[level-membership-for-pulmolory-and-respiratory-category]
Cardiac Electrophysiology
After reading this chapter, you will be able to:
• Explain the physiological significance of the resting membrane potential (RMP) of the cardiac fiber and how it is established
• Describe how the relationship between the RMP of the cardiac fiber and threshold potential affects the fiber’s tendency to depolarize
• Explain why depolarization of the cardiac fiber causes the fiber to contract
• Explain the nature of the electrochemical events that cause the cardiac muscle fiber to depolarize spontaneously
• Describe the mechanism whereby electrical charge differences across the cardiac cell membrane affect its permeability to Na+, K+, and Ca++ ions
• Explain why abnormalities in blood Ca++ and K+ concentrations affect cardiac muscle depolarization
• Correlate the activity of ion channels and gates with the graphical voltage versus time representation (action potential) of depolarization and repolarization of a single cardiac fiber
• Explain why catecholamine drugs increase cardiac contractility
• Explain why calcium channel–blocking drugs affect heart muscle contractility
• Describe the mechanisms whereby various drugs affect cardiac contractility and excitability
• Explain how ectopic foci arising from increased tissue excitability and the sinoatrial (SA) nodal block differ from each other
• Describe the purpose of the impulse transmission delay between the SA node and ventricles
• Describe the consequences of blocked atrial impulses that fail to enter the ventricles
• Explain how sympathetic and parasympathetic stimulation affects heart muscle automaticity, rhythmicity, and excitability
Membrane Potentials
Generation of Resting Membrane Potential
The electrical charge difference between two sides of a polarized myocardial cell membrane is called the resting membrane potential (RMP). The strength of RMP is measured in millivolts. Myocardial cell RMP is about −85 to −90 mV.1 The outside of the cell is assumed to have 0 (ground) potential. The RMP is therefore the difference between the cell’s inside potential and zero. The negative sign in front of the cardiac cell RMP (−90 mV) indicates the polarity of the cell’s interior.
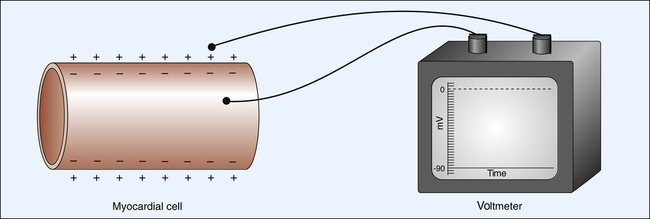
Figure 18-1 illustrates the measurement of the RMP through the placement of one microelectrode inside the cell and another outside the cell. A wire from each microelectrode is connected to a voltmeter, which measures the electrical potential difference across the membrane at about −90 mV.
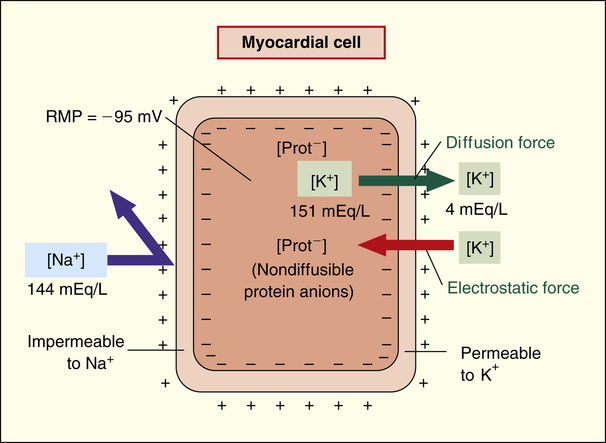
The main ions involved in generating the RMP are sodium (Na+), potassium (K+), calcium (Ca++), and large protein anions (negatively charged ions). As with all other cells in the body, K+ ion concentration inside the cardiac cell is much greater than outside the cell (about 151 mEq/L vs. 4 mEq/L).2 The opposite is true for Na+ and Ca++ ions; [Na+] is about 144 mEq/L outside the cell and about 7 mEq/L inside the cell, and [Ca++] is 5 mEq/L outside the cell and less than 1 mEq/L inside the cell.
The resting cardiac cell membrane is fairly permeable to K+ ions but only slightly permeable to Na+ and Ca++ ions. The permeability of the membrane to K+ allows potassium to diffuse out of the cell, down its high concentration gradient (Figure 18-2). Large, negatively charged protein ions and molecules cannot diffuse outward with K+ because the cell membrane is completely impermeable to them. Thus, as potassium leaves, an increasingly negative intracellular charge develops; this represents an electrostatic force that slows and eventually stops the outward diffusion of K+. The equilibrium between the outward diffusion force of K+ and the cell’s electrostatic attractive force is largely responsible for the cardiac cell RMP, altered only slightly by Na+ ion diffusion dynamics, as explained next.
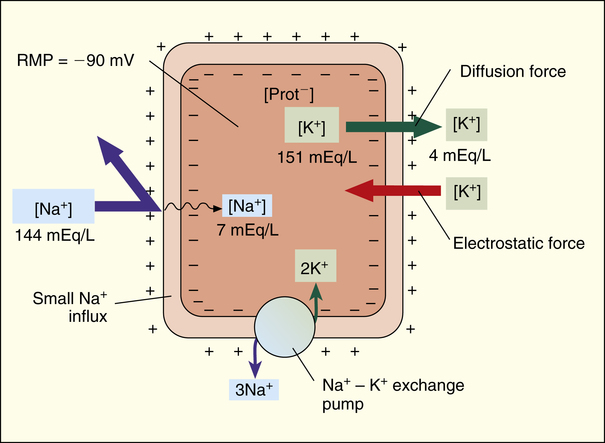
Although the cardiac cell membrane is highly impermeable to Na+, small amounts of this ion diffuse through leak channels into the cell because of its extremely high diffusion gradient (Figure 18-3).1 This diffusion alters the potassium-driven membrane potential, causing it to be slightly less negative (closer to 0) than it would be otherwise. The Na+-K+ exchange pump in the cell membrane counteracts Na+ leakage into the cell by actively pumping it out while pumping potassium back in. Three Na+ ions are pumped out of the cell for every two K+ ions pumped back in, adding to the electronegativity of the intracellular environment (see Figure 18-3). In the end, the RMP is the result of the complex interactions between (1) chemical diffusion forces, (2) electrostatic attractive forces, and (3) the Na+-K+ exchange pump.
Ion Channels and Gates
Sodium and Potassium Channels
The permeability of the myocardial cell membrane to Na+ and K+ ions is mainly controlled by voltage-gated ion channels in the membrane (Figure 18-4). These channels are formed by large protein molecules and are specific for each ion. Gatelike structures called gating proteins control these channels. Gating proteins are voltage sensitive, opening and closing in response to changes in the membrane potential. Changes in membrane potential alter the shape of the protein molecule, which opens or closes the channel gates2 (i.e., the membrane’s permeability to K+ and Na+ is voltage dependent).
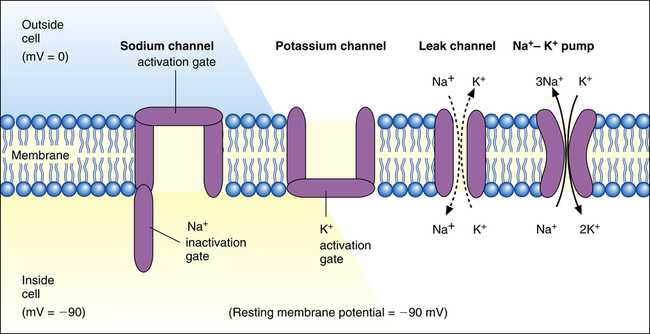
As shown in Figure 18-4, K+ and Na+ channel activation gates are closed at the cardiac cell’s RMP of −90 mV. Although both of these channels are closed, some K+ and Na+ ions diffuse across the membrane through leak channels because of their high diffusion gradients.
Calcium Channels
In addition to the Na+ and K+ channels, cardiac fibers have numerous calcium ion channels. These channels are similar to Na+ channels in that they have activation and inactivation gates. However, calcium channel gates respond 10 to 20 times more slowly than sodium channel gates to changes in membrane potential.1 Calcium channels are therefore called slow channels, whereas sodium channels are called fast channels. Although they mainly transmit Ca++ ions, calcium channels also permit the passage of some Na+ ions. The responses of sodium, potassium, and calcium channels to changes in their membrane potentials bring about an event known as the action potential. The action potential is an electrical event ultimately leading to the mechanical event of muscle fiber contraction.
Action Potential
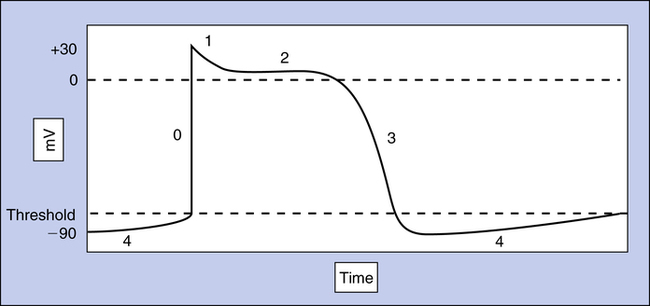
Cardiac muscle fiber depolarization occurs when the fiber RMP of −90 mV abruptly changes to 0 mV. Repolarization occurs when the RMP is again established at −90 mV. The action potential is a real-time recording of the moment-to-moment changes in the membrane potential as the cardiac fiber depolarizes and repolarizes. This recording plots voltage against time on specially designed graph paper or a monitoring screen. Figure 18-5 illustrates the action potential of a single ventricular muscle fiber. The different phases of the action potential (0 to 4) are created by Na+, K+, and Ca++ movement in and out of the cell.
Depolarization: Phase 0
The action potential of a ventricular muscle fiber (see Figure 18-5) is normally initiated by an electrical impulse originating from the sinoatrial (SA) node in the right atrium. The impulse changes the RMP to a less negative value that begins to open some of the fast sodium channel activation gates. Increased influx of sodium makes the fiber’s membrane potential even less negative, causing more activation gates to swing open. When the membrane potential reaches a critical level called the threshold potential (TP), all activation gates open instantaneously; this brings about an explosive influx of Na+, which instantaneously depolarizes the cell membrane and generates the upstroke (phase 0) of the action potential (see Figure 18-5).
Two forces cause Na+ ions to enter the cell during phase 0 of the action potential: (1) an electrostatic attraction created by the negative intracellular environment and (2) a high chemical diffusion force favoring movement of sodium into the cell (Figure 18-6, A). When the membrane potential reaches 0 mV, Na+ continues to enter the cell because a strong diffusion gradient is still present (see Figure 18-6, B and C). Consequently, the cell membrane’s polarity reverses (the membrane potential peaks at about +30 mV), creating the overshoot of the action potential (see Figures 18-5 and 18-6, D).
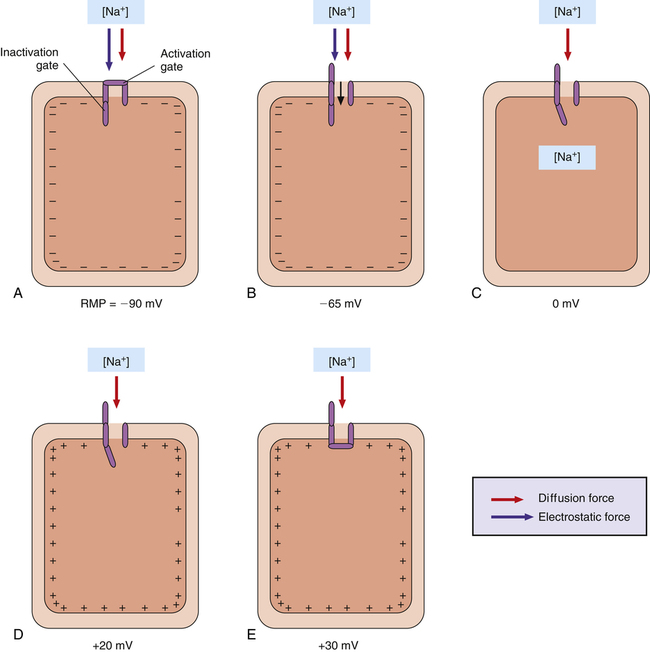
The rapid change in membrane potential from −90 mV to +30 mV causes the voltage-sensitive inactivation gates of the sodium channel to swing closed rapidly, stopping the influx of sodium (see Figure 18-6, E). These inactivation gates close only a few ten-thousandths of a second after the activation gates open—about the time the upstroke of the action potential reaches 0 mV, on its way to +30 mV.1 The inactivation gates do not reopen until the original RMP is restored; thus, the heart muscle fiber first must repolarize before it can respond to another depolarizing stimulus. That is, the heart muscle fiber is refractory to depolarizing stimuli from phase 0 through the end of phase 3 of the action potential (see Figure 18-5).
Plateau: Phases 1 and 2
Immediately after the upstroke of the action potential (phase 0), a very small, limited downstroke occurs (phase 1) just before the plateau. This brief decline in intracellular positivity reflects the sudden opening of potassium channel gates and outflow of K+ from the cell (Figure 18-7, phase 1). Almost simultaneously, slow calcium channel activation gates finally swing open, allowing Ca++ and a small amount of Na+ to enter the cell (see Figure 18-7, phase 2). This inflow of positive charges (carried by Ca++ and Na+) is counterbalanced by an equal outflow of positive charges (carried by K+). A membrane potential of about 0 mV is thus briefly maintained, creating the action potential’s plateau (see Figure 18-7, phase 2).
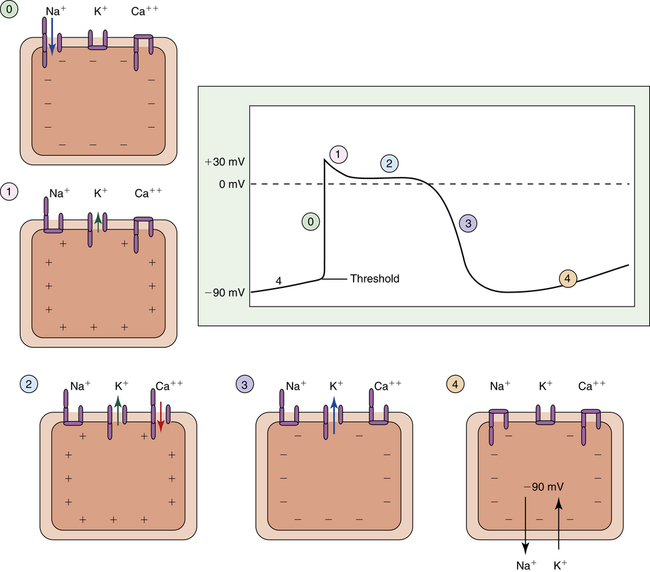
Compared with the depolarization phase (see Figure 18-7, phase 0), which lasts only a few ten-thousandths of a second, the plateau phase lasts for about 0.2 to 0.3 second.1 Therefore, cardiac muscle contraction also lasts for this relatively long time, a property of cardiac muscle that enhances its pumping effectiveness.
Ca++ ions entering the myocardial fiber during the plateau phase participate in the excitation-contraction coupling between actin and myosin fibers, as described in Chapter 17. Some drugs, such as digitalis, increase the intracellular level of Ca++, which enhances myocardial contractility during this phase of the action potential. Drugs that increase contractility are called inotropic drugs.
The equilibrium between Ca++ inflow and K+ outflow during the plateau occurs in ventricular but not in atrial myocardial fibers. K+ outflow exceeds Ca++ inflow in atrial fibers, preventing the development of a plateau.1
Repolarization: Phase 3
The outflow of K+ ions gradually increases throughout the action potential plateau. The plateau phase ends when Ca++ inactivation gates close, stopping Ca++ inflow (see Figure 18-7, phase 3). The membrane is now highly permeable to only K+ ions. Intracellular negativity is rapidly restored to the resting level as K+ explosively rushes out of the cell, down its high diffusion gradient. After repolarization is complete and the RMP is restored, excess Na+ that entered the cell during phases 0 and 2 is pumped out by the Na+-K+ exchange pump. At the same time, K+ that exited the cell during phases 2 and 3 is pumped back into the cell, restoring the original Na+ and K+ concentrations inside and outside the cell (see Figure 18-7, phase 4). The muscle fiber is then ready to accept another depolarizing stimulus. Box 18-1 summarizes the events causing the action potential.
Action Potential Propagation
Depolarization of a specific point on a cardiac fiber membrane opens sodium channels at that point. Positive electrical charges carried by the inflow of Na+ ions decrease the intracellular negativity immediately adjacent to the point of entry, bringing these areas to their threshold potentials. Sodium channels in these areas immediately open, allowing more Na+ to rush in. This new incoming Na+ brings areas farther along the fiber to their threshold potentials, and so on, instantaneously spreading the action potential along the fiber. The inflow of positively charged Na+ ions leaves a relatively negative charge on the outside fiber surface. Thus, the action potential spreads as a wave of electronegativity across the surface of the depolarizing fiber membrane (Figure 18-8). This electronegative wave is called an electrical impulse, which can be detected on the body’s surface with a special instrument called an electrocardiograph.
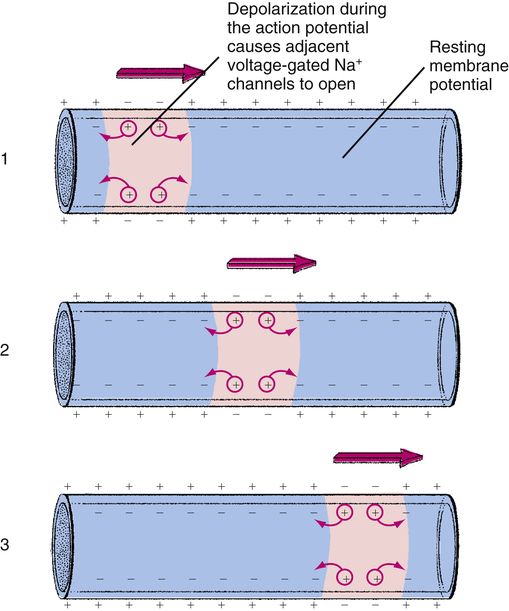
In summary, a depolarizing impulse spreads over the myocardial surface and deep into the fiber through the T-tubules (see Chapter 17). This impulse ultimately opens calcium channels, allowing Ca++ to enter the fiber and catalyze the excitation-contraction coupling of actin and myosin myofibrils. The mechanical muscle contraction event occurs only a few milliseconds after muscle fiber depolarization.
Effects of Drugs and Extracellular Ion Concentration on the Action Potential
Extracellular Ions
Potassium
An excess of potassium ions (hyperkalemia) in the extracellular fluid surrounding cardiac fibers decreases the heart rate and stroke volume. Severe hyperkalemia causes heart block, or the inability of cardiac fibers to conduct action potentials through the heart. The mechanism by which hyperkalemia causes these effects is illustrated in Figure 18-9.
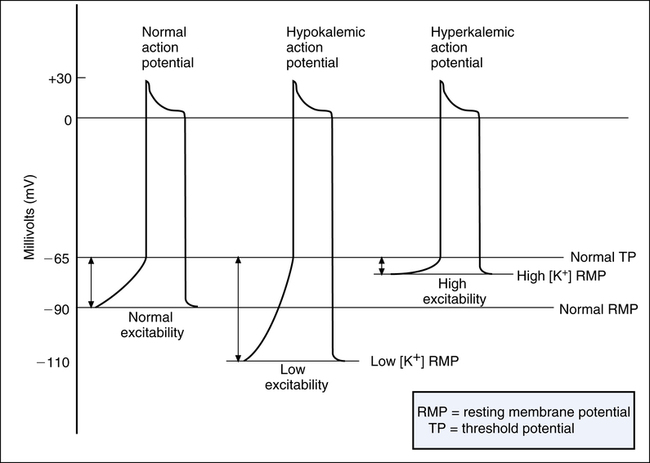
As mentioned previously, K+ diffusion out of the cell down its concentration gradient is primarily responsible for generating RMP (see Figure 18-2). High extracellular [K+] diminishes the outward diffusion gradient of K+. As a result, a smaller degree of intracellular electronegativity develops when potassium diffuses out of the cell. This means that the RMP across the cell membrane is less negative (closer to 0) and closer to the TP (see Figure 18-9). Hyperkalemia thus has a depolarizing influence on the membrane, making it more excitable (easier to depolarize). Excitability is defined as the difference between the RMP and the TP; the less the difference, the more excitable the membrane.3
Besides making the RMP less electronegative, hyperkalemia reduces the amplitude (strength) of the action potential, decreasing its rate of conduction along muscle fibers, which explains why hyperkalemia decreases the heart rate. The decreased action potential amplitude also reduces the amount of Ca++ that enters the muscle fiber during depolarization, which decreases ventricular contraction force and stroke volume. Severe hyperkalemia may depolarize the membrane to the extent that its resting potential is less (closer to 0) than its TP.3 In this case, the muscle fiber is unable to repolarize after a single action potential and is no longer excitable (i.e., the fiber is in a continual state of depolarization). This leads to cardiac muscle paralysis and is why hyperkalemia may cause heart block.
Conversely, hypokalemia increases the diffusion gradient for K+ movement out of the cell, creating a greater intracellular electronegativity than normal. As a result, the RMP is lower than normal, hyperpolarizing the membrane (see Figure 18-9); this makes the membrane less excitable or less sensitive to depolarizing stimuli. In severe hypokalemia, the heart muscle may fail to depolarize, resulting in flaccid paralysis (cardiac arrest).3 Hypokalemia may decrease the heart rate because a longer time is required for the membrane to depolarize to its threshold level. The muscle contraction force is not affected in this instance because adequate amounts of Ca++ enter the cell during the action potential.
Calcium
Ca++ entry into cardiac fibers during the plateau phase of the action potential is essential for muscle contraction (see Chapter 17). Generally, the heart muscle contractile force is directly related to extracellular [Ca++].
Calcium ions affect membrane excitability by changing the TP rather than the RMP. This phenomenon is explained by the effect of Ca++ on sodium channel activation. When extracellular [Ca++] is low (hypocalcemia), an extremely small amount of membrane depolarization above the resting potential opens the sodium channels. In other words, hypocalcemia brings the threshold at which Na+ activation gates open closer to the RMP. For example, rather than opening at a normal TP of −65 mV, Na+ gates open at −75 mV, a value much closer to the −90-mV RMP (Figure 18-10). Thus, hypocalcemia increases heart muscle excitability, causing an effect similar to hyperkalemia. Apparently, Ca++ ions bind to sodium channel protein molecules, increasing the amount of membrane depolarization that must occur to open the gate. Therefore, low extracellular [Ca++] allows the gate to open with an extremely small degree of membrane depolarization.1
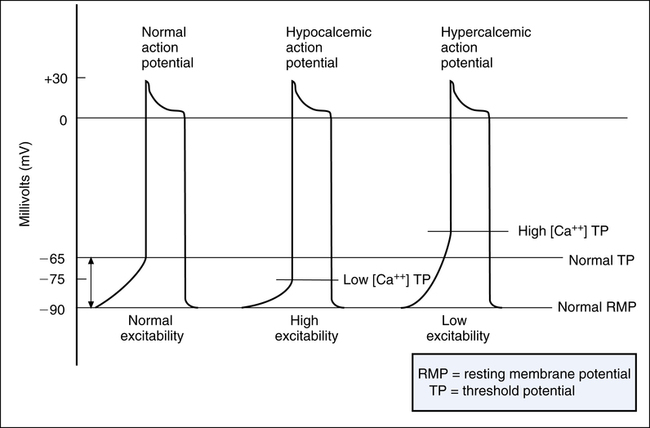
Conversely, hypercalcemia decreases membrane excitability because it raises the TP, requiring a greater amount of membrane depolarization to open the Na+ gates (see Figure 18-10). Because of its direct effect on the muscle contractile process, extremely high [Ca++] can cause the heart to develop repetitive, spastic contractions. Because blood [Ca++] is regulated within extremely narrow ranges, calcium-induced cardiac abnormalities are rarely a clinical concern.1 However, administering Ca++ to treat severe hyperkalemia may be clinically beneficial because doing so counteracts the effect of hyperkalemia on membrane excitability.3
Drugs Affecting the Action Potential
Catecholamines
Catecholamines are compounds with chemical structures similar to epinephrine and norepinephrine. Such drugs increase the influx of Ca++ into the myocardial cell during the plateau phase of the action potential. Catecholamines activate beta receptors in the cardiac cell membrane, bringing about a series of chemical events that increase intracellular levels of cyclic adenosine monophosphate (cAMP). cAMP enhances activation of the slow calcium channels, augmenting the influx of Ca++ ions. This is the main mechanism by which catecholamine drugs increase cardiac muscle contractility.2 Sympathetic nervous activity raises heart muscle contractility by the same mechanism because it raises adrenal secretion of epinephrine into the circulation.
Rhythmic Excitation of the Heart
Automatic Rhythmicity of the Cardiac Fibers
Pacemaker Function of the Sinus Node
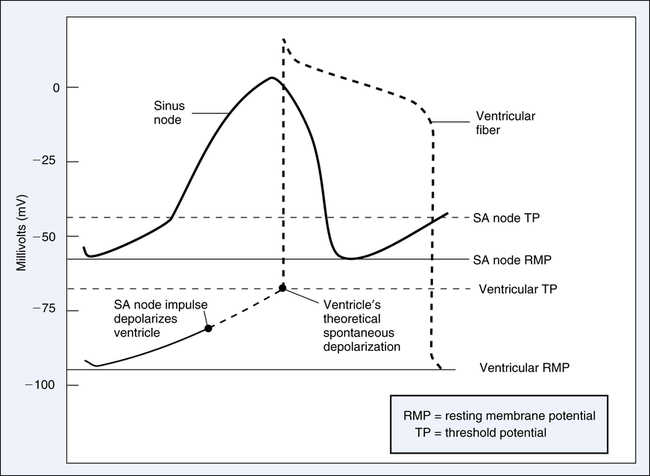
The RMP of sinus node fibers is only about −55 mV compared with −90 mV of ventricular fibers (Figure 18-11).1 Sinus node tissue is naturally leakier than ventricular fibers to sodium ions; hence, its intracellular environment is less negative. In addition, the RMP of the SA node is closer to its TP than other heart muscle; its threshold is about −40 mV, creating an RMP-TP difference of about 15 mV. In contrast, ventricular muscle RMP and TP are about −90 mV and −65 mV, for an RMP-TP difference of about 25 mV. Thus, the ventricles require more time than the SA node to reach the TP (see Figure 18-11). Hence, the SA node sets the pace and suppresses all other cardiac cell pacemaker tendencies.
Sinus Node Self-Excitation
Figure 18-11 illustrates the sinus node’s gradually rising RMP caused by the slow leak of Na+ into the cell. When its threshold of −40 mV is reached, the SA node fires (depolarizes), initiating an impulse that spreads to all heart muscle over the specialized conduction system. As soon as repolarization is achieved, RMP immediately begins to decay again toward the TP, causing the cycle to repeat itself at regular intervals. The heart rate pattern established by the SA node is known as sinus rhythm and is normally about 70 to 80 beats per minute.1
Ectopic Beats
Premature Beat
A premature beat occurs early between otherwise evenly spaced, sinus-generated beats. A premature beat is evidence that an ectopic focus has fired. In other words, an ectopic focus can initiate a beat prematurely only if it depolarizes more rapidly than the SA node. Factors that may produce such premature beats include (1) local areas of tissue hypoxia (ischemia); (2) mechanical irritation produced by calcified plaque in a diseased heart; and (3) toxic irritation caused by nicotine, caffeine, or other drugs.1 All of these factors cause the heart muscle to become more excitable and more susceptible to depolarization.
Downward Displacement of the Pacemaker
After the SA node, the atrioventricular (AV) nodal fibers have the next highest spontaneous depolarization rate. If not stimulated from some outside source, AV fibers fire at a rate of 40 to 60 times per minute. If AV impulses fail to reach the ventricles after several seconds, the Purkinje fibers begin to fire at a rate of 15 to 40 times per minute.1 These rates contrast with the intrinsic SA nodal rate of 70 to 80 beats per minute.
Lower order pacemakers not only fire at lower rates, they also tend to be more unreliable than the SA node. Ventricular pacemakers are especially unstable in this regard. If impulses from atria are totally blocked, the Purkinje cells may pause for 30 seconds before establishing a rate of 15 to 40 beats per minute. During this pause, there is no cardiac output, and the person may faint from lack of blood flow to the brain. This delayed ventricular takeover of the heartbeat is known as Stokes-Adams syndrome.1
Transmission of Impulses through the Heart
Atrial Transmission
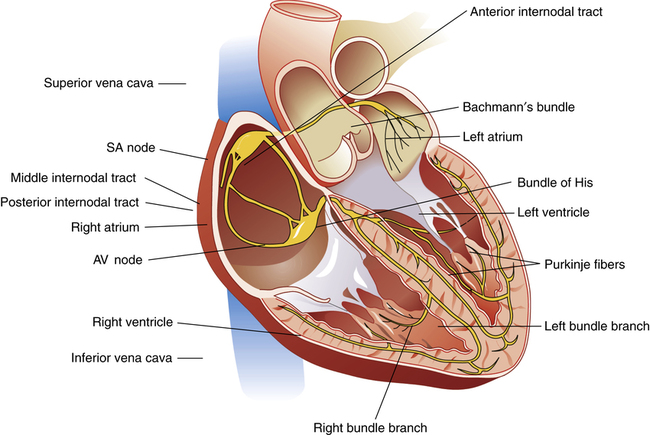
The cardiac conduction system anatomy is discussed in Chapter 17. From the SA node, the depolarizing impulse spreads rapidly over atrial muscle to the AV node. The impulse is preferentially conducted along three specialized internodal pathways that eventually converge at the AV node.1 One of these pathways, Bachmann’s bundle, conducts SA nodal impulses directly to the left atrium (Figure 18-12). The impulse velocity over these specialized conductive pathways is about 1 m per second compared with 0.3 m per second over other atrial muscle.1
Atrioventricular Conduction
The AV node is located in the septal wall of the right atrium, just behind the tricuspid valve. The SA node impulse travels across atrial muscle to the AV node in about 0.03 second; however, the impulse does not emerge from the AV node for another 0.13 second.1 About one fourth of the total delay occurs in the transitional (junctional) fibers (extremely small fibers connecting atrial muscle to nodal fibers). The conduction velocity in junctional fibers is about one twelfth of the velocity in normal cardiac muscle.1 Nodal resistance to conduction is not quite as high as junctional resistance but is still greater than that for other heart muscle.
The AV bundle, also known as the bundle of His (see Figure 18-12), is the penetrating portion of the AV nodal system. It penetrates the fibrous cardiac skeleton and is the only electrically conductive connection between atrial and ventricular muscle masses. This penetrating portion of the bundle also has greater electrical resistance than regular cardiac muscle. A unique feature of the AV bundle is its one-way conduction from atria to ventricles. Ventricular impulses cannot be conducted backward through the bundle to the atria; this prevents reentry of the cardiac impulse back to the atria.
Purkinje System Conduction
After it penetrates the fibrous cardiac skeleton, the AV bundle enters the intraventricular septum and divides into left and right bundle branches (see Figure 18-12). At the heart’s apex, smaller branches of each bundle spread throughout the ventricles back toward the base. These Purkinje fibers penetrate into the heart muscle a few millimeters and fuse with other ventricular muscle. Conduction velocity is greatly accelerated through these fibers (about 1.5 to 4 m per second). Ventricular muscle is almost instantly depolarized; about 0.03 second elapses from the time the impulse enters the bundle branches until it reaches the terminal Purkinje fibers. Conduction velocity through other ventricular muscle is only about one sixth the conduction velocity of the Purkinje fibers (0.3 to 0.5 m per second); only about 0.06 second is required for impulse transmission from bundle branches to the last ventricular muscle fiber. The total time required for impulse transmission from the SA node to the last ventricular fiber is normally about 0.22 second.1
Role of Sympathetic and Parasympathetic Nervous Control
Parasympathetic stimulation releases acetylcholine from postganglionic nerve endings. This decreases (1) SA nodal firing rate and (2) AV nodal fiber excitability, reducing the heart rate and slowing the conduction of atrial impulses to the ventricles. Strong vagal stimulation can totally block AV conduction or completely stop SA nodal firing.1 However, if this state persists, the ventricular Purkinje system takes over the pacemaker function, producing a slow rate of 15 to 40 beats per minute. Such ventricular takeover is called ventricular escape (i.e., the ventricles escape the dominance of the SA node).
Acetylcholine (from vagal stimulation) greatly increases K+ permeability of the cardiac fiber, allowing rapid outward K+ diffusion;1 this greatly increases intracellular negativity, hyperpolarizing the cardiac membrane and making depolarization more difficult. This explains why vagal stimulation causes AV heart block.
[/level-membership-for-pulmolory-and-respiratory-category][not-level-membership-for-pulmolory-and-respiratory-category]
Cardiac Electrophysiology
After reading this chapter, you will be able to:
• Explain the physiological significance of the resting membrane potential (RMP) of the cardiac fiber and how it is established
• Describe how the relationship between the RMP of the cardiac fiber and threshold potential affects the fiber’s tendency to depolarize
• Explain why depolarization of the cardiac fiber causes the fiber to contract
• Explain the nature of the electrochemical events that cause the cardiac muscle fiber to depolarize spontaneously
• Describe the mechanism whereby electrical charge differences across the cardiac cell membrane affect its permeability to Na+, K+, and Ca++ ions
• Explain why abnormalities in blood Ca++ and K+ concentrations affect cardiac muscle depolarization
• Correlate the activity of ion channels and gates with the graphical voltage versus time representation (action potential) of depolarization and repolarization of a single cardiac fiber
• Explain why catecholamine drugs increase cardiac contractility
• Explain why calcium channel–blocking drugs affect heart muscle contractility
• Describe the mechanisms whereby various drugs affect cardiac contractility and excitability
• Explain how ectopic foci arising from increased tissue excitability and the sinoatrial (SA) nodal block differ from each other
• Describe the purpose of the impulse transmission delay between the SA node and ventricles
• Describe the consequences of blocked atrial impulses that fail to enter the ventricles
• Explain how sympathetic and parasympathetic stimulation affects heart muscle automaticity, rhythmicity, and excitability
Membrane Potentials
Generation of Resting Membrane Potential
The electrical charge difference between two sides of a polarized myocardial cell membrane is called the resting membrane potential (RMP). The strength of RMP is measured in millivolts. Myocardial cell RMP is about −85 to −90 mV.1 The outside of the cell is assumed to have 0 (ground) potential. The RMP is therefore the difference between the cell’s inside potential and zero. The negative sign in front of the cardiac cell RMP (−90 mV) indicates the polarity of the cell’s interior.
Figure 18-1 illustrates the measurement of the RMP through the placement of one microelectrode inside the cell and another outside the cell. A wire from each microelectrode is connected to a voltmeter, which measures the electrical potential difference across the membrane at about −90 mV.
The main ions involved in generating the RMP are sodium (Na+), potassium (K+), calcium (Ca++), and large protein anions (negatively charged ions). As with all other cells in the body, K+ ion concentration inside the cardiac cell is much greater than outside the cell (about 151 mEq/L vs. 4 mEq/L).2 The opposite is true for Na+ and Ca++ ions; [Na+] is about 144 mEq/L outside the cell and about 7 mEq/L inside the cell, and [Ca++] is 5 mEq/L outside the cell and less than 1 mEq/L inside the cell.
The resting cardiac cell membrane is fairly permeable to K+ ions but only slightly permeable to Na+ and Ca++ ions. The permeability of the membrane to K+ allows potassium to diffuse out of the cell, down its high concentration gradient (Figure 18-2). Large, negatively charged protein ions and molecules cannot diffuse outward with K+ because the cell membrane is completely impermeable to them. Thus, as potassium leaves, an increasingly negative intracellular charge develops; this represents an electrostatic force that slows and eventually stops the outward diffusion of K+. The equilibrium between the outward diffusion force of K+ and the cell’s electrostatic attractive force is largely responsible for the cardiac cell RMP, altered only slightly by Na+ ion diffusion dynamics, as explained next.
Although the cardiac cell membrane is highly impermeable to Na+, small amounts of this ion diffuse through leak channels into the cell because of its extremely high diffusion gradient (Figure 18-3).1 This diffusion alters the potassium-driven membrane potential, causing it to be slightly less negative (closer to 0) than it would be otherwise. The Na+-K+ exchange pump in the cell membrane counteracts Na+ leakage into the cell by actively pumping it out while pumping potassium back in. Three Na+ ions are pumped out of the cell for every two K+ ions pumped back in, adding to the electronegativity of the intracellular environment (see Figure 18-3). In the end, the RMP is the result of the complex interactions between (1) chemical diffusion forces, (2) electrostatic attractive forces, and (3) the Na+-K+ exchange pump.
Ion Channels and Gates
Sodium and Potassium Channels
The permeability of the myocardial cell membrane to Na+ and K+ ions is mainly controlled by voltage-gated ion channels in the membrane (Figure 18-4). These channels are formed by large protein molecules and are specific for each ion. Gatelike structures called gating proteins control these channels. Gating proteins are voltage sensitive, opening and closing in response to changes in the membrane potential. Changes in membrane potential alter the shape of the protein molecule, which opens or closes the channel gates2 (i.e., the membrane’s permeability to K+ and Na+ is voltage dependent).
As shown in Figure 18-4, K+ and Na+ channel activation gates are closed at the cardiac cell’s RMP of −90 mV. Although both of these channels are closed, some K+ and Na+ ions diffuse across the membrane through leak channels because of their high diffusion gradients.
Calcium Channels
In addition to the Na+ and K+ channels, cardiac fibers have numerous calcium ion channels. These channels are similar to Na+ channels in that they have activation and inactivation gates. However, calcium channel gates respond 10 to 20 times more slowly than sodium channel gates to changes in membrane potential.1 Calcium channels are therefore called slow channels, whereas sodium channels are called fast channels. Although they mainly transmit Ca++ ions, calcium channels also permit the passage of some Na+ ions. The responses of sodium, potassium, and calcium channels to changes in their membrane potentials bring about an event known as the action potential. The action potential is an electrical event ultimately leading to the mechanical event of muscle fiber contraction.
Action Potential
Cardiac muscle fiber depolarization occurs when the fiber RMP of −90 mV abruptly changes to 0 mV. Repolarization occurs when the RMP is again established at −90 mV. The action potential is a real-time recording of the moment-to-moment changes in the membrane potential as the cardiac fiber depolarizes and repolarizes. This recording plots voltage against time on specially designed graph paper or a monitoring screen. Figure 18-5 illustrates the action potential of a single ventricular muscle fiber. The different phases of the action potential (0 to 4) are created by Na+, K+, and Ca++ movement in and out of the cell.
Depolarization: Phase 0
The action potential of a ventricular muscle fiber (see Figure 18-5) is normally initiated by an electrical impulse originating from the sinoatrial (SA) node in the right atrium. The impulse changes the RMP to a less negative value that begins to open some of the fast sodium channel activation gates. Increased influx of sodium makes the fiber’s membrane potential even less negative, causing more activation gates to swing open. When the membrane potential reaches a critical level called the threshold potential (TP), all activation gates open instantaneously; this brings about an explosive influx of Na+, which instantaneously depolarizes the cell membrane and generates the upstroke (phase 0) of the action potential (see Figure 18-5).
Two forces cause Na+ ions to enter the cell during phase 0 of the action potential: (1) an electrostatic attraction created by the negative intracellular environment and (2) a high chemical diffusion force favoring movement of sodium into the cell (Figure 18-6, A). When the membrane potential reaches 0 mV, Na+
[/not-level-membership-for-pulmolory-and-respiratory-category]
