Cardiac Fibroblasts and Arrhythmogenesis
Cardiac Fibroblasts
Cardiomyocytes in the heart occupy approximately 75% of the myocardial volume, but the majority of cells by number are not muscular, including fibroblasts, endothelial cells, pericytes, smooth muscle cells, macrophages, and mast and dendritic cells. Cardiac fibroblasts, the most numerous of these non-muscular cells, comprise 30% to 65% of all the cells in the healthy adult heart, but only a minor fraction of the total heart volume.1 The number of fibroblasts in the heart is not constant, but dynamically changes during development and disease and with aging.2–4 Traditionally, cardiac fibroblasts have been considered as passive cells that primarily maintain the structural and mechanical integrity of the heart through the highly regulated synthesis and degradation of extracellular matrix (ECM) proteins including collagens (mainly type I and III) and fibronectin. Recently, however, a large body of evidence has emerged suggesting the important roles of these connective tissue cells in cardiac development, function, and pathology, including cardiac arrhythmias. Because cardiac fibroblasts are highly abundant and closely interlaced with other cell types in the heart (virtually every cardiomyocyte touches one or more fibroblasts), they are in a position to regulate actively and to modify heart function by direct contact with other cells and ECM, as well as through the secretion of different cytokines, ECM proteins, and proteases.
Fibroblast Origin
Cardiac fibroblasts are the only cell type in the heart not associated with the basement membrane. They are recognized as single-nucleated, spindle-shaped cells with multiple processes and prominent endoplasmic reticulum and Golgi apparatus. Fibroblasts reside in the self-secreted extracellular matrix arranged in sheets and strands that tightly envelop cardiomyocyte fibers.1,2 Their spatial distribution in the healthy heart is mainly uniform, with the highest density found in the sinoatrial (SA) node and annulus fibrosis, where these cells provide electrical insulation to enable successful pacemaking function and coordinated activation of atria and ventricles. Developmentally, cardiac fibroblasts are derived from mesenchymal cells of a proepicardial organ, which migrate over the heart surface to form the epicardium.2,5 Through epithelial-to-mesenchymal transition, the epicardium gives rise to epicardium-derived cells that migrate into the heart wall and gradually attain a fibroblast or smooth muscle phenotype. This process is regulated by the spatially and temporally coordinated expression of different growth factors, including platelet-derived growth factor (PDGF), fibroblast growth factors (FGFs) and transforming growth factor (TGF) β.6 During prenatal growth, the number of fibroblasts in the heart steadily increases. Shortly after birth, the proliferative capacity of cardiomyocytes ceases, while the fibroblast number abruptly increases (greater than twofold) for reasons that may be related to the postnatal increase in blood pressure (and associated mechanical strains in the heart wall), oxygen tension, or both.
Phenotypic Diversity and Lack of Specific Markers
Although there is significant knowledge of the role of cardiac fibroblasts in the heart, the molecular properties of these cells remain poorly characterized, mainly because of the lack of specific and ubiquitous markers of their phenotype.7 The markers currently used are either expressed in only a fraction of fibroblasts or also label other cells (Table 30-1). For example, immunostaining for vimentin has been used widely to label cardiac fibroblasts; however, this intracellular protein is expressed in other mesoderm-derived cells, including endothelial and smooth muscle cells. Similarly, Thy-1 (CD90), a surface marker that labels cardiac fibroblasts and other mesenchymal cells also labels endothelial cells. Discoidin domain receptor 2 (DDR2), a membrane collagen-binding tyrosine kinase receptor, is expressed in a subset of cardiac fibroblasts, but is also found in endothelial and smooth muscle cells. Fibroblast-specific protein-1 (FSP1, S100A4) is another commonly used fibroblast marker that is only sparsely expressed in the normal heart, but is significantly upregulated in heart disease, where it may also label smooth muscle cells.8 Recently, Acharya et al9 reported the generation of transgenic mice in which transcription factor Tcf21 (Epicardin/Pod1/Capsulin) was selectively expressed in a subset of epicardially derived cardiac fibroblasts, but also in a multitude of other tissues.9 In general, the best existing methods to identify and isolate cardiac fibroblasts rely on the labeling with multiple positive and negative markers, such as CD31–/CD90+/DDR2+ for live cell isolation,4 or von Willebrand factor–/smooth muscle actin (SMA)–/vimentin+/DDR2+ for immunolabeling.1,10 Although genetic fate mapping methods for in situ labeling of cardiac fibroblasts in mice are extremely valuable, they require a careful interpretation that accounts for the potential pitfalls of this system.5
Table 30-1
Fibroblast and Myofibroblast Markers
| Marker | Cellular Overlap |
| Colla1 | Various cells |
| CD40 | Various antigen presenting cells |
| CD248 (TEM1) | Pericytes, endothelial cells |
| Cadherin-11 | Carcinoma and retina epithelial cells |
| FSP1/S100A4 | Smooth muscle cells, invasive carcinoma cells |
| Fibroblast surface antigen (FSA) | Monocytes/macrophages |
| Discoidin domain receptor 2 (DDR2) | Endothelial cells, smooth muscle cells |
| Fibroblast activation protein-1 (FAP1) | Activated melanocytes |
| Prolyl-4-hydroxylase | Endothelial cells, epithelial cells |
| PDGF receptor-β (PDGFRb) | Smooth muscle cells, pericytes |
| Heat shock protein-47 (HSP47) | Monocytes/macrophages, various collagen-producing cells |
| Thymus cell antigen-1 (THY1/CD90) | Leukocytes, endothelial cells, various progenitor cells |
| Vimentin | Endothelial cells, smooth muscle cells, various cells |
| Palladin 4Ig* | Smooth muscle cells |
| Periostin* | Bone and carcinoma cells |
| Cofilin* | Smooth muscle cells |
| AngII type 1 receptor (AT1R)* | Cardiomyocytes, smooth muscle cells |
| TGF-β receptor* | Various cells |
| Frizzled-2* | Smooth muscle cells |
| α-Smooth muscle actin* | Smooth muscle cells |
| Integrins (αvβ3, α1β1, α2β1, α11β1)* | Endothelial cells, various cells |
| Collagen types I, III, IV, V, VI* | Various cells |
| Lysyl oxidase* | Smooth muscle cells |
| Fibronectin ED-A* | Smooth muscle cells |
| Tenascin C* | Smooth muscle cells |
Identifying specific molecular markers of cardiac fibroblasts is additionally complicated by the diversity of their phenotype as contributed by their specific developmental origin (e.g., epicardial vs. endocardial), location in the heart (e.g., atria vs. ventricle, left vs. right heart, valves, SA node, atrioventricular groove), pathologic state (e.g., infarction, pressure overload), and aging. For example, a subset of fibroblastic cells positive for PDGF receptor-α and stem cell antigen 1 (Sca-1) was recently identified in perivascular interstitial regions of the mouse heart.11 These epicardially derived cells, termed cardiac colony-forming-unit fibroblasts, can undergo a long-term expansion for approximately 40 passages and differentiate into various cells of mesodermal lineage. The potential role of these highly proliferative cells in cardiac repair, remodeling, and fibrotic disease remains to be explored. Phenotypic diversity was also identified between canine ventricular and atrial fibroblasts, with atrial fibroblasts being more proliferative in response to a variety of growth stimuli (e.g., fetal bovine serum, PDGF, FGF-2, TGF-β1, angiotensin II [AngII], endothelin-1 [ET-1]).12 These differences were amplified in congestive heart failure and eliminated using a PDGF receptor blocker, AG1295. Different functional roles were also reported for fetal and adult mouse cardiac fibroblasts. Ieda et al4 have shown that adult fibroblasts promote cardiomyocyte hypertrophy, whereas fetal fibroblasts promote embryonic cardiomyocyte proliferation by secreting fibronectin, collagen, and heparin-binding endothelial growth factor that activate β1-integrin signaling in cardiomyocytes.4 This finding suggests the possibility that the observed reactivation of the cardiac fetal gene program during hypertrophic cardiomyopathy or heart failure may be contributed by analogous phenotypic changes in cardiac fibroblasts.
Detailed phenotypic and functional characterization of cardiac fibroblasts can be performed in vitro, but these studies must be interpreted with caution because of various confounding factors, including the possibility that enzymatic or outgrowth cell isolation selects for a particular pool of cardiac fibroblasts and that fibroblast phenotype is significantly altered by higher stiffness of the cell attachment substrate (several GPa for glass or plastic vs. tens of KPa for intact tissue),13 increased oxygen tension (21% for ambient air vs. 5% in intact tissue),14 and the lack of neurohumoral and inflammatory factors, cyclic stretch, restricted extracellular space, and vasculature. Direct culture of freshly isolated fibroblasts within a biomimetic three-dimensional environment (e.g., cyclically stretched soft hydrogel) is likely to better preserve native cell phenotype compared with the use of standard two-dimensional culture conditions.15
Cardiac Myofibroblasts
Although cardiac fibroblasts are the most dominant nonmyocyte cell type in the healthy heart, cardiac disease or myocyte loss owing to myocardial infarction, hypertension, inflammation, and other stress signals is associated with the appearance and proliferation of cardiac myofibroblasts to either replace dead myocytes with a collagenous scar (replacement fibrosis) or yield interstitial or perivascular collagen accumulation (reactive fibrosis). Myofibroblasts exhibit a contractile cell phenotype intermediate between that of fibroblasts and smooth muscle cells.16,17 Compared with fibroblasts, myofibroblasts are larger cells with increased expression of stress fibers that exhibit enhanced proliferation, migration, and secretion of ECM proteins (e.g., collagen I, collagen III, fibronectin), ECM degradation enzymes (matrix metalloproteinases [MMPs]) and their inhibitors (tissue inhibitors of metalloproteinases [TIMPs]).18 In addition, microfibroblasts show both the increased secretion of and responsiveness to various cytokines and growth factors (e.g., AngII, TGF-β, PDGF, ET-1, tumor necrosis factor [TNF] α, interleukin [IL] 1β).17,18 Although mainly absent from the normal heart (with the exception of the heart valves), myofibroblasts are found to participate actively in the cardiac wound healing response, where they support rapid tissue remodeling and the formation of a fibrous scar. The efficient scar formation initially serves to prevent harmful dilatation of the heart; however, the long-term persistence and activity of myofibroblasts in the infarct scar or other regions of the myocardium can lead to excessive collagen accumulation, stiffening of the heart, pathologic remodeling, and eventually cardiac malfunction and failure. This persistence of the activated myofibroblasts in the heart after the mature scar is formed contrasts wound-healing processes observed in other organs where myofibroblasts undergo apoptosis and disappear upon scar formation.16
Although myofibroblasts express all the known molecular markers used to label cardiac fibroblasts (DDR2, vimentin, Thy1, FSP1, periostin), no specific myofibroblast markers have been identified (see Table 30-1).16 Compared with fibroblasts, de novo myofibroblasts also express or increase the expression of various cytoskeletal (α smooth muscle actin [α-SMA], SM22α, myosin heavy chain-B, tropomyosin) and cell adhesion (paxillin, tensin, fibronectin ED-A) proteins.13,16 Furthermore, myofibroblasts and fibroblasts do not express the intermediate filament marker desmin, and they lack the expression of mature smooth muscle markers, such as smooth muscle myosin heavy chains. Regarding their origin in fibrotic disease, myofibroblasts are traditionally thought to be derived from the resident (interstitial and adventitial) fibroblasts that proliferate, migrate, and differentiate into a myofibroblast phenotype.5 This view is based on in vitro studies showing increased fibroblast proliferation, migration, and conversion to a myofibroblast phenotype in response to a variety of cytokines related to cardiac injury and disease (TGF-β, AngII, TNF-α, IL-1β, IL-6, ET-1).18 However, the need to form a collagenous scar rapidly to replace dead myocardium (thereby preventing wall rupture or dilatation after injury) would suggest that a significant portion of myofibroblasts at the injury site should be derived from nonresident or nonfibroblastic cells, rather than the activation and long-range migration of remote resident cardiac fibroblasts. In line with this reasoning, recent genetic fate mapping studies in mice have demonstrated that during acute cardiac injury, pressure overload, prolonged ischemia, or chronic AngII treatment, myofibroblasts can originate from various nonfibroblastic sources such as (1) coronary endothelium in which endothelial cells after cardiac damage undergo endothelial-to-mesenchymal transition and migrate from the microvascular bed into interstitium to become myofibroblasts,19 (2) epicardial epithelium where epicardium-derived cells formed by an epithelial-to-mesenchymal transition differentiate (potentially via Notch activation) into fibroblasts and myofibroblasts that remain to reside in the epicardium,20,21 and (3) circulating bone marrow-derived cells, such as fibrocytes, that express both markers of hematopoietic origin (CD45, CD13, CD34) and ECM proteins (collagen I and III) or monocytes that upon recruitment to the site of injury or inflammation express both myofibroblast (FSP1, α-SMA) and monocytic (CD45, CD11b, CD14) markers.5 In several studies, these non–fibroblast-derived myofibroblasts are reported to comprise a significant fraction (20% to 75%) of all myofibroblasts found in the fibrotic areas.7,19 Although some of these cells might not persist in the heart at later stages of the disease,22 their exact role in the initial and late adaptive and maladaptive fibrotic sequelae remains to be explored.
The mechanisms of fibroblast-to-myofibroblast conversion have been extensively studied in vitro despite the fact that, with time in culture, cardiac fibroblasts spontaneously attain a myofibroblast phenotype and significantly upregulate α-SMA expression.23 This spontaneous phenotypic change does not occur in the homeostatic milieu of the healthy heart, but is rather an artifact of cell culture attributed to the high stiffness of the attachment substrate or a switch to hyperoxic (ambient air) conditions.13,14 The time course and extent of this process strongly depend on the particulars of cell isolation and culture conditions, with some reports showing ubiquitous α-SMA expression as early as 1 to 2 days after fibroblast plating,24 whereas others describe little or no phenotype switch until passage 2 to 3.10 A contributing factor to this variability might be the use of different anti–α-SMA antibodies by different groups. Figure 30-1 shows passage-1 cultured ventricular fibroblasts isolated from adult human, neonatal rat, and neonatal mouse tissues stained by two commonly used anti–α-SMA antibodies, a monoclonal mouse antibody from Sigma-Aldrich and polyclonal rabbit antibody from Abcam (MA, USA). The same antibodies were used to stain paraformaldehyde-fixed adult mouse ventricular sections. The difference in the specificity of the two antibodies is obvious and suggests nonspecific staining of fibroblasts by the antibody from Sigma-Aldrich. The extent to which cultured fibroblasts and myofibroblasts faithfully represent the phenotype and function of their in vivo counterparts from healthy or diseased hearts remains unknown and certainly warrants further study.
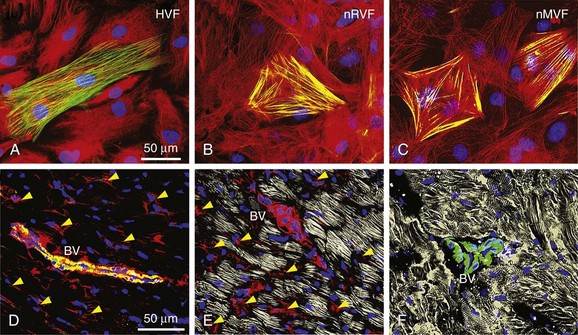
Figure 30-1 Specificity of myofibroblast labeling with anti–α-smooth muscle actin (α-SMA) antibodies. Cultured passage 2 fibroblasts from adult human (A), neonatal rat (B), and neonatal mouse (C) ventricles are shown. D-F, Ventricular tissue sections from healthy 2-month-old mice. Samples were stained for α-SMA using a monoclonal mouse antibody from Sigma-Aldrich (A2547, red) and polyclonal rabbit antibody from Abcam (ab5694, green) to label myofibroblasts and smooth muscle cells, filamentous actin (gray in E) and sarcomeric α-actinin (gray in F) to label cardiomyocytes, and 4,6-diamino-2-phenylindole (DAPI; blue) to label nuclei. Yellow arrowheads in D and E denote nonspecific, nonmyocyte labeling, which is absent in F. Anti–α-SMA images for the two antibodies were acquired using the same exposure time. BV, Blood vessel.
Fibroblast and Myofibroblast Electrophysiology
Fibroblast Voltage-sensitive Channels
Cardiac fibroblasts and myofibroblasts lack the required ion channels to initiate an action potential (AP) and are thus considered unexcitable cells. They exhibit a relatively depolarized resting membrane potential (RMP) of –50 to –20 mV, a cell capacitance from approximately 6 pF for fibroblasts to 60 pF for myofibroblasts, and an input resistance at rest of 1 to 10 GΩ.25–27 Figure 30-2, A1, shows typical current traces elicited in cultured human ventricular fibroblasts when membrane voltage in these cells was initially held at –40mV and increased by 10-mV increments from –80 to 30 mV. Lowering the initial holding potential to –100 mV increased the amplitude of both the steady-state and small time-dependent current components (see Figure 30-2, A2). Furthermore, the steady-state current-voltage (I-V) relationships of different types of fibroblasts and myofibroblasts exhibited similar shapes, typical of unexcitable cells, with a moderate outward rectification present at higher membrane potentials (see Figure 30-2, B). In a recent study, cultured fibroblasts isolated from infarcted rat ventricles exhibited a hyperpolarized resting potential and increased outward current density compared with fibroblasts isolated from healthy ventricles.25
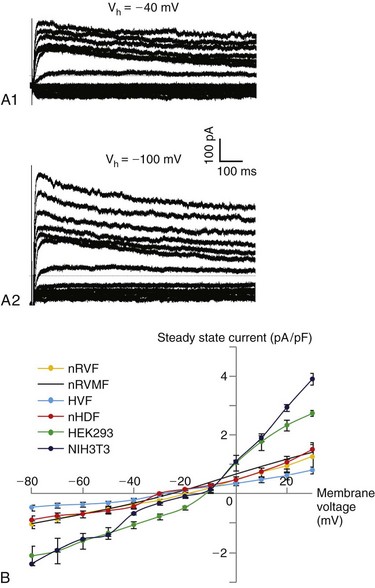
Figure 30-2 Current-voltage characteristics of various fibroblasts. A1 and A2, Representative whole-cell current traces from a human ventricular fibroblast when cell membrane voltage was held at –40 mV (A1) or –100 mV (A2) and stepped in 10-mV increments from −80 mV to 30 mV. B, Steady-state current-voltage (I-V) relationship (mean ± SEM) recorded in neonatal rat ventricular fibroblasts (nRVF; n = 5), neonatal rat ventricular myofibroblasts (nRVMF; adapted from Rohr et. al26), human ventricular fibroblasts (HVF; n = 4), neonatal human dermal fibroblasts (nHDF; n = 5), human embryonic kidney 293 fibroblasts (HEK 293; n = 3), and mouse NIH 3T3 fibroblasts (NIH3T3; n = 25).
Cardiac fibroblasts and myofibroblasts express a variety of voltage-sensitive currents and related ion channel genes and proteins.27–29 For example, inward rectifier, Ba2+-sensitive K+ current (IK1) controls RMP in freshly isolated adult rat ventricular fibroblasts (where it is likely mediated by Kir2.1 channels) and cultured commercially available (ScienCell Research Laboratory [CA, USA]) human ventricular fibroblasts (in which both Kir2.1 and Kir2.3 are expressed). Both rat and human adult ventricular fibroblasts express Ca2+-activated large conductance K+ channels (BKCa) as well as a delayed outward rectifier K+ current (IK), which is mediated by Kv1.6 channels in rat cells and by Kv1.5 and Kv1.6 in human cells. Similar slow and rapid delayed rectifier currents (IKr and IKs) have been recorded in neonatal rat ventricular fibroblasts and are presumably conducted via Kv1.2, Kv1.4, Kv1.5, and Kv2.1 channels.27 Cultured adult rat and mouse ventricular myofibroblasts express the gene encoding the Kir6.1 channel; in mouse cells, the expression of Kir6.1 along with subunits of the sulfonylurea receptor-2 (SUR2) channel generates a robust ATP-sensitive K+ current (IKATP) potentiated by pinacidil.28 Adult human and neonatal rat ventricular fibroblasts were also found to express a transient outward K+ current (Ito) that in human cells is conducted by Kv4.2 and Kv4.3 channels and in rat cells by Kv4.2 and Kv1.4. Beside K+ currents, cultured human ventricular fibroblasts express TTX-sensitive and TTX-resistant Na+ currents and swelling-induced Cl– current,29 whereas human atrial fibroblasts express voltage-gated H+ currents.27 Interestingly, a recent study has demonstrated that differentiation of human atrial fibroblasts into myofibroblasts in cell culture is associated with the de novo expression of a fast voltage-gated Na+ current predominantly carried by the α-subunit of the cardiac Na+ channel (Nav1.5).30 The potential relevance of this finding for atrial fibrotic disease and arrhythmogenesis is unknown. Endogenous expression of a functional Na+-Ca2+ exchanger (NCX1 or NCX3 isoform) and L-type Ca2+ channel α-subunit (Cav1.2) has been shown to modulate the Ca2+ inflow in cultured fibroblasts and potentially contribute to regulation of myofibroblast proliferation, migration, contraction, and collagen secretion.27,31,32 Importantly, the majority of the ion currents described in the studies mentioned here were detected in only a fraction of all the fibroblasts or myofibroblasts studied, thereby confirming the large phenotypic and functional diversity of these cells.
Fibroblast Mechanosensing and Transient Receptor Potential Channels
Both fibroblasts and myofibroblasts have been considered mechanosensitive cells, whereby their patterns of gene expression, proliferation rate, contractile and electrical properties, and sensitivity to and secretion of different soluble factors and ECM proteins are directly influenced by the mechanical state of their environment.13 The phenomenon in which cell electrical properties are altered in response to a mechanical stimulus is called mechanoelectric feedback. In cardiac fibroblasts, mechanoelectric feedback is likely mediated via Ca2+-permeable, stretch-sensitive channels of unknown molecular identity that likely belong to a family of transient receptor potential (TRP) channels.27,33,34 TRP channels are weakly sensitive to changes in membrane voltage and are regulated instead by stretch, oxidative stress, osmotic pressure, temperature, pH, or membrane receptor activation. The activity of these channels sensitizes cardiac fibroblasts to physicochemical changes in their environment. Transcripts of several TRP channels from the canonical (TRPC1,4,6), vanilloid (TRPV2,4), and melastatin (TRPM4,7) subfamilies were identified in human atrial fibroblasts, and TRMP7 (but not TRPC6 or TRPV2,4) currents were also successfully recorded in these cells using single-channel and whole-cell patch clamp. Similarly, adult rat ventricular fibroblasts were reported to express transcripts of TRPC2,3,5,6, TRPV2,4,6, and TRPM4,7 channels, and TRPV4 and TRPC6 channels were shown to mediate Ca2+ entry into these cells. Furthermore, adult rat ventricular (but not human atrial) fibroblasts were found to express nonselective cation currents likely carried by TRPC3 and TRPC6 channels, or their heteromers.34 In response to mechanical compression, cardiac fibroblasts generate membrane potential depolarizations known as mechanically induced potentials that, through capacitive or potential electrotonic coupling with cardiomyocytes, can modulate cardiac electrical properties.35 In addition to voltage-gated and TRP channels, cardiac fibroblasts and myofibroblasts express a variety of membrane-bound receptors and integrins that mediate their sensitivity to different chemical and ECM-mediated stimuli and potentially support their long-range communication and signal integration.
Arrhythmogenic Effects of Myofibroblasts
The described fibroblast-to-myofibroblast phenotype switch caused by different pathologic stimuli has a critical role in cardiac remodeling, a process that involves changes in the size, structure, and function of the heart. Upon the onset of disease, cardiac remodeling initially allows the heart to adapt to changes in its environment in order to maintain normal cardiac output. However, maladaptive cardiac remodeling (i.e., excessive cardiac fibrosis) owing to persistent myofibroblast-related activity has been implicated as a major risk factor for cardiac death by increasing the susceptibility to cardiac mechanical dysfunction and arrhythmias. In general, myofibroblasts can negatively affect cardiac electrical function through mechanisms related to their altered turnover of ECM proteins (Figure 30-3), secretion of specific soluble factors (Figure 30-4), and potential for direct contact with adjacent cardiomyocytes (Figure 30-5).
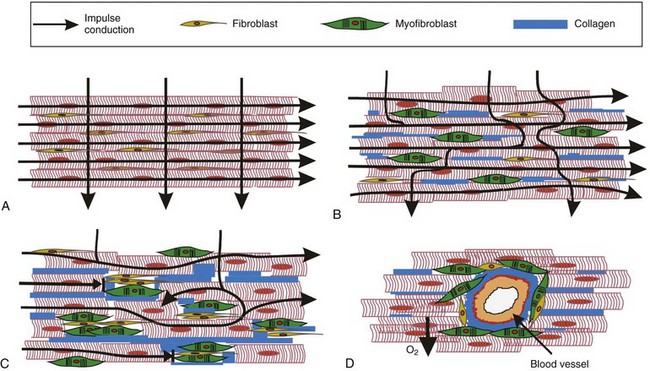
Figure 30-3 Patterns of cardiac fibrosis. A, Healthy heart with normal impulse conduction. B, Interstitial fibrosis in which lateral deposition of collagen and myofibroblasts mainly impedes transverse but not longitudinal impulse conduction. C, Patchy fibrosis where replacement of dead cardiomyocytes with fibrous tissue obstructs both longitudinal and transverse impulse conduction. D, Perivascular fibrosis in which fibrous tissue around blood vessels impairs O2 supply and conduction in surrounding myocardium.
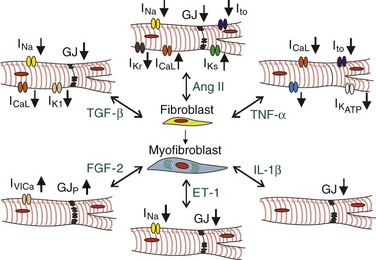
Figure 30-4 Soluble factors in fibrotic heart disease. Various growth factors and cytokines present in the fibrotic cardiac milieu mediate fibroblast-to-myofibroblast conversion and directly alter the expression of membrane ion channels and gap junctions (GJ) in cardiomyocytes. GJP, GJ phosphorylation.
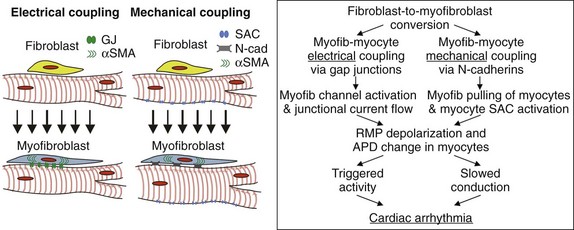
Figure 30-5 Effects of potential myofibroblast-cardiomyocyte coupling on cardiac electrophysiology. The existence of heterocellular electrical coupling via gap junctions or mechanical coupling via adherens junctions could yield cardiomyocyte depolarization and APD alterations that could create arrhythmogenic conduction slowing, triggered activity, or both.
Extracellular Matrix–Mediated Effects on Cardiac Electrical Activity
The formation of a fibrotic tissue substrate because of cardiac disease (e.g., chronic heart failure, hypertension, cardiomyopathy, myocardial infarction) or aging occurs when activated myofibroblasts respond to mechanical or oxidative stress or various inflammatory and profibrotic cytokines (e.g., AngII, TGF-β, TNF-α, IL-6, ET-1) by upregulating the secretion of ECM proteins (predominantly collagen III and I) and MMPs.16–18 The resulting degradation of old and excessive production of new ECM yields one or more of the following: (1) formation of a large collagenous scar, (2) lateral separation of cardiomyocyte bundles when collagen is deposited interstitially, (3) both longitudinal and lateral separation of cardiomyocytes when lost myocytes are replaced with fibrous tissue, or (4) perivascular fibrosis (see Figure 30-3). Based on histologic appearance, cardiac tissue fibrosis can be classified as compact, interstitial, patchy, and diffuse.36 Compact fibrosis is associated with healed infarct scars and can act as an anchor for macroscopic reentry. Interstitial fibrosis yields lateral separation of cardiomyocytes and, by selectively impeding transverse conduction, can lead to the generation of microreentrant circuits. Patchy fibrosis involves partial electrical insulation of groups of cardiomyocytes over relatively long distances (>1 mm) yielding the formation of tortuous conduction paths, increased vulnerability to block, and consequently, a highly arrhythmogenic substrate. Diffuse fibrosis is more disperse than patchy fibrosis and relatively nondisruptive to conduction; however, when excessive, it can also precipitate functional reentry. In fibrotic disease, nonconducting fibrous tissue typically creates localized alterations in the source-load ratio of upstream (excited) to downstream (unexcited) tissue areas, creating a substrate with discontinuous or nonuniform conduction. Fibrotic insulation of groups of cardiomyocytes can also facilitate the induction and propagation of early afterdepolarizations, such as those that arise during oxidative stress in aged myocardium.37 In this case, ectopic activity is likely triggered in regions with an intermediate degree of fibrosis, because too little fibrosis can prevent early afterdepolarization initiation, whereas too much fibrosis can block its propagation.
In addition to the physical impediment of AP spread, fibrotic areas in the heart can also alter (via mechanoelectric feedback) the electrical properties of bordering cardiomyocytes by stiffening their attachment matrix or by altering the mechanical load experienced by cardiomyocytes. In support of this concept, increased beta-myosine heavy chain (β-MHC) expression (characteristic of pathologic cardiac hypertrophy in mice) was predominantly detected in cardiomyocytes that bordered fibrous collagen-rich regions in pressure-overloaded and aged hearts.38 Furthermore, myofibroblast proliferation and collagen deposition can reduce the volume of cardiomyocyte extracellular space, increasing its resistance and propensity for rapid K+ accumulation during repetitive excitation, both of which are expected to increase the vulnerability to conduction slowing and block. Finally, excess myofibroblasts and collagen deposition in the perivascular space can form a diffusion barrier to oxygen and nutrients and create local ischemic environments that can significantly alter cardiomyocyte electrophysiology (see Figure 30-3).
Paracrine and Autocrine Effects on Cardiac Electrical Activity
Various cytokines and growth factors are known to promote arrhythmogenic changes in the cardiac substrate by increasing fibroblast proliferation, activation, and deposition of collagen, as well as by directly altering cardiomyocyte electrical activity (see Figure 30-4) and survival.17,18,24 The most well-studied profibrotic cytokines in the heart are TGF-β1 and AngII. TGF-β signaling is activated in response to cardiac injury, hypertrophy, or pressure overload, where it plays a central role in the generation of the profibrotic response.39,40 TGF-β in the heart is primarily expressed by fibroblasts but also by cardiomyocytes. Both cell types express TGF-β receptors that represent heteromers of type I and type II receptors. Upon TGF-β binding, the type I receptor (also known as activin-linked kinase 5) phosphorylates Smad2 and Smad3, which then bind to Smad4, and after translocation into the nucleus activate downstream pathways, including Ras/MEK/ERK, p38, and JNK. TGF-β signaling is inhibited by Smad7. Mice constitutively overexpressing TGF-β1 in cardiomyocytes show atrial but not ventricular fibrosis, confirming the higher susceptibility of atria to fibrotic disease. Treatment of cultured cardiac fibroblasts with TGF-β induces their conversion to α-SMA+ myofibroblasts and significantly enhances their ECM deposition through increased expression of ECM genes, downregulation of MMPs, and upregulation of plasminogen activator inhibitor (PAI)-1 and TIMPs.40 TGF-β can also directly alter myocyte electrical activity by reducing inward rectifier K+ current, fast Na+ and L-type Ca2+ currents, and gap junctional coupling.24
AngII is a potent vasoconstrictor and profibrotic factor abundantly expressed in hearts undergoing myocardial remodeling. Increased Ang II expression in transgenic mice by cardiac-restricted upregulation of angiotensin-converting enzyme causes atrial dilatation, focal fibrosis, and atrial fibrillation (AF).41 Ang II is produced in the injured heart by activated macrophages, cardiac myofibroblasts, and cardiomyocytes, where it acts through binding to its receptors AT1 and AT2, of which AT1 but not AT2 is expressed in cardiac fibroblasts. Binding of AngII to AT1 receptor in fibroblasts stimulates their production of profibrotic factors (TGF-β1, ET-1, FGF-2) and ECM proteins.17,18 TGF-β1 acts in an autocrine fashion on fibroblasts (and in a paracrine fashion on surrounding cardiomyocytes) to stimulate AT1 expression directly, thereby potentiating the fibrotic response. Thus, AngII and TGF-β1 act synergistically as part of an integrated signaling network to drive fibrogenic responses in the heart by augmenting the effect that TGF-β has on fibroblast proliferation, differentiation, and ECM deposition.18,41
AngII can also directly affect cardiomyocyte electrophysiology.24,42 In atrial myocytes, Ang II has been shown to upregulate the expression of L-type Ca2+ channels via protein kinase C–dependent and cyclic AMP response element-binding protein (CREB)-dependent pathways, which can lead to sarcoplasmic reticulum Ca2+ overload and triggered activity. AngII can also enhance nuclear factor κB binding to the promoter for SCN5A (the gene encoding Nav1.5), which decreases the expression of fast Na+ channels and reduces cardiomyocyte excitability. Binding of AngII to AT1 receptors in cardiomyocytes also leads to the formation and eventual internalization of a complex that includes Kv4.3 channel, thus reducing the transient outward K+ current (Ito) and potentially prolonging the cardiac action potential duration (APD). Similarly, by binding to AT1 receptors and activating the protein kinase C–dependent pathway, AngII reduces the delayed rectifier IKr/hERG current, which in turn can contribute to APD prolongation and increased arrhythmogenesis in cardiac hypertrophy and failure. On the other hand, AT1 stimulation by AngII enhances IKs in atrial cardiomyocytes, which promotes APD shortening and explains the basis for AngII-mediated increase in AF vulnerability. In addition to its effects on cardiac voltage-gated ion channels, AngII can also stimulate Ca2+ influx via activation of TRP3 and TRP6 channels leading to cardiac hypertrophy. Lastly, AngII has been shown to mediate the reduction of connexin43 in ventricular myocytes via c-Src tyrosine kinase upregulation, which could lead to increased risk of ventricular fibrillation and death.43
PDGF is another molecule that plays a significant role in cardiac fibrotic disease by stimulating fibroblast proliferation and differentiation.17,44 Recently, upregulated expression of PDGF-A and -D, and PDGF receptors -α and -β by cardiac interstitial cells including myofibroblasts was suggested as having a role in infarct scar formation and contributing interstitial fibrosis during the later stages of remodeling.45 Activation of PDGF receptors initiates signaling via mitogen-activated protein kinase, JAK/STAT, and phospholipase C pathways, leading to fibroblast hyperresponsiveness, specifically in the atria. The regional difference in PDGF receptor expression levels could also explain why the atria are more susceptible to fibrotic remodeling than the ventricles.12 PDGF can also act to promote fibrosis by elevating TGF-β levels.44
FGF-2 is primarily secreted by cardiac fibroblasts and cardiomyocytes in response to adrenergic or AngII stimulation. FGF-2 is translated into a high- and low-molecular-weight isoforms (Hi-FGF-2 and Lo-FGF-2), of which Hi-FGF-2 is predominantly expressed by fibroblasts. In the extracellular environment, FGF-2 acts in a paracrine fashion to induce fetal gene program and promote hypertrophy of cardiomyocytes and in an autocrine fashion to promote fibroblast proliferation and secretion of other hypertrophic factors, such as cardiotrophin-1 (CT-1).17,46 In addition to its profibrotic and prohypertrophic actions, FGF-2 was reported to promote the opening of non–voltage-gated Ca2+-permeable channels and stimulate Cx43 phosphorylation in cardiomyocytes.24
ET-1 is secreted by a number of cells in the heart, including endothelial cells, cardiomyocytes, and fibroblasts. TGF-β and Ang II promote ET-1 production via JNK and ERK/ROS pathways, respectively.44 ET-1 induces myocyte hypertrophy and stimulates fibroblast ECM production and differentiation to myofibroblasts.18 ET-1 also mediates electrical remodeling during heart failure by downregulating myocyte expression and phosphorylation of gap junctional proteins Cx40 and Cx43 and by reducing Nav1.5 protein expression and Na+ channel conductance.47 Similarly, exposure of cultured cardiomyocyte monolayers to ET-1 yields reduced Cx43 expression and conduction slowing.48
TNF-α in the healthy and injured heart is secreted by a number of cells, including cardiac fibroblasts in which TNF-α secretion is enhanced by hypoxia and stimulation by AngII, serotonin, or mechanical stretch.17,18 TNF-α binds to two cell receptors—TNFRI and TNFRII—both of which are expressed by cardiac fibroblasts and myocytes. TNF-α binding to its receptors stimulates fibroblast proliferation and migration, as well as the expression of MMPs and proinflammatory cytokines.18 TNF-α exerts multiple proarrhythmic changes in cardiomyocyte electrical properties, including reduction of transient outward, delayed rectifier, and ATP-sensitive K+ currents, L-type Ca2+ current,24 and an increase in sarcoplasmic reticulum Ca2+ leak.49
Upon cardiac injury, interleukin-1β (IL-1β) is produced by various cells including cardiac fibroblasts. IL-1β stimulates fibroblast migration and net ECM degradation through increased secretion of MMPs.18 IL-1β has been shown to downregulate Cx43 expression in cardiomyocyte cultures and potentially in canine cardiomyocytes, fibroblasts, and myofibroblasts within the infarct border zone.50 Reduced Cx43 expression could in turn promote cardiac fibrosis via enhanced myofibroblast activity and proliferation51,52 and result in the generation of an arrhythmogenic substrate.
In addition to studies describing the role of individual growth factors or cytokines in cardiac hypertrophy, electrophysiology, and fibrogenesis, several in vitro studies have examined how fibroblast-conditioned media affect the function of neonatal rat ventricular myocytes (NRVMs). Although most of these studies show that fibroblast paracrine factors induce hypertrophy and enhance contractile activity of NRVMs, a study by Pedrotty et al53 also described the profound effects that neonatal rat ventricular fibroblast conditioned media had on NRVM electrical properties, including increased spontaneous activity, significantly depolarized RMP, twofold conduction slowing, and twofold APD prolongation. These functional changes were accompanied by a significant reduction in the expression of Nav1.5, Kir2.1, and Kv4.3 genes, an increased β-MHC/α-MHC expression ratio, and no change in Cx43 or Cx45 gene expression. Interestingly, these deleterious effects were prevented if media conditioning by fibroblasts occurred in noncontact cocultures with cardiomyocytes, but not in the presence of cardiac conditioned media. This result suggests the existence of local paracrine cross-talk between cardiac fibroblasts and myocytes, which in physiologic conditions, when numbers of the two cell types are balanced, prevents the adverse paracrine effects to occur. However, if the number of fibroblasts was locally increased or the number of cardiomyocytes was locally decreased, such as in fibrotic disease, this protective balance would be compromised and the fibroblast paracrine action would negatively affect the electrical function of the surrounding cardiomyocytes and potentially lead arrhythmic activity. Although impulse conduction was also slowed in the NRVM monolayers exposed to media conditioned by adult myofibroblasts from healthy or infarcted rat hearts, the APD of the treated NRVMs was reduced,25 suggesting that fibroblast-secreted factors and their functional roles may be also age-dependent.
Cell Contact–Mediated Effects on Cardiac Electrical Activity
The fact that cardiac fibroblasts and myofibroblasts express various voltage-sensitive and mechanosensitive ion channels suggests the intriguing possibility that these cells could modulate cardiac electrical activity by directly coupling with cardiomyocytes through functional gap junctions (see Figure 30-5). Currently, however, there is no direct experimental evidence (except for a dye transfer study in the SA node54) that cardiac fibroblasts or myofibroblasts can functionally couple with cardiomyocytes in the intact heart or that this coupling could have a significant role in cardiac function or malfunction. In transmission electron micrographs from SA nodes or infarcted ventricles, cardiomyocytes, fibroblasts, and myofibroblasts formed close membrane appositions but still remained separated by the basement membrane.50,55 However, in the rabbit SA node,55 but not in the canine infarct border zone,50 small gap junction–like structures were observed between abutting fibroblasts. The strongest evidence for cardiomyocyte-fibroblast gap junctional coupling in situ comes from Camelliti et al54 who showed examples of Cx45+ immunostaining between fibroblasts and cardiomyocytes in the SA node. They also observed the infiltration of Cx45+/vimentin+ cells into sheep infarcts as early as a few hours after ligation, followed by the appearance of Cx43+/vimentin+ cells within the subsequent 6 to 12 days. Most recently, the same group reported that approximately 3% of Cx43+ labeling in rabbit atria and ventricles resided in cardiomyocyte-fibroblast contacts, and up to 9% of Cx40+ labeling in the atrioventricular node was heterocellular.56
In contrast to the scarce evidence for the structural and functional coupling of fibroblasts and cardiomyocytes in the intact heart, there is a general agreement that these two cell types can functionally couple in vitro. The gap junction proteins potentially involved in this heterocellular coupling vary in different reports. Rohr24 has shown strong expression of Cx43 and Cx45 but not Cx40 protein in neonatal rat ventricular myofibroblasts, but other groups have shown no Cx43 expression in these cells.57 Weak expression of Cx45 and no expression of Cx43 or Cx40 was found in neonatal rat ventricular fibroblasts10; however, cultured adult mouse fibroblasts expressed Cx40 and Cx43, but not Cx45.58 Furthermore, in recent studies, cultured myofibroblasts isolated from infarcted hearts showed increased levels of Cx43 expression compared to those isolated from healthy hearts.25,59 Although gap junctions between cultured fibroblasts/myofibroblasts and cardiomyocytes can form without doubt, the frequency of this formation and the potential that selected cellular fractions are more amenable to coupling than others still remain to be explored. Using a well-controlled setting of micropatterned cell pairs, Pedrotty et al60 found that only 9.6% of neonatal rat ventricular myocyte-fibroblast pairs showed Cx43+ staining at the approximately 90-µm-long cell-cell border, and that instead of intercellular gap junction formation, heterocellular contacts with fibroblasts predominantly yielded internalization of Cx43 in cardiomyocytes.
Despite variable reports on the type of connexins involved, most of the studies agree that a certain degree of functional electrical coupling (from very weak to relatively strong) exists between cultured fibroblasts/myofibroblasts and cardiomyocytes. Because cardiac fibroblasts are unexcitable and have a depolarized RMP, their electrical coupling with cardiomyocytes will both depolarize cardiomyocyte RMP and impose capacitive load during AP upstroke and repolarization. The specific shape of the fibroblast I-V curve (see Figure 30-2, B) relative to that of a cardiomyocyte (mostly governed by its IK1) and the strength (conductance) of their gap junctional coupling will together determine the level of cardiomyocyte RMP depolarization. Different studies report highly variable effects that fibroblast/myofibroblast coupling with cardiomyocytes has on cardiac RMP, starting from a negligible change10,61 to strong depolarization.24 In the healthy adult myocardium with strong IK1 expression and large cardiomyocyte size, one would generally expect that coupling to a fibroblast would only negligibly change the myocyte resting potential. Thus, relatively strong coupling with multiple fibroblasts or a significant increase in fibroblast/myofibroblast inward current without a change in resting potential would be necessary to induce significant cardiomyocyte depolarization. Slight depolarization of cardiomyocyte RMP can increase cardiac excitability by effectively decreasing the excitation threshold, whereas further RMP depolarization would inactivate Na+ channels and decrease the AP upstroke and conduction velocity. Even higher levels of cardiomyocyte RMP depolarization could induce triggered activity, whereas extreme RMP depolarization would render cardiomyocytes unexcitable.24,62 If the cardiomyocyte IK1 were reduced (e.g., in nodal tissues, infarction, failure, Andersen-Tawil syndrome), potential electrical coupling with fibroblasts and myofibroblasts would be more likely to induce noticeable changes in cardiac RMP. The resulting conduction slowing or triggered activity could present arrhythmogenic risks that would be further exacerbated in conditions of high oxidative stress or hypokalemia.63 However, in patients with AF and increased IK1,41 cardiomyocyte RMP depolarization owing to electrical coupling with myofibroblasts could be antiarrhythmic.
In addition to resistive (static) loading that would act to depolarize cardiomyocyte RMP, coupled fibroblasts would also impose a capacitive load on cardiomyocytes by contributing additional membrane area to be charged and discharged by cardiomyocytes, thus effectively diluting cardiomyocyte channel density. This in turn would affect any time-dependent (dynamic) change in the cardiomyocyte membrane potential, such as AP generation. In a recent study, McSpadden et al62 generated micropatterned pairs made of a neonatal rat cardiomyocyte coupled to a variable-sized Cx43-overexpressing HEK293 cell (Cx43/HEK293), used as a generic model of an unexcitable cell (see Figure 30-2, B) able to strongly couple with cardiomyocytes.62 They showed that capacitive loading by coupled fibroblasts acted to significantly slow the AP upstroke velocity in cardiomyocytes, effectively reducing cardiac excitability. For the strong HEK293-cardiomyocyte coupling studied, with an increase in HEK293 size (and capacitance), this capacitive effect on cardiomyocytes occurred before the onset of significant RMP depolarization. Accordingly, coupling of Cx43/HEK293 cells to cardiomyocyte monolayers resulted in a fivefold decrease in conduction velocity despite only a slight depolarization of cardiac RMP, signifying the capacitive loading of cardiomyocytes as a potentially powerful modulator of cardiac conduction.10,62
Besides passive (resistive and capacitive) loading of cardiomyocytes, coupled myofibroblasts could also actively modulate cardiac AP shape through the activation of their voltage-sensitive or mechanosensitive ion channels. For example, during cardiac AP, fibroblasts coupled to an activating myocyte would also undergo depolarization and, when sufficiently depolarized, activate their voltage-sensitive outward currents (see Figure 30-2), causing membrane repolarization. This in turn would tend to generate a flow of gap junctional current from the cardiomyocyte (source) into the fibroblast (sink), yielding shortening of the cardiac APD. Depending on the fibroblast RMP, magnitude and type of its voltage-sensitive currents, strength of coupling with cardiomyocytes, and the APD of unloaded cardiomyocyte, the described passive and active loading scenarios could result in different degrees of shortening or lengthening of the cardiac APD. Furthermore, fibroblast mechanosensitive channels, dynamically activated during cardiac tissue contraction, could generate inward currents in fibroblasts and in turn depolarize coupled cardiomyocytes or accelerate their firing rate (e.g., in SA node). Coupled myofibroblasts could also support passive (detrimental) impulse conduction over a distance of a few hundred micrometers and electrotonically bridge remote cardiomyocytes.24 Whether this bridging effect would be proarrhythmic or antiarrhythmic would likely depend on the specific distribution and density of such bridges in the cardiac tissue. At least in the NRVM-myofibroblast monolayers, increased myofibroblast density uniformly decreased conduction velocity and increased complexity of the reentrant activity while significantly reducing its dominant frequency. Interestingly, for a particular myofibroblast density studied, both an increase and decrease in cardiomyocyte-myofibroblast coupling strength increased the conduction velocity to a similar degree.64
In addition to electrical coupling, potential mechanical coupling between myofibroblasts and cardiomyocytes has been proposed recently as another mechanism that could contribute to the increased arrhythmogenicity observed in cardiac fibrotic disease (see Figure 30-5). In this scenario, N-cadherin–mediated “tugging” of cardiomyocyte membranes by myofibroblasts would activate mechanosensitive channels in myocytes, yielding RMP depolarization and conduction slowing.57 Consistent with this hypothesis, acute application of blebbistatin (to relax cellular prestress and prevent tugging) as well as gadolinium and streptomycin (to block stretch-sensitive channels nonspecifically) fully reverted the observed conduction slowing. Silencing of Cx43 in myofibroblasts did not alter these findings, suggesting the dominant involvement of mechanical rather than electrical coupling in these observations, although potential roles of Cx45 coupling were not addressed.57 Although direct proof for this mechanism by silencing N-cadherin expression in myofibroblasts was not provided, this study opened the possibility that, in the setting of fibrotic disease, mechanical contacts between myofibroblasts and cardiomyocytes might significantly alter cardiomyocyte electrophysiology. Naturally, the critical question remains to be determined whether myofibroblasts in the heart couple to cardiomyocytes by N-cadherin junctions and whether the strength of this coupling and mechanical properties of the surrounding three-dimensional microenvironment, which is substantially softer than the rigid substrate used for in vitro studies, are conducive to the hypothesized tugging effect. Furthermore, studies of heterocellular contact-mediated interactions are unavoidably confounded by the potential local paracrine or juxtacrine signaling by which interacting cells might also affect each other.
Other than potential functional coupling through gap or adherens junctions, recent in vitro studies have suggested the possibility that cardiomyocytes and fibroblasts can directly communicate through thin, long (up to ~20-30 micrometers) tubular structures known as tunneling nanotubes.65 It is unknown whether these structures are stable (or transiently formed secondary to cell motion) and how frequently they occur in vitro or in vivo. Regarding that the conductance of the tunneling nanotubes is in the sub-nanosiemens (sub-nS) range66 and that velocity of Ca2+ propagation through these structures appears to be approximately 1 µm/s,65 their relevance for the cardiac electrophysiology and arrhythmogenesis (assuming their presence in vivo) remains to be established. Nonetheless, the possibility that heterocellular exchange (even if transient) of genetic material, organelles, and other molecules can occur in the cardiac milieu could offer interesting opportunities for the development of new cardiac therapies.66
Based on the in vitro evidence for functional electrical coupling between cardiomyocytes and myofibroblasts, a number of recent computational studies have examined the potential effects of fibroblast number, size, passive and active membrane properties, and spatial distribution on cardiomyocyte AP shape and conduction. These studies suggest that heterogeneous spatial distribution of fibroblast-cardiomyocyte electrical coupling in the heart could yield proarrhythmic outcomes, including local conduction slowing and block, triggered activity, or spatially discordant Ca2+ alternans.67,68 Furthermore, in a realistic computational model of infarcted rabbit ventricle, moderate densities of scar myofibroblasts coupled to cardiomyocytes were found to enhance arrhythmogenicity by augmenting APD dispersion in the periinfarct zone, while high myofibroblast densities significantly depolarized myocytes, interrupted periinfarct zone conduction and prevented arrhythmia induction.69 Recently, computational studies in a simplified model of fibrotic atrial tissue suggested that complex fractionated atrial electrograms, often observed and used in the AF ablation, can form only if cardiomyocytes are electrically coupled with myofibroblasts and, unlike in previous computational studies,70 could not form because of collagen accumulation alone. Furthermore, myofibroblast-myocyte coupling facilitated AF termination by complex fractionated atrial electrogram–targeted ablation, whereas the ablation in the absence of this coupling only converted AF to sustained atrial tachycardia.71 Although potentially important, these studies belong to a realm of mere academic exercise, pending proof that the functional fibroblast-myocyte coupling in the heart is indeed a reality.
Cardiac Fibroblasts as Antiarrhythmic Targets
Recent clinical trials have identified myocardial fibrosis as an independent predictor of cardiac arrhythmias, including ventricular tachycardia and fibrillation.72 Given the important roles of cardiac fibrosis in arrhythmogenic cardiac remodeling, the attenuation or reversal of fibrogenic processes in the heart by selective pharmacologic or gene targeting could lead to effective antiarrhythmic therapies (Table 30-2). Because cardiac fibroblasts and myofibroblasts are believed to be the main mediators of extracellular collagen accumulation in myocardial fibrosis as well as the generators of various profibrotic and inflammatory cytokines, potential antifibrotic therapies for cardiac arrhythmias would likely target fibroblast proliferation, differentiation, ECM accumulation, and response to or secretion of different soluble factors. These therapies should be balanced and timely to alleviate pathologic cardiac remodeling but not interfere with the initial compensatory response to cardiac injury.73
Table 30-2
Examples of Antifibrotic Drugs
| Drug | Action |
| c-Src inhibitor | Downregulation of RAAS by AngII inhibition |
| Pirfenidone | Reduced TGF-β signaling by block of Smad nuclear translocation |
| Relaxin | Reduced TGF-β1 signaling and myofibroblast differentiation, enhanced MMP-based collagen breakdown |
| Statins | Downregulation of CTGF by RhoA inhibition, ROS inhibition, reduced MMP expression |
| Omega-3 fatty acids | Reduced TGF-β signaling by block of Smad2/3 nuclear translocation via PKG activation |
| Geranylgeranylacetone | Heat shock protein induction, TGF-β1 inhibition |
| Acetaminophen | ROS inhibition |
| β-blockers | Reduced fibroblast proliferation and IL-6 secretion |
| Losartan | AngII receptor inhibition |
Antifibrotic Drug Therapies
As previously described, upregulation of cardiac or systemic renin-angiotensin-aldosterone-system (RAAS) plays a critical role in different aspects of cardiac fibrogenesis. Furthermore, several animal models and retrospective analyses of clinical data have suggested that the downregulation of RAAS through the inhibition of AngII converting enzyme (e.g., lisinopril, imidaprilat, spironolactone), AngII receptors (e.g., eplerenone, losartan), or aldosterone (e.g., telmisartan, candesartan) is a promising strategy to reduce the incidence or persistence of atrial and ventricular arrhythmias. However, recent prospective randomized clinical trials have been inconclusive; therefore, the potential utility of RAAS inhibition for antiarrhythmic therapy remains unproven.41 Because TGF-β signaling is also a major mediator of cardiac fibrosis, its pharmacologic inhibition might exert antifibrotic and potentially antiarrhythmic actions in the heart. For example, pirfenidone, a blocker of TGF-β–induced Smad nuclear translocation, reduced atrial and left ventricular fibrosis and associated arrhythmias in models of canine heart failure and rat myocardial infarction.74 Similarly, relaxin, an inhibitor of TGF-β effects, has been shown to reduce collagen content in murine models of fibrotic cardiomyopathy.75
Through their inhibition of fibroblast proliferation, migration, differentiation, and ECM turnover,18 cholesterol-lowering statins (e.g., atorvastatin, pravastatin) exhibit strong antifibrotic effects and could aid in the clinical prevention of AF.76 Similarly, the antiinflammatory effects of omega-3 fatty acids could also translate into an antifibrotic and AF-suppressing therapy.77 Myeloperoxidase could represent yet another antiarrhythmic target because it has been shown to stimulate profibrotic signaling cascade via generation of reactive oxygen species, leading to atrial fibrosis and fibrillation.78 In addition, ET-1 receptor blockers (e.g., bosentan) aimed at the reduction of ECM production and myofibroblast differentiation could decrease the incidence of AF in patients with structural heart disease.44 Besides different profibrotic soluble factors, Ca2+ inflow through TRMP7 channels has been shown recently to regulate TGF-β–induced proliferation and differentiation of human atrial fibroblasts from AF patients33 and could potentially prove to be a novel and powerful antiarrhythmic target.
Antifibrotic Gene Therapies
Genetic modifications of fibroblast function represent another promising approach for the treatment of fibrotic heart disease and related arrhythmias. Initial proof-of-concept studies have successfully targeted the downregulation of TGF-β/AngII signaling in infarcted or pressure-overloaded rodent hearts. Recently, adeno-associated viral delivery of the small proteoglycan decorin prevented cardiac fibrosis by reducing TGF-β/Smad2/3 activation and p38 MAPK signaling in hypertensive rat hearts.79 Similarly, in vitro knockdown of myocardia-related transcription factor A and B in cardiac fibroblasts reduced their TGF-β1–induced transition to myofibroblasts, offering a potential gene therapy approach for cardiac fibrotic disease.80 Furthermore, overexpression of Fc receptors in monocytes could represent another strategy to alleviate fibrotic cardiomyopathy by preventing serum amyloid P–mediated differentiation of monocytic cells to fibroblasts.81
Recently, small noncoding RNAs (microRNAs [miRs]) have emerged as potentially important targets in the treatment of cardiac fibrosis and arrhythmias.82 For example, miR-21 expression in cardiac fibroblasts was found to be selectively upregulated in failing murine hearts where it enhanced ERK-MAP kinase activity and, by controlling fibroblast survival and cytokine secretion, contributed to the formation of interstitial fibrosis and cardiac hypertrophy. Silencing miR-21 in failing hearts could prove to be a potentially powerful antifibrotic therapy. Similarly, miR-29 family members (miR-29a, miR-29b, and miR-29c) were found to be downregulated in the border zone of murine and human infarcts, and upregulating miR-29 expression decreased the collagen secretion of fibroblasts via suppression of TGF-β signaling. Recently, the expression of miR-101a and miR-101b was found to be decreased in the periinfarct area of the rat heart, and adenoviral overexpressing of miR-101a in a model of chronic infarction decreased interstitial fibrosis via inhibition of c-Fos expression and TGF-β1 signaling.83
In addition to targeting the profibrotic function of fibroblasts, recent studies in mice revealed the exciting possibility that viral delivery of cardiac transcription factors or miRs in the heart can reprogram resident cardiac fibroblasts into functional cardiomyocytes.8,84 Provided that the reprogramming efficiency is optimized and that similar strategies can be applied to humans, this approach could open the door to a new array of powerful gene therapies for heart disease. Similarly, successful genetic targeting of endogenous fibroblasts and myofibroblasts to alter their ion currents and enhance or inhibit their coupling to surrounding cardiomyocytes could foster the development of new antiarrhythmic therapies.85
Conclusions and Future Directions
1. Fibroblast origin and diversity in the heart—are there multiple progenitors involved in the generation of cardiac fibroblasts? How does the phenotype and function of fibroblasts changes with development and aging? Are there phenotypic and functional differences among fibroblasts from different regions of the heart (left vs. right vs. septum, base vs. apex, endocardial vs. epicardial)?
2. Fibroblast roles in cardiac pathology—do fibroblasts change their phenotype with cardiac disease? What are the molecular differences between fibroblasts and myofibroblasts in situ, including the repertoire of their ion channels, coupling proteins, and paracrine factors? What is the role of resident fibroblasts versus myofibroblasts versus nonresident (blood or endothelium-derived) myofibroblasts in cardiac disease? Are myofibroblasts of different origins involved in distinct types of fibrosis (e.g., perivascular, patchy, interstitial)? When and why does the initially protective action of myofibroblasts then become deleterious?
3. Fibroblast-cardiomyocyte relationships—how do multiple paracrine factors interact in a bidirectional manner to coordinate fibroblast and myocyte function in the heart? Do potential differences in the fibroblast phenotype in different regions of the heart correlate with regional differences in cardiomyocyte electrical phenotype? Can the type and amount of secreted ECM directly change the electrical properties of the attached myocytes? Do fibroblasts and myofibroblasts electrically or mechanically couple to myocytes in the heart, and what are the consequences of this coupling for cardiac electrical function?
Acknowledgments
The authors thank Dr. Robert Kirkton for critical reading of the manuscript and for generating Figure 30-1, and Hung Nguyen for generating Figure 30-2. This work is supported in part by National Institutes of Health–Nation Heart, Lung, and Blood Institute grants HL106203, HL104326, and HL093711 to N.B.
References
1. Souders, CA, Bowers, SL, Baudino, TA. Cardiac fibroblast: The renaissance cell. Circ Res. 2009; 105:1164–1176.
2. Snider, P, Standley, KN, Wang, J, et al. Origin of cardiac fibroblasts and the role of periostin. Circ Res. 2009; 105:934–947.
3. Biernacka, A, Frangogiannis, NG. Aging and cardiac fibrosis. Aging and Disease. 2011; 2:158–173.
4. Ieda, M, Tsuchihashi, T, Ivey, KN, et al. Cardiac fibroblasts regulate myocardial proliferation through beta1 integrin signaling. Dev Cell. 2009; 16:233–244.
5. Zeisberg, EM, Kalluri, R. Origins of cardiac fibroblasts. Circ Res. 2010; 107:1304–1312.
6. Tian, Y, Morrisey, EE. Importance of myocyte-nonmyocyte interactions in cardiac development and disease. Circ Res. 2012; 110:1023–1034.
7. Krenning, G, Zeisberg, EM, Kalluri, R. The origin of fibroblasts and mechanism of cardiac fibrosis. J Cell Physiol. 2010; 225:631–637.
8. Song, K, Nam, YJ, Luo, X, et al. Heart repair by reprogramming non-myocytes with cardiac transcription factors. Nature. 2012; 485:599–604.
9. Acharya, A, Baek, ST, Huang, G, et al. The bhlh transcription factor tcf21 is required for lineage-specific emt of cardiac fibroblast progenitors. Development. 2012; 139:2139–2149.
10. McSpadden, LC, Kirkton, RD, Bursac, N. Electrotonic loading of anisotropic cardiac monolayers by unexcitable cells depends on connexin type and expression level. Am J Physiol Cell Physiol. 2009; 297:C339–351.
11. Chong, JJ, Chandrakanthan, V, Xaymardan, M, et al. Adult cardiac-resident msc-like stem cells with a proepicardial origin. Cell Stem Cell. 2011; 9:527–540.
12. Burstein, B, Libby, E, Calderone, A, et al. Differential behaviors of atrial versus ventricular fibroblasts: A potential role for platelet-derived growth factor in atrial-ventricular remodeling differences. Circulation. 2008; 117:1630–1641.
13. Hinz, B. The myofibroblast: Paradigm for a mechanically active cell. J Biomech. 2010; 43:146–155.
14. Roy, S, Khanna, S, Rink, T, et al. P21waf1/cip1/sdi1 as a central regulator of inducible smooth muscle actin expression and differentiation of cardiac fibroblasts to myofibroblasts. Mol Biol Cell. 2007; 18:4837–4846.
15. Wang, H, Haeger, SM, Kloxin, AM, et al. Redirecting valvular myofibroblasts into dormant fibroblasts through light-mediated reduction in substrate modulus. PloS One. 2012; 7:e39969.
16. van den Borne, SW, Diez, J, Blankesteijn, WM, et al. Myocardial remodeling after infarction: The role of myofibroblasts. Nat Rev Cardiol. 2010; 7:30–37.
17. Takeda, N, Manabe, I. Cellular interplay between cardiomyocytes and nonmyocytes in cardiac remodeling. Int J Inflam. 2011; 2011:535241.
18. Porter, KE, Turner, NA. Cardiac fibroblasts: At the heart of myocardial remodeling. Pharmacol Ther. 2009; 123:255–278.
19. Zeisberg, EM, Tarnavski, O, Zeisberg, M, et al. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat Med. 2007; 13:952–961.
20. Zhou, B, Honor, LB, He, H, et al. Adult mouse epicardium modulates myocardial injury by secreting paracrine factors. J Clin Invest. 2011; 121:1894–1904.
21. Russell, JL, Goetsch, SC, Gaiano, NR, et al. A dynamic notch injury response activates epicardium and contributes to fibrosis repair. Circ Res. 2011; 108:51–59.
22. van Amerongen, MJ, Bou-Gharios, G, Popa, E, et al. Bone marrow-derived myofibroblasts contribute functionally to scar formation after myocardial infarction. J Pathol. 2008; 214:377–386.
23. Santiago, JJ, Dangerfield, AL, Rattan, SG, et al. Cardiac fibroblast to myofibroblast differentiation in vivo and in vitro: Expression of focal adhesion components in neonatal and adult rat ventricular myofibroblasts. Dev Dyn. 2010; 239:1573–1584.
24. Rohr, S. Arrhythmogenic implications of fibroblast-myocyte interactions. Circ Arrhythm Electrophysiol. 2012; 5:442–452.
25. Vasquez, C, Mohandas, P, Louie, KL, et al. Enhanced fibroblast-myocyte interactions in response to cardiac injury. Circ Res. 2010; 107:1011–1020.
26. Rosker, C, Salvarani, N, Schmutz, S, et al. Abolishing myofibroblast arrhythmogeneicity by pharmacological ablation of alpha-smooth muscle actin containing stress fibers. Circ Res. 2011; 109:1120–1131.
27. Yue, L, Xie, J, Nattel, S. Molecular determinants of cardiac fibroblast electrical function and therapeutic implications for atrial fibrillation. Cardiovasc Res. 2011; 89:744–753.
28. Vasquez, C, Benamer, N, Morley, GE. The cardiac fibroblast: Functional and electrophysiological considerations in healthy and diseased hearts. J Cardiovasc Pharmacol. 2011; 57:380–388.
29. Li, GR, Sun, HY, Chen, JB, et al. Characterization of multiple ion channels in cultured human cardiac fibroblasts. PloS One. 2009; 4:e7307.
30. Chatelier, A, Mercier, A, Tremblier, B, et al. A distinct de-novo expression of nav1. 5 sodium channels in human atrial fibroblasts differentiated into myofibroblasts. J Physiol. 2012.
31. Chen, JB, Tao, R, Sun, HY, et al. Multiple ca2+ signaling pathways regulate intracellular ca2+ activity in human cardiac fibroblasts. J Cell Physiol. 2010; 223:68–75.
32. Follonier Castella, L, Gabbiani, G, McCulloch, CA, et al. Regulation of myofibroblast activities: Calcium pulls some strings behind the scene. Exp Cell Res. 2010; 316:2390–2401.
33. Du, J, Xie, J, Zhang, Z, et al. Trpm7-mediated ca2+ signals confer fibrogenesis in human atrial fibrillation. Circ Res. 2010; 106:992–1003.
34. Rose, RA, Hatano, N, Ohya, S, et al. C-type natriuretic peptide activates a non-selective cation current in acutely isolated rat cardiac fibroblasts via natriuretic peptide c receptor-mediated signalling. J Physiol. 2007; 580:255–274.
35. Kamkin, A, Kirischuk, S, Kiseleva, I. Single mechano-gated channels activated by mechanical deformation of acutely isolated cardiac fibroblasts from rats. Acta Physiol (Oxf). 2010; 199:277–292.
36. de Jong, S, van Veen, TA, van Rijen, HV, et al. Fibrosis and cardiac arrhythmias. J Cardiovasc Pharmacol. 2011; 57:630–638.
37. Karagueuzian, HS. Targeting cardiac fibrosis: A new frontier in antiarrhythmic therapy? Am J. Cardiovasc Dis. 2011; 1:101–109.
38. Pandya, K, Kim, H-S, Smithies, O. Fibrosis, not cell size, delineates beta-myosin heavy chain reexpression during cardiac hypertrophy and normal aging in vivo. Proc Natl Acad Sci U S A. 2006; 45:16864–16869.
39. Teekakirikul, P, Eminaga, S, Toka, O, et al. Cardiac fibrosis in mice with hypertrophic cardiomyopathy is mediated by non-myocyte proliferation and requires tgf-beta. J Clin Invest. 2010; 120:3520–3529.
40. Bujak, M, Frangogiannis, NG. The role of tgf-beta signaling in myocardial infarction and cardiac remodeling. Cardiovasc Res. 2007; 74:184–195.
41. Wakili, R, Voigt, N, Kaab, S, et al. Recent advances in the molecular pathophysiology of atrial fibrillation. J Clin Invest. 2011; 121:2955–2968.
42. Goette, A, Lendeckel, U. Electrophysiological effects of angiotensin ii. Part i: Signal transduction and basic electrophysiological mechanisms. Europace. 2008; 10:238–241.
43. Sovari, AA, Iravanian, S, Dolmatova, E, et al. Inhibition of c-src tyrosine kinase prevents angiotensin ii-mediated connexin-43 remodeling and sudden cardiac death. J Am Coll Cardiol. 2011; 58:2332–2339.
44. Leask, A. Potential therapeutic targets for cardiac fibrosis: Tgfbeta, angiotensin, endothelin, ccn2, and pdgf, partners in fibroblast activation. Circ Res. 2010; 106:1675–1680.
45. Zhao, W, Zhao, T, Huang, V, et al. Platelet-derived growth factor involvement in myocardial remodeling following infarction. J Mol Cell Cardiol. 2011; 51:830–838.
46. Kakkar, R, Lee, RT. Intramyocardial fibroblast myocyte communication. Circ Res. 2010; 106:47–57.
47. Mueller, EE, Momen, A, Masse, S, et al. Electrical remodelling precedes heart failure in an endothelin-1-induced model of cardiomyopathy. Cardiovasc Res. 2011; 89:623–633.
48. Reisner, Y, Meiry, G, Zeevi-Levin, N, et al. Impulse conduction and gap junctional remodelling by endothelin-1 in cultured neonatal rat ventricular myocytes. J Cell Mol Med. 2009; 13:562–573.
49. Duncan, DJ, Yang, Z, Hopkins, PM, et al. Tnf-alpha and il-1beta increase ca2+ leak from the sarcoplasmic reticulum and susceptibility to arrhythmia in rat ventricular myocytes. Cell Calcium. 2010; 47:378–386.
50. Baum, JR, Long, B, Cabo, C, et al. Myofibroblasts cause heterogeneous cx43 reduction and are unlikely to be coupled to myocytes in the healing canine infarct. Am J Physiol Heart Circ Physiol. 2012; 302:H790–H800.
51. Jansen, JA, van Veen, TA, de Jong, S, et al. Reduced cx43 expression triggers increased fibrosis due to enhanced fibroblast activity. Circ Arrhythm Electrophysiol. 2012; 5:380–390.
52. Zhang, Y, Kanter, EM, Laing, JG, et al. Connexin43 expression levels influence intercellular coupling and cell proliferation of native murine cardiac fibroblasts. Cell Commun Adhes. 2008; 15:289–303.
53. Pedrotty, DM, Klinger, RY, Kirkton, RD, et al. Cardiac fibroblast paracrine factors alter impulse conduction and ion channel expression of neonatal rat cardiomyocytes. Cardiovasc Res. 2009; 83:688–697.
54. Camelliti, P, Green, CR, LeGrice, I, et al. Fibroblast network in rabbit sinoatrial node: Structural and functional identification of homogeneous and heterogeneous cell coupling. Circ Res. 2004; 94:828–835.
55. De Maziere, AM, van Ginneken, AC, Wilders, R, et al. Spatial and functional relationship between myocytes and fibroblasts in the rabbit sinoatrial node. J Mol Cell Cardiol. 1992; 24:567–578.
56. Kohl, P, Camelliti, P. Fibroblast-myocyte connections in the heart. Heart Rhythm. 2012; 9:461–464.
57. Thompson, SA, Copeland, CR, Reich, DH, et al. Mechanical coupling between myofibroblasts and cardiomyocytes slows electric conduction in fibrotic cell monolayers. Circulation. 2011; 123:2083–2093.
58. Louault, C, Benamer, N, Faivre, JF, et al. Implication of connexins 40 and 43 in functional coupling between mouse cardiac fibroblasts in primary culture. Biochim Biophys Acta. 2008; 1778:2097–2104.
59. Zhang, Y, Kanter, EM, Yamada, KA. Remodeling of cardiac fibroblasts following myocardial infarction results in increased gap junction intercellular communication. Cardiovasc Pathol. 2010; 19:e233–240.
60. Pedrotty, DM, Klinger, RY, Badie, N, et al. Structural coupling of cardiomyocytes and noncardiomyocytes: Quantitative comparisons using a novel micropatterned cell pair assay. Am J Physiol Heart Circ Physiol. 2008; 295:H390–H400.
61. Chilton, L, Giles, WR, Smith, GL. Evidence of intercellular coupling between co-cultured adult rabbit ventricular myocytes and myofibroblasts. J Physiol. 2007; 583:225–236.
62. McSpadden, LC, Nguyen, H, Bursac, N. Size and ionic currents of unexcitable cells coupled to cardiomyocytes distinctly modulate cardiac action potential shape and pacemaking activity in micropatterned cell pairs. Circ Arrhythm Electrophysiol. 2012.
63. Nguyen, TP, Xie, Y, Garfinkel, A, et al. Arrhythmogenic consequences of myofibroblast-myocyte coupling. Cardiovasc Res. 2012; 93:242–251.
64. Zlochiver, S, Munoz, V, Vikstrom, KL, et al. Electrotonic myofibroblast-to-myocyte coupling increases propensity to reentrant arrhythmias in two-dimensional cardiac monolayers. Biophys J. 2008; 95:4469–4480.
65. He, K, Shi, X, Zhang, X, et al. Long-distance intercellular connectivity between cardiomyocytes and cardiofibroblasts mediated by membrane nanotubes. Cardiovasc Res. 2011; 92:39–47.
66. Wang, X, Gerdes, HH. Long-distance electrical coupling via tunneling nanotubes. Biochim Biophys Acta. 2012; 1818:2082–2086.
67. Xie, Y, Garfinkel, A, Camelliti, P, et al. Effects of fibroblast-myocyte coupling on cardiac conduction and vulnerability to reentry: A computational study. Heart Rhythm. 2009; 6:1641–1649.
68. Xie, Y, Garfinkel, A, Weiss, JN, et al. Cardiac alternans induced by fibroblast-myocyte coupling: Mechanistic insights from computational models. Am J Physiol Heart Circ Physiol. 2009; 297:H775–H784.
69. McDowell, KS, Arevalo, HJ, Maleckar, MM, et al. Susceptibility to arrhythmia in the infarcted heart depends on myofibroblast density. Biophys J. 2011; 101:1307–1315.
70. Jacquemet, V, Henriquez, CS. Genesis of complex fractionated atrial electrograms in zones of slow conduction: A computer model of microfibrosis. Heart Rhythm. 2009; 6:803–810.
71. Ashihara, T, Haraguchi, R, Nakazawa, K, et al. The role of fibroblasts in complex fractionated electrograms during persistent/permanent atrial fibrillation: Implications for electrogram-based catheter ablation. Circ Res. 2012; 110:275–284.
72. O’Hanlon, R, Grasso, A, Roughton, M, et al. Prognostic significance of myocardial fibrosis in hypertrophic cardiomyopathy. J Am Coll Cardiol. 2010; 56:867–874.
73. Lucas, JA, Zhang, Y, Li, P, et al. Inhibition of transforming growth factor-beta signaling induces left ventricular dilation and dysfunction in the pressure-overloaded heart. Am J Physiol Heart Circ Physiol. 2010; 298:H424–H432.
74. Nguyen, DT, Ding, C, Wilson, E, et al. Pirfenidone mitigates left ventricular fibrosis and dysfunction after myocardial infarction and reduces arrhythmias. Heart Rhythm. 2010; 7:1438–1445.
75. Du, XJ, Xu, Q, Lekgabe, E, et al. Reversal of cardiac fibrosis and related dysfunction by relaxin. Ann N Y Acad Sci. 2009; 1160:278–284.
76. Wang, Z, Zhang, Y, Gao, M, et al. Statin therapy for the prevention of atrial fibrillation: A meta-analysis of randomized controlled trials. Pharmacotherapy. 2011; 31:1051–1062.
77. Chen, J, Shearer, GC, Chen, Q, et al. Omega-3 fatty acids prevent pressure overload-induced cardiac fibrosis through activation of cyclic gmp/protein kinase g signaling in cardiac fibroblasts. Circulation. 2011; 123:584–593.
78. Friedrichs, K, Baldus, S, Klinke, A. Fibrosis in atrial fibrillation—role of reactive species and mpo. Frontiers in Physiology. 2012; 3:214.
79. Yan, W, Wang, P, Zhao, CX, et al. Decorin gene delivery inhibits cardiac fibrosis in spontaneously hypertensive rats by modulation of transforming growth factor-beta/smad and p38 mitogen-activated protein kinase signaling pathways. Hum Gene Ther. 2009; 20:1190–1200.
80. Crider, BJ, Risinger, GM, Jr., Haaksma, CJ, et al. Myocardin-related transcription factors a and b are key regulators of tgf-beta1-induced fibroblast to myofibroblast differentiation. J Invest Dermatol. 2011; 131:2378–2385.
81. Haudek, SB, Trial, J, Xia, Y, et al. Fc receptor engagement mediates differentiation of cardiac fibroblast precursor cells. Proc Natl Acad Sci U S A. 2008; 105:10179–10184.
82. Zhu, H, Fan, GC. Role of micrornas in the reperfused myocardium towards post-infarct remodelling. Cardiovasc Res. 2012; 94:284–292.
83. Pan, Z, Sun, X, Shan, H, et al. Microrna-101 inhibited postinfarct cardiac fibrosis and improved left ventricular compliance via the fbj osteosarcoma oncogene/transforming growth factor-beta1 pathway. Circulation. 2012; 126:840–850.
84. Qian, L, Huang, Y, Spencer, CI, et al. In vivo reprogramming of murine cardiac fibroblasts into induced cardiomyocytes. Nature. 2012; 485:593–598.
85. Kirkton, RD, Bursac, N. Engineering biosynthetic excitable tissues from unexcitable cells for electrophysiological and cell therapy studies. Nat Commun. 2011; 2:300.