Chapter 42 Cardiac Neurotransmission Imaging
Positron Emission Tomography
THE CARDIAC AUTONOMIC NERVOUS SYSTEM
The autonomic nervous system, referred to as the visceral nervous system, consists of two divisions: sympathetic and parasympathetic innervation. The two systems differ in their major neurotransmitters—norepinephrine and acetylcholine—which define the stimulatory and inhibitory effects of each system.1 Sympathetic and parasympathetic innervation of the heart facilitates its electrophysiologic and hemodynamic adaptation to changing cardiovascular demands. Both sympathetic and parasympathetic tone control the rate of electrophysiologic stimulation and conduction, while contractile performance is primarily modulated by sympathetic neurotransmission. This functional characterization is reflected by the anatomic distribution of nerve fibers and terminals. Sympathetic fibers include multiple nerve endings that are filled with vesicles containing norepinephrine. They travel along epicardial vascular structures and penetrate into underlying myocardium much like coronary vessels. Based on tissue norepinephrine content, the mammalian heart is characterized by dense sympathetic innervation with a gradient from atria to base of the heart and from base to apex of the ventricles.2
In contrast to sympathetic nerve fibers, parasympathetic innervation is most prevalent in the atria, the atrioventricular (AV) node, and to a lesser degree within ventricular myocardium. Parasympathetic fibers in the ventricles appear to travel close to the endocardial surface, in contrast to sympathetic innervation. The enzyme choline acetyltransferase has been used as a reliable marker of cholinergic innervation.3 Choline acetyltransferase concentration is highest in the atria and decreases sharply in the right and left ventricular myocardium.
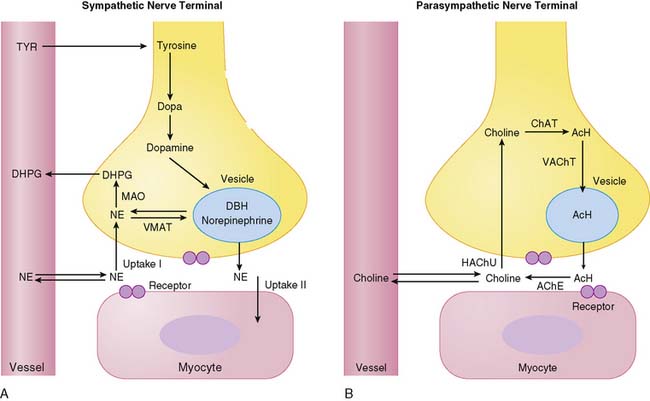
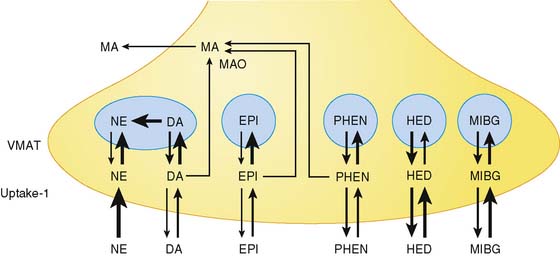
Figure 42-1 depicts neurotransmitter synthesis and turnover in the sympathetic and parasympathetic nerve terminal. Integrity and growth of both sympathetic and parasympathetic neurons are dependent on neurotrophins such as neuronal growth factor, which are secreted by myocardial target tissue and bind on nerve fibers via tyrosine kinase receptors, thereby inducing expression of growth-associated genes.4
Sympathetic Nerve Terminal
Norepinephrine, the dominant sympathetic transmitter, is synthesized from the amino acid tyrosine by several enzymatic steps. Generation of dopa from tyrosine is the rate-limiting step in catecholamine biosynthesis. Following dopa conversion to dopamine by dopa decarboxylase, dopamine is transported into storage vesicles by the energy-requiring vesicular monoamine transporter. Norepinephrine is synthesized by dopamine β-hydroxylase within storage vesicles. Nerve stimulation leads to norepinephrine release, which occurs as the vesicles fuse with the neuronal membrane and expel their content by exocytosis. Single nerve stimulation leads to exocytosis of only a small fraction of storage vesicles. The average adrenergic neuron has approximately 25,000 vesicles, with each containing approximately 250 pg of norepinephrine.5 Although most norepinephrine is thought to be released by exocytosis, nonvesicular release also occurs. Apart from neuronal stimulation, release is also regulated by a number of receptor systems. The α2-adrenergic receptors on the membrane surface provide negative feedback for exocytosis, which can thus be inhibited by presynaptic α2-agonists such as clonidine, guanabenz, and guanfacine.6 Muscarinic and adenosine receptors also have antiadrenergic effects. Neuropeptide Y is stored and released together with norepinephrine from the nerve terminal and is thought to inhibit neurotransmitter release.7 Presynaptic angiotensin II receptors and β-adrenergic receptors, on the other hand, mediate facilitation of norepinephrine release from sympathetic nerve endings so that antihypertensive agents such as angiotensin-converting enzyme (ACE) inhibitors or β-blockers can inhibit excessive norepinephrine release. The complex modulation of sympathetic neurotransmission6 obviously involves many systems, including dopamine, prostaglandin, and histamine. Only a small amount of norepinephrine release by the nerve terminal is actually available to activate receptors on the myocyte surface. Most norepinephrine undergoes reuptake into nerve terminals by the presynaptic norepinephrine transporter (uptake-1 mechanism) and recycles into vesicles or is metabolized in cytosol. The uptake-1 mechanism is characterized as saturable and sodium, energy, and temperature dependent.8 It can be inhibited by cocaine and desipramine.9 Structurally related amines such as epinephrine, guanethidine, and metaraminol are also transported by this system. In addition to neuronal uptake-1, the uptake-2 system removes norepinephrine into nonneuronal tissue.10 The uptake-2 mechanism is characterized as nonsaturable and not sodium, energy, or temperature dependent. It can be inhibited by, for example, steroids and clonidine.11 Free cytosolic norepinephrine is degraded rapidly to dihydroxyphenylglycol (DHPG) by monoamine oxidase (MAO). Only a small fraction of the released norepinephrine diffuses into vascular space, where it can be measured as norepinephrine spillover in coronary sinus vein blood.
Parasympathetic Nerve Terminal
Acetylcholine is synthesized within the parasympathetic nerve terminal. Choline is transported into the cytosol of nerve endings via a high-affinity choline uptake system,12 is then rapidly acetylated by choline acetyltransferase, and subsequently shuttled into storage vesicles. In contrast to amine uptake in sympathetic terminals, the choline uptake system is restrictive. Even close structural analogs are poor substrates for the high-affinity choline uptake system. Upon nerve stimulation, acetylcholine is released into the synaptic cleft, where it interacts with muscarinic receptors. Free acetylcholine is rapidly metabolized by acetylcholine esterase.
PET TRACERS FOR CARDIAC NEUROTRAMSMITTER IMAGING
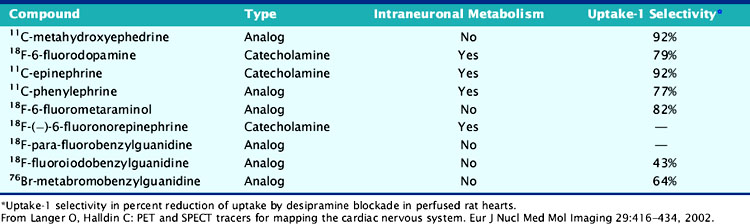
Available tracers of sympathetic innervation differ in their specificity for uptake-1 and vesicular storage. Additionally, their usefulness is defined by the low complexity of radiochemical synthesis and by the resulting specific radioactivity and chemical purity. Low specific activity and/or chemical impurity result in concomitant exposure to nonlabeled catecholamines during tracer injection, which may compete with the radiotracer for specific uptake and storage and increase the likelihood of pharmacologic adrenergic effects. Table 42-1 summarizes currently available PET tracers for imaging of presynaptic adrenergic innervation. Specific tracers, which differ in their kinetic behavior (Fig. 42-2),13 are described subsequently in an order following the amount of published clinical experience.
Carbon-11-Hydroxyephedrine
Carbon-11 (13C)-labeled metahydroxyephedrine (mHED, or simply HED) is the most frequently used PET tracer for mapping of sympathetic neurons. It is synthesized by N-methylation of metaraminol, which reliably yields HED at high specific activity.14 HED is resistant to metabolism by MAO or catechol-O-methyltransferase (COMT) and has a high affinity to uptake-1. Nonspecific myocardial uptake is low, as demonstrated in isolated perfused rat hearts following desipramine blockade15 and in denervated human hearts early after cardiac transplantation.16 Although vesicular storage seems to occur, binding inside vesicles is reduced, owing to the higher lipophilicity of HED compared to norepinephrine.17 Addition of desipramine or norepinephrine to the perfusate following application of HED in isolated rat hearts resulted in accelerated HED washout, suggesting that cardiac HED retention is dependent on reuptake by the norepinephrine transporter. HED is thus believed to undergo continuous release and reuptake by sympathetic neurons.15
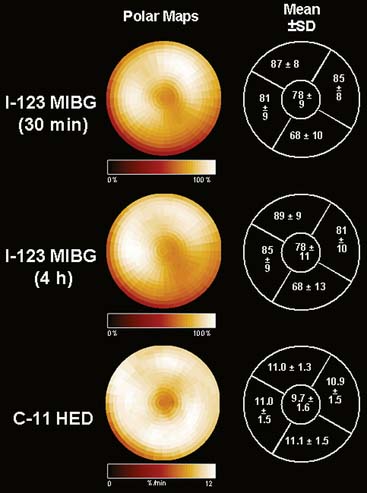
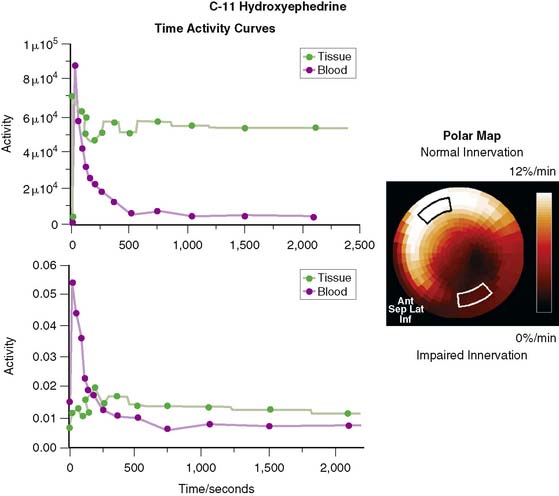
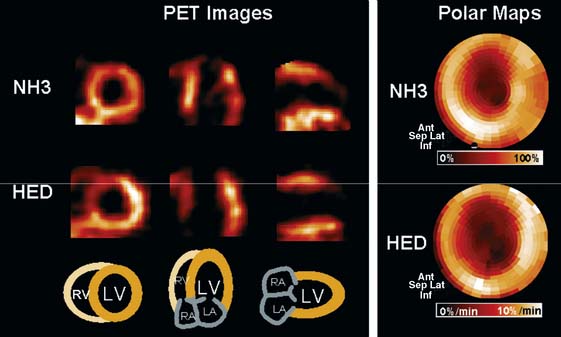
In contrast to single-photon emission computed tomography (SPECT) imaging using 123I-MIBG, where lower uptake in the inferior myocardium has been observed, distribution of HED throughout the left ventricular (LV) myocardium in healthy, normal individuals is regionally homogeneous, with high uptake in all myocardial segments (Fig. 42-3).16 For imaging, 400 to 700 MBq of 11C-HED are typically injected intravenously as a bolus, followed by a dynamic acquisition lasting 40 to 60 minutes. For quantification, a “retention index” is commonly calculated by dividing myocardial radioactivity concentration in the final image by the integral of the time-activity curve in arterial blood (which is derived from a region of interest in the LV chamber and needs correction for presence of radiolabeled plasma metabolites). Typical time-activity curves of dynamic HED PET studies are depicted in Figure 42-4. High neuronal affinity of HED makes compartmental modeling of its kinetics difficult, because in addition to norepinephrine transporter density, perfusion and transcapillary transport may significantly influence tracer uptake. An experimental, albeit complex, approach for truly quantitative kinetic modeling of HED has been introduced in the research setting18 but has not yet been transferred to human studies. In a rat model of varying degrees of cardiac sympathetic nerve denervation, semiquantitative measurement using HED retention was found to have a strong linear correlation with sympathetic nerve density, as reflected by cardiac norepinephrine transport (NET) density.19 Another experimental animal study aimed to investigate the feasibility of quantitation of 11C-HED PET retention in comparison to 123I-MIBG uptake by in vivo imaging. There was a close correlation between two quantitative parameters—myocardial 11C-HED retention fraction and the normalized 11C-HED activity (% ID/kg/g)—and 123I-MIBG uptake.20 These results would improve the understanding of the sensitivity of HED retention to denervation and aid in the interpretation of studies that use HED to investigate the effects of diseases on cardiac sympathetic innervation.
Hydroxyephedrine has been used in humans to identify alterations of sympathetic innervation in a number of pathophysiologic conditions, which are described in more detail later in this chapter. Additionally, HED has been used to determine effects of pharmacologic interventions on the sympathetic nervous system of the healthy heart. Prolonged impairments of myocardial catecholamine uptake following acute exposure to cocaine have been identified,21 and reduced myocardial HED retention was also observed in otherwise healthy chronic cocaine abusers.22 Further, the uptake-1 blocking effect of a novel antidepressant venlafaxine was characterized in a clinical trial in healthy volunteers.23
Fluorine-18-Fluorodopamine
Dopamine is a catecholamine that has high affinity for the dopamine transporter but similarly high affinity for the norepinephrine transporter. Fluorine substitution of dopamine has no significant effects on its biologic behavior,24 supporting the usefulness of fluorine-18 (18F)-fluorodopamine for evaluation of cardiac sympathetic nerves. For synthesis at sufficient specific activity, which is lower compared to, for example, 11C-HED, a complex no-carrier-added nucleophilic approach has been developed.25 Myocardial uptake of 18F-fluorodopamine occurs mainly via presynaptic uptake-1 and can be blocked by desipramine, as demonstrated in experimental26 and human studies.27 Following uptake into the neuron, fluorodopamine is sequestered into storage vesicles and β-hydroxylated to fluoronorepinephrine. Due to the half-life of 110 minutes for 18F, tracer clearance from the heart can be surveyed over a longer period than with 11C-labeled tracers (half-life, 20 minutes). Comparative studies of the kinetics of fluorodopamine and fluoronorepinephrine revealed a faster myocardial washout for fluorodopamine. These data suggested that myocardial radioactivity clearance after fluorodopamine injection can largely be attributed to inefficient β-hydroxylation and subsequent degradation by neuronal MAO.28 Additionally, rapid extraneuronal degradation has been observed. Soon after injection, radioactivity in blood corresponds mainly to radiolabeled metabolites.29
Because it permits measurement of tracer uptake and clearance, 18F-fluorodopamine has been proposed as a tracer of cardiac sympathetic innervation and function.26 It is the second tracer besides 11C-HED that has been used in a number of human studies to characterize cardiac innervation in disease conditions. For imaging, typically 35 to 140 MBq of 18F-fluorodopamine will be slowly infused over 3 minutes to avoid hemodynamic effects, then followed by dynamic PET acquisition over up to 3 hours. Because of the complex metabolic fate in myocardium and plasma, which renders quantitative kinetic modeling difficult, myocardial uptake and washout have mainly been described by basic calculations of regional radioactivity normalized to injected dose.
Carbon-11-Epinephrine
Epinephrine serves to a small degree as a physiologic neurotransmitter, but it is primarily a circulating catecholamine that is produced together with norepinephrine by the adrenal medulla and other chromaffin tissues. Epinephrine is radiolabeled with 11C by N-methylation of norepinephrine at high yield and high specific activity.3011C-epinephrine is handled in the myocardium in a manner similar to norepinephrine. It has high affinity for uptake-1, and although it is vulnerable to MAO degradation, efficient vesicular intraneuronal storage causes very slow clearance of radioactivity from the heart. In isolated perfused rat hearts, addition of desipramine to the perfusate following 11C-epinephrine administration (desipramine chase) results in much less acceleration of tracer washout compared to 11C-HED,31 suggesting that 11C-epinephrine is a tracer of neuronal uptake and vesicular storage, whereas 11C-HED largely reflects uptake-1 only. Plasma metabolism of 11C-epinephrine is considerable and needs to be taken into account for calculation of quantitative retention indices. In humans, only one study exists, that demonstrated that metabolite-corrected myocardial retention of 11C-epinephrine in healthy subjects was higher than that of 11C-HED, whereas denervated transplant recipients showed low levels of retention.32 It has thus been concluded that 11C-epinephrine may be more sensitive to neuronal abnormalities and, because of its more physiologic behavior, may be more suitable for evaluation of the cardiac presynaptic sympathetic nervous system.
Carbon-11-Phenylephrine
Phenylephrine is a sympathomimetic amine that is commonly used as a nasal decongestant. It can be labeled with carbon-11 at high specific activity.33 In contrast to HED and other catecholamine analogs used for PET imaging, phenylephrine is sensitive to MAO degradation. Studies in isolated perfused rat hearts have shown that 11C-phenylephrine is transported by uptake-1 at a slower rate than HED. Intraneuronally it is rapidly transported into storage vesicles, diffuses out of vesicles at a rate slower than HED, but is then metabolized by MAO in neuronal cytosol.34 Following injection of 11C-phenylephrine, there is thus considerable myocardial radioactivity washout, which is detectable during the imaging period (effective myocardial half-life approximately 60 minutes) and reflects MAO activity. 11C-phenylephrine may be combined with other short-lived neurotransmitter tracers such as 11C-HED and/or 11C-epinephrine to identify differential effects of disease on processes of norepinephrine uptake, vesicular storage, and metabolism in presynaptic adrenergic nerve terminals. Human experience with 11C-phenylephrine has been reported17 but remains limited.
Other Sympathoadrenergic Tracers
Similar to HED, metaraminol is a catecholamine analog that is resistant to metabolic degradation by MAO and COMT. Metaraminol can be fluorinated without altering the affinity to uptake-1. Myocardial accumulation in dogs correlated significantly with tissue norepinephrine content,35 and neuronal impairment induced by short-term ischemia was demonstrated.36 But the potent vasoactive properties of metaraminol and the limited specific activity following electrophilic labeling, which result in a mass of injected substance that is close to pharmacologic levels, have precluded clinical use of 18F-fluorometaraminol. Recently, however, efforts have been undertaken to develop alternative strategies for synthesis at higher specific activity,37 and the usefulness of fluorinated metaraminol may be reconsidered.
Efforts have also been made to label analogs of the SPECT tracer 123I-MIBG, with positron-emitting isotopes; 18F-parafluorobenzylguanidine, bromine-76 (76Br)-metabromobenzylguanidine, and 18F-fluoroiodobenzylguanidine have been synthesized successfully and tested in experimental settings.38–40 Kinetics of these tracers seemed to be comparable to those of 123I-MIBG, but affinity for uptake-1 was considerably lower than for classical PET tracers such as 11C-HED and 18F-fluorodopamine. Clinical applications of positron-emitting MIBG analogs for PET imaging have not yet emerged.
However, radiolabeled phenethylguanidines have recently been reinvestigated as a potential class of compounds with high vesicular retention. They are known to be even more potent depleters of cardiac norepinephrine stores than benzylguanidines, owing to their avid uptake and retention inside norepinephrine storage vesicles.41,42 For cardiac PET imaging studies, they can be labeled with 11C or 18F.42 Radiolabeling with 11C may offer some advantages over 18F in terms of being able to inject two to three times more activity into a patient because of 11C’s more favorable radiation dosimetry. This provides higher tissue and blood concentrations of the tracer early in the study when the kinetics of the tracer are changing most rapidly.42
Finally, the physiologic sympathetic neurotransmitter norepinephrine, has been fluorinated and used for PET imaging in baboons. It was shown that 18F-6-fluoronorepinephrine closely mimics the behavior of unlabeled norepinephrine.43 Synthesis yielded high specific activity, but experience remained limited to a few experimental studies, and the tracer has not yet entered human trials.
Tracers for Parasympathetic Innervation
Vesamicol is a compound that specifically binds to receptors on parasympathetic neuronal vesicles and inhibits storage of acetylcholine. Some vesamicol derivatives have been successfully labeled with positron-emitting radioisotopes, but most of them were tested for brain imaging; only 18F-fluoroethoxybenzovesamicol has been evaluated for cardiac imaging.44 In isolated perfused rat hearts, specific binding was low, consistent with low parasympathetic neuronal density, and nonspecific binding and washout were high. Additionally, there was considerable flow dependency of uptake, so that the usefulness of the tracer for clinical cardiac PET imaging was considered to be low.44
DISEASE-RELATED EXPERIMENTAL AND CLINICAL OBSERVATIONS
Coronary Artery Disease/Myocardial Infarction
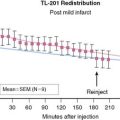
Myocardial ischemia and infarction result in nerve injury.45 The sensitivity of cardiac sympathetic nerve terminals to ischemia has been supported by early experimental observations in dogs, where short periods of coronary occlusion resulted in sustained reduction of cardiac uptake of 18F-metaraminol.36 A first study in patients early after myocardial infarction (MI) by Allman and colleagues showed that the area of reduced HED retention exceeded the perfusion defect—a finding that was especially pronounced in non-Q-wave infarcts.46 Image examples of a patient after MI are shown in Figure 42-5. The hypothesis of a higher sensitivity of sympathetic neurons to ischemia compared to cardiomyocytes is further supported by observations of regionally reduced HED retention in the absence of resting perfusion defects in patients with advanced coronary artery disease (CAD) but no evidence of MI.47 Defects in HED uptake were also demonstrated in the chronically ischemic, hibernating myocardium in a pig model.48 A recent study combined 11C-HED and 11C-CGP12177 (nonselective hydrophilic β-adrenoceptor antagonist) PET to assess presynaptic and postsynaptic sympathetic nerve functions in patients with hibernating myocardium, as assessed by magnetic resonance imaging before and after revascularization. There were significant generalized (rather than regional) reductions in norephineprine uptake-1 mechanism and β-adrenoceptor density in these patients, indicating that increased sympathetic activity is a generalized phenomenon in chronic left ventricular dysfunction associated with CAD, rather than directly related to hibernation.49 The dependency of myocardial sympathetic innervation on myocardial perfusion and coronary flow reserve (CFR) was evaluated in nondiabetic CAD patients using 11C-HED, aiming to find a relationship between the coronary vascular reactivity and uptake. The degree of 11C-HED retention in viable myocardium of CAD patients did not correlate significantly with myocardial rest perfusion over a large flow range, but there was a correlation between regional 11C-HED retention and CFR. This indicates that sympathetic innervation can be preserved even when there is major impairment of resting myocardial blood supply.50 Probably the occurrence of denervation depends on the duration and severity of ischemic episodes in the presence of reduced CFR.50 Notably, in a recent experimental study, selective regional sympathetic denervation of the left ventricle, which results in a profound reduction in 11C-HED volume of distribution, did not itself affect the baseline or hyperemic myocardial blood flow.51 Myocardial blood flow (MBF) and oxygen consumption are correlated in the normal heart, but it is unknown how this metabolism-perfusion relation is influenced by sympathetic denervation.52
Controversy exists about the occurrence of myocardial reinnervation following regional ischemic damage. Allman and colleagues observed no change of regional reduction of HED uptake until 6 months after acute infarction.46 Fallen et al., on the other hand, found a mild increase of initially impaired 18F-fluorodopamine uptake in the infarct area between 2 weeks and 3 months after the event that paralleled increased perfusion tracer uptake. They also observed a continuous reduction of fluorodopamine washout until 6 months after infarction, which was accompanied by electrophysiologic improvements of heart rate variability, and interpreted this as a partial restoration of sympathetic function.53
Whether the amount of sympathetic denervation in ischemic heart disease is linked with a higher incidence of ventricular arrhythmia and/or remodeling (and thus with an adverse outcome) needs to be evaluated in detail. PET may also be applied to describe effects of therapeutic approaches on sympathetic innervation in coronary disease. Al-Sheikh and coworkers, for example, evaluated patients with advanced coronary disease who were ineligible for percutaneous coronary angioplasty or bypass surgery before and after transmyocardial laser revascularization.54 No changes in resting and stress myocardial perfusion were observed after therapy, but epicardial induction of laser channels was associated with an increase of sympathetic denervation, identified by reduced HED retention, which may explain the relief of anginal symptoms in the absence of improved blood flow following therapy.54
Heart Failure/Cardiomyopathy
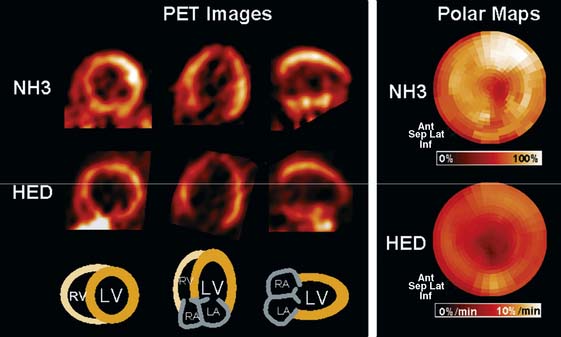
Involvement of the sympathetic nervous system in CHF is characterized by a vicious circle, where reduced cardiac output results in neurohumoral activation. The hyperadrenergic state in turn causes desensitization and down-regulation of cardiac β-adrenergic receptors and alterations of postsynaptic signal transduction, which further impair myocardial performance.55 Presynaptic cardiac sympathetic innervation is also involved in the pathophysiologic process and has been characterized in several studies using HED and PET. Hartmann et al. imaged 29 patients with dilated cardiomyopathy and moderate to severe heart failure.56 They found reduced global myocardial HED retention, which was regionally pronounced in apical and inferior segments and correlated with clinical markers of heart failure such as impaired ejection fraction and New York Heart Association (NYHA) class, as well as with elevation of plasma catecholamine levels.56 An example of HED findings in idiopathic dilated cardiomyopathy is depicted in Figure 42-6. Ungerer and coworkers then compared results of in vivo HED PET with ex vivo measurements in tissue of explanted hearts after transplantation in eight patients with terminal heart failure due to idiopathic dilated cardiomyopathy.57 Regional HED retention was significantly correlated with ex vivo tissue norepinephrine content and uptake-1 density, but not uptake-1 affinity. These data not only validated in vivo PET results but also suggested that either loss of neurons or down-regulation of uptake-1 occurs in dilated cardiomyopathy.57 At additional analysis of the same patients, regional in vivo HED uptake did not correlate with ex vivo–determined β-adrenergic receptor density, but rather with expression of the receptor-inactivating enzyme β-adrenoceptor kinase (β-ARK), giving further insights into the pathophysiology of idiopathic dilated cardiomyopathy.58 Using a combination of HED and 11C-CGP12177 PET imaging, it was shown that there was a mismatch between the presynaptic NE-1 mechanism and β-adrenergic receptor density in patients with ischemic heart failure but not in healthy controls.59,60
Although the pathogenetic mechanisms for altered presynaptic innervation in the failing cardiomyopathic heart are not yet fully elucidated, further consequences and correlates of impaired cardiac uptake-1 integrity have been identified with the help of quantitative neurotransmitter PET. In a study by Vesalainen et al. in 30 patients with mild to moderate heart failure, reduction in global myocardial HED uptake correlated with blunted vascular sympathetic effector responses, as measured by systolic pressure variability during posture-induced reflex activation and baroreflex control of heart rate, suggesting an interdependence between cardiac presynaptic innervation abnormalities and neural mechanisms important to blood pressure maintenance.61 In another study in dilated cardiomyopathy, alterations of myocardial HED retention were globally and regionally associated with impaired contractile function measured by gated blood-pool SPECT, suggesting a common pathogenetic pathway.62 Overall oxidative metabolism, determined by 11C-acetate PET, was not directly correlated with these findings,62 but a significant correlation between impaired presynaptic innervation and myocardial efficiency was identified in the failing heart.63 A recent study demonstrated a correlation between reduced HED retention and low fluorodeoxyglucose uptake in dysfunctional myocardium of patients with ischemic heart failure, suggesting a link between sympathetic nerve function and insulin resistance in the failing myocardium.64
Reduction of functioning local presynaptic catecholamine uptake sites, as demonstrated by neurotransmitter imaging, is likely to increase the myocardial overexposure to catecholamines and may facilitate progress and further deterioration of CHF. Therefore, it has been suggested that evaluation of alterations of the cardiac sympathetic innervations with noninvasive imaging methods might serve as a prognostic indicator.65,66 Maximization of the benefit of the available therapies for heart failure identification of patients at a high risk of death due to pump failure or sudden cardiac death is important.67–70 A prognostic value has been suggested for quantitative HED PET imaging in heart failure. Pietilä and colleagues found that reduced global HED retention was an independent predictor of adverse outcome in 46 patients with NYHA class II-III heart failure and an average ejection fraction of 35%.71 This observation also provides the background for using cardiac neuronal mapping as a surrogate end point to determine the effects of therapeutic approaches in the failing heart. In another study by Pietilä et al., effects of exercise training were determined in 13 heart failure patients.72 Serial measurements before and 6 months after start of the training program revealed improvement in baroreflex sensitivity and heart rate variability, which were associated with an increase of global HED retention, suggesting that physical training induces beneficial changes in cardiovascular autonomic nervous control and a shift toward normalization of the compensatory autonomic nervous imbalance.72 It has also been hypothesized that mismatch between presynaptic catecholamine uptake mechanism and postsynaptic receptor density might serve as an indicator of adverse outcomes.60 More studies are needed to determine the prognostic value of PET imaging of sympathetic nervous function in heart failure patients.
Hypertrophic cardiomyopathy is another entity of cardiac disease that may result in heart failure. Schäfers and colleagues studied alterations of the sympathetic nervous system in hypertrophic cardiomyopathy by combining HED for presynaptic assessment with the β-receptor ligand 11C-CGP12177 for postsynaptic PET measurements.73 They observed a global reduction of HED uptake as well as reduced binding of CGP12177, consistent with the hypothesis that myocardial β-receptor down-regulation is associated with impaired uptake-1 mechanism and hence increased local catecholamine levels.73 In another study, Li et al. performed regional analysis of 18F-fluorodopamine uptake in hypertrophied and nonhypertrophied regions of hypertrophic cardiomyopathy and found lower uptake in hypertrophied regions, while tracer washout was comparable.74 These data give further support for a role of the cardiac sympathetic nervous system to explain clinical features of hypertrophic cardiomyopathy.
Arrhythmogenic Disorders
Based on observations that stress and catecholamine infusion can induce ventricular arrhythmia in certain heart diseases, and based on experimental observations of supersensitivity in myocardial areas with impaired sympathetic neuronal function, a role for the cardiac sympathetic nervous system in the pathogenesis of arrhythmia has been proposed. Enhanced β-adrenoceptor stimulation is thought to be one of the possible mechanisms for lethal arrhythmias through their down-regulation and G-protein up-regulation, which leads to activation of adenylyl cyclase. This activation leads to an increased level of intracellular cyclic-AMP, inducing rise in intracellular calcium ion levels via protein kinase A.75 The heterogeneity of the local calcium transients in the myocardium may cause triggering of lethal arrhythmias by dispersion of local repolarization times.75 Also, the relationship between myocardial denervation and ventricular refractoriness has been investigated by Calkins and coworkers using 11C-HED PET and intraoperative electrophysiologic measurements during placement of an implantable defibrillator in 11 patients with a history of sustained ventricular tachycardia.76 The refractory period in areas with reduced HED retention was significantly longer than in areas with normal HED retention, documenting effects of innervation on myocardial electrophysiology and validating the usefulness of PET neurotransmitter imaging.76
Subsequently, specific arrhythmogenic diseases have been evaluated with regard to involvement of the sympathetic nervous system using PET. In nine patients with the familial long-QT syndrome, no abnormalities of global and regional ventricular HED retention could be identified by Calkins and coworkers.77 On the contrary, Mazzadi et al. recently studied 11 patients with genotyped long-QT syndrome and observed some regional reductions in HED retention using a model of 36 small segments for the LV myocardium.78 They speculated that sympathetic dysfunction could play an amplifier role for the severity of this disease, which is thought to be caused genetically by mutations in cardiac sodium and potassium channel genes.78 In right ventricular outflow tract tachycardia, Schäfers et al. used the β-receptor ligand 11C-CGP12177 and 11C-HED for combined presynaptic and postsynaptic sympathetic assessment of the left ventricle, observing a reduction of both.79 They concluded that myocardial β-adrenoceptor down-regulation occurs subsequent to increased local synaptic catecholamine levels caused by impaired catecholamine reuptake in this form of arrhythmogenic disease.79 Arrhythmogenic right ventricular cardiomyopathy was studied by the same group using the same technique. This time, only postsynaptic β-receptor density was significantly reduced, while presynaptic HED uptake tended toward reduction only. The authors speculated that a secondary β-receptor down-regulation may occur after increased local synaptic norepinephrine levels caused by increased firing rates of the efferent neurons or as result of impaired presynaptic catecholamine reuptake.80 Another lethal arrhythmogenic disorder is Brugada syndrome, which is characterized by ECG changes (right bundle branch block and persistent ST-segment elevation in leads V1-V3) and increased risk of sudden cardiac death without structural heart disease.81 Interestingly, ventricular tachyarrhythmias related to Brugada syndrome more often occur during rest or sleep when the vagal tone is predominant.81 Brugada syndrome was associated with an abnormal pattern of myocardial presynaptic recycling. Compared to the healthy subjects, nine patients with Brugada syndrome showed global increase in HED uptake but comparable β-adrenoceptor density measured with 11C-CGP12177.82 PET provided valuable quantitative measures to gain new insights into the pathophysiology of arrhythmogenic disorders, but their clinical relevance as of now remains unclear. It has been suggested that MIBG imaging may contribute to improved selection of patients for treatment of ventricular arrhythmias with an implantable cardioverter defibrillator (ICD).68 A potential prognostic value for alterations of the sympathetic nervous system as seen with PET remains to be evaluated in future studies, along with potential alleviating effects of therapeutic approaches. It is of note, for example, that the heart can be completely denervated, as found in transplant recipients early after surgery, without an increased incidence of arrhythmic events. This suggests that the entire pathophysiology of a specific disease needs to be taken into consideration for conclusions about the role of sympathetic innervation in arrhythmogenesis to be drawn.83
Some efforts have also been made to characterize innervation in supraventricular arrhythmia, although the atria are below the spatial resolution of PET scanners and cannot be imaged in vivo. Schmitt and colleagues used HED to document that radiofrequency catheter ablation, which is an effective treatment for the interruption of accessory bypass tracts in Wolff-Parkinson-White syndrome or for the modification of the AV nodal conduction system in patients with AV nodal tachycardias, does not result in impairments of ventricular sympathetic innervation.84 To study innervation of the atria, a dog model with regional phenol application for atrial denervation, followed by ex vivo PET imaging of excised atria after in vivo HED injection, was developed by Olgin et al.85 They observed regional reduction of HED uptake in phenolized segments, which was associated with increased refractory period and sustained atrial fibrillation, suggesting that heterogeneous atrial denervation creates a substrate for atrial fibrillation.85 In a follow-up study, rapid atrial pacing was performed in dogs to produce atrial fibrillation, and heterogeneity and an increase of atrial HED retention (determined at ex vivo imaging) were observed that paralleled disparate effects on atrial electrophysiology and gave further insights into the pathophysiology of atrial arrhythmia.86
Diabetes Mellitus
Diabetes mellitus may be complicated by neuropathy, often involving the autonomic nervous system. Cardiac autonomic neuropathy is traditionally determined by cardiovascular reflex testing. Using HED PET, Allman and coworkers first described a pattern of regional myocardial denervation characterizing cardiac diabetic neuropathy, which involved the apical, inferior, and lateral wall and correlated with conventional markers of autonomic dysfunction.87 Stevens et al. described the pattern of LV dysinnervation in more detail in 39 diabetic patients.88 They confirmed the regionally heterogeneous pattern of HED retention in cardiac diabetic neuropathy but also described proximal hyperinnervation complicating the presence of distal denervation. They speculated that this combination could result in potentially life-threatening myocardial electrical instability and could explain the enhanced cardioprotection from β-blockade in diabetic subjects.88 Serial assessment of the innervation pattern in diabetic subjects over a period of 3 years demonstrated that defects in LV innervation can regress or progress, depending on good or poor glycemic control, respectively.89 Additionally, observations in a rat model of streptozocin-induced diabetes not only reproduced the regional pattern of diabetic cardiac neuropathy observed in humans but also suggested a correlation between regional reduction of HED uptake and regionally reduced myocardial content of neuronal growth factor, which may thus contribute to the sympathetic neuronal impairment.90
Other studies have contributed to establishing a link between diabetic autonomic neuropathy and impairments of myocardial blood flow. Stevens and colleagues found significant reductions of global myocardial blood flow and flow reserve during adenosine-mediated vasodilation in neuropathic diabetic subjects compared to nonneuropathic diabetic and nondiabetic subjects.91 Interestingly, persistently innervated segments defined by HED PET showed the greatest impairments of regional myocardial flow.91 Di Carli et al., on the other hand, reported impaired flow response to adenosine in diabetic patients with and without PET-defined cardiac neuropathy compared to normals. The flow response to sympathetic stimulation by the endothelial-dependent cold pressor test was further reduced in diabetic patients with neuropathy compared to those without neuropathy and normal controls.92 Compared to diabetic patients without microangiopathy, diabetic patients with microangiopathy presenting as retinopathy, microalbuminuria, or early peripheral neuropathy showed more severe HED uptake reductions and, furthermore, these findings were associated with diastolic dysfunction and abnormal response of myocardial perfusion to adenosine and cold pressor test.93
These studies gave valuable insights into the pathophysiology of diabetic heart disease, but the relationship between myocardial neuropathy, flow regulation, arrhythmia, and outcome in diabetic subjects will need to be investigated in further trials to (1) determine whether neurotransmitter imaging gives incremental clinical information over conventional assessment of cardiac neuropathy and (2) provide the basis for specific pharmacologic interventions directed at individual risk reduction. Another study aimed at determining the effect of diabetes mellitus on the regenerational capacity of sympathetic neurons, using 11C-labeled physiologic neurotransmitter epinephrine PET imaging in transplanted heart. The regenerational capacity of the sympathetic nervous system of the heart was reduced but not abolished by diabetes mellitus, supporting the general link between impaired glucose handling and cardiac autonomic nerve function.94
Cardiac Transplantation
Due to transection of nerve fibers at surgery, the transplanted heart is initially completely denervated. This denervation is associated with hemodynamic alterations such as increased baseline heart rate, chronotropic incompetence during exercise, and reduced diastolic ventricular function. As a consequence, exercise capacity remains reduced compared to healthy, normal subjects.95
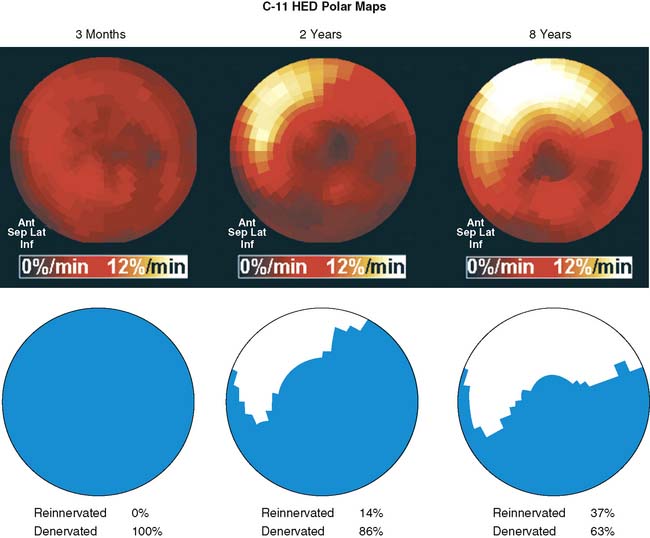
The first evidence of allograft reinnervation longer than 1 year after transplantation was derived from HED PET measurements, which showed reappearance of significant regional tracer retention in the basal anterior myocardium.96 Further studies have greatly improved the understanding of the reinnervation process. Serial assessment demonstrated a continuous increase of extent and intensity of reinnervation with time after transplantation. The area of innervation increased from the basal anterior and septal myocardium toward the apex and lateral wall, consistent with an ingrowth of nerve fibers into the territory of the left coronary artery.97Figure 42-7 shows examples of transplant reinnervation identified by HED PET. Complete restoration of sympathetic innervation was not observed until 15 years after transplantation, because inferior myocardium consistently remained denervated.97,98 Other studies have validated the results of HED PET by comparison with alternative tests of sympathetic innervation. PET-derived evidence of ventricular reinnervation correlated with invasive measurements of tyramine-induced coronary arteriovenous norepinephrine spillover99 and with electrophysiologic indexes of reinnervation derived from heart rate variability measurements.100,101 More recently, results in 77 transplant recipients were collected for a multivariate analysis, which revealed that reinnervation is determined by other factors, along with time after transplantation. Some patients remained denervated until late after transplantation. Other factors such as donor and recipient age, duration and complexity of transplant surgery, and frequency of allograft rejection were identified as independent determinants of sympathetic reinnervation.83
Because of heterogeneous reinnervation, the transplanted heart is a good model to determine the physiologic effects of sympathetic innervation in vivo by an intraindividual comparison of innervated and denervated myocardium. Di Carli and coworkers determined myocardial blood flow, flow response to the cold pressor test as an index of endothelial-dependent vasodilation, and flow response to adenosine as a composite index of endothelial-dependent and endothelial-independent vasodilation in nonrejecting, otherwise healthy reinnervated transplant recipients.102 They observed a significant improvement of flow response to cold pressor in innervated compared to denervated vascular territories, but there was no difference for the response to adenosine. These results demonstrated the importance of sympathetic innervation for regulation of endothelial-dependent vascular reactivity in general, and they also supported the physiologic relevance of reinnervation for transplant recipients.102 Other studies have focused on the effect of innervation on myocardial substrate utilization and have found higher utilization of glucose at equal rates of overall oxidative metabolism in denervated compared to reinnervated myocardium of allografts, suggesting a metabolic switch from free fatty acids to glucose under conditions of denervation.103 In another study, noninvasively determined allograft efficiency was shown to be improved in transplant recipients compared with failing hearts and was comparable to normal hearts. Differences between denervated and reinnervated allografts were not surveyed, and the dependency on loading conditions and contractility was preserved. These data suggested that normal regulatory interactions for efficiency are intact and that sympathetic tone does not play a role under resting conditions.104 Finally, the effect of reinnervation on exercise performance was determined in a group of 29 transplant recipients by HED PET and standardized exercise radionuclide angiography. Restoration of sympathetic innervation was associated with improved responses of heart rate and global as well as regional contractile function to exercise. In a recent study of Bengel et al., the increased cardiac performance after allograft cardiac transplantation was well correlated with cardiac sympathetic reinnervation as assessed using 11C-HED PET imaging. However, the beneficial effects of reinnervation on exercise capacity were abolished using acute, nonselective β-adrenergic blockade. These data suggested that β-adrenoceptors play a significant role in the functional reinnervation process; besides, the reappearance of sympathetic nerve terminals was associated with reestablishment of intact pre-/postsynaptic interaction.105 These results support the functional importance of reinnervation in transplanted hearts and suggest a clinical benefit for the transplant recipient through enhanced exercise capacity.106 It is thus worthwhile to develop approaches for supporting the process of allograft reinnervation in the future; these may be the subject of future research efforts.
Primary Neurologic Disorders
Several studies have focused on description of the involvement of cardiac innervation in primary neurologic disorders. The pure autonomic failure (PAF), Parkinson’s disease (PD), and multiple system atrophy (MSA) are the most common neurodegenerative diseases involving the central dopaminergic system as well as the cardiac and extracardiac sympathetic system.107 In idiopathic PD, this is thought to be caused primarily by postganglionic sympathetic dysfunction and in MSA by predominantly central and preganglionic degeneration. Using HED in a small group of seven patients, Berding et al. confirmed the latter hypothesis and found reduced myocardial catecholamine uptake in idiopathic PD, which was especially pronounced when the neurologic disease was accompanied by orthostatic hypotension.108 Using 18F-fluorodopamine, Goldstein and colleagues studied 41 patients with PD and observed globally reduced myocardial tracer uptake in most but not all PD patients with orthostatic hypotension, and regionally decreased uptake in 7 of 12 patients without orthostatic hypotension.109 Innervation was reduced not only in myocardium, but also in other tissues such as thyroid and renal cortex, suggesting that orthostatic hypotension reflects sympathetic neurocirculatory failure from generalized sympathetic denervation.109 A similar pattern was observed in a familial type of PD caused by mutation of the gene encoding leucine-rich repeat kinase 2.110 In another study by the same group, serial imaging over a period of 2 years confirmed progressive loss of myocardial innervation as part of the neurodegeneration of PD.111 Raffel et al. recently showed that cardiac sympathetic denervation was found to occur not only in idiopathic PD but also in other movement disorders, such as MSA and progressive supranuclear palsy. This finding implies that the scintigraphic detection of cardiac sympathetic denervation cannot be used independently to discriminate idiopathic PD from other movement disorders such as MSA and progressive supranuclear palsy.112
18F-fluorodopamine was also used to classify dysautonomias according to involvement of the cardiac sympathetic nervous system. In PAF there is an associated diffuse loss of sympathetic noradrenergic innervation, and despite the intact striatal dopaminergic neurons, there is sympathetic denervation in the cardiac and extracardiac organs.107 In one study, five patients with sympathetic neurocirculatory failure—based either on pure autonomic failure with no signs of central neurodegeneration or on classical parkinsonism—showed no myocardial 18F-fluorodopamine uptake, consistent with loss of myocardial sympathetic nerve terminals. In nine patients with sympathetic neurocirculatory failure based on the Shy-Drager syndrome (central neurodegeneration unresponsive to treatment with levodopa), increased levels of fluorodopamine uptake were identified, indicating intact sympathetic terminals and absent nerve traffic. Twelve patients with dysautonomia who did not have sympathetic neurocirculatory failure had normal levels of fluorodopamine uptake in myocardium. Based on PET and clinical observations, the authors proposed a new clinical pathophysiologic classification of dysautonomias.113 In another study, 18F-fluorodopamine was again utilized to characterize the involvement of myocardial innervation in postural tachycardia syndrome and repeated neurocardiogenic presyncope, specific diseases without persistent sympathetic neurocirculatory failure. No significant impairments compared to healthy controls were identified.114
SUMMARY AND FUTURE PERSPECTIVE
In summary, a variety of true and false neurotransmitters for the sympathetic nervous system, each with specific kinetics that reflect components of presynaptic innervation, can be labeled for cardiac PET imaging. However, all of these tracers share one common drawback: Their uptake rates into cardiac sympathetic neurons are too rapid to allow for robust and reliable compartmental modeling of their kinetics. A cardiac sympathetic nerve tracer would need two kinetic properties to be ideal for quantitative analyses: (1) a slower neuronal uptake rate than current tracers such as 11C-HED and (2) a very long neuronal retention time through trapping inside norepinephrine storage vesicles.42 The inability to use kinetic modeling methods limits the quantitative information that can be garnered from clinical studies with these tracers.42 Clinical experience is still mostly limited to the tracers 11C-HED and 18F-fluorodopamine, which have been used successfully to describe the involvement of cardiac adrenergic innervation in diseases such as ischemia, heart failure, cardiomyopathy, arrhythmia, diabetes, and other neuropathies. Using neurotransmitter PET imaging, knowledge of the physiologic and functional importance of myocardial innervation has been refined, and pathophysiologic correlates and consequences of altered innervation have been identified. For cardiac PET imaging studies, the radiolabeled phenethylguanidines were investigated as a potential class of compounds with high vesicular retention. In addition, phenethylguanidines have been shown to be more potent depleters of cardiac norepinephrine stores than benzylguanidines.42 These encouraging findings demonstrate that radiolabeled phenethylguanidines deserve further investigation as radiotracers of cardiac sympathetic innervation.
1. Randall W., Ardell J. Functional anatomy of the cardiac efferent innervation. In: Kulbertus H., Frank G., editors. Neurocardiology. New York: Futura Publishing; 1988:3-24.
2. Pierpont G.L., DeMaster E.G., Reynolds S., et al. Ventricular myocardial catecholamines in primates. J Lab Clin Med. 1985;106:205-210.
3. Schmid P.G., Greif B.J., Lund D.D., et al. Regional choline acetyltransferase activity in the guinea pig heart. Circ Res. 1975;36:547-552.
4. Thoenen H., Barde Y.A. Physiology of nerve growth factor. Physiol Rev. 1980;60:1284-1335.
5. Crout J. The uptake and release of 3H-epinephrine by the guinea pig heart in vivo. Naunyn Schmiedebergs Arch Pharmacol. 1964;248:85-92.
6. Francis G.S. Modulation of peripheral sympathetic nerve transmission. J Am Coll Cardiol. 1988;12:250-254.
7. Kilbourn M., Potter E., McCloskey D. Neuromodulation of the cardiac vagus: Comparison of neuropeptide Y and related peptides. Regul Pept. 1985;12:155-162.
8. Jaques S.Jr, Tobes M.C., Sisson J.C. Sodium dependency of uptake of norepinephrine and m-iodobenzylguanidine into cultured human pheochromocytoma cells: Evidence for uptake-one. Cancer Res. 1987;47:3920-3928.
9. Schomig A. Catecholamines in myocardial ischemia. Systemic and cardiac release. Circulation. 1990;82:II13-II22.
10. Russ H., Gliese M., Sonna J., et al. The extraneuronal transport mechanism for norepinephrine (uptake2) avidly transports 1-methyl-4-phenylpyridinium (MPP+). Naunyn Schmiedebergs Arch Pharmacol. 1992;346:158-165.
11. Salt P.J. Inhibition of norepinephrine uptake 2 in the isolated rat heart by steroids, clonidine and methoxylated phenylethylamines. Eur J Pharmacol. 1972;20:329-340.
12. Ducis I. The high-affinity choline uptake system. In: Whittaker V., editor. The cholinergic synapse. New York: Springer; 1988:400-445.
13. Raffel D.M., Wieland D.M. Assessment of cardiac sympathetic nerve integrity with positron emission tomography. Nucl Med Biol. 2001;28:541-559.
14. Rosenspire K.C., Haka M.S., Van Dort M.E., et al. Synthesis and preliminary evaluation of carbon-11-meta-hydroxyephedrine: A false transmitter agent for heart neuronal imaging. J Nucl Med. 1990;31:1328-1334.
15. DeGrado T.R., Hutchins G.D., Toorongian S.A., et al. Myocardial kinetics of carbon-11-meta-hydroxyephedrine: Retention mechanisms and effects of norepinephrine. J Nucl Med. 1993;34:1287-1293.
16. Schwaiger M., Kalff V., Rosenspire K., et al. Noninvasive evaluation of sympathetic nervous system in human heart by positron emission tomography. Circulation. 1990;82:457-464.
17. Raffel D.M., Corbett J.R., del Rosario R.B., et al. Clinical evaluation of carbon-11-phenylephrine: MAO-sensitive marker of cardiac sympathetic neurons. J Nucl Med. 1996;37:1923-1931.
18. Caldwell J.H., Kroll K., Li Z., et al. Quantitation of presynaptic cardiac sympathetic function with carbon-11-meta-hydroxyephedrine. J Nucl Med. 1998;39:1327-1334.
19. Raffel D.M., Chen W., Sherman P.S., et al. Dependence of cardiac 11C-meta-hydroxyephedrine retention on norepinephrine transporter density. J Nucl Med. 2006;47:1490-1496.
20. Nomura1 Y., Matsunari I., Takamatsu H., et al. Quantitation of cardiac sympathetic innervation in rabbits using 11C-hydroxyephedrine PET: relation to 123I-MIBG uptake. Eur J Nucl Med Mol Imaging. 2006;33:871-878.
21. Melon P.G., Nguyen N., DeGrado T.R., et al. Imaging of cardiac neuronal function after cocaine exposure using carbon-11 hydroxyephedrine and positron emission tomography. J Am Coll Cardiol. 1994;23:1693-1699.
22. Melon P.G., Boyd C.J., McVey S., et al. Effects of active chronic cocaine use on cardiac sympathetic neuronal function assessed by carbon-11-hydroxyephedrine. J Nucl Med. 1997;38:451-456.
23. Melichar J.K., Haida A., Rhodes C., et al. Venlafaxine occupation at the norepinephrine reuptake site: In vivo determination in healthy volunteers. J Psychopharmacol. 2001;15:9-12.
24. Langer O., Halldin C. PET and SPET tracers for mapping the cardiac nervous system. Eur J Nucl Med Mol Imaging. 2002;29:416-434.
25. Ding Y.S., Fowler J.S., Gatley S.J., et al. Synthesis of high specific activity 6-[18F]fluorodopamine for positron emission tomography studies of sympathetic nervous tissue. J Med Chem. 1991;34:861-863.
26. Goldstein D.S., Chang P.C., Eisenhofer G., et al. Positron emission tomographic imaging of cardiac sympathetic innervation and function. Circulation. 1990;81:1606-1621.
27. Goldstein D.S., Eisenhofer G., Dunn B.B., et al. Positron emission tomographic imaging of cardiac sympathetic innervation using 6-[18F]fluorodopamine: Initial findings in humans. J Am Coll Cardiol. 1993;22:1961-1971.
28. Ding Y.S., Fowler J.S., Gatley S.J., et al. Mechanistic positron emission tomography studies of 6-[18F]fluorodopamine in living baboon heart: Selective imaging and control of radiotracer metabolism using the deuterium isotope effect. J Neurochem. 1995;65:682-690.
29. Goldstein D.S., Holmes C. Metabolic fate of the sympathoneural imaging agent 6-[18F]fluorodopamine in humans. Clin Exp Hypertens. 1997;19:155-161.
30. Chakraborty P.K., Gildersleeve D.L., Jewett D.M., et al. High yield synthesis of high specific activity R-[11C]epinephrine for routine PET studies in humans. Nucl Med Biol. 1993;20:939-944.
31. Nguyen N.T., DeGrado T.R., Chakraborty P., et al. Myocardial kinetics of carbon-11-epinephrine in the isolated working rat heart. J Nucl Med. 1997;38:780-785.
32. Munch G., Nguyen N.T., Nekolla S., et al. Evaluation of sympathetic nerve terminals with [11C]epinephrine and [11C]hydroxyephedrine and positron emission tomography. Circulation. 2000;101:516-523.
33. Del Rosario R.B., Jung Y.W., Caraher J., et al. Synthesis and preliminary evaluation of [11C]-phenylephrine as a functional heart neuronal PET agent. Nucl Med Biol. 1996;23:611-616.
34. Raffel D.M., Wieland D.M. Influence of vesicular storage and monoamine oxidase activity on [11C]phenylephrine kinetics: Studies in isolated rat heart. J Nucl Med. 1999;40:323-330.
35. Wieland D.M., Rosenspire K.C., Hutchins G.D., et al. Neuronal mapping of the heart with 6-[18F]fluorometaraminol. J Med Chem. 1990;33:956-964.
36. Schwaiger M., Guibourg H., Rosenspire K., et al. Effect of regional myocardial ischemia on sympathetic nervous system as assessed by fluorine-18-metaraminol. J Nucl Med. 1990;31:1352-1357.
37. Langer O., Dolle F., Valette H., et al. Synthesis of high-specific-radioactivity 4- and 6-[18F]fluorometaraminol-PET tracers for the adrenergic nervous system of the heart. Bioorg Med Chem. 2001;9:677-694.
38. Vaidyanathan G., Affleck D.J., Zalutsky M.R. Validation of 4-[fluorine-18]fluoro-3-iodobenzylguanidine as a positron-emitting analog of MIBG. J Nucl Med. 1995;36:644-650.
39. Raffel D., Loc’h C., Mardon K., et al. Kinetics of the norepinephrine analog [76Br]-meta-bromobenzylguanidine in isolated working rat heart. Nucl Med Biol. 1998;25:1-16.
40. Berry C.R., Garg P.K., Zalutsky M.R., et al. Uptake and retention kinetics of para-fluorine-18-fluorobenzylguanidine in isolated rat heart. J Nucl Med. 1996;37:2011-2016.
41. Green A.L., Fielden R., Bartlett D.C., et al. New norepinephrine-depleting agents. β-Hydroxyphenethylguanidine and related compounds. J Med Chem. 1967;10:1006-1008.
42. Raffel D.M., Jung Y.W., Gildersleeve D.L., et al. Radiolabeled phenethylguanidines: Novel imaging agents for cardiac sympathetic neurons and adrenergic tumors. J Med Chem. 2007;50:2078-2088.
43. Ding Y.S., Fowler J.S., Dewey S.L., et al. Comparison of high specific activity and 6-[18F]fluoronorepinephrine and 6-[18F]fluorodopamine in baboons: Heart uptake, metabolism and the effect of desipramine. J Nucl Med. 1993;34:619-629.
44. DeGrado T.R., Mulholland G.K., Wieland D.M., et al. Evaluation of [18F]fluoroethoxybenzovesamicol as a new PET tracer of cholinergic neurons of the heart. Nucl Med Biol. 1994;21:189-195.
45. Lautamäki R., Tipre D., Bengel F. Cardiac sympathetic neuronal imaging using PET. Eur J Nucl Med Mol Imaging. 2007;34:S74-S85.
46. Allman K.C., Wieland D.M., Muzik O., et al. Carbon-11 hydroxyephedrine with positron emission tomography for serial assessment of cardiac adrenergic neuronal function after acute myocardial infarction in humans. J Am Coll Cardiol. 1993;22:368-375.
47. Bulow H.P., Stahl F., Lauer B., et al. Alterations of myocardial presynaptic sympathetic innervation in patients with multi-vessel coronary artery disease but without history of myocardial infarction. Nucl Med Commun. 2003;24:233-239.
48. Luisi A.J., Suzuki G., deKemp R., et al. Regional 11C-Hydroxyephedrine Retention in Hibernating Myocardium: Chronic Inhomogeneity of Sympathetic Innervation in the Absence of Infarction. J Nucl Med. 2005;46:1368-1374.
49. John A.S., Mongillo M., Depre C., et al. Pre- and post-synaptic sympathetic function in human hibernating myocardium. Eur J Nucl Med Mol Imaging. 2007;34:1973-1980.
50. Fricke1 E., Fricke1 H., Eckert S., et al. Myocardial sympathetic innervation in patients with chronic coronary artery disease: Is reduction in coronary flow reserve correlated with sympathetic denervation? Eur J Nucl Med Mol Imaging. 2007;34:206-211.
51. Rimoldi O.E., Drake-Holland A.J., Noble M.I.M., et al. Basal and hyperaemic myocardial blood flow in regionally denervated canine hearts: An in vivo study with positron emission tomography. Eur J Nucl Med Mol Imaging. 2007;34:197-205.
52. Alders D.J.C., Cornelussen R.N., Prinzen F.W., et al. Heart/cardiac muscle: Regional sympathetic denervation affects the relation between canine local myocardial blood flow and oxygen consumption. Exp Physiol. 2007;92:541-548.
53. Fallen E.L., Coates G., Nahmias C., et al. Recovery rates of regional sympathetic reinnervation and myocardial blood flow after acute myocardial infarction. Am Heart J. 1999;137:863-869.
54. Al-Sheikh T., Allen K.B., Straka S.P., et al. Cardiac sympathetic denervation after transmyocardial laser revascularization. Circulation. 1999;100:135-140.
55. Bristow M.R. The autonomic nervous system in heart failure. N Engl J Med. 1984;311:850-851.
56. Hartmann F., Ziegler S., Nekolla S., et al. Regional patterns of myocardial sympathetic denervation in dilated cardiomyopathy: An analysis using carbon-11 hydroxyephedrine and positron emission tomography. Heart. 1999;81:262-270.
57. Ungerer M., Hartmann F., Karoglan M., et al. Regional in vivo and in vitro characterization of autonomic innervation in cardiomyopathic human heart. Circulation. 1998;97:174-180.
58. Ungerer M., Weig H.J., Kubert S., et al. Regional pre- and postsynaptic sympathetic system in the failing human heart-regulation of beta ARK-1. Eur J Heart Fail. 2000;2:23-31.
59. Link J.M., Stratton J.R., Levy W., et al. PET measures of pre- and post-synaptic cardiac beta adrenergic function. Nucl Med Biol. 2003;30(8):795-803.
60. Caldwell J.H., Link J.M., Levy W.C., et al. Evidence for pre- to postsynaptic mismatch of the cardiac sympathetic nervous system in ischemic congestive heart failure. J Nucl Med. 2008;49:234-241.
61. Vesalainen R.K., Pietilä M., Tahvanainen K.U., et al. Cardiac positron emission tomography imaging with [11C]hydroxyephedrine, a specific tracer for sympathetic nerve endings, and its functional correlates in congestive heart failure. Am J Cardiol. 1999;84:568-574.
62. Bengel F.M., Permanetter B., Ungerer M., et al. Relationship between altered sympathetic innervation, oxidative metabolism and contractile function in the cardiomyopathic human heart: A noninvasive study using positron emission tomography. Eur Heart J. 2001;22:1594-1600.
63. Bengel F.M., Permanetter B., Ungerer M., et al. Alterations of the sympathetic nervous system and metabolic performance of the cardiomyopathic heart. Eur J Nucl Med Mol Imaging. 2002;29:198-202.
64. Mongillo M., John A.S., Leccisotti L., et al. Myocardial pre-synaptic sympathetic function correlates with glucose uptake in the failing human heart. Eur J Nucl Med Mol Imaging. 2007;34:1172-1177.
65. Higuchi T., Schwaiger M. Noninvasive imaging of heart failure: Neuronal dysfunction and risk stratification. Heart Failure Clin. 2006;2:193-204.
66. Lee D.S., Tu J.V., Juurlink D.N., et al. Risk-treatment mismatch in the pharmacotherapy of heart failure. JAMA. 2005;294(10):1240-1247.
67. Cesario D.A., Dec G.W. Implantable cardioverter-defibrillator therapy in clinical practice. J Am Coll Cardiol. 2006;47:1507-1517.
68. Nagahara D., Nakata T., Hashimoto A. Predicting the need for an implantable cardioverter-defibrillator using cardiac metaiodobenzylguanidine activity together with plasma natriuretic peptide concentration or left ventricular function. J Nucl Med. 2008;49:225-233.
69. Tomaselli G.F., Zipes D.P. What causes sudden death in heart failure? Circ Res. 2004;95(8):754-763.
70. Rubart M., Zipes D.P. Mechanisms of sudden cardiac death. J Clin Invest. 2005;115:2305-2315.
71. Pietilä M., Malminiemi K., Ukkonen H., et al. Reduced myocardial carbon-11 hydroxyephedrine retention is associated with poor prognosis in chronic heart failure. Eur J Nucl Med. 2001;28:373-376.
72. Pietilä M., Malminiemi K., Vesalainen R., et al. Exercise training in chronic heart failure: Beneficial effects on cardiac 11C-hydroxyephedrine PET, autonomic nervous control, and ventricular repolarization. J Nucl Med. 2002;43:773-779.
73. Schäfers M., Dutka D., Rhodes C.G., et al. Myocardial presynaptic and postsynaptic autonomic dysfunction in hypertrophic cardiomyopathy. Circ Res. 1998;82:57-62.
74. Li S.T., Tack C.J., Fananapazir L., et al. Myocardial perfusion and sympathetic innervation in patients with hypertrophic cardiomyopathy. J Am Coll Cardiol. 2000;35:1867-1873.
75. Higuch T., Schwaiger M. Imaging cardiac neuronal function and dysfunction. Curr Cardiol Rep. 2006;8:131-138.
76. Calkins H., Allman K., Bolling S., et al. Correlation between scintigraphic evidence of regional sympathetic neuronal dysfunction and ventricular refractoriness in the human heart. Circulation. 1993;88:172-179.
77. Calkins H., Lehmann M.H., Allman K., et al. Scintigraphic pattern of regional cardiac sympathetic innervation in patients with familial long QT syndrome using positron emission tomography. Circulation. 1993;87:1616-1621.
78. Mazzadi A.N., Andre-Fouet X., Duisit J., et al. Heterogeneous cardiac retention of 11C-hydroxyephedrine in genotyped long QT patients. A potential amplifier role for severity of the disease. Am J Physiol Heart Circ Physiol. 2003;285:H1286-H1293.
79. Schäfers M., Lerch H., Wichter T., et al. Cardiac sympathetic innervation in patients with idiopathic right ventricular outflow tract tachycardia. J Am Coll Cardiol. 1998;32:181-186.
80. Wichter T., Schäfers M., Rhodes C.G., et al. Abnormalities of cardiac sympathetic innervation in arrhythmogenic right ventricular cardiomyopathy: Quantitative assessment of presynaptic norepinephrine reuptake and postsynaptic beta-adrenergic receptor density with positron emission tomography. Circulation. 2000;101:1552-1558.
81. Brugada J., Brugada R., Brugada P. Right bundle-branch block and ST-segment elevation in leads V1 through V3: A marker for sudden death in patients without demonstrable structural heart disease. Circulation. 1998;97(5):457-460.
82. Kies P., Wichter T., Schäfers M., et al. Abnormal myocardial presynaptic norepinephrine recycling in patients with Brugada syndrome. Circulation. 2004;110(19):3017-3022.
83. Bengel F.M., Ueberfuhr P., Hesse T., et al. Clinical determinants of ventricular sympathetic reinnervation after orthotopic heart transplantation. Circulation. 2002;106:831-835.
84. Schmitt C., Meyer C., Kosa I., et al. Does radiofrequency catheter ablation induce a deterioration in sympathetic innervation? A positron emission tomography study. Pacing Clin Electrophysiol. 1998;21:327-330.
85. Olgin J.E., Sih H.J., Hanish S., et al. Heterogeneous atrial denervation creates substrate for sustained atrial fibrillation. Circulation. 1998;98:2608-2614.
86. Jayachandran J.V., Sih H.J., Winkle W., et al. Atrial fibrillation produced by prolonged rapid atrial pacing is associated with heterogeneous changes in atrial sympathetic innervation. Circulation. 2000;101:1185-1191.
87. Allman K.C., Stevens M.J., Wieland D.M., et al. Noninvasive assessment of cardiac diabetic neuropathy by carbon-11 hydroxyephedrine and positron emission tomography. J Am Coll Cardiol. 1993;22:1425-1432.
88. Stevens M.J., Raffel D.M., Allman K.C., et al. Cardiac sympathetic dysinnervation in diabetes: Implications for enhanced cardiovascular risk. Circulation. 1998;98:961-968.
89. Stevens M.J., Raffel D.M., Allman K.C., et al. Regression and progression of cardiac sympathetic dysinnervation complicating diabetes: An assessment by C-11 hydroxyephedrine and positron emission tomography. Metabolism. 1999;48:92-101.
90. Schmid H., Forman L.A., Cao X., et al. Heterogeneous cardiac sympathetic denervation and decreased myocardial nerve growth factor in streptozotocin-induced diabetic rats: Implications for cardiac sympathetic dysinnervation complicating diabetes. Diabetes. 1999;48:603-608.
91. Stevens M.J., Dayanikli F., Raffel D.M., et al. Scintigraphic assessment of regionalized defects in myocardial sympathetic innervation and blood flow regulation in diabetic patients with autonomic neuropathy. J Am Coll Cardiol. 1998;31:1575-1584.
92. Di Carli M.F., Bianco-Batlles D., Landa M.E., et al. Effects of autonomic neuropathy on coronary blood flow in patients with diabetes mellitus. Circulation. 1999;100:813-819.
93. Pop-Busui R., Kirkwood I., Schmid H., et al. Sympathetic dysfunction in type-1 diabetes association with impaired myocardial blood flow reserve and diastolic dysfunction. J Am Coll Cardiol. 2004;44:2368-2374.
94. Bengel F.M., Ueberfuhr P., Schäfer D., et al. Effect of diabetes mellitus on sympathetic neuronal regeneration studied in the model of transplant reinnervation. J Nucl Med. 2006;47:1413-1419.
95. Mancini D. Surgically denervated cardiac transplant. Rewired or permanently unplugged. Circulation. 1997;96:6-8.
96. Schwaiger M., Hutchins G.D., Kalff V., et al. Evidence for regional catecholamine uptake and storage sites in the transplanted human heart by positron emission tomography. J Clin Invest. 1991;87:1681-1690.
97. Bengel F.M., Ueberfuhr P., Ziegler S.I., et al. Serial assessment of sympathetic reinnervation after orthotopic heart transplantation. A longitudinal study using PET and C-11 hydroxyephedrine. Circulation. 1999;99:1866-1871.
98. Uberfuhr P., Ziegler S., Schwaiblmair M., et al. Incomplete sympathetic reinnervation of the orthotopically transplanted human heart: Observation up to 13 years after heart transplantation. Eur J Cardiothorac Surg. 2000;17:161-168.
99. Odaka K., von Scheidt W., Ziegler S.I., et al. Reappearance of cardiac presynaptic sympathetic nerve terminals in the transplanted heart: Correlation between PET using 11C-hydroxyephedrine and invasively measured norepinephrine release. J Nucl Med. 2001;42:1011-1016.
100. Uberfuhr P., Frey A.W., Ziegler S., et al. Sympathetic reinnervation of sinus node and left ventricle after heart transplantation in humans: Regional differences assessed by heart rate variability and positron emission tomography. J Heart Lung Transplant. 2000;19:317-323.
101. Ziegler S.I., Frey A.W., Uberfuhr P., et al. Assessment of myocardial reinnervation in cardiac transplants by positron emission tomography: Functional significance tested by heart rate variability. Clin Sci (Lond). 1996;91(Suppl):126-128.
102. Di Carli M.F., Tobes M.C., Mangner T., et al. Effects of cardiac sympathetic innervation on coronary blood flow. N Engl J Med. 1997;336:1208-1215.
103. Bengel F.M., Ueberfuhr P., Ziegler S.I., et al. Noninvasive assessment of the effect of cardiac sympathetic innervation on metabolism of the human heart. Eur J Nucl Med. 2000;27:1650-1657.
104. Bengel F.M., Ueberfuhr P., Schiepel N., et al. Myocardial efficiency and sympathetic reinnervation after orthotopic heart transplantation: A noninvasive study with positron emission tomography. Circulation. 2001;103:1881-1886.
105. Bengel F.M., Ueberfuhr P., Karja J., et al. Sympathetic reinnervation, exercise performance and effects of β-adrenergic blockade in cardiac transplant recipients. Eur Heart J. 2004;25:1726-1733.
106. Bengel F.M., Ueberfuhr P., Schiepel N., et al. Effect of sympathetic reinnervation on cardiac performance after heart transplantation. N Engl J Med. 2001;345:731-738.
107. Tipre D.N., Goldstein D.S. Cardiac and extracardiac sympathetic denervation in Parkinson’s disease with orthostatic hypotension and in pure autonomic failure. J Nucl Med. 2005;46(11):1775-1781.
108. Berding G., Schrader C.H., Peschel T., et al. [ N-methyl11C]meta-hydroxyephedrine positron emission tomography in Parkinson’s disease and multiple system atrophy. Eur J Nucl Med Mol Imaging. 2003;30:127-131.
109. Goldstein D.S., Holmes C.S., Dendi R., et al. Orthostatic hypotension from sympathetic denervation in Parkinson’s disease. Neurology. 2002;58:1247-1255.
110. Goldstein D.S., Imrich R., Peckham E., et al. Neurocirculatory and nigrostriatal abnormalities in Parkinson disease from LRRK2 mutation. Neurology. 2007;69:1580-1584.
111. Li S.T., Dendi R., Holmes C., et al. Progressive loss of cardiac sympathetic innervation in Parkinson’s disease. Ann Neurol. 2002;52:220-223.
112. Raffel D.M., Koeppe R.A., Little R., et al. PET measurement of cardiac and nigrostriatal denervation in Parkinsonian syndromes. J Nucl Med. 2006;47(11):1769-1777.
113. Goldstein D.S., Imrich R., Peckham E., et al: Neurocirculatory and nigrostriatal abnormalities in Parkinson disease from LRRK2 mutation, Neurology, 69:1580-1584, 2007. Goldstein D.S., Holmes C., Cannon R.O.III, et al. Sympathetic cardioneuropathy in dysautonomias. N Engl J Med 336:696-702, 1997.
114. Goldstein D.S., Holmes C., Frank S.M., et al. Cardiac sympathetic dysautonomia in chronic orthostatic intolerance syndromes. Circulation. 2002;106:2358-2365.