose epinephrine in adult cardiac arrest. N Engl J Med. 1992;327:1045-1050.
96 Brown C, Martin D, Pepe P, Stueven H, Cummins R, Gonzalez E, et al. Multicenter High-Dose Epinephrine Study Group. A comparison of standard-dose and high-dose epinephrine in cardiac arrest outside the hospital. New Engl J Med. 1992;327:151-155.
97 Rivers EP, Wortsman J, Rady MY, Blake HC, Mcgeorge FT, Buderer NM. The effect of the total cumulative epinephrine dose administered during human CPR on hemodynamic, oxygen transport, and utilization variables in the postresuscitation period. Chest. 1994;106:1499-1507.
98 Behringer W, Kittler H, Sterz F, Domanovits H, Schoerkhuber W, Holzer M, et al. Cumulative epinephrine dose during cardiopulmonary resuscitation and neurologic outcome. Ann Intern Med. 1998;129:450-456.
99 Kudenchuk PJ. Intravenous antiarrhythmic drug therapy in the resuscitation from refractory ventricular arrhythmias. Am J Cardiol. 1999;84:52R-55R.
100 Nolan JP, De Latorre FJ, Steen PA, Chamberlain DA, Bossaert LL. Advanced life support drugs: do they really work? Curr Opin Crit Care. 2002;8:212-218.
101 Chow MS, Kluger J, Lawrence R, Fieldman A. Lidocaine and bretylium on the defibrillation threshold during cardiac arrest and cardiopulmonary resuscitation. Proc Soc Exp Biol Med. 1986;182:63-67.
102 Kudenchuk PJ, Cobb LA, Copass MK, Cummins RO, Doherty AM, Fahrenbruch CE, et al. Amiodarone for resuscitation after out-of-hospital cardiac arrest due to ventricular fibrillation. N Engl J Med. 1999;16(341):871-878.
103 Dorian P, Cass D, Schwartz B, Cooper R, Gelaznikas R, Barr A. Amiodarone as compared with lidocaine for shock-resistant ventricular fibrillation. N Engl J Med. 2002;346:884-890.
104 Bar-Joseph G, Abramson NS, Kelsey SF, Mashiach T, Craig MT, P Safar, Brain Resuscitation Clinical Trial III (BRCT III) Study Group. Improved resuscitation outcome in emergency medical systems with increased usage of sodium bicarbonate during cardiopulmonary resuscitation. Acta Anaesthesiol Scand. 2005;49:6-15.
105 Vukmir RB, Bircher N, Safar P. Sodium bicarbonate in cardiac arrest: a reappraisal. Am J Emerg Med. 1996;14:192-206.
106 Vukmir RB, L Katz, Sodium Bicarbonate Study Group. Sodium bicarbonate improves outcome in prolonged prehospital cardiac arrest. Am J Emerg Med. 2006;24:156-161.
107 Mader TJ, Smithline HA, Gibson P. Aminophylline in undifferentiated out-of-hospital asystolic cardiac arrest. Resuscitation. 1999;41:39-45.
108 Mader TJ, Smithline HA, Durkin L, Scriver G. A randomized controlled trial of intravenous aminophylline for atropine-resistant out-of-hospital asystolic cardiac arrest. Acad Emerg Med. 2003;10:192-197.
109 Longstreth WTJr, Copass MK, Dennis LK, Rauch-Matthews ME, Stark MS, Cobb LA. Intravenous glucose after out-of-hospital cardiopulmonary arrest: a community-based randomized trial. Neurology. 1993;43:2534-2541.
110 Fatovich DM, Prentice DA, Dobb GJ. Magnesium in cardiac arrest (the MAGIC trial). Resuscitation. 1997;35:237-241.
111 Thel MC, Armstrong AL, McNulty SE, Califf RM, O’Connor CM. Randomized trial of magnesium in in-hospital cardiac arrest: Duke Internal Medicine Housestaff. Lancet. 1997;350:1272-1276.
112 Kuisma M, Alaspaa A. Out-of-hospital cardiac arrests of non-cardiac origin. Epidemiology and outcome. Eur Heart J. 1997;18:1122-1128.
113 De Vos R, Koster RW, de Haan RJ, Oosting H, van der Wouw PA, Lampe-Schoenmaeckers AJ. In-hospital cardiopulmonary resuscitation: prearrest morbidity and outcome. Arch Int Med. 1999;159:845-850.
114 Webb JG, Solankhi NK, Chugh SK, Amin H, Buller CE, Ricci DR, et al. Incidence, correlates, and outcome of cardiac arrest associated with percutaneous coronary intervention. Am J Cardiol. 2002;90:1252-1254.
115 Selker HP, Raitt MH, Schmid CH, Laks MM, Beshansky JR, Griffith JL, et al. Time-dependent predictors of primary cardiac arrest in patients with acute myocardial infarction. Am J Cardiol. 2003;91:280-286.
116 Huikuri HV, Tapanainen JM, Lindgren K, Raatikainen P, Makikallio TH, Airaksinen KEJ, et al. Prediction of sudden cardiac death after myocardial infarction in the beta-blocker age. J Am Coll Cardiol. 2003;42:652-658.
117 Chugh SS, Kelly KL, Titus JL. Sudden cardiac death with apparently normal heart. Circulation. 2000;102:649-654.
118 Spaulding CM, Joly LM, Rosenberg A, Monchi M, Weber SN, Dhainaut JF, et al. Immediate coronary angiography in survivors of out-of-hospital cardiac arrest. N Engl J Med. 1997;336:1629-1633.
119 Bulut S, Aengevaeren WRM, Luijten HJE, Verheugt FWA. Successful out-of-hospital cardiopulmonary resuscitation: what is the optimal in-hospital treatment strategy. Resuscitation. 2000;47:155-161.
120 Lai CS, Hostler D, D Cruz BJ, Callaway CW. Prevalence of troponin-T elevation during out-of-hospital cardiac arrest. Am J Cardiol. 2004;93:754-756.
121 Grubb NR, Fox KA, Cawood P. Resuscitation from out-of-hospital cardiac arrest: implications for cardiac enzyme estimation. Resuscitation. 1996;33:35-41.
122 Goktekin O, Melek M, Gorenek B, Birdane A, Kudaiberdieva G, Cavusoglu Y, et al. Cardiac troponin T and cardiac enzymes after external transthoracic cardioversion of ventricular arrhythmias in patients with coronary artery disease. Chest. 2002;122:2050-2054.
123 Keelan PC, Bunch TJ, White RD, Packer DL, Holmes DRJr. Early direct coronary angioplasty in survivors of out-of-hospital cardiac arrest. Am J Cardiol. 2003;91:1461-1463.
124 Reynolds JC, Callaway CW, El Khoudary SR, Moore CG, Alvarez RJ, Rittenberger JC. Coronary angiography predicts improved outcome following cardiac arrest: propensity-adjusted analysis. J Intensive Care Med. 2009;24:179-186.
125 Wolfrum S, Pierau C, Radke PW, Schunkert H, Kurowski V. Mild therapeutic hypothermia in patients after out-of-hospital cardiac arrest due to acute ST-segment elevation myocardial infarction undergoing immediate percutaneous coronary intervention. Crit Care Med. 2008;36:1780-1786.
126 Knafelj R, Radsel P, Ploj T, Noc M. Primary percutaneous coronary intervention and mild induced hypothermia in comatose survivors of ventricular fibrillation with ST-elevation acute myocardial infarction. Resuscitation. 2007;74:227-234.
127 Stiell IG, Wells GA, DeMaio VJ, Spaite DW, Field BJ, Munkley DP, et al. modifiable factors associated with improved cardiac arrest survival in a multicenter basic life support/defibrillation system: OPALS Study Phase I results. Ann Emerg Med. 1999;33:44-50.
128 Bruxton AE, Ellison KE, Kirk MM, Frain B, Koo C, Gandhi G, et al. Primary prevention of sudden cardiac death: trials in patients with coronary artery disease. J Interven Cardiac Electrophysiol. 2003;9:203-206.
129 Anderson JL, Hallstrom AP, Epstein AE, Pinski SL, Rosenberg Y, Nora MO, et al. Design and results of the antiarrhythmics vs implantable defibrillators (AVID) registry. The AVID Investigators. Circulation. 1999;99:1692-1699.
130 AVID Investigators. A comparison of antiarrhythmic-drug therapy with implantable defibrillators in patients resuscitated from near-fatal ventricular arrhythmias. N Engl J Med. 1997;337:1576-1583.
131 Domanski MJ, Sakseena S, Epstein AE, Hallstrom AP, Brodsky MA, Kim S, et al. Relative effectiveness of the implantable cardioverter-defibrillator and antiarrhythmic drugs in patients with varying degrees of left ventricular dysfunction who have survived malignant ventricular arrhythmias. AVID Investigators. Antiarrhythmics Versus Implantable Defibrillators. J Am Coll Cardiol. 1999;34:1090-1095.
132 Kuck KH, Cappato R, Siebels J, Ruppel R. Randomized comparison of antiarrhythmic drug therapy with implantable defibrillators in patients resuscitated from cardiac arrest : the Cardiac Arrest Study Hamburg (CASH). Circulation. 2000;102:748-754.
133 Vaagenes P, Safar P, Moossy J, Rao G, Diven W, Ravi C, et al. Asphyxiation versus ventricular fibrillation cardiac arrest in dogs. Differences in cerebral resuscitation effects–a preliminary study. Resuscitation. 1997;35:41-52.
134 Morimoto Y, Kemmotsu O, Kitami K, Matsubara I, Tedo I. Acute brain swelling after out-of-hospital cardiac arrest: pathogenesis and outcome. Crit Care Med. 1993;21:104-110.
135 Ornato JP, Ryschon TW, Gonzalez ER, Bardthauer JL. Rapid change in pulmonary vascular hemodynamics with pulmonary edema during cardiopulmonary resuscitation. Am J Emerg Med. 1985;3:137-142.
136 Hirsh DR, Ingentino EP, Goldhaber SZ. Prevalence of deep venous thrombosis among patients in medical intensive care. JAMA. 1995;274:335-337.
137 Sandler DA, Martin JF. Autopsy proven pulmonary embolism in hospital patients. Are we detecting enough deep vein thrombosis? J R Soc Med. 1989;82:203-205.
138 Courtney DM, Kline JA. Identification of prearrest clinical factors associated with outpatient fatal pulmonary embolism. Acad Emerg Med. 2001;8:1136-1142.
139 Bottiger BW, Bode C, Kern S, Gries A, Gust R, Glatzer R, et al. Efficacy and safety of thrombolytic therapy after initially unsuccessful cardiopulmonary resuscitation: a prospective clinical trial. Lancet. 2001;357:1583-1585.
140 Bottiger BW, Arntz HR, Chamberlain DA, Bluhmki E, Belmans A, Danays T, et alTROICA Trial Investigators; European Resuscitation Council Study Group. Thrombolysis during resuscitation for out-of-hospital cardiac arrest. N Engl J Med. 2008;359:2651-2662.
141 Abu-Laban RB, Christenson JM, Innes GD, van Beek CA, Wanger KP, McKnight RD, et al. Tissue plasminogen activator in cardiac arrest with pulseless electrical activity. New Engl J Med. 2002:1522-1528.
142 MacDonald JE, Struthers AD. What is the optimal serum potassium level in cardiovascular patients? J Am Coll Cardiol. 2004;43:155-161.
143 Karnik JA, Young BS, Lew NL, Herget M, Dubinsky C, Lazarus JM, et al. Cardiac arrest and sudden death in dialysis units. Kidney Int. 2001;60:350-357.
144 Isbister GK. Delayed asystolic cardiac arrest after diltiazem overdose; resuscitation with high dose intravenous calcium. Emerg Med J. 2002;19:355-357.
145 Love JN, Litovitz TL, Howell JM, Clancy C. Characterization of fatal beta blocker ingestion: a review of the American Association of Poison Control Centers data from 1985 to 1995. J Toxicol Clin Toxicol. 1997;35:353-359.
146 Tai YT, Lo CW, Chow WH, Cheng CH. Successful resuscitation and survival following massive overdose of metoprolol. Br J Clin Pract. 1990;44:746-747.
147 Antman EM, Wenger TL, Butler VPJr, Haber E, Smith TW. Treatment of 150 cases of life-threatening digitalis intoxication with digoxin-specific Fab antibody fragments. Final report of a multicenter study. Circulation. 1990;81:1744-1752.
148 Leskiw U, Weinberg GL. Lipid resuscitation for local anesthetic toxicity: is it really lifesaving? Curr Opin Anaesthesiol. 2009;22:667-671.
149 Cave G, Harvey M. Intravenous lipid emulsion as antidote beyond local anesthetic toxicity: a systematic review. Acad Emerg Med. 2009;16:815-824.
150 Kumar A, Haery C, Parrillo JE. Myocardial dysfunction in septic shock. Crit Care Clin. 2000;16:251-287.
151 Jardin F, Brun-Ney D, Auvert B, Beauchet A, Bourdarias JP. Sepsis-related cardiogenic shock. Crit Care Med. 1990;18:1055-1060.
152 Khalafi K, Ravakhah K, West BC. Avoiding the futility of resuscitation. Resuscitation. 2001;50:161-166.
153 Ebell MH. Prearrest predictors of survival following in-hospital cardiopulmonary resuscitation: a meta-analysis. J Fam Pract. 1992;34:551-558.
154 Rozenbaum EA, Shenkman L. Predicting outcome of in hospital cardiopulmonary resuscitation. Crit Care Med. 1988;16:583-586.
155 Shoemaker WC, Peitzman AB, Bellamy R, Bellomo R, Bruttig SP, Capone A, et al. Resuscitation from severe hemorrhage. Crit Care Med. 1996;24(2 Suppl):S12-S23.
156 Washington B, Wilson RF, Steiger Z, Bassett JS. Emergency thoracotomy: a four-year review. Ann Thorac Surg. 1985;40:188-191.
157 Grove CA, Lemmon G, Anderson G, McCarthy M. Emergency thoracotomy: appropriate use in the resuscitation of trauma patients. Am Surg. 2002;68:313-316.
158 Gilbert M, Busund R, Skagseth A, Nilsen PÅ, Solbø JP. Resuscitation from accidental hypothermia of 13.7°C with circulatory arrest. Lancet. 2000;355:375-376.
159 Jones AI, Swann IJ. Prolonged resuscitation in accidental hypothermia: use of mechanical cardio-pulmonary resuscitation and partial cardio-pulmonary bypass. Eur J Emerg Med. 1994;1:34-36.
160 Farstad M, Andersen KS, Koller ME, Grong K, Segadal L, Husby P. Rewarming from accidental hypothermia by extracorporeal circulation. A retrospective study. Eur J Cardiothorac Surg. 2001;20:58-64.
161 HACA—Hypothermia after Cardiac Arrest Study Group. Mild therapeutic hypothermia to improve the neurologic outcome after cardiac arrest. N Engl J Med. 2002;346:549-556.
162 Dixon SR, Whitbourn RJ, Dae MW, Grube E, Sherman W, Schaer GL, et al. Induction of mild systemic hypothermia with endovascular cooling during primary percutaneous coronary intervention for acute myocardial infarction. J Am Coll Cardiol. 2002;40:1928-1934.
163 Ireland AJ, Pathi VL, Crawford R, Colquhoun IW. Back from the dead: extracorporeal rewarming of severe accidental hypothermia victims in accident and emergency. J Accid Emerg Med. 1997;14:255-257.
164 Walpoth BH, Walpoth-Aslan BN, Mattle HP, Radanov BP, Schroth G, Schaeffler L, et al. Outcome of survivors of accidental deep hypothermia and circulatory arrest treated with extracorporeal blood warming. N Engl J Med. 1997;337:1500-1505.
165 Hall KN, Syverud SA. Closed thoracic cavity lavage in the treatment of severe hypothermia in human beings. Ann Emerg Med. 1990;19:204-206.
166 Kornberger E, Schwarz B, Lindner KH, Mair P. Forced air surface rewarming in patients with severe accidental hypothermia. Resuscitation. 1999;41:105-111.
167 Hallstrom AP, Cobb LA, Yu BH. Influence of comorbidity on the outcome of patients treated for out-of-hospital ventricular fibrillation. Circulation. 1996;93:2019-2022.
168 Ewer MS, Kish SK, Martin CG, Price KJ, Feeley TW. Characteristics of cardiac arrest in cancer patients as a predictor of survival after cardiopulmonary resuscitation. Cancer. 2001;92:1905-1912.
169 Neumar RW. Molecular mechanisms of ischemic neuronal injury. Ann Emerg Med. 2000;36:483-506.
170 White BC, Grossman LI, O’Neil BJ, DeGracia DJ, Neumar RW, Rafols JA, et al. Global brain ischemia and reperfusion. Ann Emerg Med. 1996;27:588-594.
171 Cohan SL, Mun SK, Petite J, Correia J, Tavelra Da Silva AT, Waldhorn RE. Cerebral blood flow in humans following resuscitation from cardiac arrest. Stroke. 1989;20:761-765.
172 Hicks SD, Parmele KT, DeFranco DB, Klann E, Callaway CW. Hypothermia differentially increases ERK and JNK/SAPK activation in hippocampus after asphyxial cardiac arrest. Neuroscience. 2000;98:677-685.
173 D’Cruz BJ, Fertig KC, Filiano AJ, Hicks SD, DeFranco DB, Callaway CW. Hypothermic reperfusion after cardiac arrest augments brain-derived neurotrophic factor activation. J Cereb Blood Flow Metab. 2002;22:843-851.
174 Vogel P, Putten H, Popp E, Krumnikl JJ, Teschendorf P, Galmbacher R, et al. Improved resuscitation after cardiac arrest in rats expressing the baculovirus caspase inhibitor protein p35 in central neurons. Anesthesiology. 2003;99:112-121.
175 BRCT I. A randomized clinical study of cardiopulmonary-cerebral resuscitation: design and patient characteristics. Am J Emerg Med. 1986;4:72-86.
176 BRCT II. A randomized clinical trial of calcium entry blocker administration to comatose survivors of cardiac arrest. Design, methods, and patient characteristics. The Brain Resuscitation Clinical Trial II Study Group. Control Clin Trials. 1991;12:525-545.
177 Longstreth WT, Fahrenbuch CE, Olsufka M, Walsh TR, Copass MK, Cobb LA. Randomized clinical trial of magnesium, diazepam or both after out-of-hospital cardiac arrest. Neurology. 2002;59:506-514.
178 Bernard SA, Gray TW, Buist MD, Jones BM, Silvester W, Gutteridge G, et al. Treatment of comatose survivors of out-of-hospital cardiac arrest with induced hypothermia. N Engl J Med. 2002;346:557-563.
179 Zeiner A, Holzer M, Sterz F, Schorkhuber W, Eisenburger P, Havel C, Kliegel A, et al. Hyperthermia after cardiac arrest is associated with an unfavorable neurologic outcome. Arch Intern Med. 2001;161:2007-2012.
180 Mullner M, Sterz F, Binder M, Hellwanger K, Meron G, Herkner H, et al. Arterial blood pressure after human cardiac arrest and neurologic recovery. Stroke. 1996;27:59-62.
181 Mullner M, Sterz F, Binder M, Schreiber W, Deimel A, Laggner AN. Blood glucose concentration after cardiopulmonary resuscitation influences functional neurological recovery in human cardiac arrest survivors. J Cereb Blood Flow Metab. 1997;17:430-436.
182 Gaussorgues P, Gueugniaud PY, Vedrinne JM, Salord F, Mercatello A, Robert D. Bacteremia following cardiac arrest and cardiopulmonary resuscitation. Intensive Care Med. 1988;14:575-577.
183 Bjork RJ, Snyder BD, Campion BC, Loewenson RB. Medical complications of cardiopulmonary arrest. Arch Int Med. 1982;142:500-503.
184 Marion DW, Penrod LE, Kelsey SF, Obrist WD, Kochanek PM, Palmer AM, et al. Treatment of traumatic brain injury with moderate hypothermia. N Engl J Med. 1997;336:540-546.
185 Kasner SE, Wein T, Piriyawat P, Villar-Cordova CE, Chalela JA, Krieger DW, et al. Acetaminophen for altering body temperature in acute stroke: a randomized clinical trial. Stroke. 2002;33:130-134.
186 Kuboyama K, Safar P, Oku K, Obrist W, Leonov Y, Sterz F, et al. Mild hypothermia after cardiac arrest in dogs does not affect postarrest cerebral oxygen uptake/delivery mismatching. Resuscitation. 1994;27:231-244.
187 Nolan JP, Morley PT, Vanden Hoek TL, Hickey RW, Kloeck WG, Billi J, et al. International Liaison Committee on Resuscitation. Therapeutic hypothermia after cardiac arrest: an advisory statement by the advanced life support task force of the International Liaison Committee on Resuscitation. Circulation. 2003;108:118-121.
188 Storm C, Steffen I, Schefold JC, Krueger A, Oppert M, Jörres A, Hasper D. Mild therapeutic hypothermia shortens intensive care unit stay of survivors after out-of-hospital cardiac arrest compared to historical controls. Crit Care. 2008;12:R78.
189 Don CW, Longstreth WTJr, Maynard C, Olsufka M, Nichol G, Ray T, et al. Active surface cooling protocol to induce mild therapeutic hypothermia after out-of-hospital cardiac arrest: a retrospective before-and-after comparison in a single hospital. Crit Care Med. 2009;37:3062-3069.
190 Callaway CW, Tadler SC, Lipinski CL, Katz LM, Brader E. Feasibility of external cranial cooling during resuscitation. Resuscitation. 2002;52:159-165.
191 Kim F, Olsufka M, Carlbom D, Deem S, Longstreth WTJr, et al. Pilot study of rapid infusion of 2 L of 4 degrees C normal saline for induction of mild hypothermia in hospitalized, comatose survivors of out-of-hospital cardiac arrest. Circulation. 2005;112:715-719.
192 Felberg RA, Krieger DW, Chuang R, Persse DE, Burgin WS, Hickenbottom SL, et al. Hypothermia after cardiac arrest: feasibility and safety of an external cooling protocol. Circulation. 2001;104:1799-1804.
193 Clifton GL, Choi SC, Miller ER, Levin HS, Smith KRJr, Muizelaar JP, et al. Intercenter variance in clinical trials of head trauma–experience of the National Acute Brain Injury Study: Hypothermia. J Neurosurg. 2001;95:751-755.
194 Heard KJ, Peberdy MA, Sayre MR, Sanders A, Geocadin RG, Dixon SR, et al. A randomized controlled trial comparing the Arctic Sun to standard cooling for induction of hypothermia after cardiac arrest. Resuscitation. 2010;81:9-14.
195 Hoedemaekers CW, Ezzahti M, Gerritsen A, van der Hoeven JG. Comparison of cooling methods to induce and maintain normo- and hypothermia in intensive care unit patients: a prospective intervention study. Crit Care. 2007;11:R91.
196 Pichon N, Amiel JB, François B, Dugard A, Etchecopar C, Vignon P. Efficacy of and tolerance to mild induced hypothermia after out-of-hospital cardiac arrest using an endovascular cooling system. Crit Care. 2007;11:R71.
197 Hachimi-Idrissi S, Corne L, Ebinger G, Michotte Y, Huyghens L. Mild hypothermia induced by a helmet device: a clinical feasibility study. Resuscitation. 2001;51:275-281.
198 Bernard S, Buist M, Monteiro O, Smith K. Induced hypothermia using large volume, ice-cold intravenous fluid in comatose survivors of out-of-hospital cardiac arrest: a preliminary report. Resuscitation. 2003;56:9-13.
199 Kim F, Olsufka M, Longstreth WTJr, Maynard C, Carlbom D, Deem S, et al. Pilot randomized clinical trial of prehospital induction of mild hypothermia in out-of-hospital cardiac arrest patients with a rapid infusion of 4 degrees C normal saline. Circulation. 2007;115:3064-3070.
200 Kliegel A, Janata A, Wandaller C, Uray T, Spiel A, Losert H, et al. Cold infusions alone are effective for induction of therapeutic hypothermia but do not keep patients cool after cardiac arrest. Resuscitation. 2007;73:46-53.
201 Clifton GL, Miller ER, Choi SC, Levin HS. Fluid thresholds and outcome from severe brain injury. Crit Care Med. 2002;30:739-745.
202 Abiki M, Kawaguchi S, Maekawa N. Reversible hypophosphatemia during moderate hypothermia therapy for brain-injured patients. Crit Care Med. 2001;29:1726-1730.
203 Polderman KH, Peerdeman SM, Girbes AR. Hypophosphatemia and hypomagnesemia induced nby cooling in patients with severe head injury. J Neurosurg. 2001;94:697-705.
204 Dae MW, Gao DW, Sessler DI, Chair K, Stillson CA. Effect of endovascular cooling on myocardial temperature, infarct size, and cardiac output in human-sized pigs. Am J Physiol. 2002;282:H1584-H1591.
205 Metz C, Holzschuh M, Bein T, Woertgen C, Frey A, Frey I, et al. Moderate hypothermia with severe head injury: cerebral and extracerebral effects. J Neurosurg. 1996;85:533-541.
206 Kettner SC, Sitzwohl C, Zimpfer M, Kopzek SA, Holtzer A, Spiss CK, et al. The effect of graded hypothermia (36 degrees C—32 degrees C) on hemostasis in anesthetized patients without surgical trauma. Anesth Analg. 2003;96:1772-1776.
207 Nielsen N, Hovdenes J, Nilsson F, Rubertsson S, Stammet P, Sunde K, et al. Hypothermia Network. Outcome, timing and adverse events in therapeutic hypothermia after out-of-hospital cardiac arrest. Acta Anaesthesiol Scand. 2009;53:926-934.
208 Angelos MA, Menegazzi JJ, Callaway CW. Resuscitation from prolonged ventricular fibrillation BENCH-TO-BEDSIDE. Acad Emerg Med. 2001;8:909-924.
209 Vanden Hoek TL, Shao Z, Li C, Zak R, Schumacker PT, Becker LB. Reperfusion injury on cardiac myocytes after simulated ischemia. Am J Physiol. 1996;270(4 Pt 2):H1334-H1341.
210 Buunk G, van der Hoeven JG, Frolich M, Meinders AE. Cerebral vasoconstriction in comatose patients resuscitated from a cardiac arrest. Intensive Care Med. 1996;22:1191-1196.
211 Nishizawa H, Kudoh I. Cerebral autoregulation is impaired in patients after cardiac arrest. Acta Anaesthesiol Scand. 1996;40:1149-1153.
212 Sundgreen C, Larsen FS, Herzog TM, Knudsen GM, Boesgaard S, Aldershville J. Autoregulation of cerebral blood flow in patients resuscitated from cardiac arrest. Stroke. 2001;32:128-132.
213 Rudolf J, Ghaemi M, Ghaemi M, Haupt WF, Szelies B, Heiss WD. Cerebral glucose metabolism in acute and persistent vegetative state. J Neurosurg Anesthesiol. 1999;11:17-24.
214 Schaafsma A, de Jong BM, Bams JL, Haaxma-Reiche H, Pruim J, Zijlstra JG. Cerebral perfusion and metabolism in resuscitated patients with severe post-hypoxic encephalopathy. J Neurol Sci. 2003;10:23-30.
215 Skrifvars MB, Pettila V, Rosenberg PH, Castren M. A multiple logistic regression analysis of in-hospital factors related to survival at six months in patients resuscitated from out-of-hospital ventricular fibrillation. Resuscitation. 2003;59:319-328.
216 Van den Berghe G, Wouters P, Weekers F, Verwaest C, Bruyninckx F, Schetz M, et al. Intensive insulin therapy in the critically ill patients. N Engl J Med. 2001;345:1359-1367.
217 Van den Berghe G, Wilmer A, Hermans G, Meersseman W, Wouters PJ, Milants I, et al. Intensive insulin therapy in the medical ICU. N Engl J Med. 2006;354:449-461.
218 NICE-SUGAR Study InvestigatorsFinfer S, Chittock DR, Su SY, Blair D, Foster D, Dhingra V, et al. Intensive versus conventional glucose control in critically ill patients. N Engl J Med. 2009;360:1283-1297.
219 Oksanen T, Skrifvars MB, Varpula T, Kuitunen A, Pettilä V, Nurmi J, Castrén M. Strict versus moderate glucose control after resuscitation from ventricular fibrillation. Intensive Care Med. 2007;33:2093-2100.
220 Gando S, Kameue T, Nanzaki S, Nakanishi Y. Massive fibrin formation with consecutive impairment of fibrinolysis in patients with out-of-hospital cardiac arrest. Thromb Haemost. 1997;77:278-282.
221 Bottiger BW, Martin E. Thrombolytic therapy during cardiopulmonary resuscitation and the role of coagulation activation after cardiac arrest. Curr Opin Crit Care. 2001;7:176-183.
222 Newman DH, Greenwald I, Callaway CW. Cardiac arrest and the role of thrombolytic agents. Ann Emerg Med. 2000;35:472-480.
223 Schreiber W, Gabriel D, Sterz F, Muellner M, Kuerkciyan I, Holzer M, et al. Thrombolytic therapy after cardiac arrest and its effect on neurological outcome. Resuscitation. 2002;52:63-69.
224 Adrie C, Adib-Conquy M, Laurent I, Monchi M, Vinsonneau C, Fitting C, et al. Successful cardiopulmonary resuscitation after cardiac arrest as a “sepsis-like” syndrome. Circulation. 2002;106:562-568.
225 Jaffe AS, Landau WM, Wetzel RD. Resuscitation 2000: the need for improved databases in regard to neurological outcomes. Resuscitation. 1998;37:65-66.
226 AAN—The Quality Standards Subcommittee of the American Academy of Neurology. Practice parameters: assessment and management of patients in the persistent vegetative state (summary statement). Neurology. 1995;45:1015-1018.
227 MSTFPVS. Multisociety Task Force on PVS. Medical aspects of the persistent vegetative state. New Engl J Med. 1994;330:1572-1579.
228 Giacino JT, Ashwal S, Childs N, Cranford R, Jennett B, Katz DI, et al. The minimally conscious state: definition and diagnostic criteria. Neurology. 2002;58:349-353.
229 Booth CM, Boone RH, Tomlinson G, Detsky AS. Is this patient dead, vegetative or severely neurologically impaired? Assessing outcome for comatose survivors of cardiac arrest. JAMA. 2004;291:870-879.
230 Wijdicks EF, Hijdra A, Young GB, Bassetti CL, S Wiebe, Quality Standards Subcommittee of the American Academy of Neurology. Practice parameter: prediction of outcome in comatose survivors after cardiopulmonary resuscitation (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2006;67:203-210.
231 Zandbergen EG, de Haan RJ, Stoutenbeek CP, Koelman JH, Hijdra A. Systematic review of early prediction of poor outcome in anoxic-ischaemic coma. Lancet. 1998;352:1808-1812.
232 Edgren E, Hedstrand U, Kelsey S, Sutton-Tyrrell K, Safar P. Assessment of neurological prognosis in comatose survivors of cardiac arrest. BRCT I Study Group. Lancet. 1994;343(8905):1055-1059.
233 Levy DE, Caronna JJ, Singer BH, Lapinski RH, Frydman H, Plum F. Predicting outcome from hypoxic-ischemic coma. JAMA. 1985;253:1420-1426.
234 Rossetti AO, Oddo M, Logroscino G, Kaplan PW. Prognostication after cardiac arrest and hypothermia: a prospective study. Ann Neurol. 2010;67:301-307.
235 Leithner C, Ploner CJ, Hasper D, Storm C. Does hypothermia influence the predictive value of bilateral absent N20 after cardiac arrest? Neurology. 2010;74:965-969.
236 Al Thenayan E, Savard M, Sharpe M, Norton L, Young B. Predictors of poor neurologic outcome after induced mild hypothermia following cardiac arrest. Neurology. 2008;71:1535-1537.
237 Tiainen M, Kovala TT, Takkunen OS, Roine RO. Somatosensory and brainstem auditory evoked potentials in cardiac arrest patients treated with hypothermia. Crit Care Med. 2005;33:1736-1740.
238 Gendo A, Kramer L, Hafner M, Funk GC, Zauner C, Sterz F, et al. Time-dependency of sensory evoked potentials in comatose cardiac arrest survivors. Intensive Care Med. 2001;27:1305-1311.
239 Abend NS, Topjian A, Ichord R, Herman ST, Helfaer M, Donnelly M, et al. Electroencephalographic monitoring during hypothermia after pediatric cardiac arrest. Neurology. 2009;72:1931-1940.
240 Madl C, Kramer L, Domanovits H, Woolard RH, Gervais H, Gendo A, et al. Improved outcome prediction in unconscious cardiac arrest survivors with sensory evoked potentials compared with clinical assessment. Crit Care Med. 2000;28:721-726.
241 Zingler VC, Krumm B, Bertsch T, Fassbender K, Pohlmann-Eden B. Early prediction of neurological outcome after cardiopulmonary resuscitation: a multimodal approach combining neurobiochemical and electrophysiological investigations may provide high prognostic certainty in patients after cardiac arrest. Neurology. 2003;49:79-84.
242 Rosen H, Sunnerhagen KS, Herlitz J, Blomstrand C, Rosengren L. Serum levels of the brain-derived proteins S-100 and NSE predict long-term outcome after cardiac arrest. Resuscitation. 2001;49:183-191.
243 Schoerkhuber W, Kittler H, Sterz F, Behringer W, Holzer M, Frossard M, et al. Time course of serum neuron-specific enolase. A predictor of neurological outcome in patients resuscitated from cardiac arrest. Stroke. 1999;30:1598-1603.
244 Martens P, Raabe A, Johnsson P. Serum S-100 and neuron-specific enolase for prediction of regaining consciousness after global cerebral ischemia. Stroke. 1998;29:2363-2366.
245 Tiainen M, Roine RO, Pettila V, Takkunen O. Serum neuron-specific enolase and S-100B protein in cardiac arrest patients treated with hypothermia. Stroke. 2003;34:2881-2886.
246 Pelinka LE, Hertz H, Mauritz W, Harada N, Jafarmadar M, Albrecht M, et al. Nonspecific increase of systemic neuron-specific enolase after trauma: clinical and experimental findings. Shock. 2005;24:119-123.
247 Inamasu J, Miyatake S, Tomioka H, Suzuki M, Nakatsukasa M, Maeda N, et al. Subarachnoid haemorrhage as a cause of out-of-hospital cardiac arrest: a prospective computed tomography study. Resuscitation. 2009;80:977-980.
248 Naples R, Ellison E, Brady WJ. Cranial computed tomography in the resuscitated patient with cardiac arrest. Am J Emerg Med. 2009;27:63-67.
249 Wijdicks EF, Campeau NG, Miller GM. MR imaging in comatose survivors of cardiac resuscitation. AJNR Am J Neuroradiol. 2001;22:1561-1565.
250 Grubb NR, Fox KA, Smith K, Best J, Blane A, Ebmeier KP, et al. Memory impairment in out-of-hospital cardiac arrest survivors is associated with global reduction in brain volume, not focal hippocampal injury. Stroke. 2000;31:1509-1514.
251 Pusswald G, Fertl E, Faltl M, Auff E. Neurological rehabilitation of severely disabled cardiac arrest survivors. Part II. Life situation of patients and families after treatment. Resuscitation. 2000;47:241-248.
252 Fertl E, Vass K, Sterz F, Gabriel H, Auff E. Neurological rehabilitation of severely disabled cardiac arrest survivors. Part I. Course of post-acute inpatient treatment. Resuscitation. 2000;47:231-239.
253 Shah MK, Al-Adawi S, Dorvlo ASS, Burke DT. Functional outcomes following anoxic brain injury: a comparison with traumatic brain injury. Brain. Injury. 2004;18:111-117.
254 Heiss WD, Teasel RW. Brain recovery and rehabilitation. Stroke. 2006;37:314-316.
255 Moein H, Khalili HA, Keramatian K. Effect of methylphenidate on ICU and hospital length of stay in patients with severe and moderate traumatic brain injury. Clin Neurol Neurosurg. 2006;108:539-542.
256 Saniova B, Drobny M, Kneslova L, Minarik M. The outcome of patients with severe head injuries treated with amantadine sulphate. J Neural Transm. 2004;111:511-514.
257 Nichol G, Stiell IG, Herbert P, Wells GA, Vandembeen K, Laupacis A. What is the quality of life for survivors of cardiac arrest? A prospective study. Acad Emerg Med. 1999;6:95-102.
258 De Vos R. Quality of life after cardiopulmonary resuscitation. Resuscitation. 1997;35:231-236.
