16 Cardiac Physiology and Pharmacology
A thorough understanding of the fetal circulation, the changes at birth, and the age-specific characteristics is important for the safe management of neonates, infants, and especially the growing number of preterm and small-for-gestational-age (SGA) babies who come to our diagnostic suites and operating rooms. Given the complex embryology and difficult transition from fetal to extrauterine life, it is amazing that more than 90% of neonates are delivered without any special interventions and that congenital heart defects occur in only 7 to 10 of every 1000 live births.1 (A detailed discussion of the embryologic development is beyond the scope of this chapter; the interested reader is referred to the excellent review by Van Praagh2 or Langman’s classic embryology textbook.3)
With recent advances in surgical techniques, critical care, and anesthesia management, 85% of all infants with CHD are now expected to reach adulthood. Anesthesiologists will increasingly encounter children with “repaired” or “palliated” CHD presenting for noncardiac procedures. Chapter 21 addresses specific long-term problems and anesthetic considerations for various repaired heart defects, but a few conditions deserve additional discussion: the basic changes in the exercise physiology of repaired heart defects, the characteristics of the so-called Fontan physiology after single ventricle palliation, and the altered physiologic responses in the transplanted heart.
Cardiovascular Physiology
Fetal Circulation
In utero, the placental gas exchange provides the fetus with relatively oxygenated blood; the partial pressure of oxygen (Po2) in the umbilical vein is approximately 30 mm Hg, and in the umbilical arteries it is approximately 16 mm Hg. The fetal lungs are fluid filled and only minimally perfused (10% to 15% of the cardiac output). The normal postnatal circulation can be principally described as a serial circuit: Two pumps, the right ventricle [RV] and left ventricle [LV], support two different resistance systems, the pulmonary and systemic vasculatures, one after the other. In contrast, the fetal circulation is better explained by the concept of a parallel circuit: Both ventricles provide systemic blood flow and a variety of fetal shortcuts or connections allow for mixing of oxygenated and deoxygenated blood (Fig. 16-1).4,5 Oxygenated blood from the placenta returns via the umbilical vein to the portal venous system, where 30% to 50% of the blood flow is shunted across the ductus venosus to the inferior vena cava (IVC), bypassing the liver and thereby maintaining higher oxygenation and velocity. The rest of the umbilical venous blood passes through the hepatic microcirculation into the suprahepatic IVC.

Most of the venous return from the superior vena cava (SVC) and about 20% of the IVC blood flow (mainly the low-velocity, deoxygenated part) reach the RV and are pumped into the pulmonary artery (PA), where the high pulmonary resistance in the nonexpanded lung redirects 90% of the blood flow into the descending aorta via the ductus arteriosus. The bulk of the blood flow in the descending aorta is generated by the RV, with minor contributions from the LV. The blood has a saturation of only 55% to 60%; two thirds of it returns to the placenta for oxygenation, and the rest is distributed to the intestines, the kidneys, and the lower part of the body (Fig. 16-2).

The fetal circulation has to support a growing fetus in a relatively cyanotic atmosphere (highest oxygen saturation, 65% to 70%). This difficult task is further complicated by the parallel circuit, which creates increased workload for the RV, and the limitations of the fetal shortcuts, which add additional volume load by incomplete shunting of oxygenated and deoxygenated blood. Initially, understanding of the fetal circulation was based mainly on experimental animal data, but recent advances in ultrasound technology have made it possible to assess and monitor fetal cardiovascular parameters, especially stroke volume and cardiac output, under various conditions throughout the gestational period. RV stroke volume has been found to increase from about 0.7 mL at 20 weeks to 7.6 mL at 40 weeks, and LV stroke volume increases from 0.7 mL to 5.2 mL. The combined fetal cardiac output of both ventricles is estimated to be 400 to 425 mL/kg/min, with an RV dominance due to the increased volume load. At 38 weeks, the RV provides approximately 60% of the combined cardiac output (E-Table 16-1).6–8 Intrauterine growth restriction and placental compromise are associated with redistribution of cardiac output and relative changes in the size of the foramen ovale.9 A functional placenta, the fetal cardiovascular high-output state, greater hemoglobin concentrations, and additional alterations in oxygen binding and release (hemoglobin F, increased 2,3-diphosphoglycerate [2,3-DPG]) are all necessary to provide adequate tissue oxygenation for the developing fetus.

E-TABLE 16-1 Combined Cardiac Output and Distribution in Human Fetuses during the Second Half of Pregnancy

Until recently, CHD was thought to be relatively well tolerated in utero, but growing evidence suggests that fetal cardiovascular defects can induce intrinsic autoregulatory changes in cerebral perfusion and thereby compromise brain development.10,11 Ultrasound and magnetic resonance imaging demonstrate that 30% to 50% of neonates with CHD have neurologic abnormalities before any surgical intervention.12
Transitional Circulation
At birth, a variety of humoral, biochemical, and physiologic changes occur abruptly. First, the placental circulation is eliminated shortly after the lungs expand. Second, expansion of the lungs to a normal functional residual capacity (FRC) results in an optimal geometric relationship of the pulmonary microvasculature. Third, air entering the lungs causes the alveolar Pco2 to decrease and the alveolar Po2 to increase. These three factors act in concert to markedly reduce pulmonary vascular resistance (PVR).5,13,14 The net effect is a considerable increase in pulmonary blood flow, which augments pulmonary venous return to the left heart. Along with elimination of the placenta and the low-resistance umbilical circulation, the LV is suddenly subjected to increased volume and afterload (Table 16-1). Typically, LV end-diastolic pressure, and thus LA pressure, increases enough to exert hydrostatic pressure on the septum primum, resulting in functional closure of the foramen ovale. In contrast to the increased stress for the LV, the RV is relatively unloaded by the transition to extrauterine life.
| Right Ventricle | Left Ventricle |
|---|---|
| Decreased afterload: | Increased afterload: |
| Decreased pulmonary vascular resistance | Placenta eliminated |
| Ductal closure | Ductal closure |
| Decreased volume load: | Increased volume load: |
| Eliminated umbilical vein return | Increased pulmonary venous return |
| Output diminished 25% | Output increased almost 50% |
| Transient left-to-right shunt at ductus |
The three fetal connections (ductus arteriosus, ductus venosus, and foramen ovale) close over a variable period. The ductus arteriosus has functionally (but not anatomically) closed in 58% of normal full-term infants by 2 days of life and in 98% by day 4.15 Although many substances such as eicosanoids have been implicated, initial constriction probably occurs primarily in response to the increased arterial oxygen tension16,17 and the reduction in circulating prostaglandins that follow separation of the placenta.18 The response to oxygen is age dependent: Term neonates usually demonstrate effective constriction of the smooth muscles in the ductal tissue when exposed to oxygen, whereas preterm infants poorly respond and often require medical (prostaglandin inhibitor) or even surgical therapy. Additional catecholamine-induced changes in PVR and systemic vascular resistance (SVR) and other substances such as acetylcholine contribute to ductal closure. Within 2 to 3 weeks, functional constriction is followed by a process of ductal fibrosis, leaving a band-like structure, the ligamentum arteriosum.19,20 With ligation of the umbilical vein, the portal pressure falls, triggering functional closure of the ductus venosus. This process rarely requires more than 1 to 2 weeks; by 3 months only fibrous tissue, the ligamentum venosum, is left.
The foramen ovale is functionally closed when the LA pressure exceeds the pressure in the RA, but it remains anatomically patent in most infants, in 50% of children younger than 5 years of age, and in 25% to 30% of adults.21 Echocardiographic studies have confirmed right-to-left shunting via the foramen ovale in healthy infants emerging from general anesthesia, and this can be a significant cause of persistent arterial desaturation at that time despite ventilation with 100% oxygen.22
Neonatal Cardiovascular System
Compared with the adult, the neonatal myocardium is immature and incompletely developed (Table 16-2). Differences in cytoarchitecture and metabolism account for many of the functional limitations. The neonatal heart contains fewer muscle cells and more connective tissue than the adult myocardium. Contractile elements add up to only 30% of the total cardiac mass, in contrast to 60% in the adult.23 The ratio of surface area to mass and that of water to collagen content are larger. There are fewer myofibrils within the muscle cells, and they tend to be less organized (i.e., not parallel to the long axis of the cell). The sarcoplasmic reticulum and the T-tubule network, both important components of rapid and effective calcium regulation, are incompletely developed, and the immature myocardium relies substantially on the calcium flux through the sarcolemma to initiate and terminate contraction.24–26 One likely practical consequence is a greater degree of contractile dysfunction in the infant exposed to substances that decrease extracellular ionized calcium, such as citrate (blood products) and albumin; there is also increased sensitivity to volatile anesthetics and calcium channels blockers.
TABLE 16-2 Characteristic Differences between the Immature and the Adult Myocardium
| Immature Myocardium | Adult Myocardium | |
|---|---|---|
| Cytoarchitecture |
ATP, Adenosine triphosphate; CO, cardiac output; SR, sarcoplasmic reticulum.
Data from Mossad EB, Farid I. Vital organ preservation during surgery for congenital heart disease. In: Lake CL, Booker PD, editors. Pediatric cardiac anesthesia. 4th ed. Philadelphia: Lippincott, Williams & Wilkins; 2005, p. 266-90; and DiNardo J, Zwara DA. Congenital heart disease. In: DiNardo J, Zwara DA, editors. Anesthesia for cardiac surgery. 3rd ed. Malden, Mass.: Blackwell Publishing; 2008. p. 167-251.
Reduced numbers of underdeveloped mitochondria and maturational differences in various signaling pathways and related messenger systems are also characteristic of the neonatal myocardium. Immature mitochondrial enzyme activity for fatty acid transport may explain the primary use of carbohydrates and lactates as energy sources and might be a reason for the greater anaerobic tolerance and faster recovery after periods of ischemia. A variety of developmental changes in contractile proteins occur from fetal through early postnatal life, including changes in pH, calcium sensitivity, and adenosine triphosphate (ATP) hydrolyzing activity. The key features of the immature cardiac function are summarized in Table 16-2.
The increased amount of non-contractile tissue in the neonate results in poor ventricular compliance and limited response to increased preload. Compliance of both ventricles progressively increases during fetal life and the postnatal period so that maximal stroke volume occurs at a significantly reduced atrial pressure in the neonate compared with the fetus (Figs. 16-3 and 16-4).27–29 The extraordinarily high metabolic rate of the neonate (oxygen consumption, 6 to 8 mL/kg/min, compared with 2 to 3 mL/kg/min in the adult) requires a proportional increase in cardiac output. The neonatal heart functions at close to maximal rate and stroke volume just to meet the basic demands for oxygen delivery.30,31 The cardiac output is commonly described as being primarily heart rate (HR) dependent due to a fixed stroke volume, but echocardiographic studies in human fetuses and neonates clearly demonstrate the capacity to increase stroke volume (Fig. 16-5).32 In fact, the neonate employs both tachycardia and stroke volume adjustments simply to meet metabolic demand. On the other hand, neonates exhibit exquisite sensitivity to pharmacologic agents that produce negative inotropic or chronotropic effects. At birth both ventricles are equal in mass and connected via a common septum. Increased pressures in one ventricle lead to a septal shift and decreased compliance of the opposite ventricle, causing a reduction in cardiac output. Neonates and infants often present with biventricular failure as a result of this interventricular dependence.



Immature autonomic regulation of cardiac function persists throughout the neonatal period. Both sympathetic and parasympathetic innervation of the heart can be demonstrated at birth. However, evidence suggests that development of the sympathetic nervous system is incomplete at both the postganglionic nerve-receptor level and the receptor-effector level.33 The sympathetic system reaches maturity by early infancy, whereas the parasympathetic system reaches maturity within a few days after birth.34 The relative imbalance of these two components of the autonomic nervous system at birth may account for the clinical observation that neonates are predisposed to exhibit marked vagal responses to a variety of stimuli.
Pulmonary Vascular Physiology
At birth, pulmonary vascular development is incomplete. Lung sections demonstrate diminished numbers of arterioles, and the arterioles exhibit thick medial muscularization (Fig. 16-6).35–37 The pulmonary vasculature matures during the first few years of life. During this period, arterioles proliferate faster than alveoli, and the medial smooth muscle thins and extends more distally in the vascular tree. PVR continues to decrease as long as pulmonary mechanics and alveolar gas composition remain favorable, with a significant decrease occurring immediately after birth due to lung expansion and oxygenation. Progressive remodeling of the pulmonary vasculature facilitates further decreases in PVR (assuming normal physiology) during the first 2 to 3 months of life; by 6 months of age, the PVR has almost reached the level seen in healthy adults.37

The fetal pulmonary vasculature is extremely reactive to a number of stimuli. Hypoxia, acidosis, increased levels of leukotrienes, and mechanical stimulation can cause significant and prolonged increases in PVR (e.g., reactive pulmonary hypertension). On the other hand, acetylcholine, histamine, bradykinin, prostaglandins, β-adrenergic catecholamines, and nitric oxide are strong vasodilators.35 In the first days of life, many pathophysiologic conditions can trigger severe and sustained increases in PVR38,39 and prevent the normal adjustment to extrauterine life (E-Table 16-2). The acute load imposed on the RV can induce diastolic dysfunction and promote right-to-left shunting via the foramen ovale. Once PVR exceeds the SVR, a right-to-left shunt develops via the ductus arteriosus and patent foramen ovale. This situation is called persistent fetal circulation,40 and it can result in a life-threatening hypoxemia that may require inhaled nitric oxide,41–45 sildenafil,46 or extracorporeal support (i.e., extracorporeal membrane oxygenation)47,48 (see Chapter 19) to provide oxygenation and sustain life.

Pulmonary vascular occlusive disease (PVOD) is a term used to describe structural changes in the pulmonary vasculature after longstanding exposure to abnormal pressures and flow patterns in utero and after birth. Lung biopsies demonstrate thickened muscle layers in the small pulmonary arteries, intimal hyperplasia, scarring, and thrombosis as well as a decreased number of distal (intraacinar) arteries.36 Over time, these changes lead to progressive and finally irreversible obstruction of pulmonary blood flow with increases in PVR and PA pressures. The highly muscularized pulmonary arteries are also extremely reactive to pulmonary vasoconstrictors, which can easily trigger a pulmonary hypertensive crisis.
 Exposure of the pulmonary vasculature to systemic arterial pressures and high flow: The classic example is a large, nonrestrictive ventricular septal defect (VSD) with rapid progression of PVOD.
Exposure of the pulmonary vasculature to systemic arterial pressures and high flow: The classic example is a large, nonrestrictive ventricular septal defect (VSD) with rapid progression of PVOD.


The muscle tone in the pulmonary arteries is regulated by numerous factors, and various therapeutic interventions can be used to manipulate the PVR (Table 16-3)14:
 Arterial oxygen tension (Pao2): Alveolar as well as arterial hypoxia increases PVR. A Pao2 value lower than 50 mm Hg, especially when associated with an acidic pH (<7.4), leads to significant pulmonary vasoconstriction. On the other hand, increased inspired oxygen can lead to pulmonary vasodilation.
Arterial oxygen tension (Pao2): Alveolar as well as arterial hypoxia increases PVR. A Pao2 value lower than 50 mm Hg, especially when associated with an acidic pH (<7.4), leads to significant pulmonary vasoconstriction. On the other hand, increased inspired oxygen can lead to pulmonary vasodilation.

 pH: Respiratory and metabolic acidosis increase PVR; alkalosis reduces PVR.
pH: Respiratory and metabolic acidosis increase PVR; alkalosis reduces PVR.


 Vasodilators: Most intravenous (IV) agents used for pulmonary vasodilation also affect the systemic circulation and induce hypotension. Alternatively, inhaled substances such as nitric oxide (NO) or prostacyclin can provide a more selective pulmonary vasodilation (see “Cardiovascular Pharmacology”).
Vasodilators: Most intravenous (IV) agents used for pulmonary vasodilation also affect the systemic circulation and induce hypotension. Alternatively, inhaled substances such as nitric oxide (NO) or prostacyclin can provide a more selective pulmonary vasodilation (see “Cardiovascular Pharmacology”).
TABLE 16-3 Manipulations of Pulmonary Vascular Resistance (PVR)
| Increasing PVR | Decreasing PVR |
|---|---|
Fio2, Fraction of inspired oxygen; FRC, functional residual capacity; PEEP, positive end-expiratory pressure.
Incidence and Prevalence of Congenital Heart Disease
CHD can be defined as “a gross structural abnormality of the heart or intrathoracic great vessels that is actually or potentially of functional significance.”49 This definition covers a wide array of defects, which are among the most common congenital malformations. However, the precise incidence of CHD, both collectively and by individual anatomic subset, varies depending on definition, method of case identification, and epoch (E-Table 16-3). Including all categories of CHD, large epidemiologic surveys place the prevalence anywhere between 4 and 50 cases per 1000 live births.50–53 When stratified according to trivial, moderate, and severe forms, the incidence for moderate and severe forms of CHD is relatively consistent at about 6 per 1000 live births.


Anatomic diagnoses within the population of infants with CHD vary according to the method used to identify cases. In 2002, Hoffman and colleagues compiled 62 epidemiologic studies published after 1955 and investigated the potential causes for the wide variability in the reported incidence of CHD.54 More recent studies based mainly on prenatal and postnatal echocardiographic screening data often include a large number of trivial lesions (e.g., tiny VSDs, nonstenotic bicuspid aortic valve, “silent” PDA) for which no interventions may be required; other data collections, such as the New England Regional Infant Cardiac Program (NERICP), a registry of children with CHD who died or required catheterization or surgery during the first year of life, are clearly biased toward more severe forms of CHD.1
The increasing availability of prenatal diagnostic methods may exert an impact on the relative prevalence of reported lesions as well as their outcome. When fetal echocardiography is used, the apparent shift toward more complex lesions may reflect technical limitations in identifying simple defects.55 In addition, evaluation in utero skews the results because it includes fatally malformed fetuses that will not survive to term. The prevalence of CHD among spontaneous abortions reaches 20% and remains as large as 10% among stillborn infants.56 In one study, 50% of women whose children were given a prenatal diagnosis of CHD elected to terminate the pregnancy, particularly when presented with complex heart lesions.55
On the other hand, female infants with severe CHD have a 5% lower mortality rate than similarly affected male infants,1 and with increased survival rates, more females will reach childbearing age. The recurrence risk of CHD for their offspring is about 3% to 4%.57,58
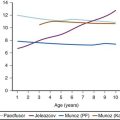
A study from Canada examined the changing epidemiology of CHD with respect to prevalence and age distribution in the general population between 1985 and 2000.59 The prevalence of all categories of CHD in 2000 was 11.89 per 1000 in children (<18 years of age), 4.09 per 1000 in adults, and 5.78 per 1000 in the general population. For the subcategory of severe CHD, the prevalence was 1.45 per 1000 in children and 0.38 per 1000 in adults. In 2000, 49% of all patients with severe CHD were adults, compared with 35% in 1985, and females accounted for 57% of the adult CHD population. The prevalence of CHD increased for both children and adults between 1985 and 2000, although the increase for severe CHD was 85% in adults compared with 22% in children. The median age of all patients with severe CHD was 11 years in 1985 and 17 years in 2000, reflecting the fact that more children with CHD were surviving to adulthood (E-Fig. 16-1). Improved survival may be attributed to improved prenatal care, early diagnostic imaging, and major advances in pediatric cardiac care, particularly for those with severe CHD; these improved outcomes will continue to influence the future demographic profile. The growing number of adolescents and adults with CHD will require long-term follow-up with experienced cardiologists and access to specialized care facilities; this will require a thorough understanding of their underlying pathophysiology by all members of the adult care team, including anesthesiologists.

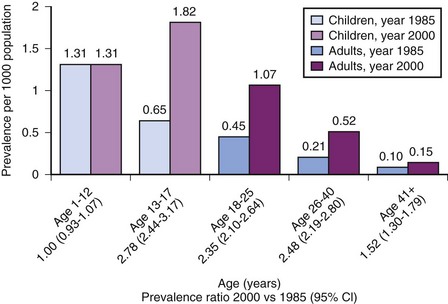
E-FIGURE 16-1 Change in prevalence of congenital heart disease (CHD) from 1985 to 2000 among patients of different age groups.
(From Marelli AJ, Mackie AS, Ionescu-Ittu R, Rahme E, Pilote L. Congenital heart disease in the general population: changing prevalence and age distribution. Circulation 2007;115:163-72.)
Pathophysiologic Classification of Congenital Heart Disease
CHD consists of an almost endless array of anatomic and functional variants. Many different classification systems have been introduced, some using a segmental approach to anatomic features, others by examining the amount of pulmonary blood flow (cyanotic versus acyanotic) or the common physiologic characteristics (e.g., volume versus pressure overload).60–70 Several of these classifications are discussed in Chapter 14. However, certain defects are better described using the concepts of shunting (physiologic, anatomic, simple or complex), intercirculatory mixing, and single ventricle physiology, which are presented in the following sections.
Shunting
Anatomic shunts are communications between the two circulatory systems, either within the heart or at the level of the great vessels. They can be divided into simple and complex shunts, depending on the presence of additional outflow obstructions. In simple shunts without any additional outflow obstruction, the size of the communication (the so-called shunt orifice) determines the flow characteristics. For small orifices (restrictive shunts) with large pressure gradients across the communication, the size of the opening essentially regulates the amount of shunting. Changes in SVR or PVR have little influence. In contrast, for large orifices or nonrestrictive shunts (also classified as dependent shunts), the quantity and direction of blood flow are controlled by the respective outflow resistances (i.e., the ratio of SVR to PVR) (Table 16-4 and Fig. 16-7).
TABLE 16-4 Characteristics of Simple Shunts (without Additional Outflow Obstruction)
| Restrictive (Small Shunt Orifice) |
Nonrestrictive (Large Shunt Orifice) |
|
|---|---|---|
| Examples | Small ASD, VSD, or PDA; modified Blalock-Taussig shunt | Large VSD, PDA, CAVC |
| Pressure gradient across shunt | Large | Small or none |
| Direction and magnitude of shunt | Independent of PVR/SVR | PVR/SVR dependent |
| Influence of pharmacologic and ventilatory interventions | Minimal | Large |
ASD, Atrial septal defect; CAVC, common atrioventricular canal; PDA, patent ductus arteriosus; PVR, pulmonary vascular resistance; SVR, systemic vascular resistance; VSD, ventricular septal defect.
Modified from DiNardo J, Zwara DA. Congenital heart disease. In: DiNardo J, Zwara DA, editors. Anesthesia for cardiac surgery. 3rd ed. Malden, Mass.: Blackwell Publishing; 2008. p. 167-251.

Complex shunts are defined by an additional outflow obstruction, which can be at various levels within the ventricle, valves, or great vessels and is often described as subvalvular, valvular, or supravalvular. These obstructions can be fixed (e.g., valvular stenosis) or variable (e.g., dynamic infundibular obstruction by muscle bundles). Shunt flow and direction are determined by the combined resistance across the outflow obstruction and the pulmonary/systemic vascular beds. For severe obstructions downstream, SVR or PVR will have little influence on the shunt. Tetralogy of Fallot is a good example of a complex shunt lesion. The amount of right-to-left shunt and therefore the amount of cyanosis are influenced by the degree and type of right ventricular outflow tract obstruction (RVOTO). This is especially evident in the setting of a dynamic infundibular obstruction, where changes in preload, contractility, and HR can lead to significant decreases in pulmonary blood flow and increased shunting (Table 16-5).
TABLE 16-5 Characteristics of Complex Shunts (with Additional Outflow Obstruction)
| Partial Outflow Obstruction | Complete Outflow Obstruction | |
|---|---|---|
| Examples | TOF, VSD/PS, VSD/coarctation | Tricuspid or mitral atresia Pulmonary or aortic atresia |
| Shunt magnitude and direction | Relatively fixed | Totally fixed |
| Dependence on PVR/SVR ratio | Inversely related to obstruction | Independent |
| Pressure gradient across shunt | Dependent on shunt orifice and degree of obstruction | Dependent only on shunt orifice |
PS, Pulmonary stenosis; PVR, pulmonary vascular resistance; SVR, systemic vascular resistance; TOF, tetralogy of Fallot; VSD, ventricular septal defect.
Modified from DiNardo J, Zwara DA. Congenital heart disease. In: DiNardo J, Zwara DA, editors. Anesthesia for cardiac surgery. 3rd ed. Malden, Mass.: Blackwell Publishing; 2008, p. 167-251.
Intercirculatory Mixing
The concept of intercirculatory mixing is often used to explain the unique physiology in children with transposition of the great arteries (TGA). In this cardiac defect, the aorta arises from the RV, transporting deoxygenated blood back to the right heart, and the PA originates from the LV, returning oxygenated blood to the pulmonary circulation (see Fig. 15-6). Unless there is some mixing of blood via an ASD, VSD, or PDA, this defect will result in a complete separation of the two systems, a parallel circulation with 100% physiologic shunting or recirculation of oxygenated and deoxygenated blood that is incompatible with life once the fetal ductus arteriosus has closed. Effective pulmonary blood flow (i.e., deoxygenated blood reaching the pulmonary vascular bed for oxygenation) has to be provided by some form of right-to-left shunt; effective systemic blood flow (i.e., oxygenated blood returning to the systemic circulation) must be achieved by a left-to-right shunt. Intercirculatory mixing is the combined systemic and pulmonary effective blood flow and is only a small portion of the total blood flow. The bulk of the respective systemic and pulmonary total blood flows consists of recirculated blood (Fig. 16-8). Usually the total blood flow and the volume in the pulmonary system are two to three times greater than in the systemic circulation.
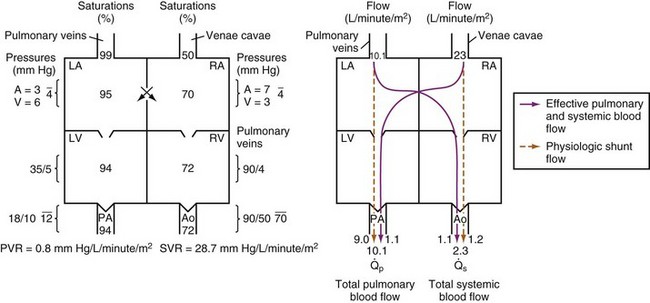
FIGURE 16-8 Depiction of saturations, pressures, and blood flows in transposition of the great arteries with a nonrestrictive atrial septal defect134 and a small left ventricular (LV) outflow tract gradient. Intercirculatory mixing occurs at the atrial level. Effective pulmonary and effective systemic blood flows are equal (1.1 L/min/m2) and are the result of a bidirectional anatomic shunt at the atrial level. The physiologic left-to-right shunt is 9.0 L/min/m2; this represents blood recirculated from the pulmonary veins to the pulmonary artery (PA). The physiologic right-to-left shunt is 1.2 L/min/m2; this represents blood recirculated from the systemic veins to the aorta (Ao). Total pulmonary blood flow ( = 10.1 L/min/m2) is almost five times higher than the total systemic blood flow (
= 10.1 L/min/m2) is almost five times higher than the total systemic blood flow ( = 2.3 L/min/m2). The bulk of pulmonary blood flow is recirculated pulmonary venous blood. In this depiction, pulmonary vascular resistance (PVR) is low (approximately 1/35 of systemic vascular resistance [SVR]) and there is a small (17 mm Hg peak to peak) gradient from the LV to the PA. These findings are compatible with the high pulmonary blood flow depicted. LA, Left atrium; RA, right atrium; RV, right ventricle.
= 2.3 L/min/m2). The bulk of pulmonary blood flow is recirculated pulmonary venous blood. In this depiction, pulmonary vascular resistance (PVR) is low (approximately 1/35 of systemic vascular resistance [SVR]) and there is a small (17 mm Hg peak to peak) gradient from the LV to the PA. These findings are compatible with the high pulmonary blood flow depicted. LA, Left atrium; RA, right atrium; RV, right ventricle.
(From DiNardo J, Zwara DA. Congenital heart disease. In: DiNardo J, Zwara DA, editors. Anesthesia for cardiac surgery. 3rd ed. Malden, Mass.: Blackwell Publishing; 2008. p. 167-251.)

Single Ventricle Physiology
Single ventricle physiology defines the circulation present in a wide variety of complex cardiac defects. It is characterized by complete mixing of systemic and pulmonary venous blood return at either the atrial or the ventricular level; the mixed blood is then distributed to both systemic and pulmonary circulations in parallel. The defects can consist of one anatomic single ventricle with severe hypoplasia and inflow or outflow obstruction of the other one (hypoplastic left heart syndrome [HLHS] or pulmonary atresia with intact ventricular septum) or even two well-developed ventricles with atresia of the outflow tract or severe obstruction (tetralogy of Fallot with pulmonary atresia, interrupted aortic arch). In some lesions, a PDA is the only source of systemic or pulmonary blood flow; these are called duct-dependent circulations. In others, intracardiac communications provide adequate blood flow to both circulations (Table 16-6).
| Congenital Heart Defect | Aortic Blood Flow from | Pulmonary Blood Flow from |
|---|---|---|
| Hypoplastic left heart syndrome | PDA | RV |
| Neonatal critical aortic stenosis | PDA | RV |
| Interrupted aortic arch | Proximal LV, distal PDA | RV |
| Tetralogy of Fallot with pulmonary atresia | LV | PDA, MAPCAs |
| Pulmonary atresia with intact septum | LV | PDA |
| Tricuspid atresia 1B (VSD and PS) | LV | LV through VSD to RV |
| Truncus arteriosus | LV and RV | Aorta |
| Double inlet left ventricle, no TGA | LV | LV through VSD to bulboventricular foramen |
LV, Left ventricle; MAPCAs, major aortopulmonary collateral arteries; PDA, patent ductus arteriosus; PS, pulmonary stenosis; RV, right ventricle; TGA, transposition of the great arteries; VSD, ventricular septal defect.
Modified from DiNardo J, Zwara DA. Congenital heart disease. In: DiNardo J, Zwara DA, editors. Anesthesia for cardiac surgery. 3rd ed. Malden, Mass.: Blackwell Publishing; 2008, p. 167-251.

 The ratio of pulmonary to systemic blood flow (
The ratio of pulmonary to systemic blood flow ( ). With high
). With high 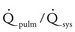 , a greater percentage of the blood in the ventricle (or ventricles) is oxygenated because more fully saturated pulmonary venous blood is entering the heart to mix with desaturated systemic venous return. Saturations greater than 85% can be achieved only by significant pulmonary overcirculation.
, a greater percentage of the blood in the ventricle (or ventricles) is oxygenated because more fully saturated pulmonary venous blood is entering the heart to mix with desaturated systemic venous return. Saturations greater than 85% can be achieved only by significant pulmonary overcirculation. 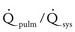 can be influenced by careful manipulations of the PVR/SVR ratio.
can be influenced by careful manipulations of the PVR/SVR ratio.

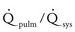 and pulmonary venous saturation (Spulmv
and pulmonary venous saturation (Spulmv
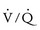 mismatch, or large intrapulmonary shunts can cause significant desaturations.
mismatch, or large intrapulmonary shunts can cause significant desaturations. 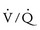 mismatch usually responds to therapy with increased inspired oxygen, whereas intrapulmonary shunts are refractory to oxygen therapy. Pulmonary venous desaturation will decrease arterial saturations.
mismatch usually responds to therapy with increased inspired oxygen, whereas intrapulmonary shunts are refractory to oxygen therapy. Pulmonary venous desaturation will decrease arterial saturations.Special Situations
Exercise Physiology in the Child with Repaired Congenital Heart Disease
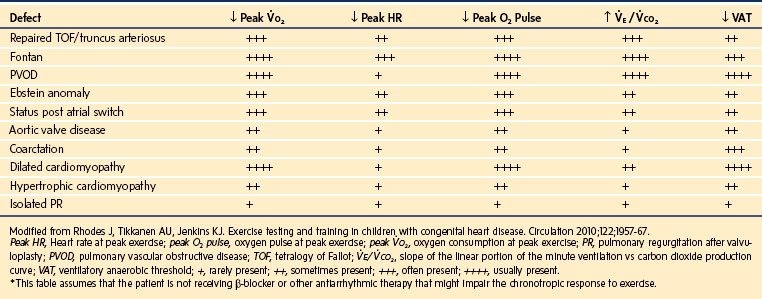
As summarized in E-Table 16-4, children with CHD, including those with lesions considered repaired, exhibit an array of abnormalities elicited during exercise testing consistent with reduced exercise capacity. It is worthwhile to review the various exercise testing abnormalities in order to gain insight into the limitations imposed by the presence of congenital heart lesions.

E-TABLE 16-4 Cardiopulmonary Exercise Testing Abnormalities in Pediatric Patients with Various Forms of Congenital Heart Disease*
Oxygen consumption (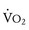 ) is equal to cardiac output times O2 extraction. O2 extraction is equal to the arterial–venous oxygen content difference. Peak
) is equal to cardiac output times O2 extraction. O2 extraction is equal to the arterial–venous oxygen content difference. Peak 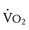 is the greatest measure of
is the greatest measure of 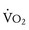 obtained during a progressively more difficult exercise test.
obtained during a progressively more difficult exercise test. 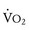 at rest is defined as 1 metabolic equivalent energy expenditure unit or 1 MET (approximately 3.5 mL O2/kg/min).71 A typical elite endurance athlete can reach 20 to 22 METs, or 70 to 77 mL O2/kg/min, at peak exercise. Activities of daily living require at least 4 METs or 14 mL O2/kg/min. Peak
at rest is defined as 1 metabolic equivalent energy expenditure unit or 1 MET (approximately 3.5 mL O2/kg/min).71 A typical elite endurance athlete can reach 20 to 22 METs, or 70 to 77 mL O2/kg/min, at peak exercise. Activities of daily living require at least 4 METs or 14 mL O2/kg/min. Peak 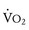 is the best overall assessment of the capabilities of the cardiovascular system, but determination of normal values is difficult due to the effects of age, gender, effort, and lean body mass versus adipose tissue on peak
is the best overall assessment of the capabilities of the cardiovascular system, but determination of normal values is difficult due to the effects of age, gender, effort, and lean body mass versus adipose tissue on peak 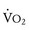 . Nonetheless, peak
. Nonetheless, peak 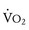 has been demonstrated to be a reliable predictor of hospitalization and mortality in patients with a wide variety of congenital heart lesions.72
has been demonstrated to be a reliable predictor of hospitalization and mortality in patients with a wide variety of congenital heart lesions.72
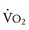 . Normal peak HR is generally defined (in beats/min) as 220 minus age in years. In children with chronotropic incompetence, which is defined as the inability to increase HR to greater than 80% of the predicted value at peak exercise, the relationship between HR and
. Normal peak HR is generally defined (in beats/min) as 220 minus age in years. In children with chronotropic incompetence, which is defined as the inability to increase HR to greater than 80% of the predicted value at peak exercise, the relationship between HR and 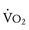 is depressed. Chronotropic incompetence is an indicator of poor prognosis and is most commonly the result of sinus node dysfunction. By comparison, well-trained endurance athletes have a normal peak HR and a depressed HR/
is depressed. Chronotropic incompetence is an indicator of poor prognosis and is most commonly the result of sinus node dysfunction. By comparison, well-trained endurance athletes have a normal peak HR and a depressed HR/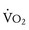 relationship, because they can generate a larger-than-normal stroke volume increase as exercise progresses. The inability to increase stroke volume (discussed later) during exercise results in an increased HR/
relationship, because they can generate a larger-than-normal stroke volume increase as exercise progresses. The inability to increase stroke volume (discussed later) during exercise results in an increased HR/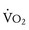 relationship as a compensatory mechanism.
relationship as a compensatory mechanism.The oxygen pulse is the quantity of oxygen delivered per heartbeat. The peak O2 pulse is calculated by dividing the peak 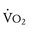 by the peak HR. Because peak
by the peak HR. Because peak 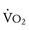 = cardiac output × O2 extraction and because O2 extraction remains remarkably constant over a wide range of exercise, O2 pulse is proportional to stroke volume. Determination of normal peak O2 pulse is hampered by the same factors that confound determination of normal peak
= cardiac output × O2 extraction and because O2 extraction remains remarkably constant over a wide range of exercise, O2 pulse is proportional to stroke volume. Determination of normal peak O2 pulse is hampered by the same factors that confound determination of normal peak 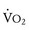 . In addition, O2 pulse overestimates stroke volume in the presence of erythrocytosis and underestimates it in the presence of anemia or reduced arterial O2 saturation. O2 pulse is reduced in patients with impaired ventricular function, severe valvular regurgitation, or pulmonary vascular disease.72 It is also uniformly reduced in those with Fontan physiology as a consequence of the inability of this circulation to augment systemic ventricular preload during exercise.73
. In addition, O2 pulse overestimates stroke volume in the presence of erythrocytosis and underestimates it in the presence of anemia or reduced arterial O2 saturation. O2 pulse is reduced in patients with impaired ventricular function, severe valvular regurgitation, or pulmonary vascular disease.72 It is also uniformly reduced in those with Fontan physiology as a consequence of the inability of this circulation to augment systemic ventricular preload during exercise.73
The respiratory exchange ratio (RER) is defined as the ratio 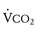 /
/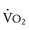 (ratio of the volume of CO2 produced per minute to the volume of oxygen consumed per minute). A normal resting RER is between 0.67 and 1.0, depending on the precise composition of protein, carbohydrates, and fat in the diet. As exercise intensifies, anaerobic metabolism commences and the lactate threshold is reached; buffering of lactic acid with bicarbonate causes the carbon dioxide production (
(ratio of the volume of CO2 produced per minute to the volume of oxygen consumed per minute). A normal resting RER is between 0.67 and 1.0, depending on the precise composition of protein, carbohydrates, and fat in the diet. As exercise intensifies, anaerobic metabolism commences and the lactate threshold is reached; buffering of lactic acid with bicarbonate causes the carbon dioxide production (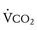 ) to increase out of proportion to oxygen consumption (
) to increase out of proportion to oxygen consumption (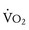 ), resulting in an increased RER. An RER of 1.09 or greater is thought to indicate the onset of anaerobic metabolism and to be consistent with a good effort.71,72 Because RER increases only if anaerobic metabolism occurs, exercise limitation and low
), resulting in an increased RER. An RER of 1.09 or greater is thought to indicate the onset of anaerobic metabolism and to be consistent with a good effort.71,72 Because RER increases only if anaerobic metabolism occurs, exercise limitation and low 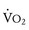 due to musculoskeletal problems or poor effort are associated with an RER below this threshold.
due to musculoskeletal problems or poor effort are associated with an RER below this threshold.
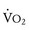 and is relatively effort and motivation independent. As aerobic exercise progresses, minute ventilation (
and is relatively effort and motivation independent. As aerobic exercise progresses, minute ventilation (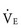 ) increases in direct proportion to
) increases in direct proportion to 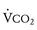 and
and 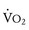 . When anaerobic metabolism commences and CO2 production increases as lactic acid is buffered,
. When anaerobic metabolism commences and CO2 production increases as lactic acid is buffered, 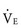 increases accordingly. VAT is the point at which
increases accordingly. VAT is the point at which 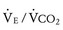 and
and 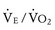 diverge, with
diverge, with 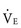 rising in proportion to
rising in proportion to 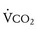 but out of proportion to
but out of proportion to 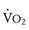 . An important characteristic of successful endurance athletes is the ability to reach and sustain effort at an anaerobic threshold that is a large percentage (80% to 85%) of peak
. An important characteristic of successful endurance athletes is the ability to reach and sustain effort at an anaerobic threshold that is a large percentage (80% to 85%) of peak 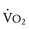 .
.Ventilation efficiency can be assessed with the use of the 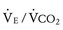 slope. This relationship is defined as 863 •
slope. This relationship is defined as 863 • 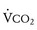 / [Paco2 • (1 − VD/VT)], where VD/VT is the ratio of physiologic dead space to tidal volume.75 The
/ [Paco2 • (1 − VD/VT)], where VD/VT is the ratio of physiologic dead space to tidal volume.75 The 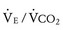 slope can be thought of as the number of liters of ventilation required to eliminate 1 L of CO2. Normal children have a
slope can be thought of as the number of liters of ventilation required to eliminate 1 L of CO2. Normal children have a 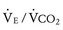 slope of less than 28.72 In order to maintain a normal Paco2 during exercise, children with increased VD/VT and reduced ventilatory efficiency have a greater than normal increase in
slope of less than 28.72 In order to maintain a normal Paco2 during exercise, children with increased VD/VT and reduced ventilatory efficiency have a greater than normal increase in 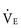 and therefore a steeper
and therefore a steeper 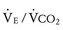 slope. Increased VD/VT is the consequence of either reduced VT in the setting of a normal VD or pulmonary flow maldistribution and subsequent
slope. Increased VD/VT is the consequence of either reduced VT in the setting of a normal VD or pulmonary flow maldistribution and subsequent 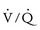 mismatch that increases VD. The latter is the major source of inefficient ventilation and steepening of the
mismatch that increases VD. The latter is the major source of inefficient ventilation and steepening of the 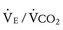 slope in children with cardiac disease.
slope in children with cardiac disease.
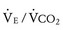 slope is associated with increased mortality. When pulmonary stenosis is corrected in children with tetralogy of Fallot, the
slope is associated with increased mortality. When pulmonary stenosis is corrected in children with tetralogy of Fallot, the 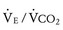 slope and peak
slope and peak 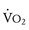 improve.
improve.Children with Fontan physiology also exhibit an increase in 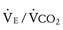 slope. These children have inherent nonhomogeneous pulmonary perfusion at rest due to the lack of pulsatile pulmonary blood flow. In addition, there is poor recruitment of distal pulmonary vasculature during exercise. The presence of a Fontan fenestration further contributes to this increase in the
slope. These children have inherent nonhomogeneous pulmonary perfusion at rest due to the lack of pulsatile pulmonary blood flow. In addition, there is poor recruitment of distal pulmonary vasculature during exercise. The presence of a Fontan fenestration further contributes to this increase in the 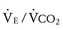 slope by allowing mixed venous blood high in CO2 to be shunted into the systemic circulation. This produces, via central chemoreceptor stimulation, an increase in
slope by allowing mixed venous blood high in CO2 to be shunted into the systemic circulation. This produces, via central chemoreceptor stimulation, an increase in 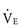 out of proportion to
out of proportion to 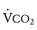 .76 Fontan fenestration closure eliminates this right-to-left shunt and reduces the
.76 Fontan fenestration closure eliminates this right-to-left shunt and reduces the 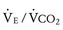 slope but does not improve peak
slope but does not improve peak 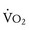 .76 The reason is that the primary limitation to increases in
.76 The reason is that the primary limitation to increases in 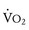 during exercise in Fontan patients is the inherent inability of the pulmonary vascular bed to substantially increase surface area, flow, and preload delivery to the systemic ventricle.
during exercise in Fontan patients is the inherent inability of the pulmonary vascular bed to substantially increase surface area, flow, and preload delivery to the systemic ventricle.
Fontan Physiology
Francis Fontan, a French cardiac surgeon, described a new treatment for complex cardiac malformations with only one ventricle in 1971.77 To decrease the chronic volume overload for the single ventricle and normalize oxygenation, he separated the systemic and pulmonary circulations by directly connecting the systemic venous return (SVC and IVC) to the PA, without a pumping chamber. This created a circulation wherein pulmonary blood was driven solely by a nonpulsatile pressure gradient across the pulmonary vascular bed, with the single ventricle being the sole source of kinetic energy. All other shunt connections were interrupted. The original indication was tricuspid atresia, but over the years the classic Fontan technique has been modified in many ways and is now used for various complex cardiac lesions with single ventricle physiology, such as HLHS, double-inlet RV, and pulmonary atresia with intact septum (see also Chapters 15 and 21).78–82
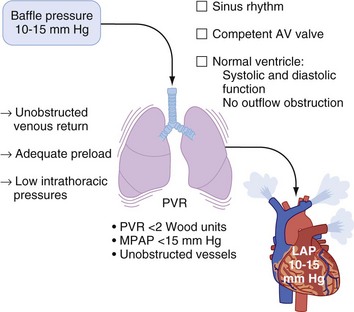
It is impossible to create a Fontan circulation at birth; high PVR and small vessel sizes prevent adequate pulmonary blood flow. In the neonatal period, palliative procedures such as stage I Norwood operation with aortic arch reconstruction, atrial septostomy, and aortopulmonary shunts (modified Blalock-Taussig shunt) or the Sano modification of the Norwood procedure (RV-to-PA conduit) aim for balanced systemic and pulmonary blood flows, allowing the baby to grow for several months despite cyanosis and volume load on the ventricle. At the age of 3 to 6 months, an intermediate procedure called the bidirectional Glenn operation or superior cavopulmonary anastomosis, is performed. The SVC is connected directly to the PA, providing nonpulsatile pulmonary blood flow, whereas the IVC remains connected to the heart. As a result, the volume load on the ventricle is significantly reduced, but oxygenated and deoxygenated blood still mix and the saturations remain in the low 80% region. By the age of 1 to 5 years, most of these children are ready for the Fontan circulation. With adequate growth and maturation of the pulmonary vascular bed, the resistance should be low enough to allow the complete separation of the systemic and pulmonary flows. The IVC is now also connected to the PA, most often via a lateral tunnel in the atrium or an extracardiac conduit, with or without a small fenestration (small opening in the baffle or conduit connecting the systemic venous return with the common atrium of the single ventricle). The fenestration can provide a residual right-to-left shunt in case of sudden increases in PVR, maintaining ventricular preload and function. This seems to facilitate the adaptation to the new loading conditions, shorten the recovery time, and decrease the incidence of early complications. The fenestration often occludes spontaneously, or it is closed during a cardiac catheterization and hemodynamic evaluation with a special device (Fig. 16-9; see also Figs. 15-11 through 15-13).83–86
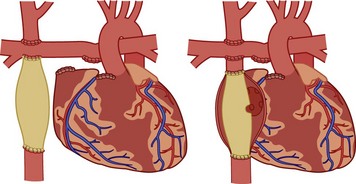
FIGURE 16-9 Fontan modifications: extracardiac conduit (left) and lateral tunnel with fenestration (right).
(From the Children’s Hospital Boston web site. Available at http://www.childrenshospital.org/cfapps/mml/index.cfm?CAT=media&MEDIA_ID=1837)
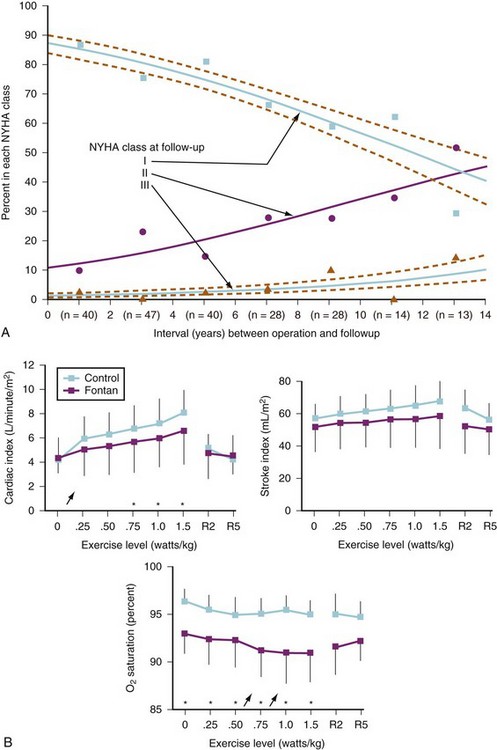
The Fontan operation has dramatically improved the mortality rates for children with single ventricles, but the success comes at a price: Chronic systemic venous hypertension and congestion has been implicated in a multitude of potential early and long-term complications, including arrhythmias, residual right-to-left shunts, coagulopathies with increased risk for thrombosis and stroke, lymphatic dysfunction with pleural effusions, and protein-losing enteropathy.87–92 Late cardiac failure and poor functional outcome remain risks for patients with Fontan circulations. The anatomy of the single ventricle and the type of Fontan connection influence the duration of freedom from complications. Children with systemic RVs and the classic atriopulmonary Fontan (RA directly anastomosed to the PA) tend to have a shorter duration of freedom from complications than those with systemic LVs and newer Fontan modifications (E-Figs. 16-2 and 16-3).93

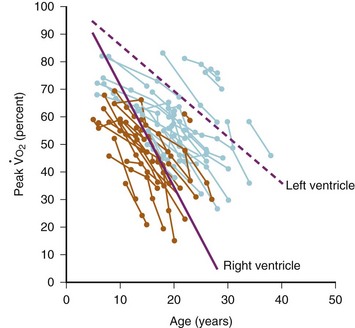
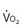 over time in individual Fontan patients according to the presence of a morphologically right ventricle (orange circles) or left ventricle (blue circles). Solid and dashed lines indicate the average of the individual slopes of decrease of peak
over time in individual Fontan patients according to the presence of a morphologically right ventricle (orange circles) or left ventricle (blue circles). Solid and dashed lines indicate the average of the individual slopes of decrease of peak 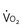 in patients with morphologically right versus left ventricle, respectively.
in patients with morphologically right versus left ventricle, respectively.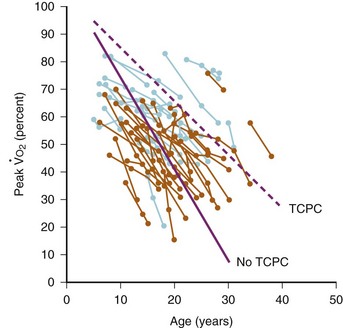
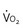 over time in individual Fontan patients according to the type of Fontan surgery. Orange circles indicate atriopulmonary or atrioventricular connections; blue circles refer to intracardiac or extracardiac total cavopulmonary connection (TCPC). The dashed and solid lines, respectively, indicate the average of the individual slopes of decrease of peak
over time in individual Fontan patients according to the type of Fontan surgery. Orange circles indicate atriopulmonary or atrioventricular connections; blue circles refer to intracardiac or extracardiac total cavopulmonary connection (TCPC). The dashed and solid lines, respectively, indicate the average of the individual slopes of decrease of peak 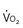 in patients with and without a TCPC.
in patients with and without a TCPC.Inherent limitations of the Fontan circulation, such as altered control of cardiac output with decreased hemodynamic response to stress and reduced exercise tolerance, have been documented (Fig. 16-10).94–102 Even at rest, cardiac output is usually only 70% (range, 50% to 80%) of normal for body surface area. Cardiac output is classically determined by four factors: preload, contractility, HR, and afterload. Over a physiologic range, cardiac output improves with increased preload, contractility, and HR and with decreased afterload. For the Fontan circulation, the determinants of CO are more complex (Fig. 16-11).97,103 The classic determinants of cardiac output are less effective, while other factors, such as transpulmonary gradient and PVR, must be considered. The following mechanisms regulate cardiac output in children with Fontan physiology.
 Preload: The RV usually provides the kinetic energy to distend the pulmonary vasculature and create a preload reservoir for the LV, thereby enabling an increase in cardiac output up to fivefold or greater with exercise.103,104 The lack of a pre-pulmonary pump leads to a significant decrease in available pulmonary blood volume and, consequently, reduced or absent LV preload reserve.97,105,106
Preload: The RV usually provides the kinetic energy to distend the pulmonary vasculature and create a preload reservoir for the LV, thereby enabling an increase in cardiac output up to fivefold or greater with exercise.103,104 The lack of a pre-pulmonary pump leads to a significant decrease in available pulmonary blood volume and, consequently, reduced or absent LV preload reserve.97,105,106
 Contractility: During the staged palliation, the single ventricle typically develops from a volume-overloaded and dilated ventricle to a hypertrophied, underfilled ventricle.107,108 Although the contractile response to β-adrenergic stimulation seems to be preserved, the resulting increase in cardiac output is diminished, most likely due to limited preload reserve.100,106
Contractility: During the staged palliation, the single ventricle typically develops from a volume-overloaded and dilated ventricle to a hypertrophied, underfilled ventricle.107,108 Although the contractile response to β-adrenergic stimulation seems to be preserved, the resulting increase in cardiac output is diminished, most likely due to limited preload reserve.100,106
 Heart rate and rhythm: Within the physiologic range, atrial pacing at different HRs does not alter cardiac output because there is a simultaneous decrease in stroke volume.109 Normalization of HR increases the reduced cardiac output associated with severe bradycardia or tachycardia.110 During exercise testing, Fontan patients demonstrate chronotropic incompetence, a blunted HR response to exercise. This is likely the result of autonomic dysfunction or abnormal reflex control. In contrast to the HR, cardiac rhythm is of utmost importance. Ectopy or loss of atrioventricular (AV) synchronization compromises ventricular filling and decreases the transpulmonary gradient.111
Heart rate and rhythm: Within the physiologic range, atrial pacing at different HRs does not alter cardiac output because there is a simultaneous decrease in stroke volume.109 Normalization of HR increases the reduced cardiac output associated with severe bradycardia or tachycardia.110 During exercise testing, Fontan patients demonstrate chronotropic incompetence, a blunted HR response to exercise. This is likely the result of autonomic dysfunction or abnormal reflex control. In contrast to the HR, cardiac rhythm is of utmost importance. Ectopy or loss of atrioventricular (AV) synchronization compromises ventricular filling and decreases the transpulmonary gradient.111
 Afterload: The Fontan circulation is characterized by increased afterload, which is a physiologic response to decreased cardiac output and occurs because a single ventricle is ejecting into two large resistance beds (systemic and pulmonary vascular) arranged in series.97,106,112,113 Autonomic regulation and activation of various endocrine systems increase the systemic venous resistance and help to maintain adequate perfusion pressures and venous tone. Because of the limited preload reserve, attempts at afterload reduction often result in significant hypotension. On the other hand, excessive afterload, such as that which occurs with residual aortic arch obstruction, is poorly tolerated.
Afterload: The Fontan circulation is characterized by increased afterload, which is a physiologic response to decreased cardiac output and occurs because a single ventricle is ejecting into two large resistance beds (systemic and pulmonary vascular) arranged in series.97,106,112,113 Autonomic regulation and activation of various endocrine systems increase the systemic venous resistance and help to maintain adequate perfusion pressures and venous tone. Because of the limited preload reserve, attempts at afterload reduction often result in significant hypotension. On the other hand, excessive afterload, such as that which occurs with residual aortic arch obstruction, is poorly tolerated.
 Transpulmonary flow: Transpulmonary flow is directly proportional to the gradient between the systemic venous pressure (usually between 10 and 15 mm Hg, rarely greater than 20 mm Hg) and the preventricular atrial pressure, which is determined by the functional status of the AV valve, the ventricle, the rhythm, and the potential presence of outflow obstruction. Transpulmonary flow is inversely proportional to the resistance over the Fontan circuit. This resistance is largely determined by PVR, but mechanical obstruction such as stenosis or thrombosis may also play a role. The geometry of the cavopulmonary connections are also important in that turbulent flow produces energy loss and a reduction in effective driving pressure. It has been suggested that PVR is the key determinant of transpulmonary flow, delivery of pulmonary venous flow to the systemic ventricle, and, consequently, cardiac output (Fig. 16-12).97,114–124
Transpulmonary flow: Transpulmonary flow is directly proportional to the gradient between the systemic venous pressure (usually between 10 and 15 mm Hg, rarely greater than 20 mm Hg) and the preventricular atrial pressure, which is determined by the functional status of the AV valve, the ventricle, the rhythm, and the potential presence of outflow obstruction. Transpulmonary flow is inversely proportional to the resistance over the Fontan circuit. This resistance is largely determined by PVR, but mechanical obstruction such as stenosis or thrombosis may also play a role. The geometry of the cavopulmonary connections are also important in that turbulent flow produces energy loss and a reduction in effective driving pressure. It has been suggested that PVR is the key determinant of transpulmonary flow, delivery of pulmonary venous flow to the systemic ventricle, and, consequently, cardiac output (Fig. 16-12).97,114–124
Physiology of the Transplanted Heart
According to the International Society of Heart and Lung Transplantation (ISHLT), children younger than 18 years of age account for about 12.5% of all heart transplantations. Every year, approximately 450 new pediatric cardiac transplantation cases are reported to this voluntary registry, mainly from centers in Europe and North America.125 Major indications are cardiomyopathies, CHD, and a growing number of re-transplantations, especially in older children. The average survival time is currently 18 years for infants, 15 years for children (aged 1 to 10 years), and 11 years for teenagers. Ninety-two percent of transplant recipients describe a normal functional status with no limitations during physical activity.126,127
Physiology of the Denervated Heart
After transplantation, the function of the surgically denervated heart is primarily dependent on an intact Frank-Starling mechanism and stimulation from circulating catecholamines. The classic Frank-Starling mechanism describes the ability of the cardiac muscle to increase contractility in response to stretch or tension (e.g., increasing cardiac output with increases in venous return). Afferent and efferent denervation has multiple effects on circulatory control mechanisms and leads to significant physiologic changes, including an increase in the resting HR and a blunted response to stress and exercise. Despite excellent physical activity, exercise testing easily demonstrates that heart transplant recipients can usually achieve only 60% to 70% of normal capacity. In the transplanted heart, exercise-induced increase in cardiac output is initially caused by increase in stroke volume, a highly preload dependent process. Tachycardia occurs only later, in response to circulating catecholamines.128 Further details of the altered physiology are summarized in Table 16-7.128 The incidence, timing, and extent of sympathetic reinnervation are still being investigated, but the positive effects on cardiac performance have been clearly demonstrated.129 During standardized exercise testing, transplant recipients with evidence of reinnervation show improved endurance with greater peak HRs and better contractile function.
TABLE 16-7 Physiology of the Transplanted Heart
AV, Atrioventricular; LVEDP, left ventricular end-diastolic pressure.
Data from Schure AY, Kussman BD. Pediatric heart transplantation: demographics, outcomes, and anesthetic implications. Paediatr Anaesth 2011;21:594-603; and Cotts WG, Oren RM. Function of the transplanted heart: unique physiology and therapeutic implications. Am J Med Sci 1997;314:164-72.
Chronic denervation also causes an altered response to many medications. Atropine, glycopyrrolate, digoxin, and pancuronium have no chronotropic effect on the denervated heart. Sympathomimetics that act indirectly, such as ephedrine and dopamine, have a blunted response, whereas direct-acting adrenergic agents such as epinephrine, isoproterenol, and dobutamine can cause exaggerated effects and should be carefully titrated.130 Several case reports have described profound bradycardia and even cardiac arrest after neostigmine used for reversal of neuromuscular blockade.131–134 Neostigmine has been shown to produce an atropine-sensitive, dose-dependent bradycardia in both recent (<6 months) and remote (>6 months) cardiac transplants. Direct stimulation of postganglionic nicotinic cholinergic receptors with denervation hypersensitivity, a direct effect of the old sinoatrial node on the pacemaker cell of the new sinoatrial node, and parasympathetic reinnervation have been postulated as potential mechanisms.135–137 Avoidance of neuromuscular blockade, use of short-acting neuromuscular blocking agents without reversal, and use of edrophonium for reversal have all been suggested.133,134 Edrophonium seems to have less effect on the HR in this population than neostigmine.138
The denervated heart is also extremely sensitive to adenosine. The magnitude and duration of the effect on the AV node is three to five times greater, and the dose should be reduced by 50%.139 Calcium channel blockers and β-blockers are associated with exaggerated bradycardia and hypotension. The lack of reflex tachycardia can also lead to profound hypotension with the use of direct vasodilators such as nitroglycerine, nitroprusside, or hydralazine.
Transplant Morbidity
Children who have undergone cardiac transplantation continue to experience significant morbidity associated with immunosuppressive therapy. Rehospitalization for treatment of infections and episodes of rejection are common, especially during the first year (50%). Acute rejection episodes are a major threat. The incidence decreases from 30% during the first year to 12% after 5 years. Rejection is thought to be associated with the development of cardiac allograft vasculopathy (CAV) or coronary artery disease, which is a major cause of morbidity and graft failure. Indeed, 10 years after transplantation, 34% of children have CAV.127 Most centers include annual coronary angiography or intravascular echocardiography as a part of regular rejection surveillance. Children with CAV present the same anesthetic challenges as adults with severe coronary artery disease and ischemic heart disease. Aggressive immunosuppressive therapy with induction and maintenance regimens carries its own risks. A detailed discussion is beyond the scope of this chapter, but in general, monoclonal or polyclonal T-cell antibodies (OKT, ATG) and specific interleukin 2 receptor antagonists (basiliximab, daclizumab) are used for the induction phase and various combinations of corticosteroids, calcineurin inhibitors (cyclosporine, tacrolimus, FK506), antiproliferative agents (azathioprine, mycophenolate mofetil), and target of rapamycin inhibitors (sirolimus) for maintenance. Side effects are common and include neurotoxicity with seizures, hypertension, liver and renal dysfunction, hyperlipidemia, diabetes, gingival hypertrophy, hypertrichosis, bone marrow suppression, and posttransplantation lymphoproliferative disease (PTLD).140
Several excellent review articles describe the anesthetic management of children with heart transplants (see also Chapter 21).141–143 A thorough preoperative evaluation with attention to episodes of rejection, presence of coronary artery disease, and organ dysfunction; a detailed medication history with investigation of major side effects; consideration of the denervated physiology; and appropriate choice of anesthetic drugs and other medications are essential components of a sensible anesthetic plan for those with a transplanted heart.
Cardiovascular Pharmacology
Rational Use of Vasoactive Drugs
Catecholamines and catecholamine-like agents remain the most commonly used inotropic and vasoconstrictor drugs. It is likely that improvements in cardiac output in neonates in response to drugs such as dopamine or dobutamine are the result of increases in both HR and contractility. Some evidence exists in infants and young children after cardiac surgery that the increases in cardiac output produced by dopamine and dobutamine may be more related to a positive chronotropic effect than to an increase in the intrinsic contractile state.144–146 With few exceptions, drugs that primarily increase afterload, such as α-adrenergic agonists, have limited use in children. Large increases in afterload without corresponding improvements in contractile state are often poorly tolerated by infants and children, particularly in the context of significant underlying contractile dysfunction.
Practical Considerations for the Use of Vasoactive Agents
Commonly used drugs, their doses, and a summary of their effects on selected cardiac functions are presented in Table 16-8. Most of this information has been empirically derived from studies in adults. Limited direct information regarding the effects of commonly used vasoactive drugs in children at different ages and in various pathophysiologic states is available. Neonates, infants, and small children demonstrate unique responses to inotropic and vasoactive drugs due mainly to age-specific pharmacokinetics; differences in receptor types, number, and function; and a high variability in drug delivery. Substantial variations in volume of distribution and measured plasma concentrations have been observed in children receiving inotropic agents. As much as a 10-fold range in plasma concentration has been reported for a given infusion rate.147–149
| Agent | Intravenous Dose | Comments |
|---|---|---|
| Dopamine | 2-20 µg/kg/min infusion | Primary effects at β1, β2, and dopamine receptors, somewhat related to dose; lower doses (2-5 µg/kg/min) can increase contractility and can also have a direct dopaminergic receptor effect to increase splanchnic and renal perfusion; increasing doses increase contractility via β-effects and also increase likelihood of α-mediated vasoconstriction; effects depend on endogenous catecholamine stores. |
| Dobutamine | 2-20 µg/kg/min infusion | Relatively selective β1 stimulation; also potential β2 stimulation, tachycardia, and vasodilation, especially at higher doses (>10 µg/kg/min); may be less potent than dopamine, especially in immature myocardium; no significant α-adrenergic effects; tachydysrhythmias perhaps more likely than with dopamine; effects are independent of endogenous catecholamine stores. |
| Epinephrine | 0.02-2.0 µg/kg/min infusion | Primary β-effects to increase contractility and vasodilation at lower doses (0.02-0.10 µg/kg/min); increasing doses (>0.1 µg/kg/min) are accompanied by increased contractility and also increased α-mediated vasoconstriction; may be best choice to augment contractility and perfusion, especially in situations of severely compromised ventricular function, shock, or anaphylaxis. |
| Isoproterenol | 0.05-2.0 µg/kg/min infusion | Pure, nonselective β-agonist; significant inotropic, chronotropic (β1 and β2) and vasodilatory (β2) effects; may be an effective pulmonary vasodilator in some children; tachycardia and increased myocardial oxygen consumption may be dose limiting; tachydysrhythmias may also occur; bronchodilator. |
| Phenylephrine | 1-10 µg/kg bolus, 0.1-0.5 µg/kg/min infusion
Preterm and term infants may require as much as 30 µg/kg (see Chapter 15). |
Pure α-mediated vasoconstriction; no increase in contractility. |
| Amrinone | 0.75-1 mg/kg repeated twice, maximum 3 mg/kg. Neonates and infants may require loading doses of 2-4 mg/kg and infusions of 10 µg/kg/min. |
Increases cyclic adenosine monophosphate by phosphodiesterase inhibition; positive inotropy, positive lusitropy, and smooth muscle vasorelaxation; hypotension; reversible thrombocytopenia. |
| Milrinone | 50-75 µg/kg loading dose, 0.5-1.0 µg/kg/min infusion | Similar to amrinone (antiplatelet effects may be less). |
| Calcium chlorideCalcium gluconate | 10-20 mg/kg/dose (slowly)30-60 mg/kg/dose (slowly) | Positive inotropic and direct vasoconstricting effects; inotropy is significant only if ionized calcium is low and/or ventricular function is depressed by other agents; can slow sinus node; increases electrophysiologic abnormalities from hypokalemia and digoxin. |
| Digoxin | Total digitalizing dose (TDD): |
 TDD followed by
TDD followed by 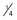 TDD q8-12hr × 2 increases cardiac contractility; slows sinus node and decreases atrioventricular (AV) node conduction; long half-life (24-48 hours) that is prolonged by renal dysfunction; numerous drug interactions; toxicity includes supraventricular tachycardia, AV block, ventricular dysrhythmias; symptoms include drowsiness, nausea, vomiting; toxicity exacerbated by hypokalemia.
TDD q8-12hr × 2 increases cardiac contractility; slows sinus node and decreases atrioventricular (AV) node conduction; long half-life (24-48 hours) that is prolonged by renal dysfunction; numerous drug interactions; toxicity includes supraventricular tachycardia, AV block, ventricular dysrhythmias; symptoms include drowsiness, nausea, vomiting; toxicity exacerbated by hypokalemia.Substantial pharmacodynamic variability (i.e., variability in the serum concentration required to produce the desired effect) can also be observed. Some of these differences are related to receptor maturation and function. For example, it appears that β-adrenergic receptors have a high density in the term neonate and young infant but their coupling to adenyl cyclase may be incomplete.33,150 In addition to developmental changes, which are to some extent controlled by thyroid hormone, β-receptor and adenyl cyclase activities are diminished in response to sustained administration of exogenous β-agonists and also as a result of increased endogenous catecholamine concentrations, which are often seen as a complication of moderate to severe heart failure and other forms of severe stress (e.g., sepsis).151–153
In the neonatal myocardium, chronic catecholamine exposure may upregulate adrenergic receptor number or function, or both, perhaps mimicking the normal developmental program of increasing sympathetic nervous system activity as term approaches.154 With further maturation in early postnatal life, β-adrenergic receptor density declines. The impacts of various pathophysiologic states on these processes have been incompletely identified. For example, congestive heart failure, cardiopulmonary bypass, and ischemic reperfusion all lead to decreased β-receptor and adenyl cyclase expression and activity.155–158 On the other hand, the myocardium of infants with tetralogy of Fallot exhibits increased β-receptor density and greater receptor-stimulated adenyl cyclase activity with increased gene and protein expression.159,160
One must pay particular attention to technical issues when administering vasoactive infusions to infants. Infusions are often specifically prepared as very concentrated, nonstandardized solutions to minimize the amount of volume infused; hence, the potential for dose or concentration error is substantial. One study at a tertiary care children’s hospital demonstrated that the actual concentration of prepared solutions varied significantly.161 Because of the high concentration of these drugs relative to the child’s size, small errors (either in calculation or in infusion pump flow rate) can have a large impact on the actual amount of drug delivered. The extremely small infusion rates can also lead to a delay in drug effect (see Chapter 51). Confirming that the pump drive mechanism is actually delivering drug at the distal end of the infusion tubing, connecting the infusion tubing as close to the child as possible, and using a carrier infusion to “push” the medication at a constant rate are important steps to ensure the safety and efficacy of drug infusions. The rate of the carrier infusion is also crucial. Most standard infusion setups require rates in excess of 5 mL/hr to effect rapid (less than about 10 minutes) changes in the concentration of drug delivered to the infant and therefore preclude all attempts of fluid limitation.
Vasoactive Drugs
Dopamine
Dopamine continues to be the most frequently used inotropic agent in neonates, infants, and children. It has activity at α-, β-, and dopaminergic receptors. Dopamine augments cardiac contractility through two mechanisms. First, it directly stimulates cardiac β1-receptors and provokes norepinephrine release from cardiac sympathetic nerve terminals. Second, circulating concentrations of endogenous epinephrine and norepinephrine increase significantly during dopamine infusion, leading to the suggestion that at least some of the effects of a dopamine infusion are indirectly mediated via induced release of endogenous catecholamines.162 Because of its indirect effects, particularly the release of myocardial norepinephrine stores, the response to dopamine may be diminished in children with congestive heart failure or other relatively long-standing forms of hemodynamic stress.
Activity at dopaminergic receptors in the kidney and gastrointestinal tract can lead to improved perfusion of these organ systems. The evidence that dopamine specifically and selectively improves renal perfusion via stimulation of renal dopaminergic receptors (i.e., as opposed to a nonspecific and generalized improvement in cardiac output that might occur with any positive inotrope) is conflicting.163–167 Regardless of the mechanism, most evidence indicates that renal blood flow and perfusion are increased by dopamine, even at very large doses.
Similar to the situation with other inotropes, pharmacokinetic studies of dopamine have shown wide variability in serum concentration in neonates and children.168,169 Because of this variability in plasma concentration for a given infusion rate, as well as the wide range of serum concentrations necessary to produce a given effect, doubling or halving a given dopamine infusion rate may be a logical way to approach alterations in therapy. The frequent practice of increasing or decreasing the infusion rate by small proportions (i.e., 5% to 10%) may not be consistent with what is currently known about the pharmacokinetics and pharmacodynamics of most inotropes.
Neonates have classically been considered to have a greater dependence on HR, a reduced myocardial compliance, and a relative resistance to inotropic effects of exogenous catecholamines. Nonetheless, there is substantial echocardiographic evidence of increased myocardial contractility occurring at small dopamine infusion rates (≤5 µg/kg/min) and before significant increases in HR.170,171 Evidence about the effects of dopamine in sick preterm infants is also somewhat controversial.164,172–176 There may be a relative dissociation between its effects on the renal and mesenteric beds in these infants, such that a portion of the increase in arterial blood pressure seen with a dopamine infusion is the result of mesenteric vasoconstriction and an actual decrease in mesenteric blood flow.
Although it is generally believed that vasoconstriction is significant when large infusion rates (>10 to 15 µg/kg/min) of dopamine are employed, studies have shown evidence of improved cardiac output and renal blood flow even at very large doses (≥20 µg/kg/min) in neonates and infants.171a
The effects of dopamine on PVR are variable. Both minimal effect and increased PVR have been found.146,177–183 The effects of dopamine on PVR most likely depend on the dose as well as the underlying state of the vascular endothelium and smooth muscle. Vasoconstriction may be more likely after ischemia-reperfusion and in the presence of hypoxia. Conversely, the presence of vasodilators, such as nitroprusside, or α-adrenergic blockers, such as phenoxybenzamine, can prevent increased PVR in response to dopamine.184,185 Overall, dopamine remains the drug of choice in most infants and children, owing to its beneficial effects on mesenteric and renal blood flow, lesser chronotropic effects than some other agents, and a somewhat reduced arrhythmogenic potential.
Dobutamine
Dobutamine is a structural analogue of isoproterenol. It was developed to provide relatively selective β-adrenergic receptor stimulation. The inotropic effects are somewhat less potent than those of dopamine, and it has considerably less overall α-adrenergic potential compared with dopamine. Dobutamine does possess significant β2-adrenergic receptor agonist properties, and for this reason it can cause peripheral vasodilation. Substantial vasodilation and tachycardia occur at larger infusion rates (≥10 µg/kg/min).145,186–189 The tendency toward significant tachycardia and tachyarrhythmia may be greater in neonates than in older children or adults. There is some evidence that the efficacy of dobutamine is reduced in immature animals, perhaps because of greater circulating catecholamine concentrations and alterations in β-receptor expression and function in these immature animal models.144,145
Because the actions of dobutamine do not depend on endogenous catecholamine stores, the drug may be more effective in increasing cardiac output in patients with severe congestive heart failure or cardiogenic shock.190,191 In children with normal LV function, dobutamine has been shown to significantly increase LV relaxation. A decrease in end-systolic wall stress likely contributes to the improved diastolic relaxation caused by dobutamine.192 As with dopamine, questions of a relative resistance in neonates have arisen, although evidence indicates that dobutamine improves LV contractility in neonates with LV dysfunction.193 Dobutamine does not selectively improve renal or mesenteric blood flow independently of its effect of increasing cardiac output. The improvement in cardiac output seen with dobutamine is related to both increased contractility and decreased SVR via vasodilation. Pulmonary vasodilation in the presence of increased PVR may also occur.194
As was discussed with dopamine, exponential increases in serum concentrations are required to produce linear improvements in cardiac index. There is also substantial pharmacokinetic variability in dobutamine plasma concentrations.148,195,196 Tolerance may occasionally develop.197In one animal study, high-dose dobutamine infusion was associated with significant dysfunction of platelet aggregation after hypoxia and reoxygenation.198
Isoproterenol
Isoproterenol is a pure, nonselective β-adrenergic agonist.199 It increases HR and contractility and causes vasodilation in mesenteric, renal, and skeletal muscle tissue beds. Isoproterenol is also a fairly effective vasodilator of the pulmonary circulation.200 Significant tachycardia almost always accompanies its use. The tachycardia and greater contractility cause a significant increase in myocardial oxygen consumption, which is usually well tolerated. However, these changes may be limiting in compromised hearts. The pulmonary vasodilation produced by isoproterenol may be useful in settings in which tachycardia is either unimportant or somewhat beneficial.201 Systemic vasodilation induced by isoproterenol can be sufficiently profound as to cause systemic hypotension.182,202 The positive chronotropic effects of isoproterenol may be useful in children with bradycardia.203 The drug is increasingly used in electrophysiology suites to facilitate the detection of abnormal conduction pathways in infants and children under general anesthesia.204 Isoproterenol is also a potent bronchodilator. Prolonged use or high doses of isoproterenol and other catecholamines may be associated with the development of myocardial fibrosis.205
Epinephrine
Epinephrine has α-, β1-, and β2-adrenergic agonist effects. Data derived mainly from studies in adults indicate that lower doses of 0.02 to 0.1 µg/kg/min are associated with predominantly β-adrenergic effects. In this range, increases in HR and systolic blood pressure and reduced diastolic blood pressure due to skeletal muscle vasodilation predominate. Doses between 0.1 and 0.2 µg/kg/min have mixed α- and β-effects. At larger doses, α-adrenergic–induced vasoconstriction is significant, and hence there is reduced skin, muscle, renal, and mesenteric blood flow. Compared with pure α-agonists, epinephrine provides significant inotropic effect. The effects of epinephrine do not depend on endogenous tissue catecholamine stores. Based on experience, epinephrine seems to be effective in children who do not respond to dopamine or dobutamine, particularly those with significant dysfunction of the systemic ventricle in the immediate postoperative period. The addition of moderate vasoconstriction to increased contractility may be advantageous to maintain myocardial perfusion and may also increase both systemic and pulmonary blood flow in children with shunt-dependent circulations. Important adverse effects include dysrhythmias (usually ventricular) and, at larger doses, regional ischemia and hypoperfusion due to vasoconstriction. Pharmacokinetic studies have shown a linear relationship between serum concentration and infusion rate, but again there is significant variability in the individual response to a specific concentration.206
Phenylephrine
Phenylephrine is a pure α-adrenergic agonist. As such, its major function is to cause peripheral vasoconstriction. It has no β-adrenergic or inotropic effect and therefore does not increase contractility. It may be temporarily used to improve afterload, systemic blood pressure, and, therefore, critical organ blood flow. But without concurrent inotropic support, an isolated acute increase in afterload is often poorly tolerated, particularly by a compromised ventricle. There are a few situations in which phenylephrine can be extremely useful. For example, it can be given to increase systemic afterload and thereby decrease right-to-left shunting in children with tetralogy of Fallot and dynamic RVOTO (“Tet-spell”). Any additional increase in contractility would worsen the outflow obstruction in this situation. Phenylephrine is also beneficial in cyanotic children who depend on a systemic-to-PA shunt for pulmonary blood flow and adequate oxygenation. The increased afterload may increase flow across the shunt and improve pulmonary blood flow. Treatment of acute hypotension in children with hypertrophic obstructive cardiomyopathy or critical aortic stenosis is another potential indication.207–210
Vasopressin
In adults, vasopressin has been shown to be beneficial in the treatment of vasodilatory shock and during cardiopulmonary resuscitation.211 A few pediatric case reports and small observational studies have demonstrated improved blood pressure and accelerated weaning of inotropic support with low-dose vasopressin.212–215 However, a multicenter randomized, controlled trial in children with vasodilatory shock could not confirm these findings. Low-dose vasopressin (0.0005-0.002 U/kg/min) had no beneficial effects compared with placebo; there was even a suggestion of increased mortality.216 Further studies are necessary to establish the effectiveness and safety of vasopressin in children. Currently its role as a “rescue” medication for the treatment of catecholamine-resistant vasodilation during or after congenital cardiac surgery217,218 and for refractory hypotension in infants with extremely low birth weight219 is being investigated. A relative deficiency of arginine vasopressin has been reported in some children with CHD who underwent open heart surgery. In a subgroup of children, baseline concentrations of arginine vasopressin were reduced and remained reduced for up to 48 hours after cardiopulmonary bypass. Interestingly, only children with reduced concentrations of vasopressin responded to vasopressin therapy.220 Extrapolated from adult data, pediatric dosing algorithms currently range from 0.0003 to 0.002 U/kg/min. Careful attention to the infusion rate is important, especially when programming syringe pumps. The literature and common reference tools often cite the doses in U/min, U/kg/hr, U/kg/min or even mU/kg/min, which can be quite confusing and can lead to potential errors.
Phosphodiesterase Inhibitors
Phosphodiesterase inhibitors, which include amrinone, milrinone, and enoximone, are the most commonly used non–catecholamine-mediated inotropic agents. Their mechanism of action is also relatively straightforward. Phosphodiesterases degrade cyclic adenosine monophosphate (cAMP) to 5′-AMP. Phosphodiesterase inhibitors prevent this degradation and therefore increase levels of cyclic nucleotides, primarily cAMP. The increased concentration of this secondary messenger leads to an increase in calcium availability and thus increased contractility. Because the response is related to an increase in cAMP and not purely to inhibition of phosphodiesterase, the greatest effect occurs if initial levels of cAMP exceed normal values. In this way, synergy exists with β-agonists.221 The absence of adrenergic stimulation minimizes effects on HR, rhythm, and dependency on endogenous tissue catecholamine stores. In addition to positive inotropic effects, these drugs also have significant lusitropic properties (i.e., diastolic relaxation) and promote peripheral vasodilation.222–224 Phosphodiesterase drugs may also have substantial antiinflammatory properties that are not well understood at present.225–227
Amrinone
As with all phosphodiesterase inhibitors, there has been an ongoing debate about the relative contribution of systemic vasodilation and afterload reduction versus increased cardiac contractility as the underlying mechanism for improving cardiac output. The bulk of evidence currently indicates that amrinone produces significant increases in myocardial contractility and reduces ventricular afterload.223,228,229 Evidence of amrinone-induced improvements in cardiac performance in neonates and infants after cardiac surgery has been demonstrated in a number of situations, including postoperatively in children who have undergone the arterial switch procedure and in older children who have undergone the Fontan operation.224,228,230 Pharmacokinetic data in children suggest that the loading and infusion doses for amrinone need to be approximately twice those reported for adults.231 In addition to differences in volume of distribution and clearance (both greater in infants), binding of amrinone to the oxygenator membrane may be another factor that needs to be considered if the loading dose is administered during cardiopulmonary bypass.232 Overall, these data suggest that loading doses of 2 to 4 mg/kg and infusion rates starting in the range of 10 µg/kg/min may be indicated in the neonate and infant.232 Large bolus doses of amrinone can cause significant systemic hypotension, particularly in the period immediately after cardiac surgery. From a practical standpoint, it may be best to administer the loading dose of amrinone slowly over the period of 1 hour. Caution is also indicated because the elimination half-life of amrinone is large (3 to 15 hours). Other side effects include thrombocytopenia that is reversible, occasional drug-related fever, and increased hepatic enzymes.
Milrinone
Several studies have demonstrated improved cardiac output and overall efficacy of milrinone after cardiac surgery in neonates and infants and with other states associated with ventricular dysfunction.233–238 Milrinone has a larger volume of distribution and greater rate of clearance in infants and children compared with adults; adjustments to bolus dosing and infusion rates have therefore been recommended.233,238 Unlike amrinone, milrinone does not appear to bind to the cardiopulmonary bypass circuit and has less deleterious effect on platelet function. Loading doses of 50 to 100 µg/kg (typically 75 µg/kg) and initial infusion rates of 0.50 to 1.0 µg/kg/min have been recommended.233,236 Neonates with HLHS who underwent stage I palliation exhibited reduced renal clearance in the immediate postoperative period, and dose adjustments should be considered (i.e., infusion rates of 0.2 µg/kg/min).239 Milrinone increases cardiac output, reduces cardiac filling pressures, and reduces afterload; its effects are typically not associated with tachyphylaxis and are independent of β-adrenergic receptor density or activity. A recent multicenter, double-blind, placebo-controlled trial demonstrated that prophylactic use of high-dose milrinone significantly reduced the development of low cardiac output syndrome relative to placebo after cardiac surgery in high-risk pediatric subjects.235 Milrinone is also increasingly used to improve oxygenation in neonates with persistent pulmonary hypertension who are unresponsive to therapy with nitric oxide.240 Animal studies have shown that IV or inhaled milrinone enhances the response of the pulmonary vasculature to iloprost and prostacyclin.241,242
Enoximone
Enoximone is a phosphodiesterase inhibitor that has been used extensively in Europe but is not currently available in the United States. Its properties are similar to those of the other members of its class.222,243 Improvements in indirect indices of cardiac function, such as mixed venous oxygen saturation, ventricular filling pressures, and systemic arterial blood pressure, were observed in infants treated with enoximone after cardiac surgery. In addition, the length of hospital stay was reduced.244 Enoximone was also useful to support cardiac function and potentially reduce PVR in children after cardiac transplantation.245
Digoxin
Digoxin remains the most frequently administered oral inotropic agent. Its efficacy in children with congestive heart failure due to large left-to-right shunts has been questioned. The clinical picture in such children often improved without echocardiographic evidence of increased contractility, and frequently progressive ventricular dilatation was observed.246,247 Use of digoxin to improve RV dysfunction due to pulmonary hypertension or as part of a multimodel therapy during the interstage phase for infants with single ventricle physiology has also been debated.248
Because digoxin has a slow distribution phase after oral administration and a long elimination half-life (up to 1 to 2 days in neonates and young infants), a loading dose is usually administered (see Table 16-8). Renal dysfunction can significantly prolong the elimination half-life. Therapeutic digoxin levels are between 0.5 and 2.0 ng/mL. IV and oral dosing regimens are the same, although the onset of electrophysiologic effects from IV dosing is much more rapid (5 to 20 minutes).
Many drugs interact with digoxin and influence its pharmacokinetics. It should be assumed that almost any drug administered along with digoxin can affect the absorption and clearance, usually necessitating a reduction in dose.249,250 The likelihood of digoxin toxicity increases with serum concentrations greater than 3 ng/mL.251,252 Symptoms of toxicity include drowsiness, nausea, and vomiting. Various conduction abnormalities and SVT are the most frequent cardiac rhythm manifestations of digoxin toxicity in infants and young children. Older children and adults are more likely to experience AV block, ventricular dysrhythmias, junctional tachycardia, and premature ventricular contractions. Hypokalemia, specifically intracellular potassium depletion (often a result of longstanding diuretic use), exacerbates the proarrhythmogenic effects of digoxin.
Calcium
The role, mechanisms of action, and potential for deleterious consequences of IV calcium continue to be controversial.253 It is the ionized calcium concentration that is important for myocardial function. Calcium is a positive inotrope, particularly when it is administered in the presence of ionized hypocalcemia. It may also improve ventricular contractility when LV function is depressed by halothane, β-adrenergic blockade, or disease (e.g., sepsis).254,255 In the presence of a normal myocardium and normal ionized calcium concentrations, the effects of IV calcium on contractility are much more modest.256 Extracellular calcium levels also play an important role in the regulation of peripheral vascular resistance. A calcium-sensing receptor (CaSR) has been identified on vascular cell walls.257
There is evidence in adults that the primary effect of calcium administered after cardiac surgery is to increase SVR and mean arterial pressure with little or no effect on intrinsic myocardial contractility.253,258 In fact, the increase in afterload, if not accompanied by a corresponding increase in contractility, may only serve to decrease stroke volume and cardiac output. Calcium may cause or exacerbate reperfusion injury and cellular damage by mechanisms that include activation of calcium-dependent proteases and phospholipases and organelle damage due to cellular calcium overload.259,260 These concerns are particularly relevant in children immediately after cardiac surgery. IV calcium may also attenuate the β-adrenergic effects of concurrently administered epinephrine.253
The role of IV calcium administration in neonates and young infants, both alone and after cardiac surgery, is more complicated. Preterm and term neonates have erratic calcium handling and are prone to ionized hypocalcemia.261,262 The neonatal myocardium is more sensitive to ionized hypocalcemia than the adult myocardium, owing to reduced intracellular calcium stores, immaturity of sarcoplasmic reticulum calcium-handling mechanisms, and greater dependency on transmembrane calcium flux for excitation-contraction coupling.263 Furthermore, the need to administer substantial volumes of citrated and albumin-containing blood products (both of which bind calcium) and other fluids after cardiopulmonary bypass increases the likelihood of ionized hypocalcemia.264 The most prudent approach includes awareness of the greater dependency of the immature myocardium on extracellular calcium, monitoring of ionized calcium concentrations, and careful administration to maintain normal, or at most mildly increased, ionized calcium concentrations. This approach is particularly needed in neonates and in those with diminished LV function. Administration of large bolus doses of calcium immediately on reperfusion of the heart after a period of ischemia is probably ill advised because of the potential to exacerbate reperfusion injury and even to cause myocardial contracture.
Extravasation of calcium can cause local venous irritation and significant tissue necrosis. Although it has been suggested that calcium gluconate may cause less harm in this regard than calcium chloride, we recommend that both forms be administered via a centrally positioned catheter whenever possible. Both forms of calcium increase ionized calcium concentrations similarly when equal amounts of elemental calcium are administered.265 Calcium may cause significant slowing of AV conduction and should be administered cautiously in children with sinus bradycardia or junctional rhythm. Care must also be exercised when administering calcium to children who are receiving digoxin, particularly in the presence of concurrent hypokalemia, because IV calcium exacerbates the potential for digoxin-induced dysrhythmias in this setting.
Triiodothyronine
Triiodothyronine hormone (T3) is essential for the maturation of sarcolemmal calcium channels, myosin, actin, and troponin. In addition, hypothyroid rats demonstrate reduced numbers of β-receptors and reduced density of stimulatory secondary messenger protein with an increase in inhibitory secondary messenger protein density. T3 is mostly produced by monodeiodination of thyroxine. This process is inhibited by surgery, hypothermia, catecholamines, propranolol, and amiodarone; therefore, postoperative T3 levels are often reduced.266–268
Endocrine function is compromised after cardiac surgery. Infants younger than 3 months of age with low T3 concentrations on intensive care admission after cardiac surgery have a more complicated intensive care course. Low cortisol concentration is common in the early postoperative period but is not associated with postoperative complications.269 A randomized, double-blind, placebo-controlled study of T3 administration in children undergoing simple or complex cardiac surgery demonstrated that myocardial function was better and length of stay in the intensive care unit was decreased in the T3 group.270 T3 improved contractility without any associated increase in oxygen consumption. In addition, the T3 group demonstrated no delay in recovery of thyroid function secondary to exogenous administration. The dose of T3 used was 2 µg/kg on day 1 followed by 1 µg/kg on days 2 through 12. Many of the follow-up studies have been flawed by small numbers and significant patient heterogenicity, and routine postoperative T3 replacement therapy remains controversial.271–274
Calcium-Sensitizing Agents
Levosimendan
Levosimendan has also been shown to stimulate membrane and mitochondrial potassium-sensitive ATP (KATP) channels. The former dilates both the coronary and the peripheral vasculature. Opening mitochondrial KATP channels is likely to be an important mechanism of pharmacologic (and anesthetic) preconditioning and potential cytoprotection. Interestingly, and for reasons that are not entirely clear, levosimendan has either no effect or a positive effect on lusitropy (diastolic relaxation). At much larger doses than clinically used, it does inhibit phosphodiesterase III. Compared with other inotropic agents (e.g., dopamine, amrinone, milrinone), its efficacy as a positive inotrope is maintained in the depressed myocardium. Both its mechanism of action and experience thus far indicate limited, if any, potential to stimulate arrhythmias.275
Clinical effects include improved cardiac output, reduced ventricular filling pressures, and decreased PVR during the acute treatment of adult patients with either stable or decompensated heart failure.276–279 However, mortality studies failed to show any improvements in short- or long-term prognosis for acute heart failure with levosimendan compared to dobutamine or placebo.280 Other investigations demonstrated improved cardiac performance after cardiotomy and bypass, including beneficial responses in adult patients who appeared to be poorly responsive to other inotropes.281–286 Beneficial effects were also found with levosimendan pretreatment directly before bypass.287
Current recommendations suggest a 6- to 12-µg/kg loading dose followed by an infusion of 0.05 to 0.2 µg/kg/min. The drug’s elimination half-life is approximately 1 hour. There is at least one metabolite that has prolonged (approximately 80 hours) effects, and this may, in part, account for observations of sustained benefit after discontinuation of the drug.288 There are limited data available regarding its use in children or immature animal preparations.289–297 One study in a relatively small, heterogeneous group of inotrope-dependent children with acute or end-stage heart failure demonstrated significant reductions in the use of inotropes in both groups and improved ejection fraction in those with acute heart failure.294 Another study compared milrinone and levosimendan in children after congenital cardiac surgery and concluded that levosimendan is at least as effective as milrinone.291 These early data and the drug’s mechanism of action suggest the need for further study in children and a likely indication for use in infants and children with decreased myocardial performance from a variety of causes, including cardiac surgery, myocarditis, and sepsis.298
B-Type Natriuretic Peptide and Nesiritide
B-type natriuretic peptide (BNP) and its N-terminal precursor (NTpBNP) are members of the natriuretic peptide family. These peptides are released from the heart in response to pressure and volume overload and play an important role in maintaining fluid balance and hemodynamic stability. BNP is secreted from cardiac ventricles in response to increased stimulation of cardiac stretch receptors and increased wall tension. It acts mainly via natriuretic peptide receptors (NPRs) that are present in large vessels and kidneys. Once stimulated, NPRs promote diuresis, natriuresis, and vasodilation and inhibit the renin-angiotensin-aldosterone system. BNP is used as a marker for heart failure in adults and as a monitor for the response to anticongestive heart failure therapies.299 These markers are also increasingly used to diagnose cardiovascular disease in neonates, infants, and children. The concentrations of NTpBNP are often markedly increased immediately after birth but decrease during the first week of life. Age-adjusted cut points and reference values have been suggested.300,301 Currently, it is unclear whether these biomarkers can significantly improve diagnostic accuracy or even serve as a prognostic tool.302
Nesiritide is a recombinant form of BNP and therefore acts via NPRs to promote diuresis, natriuresis, and vasodilation. Early investigations in the adult population suggested that nesiritide may be beneficial for patients with decompensated heart failure; it seemed to improve cardiac output, reduce pulmonary capillary occlusion pressure, and dilate arterial and venous vessels with only minimal increase in HR or myocardial oxygen consumption.303–305 However, a recent multicenter study found no significant advantages of nesiritide and only recommended its use as an individualized case-based therapy.306 Although nesiritide reduces mean arterial pressure in children after cardiac surgery, more extensive studies are required before its usefulness and safety in children can be determined.307–313
β-Blocking Agents
There are several indications for the use of β-blockers in children, including control of hypertension (both acutely in a perioperative period and chronically), treatment of cyanotic spells and RVOTO in tetralogy of Fallot, reduction of LVOTO in hypertrophic cardiomyopathy, control of HR in thyrotoxicosis and pheochromocytoma, and control of SVT (see later discussion).314–318 In contrast to the situation in adults, the use of β-blockers to treat chronic heart failure in children is controversial.319 Important distinctions include β-receptor subtype selectivity, variability in half-life and metabolism, and intrinsic sympathomimetic activity. Even “selective” β-blockers lose their selectivity at increased plasma concentrations.
Propranolol
Propranolol is one of the most frequently used β-blockers in children. Typical oral doses start at 0.25 to 0.5 mg/kg every 6 hours, titrated every 3 to 5 days; the usual dose is 2 to 4 mg/kg/day. A sustained-release form is available for older children who are able to swallow pills. IV propranolol is administered at doses of 0.01 to 0.1 mg/kg over several minutes; this may be increased if necessary (maximum dose 1 mg in infants, 3 mg in children). Sinus bradycardia and hypotension can be serious complications, particularly in infants or after IV administration. Propranolol may also cause conduction disturbances at the level of AV node and worsen pump function in congestive heart failure. Other important adverse effects include fatigue, depression, and lethargy. Interactions with the β2-receptor may exacerbate bronchospasm and predispose children to hypoglycemia.320 Propranolol is primarily metabolized in the liver. Significant population variability in its kinetics has been noted. Metabolism is also affected by factors that alter hepatic blood flow and hepatic metabolic enzyme activity. Its major metabolite, 4-hydroxypropranolol, is also active.315
Atenolol
The use of atenolol has been increasing in children.318,321,322 Compared with propranolol, it is more selective for the β1-adrenergic receptor subtype; the elimination half-life is 8 to 12 hours. There is little hepatic biotransformation, and there are no active metabolites. The typical starting dose is 0.8 to 1.5 mg/kg/day in one or two doses daily, with an upper limit in the range of 2 mg/kg/day. No IV form is available. Atenolol does not cross the blood-brain barrier, so some of the limiting adverse effects common to propranolol are absent. At large doses, β1 selectivity is probably lost, leading to the potential to exacerbate bronchospasm and hypoglycemia.
Esmolol
Esmolol is a relatively selective β1-adrenergic blocker with several unique features. Its onset is fast, it can easily be titrated to a desired end point, and its effects are rapidly terminated via metabolism by red blood cell and plasma esterases.323,324 The drug has been particularly useful for the acute control of perioperative hypertension and for treatment of supraventricular tachyarrhythmia (see later discussion). Loading doses between 100 and 500 µg/kg given over 1 to 5 minutes are followed by maintenance infusions of 50 to 100 µg/kg/min. If the desired response is not achieved, the infusion rate is then typically doubled every 5 minutes until a desired response is achieved.
Specific data on pediatric dosing are limited at present.325 One study investigated the pharmacokinetics of esmolol therapy after an episode of stimulated or spontaneous SVT. The results were similar to the findings in the adult population.326 A maximum loading dose of 500 µg/kg and infusion rates of 250 to 300 µg/kg/min are currently suggested. A major potential adverse effect of esmolol is hypotension, particularly during bolus therapy. As noted earlier, esmolol rapidly distributes and has a very small elimination half-life (7 to 10 minutes) that is unaffected by organ blood flow or disease. Therefore, hypotension is usually short-lived, but therapy with vasopressors may occasionally be required until it resolves.327
Labetalol
Labetalol has nonselective β-adrenergic blocking properties and is also a selective α-adrenergic receptor blocker. The ratio of α- to β-blockade efficiency is 1 : 3 and 1 : 7 after oral and IV administration, respectively. The primary use of labetalol in children is to control hypertension. The drug has been given intravenously to treat hypertensive crisis, to control hypertension after aortic coarctation repair, and as an adjunct to induce controlled hypotension during surgery.328–331 Typical doses are 0.1 to 0.4 mg/kg given every 5 to 10 minutes until the desired effect is achieved with infusions of 0.25 to 1 mg/kg/hr. The elimination half-life of labetalol is 3 to 5 hours.
Carvedilol
Carvedilol is a newer nonselective β-blocker with additional vasodilatory and also some antioxidant properties.332,333 The ratio β1 to α1 blockade is 1.7 : 1. It is primarily used for the treatment of heart failure. In adults, significant benefit has been demonstrated, including reduced mortality, reduced hospital stay, improved New York Health Association (NYHA) functional class, and a somewhat reduced progression of the clinical disease.334 However, a randomized, double-blind, placebo-controlled, multicenter study of children and adolescents with symptomatic systolic heart failure found no significant differences in outcome between carvedilol and placebo during an 8-month follow-up period.335 Carvedilol may have differential effects based on ventricular morphology, and further studies are necessary.336–339 A retrospective review of the initial experience with carvedilol therapy in children showed that adverse effects—mainly dizziness, headaches, and hypotension—were common (>50%) but well tolerated.340 The ideal dose in children has not yet been determined. Pharmacokinetic simulation studies suggest that larger doses341 are appropriate, although clinical experience in small patient populations favors smaller doses.339
Vasodilators
Vasodilators can be divided in several different pharmacologic groups. Direct-acting nitrosovasodilators such as sodium nitroprusside and nitroglycerin are among the most commonly used cardiovascular medications. These drugs directly relax vascular smooth muscle to cause vasodilation. Hydralazine is another direct-acting smooth muscle vasodilator that is occasionally given to children to reduce blood pressure. Selective α-adrenergic blockers, such as phentolamine and phenoxybenzamine, are occasionally used to reduce blood pressure and SVR in the perioperative period. The classic indication is the treatment of pheochromocytoma, but they are also frequently given during hypothermic cardiopulmonary bypass. Ventricular “remodeling” and long-term blood pressure control is often achieved with angiotensin-converting enzyme (ACE) inhibitors. Prostaglandin E1 (PGE1) is a direct-acting vasodilator primarily used to maintain the patency of the ductus arteriosus in duct-dependent circulations. In contrast, prostacyclin and inhaled nitric oxide are vasodilators with relatively selective effects on the pulmonary vasculature. Commonly used vasodilators and antihypertensive agents are summarized in Table 16-9.
| Drug | Intravenous Dose | Comments |
|---|---|---|
| Propranolol | 0.01-0.1 mg/kg slowly | Nonselective β-blockade; bradycardia, hypotension, worsening of myocardial pump function; atrioventricular block; hypoglycemia; bronchospasm; depression; fatigue |
| Labetalol | 0.1-0.4 mg/kg per dose; 0.25-1.0 mg/kg/hr infusion | Nonselective β-blockade; selective α-blockade; ratio of α- to β-blockade is 1 : 7 for intravenous form; doses (0.1 mg/kg) can be repeated every 5-10 minutes until desired effect is achieved; side effects are similar to those of propranolol. |
| Esmolol | 100-500 µg/kg loading dose (over 5 minutes); 50-250 µg/kg/min infusion | Relatively selective β-blockade; short elimination half-life (7-10 minutes); hypotension, especially during bolus administration; if less than desired response after 5 minutes, can repeat or double bolus dose, followed by doubling infusion rate; non–organ-based metabolism by plasma and red blood cell esterases; infusion concentrations >10 mg/mL may predispose to venous sclerosis; dilute infusion at high rates increases risk of volume overload. |
| Sodium nitroprusside | Start at 0.5-1.0 µg/kg/min infusion; maximum 6-10 µg/kg/min | Potent direct smooth muscle relaxation; dilates both arteriolar resistance and venous capacitance vessels; hypotension potentiated by hypovolemia, inhalation anesthetics, other antihypertensives; variable pulmonary vasodilation; potential cyanide toxicity; reflex tachycardia; check cyanide and thiocyanate levels if >4 µg/kg/min is infused or drug is used longer than 2-3 days. |
| Nitroglycerin | 0.5-10 µg/kg/min infusion | Direct smooth muscle relaxation; predominantly dilates venous capacitance vessels, modest effects on arterial resistance at larger doses; weak antihypertensive effects; variable pulmonary vasodilation; used to facilitate cooling and rewarming during cardiopulmonary bypass. |
| Phentolamine | 0.05-0.1 mg/kg dose; 0.5-5 µg/kg/min infusion | Selective α-blocker, produces mainly arteriolar vasodilation; some direct vasodilation with mild venodilation. |
| Enalaprilat | 5-10 µg/kg per dose q8-24hr | Long duration of effect; angioedema, renal failure, hyperkalemia; potential problematic hypotension with anesthetic agents (see text) |
| Hydralazine | 0.1-0.2 mg/kg bolus q6hr | Maximum 20 mg/dose; direct-acting smooth muscle (predominantly arteriolar) vasodilation; long effective half-life; tachyphylaxis; reflex tachycardia; lupus-like syndrome; drug fever; thrombocytopenia |
| Prostaglandin E1 | 0.05-0.1 µg/kg/min infusion | Direct smooth muscle relaxation, relatively specific for ductus arteriosus; variable pulmonary and systemic vasodilation; apnea in neonates |
*All drugs should be started in the lower dose range and titrated to effect.
Sodium Nitroprusside
The primary indication for sodium nitroprusside is the rapid and reliable reduction of afterload and blood pressure before, during, and after a wide variety of procedures. For example, it is used to control intraoperative and postoperative hypertension in children with aortic coarctation and other forms of LVOTO. The reduction in afterload may improve performance of a dysfunctional ventricle, particularly in combination with a positive inotropic agent.342,343 The ability of nitroprusside to successfully treat pulmonary hypertension is variable and may be age dependent.344–348
Sodium nitroprusside is an extremely potent vasodilator that acts directly on smooth muscle to cause dilation.349,350 Its effects reduce cardiac preload as well as afterload. Onset of effect is fast (within minutes), and offset is similarly rapid; the effect ends within 1 to 2 minutes after termination of the infusion. Because of its potency, it should always be administered via an infusion pump in conjunction with continuous direct arterial pressure monitoring. The starting dosage is 0.5 to 1 µg/kg/min. This can be increased to achieve the desired effect. The hypotensive effects of nitroprusside are potentiated by hypovolemia, inhalation anesthetics, and drugs that inhibit the reflex responses to direct vasodilation (e.g., increase in sympathetic tone, renin release), such as propranolol and ACE inhibitors.
Adverse effects of sodium nitroprusside include cyanide and thiocyanate toxicities, rebound hypertension, inhibition of platelet function, and increased intrapulmonary shunting. Rebound hypertension is most likely caused by activation of the aforementioned reflex mechanisms. It can usually be avoided by slowly tapering the infusion rather than abruptly discontinuing it. Toxicity may occur when more than 10 µg/kg/min of sodium nitroprusside is administered, if tachyphylaxis develops within 30 minutes, or if there is immediate resistance to the drug. A blood cyanide concentration of approximately 500 µg/dL has been associated with death in a child.351 Cyanide and thiocyanate toxicities are rare but may be more likely in neonates and young infants and in those with impaired hepatic or renal function.351,352
Cyanide is produced from the metabolism of sodium nitroprusside. Free cyanide is then conjugated with thiosulfate by rhodanase in the liver to produce thiocyanate. A major mechanism of cyanide toxicity is binding to cytochrome oxidase in the mitochondrial electron transport chain, which prevents mitochondrial respiration and ATP production. Signs of toxicity include tachyphylaxis and an increase in mixed venous oxygen saturation and metabolic acidosis. In children who have received prolonged (>24 hours) or large-dose infusions of nitroprusside and in those with organ dysfunction, it may be advisable to measure blood cyanide concentrations.353–357 Serum thiocyanate concentrations may also be measured. Thiocyanate concentrations may increase if renal function is abnormal. Central nervous system (CNS) dysfunction can occur when thiocyanate concentrations reach 5 to 10 mg/dL. Treatment of cyanide toxicity consists of IV infusion of sodium nitrite, 6 mg/kg (maximum dose 300 mg) over 5 minutes, and sodium thiosulfate, 250 mg/kg or 7 g/m2 (maximum dose 12.5 g) over 15 minutes. In children with abnormal renal function in whom stimulating the production of thiocyanate from thiosulfate may be contraindicated, administration of hydroxocobalamin has been recommended.357a,357b
Nitroglycerin
Nitroglycerin is primarily a venodilator that acts on venous capacitance vessels. It has a substantially smaller effect on arteriolar smooth muscle, and its ability to attenuate an increased PVR is variable. Compared with nitroprusside, it is a poor antihypertensive agent. It has a short half-life and no significant toxic metabolites. Similar to nitroprusside, it may increase intrapulmonary shunting and cause platelet dysfunction. Nitroglycerin is typically administered in doses of 0.5 to 3.0 µg/kg/min. Effects occur within 2 minutes after nitroglycerin is started and resolve within 5 minutes after discontinuation. Mild decreases in blood pressure may be observed at doses exceeding 2 to 3 µg/kg/min. In case of prolonged simultaneous infusions of nitroglycerin and nitroprusside, methemoglobin and cyanmethemoglobin levels can accumulate.358
Nitroglycerin is frequently used during cardiopulmonary bypass to facilitate rapid and effective cooling and rewarming and to improve tissue blood flow. Nitroprusside and nitroglycerin differ substantially with regard to their effects on the microcirculation. Because nitroprusside primarily reduces arteriolar tone and dilates precapillaries, it decreases microvascular blood flow and tissue perfusion more than nitroglycerin does, particularly in the presence of reduced arterial blood pressure (e.g., during cardiopulmonary bypass).359 In contrast, nitroglycerin dilates precapillaries and postcapillaries with equal efficacy, thereby maintaining stable or enhanced capillary perfusion.360,361
Phentolamine and Phenoxybenzamine
Both phentolamine and phenoxybenzamine are α-adrenergic blocking agents with little selectively for α-receptor subtypes. Their primary effect is to decrease resistance on the arterial side of the circulation, although both possess weak venodilating capabilities. Phentolamine is usually administered by infusion at 0.5 to 5 µg/kg/min, whereas phenoxybenzamine is administered orally initially 0.2 mg/kg once daily, then slowly increased every 4 days by 0.2 mg/kg/day. The usual maintenance dose is 0.4 to 1.2 mg/kg/day divided in three doses. The elimination half-life of phenoxybenzamine is much greater than that of phentolamine. Some cardiovascular centers have found the potent arteriolar dilating effects of phenoxybenzamine and its prolonged elimination half-life to be advantageous, especially to provide adequate vasodilation during deep hypothermic cardiopulmonary bypass.362–366
Angiotensin-Converting Enzyme Inhibitors
ACE inhibitors are administered to children with increasing frequency.367–369 In the perioperative setting, they are given to control blood pressure after aortic coarctation repair or to relieve LVOTO. In addition, ACE inhibitors are given on a more long-term basis to reduce afterload on the systemic ventricle and to improve ventricular performance in children with congestive heart failure or single ventricle physiology.248,370 Among the growing number of ACE inhibitors, captopril, enalapril, and lisinopril are most often used in children, although data in children are scant. Adverse effects common to all ACE inhibitors include angioedema, acute renal failure, and hyperkalemia; case reports of significant complications have been published.371,372 Cyanosis and coadministration of furosemide were shown to be independent risk factors for acute kidney injury in children undergoing cardiac surgery who were treated with ACE inhibitors.373
The contribution of ACE inhibitors to anesthetic-induced hypotension remains controversial.374–377 Angiotensin receptor—blocking agents such as losartan and lisinopril can produce significant and refractory hypotension with standard anesthetic induction techniques.378 Because of the potential risk of significant and refractory hypotension, which is usually unresponsive to volume expansion and requires substantial vasoconstrictor treatment, it is our practice to discontinue long-acting ACE inhibitors 1 day before surgery.
Captopril
Captopril has a relatively brief elimination half-life (<2 hours); it is metabolized in the liver and then excreted by the kidney.379 Oral dosing in neonates is 0.05 to 0.1 mg/kg every 8 to 24 hours, titrated up to 0.5 mg/kg every 6 to 24 hours. Infants initially receive 0.15 to 0.3 mg/kg every 6 to 8 hours. This can be titrated toward a maximum dose of 6 mg/kg/day in four divided doses. Older children may receive 0.3 to 0.5 mg/kg every 6 to 12 hours. The brief duration of effect, necessitating more frequent dosing, has led to increased use of the longer-acting ACE inhibitors (enalapril and lisinopril) in children.
Enalapril
Enalapril is metabolized in the liver to its active form, enalaprilat. Enalapril is the only ACE inhibitor currently available in the United States that has an IV formulation. It can be given orally, once or twice daily, with daily doses ranging between 0.1 and 0.5 mg/kg. The IV dose is 0.005 to 0.01 mg/kg/dose, one to three times a day.367 Both enalapril and lisinopril are eliminated by the kidney. The duration of their hypotensive actions averages 24 hours but can extend to 30 hours.
Hydralazine
Hydralazine was frequently used in the past for long-term blood pressure control in children, but has been largely replaced by ACE inhibitors. In contrast to its effect in adults, its ability to decrease pulmonary hypertension in children was disappointing.384 Hydralazine directly relaxes smooth muscle without known effects on receptors. It reduces cardiac afterload but may cause significant reflex tachycardia. With long-term use, it may also cause fluid retention, requiring concurrent administration of a diuretic. Oral dosing is in the range of 0.75 to 1 mg/kg/day divided in two to four doses and is slowly increased over 3 to 4 weeks to a maximum dose of 5 mg/kg/day for infants and 7.5 mg/kg/day for children. In the perioperative setting, it is occasionally used intravenously to control blood pressure and reduce afterload. IV doses are administered as a bolus of 0.1 to 0.2 mg/kg not to exceed 20 mg. The effects of IV hydralazine on PVR are variable.384,385 Tachyphylaxis to the antihypertensive effects of IV hydralazine may occur. Important side effects include a drug-related fever, rash, pancytopenia, and lupus-like syndrome. The elimination half-life of the drug is approximately 4 hours, but the effective biologic half-life may be substantially longer due to significant binding of the drug to vascular smooth muscle.386
Prostaglandin E1
The major indication for PGE1 is to establish or maintain patency of the ductus arteriosus in infancy. It is best able to reopen a closing ductus in neonates up to 1 to 2 weeks of age but may occasionally be effective even in older infants.19,387
Ductal patency is important and often lifesaving for duct-dependent circulations—those in which either the lower body is supplied by right-to-left ductal flow (e.g., interrupted aortic arch, critical aortic stenosis, HLHS) or the PDA is the sole provider of pulmonary blood flow (e.g., pulmonary atresia, tricuspid atresia, severe tetralogy of Fallot). Adverse effects of PGE1 include systemic hypotension, apnea, increased risk of infection, leukocytosis, gastric outlet obstruction, and CNS irritability.388–390 PGE1 infusions are usually begun at 0.05 µg/kg/min and may be increased to 0.1 µg/kg/min or more. The risk of apnea may be related to the infusion rate. Tracheal intubation and ventilation are often required with infusion rates greater than 0.05 µg/kg/min.391,392 Prophylactic treatment with aminophylline was found to be effective in reducing the apnea risk.393 PGE1 has also been used to treat primary or acquired pulmonary hypertension with varying degrees of success.394–397
Inhaled Nitric Oxide
An important development in the treatment of pulmonary hypertension is inhaled nitric oxide gas, which can be delivered directly to the pulmonary circulation. Nitric oxide is an endothelium-derived relaxing factor that acts on guanylate cyclase in vascular smooth muscle.398 Endogenous nitric oxide is produced by endothelial cell nitric oxide synthase. Nitric oxide synthases convert the amino acid l-arginine into nitric oxide and the byproduct l-citrulline. Nitric oxide then diffuses into the subjacent vascular smooth muscle. It produces relaxation by acting on smooth muscle guanylate cyclase to produce cyclic guanosine monophosphate (cGMP), which acts on a series of protein kinases and reduces intracellular calcium levels to inhibit muscle contraction (see Fig. 36-1 in Chapter 36). Nitric oxide diffusing in the other direction from the endothelial cell into the blood vessel lumen can decrease the adhesiveness of white blood cells and platelets. Nitric oxide in the blood is rapidly bound by oxyhemoglobin, which is then oxidized to methemoglobin. From this reaction, nitric oxide is inactivated and nitrite and nitrate are released in the blood. Red blood cell methemoglobin is subsequently reduced back to hemoglobin. The rapid binding and inactivation of nitric oxide in the blood means that inhaled nitric oxide has a minimal effect on the systemic circulation and functions as a very specific pulmonary vasodilator.
Significant reductions in PVR from inhaled nitric oxide have been demonstrated in adults with mitral stenosis, in neonates with persistent pulmonary hypertension of the neonate, in lung transplant recipients, and in children after surgical repair of a variety of CHDs.399–403 The efficacy of inhaled nitric oxide is in large part related to the ability to deliver it into the alveolus, which is in close proximity to the pulmonary vascular smooth muscle.
Inhaled nitric oxide has found several indications in children with CHD. In the cardiac catheterization laboratory, it is used to assess the reactivity of the pulmonary vasculature in children with pulmonary hypertension. In this use, it can help distinguish between children with fixed pulmonary vascular obstructive disease and those with a reversible component to pulmonary hypertension, thereby facilitating therapeutic management and operative planning.404–406
In the postoperative period after the repair of CHD, nitric oxide can be used to reduce PVR and improve cardiopulmonary performance.407,408 Experience thus far suggests that children with two ventricles who have increased LA pressure or its pathophysiologic equivalent (e.g., mitral stenosis, severe congestive heart failure, cardiomyopathy, large left-to-right shunt, TAPVR) are more likely to respond to nitric oxide in the postoperative period. On the other hand, children with single ventricle physiology often show no or only minimal improvements in pulmonary blood flow and oxygenation after nitric oxide. Some children who do not respond to nitric oxide immediately after cardiopulmonary bypass in the operating room demonstrate significant reductions in PVR with nitric oxide several hours later. Nitric oxide is administered in concentrations of 1 to 80 ppm in oxygen via a special delivery device attached to the ventilator or oxygen delivery system. Inspired gas is monitored for toxic nitrogen oxides, and during long-term therapy with nitric oxide, blood methemoglobin concentrations should be assessed on a regular basis.409
Prostanoids
Members of the prostacyclin and prostaglandin families are often classified as prostanoids. All prostanoids are potent vasodilators and inhibitors of platelet aggregation. Since the 1980s, they have been used in the treatment of PA hypertension and are part of the official treatment guidelines,410,411 even though they are not selective pulmonary vasodilators. Common side effects include flushing, hypotension, headache, jaw pain, skin rash, nausea and diarrhea, and nonspecific musculoskeletal pains. Tolerance can develop over time, requiring increasing doses.412 In the United States, only three prostanoids are currently approved by the Food and Drug Administration (FDA): epoprostenol (Flolan), treprostinil (Remodulin), and iloprost (Vantavis). Beroprost is an oral prostacyclin analogue that is licensed in Japan and still being investigated.
Epoprostenol (Flolan)
Continuous IV infusion of epoprostenol has been used effectively for many years in children with PA hypertension of any cause. It improves hemodynamics by reducing PA pressure, increasing cardiac output, increasing oxygen transport, and improving symptoms such as exercise capacity and dyspnea.413 It has been used with excellent results in the treatment of primary pulmonary hypertension and irreversible acquired pulmonary hypertension in CHD; in children awaiting heart, lung, or heart-lung transplantation; in children with primary pulmonary hypertension; in neonates with persistent pulmonary hypertension; and in pulmonary hypertensive crises.414–420 The fact that children who were initially nonresponders may respond after prolonged use of nitric oxide suggests that its mechanism of action involves a degree of remodeling, although no absolute mechanism has been elucidated.
Unfortunately, epoprostenol is chemically unstable at room temperature and has an elimination half-life of only 1 to 2 minutes; it requires continuous IV infusion via a central venous catheter and a specific delivery system. Rapid and unintended decreases in the rate, dislodgments or occlusions of the central venous catheter, or pump malfunctions can lead to severe and life-threatening rebound pulmonary hypertension.421,422 The infusion is usually started at 1 to 2 ng/kg/min and gradually increased over several months to doses between 30 and 80 ng/kg/min. In patients with pulmonary venous disease, IV epoprostenol can worsen the pulmonary edema; it can also increase the ventilation/perfusion mismatch in patients with pneumonia and thereby negatively affect oxygenation. In the acute setting, epoprostenol is increasingly used in the inhaled form to benefit from the selective pulmonary vasodilation and reduction of ventilation/perfusion mismatch.412,423–425 Some centers have used inhaled epoprostenol as an alternative to the rather expensive nitric oxide, although the currently available delivery systems are far from ideal and may alter the delivered tidal volumes.412
Treprostinil (Remodulin)
Treprostinil was first introduced in 2002, initially only for continuous subcutaneous infusion, but later also for IV infusion in patients who could not tolerate the pain at the subcutaneous infusion site. The hemodynamic effects are similar to those of epoprostenol, with fewer adverse effects.426–429 IV infusions of treprostinil are started at 1 to 2 ng/kg/min and slowly increased over several weeks to 40 ng/kg/min, occasionally even higher doses (80-120 ng/kg/min). One pediatric study confirmed the effectiveness of IV treprostinil in children.430 Since 2009, inhaled treprostinil has been used for long-term outpatient management. Because of the relative short half-life, it must be administered every 6 hours.431–434
Iloprost (Ventavis)
Iloprost, approved by the FDA in 2004, is another prostaglandin I2 analogue that can be delivered intravenously or via an ultrasonic nebulizer. It has a very brief elimination half-life of 15 to 20 minutes and requires frequent nebulized treatments (six to nine times each day). In adult studies, iloprost was shown to be beneficial in patients with PA hypertension of any cause, idiopathic PA hypertension, or chronic thromboembolic pulmonary hypertension. These patients demonstrated improvements in hemodynamics and in subjective parameters such as quality of life scores.435,436 Studies in children are scarce and have involved only small numbers of patients, although numerous case reports are encouraging.412,437–443 When iloprost and nitric oxide were compared in children with CHD, the two agents produced similar effects.444 In one study, combination therapy using both systemic and inhaled prostacyclin analogues showed promise.445 An animal study in lambs demonstrated an enhanced effect of prostacyclin and iloprost when combined with milrinone.242
Beroprost
Beraprost sodium is an oral prostacyclin analogue that is chemically more stable and has a prolonged elimination half-life but nevertheless requires dosing three to four times per day, reaching its peak blood concentration at 30 minutes. Two double-blind studies showed no significant long-term benefit, but there may still be a role for this drug in combination therapy, because improvements have been observed in the exercise capacity of children with idiopathic PA hypertension.446,447 Case reports of successful long-term treatment with beraprost and of combination therapy using oral beraprost and inhaled prostacyclin have been published.445,448–450