Chapter 41 Cardiac Neurotransmission Imaging
Single-Photon Emission Computed Tomography
INTRODUCTION
Afferent tracts arising from myocardial nerve terminals and reflex receptors (e.g., baroreceptors) are integrated centrally within hypothalamic and medullary cardiostimulatory and cardioinhibitory brain centers, and on central modulation of sympathetic and parasympathetic outflow at the level of the spinal cord and within cervical and thoracic ganglia. There are additional levels of intricate processing within the extraspinal cervical and thoracic ganglia and within the cardiac ganglionic plexus, where interneurons provide noncentral integration.1
The SNS is dominant in the heart, principally in the ventricles. Sympathetic nerve fibers travel along the vascular structures, penetrating into the myocardium from the epicardium toward the endocardium. There is a gradient distribution of nerve terminals from the base to the apex of the left ventricle (LV).2
Most of the released NE undergoes reuptake in the presynaptic neurons by the NE transporter (NET), a saturable and sodium-, energy-, and temperature-dependent transport protein, cocaine and desipramine sensitive, with high affinity to catecholamines and catecholamine analogs, in a process known as uptake-1.3 In addition, there is NE uptake by a second, corticosterone- and clonidine-sensitive, low-affinity, high-capacity, extraneuronal transport system known as uptake-2.4 Uptake-1 predominates at low concentrations of catecholamines, whereas uptake-2 predominates at higher concentrations,5 but its contribution in humans is low.6
Once NE is again inside the nerve terminal, it is either metabolized by monoamine oxidase (MAO) or stored in vesicles by the vesicular monoamine transporter (VMAT), a proton-dependent transport protein localized in the vesicle membrane. Neuronal uptake-1 regulates the concentration of adrenergic neurotransmitters in the synaptic cleft, playing important physiologic and pathophysiologic roles in modifying signal transduction and extraneuronal catecholamine concentration. It is of paramount importance in protecting the heart from the deleterious effects of elevated levels of circulating catecholamines.7,8
METAIODOBENZYLGUANIDINE (See Chapter 30)
MIBG is an iodinated aromatic analog of the hypotensive false neurotransmitter, guanethidine, which in its turn, is an analog of NE. MIBG and NE have similar molecular structures, and both utilize the same uptake, storage, and release mechanisms in the sympathetic nerve endings. However, MIBG is neither metabolized at nor interacts with postsynaptic receptors. Owing to these characteristics, the labeling of MIBG with iodine-123 (123I) enables in vivo scintigraphic visualization of the sympathetic postganglionic presynaptic fibers,9–14 thus allowing the assessment of both anatomic integrity12,13 and function of the nerve terminals.13 The ability of sympathetic nerves to take up radiolabeled catecholamines was shown to be a more sensitive indicator of intact neuronal function than cardiac NE content.15
MIBG, like catecholamines, is primarily removed from the circulation by the uptake-1 system. Blocking experiments have shown that uptake-2 is responsible for up to 61% of cardiac MIBG uptake.11–13 For clinical studies, the production of 123I-MIBG involves isotopic exchange, which results in a low specific activity (lower ratio of radiolabeled to nonradiolabeled MIBG), with considerable amount of carrier in the final product. Since uptake-2 predominates at higher concentrations of MIBG,5 scintigraphic images do not improve by increasing the dose of 123I-MIBG, which might even lead to saturation of the uptake-1. Reduction of the total amount of MIBG in combination with a higher specific activity (no-carrier-added MIBG) improves myocardial uptake of MIBG through uptake-1, leading to better contrast between specific and nonspecific MIBG uptake as compared with carrier-added MIBG.16
Planar and SPECT Cardiac Imaging with 123I-MIBG
Before the administration of the radiotracer, it is necessary to withdraw medications known to interfere with the accumulation of MIBG (Table 41-1), taking into account their respective blood half-lives.17
Table 41-1 Drugs Known or Expected to Reduce MIBG Neuronal Accumulation
| Drugs | Mechanism of Interference |
|---|---|
| Tricyclic antidepressants, cocaine, labetalol | Inhibition of uptake-1 |
| Reserpine, tetrabenazine | Inhibition of vesicular uptake |
| Norepinephrine, serotonin, guanethidine | Competition for vesicular uptake |
| Reserpine, guanethidine, labetalol, sympathicomimetic amines (e.g., phenylpropanolamines, anorectics) | Depletion of content from storage vesicles |
| Calcium antagonists | Calcium mediated |
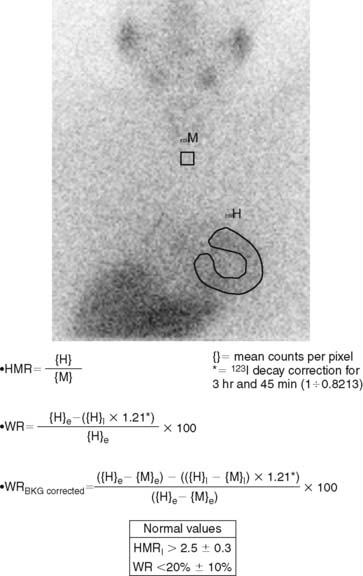
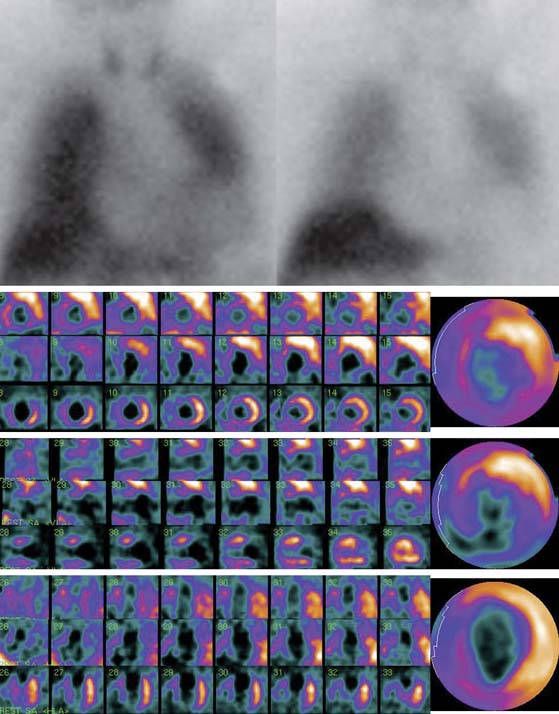
123I-MIBG uptake is semiquantified by calculating a heart-to-mediastinum ratio (HMR)18–20 after drawing regions of interest (ROIs) over the heart (including or not including the cavity) and the upper mediastinum (avoiding the thyroid gland) in the planar anterior view. Average counts per pixel in the myocardium are divided by average counts per pixel in the mediastinum.21 The myocardial washout rate (WR) from initial to late images is also calculated; WR is expressed in percentage as the rate of decrease in myocardial counts over time between early and late imaging (normalized to mediastinal activity) (Fig. 41-1). The late HMR reflects the relative distribution of sympathetic nerve terminals, offering the global information about neuronal function resulting from uptake, storage, and release.22 The WR reflects the neuronal integrity or sympathetic tone, mainly representing the uptake-1.22 More studies are needed to establish the differences in early HMR, late HMR, and WR. Intraobserver and interobserver variabilities of these calculations are less than 5%.21 Normal values for late HMR and WR are ≥ 2.5 ± 0.3 and ≤ 20% ± 10, respectively,23 but vary related to age (late HMR inversely, WR directly).20,24 Moreover, these parameters fluctuate significantly due to lack of validation and standardization of acquisition parameters such as acquisition duration and type of collimation used (in relation to the additional photo peak of 123I at 529 keV, capable of septal penetration when using the common low-energy collimators). Improved standardization of cardiac 123I-MIBG imaging parameters would contribute to increased clinical applicability for this procedure.25
SPECT images can be scored using a point scale for visual evaluation of 123I-MIBG concentration in given cardiac segments, comparable to a myocardial perfusion imaging scoring approach. Careful interpretation should be performed, with knowledge of normal variants and potential artifacts. Normal cardiac 123I-MIBG distribution includes a relatively low uptake in the inferior wall,26 which is more pronounced in the elderly (Figs. 41-2 and 41-3).20 In addition, there may be substantial 123I-MIBG uptake in the liver, which overlaps the inferior LV wall. Moreover, scattering from the lung field to the lateral LV wall may also occur. Polar maps (see Fig. 41-3)27 can be generated from SPECT data and compared with those of normal individuals. Scores of the extension and severity of 123I-MIBG eventual defects and calculation of the mean global and regional WR of the LV are feasible. However, it has to be taken into account that in some pathophysiologic conditions, cardiac 123I-MIBG uptake may be severely reduced, hampering the acquisition and processing of the tomographic slices (Fig. 41-4).
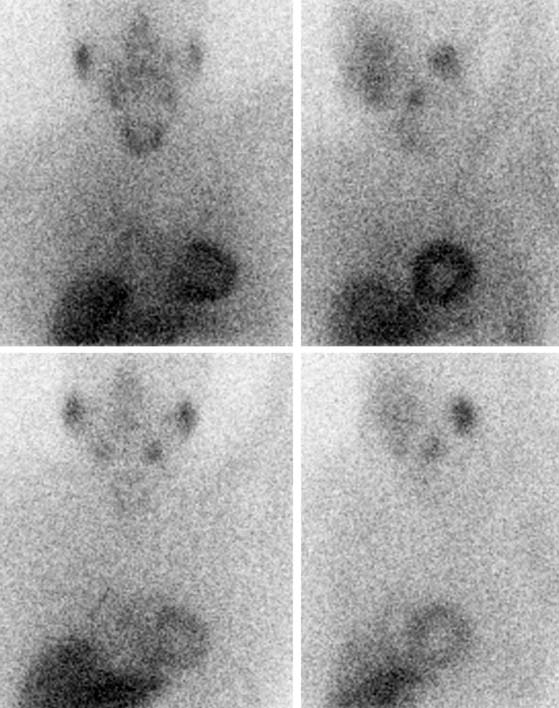
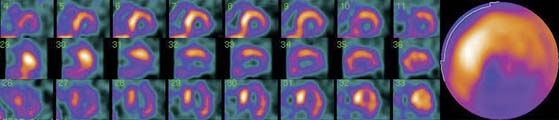
Figure 41-3 Late 123I-MIBG SPECT images of the subject shown in Figure 41-2. Both the SPECT slices and the polar map show reduced tracer uptake in the inferior wall and apex, which is more apparent than on planar images and should be considered a normal variant in the elderly. Upper row: short-axis slices (extending from apex to base). Middle row: vertical long-axis slices (extending from septum to lateral wall). Bottom row: horizontal long-axis slices (extending from inferior wall to anterior wall). 123I-MIBG, iodine-123-labeled metaiodobenzylguanidine; SPECT, single-photon emission computed tomography.
123I-MIBG Imaging in Coronary Artery Disease
The sympathetic nervous tissue is more sensitive to the effects of ischemia than the myocardial tissue.28–30 It has been shown that the uptake of 123I-MIBG is significantly reduced in areas of myocardial infarction (MI)31–33 and adjacent noninfarcted regions (see Fig. 41-4),34,35 as well as in areas with acute and chronic ischemia.36,37 It is likely that ischemia induces damage to sympathetic neurons, which may take a long time to regenerate, and that episodes of ischemia result in decreased 123I-MIBG uptake.38 Gaudino et al.39 provided evidence of good correspondence between 123I-MIBG imaging and the presence or absence of sympathetic cardiac nerves by direct immunohistochemical staining in patients with LV aneurysms due to long-lasting anterior MI.
Reinnervation late after MI in periinfarct regions has been demonstrated by reappearance of 123I-MIBG uptake, which may be in part responsible for the improvement of function. However, reinnervation may be incomplete as late as 3 months after acute MI.40 Hartikainen et al.41 examined 123I-MIBG uptake at 3 and 12 months after a first MI and found no difference in 123I-MIBG activity over time within the infarcted zone but an increase in activity in the peri-infarcted region, without a change in perfusion.
Dissociation between recovery of myocardial perfusion after an ischemic event and myocardial innervation, as determined with 123I-MIBG SPECT, was reported by Matsunari et al.42 Despite considerable myocardial salvage following coronary artery reperfusion, 123I-MIBG images obtained a mean of 11 days after MI and reperfusion demonstrated a persistent area of myocardial denervation within the LV. This area was comparable to the area of ischemic myocardium at risk, as determined by myocardial perfusion SPECT during the acute ischemic event.42 Such modifications of cardiac neuronal function may have an important role in the pathophysiology of HF and arrhythmias, but further studies are warranted to determine the clinical value of innervation imaging in ischemic heart disease.
Concordance between the extent of 123I-MIBG defect at rest and perfusion defect at exercise has been shown in patients with coronary artery disease (CAD).43 This concordance suggests that resting imaging with 123I-MIBG combined with resting myocardial perfusion imaging may be useful as a marker of reversible ischemia in patients incapable of exercise and with contraindications to pharmacologic stress.43 Another potential role for 123I-MIBG imaging at rest is the detection of sporadic transient ischemic attacks such as those of vasospastic angina, otherwise difficult to identify with myocardial perfusion imaging. Watanabe and coworkers44 studied the clinical implications of fatty acid metabolic imaging with iodine-123-labeled β-methyl-iodophenyl pentadecanoic acid (123I-BMIPP) and of cardiac sympathetic nerve functional imaging with 123I-MIBG in patients with vasospastic angina. 123I-BMIPP imaging showed superior specificity but inferior sensitivity compared to 123I-MIBG imaging for the identification of vasospastic regions. The combination of fatty acid and sympathetic imaging was highly accurate for determining the presence and location of vasospasm and superior to the visualization of wall-motion abnormalities by left ventriculography. The mechanism for the decreased 123I-MIBG uptake in coronary vasospastic angina is thought to be memorization of repeated ischemia, but in its turn, the abnormal sympathetic function detected may also be involved in the pathogenesis of the coronary spasms. The property of “ischemic memory” for resting 123I-MIBG imaging also has promise in the evaluation of the area at risk in the subacute phase of acute coronary syndromes by revealing more extensive defects than myocardial perfusion imaging with technetium-labeled agents.45
Although association of 123I-MIBG defects with clinical angina can be explained as a consequence of ischemia, it is intriguing that the perception of pain occurs from the area that lacks sympathetic innervation.34 However, since 123I-MIBG defects in the ischemic territory are not absolute, partial neuronal innervation may allow perception of chest pain.
The number of patients with end-stage ischemic heart disease who cannot be revascularized because of either previous surgery or poor vessels for grafting, angioplasty, or stenting is increasing. When medical therapy fails, these patients are left with disabling angina and/or ischemic HF. New methods to revascularize the heart are under investigation. One method consists of the placement of laser channels into the myocardium using either transmural or endocardial approaches, which would connect cavitary blood with the myocardium to offer nutrient perfusion. Patients treated with either transmural or endocardial laser experience relief of anginal symptoms within days of the procedure, which has been attributed to denervation. Nevertheless, Johnson et al.46 demonstrated that there is negligible regional denervation 3 days after placement of endomyocardial laser channels using autoradiography with 125I-MIBG and hematoxylin-eosin staining in a swine model. Muxi et al,47 studying patients with CAD who underwent transmyocardial laser revascularization, reported significant improvement in stress myocardial perfusion scintigraphy in revascularized areas after 3 and 12 months of the intervention. At 3 months after the procedure, a significant worsening of myocardial innervation was observed in such areas by means of 123I-MIBG, which recovered partially at 12 months. Thus, transmyocardial laser revascularization may involve both perfusion improvement and denervation, mainly at 3 months, the latter partially recovering at 12 months.
123I-MIBG Imaging in Arrhythmogenesis
Cardiovascular disease is the single most common cause of natural death in developed countries. SCD accounts for 50% of all cardiovascular deaths. The most common primary electrical event at the time of SCD is ventricular tachycardia (VT), which degenerates first to ventricular fibrillation (VF) and later to asystole. Bradyarrhythmia or electromechanical dissociation are also frequently documented, mainly in patients with advanced heart disease.48
SCD is usually attributed to structural heart disease, principally in the setting of acute or chronic MI (80%). The cardiomyopathies account for another 10% to 15% of all SCDs. The remaining 5% to 10% of SCDs occur in other cardiac disorders, such as valvular or congenital heart diseases, acquired infiltrative diseases, and ion channelopathies.48 Despite all these epidemiologic data, most SCDs occur among subjects with no previous cardiovascular history, with an apparent need for strategies to allow screening for markers of increased risk of death from arrhythmia among those with low- and intermediate-risk profiles48,49 and better therapy and outcome for these individuals.
Current evidence underscores the importance of the autonomic influences in triggering and sustaining arrhythmias in patients with susceptible substrate,50 and provides important mechanistic insights into the ionic and cellular mechanisms involved.1
Heterogeneity of sympathetic innervation in response to injury is highly arrhythmogenic. Sequential changes consisting of nerve degeneration followed by neurilemma cell proliferation and axonal regeneration have been described after episodes of MI, rapid pacing, radiofrequency ablation, hypercholesterolemia, and stem-cell transplantation.51 According to this “nerve-sprouting” hypothesis, it is possible that the resulting sympathetic hyperinnervation might increase the propensity for cardiac arrhythmia. The coexistence of denervated and hyperinnervated areas in the diseased myocardium could result in increased electrophysiologic heterogeneity during sympathetic activation, leading to ventricular arrhythmia and SCD.51 Ventricular tachyarrhythmias can be provoked in these patients with heterogeneous remodeling of sympathetic innervation, attributable to nerve growth and degeneration by physical or mental stress or by catecholamine application. On the other hand, adrenergic denervation of viable myocardium may also result in denervation supersensitivity, with exaggerated response of myocardium to sympathetic stimulation and increased vulnerability to ventricular arrhythmias.52,53 Therefore, when assessing the arrhythmogenic potential, variables such as the presence of denervated but viable myocardium, the severity of denervation, the underlying level of sympathetic tone, and the subsequent influences on local ventricular repolarization should be taken into account.
While prevention of arrhythmic deaths is generally ineffective with pharmacologic treatment,54 implantable cardioverter defibrillators (ICDs) reduce the mortality rate in subgroups of patients thought to be at risk.55,56 However, identification of patients who most benefit from these devices remains difficult, and implantation of the device in patients who will not benefit leads to unnecessary morbidity, with increased medical costs.57,58 Current available methods for precise identification of individuals at risk for SCD, such as conventional coronary risk factors, functional impairment, degree of LV dysfunction, and cardiovascular testing analyzing electrocardiographic (ECG) variables and Holter monitoring (frequency of premature ventricular depolarizations, nonsustained and sustained VT) have low accuracy. Additionally, heart rate variability (HRV) and baroreflex sensitivity still have an unknown predictive value. On the other hand, electrophysiologic testing has relatively low sensitivity and positive predictive value.48 Arora et al.59 evaluated the use of 123I-MIBG cardiac imaging (as means of local myocardial sympathetic innervation) and spectral analysis of HRV (as means of central autonomic tone) in patients with ICDs. They studied 17 patients who had ICDs for various indications. The combined use of 123I-MIBG scintigraphy and HRV analysis correlated with the occurrence of an appropriate ICD discharge. Patients with ICD discharges had lower early HMR, more extensive 123I-MIBG defects, and more 123I-MIBG/99mTc-sestamibi (technetium-99m-labeled sestamibi) mismatch segments (denervation in areas of myocardial viability) compared to patients without previous ICD discharge. In addition, the ICD discharge group had reduced values for HRV, suggesting abnormally increased sympathetic tone, when compared with patients without previous ICD discharge. Therefore, the combined noninvasive evaluation of local cardiac autonomic innervation and systemic autonomic function by means of 123I-MIBG and HRV allowed identification of patients at risk for potentially fatal arrhythmias and SCD, who were most likely to benefit from an ICD.
Schäfers et al.60 reported the use of 123I-MIBG SPECT in 25 patients with IVF, demonstrating presynaptic innervation defects in these patients. The same group of investigators subsequently reported the long-term follow-up in 20 of these patients,61 13 of whom showed abnormal 123I-MIBG uptake. During a follow-up period of 7.2 ± 1.5 years, 18 episodes of VF/fast polymorphic ventricular tachycardias occurred in 4 IVF patients with abnormal 123I-MIBG uptake, while only 2 episodes of monomorphic ventricular tachycardia (and no VF) occurred in a single IVF patient with normal 123I-MIBG uptake. Therefore, impaired 123I-MIBG uptake may indicate a higher risk of future recurrent episodes of life-threatening ventricular tachyarrhythmias in patients with IVF.
Simões et al.62 subsequently evaluated 67 consecutive patients within 14 days after acute MI by means of resting myocardial perfusion imaging, 123I-MIBG cardiac imaging, and electrophysiologic parameters. They reported a significant correlation between the extent of sympathetically denervated but viable myocardium and prolonged repolarization, defined by QTc interval in resting ECG, and indexes of delayed depolarization from signal-averaged ECG. However, a significant relation between the presence and frequency of ventricular arrhythmias and the extent of denervated but viable myocardium could not be demonstrated after 4.3 ± 1 years of follow-up. This could be related to the low incidence of ventricular arrhythmias in the study population, and possibly the fact that patients with severely depressed LV function were not included. In a phase 2, open-label, multicenter study that enrolled 50 patients with LV dysfunction and previous myocardial infarction, Bax et al.63 found that late 123I-MIBG SPECT defect score was the only variable which showed a significant difference between patients with and without positive electrophysiological studies. Further studies should demonstrate clinically relevant risk stratification of 123I-MIBG imaging, such as high negative predictive value for life-threatening ventricular arrhythmias, especially in patients with ischemic heart disease and depressed LVEF.
Increase of the electrical heterogeneities of ventricular repolarization contributes to the electrocardiographic phenotype and arrhythmogenicity of ion channelopathies such as the Brugada and long-QT syndromes, despite structurally normal hearts. The ANS plays a prominent role in unmasking these syndromes and precipitating life-threatening ventricular tachyarrhythmias. Both sympathetic and parasympathetic influences on ion channel activity have been found to accentuate electrical heterogeneities, thus contributing to arrhythmogenesis in the long-QT and Brugada syndromes.1
123I-MIBG studies have demonstrated reduced and heterogeneous uptake associated with increased regional WR in patients with long-QT syndrome.64,65 Likewise, Wichter et al.66 reported reduced 123I-MIBG uptake in the inferior and septal LV walls in 47% of patients with Brugada syndrome compared with control subjects, which pointed to presynaptic sympathetic dysfunction of the heart in a large proportion of patients with Brugada syndrome. The pathophysiologic implication of these findings requires further investigation.
Ventricular loss of myocytes and fatty or fibrofatty tissue replacement, resulting in regional or global abnormalities, are the main structural abnormality in arrhythmogenic right ventricular cardiomyopathy (ARVC).67 It has been shown with 123I-MIBG SPECT and 11C-hydroxyephedrine PET that although the LV is not involved in the disease, there is evidence of global and regional denervation in presynaptic catecholamine reuptake and storage of the LV, as well as a reduction in the postsynaptic β-adrenoceptor density assessed by 11C-CGP PET.68–70 Correlation of observed regional abnormalities of 123I-MIBG uptake in the basal posteroseptal regions of the LV with the site of origin of VT (as demonstrated by electrophysiologic testing) has been reported.68,69 These findings suggest a reduced activity of the NE transporter (uptake-1), with subsequent β-adrenoceptor down-regulation, and have potential impact on diagnostic evaluation and therapeutic management of patients with ARVC.
123I-MIBG Imaging in Heart Failure (See Chapter 30)
HF is a complex clinical syndrome characterized by dyspnea, fatigue, and fluid retention, which results from any structural or functional cardiac disorder that impairs the ability of the LV to fill with or eject blood. It is a common, costly, disabling, and potentially fatal disorder. The prevalence and incidence of HF are increasing rapidly in the Western world because of the aging of the population and an ever-increasing number of acute coronary syndrome survivals (CAD is the most prevalent underlying etiology, affecting about 70% of cases), despite advances in different pharmacologic and nonpharmacologic therapies.71
The development of HF initiates with some injury to, or stress on, the myocardium, which usually produces progressive changes in the geometry and structure of the LV with pathologic hypertrophic growth. This pathologic remodeling involves a shift toward glycolytic metabolism, disorganization of the sarcomere, alterations in calcium handling, changes in contractility, loss of myocytes, fibrotic replacement, LV dilation, systolic or diastolic dysfunction, and electrical remodeling (i.e., alterations in the expression or function of ion-transporting proteins, or both) with propensity to malignant ventricular arrhythmia.72 Although several factors can accelerate this process, there is evidence of the important role played by the activation of endogenous neurohormonal systems. Patients with HF have elevated circulating or tissue levels of NE, angiotensin II, aldosterone, endothelin, vasopressin, and cytokines, which can adversely affect the structure and function of the heart.71,72 These neurohormonal factors not only increase the hemodynamic stresses on the LV by causing sodium retention and peripheral vasoconstriction but may also exert direct noxious effects on cardiomyocytes (apoptosis and regression to a fetal phenotype) and changes in the nature of the extracellular matrix (stimulation of myocardial fibrosis), which can further alter the architecture and impair the performance of the failing heart.73,74
The hyperadrenergic state present in HF results in down-regulation and uncoupling of cardiac β-adrenergic receptors, which contributes to progressive impairment of LV systolic function by altering postsynaptic signal transduction. Increased sympathetic tone in HF is directly linked to disease progression, prognosis, and risk of SCD.75 Therefore, noninvasive strategies to determine the state of cardiac autonomic regulation are of significant interest.
The most recent definition of cardiomyopathies makes reference to the presence of HF or electrical disorder. Specifically, they are defined as “a heterogeneous group of diseases of the myocardium associated with mechanical and/or electrical dysfunction that usually (but not invariably) exhibit inappropriate ventricular hypertrophy or dilation and are due to a variety of causes that frequently are genetic. Cardiomyopathies either are confined to the heart or are part of generalized systemic disorders, often leading to cardiovascular death or progressive HF-related disability.”76 Several studies have shown reduced cardiac 123I-MIBG uptake and increased WR in different cardiomyopathies, which could reflect the contribution of the altered cardiac sympathetic nervous function to the development of the myocardial disorder (see Fig. 41-4).
In patients with dilated cardiomyopathy (DCM) and hypertrophic cardiomyopathy (HCM), Zhao et al.77 found significant correlations of LV function and perfusion with cardiac sympathetic nervous function by means of cardiac 123I-MIBG and 99mTc-tetrofosmin imaging. WR and early uptake of 123I-MIBG resulted in the most significant factors for predicting LV function and LV perfusion, respectively. These investigators indicated that impairment of neuronal uptake function might not be dominant in HCM, since early 123I-MIBG uptake in the nondilated phase of HCM was preserved and correlated significantly with blood flow. On the other hand, in DCM, early 123I-MIBG uptake was decreased, but it was doubtful whether this phenomenon was caused by the low perfusion other than the impaired neuronal uptake function. In patients with DCM, decreased early 123I-MIBG uptake was closely associated with low perfusion. These findings suggest that the kinetics of 123I-MIBG might be different in these two types of cardiomyopathies. It is still unclear whether the decrease of 123I-MIBG uptake is due to sympathetic denervation or to the impairment of neuronal uptake function. 123I-MIBG WR could reflect cardiac functional impairment, whereas early 123I-MIBG uptake might be determined by myocardial perfusion.
In patients with HCM, Shimizu et al.78 analyzed early and late HMR and WR of 123I-MIBG in planar images, as well as the dispersion and standard deviation (SD) of uptake and the WR from the polar maps of the LV obtained by the SPECT. The late HMR was significantly lower and the WR of the whole heart was significantly higher in patients with HCM than those in the control group. In patients with HCM, the late HMR, early uptake dispersion, and SD of early uptake showed good correlation with the LV end-diastolic and end-systolic dimensions and the percentage of fractional shortening. In other words, patients with larger regional variations of early 123I-MIBG had larger LV volumes and a lower percentage of fractional shortening. The SD of early uptake was a powerful determinant for the percentage of fractional shortening in patients with HCM. Therefore the heterogeneity of regional cardiac sympathetic nerve activity may be correlated with cardiac dysfunction in patients with HCM. Sipola et al.79 studied patients with HCM due to the same gene mutation and have described that the degree of LV hypertrophy is related to 123I-MIBG WR, suggesting that cardiac adrenergic activity may modify phenotypic expression in HCM. Global 123I-MIBG WR correlated with the LV mass and LV maximal wall thickness. The mean 123I-MIBG WR was higher in LV segments equal to or greater than 15 mm thick than in LV segments less than 15 mm thick. It has been recently suggested that the primary abnormality in the pathogenesis of HCM is a decreased contractility of cardiac myocytes, producing increased cell stress and expression of stress-responsive trophic factors, including cardiac adrenergic activation, which in turn induces LV hypertrophy.80 Accordingly, the findings of Sipola et al.79 suggest that accelerated myocardial adrenergic activity is an important factor contributing to LV hypertrophy in HCM.
Terai et al.81 evaluated the relationship between cardiac sympathetic nerve activity and the occurrence of VT in patients with HCM. The global WR of 123I-MIBG was significantly higher in patients with VT than in those without VT, and it was the most powerful predictor of VT. In contrast, indices of regional 123I-MIBG uptake were not different between the two groups. Thus, the occurrence of malignant VT in HCM may be associated with global cardiac sympathetic nerve activity rather than with the heterogeneity of such activity.
Resting cardiac 123I-MIBG imaging can noninvasively evaluate LV functional reserve in patients with nonobstructive HCM, as reported by Isobe and colleagues.82 The percentage of increase from rest to exercise in LV isovolumic contraction (LV dP/dtmax) and the percentage shortening of LV pressure half-time (T1/2) as an index of isovolumic relaxation were significantly less in patients with decreased late HMR (= 1.8) than in patients with HMR greater than 1.8. The percentage increase of LV dP/dtmax and T1/2 significantly correlated with both early and late HMR. Similarly, in another study in patients with DCM, a relation between decreased late HMR, impaired LV contractile reserve (determined by LV dP/dtmax), and down-regulation of SERCA2 mRNA was reported by Ohshima et al.,83 establishing possibilities for a new noninvasive assessment of myocardial contractile reserve in patients with DCM.
Stress cardiomyopathy (takotsubo syndrome, or “apical ballooning”) is a clinical entity characterized by acute but rapidly reversible LV systolic dysfunction, with midventricular wall-motion abnormalities, apical akinesia or dyskinesia, and preserved or hyperkinetic contractile function of the basal LV segments in the absence of atherosclerotic CAD, triggered by acute emotional or physical stress.84 Several hypotheses have been advocated as possible pathophysiologic mechanisms, including catecholamine-mediated multivessel epicardial spasm, microvascular coronary spasm, and direct catecholamine-mediated myocyte toxicity. However, it remains unclear whether these abnormalities are the primary cause of the disorder or secondary events. Since the entity is much more common in postmenopausal women, important influences of sex hormones on the sympathetic neurohormonal axis and on coronary vasoreactivity have been suggested. 123I-MIBG SPECT imaging has shown reduced uptake in the akinetic LV apex, with normal or only mildly reduced perfusion within this region.85
Assessment of Prognosis
Currently, long-term prognosis of patients with HF remains poor, with a 5-year mortality rate of 59% for men and 45% for women.86 Furthermore, prognosis in HF can be determined reliably only in populations and not in individuals. To improve survival, adequate risk stratification is needed. The most significant predictors of survival in clinical management include: decreasing left ventricular ejection fraction (LVEF), worsening New York Heart Association (NYHA) functional status, degree of hyponatremia, decreasing peak exercise oxygen uptake (VO2), decreasing hematocrit, widened QRS on 12-lead ECG, chronic hypotension, resting tachycardia, renal insufficiency, intolerance to conventional therapy, and refractory volume overload. Routine use of ambulatory ECG monitoring, T-wave alternans analysis, HRV measurement, and signal-averaged electrocardiography do not show incremental value in assessing overall prognosis, although ambulatory ECG monitoring and T-wave alternans can be useful in decision making regarding placement of ICDs.71,87,88
Impaired cardiac adrenergic innervation as assessed by 123I-MIBG imaging is strongly related to mortality in patients with HF,18,89,90 independently of its cause.22 Merlet et al.18 studied the prognostic value of 123I-MIBG scintigraphy compared with that of other noninvasive cardiac imaging indices in 90 patients suffering from either ischemic (n = 24) or idiopathic (n = 66) cardiomyopathy. During a follow-up period of 1 to 27 months, 10 patients underwent cardiac transplantation, and 22 died. Among all clinical and imaging variables (cardiac 123I-MIBG uptake, radionuclide LVEF, x-ray cardiothoracic ratio, and echographic end-diastolic diameter), the late HMR was the best predictor of event-free survival. These authors90 subsequently evaluated 112 patients with HF and DCM (NYHA classes II-IV, LVEF < 40%, LV end-diastolic diameter 70 ± 8 mm, and pulmonary capillary wedge pressure 19 ± 8 mm Hg). Among all variables (cardiac 123I-MIBG uptake, circulating NE concentration, LVEF, peak VO2, x-ray cardiothoracic ratio, M-mode echographic end-diastolic diameter, and right-sided heart catheterization parameters), only the late HMR and LVEF were independent predictors for mortality (mean follow-up 27 ± 20 months). In addition, 123I-MIBG uptake and circulating NE concentration were the only independent predictors for life duration. In both studies,18,90 a late HMR of 1.2 was used to identify reduced 123I-MIBG uptake.
In a study of 93 HF patients (LVEF < 45%), Cohen-Solal et al.91 showed that late HMR was reduced and correlated with other predictors of prognosis such as LVEF, cardiac index, pulmonary wedge pressure, and peak VO2. Moreover, a late HMR less than or equal to 1.2 and peak VO2 were predictive of death or cardiac transplantation over 10 ± 8 months follow-up, whereas the early HMR and WR were not. Nakata et al.92 studied 414 patients (42% with symptomatic HF) with 123I-MIBG scintigraphy. Over a mean follow-up of 22 ± 7 months, 37 cardiac deaths occurred. Late HMR was the most powerful predictor of cardiac mortality among the variables. A late HMR less than or equal to 1.74, age older than 60 years, a history of MI, and NYHA class III or IV strongly indicated poor clinical outcomes.
Wakabayashi et al.22 reported 123I-MIBG imaging as the most powerful independent long-term prognostic value for both ischemic (n = 76) and idiopathic (n = 56) cardiomyopathy patients, which hints at cardiac autonomic dysfunction as a common end point leading to cardiac death, regardless of the underlying etiology of the cardiac disease. Late HMR was the most powerful independent predictor of cardiac mortality in both groups of patients (superior to early HMR and WR) and had an identical threshold (1.82) for both groups for identifying patients at risk of cardiac death. Nevertheless, when analyzing patients with LVEF less than 40%, the upper cutoff value of late HMR was 1.50 for ischemic patients and 2.02 for idiopathic patients, which may suggest that the underlying etiology of HF relates that the threshold of the late HMR for the differentiation of high-risk patients. This study was designed, however, before optimal up-to-date HF treatment, and derived results may not translate to current cohorts of HF patients.
Kyuma et al.93 reported that plasma brain natriuretic peptide (BNP) level significantly but roughly correlated with cardiac sympathetic nerve innervation in 158 patients with HF. Univariate analysis identified BNP level, HMR, chronic renal dysfunction, diabetes mellitus, age, and use of nitrates as significant predictors of fatal pump failure, and multivariate Cox analysis showed that BNP level was the most powerful predictor cardiac death. Patients with both plasma BNP level of equal to or greater than 172 pg/mL and late HMR less than or equal to 1.74 had a greater annual rate of fatal pump failure than had those without. The hazard ratio of BNP level (7.2) or cardiac 123I-MIBG activity (10.1) increased to 34.4 when both variables were used, and prevalence of fatal pump failure significantly increased from 22% to 62.5% when diabetes mellitus and chronic renal dysfunction were present with a higher BNP level and low cardiac 123I-MIBG activity.
All these results suggest that the late HRM may be the best prognostic parameter that can be obtained from planar 123I-MIBG imaging.94 The results of a recent retrospective study with the participation of six centers from five European countries confirm the strong prognostic value of late HMR in patients with HF.95 Blind review and prospective quantitative reanalysis of the late HMR of 290 patients with ischemic and nonischemic HF (NYHA class II-IV, 262 patients with LVEF < 50%) with follow-up data for 2 years permitted the identification of potential late HMR threshold values for defining groups with high and very low likelihood of major cardiac events. The mean HMR was significantly different between patients with and without events (1.51 versus 1.97). Based on receiver operating characteristic (ROC) curve analysis, a threshold value for MHR of 1.75 yielded a sensitivity of 84% with a specificity of 60% to predict events. Based on this threshold value, the 2-year event-free survival was 62% for late HMR less than 1.75, versus 95% for late HMR of 1.75. Logistic regression showed late HMR and LVEF as the only significant predictors of major cardiac events. When the late HMRs were divided into quartiles, the 2-year event-free survival rates in the lowest quartile (1.45) and the highest quartile (2.17) were 52% and 98%, respectively.95 Further and larger studies with standardization of data acquisition and data analysis of 123I-MIBG imaging are needed to obtain the optimal threshold value of late HMR for accurate risk stratification of patients with HF.
Besides the late HMR, the prognostic value of other 123I-MIBG parameters has also been reported. Ogita and coworkers96 evaluated 79 HF patients with LVEF less than 40% with planar 123I-MIBG imaging during a follow-up period between 1 and 52 months. A WR less than 27% was a strong predictor of survival. Kioka et al.97 studied 97 outpatients with HF (mean LVEF 29%) by means of cardiac 123I-MIBG. Forty-eight patients had abnormal WR (=27%), whereas the remaining 49 patients had normal WR (<27%). During a mean follow-up of 65 ± 29 months, 12 patients with abnormal WR and 2 patients with normal WR died suddenly. SCD was significantly more frequent in patients with abnormal WR than those with normal WR. 123I-MIBG WR and both early and late HMR were significantly associated with SCD.
Yamada et al.98 prospectively compared the prognostic value of cardiac 123I-MIBG imaging with that of time and frequency domain parameters of HRV in patients with mild to moderate chronic HF (65 outpatients with a radionuclide LVEF < 40%). WR, late HMR, and normalized very low-frequency power (n-VLFP) showed a significant association with the cardiac events after a follow-up of 34 ± 19 months on univariate analysis. Multivariate analysis showed that WR was the only independent predictor of cardiac events, although the predictive accuracy for the combination of abnormal WR and n-VLFP significantly increased, so such a combination would identify a higher-risk subset of patients for cardiac events in chronic HF. In contrast to previous studies that showed a higher predictive value of HMR over WR, these results may be due to differences in the method used to calculate the WR, such as the application of background subtraction. Anastasiou-Nana et al.99 studied 52 HF patients with LVEF less than 40% and demonstrated that the early HMR was the best predictor for long-term (2-year) outcome. Importantly, the early HMR was obtained at 1 hour after tracer injection, whereas most studies used a 10- to 20-minute interval to obtain the early images.
Recently, Tamaki et al.100 prospectively compared the predictive value of cardiac 123I-MIBG imaging for SCD with that of the signal-averaged electrocardiogram (SAECG), HRV, and QT dispersion in 106 consecutive outpatients with mild-to moderate stable HF due to ischemic heart disease in 55 patients and idiopathic DCM in 51 patients. During a follow-up period of 65 ± 31 months, 18 of 106 patients died suddenly. A multivariate Cox analysis revealed that only WR and LVEF were significantly and independently associated with SCD. Patients with an abnormal WR (>27%) had a significantly higher risk of SCD (adjusted hazard ratio: 4.79, 95% CI: 1.55 to 14.76). Even when confined to the patients with LVEF >35%, SCD was significantly more frequently observed in the patients with than without an abnormal WR.
A recent systematic review by Verberne et al.101 on 18 published studies regarding survival in 1755 patients with HF stratified by semiquantitative 123I-MIBG myocardial parameters, showed that patients with HF and decreased cardiac 123I-MIBG uptake or increased WR have a worse prognosis compared with those with normal 123I-MIBG parameters. The pooled hazard ratio estimates for cardiac death and cardiac events associated with WR showed no significant heterogeneity and were 1.72 (95% CI: 1.72−2.52; P = 0.006) and 1.08 (95% CI: 1.03−1.12; P < 0.001), respectively. The pooled hazard ratio estimates for cardiac death and cardiac events associated with early and late HMR showed significant heterogeneity (I2>75%). Limiting the pooling to the qualitative best three studies rendered I2 insignificant (I2 = 0) and resulted in a pooled hazard ratio of late HMR for cardiac death of 1.82 (95% CI: 0.80−4.12; P = 0.15) and for cardiac events of 1.98 (95% CI: 1.57−2.50; P < 0.001).
In the near future, potential valuable data may be obtained from two currently ongoing identical prospective open-label, multicenter international, phase 3, industry sponsored studies, evaluating the prognostic usefulness of cardiac 123I-MIBG imaging for identifying subjects with HF (NYHA class II and III and LVEF less than or equal to 35%) who will experience a major adverse cardiac event.102 In these so called ADMIRE-HF trials, subjects are being monitored on a regular basis for 2 years. Time to first occurrence of one of the following NYHA class progression, potentially life-threatening arrhythmic event (including ICD discharge), or cardiac death will be analyzed in comparison to quantitative parameters derived from 123I-MIBG imaging.
Assessment of Treatment
Cardiac 123I-MIBG imaging can detect drug-induced changes in cardiac adrenergic activity. Somsen et al.103 reported that enalapril improves cardiac sympathetic uptake function but did not affect plasma NE levels in a group of patients with HF, supporting the concept that a restoration of cardiac neuronal uptake of NE is one of the beneficial effects of enalapril treatment in these patients.
Characterization of the sympathetic denervation of the heart with 123I-MIBG could also be useful for selecting HF patients for β-blocker therapy. Fukuoka et al. observed that patients with DCM who showed improvement of LVEF equal to 5% after 3 months of treatment with metoprolol had previously shown decrease in regional WR of 123I-MIBG (at 1-month of β-blocker therapy).104 Watanabe and colleagues105 studied the cardioprotective features of long-term treatment with carvedilol in a rat model of DCM after autoimmune myocarditis. They measured cardiac uptake of 125I-MIBG in the LV by means of autoradiography and found that late uptake of 125I-MIBG increased after treatment, while the WR decreased. These changes appeared together with a decrease in heart weight, myocardial fibrosis, and LV end-diastolic pressure, supporting the beneficial effects of the drug and the protection of cardiac adrenergic neurons in DCM.
Gerson and coworkers106 studied the effect of chronic carvedilol treatment in patients with HF and cardiac sympathetic nerve dysfunction of varying severity due to idiopathic cardiomyopathy. Most patients showed a favorable response in LV function to the treatment, regardless of the baseline level of cardiac sympathetic nervous system function, as assessed by cardiac 123I-MIBG imaging. Patients with relatively advanced cardiac sympathetic dysfunction (baseline late HMR < 1.40 in 123I-MIBG studies) were the most likely to show evidence of improved cardiac sympathetic nervous system function in response to carvedilol treatment. Conversely, Suwa et al.107 in a study with patients treated with bisoprolol (β1-specific blocker) reported that only those patients who showed a late HMR greater than 1.7 had a significant improvement in LV size and clinical status (sensitivity of 91% and specificity of 92% for predicting response to β-blocker therapy). Other studies showing improvement of cardiac 123I-MIBG uptake in response to β-blocker treatment did not show any relationship between the severity of baseline cardiac 123I-MIBG uptake and subsequent improvement in adrenergic function.108,109 The discordance between these studies may reflect the different properties of various β-blockers. Similar increase in cardiac 123I-MIBG uptake has been observed after treatment with ACEIs103,110 and ARBs111 in patients with chronic HF.
Aldosterone prevents the uptake of NE and promotes structural remodeling of the heart. Spironolactone is an aldosterone receptor blocker that improves LV remodeling in patients with DCM. Kasama and coworkers112 assessed the influence of aldosterone treatment on cardiac sympathetic nerve activity. These investigators compared two groups of patients with DCM treated with an ACEI and a loop diuretic, but differing in the addition of spironolactone only in one group. After 6 months of treatment with spironolactone, the late HMR of 123I-MIBG and LVEF significantly increased, and the late total defect score as well as the WR of 123I-MIBG significantly decreased, with parallel reduction of the LV end-diastolic volume. There were no significant changes in these parameters in the control group. Moreover, significant correlation between changes in the 123I-MIBG kinetics and changes in LV end-diastolic volume with spironolactone treatment was found. The functional class improved in both groups but showed a greater improvement in the spironolactone group than in the control group, indicating the beneficial effect of spironolactone on cardiac sympathetic activity and LV remodeling.
Approximately 40% of patients with severe HF die suddenly, probably of arrhythmia.113 In patients with HF and frequent episodes of tachyarrhythmias the association of β-blockers and amiodarone is recommended.71 It is unclear how amiodarone exerts its effects on LV remodeling and cardiac sympathetic nerve function in HF. Amiodarone, unlike most antiarrhythmic agents, has neither cardiodepressant nor proarrhythmic effects114 and has been shown not to adversely affect survival of patients with HF. However, association of β-blockers and amiodarone may cause bradyarrhythmia. This is important, since it has been recently suggested that SCD in these patients may come from bradyarrhythmia or electrical-mechanical dissociation. Toyama et al.115 described a prospective 1-year cohort study comparing amiodarone to β-blockers in the treatment of patients with idiopathic DCM. The authors reported similar improvement in cardiac symptoms, function, and sympathetic nerve activity with both drugs, raising the possibility of substituting β-blockers with amiodarone in the treatment of patients with DCM, avoiding the induction of bradyarrhythmias while having comparable beneficial effects. Further and larger clinical studies using the standard doses of amiodarone and with different severity of baseline cardiac 123I-MIBG uptake are needed to confirm these auspicious data. Tachikawa et al.116 reported that long-term amiodarone treatment on a rat model of DCM (healed cardiac myosin-induced autoimmune myocarditis) prevented LV remodeling, improved cardiac function, and restored cardiac sympathetic tone (late HMR of 123I-MIBG increased, and WR decreased) to hold NE in the heart.
Recently, Kasama and colleagues,117 taking into account that 123I-MIBG imaging improves by the current medical treatment for HF, analyzed the usefulness of serial 123I-MIBG studies for prognostication in 208 patients with stabilized mild to moderate HF and LVEF less than 45% of both ischemic and nonischemic origin. 123I-MIBG and echocardiographic studies were performed once patients were stabilized and after 6 months of treatment, which included ACEIs, ARBs, β-blockers, loop diuretics, and spironolactone. Treatment did not change during the follow-up. Fifty-six patients experienced fatal cardiac events during the study period (13 died from SCD). Clinical characteristics were similar in both noncardiac death and cardiac death groups. With respect to pharmacotherapy in the two groups, only the use of β-blockers in the noncardiac death group was significantly higher than in the cardiac death group. The variation in the WR between the sequential 123I-MIBG (Δ-WR) was the only independent predictor of cardiac death. The Δ-WR was significantly lower in the noncardiac death group (less than −5%) than that in the cardiac death group (more than or equal to −5%). Moreover, this parameter was also useful for predicting SCD in patients with HF, indicating that serial 123I-MIBG imaging is useful for predicting cardiac death and SCD in stabilized patients with HF.
123I-MIBG Imaging in Heart Transplantation
Lack of autonomic nerve supply is associated with major physiologic alterations such as increased baseline heart rate, chronotropic incompetence during exercise (the sinus node is denervated), and reduced diastolic ventricular function. As a result, exercise capacity is reduced compared with that in healthy, normal subjects.118 In addition, the decreased ability to perceive pain may not allow symptomatic recognition of accelerated allograft vasculopathy, and heart transplant patients often develop acute ischemic events or LV dysfunction or die suddenly. Furthermore, loss of vasomotor tone may adversely affect the physiologic adaptations in blood flow in different circumstances. Scintigraphic uptake of 123I-MIBG supports the concept of spontaneous re-innervation taking place after transplantation.6,119,120 All studies performed up to 5 years after heart transplantation suggest that re-innervation is likely to be a slow process and partially occurs only after 1 year posttransplantation.121
Sympathetic re-innervation, measured by regional distribution and intensity of cardiac 123I-MIBG uptake, increases with time after transplantation, with a positive correlation between HMR and time after transplantation. Serial 123I-MIBG studies over time show that re-innervation begins from the base of the heart and spreads towards the apex. 123I-MIBG uptake is seen primarily in the anterior, anterolateral, and septal regions, and it is usually not apparent in the inferior wall (except for some uptake in basal segments). Complete re-innervation of the transplanted heart is not seen on scintigraphic studies, even up to 12 years posttransplantation. Therefore, sympathetic re-innervation after cardiac transplantation is not simply a function of time. It has been observed by means of PET with the catecholamine analog 11C-hydroxyephedrine (11C-HED) that re-innervation is more likely with young age of the donor and recipient, fast and uncomplicated surgery, and low rejection frequency.122
123I-MIBG Imaging in Dysautonomias
Diabetes Mellitus
Development of diabetic autonomic neuropathy is associated with the advent of orthostatic hypotension, resting tachycardia, exercise intolerance, derangement in myocardial blood flow regulation, LV dysfunction, and silent myocardial ischemia/infarction. Thus, development of autonomic neuropathy is associated with an increased rate of morbidity and mortality in diabetic patients. Kim et al.123 reported decreased 123I-MIBG uptake associated with autonomic dysfunction in diabetic patients and have correlated decreased cardiac 123I-MIBG uptake with increased mortality. It seems that 123I-MIBG scintigraphy is more sensitive than autonomic nervous function tests for the detection of dysautonomia in diabetes, particularly in early stages of the disease. Atherosclerosis of the large coronary arteries is not a prerequisite cause for these findings, since 123I-MIBG defects are present even in the absence of CAD. However, the role of microvascular disease in adrenergic dysfunction has not been well defined.
Scognamiglio et al.124 showed that in diabetic patients the impairment of cardiac sympathetic innervation, as shown by 123I-MIBG studies, correlates with abnormal response to exercise and may contribute to LV dysfunction before the appearance of irreversible damage and overt HF. Hattori et al.125 reported that diabetic patients in whom there is scintigraphic evidence of severe myocardial denervation present with hyperreaction to dobutamine stress, which may in part explain the high incidence of SCD in patients with advanced diabetes mellitus.
Increased incidence of SCD has been related to QT-interval prolongation and QT dispersion, particularly in patients with diabetes. These abnormalities indicate variation in repolarization in different myocardial regions and may reflect inhomogeneities in LV sympathetic innervation. They are more pronounced in patients with diabetic dysautonomia than in healthy control subjects, but there are no consistent differences between diabetic patients with and without dysautonomia. Most studies have failed to show a relationship between 123I-MIBG uptake and the presence or absence of QT abnormalities.126,127
Neurodegenerative Disorders
In PD, autonomic failure is caused by damage and Lewy body (intraneuronal cytoplasmic inclusions that stain with periodic acid–Schiff and ubiquitin) deposition in the postganglionic part of the autonomic nervous system,128 whereas in MSA, degeneration of preganglionic and central autonomic neurons is revealed histopathologically. The assumption that selective investigation of postganglionic cardiac neurons possibly enables a safe differentiation between both entities is supported by a study of Braune et al.129 in which patients with PD showed reduced late HMR of 123I-MIBG uptake independent of duration and severity of autonomic and parkinsonian symptoms, whereas it was normal in patients MSA. Similarly, Yoshita et al.130 described that the mean value of HMR in patients with PD was significantly lower than those with SND, PSP, or no disease, regardless of disease severity or intensity of antiparkinsonian treatment. The mean value of HMR in patients with SND and orthostatic hypotension was lower than that in patients with SND but without orthostatic hypotension. Although the mean value of HMR in PSP with amitriptyline treatment was significantly lower than that in PSP patients without this treatment, there was no significant difference between the mean value of HMR in PSP patients without amitriptyline treatment and that in control patients. Takatsu and colleagues131 also described reduced early and late cardiac 123I-MIBG uptake in PD, even at the earlier stages of physical activity or disease duration, and also in patients with normal blood pressure response and normal circadian patterns of variation in blood pressure. Additionally, they observed that cardiac 123I-MIBG uptake in PD patients with normal blood pressure patterns was lower than that in patients with MSA and also normal blood pressure response and variation, which suggests that the decrease in cardiac 123I-MIBG accumulation may be specific for PD among the akinetic-rigid syndromes, independent of clinical stage, disease duration, and systemic sympathetic dysfunction. Furthermore, these investigators determined 125I-MIBG accumulation in an experimental murine model of PD in which the animals were pretreated with the standard dose of 1-methyl-4-phenyl 1,2,3,6-tetrahydropyridine (MPTP) used to destroy dopaminergic neurons. The MPTP significantly reduced cardiac 125I-MIBG accumulation. However, the decrease of 125I-MIBG uptake was still significant with greatly reducing MPTP dose, which may reflect early damage of the postganglionic sympathetic nerves in PD, probably because dopaminergic neurons and sympathetic nerves are substantially similar in their plasma membrane transporters. Therefore, cardiac scintigraphy with 123I-MIBG may be used in the diagnosis and characterization of akinetic-rigid syndromes, especially PD.
In a meta-analysis that included 246 cases of PD and 45 cases of MSA, Braune132 reported that quantification of cardiac 123I-MIBG uptake is a valuable tool to identify patients with PD and to discriminate PD from MSA early in the course of the disease, with an overall sensitivity of 89.7% and specificity of 94.6%. However, in another study that included 391 outpatients with parkinsonian-like symptoms (PD 122, MSA 14, dementia with Lewy bodies [DLB] 5, PSP 7, senile dementia of Alzheimer type [AD] 15, cerebrovascular disease 129, other disorders 81, and unknown diagnosis 18), the sensitivity and specificity of 123I-MIBG scintigraphy for detecting PD were 87.7% and 37.4%, respectively.133 Therefore, reduced uptake of 123I-MIBG can be present in autonomic failure disorders other than PD, although the reductions reported are smaller than those in PD.133,134 Recently, Raffel et al.,135 using PET and 11C-HED, showed that cardiac sympathetic denervation occurs not only in PD but also in MSA and PSP, thus implying that 123I-MIBG scintigraphy may not be used independently to discriminate PD from other movement disorders such as MSA and PSP. These authors also investigated the relationship between cardiac denervation (with 11C-HED) and nigrostriatal denervation by measuring striatal presynaptic monoaminergic nerve density with PET and 11C-dihydrotetrabenazine (11C-DTBZ), a radioligand for the VMAT, to determine whether the central and peripheral nervous system degenerative processes occur in parallel. Cardiac sympathetic denervation was not correlated with striatal denervation, suggesting that the pathophysiologic processes underlying cardiac denervation and striatal denervation occur independently in patients with parkinsonian syndromes. These findings provide novel information about central and peripheral denervation in patients with neurodegenerative disorders and should be confirmed by additional and larger clinical studies.
Dementia with Lewy bodies is the second most common cause of dementia after AD. Differential diagnosis between these two degenerative senile dementias is clinically challenging. Cardiac 123I-MIBG imaging has been advocated to be useful in this setting because of the similitude between PD and DLB.136,137 Decreased cardiac 123I-MIBG uptake in DLB has been reported, whereas the uptake is normal or near normal in AD. Moreover, these findings are not related to the presence of autonomic failure or parkinsonism.137,138 An advantage of cardiac 123I-MIBG imaging over other functional studies is the short acquisition time and comfortable planar imaging, which is appreciated by patients and their caregivers.139 Randomized controlled multicenter clinical trials should confirm these results.
123I-MIBG Imaging in Drug-Induced Cardiotoxicity
123I-MIBG studies have been used to assess adrenergic innervation impairment due to anthracycline cardiotoxicity. Decreased cardiac 123I-MIBG uptake has been observed after doxorubicin administration, with limited morphologic damage and preceding LVEF deterioration.140 Valdés Olmos et al.141 reported decreased cardiac 123I-MIBG uptake in patients with severely decreased LVEF after doxorubicin administration. Correlation with parameters derived from radionuclide angiocardiography suggested a global process of cardiac adrenergic derangement.
A decrease in cardiac 123I-MIBG uptake has also been reported regardless of the patient’s functional status. The decrease in cardiac 123I-MIBG accumulation parallels the evolution of LVEF.142 At intermediate cumulative doses of 240 to 300 mg/m2, 25% of patients present with some decrease in 123I-MIBG uptake. At maximal cumulative doses, there is a significant decrease in 123I-MIBG uptake consistent with impaired cardiac adrenergic activity. A moderate to marked decrease in cardiac 123I-MIBG uptake at high cumulative doses was observed in all but one patient with decrease in LVEF more than or equal to 10%.142 These data suggest that the assessment of drug-induced sympathetic damage could be used to select patients at risk of severe functional impairment and who might benefit from cardioprotective agents or changes in the schedule or administration technique of antineoplastic drugs.
CONCLUSIONS
Further studies assessing the role of pharmacologic intervention on 123I-MIBG cardiac imaging are warranted. Identification of different responses to pharmacologic challenge may unmask subclinical neuronal dysfunctional states.143 In addition, large-scale clinical trials are required to ascertain the clinical value of cardiac neurotransmission imaging in various specific indications, such as the assessment of prognosis and prediction of the effectiveness of therapy in patients with HF. Progress in the prevention and treatment of HF is now accelerating rapidly. Expensive therapies for HF (e.g., biventricular pacemakers, ICDs, and ventricular assist devices) are currently being used, and cardiac neurotransmission imaging could be one of the means to identify those patients who would benefit the most.
1. Verrier R.L., Antzelevitch C. Autonomic aspects of arrhythmogenesis: the enduring and the new. Curr Opin Cardiol. 2004;19:2-11.
2. Kawano H., Okada R., Yano K. Histological study on the distribution of autonomic nerves in the human heart. Heart Vessels. 2003;18:32-39.
3. Pacholczyk T., Blakely R.D., Amara S.G. Expression cloning of a cocaine- and antidepressant-sensitive human noradrenaline transporter. Nature. 1991;350:350-354.
4. Iversen L.L. The uptake of catecholamines at high perfusion concentrations in the rat isolated heart: a novel catecholamine uptake process. Br J Pharmacol. 1965;25:18-33.
5. DeGrado T.R., Zalutsky M.R., Vaidyanathan G. Uptake mechanisms of meta-[123I]iodobenzylguanidine in isolated rat heart. Nucl Med Biol. 1995;22:1-12.
6. Dae M.W., De-Marco T., Botvinick E.H., et al. Scintigraphic assessment of MIBG uptake in globally denervated human and canine hearts: implications for clinical studies. J Nucl Med. 1992;33:1444-1450.
7. Bristow M.R., Minobe W., Rasmussen R., et al. Beta-adrenergic neuroeffector abnormalities in the failing human heart are produced by local rather than systemic mechanisms. J Clin Invest. 1992;89:803-815.
8. Bristow M.R., Anderson F.L., Port J.D., et al. Differences in beta-adrenergic neuroeffector mechanisms in ischemic versus idiopathic dilated cardiomyopathy. Circulation. 1991;84:1024-1039.
9. Wieland D.M., Wu J., Brown L.E., et al. Radiolabeled adrenergic neuron-blocking agents: Adrenomedullary imaging with 123I-iodobenzylguanidine. J Nucl Med. 1980;21:349-353.
10. Wieland D.M., Brown L.E., Rogers W.L., et al. Myocardial imaging with a radioiodinated norepinephrine storage analog. J Nucl Med. 1981;22:22-31.
11. Sisson J.C., Shapiro B., Meyers L., et al. Metaiodobenzylguanidine to map scintigraphically the adrenergic nervous system in man. J Nucl Med. 1987;28:1625-1636.
12. Dae M.W., O’Connell J., Botvinick E.H., et al. Scintigraphic assessment of regional cardiac innervation. Circulation. 1989;79:634-644.
13. Sisson J.C., Wieland D.M., Sherman P., et al. Metaiodobenzylguanidine as an index of adrenergic nervous system integrity and function. J Nucl Med. 1987;28:1620-1624.
14. Sisson J.C., Bolgos G., Johnson J. Measuring acute changes in adrenergic nerve activity of the heart in the living animal. Am Heart J. 1991;121:1119-1123.
15. Kaye M.P., Tyce G.M. Norepinephrine uptake as an indicator of cardiac reinnervation in dogs. Am J Physiol. 1978;235:H289-H294.
16. Verberne H.J., de Bruin K., Habraken J.B.A., et al. No-carrier-added versus carrier-added 123I-metaiodobenzylguanidine for the assessment of cardiac sympathetic nerve activity. Eur J Nucl Med Mol Imaging. 2006;33:483-490.
17. Solanki K.K., Bomanji J., Moyes J., et al. A pharmacological guide to medicines which interfere with the biodistribution of radiolabelled meta-iodobenzylguanidine (MIBG). Nucl Med Commun. 1992;13:513-521.
18. Merlet P., Valette H., Dubois-Rande J.L., et al. Prognostic value of cardiac metaiodobenzylguanidine imaging in patients with heart failure. J Nucl Med. 1992;33:471-477.
19. Flotats A., Carrió I. Cardiac neurotransmission SPECT imaging. J Nucl Cardiol. 2004;11:587-602.
20. Estorch M., Carrio I., Berna L., et al. Myocardial iodine-labeled metaiodobenzylguanidine uptake relates to age. J Nucl Cardiol. 1995;2:126-132.
21. Somsen G.A., Verberne H.J., Fleury E., Righetti A. Normal values and within-subject variability of cardiac I-123 MIBG scintigraphy in healthy individuals: implications for clinical studies. J Nucl Cardiol. 2004;11:126-133.
22. Wakabayashi T., Nakata T., Hashimoto A., et al. Assessment of underlying etiology and cardiac sympathetic innervation to identify patients at high risk of cardiac death. J Nucl Med. 2001;42:1757-1767.
23. Patel A.D., Iskandrian A.E. MIBG imaging. J Nucl Cardiol. 2002;9:75-94.
24. Chen W., Botvinick E.H., Alavi A., et al. Age-related decrease in cardiopulmonary adrenergic neuronal function in children as assessed by I-123 metaiodobenzylguanidine imaging. J Nucl Cardiol. 2008;15:73-79.
25. Verberne H.J., Habraken J.B., van Eck-Smit B.L., et al. Variations in (123)I-metaiodobenzylguanidine (MIBG) late heart mediastinal ratios in chronic heart failure: a need for standardisation and validation. Eur J Nucl Med Mol Imaging. 2008;35:547-553.
26. Gill J.S., Hunter G.J., Gane G., Camm A.J. Heterogeneity of the human myocardial sympathetic innervation: in vivo demonstration by iodine 123-labeled metaiodobenzylguanidine scintigraphy. Am Heart J. 1993;126:390-398.
27. Yamazaki J., Muto H., Ishiguro K., et al. Quantitative scintigraphic analysis of 123I-MIBG by polar map in patients with dilated cardiomyopathy. Nucl Med Commun. 1997;3:219-229.
28. Dae M.W., O’Connell J.W., Botvinick E., Chin M.C. Acute and chronic effects of transient myocardial ischemia on sympathetic nerve activity, density, and norepinephrine content. Cardiovasc Res. 1995;30:270-280.
29. Agostini D., Lecluse E., Belin A., et al. Impact of exercise rehabilitation on cardiac neuronal function in heart failure: an iodine-123 metaiodobenzylguanidine scintigraphy study. Eur J Nucl Med. 1998;25:235-241.
30. Estorch M., Flotats A., Serra-Grima R., et al. Influence of exercise rehabilitation on myocardial perfusion and sympathetic heart innervation in ischemic heart disease. Eur J Nucl Med. 2000;3:333-339.
31. Fagret D., Wolf J.E., Comet M. Myocardial uptake of meta-[123I]-iodobenzylguanidine ([123I]-MIBG) in patients with myocardial infarct. Eur J Nucl Med. 1989;15:624-628.
32. McGhie A.I., Corbett J.R., Akers M.S., et al. Regional cardiac adrenergic function using I-123 meta-iodobenzylguanidine tomographic imaging after acute myocardial infarction. Am J Cardiol. 1991;67:236-242.
33. Nishimura T., Oka H., Sago M., et al. Serial assessment of denervated but viable myocardium following acute myocardial infarction in dogs using iodine-123 MIBG and thallium-201 chloride myocardial single photon emission tomography. Eur J Nucl Med. 1992;19:25-29.
34. Hartikainen J., Mäntysaari M., Kuikka J., et al. Extent of cardiac autonomic denervation in relation to angina on exercise test in patients with recent acute myocardial infarction. Am J Cardiol. 1994;74:760-763.
35. Kramer C.M., Nicol P.D., Rogers W.J., et al. Reduced sympathetic innervation underlies adjacent noninfarcted region dysfunction during left ventricular remodelling. J Am Coll Cardiol. 1997;30:1079-1085.
36. Hartikainen J., Mustonen J., Kuikka J., et al. Cardiac sympathetic denervation in patients with coronary artery disease without previous myocardial infarction. Am J Cardiol. 1997;80:273-277.
37. Nakata T., Nagao K., Tsuchihashi K., et al. Regional cardiac sympathetic nerve dysfunction and the diagnostic efficacy of metaiodobenzylguanidine tomography in stable coronary artery disease. Am J Cardiol. 1996;78:292-297.
38. Minardo J.D., Tuli M.M., Mock B.H., et al. Scintigraphic and electrophysiological evidence of canine myocardial sympathetic denervation and reinnervation produced by myocardial infarction or phenol application. Circulation. 1988;78:1008-1019.
39. Gaudino M., Giordano A., Santarelli P., et al. Immunohistochemical-scintigraphic correlation of sympathetic cardiac innervation in postischemic left ventricular aneurysms. J Nucl Cardiol. 2002;9:601-607.
40. Simula S., Lakka T., Kuikka J., et al. Cardiac adrenergic innervation within the first 3 months after acute myocardial infarction. Clin Physiol. 2000;20:366-373.
41. Hartikainen J., Kuikka J., Mäntysaari M., Länsimies E., Pyörälä K. Sympathetic reinnervation after acute myocardial infarction. Am J Cardiol. 1996;77:5-9.
42. Matsunari I., Schricke U., Bengel F.M., et al. Extent of cardiac sympathetic neuronal damage is determined by the area of ischemia in patients with acute coronary syndromes. Circulation. 2000;22:2579-2585.
43. Estorch M., Narula J., Flotats A., et al. Concordance between rest MIBG and exercise tetrofosmin defects: possible use of rest MIBG imaging as a marker of reversible ischaemia. Eur J Nucl Med. 2001;28:614-619.
44. Watanabe K., Takahashi T., Miyajima S., et al. Myocardial sympathetic denervation, fatty acid metabolism, and left ventricular wall motion in vasospastic. J Nucl Med. 2002;43:1476-1481.
45. Tsutsui H., Ando S., Fukai T., et al. Detection of angina-provoking coronary stenosis by resting iodine 123 metaiodobenzylguanidine scintigraphy in patients with unstable angina pectoris. Am Heart J. 1995;129:708-715.
46. Johnson L.L., Thambar S., Donahay T., et al. Effect of endomyocardial laser channels on regional innervation shown with 125I-MIBG and autoradiography. J Nucl Med. 2002;43:551-555.
47. Muxi A., Magrina J., Martin F., et al. Technetium 99m-labeled tetrofosmin and iodine 123-labeled metaiodobenzylguanidine scintigraphy in the assessment of transmyocardial laser revascularization. J Thorac Cardiovasc Surg. 2003;125:1493-1498.
48. Huikuri H.V., Castellanos A., Myerberg R.J. Sudden death due to cardiac arrhythmias. N Engl J Med. 2001;345:1473-1482.
49. Myerburg R.J., Mitrani R., Interian A.Jr, Castellanos A. Interpretation of outcomes of antiarrhythmic clinical trials: design features and population impact. Circulation. 1998;97:1514-1521.
50. Schwartz P.J., La Rovere M.T., Vanoli E. Autonomic nervous system and sudden cardiac death. Experimental basis and clinical observations for post-myocardial infarction risk stratification. Circulation. 1992;85(Suppl 1):I77-1191.
51. Chen L.S., Zhou S., Fishbein M.C., Chen P.S. New perspectives on the role of autonomic nervous system in the genesis of arrhythmias. J Cardiovasc Electrophysiol. 2007;18:123-127.
52. Inoue H., Zipes D.P. Results of sympathetic denervation in the canine heart: supersensitivity that may be arrhythmogenic. Circulation. 1987;75:877-887.
53. Kammerling J.J., Green F.J., Watanabe A.M., et al. Denervation supersensitivity of refractoriness in noninfarcted areas apical to transmural myocardial infarction. Circulation. 1987;76:383-393.
54. Singh S.N., Fletcher R.D., Fisher S.G., et al. Amiodarone in patients with congestive heart failure and asymptomatic ventricular arrhythmia. Survival Trial of Antiarrhythmic Therapy in Congestive Heart Failure. N Engl J Med. 1995;333:77-82.
55. Kuck K.H., Cappato R., Siebels J., Ruppel R. Randomized comparison of antiarrhythmic drug therapy with implantable defibrillators in patients resuscitated from cardiac arrest : the Cardiac Arrest Study Hamburg (CASH). Circulation. 2000;102:748-754.
56. Moss A.J., Zareba W., Hall W.J., et al. Multicenter Automatic Defibrillator Implantation Trial II Investigators. Prophylactic implantation of a defibrillator in patients with myocardial infarction and reduced ejection fraction. N Engl J Med. 2002;346:877-883.
57. Bigger J.T. Expanding indications for implantable cardiac defibrillators. N Engl J Med. 2002;346:931-932.
58. Hallstrom A.P., McAnulty J.H., Wilkoff B.L., et al. Antiarrhythmics Versus Implantable Defibrillator (AVID) Trial Investigators. Patients at lower risk of arrhythmia recurrence: a subgroup in whom implantable defibrillators may not offer benefit. Antiarrhythmics Versus Implantable Defibrillator (AVID) Trial Investigators. Circulation. 2001;37:1093-1099.
59. Arora R., Ferrick K.J., Nakata T., et al. 123I-MIBG imaging and heart rate variability analysis to predict the need for an implantable cardioverter defibrillator. J Nucl Cardiol. 2003;10:121-131.
60. Schäfers M., Wichter T., Lerch H., et al. Cardiac 123I-MIBG uptake in idiopathic ventricular tachycardia and fibrillation. J Nucl Med. 1999;40:1-5.
61. Paul M., Schafers M., Kies P., et al. Impact of sympathetic innervation on recurrent life-threatening arrhythmias in the follow-up of patients with idiopathic ventricular fibrillation. Eur J Nucl Med Mol Imaging. 2006;33:866-870.
62. Simões M.V., Barthel P., Matsunari I., et al. Presence of sympathetically denervated but viable myocardium and its electrophysiologic correlates after early revascularised, acute myocardial infarction. Eur Heart J. 2004;25:551-557.
63. Bax J.J., Kraft O., Buxton A.E., et al. 123I-mIBG Scintigraphy to predict inducibility of ventricular arrhythmias on cardiac electrophysiology testing: a prospective multicenter pilot study. Circ Cardiovasc Imaging. 2008;1:131-140.
64. Muller K.D., Jakob H., Neuzner J., et al. 123I-metaiodobenzylguanidine scintigraphy in the detection of irregular regional sympathetic innervation in long QT syndrome. Eur Heart J. 1993;14:316-325.
65. Yamanari H., Nakayama K., Morita H., et al. Effects of cardiac sympathetic innervation on regional wall motion abnormality in patients with long QT syndrome. Heart. 2000;83:295-300.
66. Wichter T., Matheja P., Eckardt L., et al. Cardiac autonomic dysfunction in Brugada syndrome. Circulation. 2002;105:702-706.
67. Basso C., Thiene G., Corrado D., et al. Arrhythmogenic right ventricular cardiomyopathy. Dysplasia, dystrophy, or myocarditis. Circulation. 1996;94:983-991.
68. Lerch H., Bartenstein P., Wichter T., et al. Sympathetic innervation of the left ventricle is impaired in arrhythmogenic right ventricular disease. Eur J Nucl Med. 1993;20:207-212.
69. Wichter T., Hindricks G., Lerch H., et al. Regional myocardial sympathetic dysinnervation in arrhythmogenic right ventricular cardiomyopathy. An analysis using 123I-MIBG scintigraphy. Circulation. 1994;89:667-683.
70. Wichter T., Schafers M., Rhodes C.G., et al. Abnormalities of cardiac sympathetic innervation in arrhythmogenic right ventricular cardiomyopathy: quantitative assessment of presynaptic norepinephrine reuptake and postsynaptic beta-adrenergic receptor density with positron emission tomography. Circulation. 2000;101:1552-1558.
71. Hunt S.A., Abraham W.T., Chin M.H., et al. ACC/AHA 2005 guideline update for the diagnosis and management of chronic heart failure in the adult: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Update the 2001 Guidelines for the Evaluation and Management of Heart Failure). American College of Cardiology Web Site. Circulation. 2005;20(112):e154-e235. Available at: http://www.acc.org/clinical/guidelines/failure//index.pdf
72. Hill J.A., Olson E.N. Cardiac plasticity. N Engl J Med. 2008;358:1370-1380.
73. Ungerer M., Bohm M., Elce J., et al. Altered expression of beta-adrenergic receptor kinase and beta-adrenergic receptors in the failing human heart. Circulation. 1993;87:454-463.
74. Henderson E.B., Kahn J.K., Corbet J.R., et al. Abnormal I-123-MIBG myocardial wash-out and distribution may reflect myocardial adrenergic derangement in patients with congestive cardiomyopathy. Circulation. 1988;78:1192-1199.
75. Cohn J.N., Levine T.B., Olivari M.T., et al. Plasma norepinephrine as a guide to prognosis in patients with chronic congestive heart failure. N Engl J Med. 1984;311:819-823.
76. Maron B.J., Towbin J.A., Thiene G., et al. Contemporary definitions and classification of the cardiomyopathies: an American Heart Association Scientific Statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation. 2006;113:1807-1816.
77. Zhao C., Shuke N., Yamamoto W., et al. Comparison of cardiac sympathetic nervous function with left ventricular function and perfusion in cardiomyopathies by 123I-MIBG SPECT and 99mTc-tetrofosmin electrocardiographically gated SPECT. J Nucl Med. 2001;42:1017-1024.
78. Shimizu M., Ino H., Yamaguchi M., et al. Heterogeneity of cardiac sympathetic nerve activity and systolic dysfunction in patients with hypertrophic cardiomyopathy. J Nucl Med. 2002;43:15-20.
79. Sipola P., Vanninen E., Aronen H.J., et al. Cardiac adrenergic activity is associated with left ventricular hypertrophy in genetically homogeneous subjects with hypertrophic cardiomyopathy. J Nucl Med. 2003;44:487-493.
80. Marian A.J. Pathogenesis of diverse clinical and pathological phenotypes in hypertrophic cardiomyopathy. Lancet. 2000;355:58-60.
81. Terai H., Shimizu M., Ino H., et al. Cardiac sympathetic nerve activity in patients with hypertrophic cardiomyopathy with malignant ventricular tachyarrhythmias. J Nucl Cardiol. 2003;10:304-310.
82. Isobe S., Izawa H., Iwase M., et al. Cardiac 123I-MIBG reflects left ventricular functional reserve in patients with nonobstructive hypertrophic cardiomyopathy. J Nucl Med. 2005;46:909-916.
83. Ohshima S., Isobe S., Izawa H., et al. Cardiac sympathetic dysfunction correlates with abnormal myocardial contractile reserve in dilated cardiomyopathy patients. J Am Coll Cardiol. 2005;46:2061-2068.
84. Gianni M., Dentali F., Grandi A.M., et al. Apical ballooning syndrome or takotsubo cardiomyopathy: a systematic review. Eur Heart J. 2006;27:1523-1529.
85. Burgdorf C., von Hof K., Schunkert H., Kurowski V. Regional alterations in myocardial sympathetic innervation in patients with transient left-ventricular apical ballooning (Tako-Tsubo cardiomyopathy). J Nucl Cardiol. 2008;15:65-72.
86. Levy D., Kenchaiah S., Larson M.G., et al. Long-term trends in the incidence of and survival with heart failure. N Engl J Med. 2002;347:1397-1402.
87. Zipes D.P., Camm A.J., Borggrefe M., et al. ACC/AHA/ESC 2006 guidelines for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death-executive summary: a report of the American College of Cardiology/American Heart Association Task Force and the European Society of Cardiology Committee for Practice Guidelines (Writing Committee to Develop Guidelines for Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death). Eur Heart J. 2006;27:2099-2140.
88. Chow T., Kereiakes D.J., Bartone C., et al. Microvolt T-wave alternans identified patients with ischemic cardiomyopathy who benefit from implantable cardioverter-defibrillator therapy. J Am Coll Cardiol. 2007;49:50-58.
89. Nakata T., Miyamoto K., Doi A., et al. Cardiac death prediction and impaired cardiac sympathetic innervation assessed by metaiodobenzylguanidine in patients with failing and non-failing hearts. J Nucl Cardiol. 1998;5:579-590.
90. Merlet P., Benvenuti C., Moyse D., et al. Prognostic value of MIBG imaging in idiopathic dilated cardiomyopathy. J Nucl Med. 1999;40:917-923.
91. Cohen-Solal A., Esanu Y., Logeart D., et al. Cardiac metaiodobenzylguanidine uptake in patients with moderate chronic heart failure: relationship with peak oxygen uptake and prognosis. J Am Coll Cardiol. 1999;33:759-766.
92. Nakata T., Miyamoto K., Doi A., et al. Cardiac death prediction and impaired cardiac sympathetic innervation assessed by MIBG in patients with failing and nonfailing hearts. J Nucl Cardiol. 1998;5:579-590.
93. Kyuma M., Nakata T., Hashimoto A., et al. Incremental prognostic implications of brain natriuretic peptide, cardiac sympathetic nerve innervation, and noncardiac disorders in patients with heart failure. J Nucl Med. 2004;45:155-163.
94. Bax J.J., Boogers M., Henneman M.M. Can cardiac iodine-123 metaiodobenzylguanidine imaging contribute to risk stratification in heart failure patients? Eur J Nucl Med Mol Imaging. 2008;35:352-354.
95. Agostini D., Verberne H.J., Burchert W., et al. I-123-mIBG myocardial imaging for assessment of risk for a major cardiac event in heart failure patients: insights from a retrospective European multicenter study. Eur J Nucl Med Mol Imaging. 2008;35:535-546.
96. Ogita H., Shimonagata T., Fukunami M., et al. Prognostic significance of cardiac (123)I metaiodobenzylguanidine imaging for mortality and morbidity in patients with chronic heart failure: a prospective study. Heart. 2001;86:656-660.
97. Kioka H., Yamada T., Mine T., et al. Prediction of sudden death in patients with mild to moderate chronic heart failure by using cardiac iodine-123 metaiodobenzylguanidine imaging. Heart. 2007;93:1213-1218.
98. Yamada T., Shimonagata T., Fukunami M., et al. Comparison of the prognostic value of cardiac iodine-123 metaiodobenzylguanidine imaging and heart rate variability in patients with chronic heart failure. A prospective study. J Am Coll Cardiol. 2003;41:231-238.
99. Anastasiou-Nana M.I., Terrovitis J.V., et al. Prognostic value of iodine-123-metaiodobenzylguanidine myocardial uptake and heart rate variability in chronic congestive heart failure secondary to ischemic or idiopathic dilated cardiomyopathy. Am J Cardiol. 2005;96:427-431.
100. Tamaki S., Yamada T., Okuyama Y., et al. Cardiac iodine-123 metaiodobenzylguanidine imaging predicts sudden cardiac death independently of left ventricular ejection fraction in patients with chronic heart failure and left ventricular systolic dysfunction. J Am Coll Cardiol. 2009;53:426-435.
101. Verberne H.J., Brewster L.M., Somsen G.A., et al. Prognostic value of myocardial 123I-metaiodobenzylguanidine (MIBG) parameters in patients with heart failure: a systematic review. Eur Heart J. 2008;29:1147-1159.
102. Jacobson A.F., Lombard J., Banerjee G., Camici P.G. 123I-mIBG scintigraphy to predict risk for adverse cardiac outcomes in heart failure patients: Design of two prospective multicenter international trials. J Nucl Cardiol. 2009;16:113-121.
103. Somsen G.A., van Vlies B., de Milliano P.A., et al. Increased myocardial [123-I]-metaiodobenzylguanidine uptake after enalapril treatment in patients with chronic heart failure. Heart. 1996;76:218-222.
104. Fukuoka S., Hayashida K., Hirose Y. Use of iodine-123 metaiodobenzylguanidine myocardial imaging to predict the effectiveness of beta-blocker therapy in patients with dilated cardiomyopathy. Eur J Nucl Med. 1997;24:523-529.
105. Watanabe K., Takahashi T., Nakazawa M., et al. Effects of carvedilol on cardiac function and cardiac adrenergic neuronal damage in rats with dilated cardiomyopathy. J Nucl Med. 2002;43:531-535.
106. Gerson M.C., Craft L.L., McGuire N., et al. Carvedilol improves left ventricular function in heart failure patients with idiopathic dilated cardiomyopathy and a wide range of sympathetic nervous system function as measured by iodine123 metaiodobenzylguanidine. J Nucl Cardiol. 2002;9:608-615.
107. Suwa M., Otake Y., Moriguchi A., et al. Iodine-123 metaiodobenzylguanidine myocardial scintigraphy for prediction of response to beta-blocker therapy in patients with dilated cardiomyopathy. Am Heart J, 133:353-358, 1997. Erratum in: Am Heart J. 134:1141, 1997.
108. Agostini D., Belin A., Amar M.H., et al. Improvement of cardiac neuronal function after carvedilol treatment in dilated cardiomyopathy: a 123I-MIBG scintigraphic study. J Nucl Med. 2000;41:845-851.
109. Lotze U., Kaepplinger S., Kober A., et al. Recovery of the cardiac adrenergic nervous system after long-term beta blocker therapy in idiopathic dilated cardiomyopathy: assessment by increase in myocardial 123I-metaiodobenzylguanidine uptake. J Nucl Med. 2001;42:49-54.
110. Gilbert E.M., Sandoval A., Larrabee P., et al. Lisinopril lowers cardiac adrenergic drive and increases beta-receptor density in the failing human heart. Circulation. 1993;88:472-480.
111. Kasama S., Toyama T., Kumakura H., et al. Effects of candesartan on cardiac sympathetic nerve activity in patients with congestive heart failure and preserved left ventricular ejection fraction. J Am Coll Cardiol. 2005;45:661-667.
112. Kasama S., Toyama T., Kumakura H., et al. Effect of spironolactone on cardiac sympathetic nerve activity and left ventricular remodeling in patients with dilated cardiomyopathy. J Am Coll Cardiol. 2003;41:574-581.
113. Francis G. Development of arrhythmias in the patient with congestive heart failure: pathophysiology, prevalence and prognosis. Am J Cardiol. 1986;57:3B-7B.
114. Packer M. Hemodynamic consequences of antiarrhythmic drug therapy in patients with chronic heart failure. J Cardiovasc Electrophysiol. 1991;2:S240-S247.
115. Toyama T., Hoshizaki H., Seki R., et al. Efficacy of amiodarone treatment on cardiac symptom, function and sympathetic nerve activity in patients with dilated cardiomyopathy: comparison with beta-blocker therapy. J Nucl Cardiol. 2004;11:134-141.
116. Tachikawa H., Kodama M., Watanabe K., et al. Amiodarone improves cardiac sympathetic nerve function to hold norepinephrine in the heart, prevents left ventricular remodeling, and improves cardiac function in rat dilated cardiomyopathy. Circulation. 2005;111:894-899.
117. Kasama S., Toyama T., Sumino H., et al. Prognostic value of serial cardiac 123I-MIBG imaging in patients with stabilized chronic heart failure and reduced left ventricular ejection fraction. J Nucl Med. 2008;49:907-914.
118. Mancini D. Surgically denervated cardiac transplant. Rewired or permanently unplugged. Circulation. 1997;96:6-8.
119. De Marco T., Dae M., Yuen -Green M.S., et al. Iodine-123 MIBG scintigraphic assessment of the transplanted human heart: evidence for late reinnervation. J Am Coll Cardiol. 1995;25:927-931.
120. Schwaiger M., Hutchins G.B., Kalff V. Evidence for regional catecholamine uptake and storage sites in the transplanted human heart by positron emission tomography. J Clin Invest. 1991;87:1681-1690.
121. Estorch M., Camprecios M., Flotats A., et al. Sympathetic reinnervation of cardiac allografts evaluated by 123I-MIBG imaging. J Nucl Med. 1999;40:911-916.
122. Bengel F.M., Ueberfuhr P., Hesse T., et al. Clinical determinants of ventricular sympathetic reinnervation after orthotopic heart transplantation. Circulation. 2002;106:831-835.
123. Kim S.J., Lee J.D., Ryu Y.H., et al. Evaluation of cardiac sympathetic neuronal integrity in diabetic patients using iodine-123 MIBG. Eur J Nucl Med. 1996;23:401-406.
124. Scognamiglio R., Avogaro A., Casara D., et al. Myocardial dysfunction and adrenergic cardiac innervation in patients with insulin-dependent diabetes. J Am Coll Cardiol. 1998;31:404-412.
125. Hattori N., Tamaki N., Hayashi T., et al. Regional abnormality of iodine-123-MIBG in diabetic hearts. J Nucl Med. 1996;37:1985-1990.
126. Bellavere F., Ferri M., Guarini L., et al. Prolonged QT period in diabetic autonomic neuropathy: a possible role in sudden cardiac death? Br Heart J. 1989;59:379-383.
127. Wei K., Dorian P., Newman D., Langer A. Association between QT dispersion and autonomic dysfunction in patients with diabetes mellitus. J Am Coll Cardiol. 1995;26:859-863.
128. Wakabayashi K., Takahashi H. Neuropathology of autonomic nervous system in Parkinson’s disease. Eur Neurol. 1997;38(Suppl 2):2-7.
129. Braune S., Reinhardt M., Schnitzer R., Riedel A., Lücking C.H. Cardiac uptake of [123I]MIBG separates Parkinson’s disease from multiple system atrophy. Neurology. 1999;53:1020-1025.
130. Yoshita M. Differentiation of idiopathic Parkinson’s disease from striatonigral degeneration and progressive supranuclear palsy using iodine-123 meta-iodobenzylguanidine myocardial scintigraphy. J Neurol Sci. 1998;155:60-67.
131. Takatsu H., Nishida H., Matsuo H., et al. Cardiac sympathetic denervation from the early stage of Parkinson’s disease: Clinical and experimental studies with radiolabeled MIBG. J Nucl Med. 2000;41:71-77.
132. Braune S. The role of cardiac metaiodobenzylguanidine uptake in the differential diagnosis of parkinsonian syndromes. Clin Auton Res. 2001;11:351-355.
133. Nagayama H., Hamamoto M., Ueda M., et al. Reliability of MIBG myocardial scintigraphy in the diagnosis of Parkinson’s disease. J Neurol Neurosurg Psychiatry. 2005;76:249-251.
134. Hirayama M., Hakusui S., Koike Y., et al. A scintigraphical qualitative analysis of peripheral vascular sympathetic function with meta-[123I]iodobenzylguanidine in neurological patients with autonomic failure. J Auton Nerv Syst. 1995;53:230-234.
135. Raffel D.M., Koeppe R.A., Little R., et al. PET measurement of cardiac and nigrostriatal denervation in parkinsonian syndromes. J Nucl Med. 2006;47:1769-1777.
136. Watanabe H., Ieda T., Katayama T., et al. Cardiac 123I-meta-iodobenzylguanidine (MIBG) uptake in dementia with Lewy bodies: comparison with Alzheimer’s disease. J Neurol Neurosurg Psychiatry. 2001;70:781-783.
137. Yoshita M., Taki J., Yokoyama K., et al. Value of 123I-MIBG radioactivity in the differential diagnosis of DLB from AD. Neurology. 2006;66:1850-1854.
138. Kashihara K., Ohno M., Kawada S., Okumura Y. Reduced cardiac uptake and enhanced washout of 123I-MIBG in pure autonomic failure occurs conjointly with Parkinson’s disease and dementia with Lewy bodies. J Nucl Med. 2006;47:1099-1101.
139. Estorch M., Camacho V., Paredes P., et al. Cardiac (123)I-metaiodobenzylguanidine imaging allows early identification of dementia with Lewy bodies during life. Eur J Nucl Med Mol Imaging. 2008. [Epub ahead of print]
140. Wakasugi S., Fischman A.J., Babich J.W., et al. Metaiodobenzylguanidine: evaluation of its potential as a tracer for monitoring doxorubicin cardiomyopathy. J Nucl Med. 1993;34:1283-1286.
141. Valdés Olmos R.A., ten Bokkel Huinink W.W., ten Hoeve R.F., et al. Assessment of anthracycline-related myocardial adrenergic derangement by 123I-MIBG scintigraphy. Eur J Cancer. 1995;31A:26-31.
142. Carrio I., Estorch M., Berna L., et al. 111In-antimyosin and 123I-MIBG studies in the early assessment of doxorubicin cardiotoxicity. J Nucl Med. 1995;36:2044-2049.
143. Estorch M., Carrió I., Mena E., et al. MIBG imaging and pharmacologic intervention: effect of a single dose of oral amitriptyline on regional cardiac MIBG uptake. Eur J Nucl Med Mol Imaging. 2004;31:1575-1580.