[level-membership-for-cardiovascular-category]2
CARDIAC MUSCLE STRUCTURE AND FUNCTION
Cardiac muscle
In skeletal muscle, fibres can be activated individually or in groups to vary the strength of the contraction. In the heart, the coordinated contraction resulting from the spread of activity across the atria and the ventricles requires that the cardiac muscle cells are electrically connected through gap junctions. In skeletal muscle the strength of contraction of individual fibres can be varied by changing the frequency of action potentials, but this is not an option for the heart where rhythmic contractions involve all of the cardiac muscle cells. Instead, the strength of contraction of cardiac muscle is regulated, as in smooth muscle, by varying the intracellular calcium concentration during activation of the cells. This provides a target site for drugs which affect the strength of cardiac contraction and hence cardiac output. The actions of these ‘inotrope’ drugs are discussed in Chapter 4.
Structure of cardiac muscle
The fundamental contractile unit in both skeletal and cardiac muscle is the sarcomere (Fig. 2.1). These units are about 2 μm long and are defined at each end by the Z line which is formed from the protein α-actinin. Attached to the Z lines are the thin filaments made from F-actin which in turn consist of G-actin monomers joined together, in a structure sometimes said to resemble a helical string of beads, to form the thin filament (Fig. 2.2). These actin thin filaments are arranged in a parallel sandwich structure with the protein myosin (thick filament). Under polarized light microscopy the arrangement of the actin and myosin protein creates a striated (striped) appearance in which the A band is generated by the myosin filaments and the I band is composed mainly of actin (Fig. 2.1).
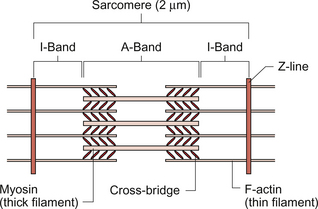
Parallel bundles of sarcomeres are joined end to end to make up a myofibril. The myocytes primarily contain bundles of myofibrils together with mitochondria and the cell nucleus, which is displaced to one side of the cell. Unlike skeletal muscle the myocytes of the atria and ventricles are not attached to specific skeletal insertion points by tendons but instead they are joined together in a branched meshwork to form a muscular bag in which the cells contract against their attachment to adjacent cells. Each individual myocyte in the adult human heart is about 50–100 μm in length and about 10–20 μm in diameter. In order to work as an effective contractile unit and to permit electrical activity to spread across heart muscle the individual muscle cells must be both physically joined together and electrically connected. This is achieved by the presence of ‘intercalated discs’—sites of apposition and thickening of the sarcolemma of adjacent cells. The intercalated discs contain both high conductance gap junctions (connexons) which provide for electrical continuity and desmosomes containing the protein cadherin to form a junction with adequate physical strength.
Contractile mechanism in cardiac muscle
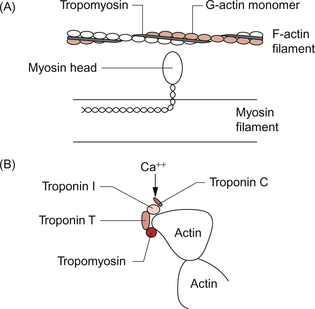
An increase in intracellular [Ca++] causes contraction of the myocardial cell by a sliding filament mechanism similar to that in skeletal muscle. The actin and myosin filaments pass over each other as a result of the breaking and reforming of cross-bridges between the filaments (Fig. 2.2). The cross-bridges are formed between the heads of the myosin-filaments and the actin filaments. When the [Ca++i] (the intracellular calcium ion concentration) rises the calcium ions bind to troponin C, a part of the three protein troponin complex. Troponin C is attached to tropomyosin, a protein which in the resting state shields a specific myosin binding region on the actin filament. The resulting ‘cross-bridge’ between myosin and actin undergoes a structural change which moves the actin filament over the myosin filament producing a small contraction. The myosin head then disengages and the process is repeated causing the actin to ‘walk’ down the myosin filament. The force of contraction depends on the number of cross-bridges formed, a parameter which in turn depends on the [Ca++] inside the muscle cell. Under resting conditions only a relatively small proportion of the potential total cross-bridge formation actually occurs. This means that physiological stimulation, via sympathetic nervous system activation, and drugs which increase intracellular [Ca++] can generate a more forceful cardiac muscle contraction than occurs at resting levels.
Each cycle of cross-bridge formation involves the hydrolysis of an ATP molecule to alter the configuration of the myosin head as part of the contraction process. Cardiac muscle cells are continually contracting and require substantial amounts of energy. Metabolically they are similar to ‘slow’ skeletal muscle fibres in that they derive their energy from ATP generated by oxidative phosphorylation and the myocytes thus contain large numbers of mitochondria. It must be appreciated that, like any muscle, the contraction of cardiac muscle, particularly in the left ventricle, impedes the flow of blood through the coronary blood vessels and thus the heart muscle is only effectively perfused during its relaxation (diastolic) phase (see Chapter 5). A period of powerful cardiac contractions at a rapid rate such as might occur during exercise may result in the oxygen supply to the myocardium being insufficient to meet the metabolic demand. Responses to exercise are discussed in Chapter 13.
Regulation of intracellular [Ca++] in cardiac muscle
As previously noted, the force of cardiac muscle contraction depends on the intracellular [Ca++]. Opposite each Z line there is a tubular structure, the T tubule, running at right angles to the plasma membrane of the cell (Fig. 2.3). The T tubules help to spread electrical excitation rapidly into the cell and they run close to the sarcoplasmic reticulum (SR) in which Ca++ ions are stored. Ca++ is pumped into the stores by using Ca++ ATPase pumps which are regulated by the inhibitory protein phospholamban. The Ca++ used to trigger contraction of cardiac muscle therefore comes from two sources, the SR (about 75% of the total) and also transmembrane flux of Ca++ from the extracellular fluid (about 25% of the total). This is in contrast to skeletal muscle which only uses SR stores of Ca++ for contraction.
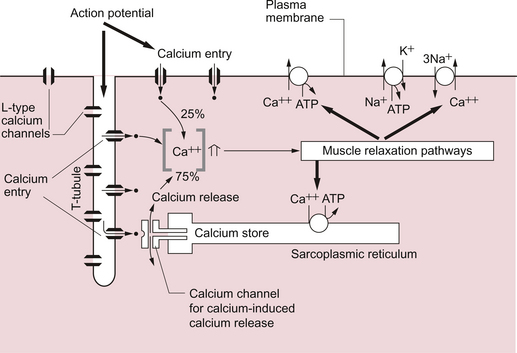
The resting intracellular [Ca++] is about 0.1 μmol/L and when an action potential (see p. 21) occurs in a cardiac muscle cell it triggers an initial increase in the intracellular calcium ion [Ca++i] concentration. The action potential results in an inward flow of calcium from the extracellular fluid where the ionized calcium concentration is about 1.2 mmol/L. This takes place through L-type calcium channels located in the T tubules and in the plasma membrane. The initial small increase in [Ca++i] causes the release of further calcium ions from the SR stores—the so-called calcium-induced calcium release. This is mediated by a Ca++ binding site on the SR which is part of a calcium channel protein often referred to as a ‘ryanodine-sensitive receptor’ or as a ‘foot protein’ (Fig. 2.3). As a result of calcium release from the SR the [Ca++i] increases, normally to about 0.5–2 μmol/L. In heart failure (see Chapter 6) there are significant alterations in how myocyte [Ca++] is regulated.
During relaxation some Ca++ has to be exported back out of the cell and some replaced into the SR. Ca++ is predominantly expelled from the myocyte via a 3Na+−Ca++ exchanger which uses the inward ‘downhill’ movement of the 3Na+ to move Ca++ out of the cell (Fig. 2.3). This mechanism per se does not consume ATP although the Na+/K+-ATPase actively expels Na+ across the plasma membrane in order to maintain the electrochemical gradient for Na+. A portion of the Ca++ is actively expelled from the cell across the plasma membrane by Ca ATPases. ATP is also used to pump Ca++ back into the SR stores. Within the SR much of the calcium is stored as ionized Ca++. However some is attached to calcium binding proteins of which calsequestrin is one of the most important.
Physiological stimulation of sympathetic nerves to the heart results in an increased force of contraction (see Chapter 4). The β1-adrenoceptor activation leads to a rise in intracellular cyclic AMP (see Fig. 4.6), a second messenger which activates several protein kinases. Subsequent phosphorylation of the protein phospholamban accelerates transport of Ca++ into the SR thus favouring retention of Ca++ in the SR at the expense of efflux back across the plasma membrane. Contractility of the heart is therefore increased by raising the amount of Ca++ stored in the SR. The rate of relaxation of cardiac muscle is also increased as the Ca++ re-enters the SR more quickly. The effects of cAMP in these events can be manipulated by drugs such as milrinone and caffeine which act as phosphodiesterase inhibitors and hence prolong the half-life of cAMP.
Cardiac electrical activity
Resting potential of ventricular muscle cells
The resting potential of a cardiac muscle cell is about −85 mV and, as in other excitable cells, this occurs as a result of the ionic concentration gradients maintained by the action of the Na+/K+-ATPase (see Chapter 1). The intracellular [K+i] is about 140 mmol/L whilst the extracellular [K+o] is about 4 mmol/L. We can consider a theoretical cell with such a concentration gradient for K+ and, initially, an equal number of positive and negative charges inside the cell. There is a diffusion gradient for positively charged potassium ions to move out of this theoretical cell and thus create a charge imbalance (potential difference) across the cell membrane with the inside of the cell negatively charged. The negative charge inside cells is mainly in the form of organic phosphates and ionizable groups on proteins, molecules which are too large to follow the K+ across the cell membrane. Eventually a situation is reached where the tendency for K+ ions to move out of the cell down the concentration gradient is balanced by the electrical gradient which will tend to move K+ ions back into the cell. This concept of the balance between the diffusive gradient and the electrical gradient is the basis for the derivation of the Nernst equation.
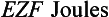
where E = the potential difference in volts, Z = the valency of the ion (i.e. 1, in the case of Na+ and K+) and F = the number of charges in one mole of ions (the Faraday).
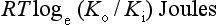
where R = the gas constant, T = temperature in degrees Kelvin, Ko = potassium concentration outside the cell and Ki = potassium concentration inside the cell.
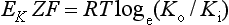
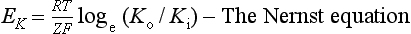
The equilibrium potential for K+ in cardiac muscle cells (−94 mV) is a little more negative than the actual resting potential (RP) of the cells which is about −85 mV.This is because the resting potential is also partly determined by the movement of ions other than K+. However because the membrane is relatively permeable to K+ and there is a substantial K+ gradient maintained by the Na+/K+-ATPase, potassium normally has the greatest influence on the magnitude of the resting potential. In practice the membrane is also a little permeable to Na+. It is possible to calculate the true resting potential by combining the concentration gradients of K+, Na+ and other ions weighted according to their relative permeabilities. Negatively charged ions have their concentration gradient reversed. The resulting equation is called the Goldman equation (see Box 2.1 for further explanation).
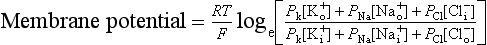
Cardiac action potential in ventricular muscle
The ventricular muscle action potential has five phases (Fig. 2.4). In phase 0 the sodium gates open and the permeability to sodium (PNa) increases about a hundred-fold which makes the sodium permeability much greater than that for other ions, including K+. As a result the membrane potential rises to between +20 and +30 mV, that is approaching the equilibrium potential for sodium (ENa) which is about +40 mV. At this point the sodium gates are ‘inactivated’ by the electric charge distribution across the cell membrane and the sodium permeability falls.
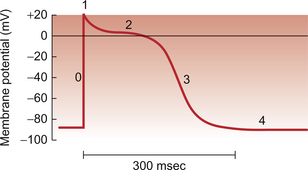
In phase 1 the K+ permeability (PK) begins to increase and K+ leaves the cell at an increased rate down both a favourable concentration and electrical gradient. However the membrane potential does not immediately fall to EK because there is a simultaneous opening of L-type voltage-gated Ca++ channels and an inward flow of Ca++ ions from outside the cell (phase 2). This calcium current results in a plateau phase of the membrane potential which lasts for as long as the calcium current flows. As described earlier, these events cause the release of a larger quantity of Ca++ from the sarcoplasmic reticulum which generates myocyte contraction (Fig. 2.3). Eventually the calcium channels are inactivated partly as a direct result of the rise in intracellular [Ca++]. The membrane potential, under the influence of increased K+ channel opening, falls (phase 3) to a value close to the potassium equilibrium potential (EK). At this stage the cycle recommences from the resting potential (phase 4).
As described, the Na+ channels in the muscle cell close at the peak of the action potential. They clearly have to be returned to a state where they can be stimulated to re-open before another action potential can be produced. The Na+ channels remain closed during the plateau phase of the action potential (Fig. 2.4) and stimulation of the muscle during this phase cannot produce a further action potential. This is the ‘absolute refractory’ period of the myocytes. During repolarization (phase 3) many, but not all, of the Na+ channels have re-opened by the time the membrane potential reaches −50 mV. Between −50 mV and complete repolarization a further action potential can be generated but this requires a greater than normal stimulation. This is the ‘relative refractory period’. This has an important consequence in clinical medicine. Hyperkalaemia, a rise in plasma [K+], is a frequent consequence of acidosis or inadequate excretion of K+ from the body, a process normally regulated by aldosterone effects on the kidney. Hyperkalaemia may become life threatening because it will lead to depolarization of cardiac myocytes, that is a rise in the resting potential towards zero. This will mean that return to a sufficiently low (negative) membrane potential to ensure opening of all populations of Na+ channels will not take place. Cardiac arrest may be the consequence.
Pacemaker tissue
Cardiac muscle differs from skeletal muscle and most neurones in that in some areas of the heart the ‘resting potential’ is particularly unstable. After an action potential in these areas the membrane potential gradually drifts upwards (depolarizes) until a threshold potential is reached where the opening of sodium ion channel gates is triggered and ion permeability rises rapidly producing another action potential. Cardiac muscle therefore has the property of producing rhythmic depolarizations which, in turn, result in rhythmic contractions. The mammalian heart is said to be capable of myogenic activity. This can be seen when a piece of cardiac muscle is removed from a living heart. If it is kept warm and oxygenated in an artificial extracellular fluid environment it will continue to contract and relax spontaneously for some time. The resting and action potentials generated in the sinoatrial node are illustrated in Fig. 2.5.

The rate of contraction of any given piece of muscle will depend upon the rate of depolarization of the resting potential in the muscle cells. The part of the heart with the fastest rate of drift of the resting potential will have the fastest intrinsic rhythm. In the human heart this is the sinoatrial node (SAN), which is a band of tissue in the right atrium close to the junction with the superior vena cava (Fig. 2.6). In the SAN the K+ permeability is lower and hence the initial resting potential (−60 mV) is less negative than elsewhere in the heart mainly because of the absence of one type of K+ channel (the ‘inward rectifier’ potassium channel). The membrane potential drifts upwards (depolarizes) faster than in other parts of the heart and is called a pacemaker potential. The ionic events which contribute to the pacemaker potential are complex and include inward movement of Na+ ions, an outward movement of K+ ions which decays with time and, once the pacemaker potential has depolarized past −55 mV, there is an inward Ca++ current. The latter ion movement accelerates the rate of depolarization towards the threshold potential of between −55 mV and −40 mV at which an action potential is triggered (Fig. 2.5). In the normal heart the SAN serves as the cardiac primary pacemaker and determines the rate at which the whole heart beats. The intrinsic rate of the human SAN is about 110–120 beats per minute (bpm) but at rest it is normally under tonic parasympathetic inhibition so that the resting heart rate is typically about 70 bpm.
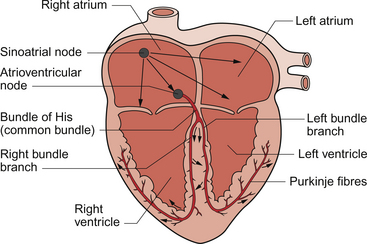
If, for some reason, the SAN fails to function or becomes electrically isolated from the rest of the heart then the area with the next fastest intrinsic rhythm is the atrioventricular node (AVN) which has an intrinsic rate of about 50 bpm. If the conduction of activity to the ventricle through the AVN is interrupted (‘complete heart block’) then the ventricles will beat at their own rate of about 30–40 bpm driven by the intrinsic depolarization rate of the Purkinje fibres.
The control of heart rate is discussed in Chapter 4. Sympathetic nerve stimulation at the SAN increases the rate of phase 4 depolarization and therefore the threshold potential is reached more quickly and heart rate increases. Parasympathetic nerve stimulation via the vagus nerve slows the heart rate by a combination of two mechanisms. The SAN cells are hyperpolarized and the rate of rise of the phase 4 resting potential is also reduced. Both of these effects mean that it takes longer for the resting potential to reach the threshold potential.
An outline of a case history of a child with a disturbance of the cardiac conduction system is described in Case 2.1:1.
The transmission of the cardiac action potential
The cardiac action potential normally originates at the SAN because it is the region of the heart with the fastest intrinsic contractile rate. The action potential then spreads across the left and right atria. The atrial action potential is similar in shape to that of the ventricle although the plateau phase is shorter. There is a region of fibrous tissue called the annulus fibrosis, part of the fibrous skeleton of the heart, which effectively provides an area of insulation between the atria and the ventricles so that excitation must normally pass through the AVN in order to reach the ventricles. If there is an electrical ‘leak’ in the ring of fibrous tissue separating the atria and the ventricles this will be an alternative, direct route for excitation to spread from atria to ventricles. When this occurs it is called the Wolff–Parkinson–White (WPW) or pre-excitation syndrome and it generates a characteristic arrhythmia (see Chapter 7).
The depolarization passes from the AVN into the ‘bundle of His’ (Fig. 2.6) which consists of specialized conducting tissue known as Purkinje fibres. These fibres are modified ventricular muscle cells and are grouped together in left and right bundles. They have a relatively large diameter and they are the largest of all the cells in the heart. This means they have a high conduction velocity. Purkinje fibres, together with the AVN tissue, have the longest refractory period of any cardiac cells. The functional importance of this is that is protects the heart against ‘re-entry’ excitation from adjacent myocytes back into the conducting tissue which could potentially spread back to the atria and cause dangerous arrhythmias. Other characteristics of the cells which comprise the conducting system are that although they are modified cardiac myocytes they have few myofibrils and so do not contract significantly when they are depolarized.
The Purkinje fibres conduct excitation quickly down each side of the septum before spreading out over the ventricles. As a result, depolarization of the ventricles occurs in a prescribed sequence starting with the papillary muscles and the septum and then spreading to the endocardial (inner) part of the ventricular muscle and out towards the epicardial (outer) surface. This coordinated spread of the electrical activity through the heart is responsible for the shape of the ECG (see Chapter 7). Repolarization of the muscle proceeds from epicardial to endocardial surface, the opposite direction to depolarization. This is the basis for the fact that the QRS complex (depolarization) and the T wave (repolarization) are both upwards deflections in a normal ECG, opposite polarity currents moving in opposite directions.
Drugs which act on the heart
The conducting pathway of the cardiac impulse and the electrical properties of the cardiac myocytes provide important therapeutic targets for drug actions. There are three main types of drugs which act upon the heart, those which are responsible for modifying heart rate, those which are used to regulate the rhythm of the heart and those which are used to regulate the force of cardiac contractions. The first group which broadly mimic or block the actions of the sympathetic and parasympathetic nerves controlling heart rate and the latter group (positive and negative inotropes) are discussed in Chapter 4.
Arrhythmias
The beating of the heart is normally driven by the spontaneous activity of the SAN and the heart is thus said to be in sinus rhythm. However there are occasions when this is not the case and an arrhythmia (the more scientifically rigorous term dysrhythmia is preferable but sadly it is not in widespread use) may be present. Arrhythmias may result from abnormal depolarization of cardiac tissue such that the cardiac rhythm is not being generated by the SAN or they may occur because cardiac excitation originates at the SAN but its conduction through the heart is abnormal (see Chapter 7).
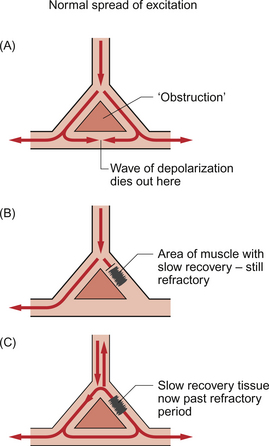
If a piece of cardiac muscle is damaged and unable to conduct activity then the wave of excitation must go around it much like a crowd of people leaving a football match might move to either side of a tree. If one pathway is much longer than the other then, when the activity rejoins the healthy tissue, it may do so after the impulse which took the shorter route has gone and the cells are past their refractory period and capable of being stimulated again. As a result an action potential will be set up which will be conducted in the conventional direction but also backwards (retrogradely) along the faster route (Fig. 2.7). This activity may collide with the next impulse passing down the faster route and negate it, or it may carry on around the loop producing what is called a re-entry arrhythmia. In circumstances where the ventricular rate is irregular, not all ventricular contractions may result in sufficient ejection of blood to allow a pulse to be palpated at, for example, the radial or brachial artery. As a result the pulse rate may not be the same as the ventricular rate recorded on an ECG.
On occasions, the ability of the wave of depolarization originating from the SAN to successfully pass through the AVN is impaired and this will result in various degrees of ‘heart block’. In first-degree heart block there is a prolongation of the P–R interval of the ECG to greater than 0.2 s (see Chapter 7) as transmission through the AVN is slowed. In second-degree heart block one may see a situation where not every wave originating at the SAN is able to pass through the AVN. This usually occurs in a repetitive manner such that perhaps every third or fourth atrial depolarization wave fails to produce ventricular activity. Missed beats are often accompanied by a progressive lengthening of the P–R interval in the two or three preceding beats. Finally, in complete heart block there is no synchronicity between atrial and ventricular activity at all. In patients with complete heart block atrial systole will occasionally coincide with ventricular systole and thus contraction of the right atrium is attempting to move blood against a closed tricuspid valve (see Chapter 3). When this occurs blood from the right atrium flows back up the neck in the jugular vein producing what are known as cannon waves.
The bundle of His has two main branches, right and left. Blockade of conduction through only one branch is called bundle branch block. This is further discussed in Chapter 7 in the context of the characteristic ECG patterns.
Antiarrhythmic drugs
There is a widely used classification of antiarrhythmic drugs—the Vaughan Williams classification.
Class I
These drugs act on phase 0 of the action potential and slow the rate of rise of the action potential by inhibiting fast sodium channels. Class I includes a range of drugs with differing actions on ion channels. They are subdivided into three categories on the basis of their effects on the duration of the action potential: 1a, increases duration (disopyramide, procainamide and quinidine); 1b, decreases duration (lidocaine (lignocaine) and mexiletine); 1c, has no effect on duration (flecainide and propafenone).
Bers, D. M. Altered cardiac myocyte Ca regulation in heart failure. Physiol.. 2006; 21:380–387.
DiFrancesco, D. Pacemaker mechanisms in cardiac tissue. Annu. Rev. Physiol.. 1993; 55:451–472.
Irisawa, H., Brown, H. F., Giles, W. Cardiac pacemaking in the sino atrial node. Physiol. Rev.. 1993; 73:197–227.
Katz, A. M. Physiology of the Heart, second ed. New York: Raven; 1992.
Levick, J. R. An Introduction to Cardiovascular Physiology, fifth ed. London: Arnold; 2009.
Noble, D. The Initiation of the Heart Beat. Oxford: Clarendon Press; 1979.
Sanguinetti, M. C., Keating, M. T. Role of delayed rectifier potassium channels in cardiac repolarization and arrhythmias. News Physiol. Sci.. 1997; 12:152–157.
Sommer, I. R., Johnson, E. A., Ultrastructure of cardiac muscleBerne, R. M., eds. Handbook of Physiology, Cardiovascular System, Vol 1. The Heart. American Physiological Society, Bethesda, 1979:113–186.
Waller, D. G., Renwick, A. G., Hillier, K. Medical Pharmacology and Therapeutics, third ed. Edinburgh: WB Saunders; 2009.
[/level-membership-for-cardiovascular-category][not-level-membership-for-cardiovascular-category]2
CARDIAC MUSCLE STRUCTURE AND FUNCTION
Cardiac muscle
In skeletal muscle, fibres can be activated individually or in groups to vary the strength of the contraction. In the heart, the coordinated contraction resulting from the spread of activity across the atria and the ventricles requires that the cardiac muscle cells are electrically connected through gap junctions. In skeletal muscle the strength of contraction of individual fibres can be varied by changing the frequency of action potentials, but this is not an option for the heart where rhythmic contractions involve all of the cardiac muscle cells. Instead, the strength of contraction of cardiac muscle is regulated, as in smooth muscle, by varying the intracellular calcium concentration during activation of the cells. This provides a target site for drugs which affect the strength of cardiac contraction and hence cardiac output. The actions of these ‘inotrope’ drugs are discussed in Chapter 4.
Structure of cardiac muscle
The fundamental contractile unit in both skeletal and cardiac muscle is the sarcomere (Fig. 2.1). These units are about 2 μm long and are defined at each end by the Z line which is formed from the protein α-actinin. Attached to the Z lines are the thin filaments made from F-actin which in turn consist of G-actin monomers joined together, in a structure sometimes said to resemble a helical string of beads, to form the thin filament (Fig. 2.2). These actin thin filaments are arranged in a parallel sandwich structure with the protein myosin (thick filament). Under polarized light microscopy the arrangement of the actin and myosin protein creates a striated (striped) appearance in which the A band is generated by the myosin filaments and the I band is composed mainly of actin (Fig. 2.1).
Parallel bundles of sarcomeres are joined end to end to make up a myofibril. The myocytes primarily contain bundles of myofibrils together with mitochondria and the cell nucleus, which is displaced to one side of the cell. Unlike skeletal muscle the myocytes of the atria and ventricles are not attached to specific skeletal insertion points by tendons but instead they are joined together in a branched meshwork to form a muscular bag in which the cells contract against their attachment to adjacent cells. Each individual myocyte in the adult human heart is about 50–100 μm in length and about 10–20 μm in diameter. In order to work as an effective contractile unit and to permit electrical activity to spread across heart muscle the individual muscle cells must be both physically joined together and electrically connected. This is achieved by the presence of ‘intercalated discs’—sites of apposition and thickening of the sarcolemma of adjacent cells. The intercalated discs contain both high conductance gap junctions (connexons) which provide for electrical continuity and desmosomes containing the protein cadherin to form a junction with adequate physical strength.
Contractile mechanism in cardiac muscle
An increase in intracellular [Ca++] causes contraction of the myocardial cell by a sliding filament mechanism similar to that in skeletal muscle. The actin and myosin filaments pass over each other as a result of the breaking and reforming of cross-bridges between the filaments (Fig. 2.2). The cross-bridges are formed between the heads of the myosin-filaments and the actin filaments. When the [Ca++i] (the intracellular calcium ion concentration) rises the calcium ions bind to troponin C, a part of the three protein troponin complex. Troponin C is attached to tropomyosin, a protein which in the resting state shields a specific myosin binding region on the actin filament. The resulting ‘cross-bridge’ between myosin and actin undergoes a structural change which moves the actin filament over the myosin filament producing a small contraction. The myosin head then disengages and the process is repeated causing the actin to ‘walk’ down the myosin filament. The force of contraction depends on the number of cross-bridges formed, a parameter which in turn depends on the [Ca++] inside the muscle cell. Under resting conditions only a relatively small proportion of the potential total cross-bridge formation actually occurs. This means that physiological stimulation, via sympathetic nervous system activation, and drugs which increase intracellular [Ca++] can generate a more forceful cardiac muscle contraction than occurs at resting levels.
Each cycle of cross-bridge formation involves the hydrolysis of an ATP molecule to alter the configuration of the myosin head as part of the contraction process. Cardiac muscle cells are continually contracting and require substantial amounts of energy. Metabolically they are similar to ‘slow’ skeletal muscle fibres in that they derive their energy from ATP generated by oxidative phosphorylation and the myocytes thus contain large numbers of mitochondria. It must be appreciated that, like any muscle, the contraction of cardiac muscle, particularly in the left ventricle, impedes the flow of blood through the coronary blood vessels and thus the heart muscle is only effectively perfused during its relaxation (diastolic) phase (see Chapter 5). A period of powerful cardiac contractions at a rapid rate such as might occur during exercise may result in the oxygen supply to the myocardium being insufficient to meet the metabolic demand. Responses to exercise are discussed in Chapter 13.
Regulation of intracellular [Ca++] in cardiac muscle
As previously noted, the force of cardiac muscle contraction depends on the intracellular [Ca++]. Opposite each Z line there is a tubular structure, the T tubule, running at right angles to the plasma membrane of the cell (Fig. 2.3). The T tubules help to spread electrical excitation rapidly into the cell and they run close to the sarcoplasmic reticulum (SR) in which Ca++ ions are stored. Ca++ is pumped into the stores by using Ca++ ATPase pumps which are regulated by the inhibitory protein phospholamban. The Ca++ used to trigger contraction of cardiac muscle therefore comes from two sources, the SR (about 75% of the total) and also transmembrane flux of Ca++ from the extracellular fluid (about 25% of the total). This is in contrast to skeletal muscle which only uses SR stores of Ca++ for contraction.
The resting intracellular [Ca++] is about 0.1 μmol/L and when an action potential (see p. 21) occurs in a cardiac muscle cell it triggers an initial increase in the intracellular calcium ion [Ca++i] concentration. The action potential results in an inward flow of calcium from the extracellular fluid where the ionized calcium concentration is about 1.2 mmol/L. This takes place through L-type calcium channels located in the T tubules and in the plasma membrane. The initial small increase in [Ca++i] causes the release of further calcium ions from the SR stores—the so-called calcium-induced calcium release. This is mediated by a Ca++ binding site on the SR which is part of a calcium channel protein often referred to as a ‘ryanodine-sensitive receptor’ or as a ‘foot protein’ (Fig. 2.3). As a result of calcium release from the SR the [Ca++i] increases, normally to about 0.5–2 μmol/L. In heart failure (see Chapter 6) there are significant alterations in how myocyte [Ca++] is regulated.
During relaxation some Ca++ has to be exported back out of the cell and some replaced into the SR. Ca++ is predominantly expelled from the myocyte via a 3Na+−Ca++ exchanger which uses the inward ‘downhill’ movement of the 3Na+ to move Ca++ out of the cell (Fig. 2.3). This mechanism per se does not consume ATP although the Na+/K+-ATPase actively expels Na+ across the plasma membrane in order to maintain the electrochemical gradient for Na+. A portion of the Ca++ is actively expelled from the cell across the plasma membrane by Ca ATPases. ATP is also used to pump Ca++ back into the SR stores. Within the SR much of the calcium is stored as ionized Ca++. However some is attached to calcium binding proteins of which calsequestrin is one of the most important.
Physiological stimulation of sympathetic nerves to the heart results in an increased force of contraction (see Chapter 4). The β1-adrenoceptor activation leads to a rise in intracellular cyclic AMP (see Fig. 4.6), a second messenger which activates several protein kinases. Subsequent phosphorylation of the protein phospholamban accelerates transport of Ca++ into the SR thus favouring retention of Ca++ in the SR at the expense of efflux back across the plasma membrane. Contractility of the heart is therefore increased by raising the amount of Ca++ stored in the SR. The rate of relaxation of cardiac muscle is also increased as the Ca++ re-enters the SR more quickly. The effects of cAMP in these events can be manipulated by drugs such as milrinone and caffeine which act as phosphodiesterase inhibitors and hence prolong the half-life of cAMP.
Cardiac electrical activity
Resting potential of ventricular muscle cells
The resting potential of a cardiac muscle cell is about −85 mV and, as in other excitable cells, this occurs as a result of the ionic concentration gradients maintained by the action of the Na+/K+-ATPase (see Chapter 1). The intracellular [K+i] is about 140 mmol/L whilst the extracellular [K+o] is about 4 mmol/L. We can consider a theoretical cell with such a concentration gradient for K+ and, initially, an equal number of positive and negative charges inside the cell. There is a diffusion gradient for positively charged potassium ions to move out of this theoretical cell and thus create a charge imbalance (potential difference) across the cell membrane with the inside of the cell negatively charged. The negative charge inside cells is mainly in the form of organic phosphates and ionizable groups on proteins, molecules which are too large to follow the K+ across the cell membrane. Eventually a situation is reached where the tendency for K+ ions to move out of the cell down the concentration gradient is balanced by the electrical gradient which will tend to move K+ ions back into the cell. This concept of the balance between the diffusive gradient and the electrical gradient is the basis for the derivation of the Nernst equation.
[/not-level-membership-for-cardiovascular-category]
